

Abstract
Cadmium (Cd), a toxic environment contaminant, induces reactive oxygen species (ROS)-mediated neuronal apoptosis and consequential neurodegenerative disorders. Metformin, an anti-diabetic drug, has recently received a great attention owing to its protection against neurodegenerative diseases. However, little is known regarding the effect of metformin on Cd-induced neurotoxicity. Here we show that metformin effectively prevented Cd-evoked apoptotic cell death in neuronal cells, by suppressing Cd activation of c-Jun N-terminal kinases (JNK), which was attributed to blocking Cd inactivation of protein phosphatase 5 (PP5) and AMP-activated protein kinase (AMPK). Inhibition of JNK with SP600125, knockdown of c-Jun, or overexpression of PP5 potentiated metformin’s inhibitory effect on Cd-induced phosphorylation of JNK/c-Jun and apoptosis. Activation of AMPK with AICAR or ectopic expression of constitutively active AMPKα strengthened the inhibitory effects of metformin on Cd-induced phosphorylation of JNK/c-Jun and apoptosis, whereas expression of dominant negative AMPKα weakened these effects of metformin. Metformin repressed Cd-induced ROS, thereby diminishing cell death. N-acetyl-L-cysteine enhanced the inhibitory effects of metformin on Cd-induced ROS and apoptosis. Moreover, using Mito-TEMPO, we further demonstrated that metformin attenuated Cd-induced cell death by suppressing induction of mitochondrial ROS. Taken together, these results indicate that metformin prevents mitochondrial ROS inactivation of PP5 and AMPK, thus attenuating Cd-induced JNK activation and apoptosis in neuronal cells. Our data highlight that metformin may be a promising drug for prevention of Cd-induced oxidative stress and neurodegenerative diseases.
Keywords: Metformin, Cadmium, Neuronal cells, ROS, PP5, AMPK, JNK
1. Introduction
Heavy metal contamination, one of the greatest global problems, not only negatively affects environment but also endangers humans and animals. Cadmium (Cd), an occupationally and environmentally heavy metal pollutant, can accumulate in multiple organs and tissues due to its long half-life (15~20 years) in the human body, thus resulting in a high incidence of related diseases, including impaired renal function, decreased bone mineral density, increased calcium excretion, reproductive toxicity, and neurotoxicity (Akesson et al., 2006; Johri et al., 2010; Wang and Du, 2013; Xu et al., 2017). Upon absorption into the bloodstream, Cd can easily traverse the blood-brain barrier and accumulate in the brain, manifesting with symptoms including headache and vertigo, olfactory dysfunction, slowed vasomotor function, peripheral neuropathy, decreased equilibrium, decreased ability to concentrate, and learning disabilities (Fowler, 2009; Okuda et al., 1997; Satarug and Nazar, 2010). Merging data have demonstrated that Cd exposure can induce increased levels of reactive oxygen species (ROS) and trigger neuronal apoptosis (Chen et al., 2011; Di Carlo et al., 2012; Yang et al., 2007). Excessive ROS induced by Cd have been recognized as an important pathogenic factor in the development of neuronal cell death and consequential neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) (Ben Mimouna et al., 2019; Mendez-Armenta and Rios, 2007; Okuda et al., 1997; Peng et al., 2017; Rigon et al., 2008; Williams et al., 2010).
Mitogen-activated protein kinases (MAPKs), a group of evolutionarily highly conserved cascade of dual (tyrosine and serine/threonine) protein kinases, can be activated by different extracellular stimuli such as cytokines, neurotransmitters, hormones and cellular stresses, and play a crucial role in signal transduction and regulate numerous cellular events such as cell growth and survival (Kyriakis and Avruch, 2012). MAPKs in mammalian cells possess at least three distinct groups, including the extracellular signal-regulated kinases (Erk1/2, Erk3/4, Erk5, and Erk7/8), the c-Jun N-terminal kinases (JNK1/2/3), and the p38 MAPKs (p38α/β/γ/δ) (Kyriakis and Avruch, 2012). It is well known that the phosphorylation of MAPKs is balanced by specific MAPK kinases and phosphatases (Kim and Choi, 2010; Kyriakis and Avruch, 2001). The protein phosphatase 5 (PP5) negatively regulates the JNK pathway involved in oxidative stress-induced apoptosis (Chen et al., 2008a; Chen et al., 2009; Han et al., 2012; Morita et al., 2001). AMP-activated protein kinase (AMPK) is a key regulator of energy homeostasis and cell survival in response to inflammation and oxidative stress (Chiang et al., 2018). Studies have documented that AMPK inactivation or activation due to oxidative stress is involved in neurodegenerative diseases (Chiang et al., 2018; Jiang et al., 2013; Vingtdeux et al., 2011). Our group has observed that Cd activates JNK pathway contributing to cell death by inducing ROS inactivation of PP5 in neuronal cells (Chen et al., 2008a; Xu et al., 2017). We have also demonstrated that Cd triggers ROS-dependent inactivation of AMPK leading to apoptosis of neuronal cells (Chen et al., 2011; Zhang et al., 2017). Hence, we postulated that a compound that can regulate ROS-dependent PP5-JNK and/or AMPK pathways might be useful to prevent Cd neurotoxicity.
Several studies have shown resveratrol, celastrol or nobiletin protection against Cd neurotoxicity by blocking ROS, involving AMPK or JNK pathways (Chen et al., 2014a; Qu et al., 2018; Shati, 2019; Zhang et al., 2017). Anti-diabetic drug metformin (N, N-dimethyl biguanide), a well-known AMPK activator, results in clear benefits in relation to glucose metabolism and diabetes mellitus-related complications (Barzilai et al., 2016; Lu et al., 2016; Singh et al., 2016). It is emerging that metformin may be also an effective drug to protect neurons from injuries and improve behavior in neurodegenerative diseases, such as memory function in a mouse model of Alzheimer’s disease, the risk of Parkinson’s disease in diabetes and MPTP-induced Parkinson’s disease in mice (Patil et al., 2014; DiTacchio et al., 2015; El-Mir et al., 2008; Meng et al., 2016). Metformin has been demonstrated to exert protective effects against several aging-related diseases in humans by decreasing insulin levels, activating AMPK, inhibiting the mammalian target of rapamycin (mTOR), inhibiting mitochondrial complex 1 in the electron transport chain, reducing endogenous ROS production (Barzilai et al., 2016; Duca et al., 2015; Kickstein et al., 2010; Rotermund et al., 2018; Zheng et al., 2012). This prompted us to investigate whether and how metformin protects against Cd-induced neuronal apoptosis. Here, for the first time, we show that metformin ameliorates Cd-evoked neuronal apoptosis via blocking ROS-dependent PP5/AMPK-JNK signaling pathway. The results improve our understanding of the molecular mechanism by which metformin has a potential for the prevention of oxidative stress and neurodegeneration promoted by Cd toxicity.
2. Materials and methods
2.1. Reagents
Cadmium chloride, metformin, 4’,6-diamidino-2-phenylindole (DAPI), poly-D-lysine (PDL), N-acetyl-L-cysteine (NAC), SP600125 and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA), whereas 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was purchased from MP Biomedicals (Solon, OH, USA). Mito-TEMPO was acquired from ALEXIS Biochemicals (San Diego, CA, USA). 5-amino-4-imidazolecarboxamide ribose (AICAR) was from Enzo Life Sciences (Farmingdale, NY, USA). CellTiter 96®AQueous One Solution Cell Proliferation Assay kit was from Promega (Madison, WI, USA). Enhanced chemiluminescence solution was from Sciben Biotech Company (Nanjing, China). Dulbecco’s modified Eagle medium (DMEM), 0.05% Trypsin–EDTA, NEUROBASAL™ Media and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
2.2. Cell culture
Rat pheochromocytoma (PC12) and human neuroblastoma SH-SY5Y cell lines were from American Type Culture Collection (ATCC) (Manassas, VA, USA), which were seeded in a 6-well plate or 96-well plate pre-coated with (for PC12) or without (for SH-SY5Y) PDL (0.2 μg/ml). PC12 cells were cultured in antibiotic-free DMEM supplemented with 10% horse serum and 5% FBS, whereas SH-SY5Y cells were grown in antibiotic-free DMEM supplemented with 10% FBS. Cells were maintained in a humid incubator (37°C, 5% CO2). Primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in female ICR mice (being pregnant) as described (Chen et al., 2009; Chen et al., 2010), and seeded in a 6-well plate or 96-well plate coated with 10 μg/mL PDL for experiments after 6 days of culture. All procedures used in this study were approved by the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
2.3. Recombinant adenoviral constructs and infection of cells
The recombinant adenoviruses expressing HA-tagged wild-type (wt) human PP5 (Ad-PP5) (Morita et al., 2001) (a gift from Dr. Hidenori Ichijo, University of Tokyo, Tokyo, Japan), FLAG-tagged dominant negative c-Jun (FLAG-Δ169) (Ad-dn-c-Jun) (Whitfield et al., 2001) (a gift from Dr. Jonathan Whitfield, Eisai London Research Laboratories, University College London, London, UK), myc-tagged constitutively active mutant of rat AMPKα1 (T172D) (Ad-AMPKα-ca) (Zang et al., 2004) (a gift from Dr. Kenneth Walsh, Boston University School of Medicine, Boston, MA, USA), HA-tagged dominant-negative mutant of human AMPKα1 (D159A) (Ad-dn-AMPKα) (Lu et al., 2008) (a gift from Dr. Nicholas J.G. Webster, University of California, San Diego, CA, USA), and the control virus expressing the green fluorescent protein (GFP) (Ad-GFP) were described previously (Chen et al., 2008a; Chen et al., 2008b; Chen et al., 2010; Liu et al., 2010). For experiments, PC12 cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Afterwards, cells were used for experiments. Cells infected with Ad-GFP served as a control. Expression of HA-tagged PP5 or dn-AMPKα, FLAG-tagged dn-c-Jun, or myc-tagged AMPKα-ca was assessed by Western blot analysis with antibodies to HA, FLAG or myc.
2.4. Drugs administration and Cd treatments
Experimental treatments and time line were shown in Fig. 1. In brief, PC12 cells, SH-SY5Y cells and/or primary neurons, or PC12 cells infected with Ad-PP5, Ad-dn-c-Jun, Ad-dn-AMPKα, Ad-AMPKα-ca or Ad-GFP, respectively, were seeded in a PDL-uncoated or -coated 6-well plate (1 × 106 cells/well for Western blotting) or 96-well plate (1 × 104 cells/well for MTS assay and caspase-3/7 activity assay), or at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-uncoated or -coated glass coverslip per well (for DAPI/TUNEL staining, Immunofluorescence staining and ROS imaging). The next day, cells were treated with/without Cd (10 and/or 20 μM) for 4 h or 24 h following pre-incubation with/without metformin (1 mM) for 24 h and/or with/without a JNK inhibitor SP600125 (20 μM) (Chen et al., 2008b; Chen et al., 2009), an AMPK activator AICAR (2 mM) (Chen et al., 2011), an antioxidant and ROS scavenger NAC (5 mM) (Chen et al., 2008a; Chen et al., 2011) or a mitochondria-targeted antioxidant Mito-TEMPO (10 μM) (Yeh et al., 2014) for 1 h with 5 replicates of each treatment. Subsequently, cells were conducted for the below analysis.
Fig. 1.

Experimental treatments and time line in vitro. PC12 cells, SH-SY5Y cells and/or primary neurons, or PC12 cells infected with Ad-PP5, Ad-dn-c-Jun, Ad-dn-AMPKα, Ad-AMPKα-ca or Ad-GFP, respectively, were treated with/without Cd for 4 h or 24 h following pre-incubation with/without metformin for 24 h and/or with/without SP600125, AICAR, NAC or Mito-TEMPO for 1 h. Subsequently, cells were conducted for the analysis.
2.5. Assays for cell viability and caspase-3/7 activity
The indicated cells, after treatments, were incubated with MTS reagent (one solution reagent) (20 μl/well) for 4 h, and then the viability of the cells was assayed by measuring the optical density (OD) at 490 nm using a Victor X3 Light Plate Reader (PerkinElmer, Waltham, MA, USA). Caspase-3/7 activity was determined according to the supplier’s instructions of Caspase-Glo® 3/7 Assay Kit (Promega, Madison, WI, USA).
2.6. DAPI and TUNEL staining
For the indicated cells, after treatments, the cells with fragmented and condensed nuclei were determined using DAPI staining as described (Chen et al., 2008b). For the cells using DAPI/TUNEL staining, following DAPI staining, TUNEL staining was conducted according to the manufacturer’s protocols of In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). Finally, photographs for all stained samples were recorded under a fluorescence microscope (200×) (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. For quantitative analysis of the fluorescence intensity using TUNEL staining, the integral optical density (IOD) was determined by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.7. Immunofluorescence and imaging
The indicated cells on the coverslips, after treatments, were fixed with 4% paraformaldehyde and incubated with 3% normal goat serum to block non-specific binding. Next, the cells were incubated with rabbit anti-phospho-JNK (Thr183/Tyr185) antibody (Cell Signaling, Danvers, MA, USA, 1:50, diluted in PBS containing 1% BSA) overnight at 4°C, washed 3 times with PBS, and further incubated with FITC-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500, diluted in PBS containing 1% BSA) for 1 h at room temperature. The cells were then washed 3 times with PBS. Finally, slides were mounted in glycerol/PBS (1:1, v/v) containing 2.5% 1,4-diazabiclo-(2,2,2) octane. Cell images and IOD for fluorescence intensity were captured under a fluorescence microscope (200×) and quantitatively analyzed as described above, respectively.
2.8. Cell ROS imaging
For the indicated cells, after treatments, intracellular ROS fluorescence was imaged using an oxidant-sensitive probe, CM-H2DCFDA, and quantitatively analyzed as described (Xu et al., 2016).
2.9. Western blot analysis
The indicated cells, after treatments, were lysed, followed by western blotting, as described previously (Chen et al., 2010). The blots for detected protein were semi-quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA). The following antibodies were used: phospho-AMPKα (p-AMPKα) (Thr 172), phospho-acetyl-CoA carboxylase (p-ACC) (Ser79), ACC, JNK, cleaved-caspase-3, poly (ADP-ribose) polymerase (PARP) (Cell Signaling Technology, Danvers, MA, USA), p-c-Jun (Ser63), PP5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-JNK (Thr183/Tyr185), c-Jun, AMPKα (Epitomics, Burlingame, CA, USA), FLAG, HA, myc epitope, β-tubulin, goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Sciben Biotech Company, Nanjing, China).
2.10. Statistical analysis
Results were expressed as mean ± SEM. The normality of data was determined using Shapiro-Wilk tests. Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability was compared using one-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. Significance was accepted at p < 0.05.
3. Results
3.1. Metformin attenuates Cd-induced apoptotic cell death in neuronal cells
Recent reports have suggested that metformin possesses protective effects against several aging-related diseases in humans (Barzilai et al., 2016; Duca et al., 2015; Kickstein et al., 2010; Zheng et al., 2012). Metformin, in clinically relevant concentrations, at 0.5–2 mM, substantially protected neuronal PC12 cells from hydrogen peroxide (H2O2)-induced cell death (Zhao et al., 2019). Based on the above finding and our pre-experiment results in PC12 cells, SH-SY5Y cells and primary neurons pretreated with metformin (0–1.5 mM) for 24 h and then exposed to Cd (10 and 20 μM) for 24 h (data not shown), we selected 1 mM metformin as an appropriate concentration for this research. Here we observed that pretreatment of neuronal cells (PC12 cells, SH-SY5Y cells and primary neurons) with metformin (1 mM) for 24 h attenuated apoptotic cell death induced by 24-h exposure with Cd (10 and 20 μM). This is evidenced by the following findings. 1) Cell viability in the Cd/metformin group was significantly higher than that in the Cd alone group (Fig. 2A), as determined by MTS assay. 2) Pretreatment with metformin for 24 h dramatically diminished the percentage of the cells with nuclear fragmentation and condensation (arrows) and the number of TUNEL-positive cells with fragmented DNA (in green) in PC12 cells, SH-SY5Y cells and primary neurons exposed to Cd (Fig. 2B–D), as imaged and quantified by DAPI and TUNEL staining. 3) Treatment with Cd for 4 h resulted in robust cleavages of caspase-3 and PARP in PC12 cells, SH-SY5Y cells and primary neurons, which were strikingly attenuated by addition of metformin (Fig. 2E and F). 4) Metformin profoundly decreased Cd-elicited activation of caspases-3/7 in the cells (Fig. 2G), as detected by the caspase-3/7 activity assay. Therefore, our results strongly support the notion that metformin can prevent against Cd-induced neuronal cell death.
Fig. 2.

Metformin attenuates Cd-induced apoptotic cell death in neuronal cells. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h (for Western blotting) or 24 h (for MTS assay, DAPI/TUNEL staining and caspase-3/7 activity assay). A) Relative cell viability was evaluated by MTS assay. B) Apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining (upper panel) and concurrently by in situ detection of fragmented DNA (in green) using TUNEL staining (lower panel). Scale bar: 20 μm. C and D) The percentages of cells with fragmented nuclei and the number of TUNEL-positive cells were quantified. E) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. F) The relative densities for cleaved-caspase-3, cleaved-PARP to β-tubulin were semi-quantified using NIH image J. G) Caspase-3/7 activities were detected using Caspase-3/7 Assay Kit. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with 10 μM Cd group.
3.2. Metformin suppresses Cd-induced activation of JNK pathway, preventing apoptosis of neuronal cells
Numerous studies have reported that metformin may alter the activity of JNK under various conditions, thereby affecting cell survival and apoptosis (Chen et al., 2016; Ota et al., 2007; Wu et al., 2011; Zhu et al., 2016). We have recently demonstrated that Cd induces neuronal apoptosis in part through activation of JNK signaling pathway (Xu et al., 2017). Therefore, we reasoned that metformin might ameliorate Cd-evoked neuronal apoptosis by blocking activation of JNK pathway. In this connection, PC12 cells, SH-SY5Y cells and primary neurons were pretreated with metformin (1 mM) for 24 h, and then exposed to Cd (10 and 20 μM) for 4 h, followed by Western blot analysis. As predicted, pretreatment with metformin effectively inhibited Cd-induced phosphorylation of JNK (Thr183/Tyr185) and its downstream molecule, protein expression and phosphorylation of c-Jun (Ser63) in the cells (Fig. 3A and B). To confirm this, we next conducted phospho-JNK (p-JNK) (Thr183/Tyr185) immunofluorescence staining in PC12 cells, SH-SY5Y cells and primary neurons, respectively. Treatment with Cd (10 and 20 μM) for 24 h elicited a remarkable increase in p-JNK (Thr183/Tyr185) (in green), which was significantly hindered by metformin pretreatment (Fig. 3C and D).
Fig. 3.

Metformin prevents Cd-induced activation of JNK pathway. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h (for Western blotting) or 24 h (for immunofluorescence staining). A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B) The relative densities for p-JNK to JNK, and p-c-Jun to β-tubulin were semi-quantified using NIH image J. C and D) Expression of p-JNK (Thr183/Tyr185) was stained and imaged using immunofluorescence, showing that treatment of the cells with Cd for 24 h resulted in lower p-JNK expression (in green), which was obviously reversed by metformin. Scale bar: 20 μm. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with 10 μM Cd group.
To corroborate the observation that metformin’s inhibition of Cd-induced neuronal apoptosis is attributed to its blockage of JNK/c-Jun activation, SP600125, a JNK inhibitor (Chen et al., 2008b; Chen et al., 2009), was employed. When PC12 cells and primary neurons were treated with SP600125 (20 μM) for 1 h alone, or co-treated with metformin (1 mM) for 24 h in the presence or absence of SP600125 (20 μM) for 1 h, and then exposed to Cd (10 μM) for 4 h or 24 h, we found that SP600125 or metformin alone obviously suppressed the phosphorylation of JNK (Thr183/Tyr185) and c-Jun (Ser63), as well as the cleavage of caspase-3 in the cells in response to Cd (Fig. 4A and B). Especially, co-treatment with metformin/SP600125 exhibited a stronger inhibitory effect on Cd-induced p-JNK, p-c-Jun and cleaved-caspase-3 (Fig. 4A and B). Consistently, the combination of metformin with SP600125 also demonstrated more potent suppression of Cd-evoked apoptosis than metformin or SP600125 alone, as evidenced by the decreased percentage of cells with fragmented nuclei (Fig. 4C).
Fig. 4.

Pharmacological inhibition of JNK or expression of dominant negative c-Jun strengthens metformin’s inhibition of Cd-induced apoptosis in neuronal cells. PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-GFP (as control), respectively, were pretreated with/without SP600125 (20 μM) for 1 h and then metformin (1 mM) for 24 h, or pretreated with/without metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for DAPI staining). A and D) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and E) The relative densities for p-JNK to JNK, and p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. C and F) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with no SP600125 group; Using Student’s t-test, cp < 0.05 for Ad-dn-c-Jun group versus Ad-GFP group.
In addition, we verified the above finding using a genetic approach. PC12 cells, infected with Ad-dn-c-Jun and Ad-GFP (as control), respectively, were pretreated with metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h or 24 h. The results revealed that ectopic expression of dn-c-Jun, but not GFP, almost completely blocked Cd-induced p-c-Jun and cleavage of caspase-3 (Fig. 4D and E). Of importance, overexpression of dn-c-Jun significantly potentiated the inhibitory effect of metformin on Cd-induced apoptosis in PC12 cells (Fig. 4F). Collectively, our data underline the concept that metformin suppresses Cd activation of JNK/c-Jun-dependent neuronal apoptosis.
3.3. Metformin prevents Cd activation of JNK/c-Jun-dependent apoptosis by blocking Cd inactivation of PP5 in neuronal cells
PP5, as a physiological inhibitor of ASK1-JNK signaling, is well-known to negatively regulate JNK pathway, involved in stress response, cell survival and apoptosis (Chen et al., 2008a; Chen et al., 2009; Han et al., 2012; Morita et al., 2001; Xu et al., 2017). Our previous research has demonstrated that Cd activates JNK pathway leading to apoptosis in part by inhibition of PP5 in neuronal cells (Chen et al., 2008a), and this study has found that metformin inhibited Cd activation of JNK pathway contributing to neuronal cell death (Fig. 3 and Fig. 4). Therefore, we postulated that metformin suppresses Cd activation of JNK pathway by preventing Cd from inactivating PP5. To this end, PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, and then exposed to Cd (10 and 20 μM) for 4 h, followed by Western blot analysis. As shown in Fig. 5A and B, Cd-reduced PP5 expression was indeed partly rescued by metformin in PC12 cells, SH-SY5Y cells and primary neurons. To define the role of PP5 in metformin’s blockage of Cd-induced JNK activation and neuronal apoptosis, PC12 cells, infected with recombinant adenovirus expressing HA-tagged wild-type human PP5 (Ad-PP5) or Ad-GFP (as control), were pretreated with metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h or 24 h. By Western blot analysis, we observed over-expression of HA-tagged PP5 in the cells infected with Ad-PP5 (Fig. 5C and D). In line with our previous findings (Chen et al., 2008a), exposure to Cd decreased PP5 expression, and correspondingly increased the phosphorylation of JNK/c-Jun in the control cells infected with Ad-GFP (Lane 3 vs. Lane 1) (Fig. 5C and D). Overexpression of PP5 partially blocked Cd-induced p-JNK/p-c-Jun (Lane 7 vs. Lane 3) (Fig. 5C and D). Moreover, overexpression of PP5 also reinforced the protective effect of metformin against Cd-induced cleavage of caspase-3 (Fig. 5C and D). Using MTS assay and DAPI staining, we further exhibited that overexpression of PP5 in part reversed Cd-induced cell viability reduction and apoptosis, and strengthened the protective activity of metformin (Fig. 5E and F). These results indicate that metformin prevents Cd activation of JNK/c-Jun-dependent neuronal apoptosis in part by blocking Cd inactivation of PP5.
Fig. 5.

Metformin blocks Cd activation of JNK/c-Jun-dependent neuronal apoptosis by suppressing Cd inactivation of PP5. PC12 cells, SH-SY5Y cells and primary neurons, or PC12 cells infected with Ad-PP5 or Ad-GFP (as control), respectively, were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h (for Western blotting) or 24 h (for MTS assay and DAPI staining). A and C) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and D) The relative densities for p-JNK to JNK, and PP5, p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. E) Relative cell viability was evaluated by MTS assay. F) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with 10 μM Cd group; Using Student’s t-test, cp < 0.05 for Ad-PP5 group versus Ad-GFP group.
3.4. AMPKα-mediated JNK pathway is involved in metformin’s prevention of Cd-induced apoptosis in neuronal cells
We have recently shown that Cd induces neuronal apoptosis through inactivation of AMPKα (Chen et al., 2011; Zhang et al., 2017). Metformin is a well-known AMPK activator (Singh et al., 2016). Therefore, we reasoned that metformin prevents Cd-induced neuronal apoptosis partly by blocking Cd inactivation of AMPKα. To this end, PC12 cells, SH-SY5Y cells and primary neurons were exposed to Cd (10 and 20 μM) for 4 h following pretreatment with/without metformin (1 mM) for 24 h. Western blotting results showed that metformin markedly attenuated Cd-reduced phosphorylation of AMPKα (Thr172) and its substrate ACC (Ser79) (Fig. 6A and B). Next, we determined whether metformin inhibits Cd-induced neuronal apoptosis by preventing Cd inactivation of AMPKα. For this, PC12 cells and primary neurons were treated with/without Cd (10 μM) for 4 h or 24 h following pre-incubation with/without metformin (1 mM) for 24 h and/or with/without AICAR (2 mM), an AMPK activator (Chen et al., 2011), for 1 h. In line with our previous finding (Chen et al., 2011), Cd markedly evoked the decline of p-AMPKα and p-ACC, and the elevation of cleaved-caspase-3, which was attenuated by AICAR (Fig. 6C and D). Interestingly, co-treatment with metformin/AICAR rescued the cells from Cd-induced caspase-3 cleavage, viability reduction and apoptosis more potently than metformin or AICAR alone (Fig. 6C–F). Our observation that AICAR powerfully reinforced the inhibitory effect of metformin on Cd-elicited events supports the idea that metformin ameliorates Cd-induced cell death in part by activating AMPK, which plays a significant role in various aspects in neuronal cells. In addition, we found that Cd-induced phosphorylation of JNK was, to some extent, inhibited by pretreatment with AICAR or metformin alone, and more potently inhibited by co-treatment with metformin/AICAR (Fig. 6C and D). These results imply that AMPKα-mediated JNK pathway may be involved in metformin’s prevention of Cd-induced neuronal cell death.
Fig. 6.

Metformin prevents Cd activation of JNK/c-Jun-dependent neuronal apoptosis by repressing Cd inactivation of AMPK. PC12 cells, SH-SY5Y cells and/or primary neurons, respectively, were pretreated with/without metformin (1 mM) for 24 h, or pretreated with/without AICAR (2 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h (for Western blotting) or 24 h (for MTS assay and DAPI staining). A and C) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and D) The relative densities for p-AMPKα to AMPK, p-ACC to ACC, p-JNK to JNK, and p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. E) Relative cell viability was evaluated by MTS assay. F) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with 10 μM Cd group, cp < 0.05 for difference with no AICAR group.
To confirm the above findings, PC12 cells, infected with recombinant adenoviruses expressing HA-tagged dominant negative AMPKα1 (Ad-dn-AMPKα), myc-tagged continuous-active AMPKα (Ad-AMPKα-ca) and control virus encoding GFP alone (Ad-GFP), respectively, were exposed to Cd (10 μM) for 4 h or 24 h following pretreatment with/without metformin (1 mM) for 24 h. We observed a high level of HA-tagged dn-AMPKα in Ad-dn-AMPKα-infected cells and that of myc-tagged AMPKα-ca in Ad-AMPKα-ca-infected cells, but not that in Ad-GFP-infected cells (control) (Fig. 7A and D). As expected, ectopic expression of dn-AMPKα decreased the AMPK activity, while expression of AMPKα-ca increased the AMPK activity, as evidenced by the decreased/increased basal or Cd-induced p-AMPKα and p-ACC levels detected by Western blotting (Fig. 7A, B, D, and E). Of note, expression of dn-AMPKα profoundly enhanced the basal or Cd-increased p-JNK, p-c-Jun, c-Jun, cleaved-caspase-3 and cell apoptosis, and obviously attenuated the inhibitory effect of metformin on Cd-induced events (Figure 7A–C). In contrast, expression of AMPKα-ca remarkably potentiated the inhibitory activity of metformin (Fig. 7D–F). These results demonstrate the importance of metformin’s prevention of Cd-inactivated AMPKα in blocking Cd activation of JNK/c-Jun-dependent apoptosis in neuronal cells.
Fig. 7.

Ectopic expression of dominant negative AMPKα or continuous-active AMPKα affects metformin preventing Cd activation of JNK pathway and apoptosis in neuronal cells. PC12 cells, infected with Ad-dn-AMPKα, Ad-AMPKα-ca or Ad-GFP (as control), respectively, were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for DAPI staining). A and D) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and E) The relative densities for p-AMPKα to AMPK, p-ACC to ACC, p-JNK to JNK, and p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. C and F) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results are presented d as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group; Using Student’s t-test, bp < 0.05 for Ad-dn-AMPKα group versus Ad-GFP group, cp < 0.05 for Ad-AMPKα-ca group versus Ad-GFP group.
3.5. Metformin ameliorates Cd-induced neuronal apoptosis by suppressing ROS-dependent PP5/AMPK-JNK signaling pathway.
It has been documented that Cd induces excessive ROS contributing to neuronal apoptosis (Chen et al., 2011; Di Carlo et al., 2012; Yang et al., 2007). AMPK inactivation/activation due to oxidative stress is associated with neurodegenerative diseases (Chiang et al., 2018; Jiang et al., 2013; Vingtdeux et al., 2011). Our group has demonstrated that Cd induction of ROS results in inactivation of PP5 and AMPK leading to apoptosis of neuronal cells (Chen et al., 2008a; Chen et al., 2011; Xu et al., 2017; Zhang et al., 2017). Next, we wondered whether metformin suppresses Cd-induced inactivation of PP5/AMPK, activation of JNK/c-Jun, and neuronal apoptosis by reducing Cd-induced ROS level. For this, ROS assay was performed. We observed that pretreatment with metformin (1 mM) for 24 h significantly attenuated Cd-elevated intracellular ROS levels in PC12 cells, SH-SY5Y cells and primary neurons (Fig. 8A and B), as measured by imaging and quantifying using an oxidant-sensitive probe, CM-H2DCFDA. This was consistent with the observations that Cd induced cell viability reduction and apoptosis (Fig. 2). Further, pretreatment with NAC, an antioxidant and ROS scavenger (Chen et al., 2008a; Chen et al., 2011), clearly repressed Cd-evoked ROS levels (Fig. 8C) and markedly strengthened metformin’s intervention in Cd-triggered abnormal ROS accumulation in PC12 cells, SH-SY5Y cells and primary neurons (Fig. 8C). In line with this, co-treatment with metformin/NAC diminished the percentage of cells with fragmented nuclei more potently than treatment with NAC or metformin alone (Fig. 8D). Further, we exhibited that NAC alone slightly promoted the phosphorylation of AMPKα and ACC (Fig. 8E and F). NAC alone also, to some degree, prevented Cd-induced inactivation of PP5/AMPK, activation of JNK, and cleavage of caspase-3 (Fig. 8E and F). These findings suggest that metformin suppresses Cd elevation of ROS, which links to its prevention of Cd-induced inactivation of PP5/AMPK, activation of JNK, and apoptosis in neuronal cells.
Fig. 8.

NAC potentiates metformin’s protection against Cd-induced ROS-dependent PP5/AMPK inactivation, JNK activation and apoptosis in neuronal cells. PC12 cells, SH-SY5Y cells and/or primary neurons, respectively, were pretreated with/without metformin (1 mM) for 24 h, or pretreated with/without NAC (5 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h (for Western blotting) or 24 h (for cell ROS imaging and DAPI staining). A-C) Cell ROS was imaged and quantified using an oxidant-sensitive probe CM-H2DCFDA, showing that Cd-evoked abnormal ROS fluorescence (in green) was potently suppressed by metformin in the cells. Scale bar: 20 μm. D) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. E) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. F) The relative densities for p-AMPKα to AMPK, p-ACC to ACC, p-JNK to JNK, and PP5, p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with 10 μM Cd group, cp < 0.05 for difference with no NAC group.
Since metformin induces mitochondrial biogenesis (Singh et al., 2016), whereas Cd induces mitochondrial ROS, we next sought to validate whether metformin protection from Cd-induced neuronal apoptosis is dependent on mitochondrial ROS change. For this, PC12 cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) in the presence or absence of Mito-TEMPO (10 μM), a mitochondria-targeted antioxidant (Yeh et al., 2014). As expected, Mito-TEMPO significantly repressed Cd-induced ROS levels (Fig. 9A) and cell apoptosis (Fig. 9B), and dramatically strengthened the inhibitory effects of metformin on Cd-induced events in PC12 cells and primary neurons (Fig. 9A and B). In addition, we noticed that Mito-TEMPO reversed the expression of PP5, p-AMPKα, p-ACC, p-JNK, p-c-Jun, c-Jun, as well as cleavage of caspase-3 in the cells in response to Cd (Fig. 9C and D). Especially, co-treatment with metformin/Mito-TEMPO exhibited a stronger intervention for Cd-evoked events in the cells (Fig. 9C and D). Taken together, we conclude that metformin attenuates Cd-induced mitochondrial ROS, thereby preventing Cd inactivation of PP5/AMPKα and activation of JNK pathway, and ultimately protecting against apoptosis in neuronal cells.
Fig. 9.

Metformin prevents Cd-induced mitochondrial ROS-dependent PP5/AMPK inactivation, JNK activation and apoptosis in neuronal cells. PC12 cells and primary neurons were pretreated with/without Mito-TEMPO (10 μM) for 1 h and then metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting) or 24 h (for cell ROS imaging and DAPI staining). A) Cell ROS was imaged and quantified using an oxidant-sensitive probe CMH2DCFDA. B) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. C) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. D) The relative densities for p-AMPKα to AMPK, p-ACC to ACC, p-JNK to JNK, and PP5, p-c-Jun, cleaved-caspase-3 to β-tubulin were semi-quantified using NIH image J. Results are presented as mean ± SEM (n = 3–5). Using one-way ANOVA, *p < 0.05 for difference with no metformin group, ap < 0.05 for difference with no Cd group, bp < 0.05 for difference with no Mito-TEMPO group.
4. Discussion
Abundant evidence has shown that smelting, burning of fossil fuels and municipal wastes, refining of metals, and cigarette smoking are the main sources of Cd pollution in water, air and soil (Chen et al., 2008a; Chen et al., 2011; Wang and Du, 2013). So people can easily expose to Cd through daily life. Especially, after entering human body, Cd can readily pass through the blood-brain barrier and accumulate in the brain, hinder the function of the nervous system by its induction of neuronal apoptosis, and thereby contributing to neurodegenerative diseases, such as PD, AD and HD (Ben Mimouna et al., 2019; Mendez-Armenta and Rios, 2007; Okuda et al., 1997; Peng et al., 2017; Rigon et al., 2008; Williams et al., 2010). Our group has shown that Cd evokes ROS overproduction and consequent cell death in neuronal cells (Chen et al., 2008a; Chen et al., 2011; Chen et al., 2014b; Xu et al., 2017; Zhang et al., 2017). Therefore, it is of great importance to find a novel therapeutic strategy to prevent Cd-induced ROS-dependent neuronal apoptosis. Metformin, a well-known antidiabetic drug, has been investigated and proposed to possess neuroprotective activity in the models of neurodegenerative disorders (Foretz et al., 2014; Kickstein et al., 2010; Ma et al., 2007; Singh et al., 2016). Some studies have revealed that metformin acts in protection against several aging-related diseases in humans by activating AMPK and reducing endogenous ROS production (Barzilai et al., 2016; Duca et al., 2015; Kickstein et al., 2010; Zheng et al., 2012). However, little is known about whether and how metformin prevents Cd neurotoxicity. Here, for the first time, we provide evidence that metformin prevents Cd-elicited apoptotic cell death in neuronal cells, by repressing mitochondrial ROS inactivation of PP5 and AMPK, leading to suppression of JNK cascade.
It has been shown that metformin may alter the activity of JNK under various conditions, and thus affecting cell survival and apoptosis (Chen et al., 2016; Ota et al., 2007; Wu et al., 2011; Zhu et al., 2016). Our previous studies have identified that Cd activates JNK cascade leading to neuronal apoptosis (Chen et al., 2008a; Chen et al., 2008b). In the current study, we found that metformin inhibited Cd-induced phosphorylation of JNK, including protein expression and phosphorylation of c-Jun, the substrate of JNK, in PC12 cells, SH-SY5Y cells and primary neurons (Fig. 3). This was further verified by using pharmacological JNK inhibitor SP600125 or genetic manipulation of c-Jun (Fig. 4).
PP5 negatively regulates JNK pathway, involved in stress response, cell survival and apoptosis (Chen et al., 2008a; Chen et al., 2009; Han et al., 2012; Morita et al., 2001; Xu et al., 2017). Here we revealed that metformin alleviated Cd-induced activation of JNK/c-Jun-dependent neuronal apoptosis through preventing Cd from inhibition of PP5. This is supported by the findings that overexpression of PP5 partially prevented Cd-induced activation of JNK/c-Jun, cleaved-caspase-3 and cell death, and potentiated the inhibitory activity of metformin for Cd-elicited the events (Fig. 5). Based on the entire data from our observations and other literatures, to our knowledge, this is the first report presenting that metformin protects against Cd neurotoxicity partly by rescuing PP5-dependent suppression of JNK pathway.
AMPK activation/phosphorylation is a key biochemical event responsible for cell survival and apoptosis, which is involved in aging and neurodegenerative diseases (Chiang et al., 2018; Jiang et al., 2013; Vingtdeux et al., 2011; Zhao et al., 2019). It is known that metformin is a widely accepted AMPK activator, which can accelerate AMPK phosphorylation at Thr172 site (Barzilai et al., 2016; Lu et al., 2016; Singh et al., 2016). In this study, we observed that metformin obviously blocked Cd-induced decline of p-AMPKα (Thr172) and its substrate p-ACC (Ser79) in PC12 cells, SH-SY5Y cells and primary neurons (Fig. 6), implying that metformin prevents Cd inactivation of AMPKα. Of note, the neuroprotective effect of metformin in primary neurons was found to be more profound at low doses of Cd (10 μM), probably because of the higher toxicity of 20 μM Cd. Using AMPKα activator (AICAR) and overexpression of constitutively activated AMPKα (AMPKα-ca) or dominant negative AMPKα (dn-AMPKα), we further demonstrated that metformin inhibited Cd activation of JNK/c-Jun-dependent neuronal apoptosis by preventing Cd inactivation of AMPKα (Fig 6 and 7). Overall, these observations support that metformin protects against Cd neurotoxicity partly by restraining Cd inactivation of AMPK, thereby preventing activation of JNK/c-Jun pathway.
Under pathological conditions, excessive ROS in the mitochondria reduces mitochondrial biogenesis, which, in turn, further promotes ROS induction and mitochondrial dysfunction, and thus commits the cell to undergo apoptosis (Lu et al., 2012; Seo et al., 2010; Zhang et al., 2017). Some reports have shown that mitochondrion is the primary target of metformin within the cell in which metformin transiently inhibits mitochondrial complex 1 in the electron transport chain, promotes mitochondrial biogenesis (Singh et al., 2016; Vancura et al., 2018). Recent evidence exhibits that metformin protects PC12 cells and hippocampal neurons from H2O2-induced oxidative insult via activating AMPK pathway, implying metformin resistance to oxidative stress as an anti-oxidative compound (Zhao et al., 2019). In this study, when metformin-pretreated PC12 cells, SH-SY5Y cells and primary neurons were exposed to Cd for 24 h, intracellular abnormal ROS accumulation was significantly suppressed compared to the vehicle-pretreated cells (Fig. 8A and B). This is in line with the finding that metformin possesses antioxidant activity (Zhao et al., 2019). Using NAC, an antioxidant and ROS scavenger (Chen et al., 2008a; Chen et al., 2011), and Mito-TEMPO, a mitochondria-targeted antioxidant (Yeh et al., 2014), we further observed that addition of NAC or Mito-TEMPO obviously reinforced metformin inhibition of Cd-evoked ROS elevation and apoptosis, in PC12 cells and primary neurons (Fig. 8 and 9). Subsequently, we unveiled that Cd-induced inactivation of PP5/AMPK and consequential activation of JNK/c-Jun and caspase-3 were more powerfully ameliorated in the cells co-treated with metformin/NAC or metformin/Mito-TEMPO than in those treated with metformin, NAC or Mito-TEMPO alone (Fig. 8 and 9). Together, the findings support that metformin prevents Cd-induced apoptosis in neuronal cells by suppressing mitochondrial ROS-dependent PP5/AMPK-JNK signaling pathway.
Given the potential therapeutic effects of metformin, trials have been invested to study counteraction of metformin on neurodegenerative diseases in animals and humans, but its therapeutic use is not yet accepted since the results are often conflicting, such as divergent effects of oral metformin on an inflammatory model of Parkinson’s disease in rats, oral metformin’s protection on various brain tissues as cortical neurons through activation of AMPK, and metformin’s reduction of neurological symptoms in some patients and disease phenotypes in animal and cell models (Rotermund et al., 2018; Tayara et al., 2018). Acute metformin (10 mg/kg, i.p. for 24 h) preconditioning confers neuroprotection against focal cerebral ischemia by activation of AMPK-dependent autophagy in rats (Jiang et al., 2014), and chronic metformin (50 mg/kg daily, i.p. for 3 weeks) preconditioning provides neuroprotection in rats with permanent cerebral ischemia (Zhu et al., 2015). At the molecular level the findings vary depending on the doses of metformin used and duration of treatment, with clear differences between acute and chronic administration (Rotermund et al., 2018; Vancura et al., 2018; Zhang et al., 2016). Metformin’s bioavailability in brain has not been reported, but the neuroprotective effects of metformin on neurodegenerative diseases imply that metformin could cross the blood brain barrier. In the last 10 years, the researchers have moved from a simple picture, that metformin improves glycemia by acting on the liver via AMPK activation, to a much more complex picture reflecting its multiple modes of action. More studies on the in vivo metformin’s prevention of Cd toxicity in animal models are required to truly understand how metformin works on Cd neurotoxicity associated with aging and neurodegenerative diseases.
In conclusion, we have identified that metformin attenuated Cd-elicited neuronal apoptosis by preventing mitochondrial ROS inactivation of PP5 and AMPK, thus suppressing activation of JNK pathway (Fig. 10). Our results suggest that metformin is a promising drug for prevention of Cd-induced oxidative stress and neurodegenerative diseases.
Fig. 10.
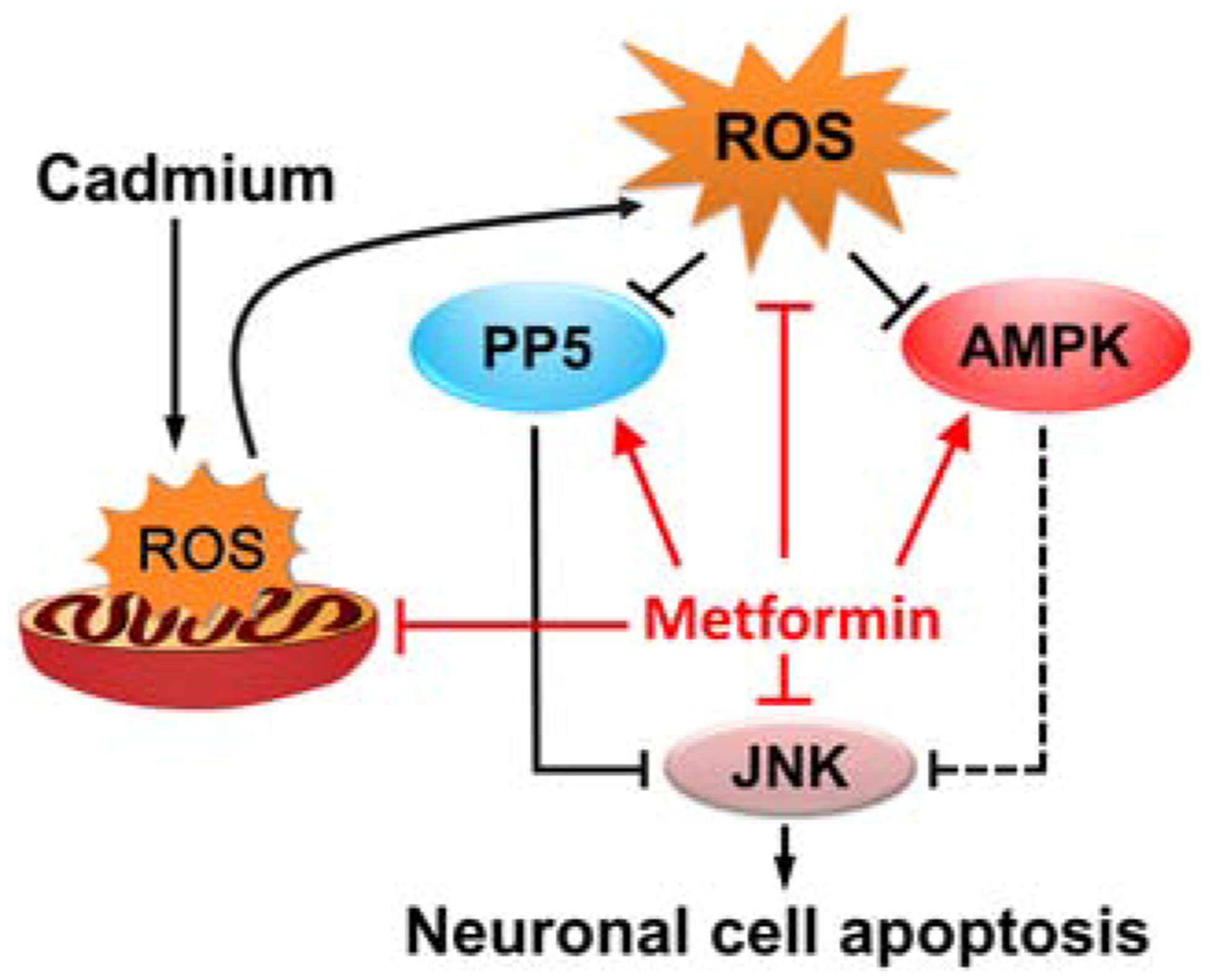
A graphical model of how metformin protects against Cd-induced neuronal apoptosis. Metformin attenuates Cd-elicited neuronal apoptosis by preventing mitochondrial ROS inactivation of PP5 and AMPK, thus suppressing activation of JNK pathway.
Supplementary Material
Supplementary Fig. 1. Untruncated images of blots in Fig. 1E. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 2. Untruncated images of blots in Fig. 2A. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 3. Untruncated images of blots in Fig. 3A and D. PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-GFP (as control), respectively, were pretreated with/without SP600125 (20 μM) for 1 h and then metformin (1 mM) for 24 h, or pretreated with/without metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting). Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 4. Untruncated images of blots in Fig.4A and C. PC12 cells, SH-SY5Y cells and primary neurons, or PC12 cells infected with Ad-PP5 or Ad-GFP (as control), respectively, were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 5. Untruncated images of blots in Fig. 5A. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 6. Untruncated images of blots in Fig. 5C. PC12 cells and primary neurons were pretreated with/without AICAR (2 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 7. Untruncated images of blots in Fig. 6A and D.PC12 cells, infected with Ad-dn-AMPK?, Ad-AMPK?-ca or Ad-GFP (as control), were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 8. Untruncated images of blots in Fig. 7E. PC12 cells and primary neurons were pretreated with/without NAC (5 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 9. Untruncated images of blots in Fig. 8C. PC12 cells and primary neurons were pretreated with/without Mito-TEMPO (10 μM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Highlights.
Metformin attenuates Cd-evoked apoptotic cell death in neuronal cells.
Metformin suppresses Cd-activated JNK pathway, preventing neuronal apoptosis.
Metformin prevents Cd-elicited mitochondrial ROS inactivation of PP5 and AMPK, thereby suppressing activation of JNK pathway.
Metformin may be a promising drug for prevention of Cd-induced oxidative stress and neurodegenerative diseases
Acknowledgments
This work was supported in part by the grants from National Natural Science Foundation of China (No. 81873781; L.C.), NIH (CA115414; S.H.), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010; L.C.), and American Cancer Society (RSG-08-135-01-CNE; S.H.).
Abbreviations:
- ACC
acetyl-CoA carboxylase
- AD
Alzheimer disease
- AICAR
5-amino-4-imidazolecarboxamide ribose
- AMPK
AMP-activated protein kinase
- Cd
cadmium
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate; DAPI, 4′, 6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
fetal bovine serum
- HD
Huntington’s disease
- H2O2
hydrogen peroxide
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- NAC
N-acetyl-L-cysteine
- PBS
phosphate buffered saline
- PD
Parkinson disease
- PDL
poly-D-lysine
- PP5
protein phosphatase 5
- ROS
reactive oxygen species
- TUNEL
the terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- Akesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M, 2006. Cadmium-induced effects on bone in a population-based study of women. Environ. Health Perspect 114(6), 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA, 2016. Metformin as a Tool to Target Aging. Cell Metab. 23(6), 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mimouna S, Le Charpentier T, Lebon S, Van Steenwinckel J, Messaoudi I, Gressens P, 2019. Involvement of the synapse-specific zinc transporter ZnT3 in cadmium-induced hippocampal neurotoxicity. J. Cell. Physiol 234(9), 15872–15884. [DOI] [PubMed] [Google Scholar]
- Chen B, Teng Y, Zhang X, Lv X, Yin Y, 2016. Metformin Alleviated Abeta-Induced Apoptosis via the Suppression of JNK MAPK Signaling Pathway in Cultured Hippocampal Neurons. Biomed. Res. Int 2016, 1421430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S, 2008a. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic. Biol. Med 45(7), 1035–1044. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Luo Y, Huang S, 2008b. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J. Neurochem 105(1), 251–261. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Yin J, Luo Y, Huang S, 2009. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol 41(6), 1284–1295. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S, 2010. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab. Invest 90(5), 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S, 2011. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic. Biol. Med 50(5), 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q, Guo M, Huang S, Chen L, 2014a. Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J. Neurochem 128(2), 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y, Guo M, Ji H, Xu C, Gu C, Gao W, Huang S, Chen L, 2014b. N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol. Appl. Neurobiol 40(6), 759–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Nicol CJ, Cheng YC, 2018. Resveratrol activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced inflammation and oxidative stress. Neurochem. Int 115, 1–10. [DOI] [PubMed] [Google Scholar]
- Di Carlo M, Giacomazza D, Picone P, Nuzzo D, San Biagio PL, 2012. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radic. Res 46(11), 1327–1338. [DOI] [PubMed] [Google Scholar]
- DiTacchio KA, Heinemann SF, Dziewczapolski G, 2015. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J. Alzheimers Dis 44(1),43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duca FA, Cote CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, Lam TK, 2015. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med 21(5), 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mir MY, Detaille D, R-Villanueva G, Delgado-Esteban M, Guigas B, Attia S, Fontaine E, Almeida A, Leverve X, 2008. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci 34(1), 77–87. [DOI] [PubMed] [Google Scholar]
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B, 2014. Metformin: from mechanisms of action to therapies. Cell Metab. 20(6), 953–966. [DOI] [PubMed] [Google Scholar]
- Fowler BA, 2009. Monitoring of human populations for early markers of cadmium toxicity: a review. Toxicol. Appl. Pharmacol 238(3), 294–300. [DOI] [PubMed] [Google Scholar]
- Han X, Xu B, Beevers CS, Odaka Y, Chen L, Liu L, Luo Y, Zhou H, Chen W, Shen T, Huang S, 2012. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis 33(4), 868–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HL, Jin JZ, Wu D, Xu D, Lin GF, Yu H, Ma DY, Liang J, 2013. Celastrol exerts synergistic effects with PHA-665752 and inhibits tumor growth of c-Met-deficient hepatocellular carcinoma in vivo. Mol. Biol. Rep 40(7), 4203–4209. [DOI] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, Zhang QQ, Gao L, Shi JQ, Zhang YD, Tan L, (2014. Acute metformin preconditioning confers neuroprotection against focal cerebral ischemia by pre-activation of AMPK-dependent autophagy. Br. J. Pharmacol 171(13), 3146–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri N, Jacquillet G, Unwin R, 2010. Heavy metal poisoning: the effects of cadmium on the kidney. Biometals 23(5), 783–792. [DOI] [PubMed] [Google Scholar]
- Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H, Schneider R, Sutherland C, Schweiger S, 2010. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. U. S. A 107(50), 21830–21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ, 2010. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802(4), 396–405.20079433 [Google Scholar]
- Kyriakis JM, Avruch J, 2001. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev 81(2), 807–869. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J, 2012. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev 92(2), 689–737. [DOI] [PubMed] [Google Scholar]
- Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, Zhou H, Han X, Huang S, 2010. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J. Biol. Chem 285(49), 38362–38373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF, 2012. Purple sweet potato color attenuates domoic acid-induced cognitive deficits by promoting estrogen receptor-alpha-mediated mitochondrial biogenesis signaling in mice. Free Radic. Biol. Med 52(3), 646–659. [DOI] [PubMed] [Google Scholar]
- Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G, 2016. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol 19(9), pii: pyw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Tang Q, Olefsky JM, Mellon PL, Webster NJ, 2008. Adiponectin activates adenosine monophosphate-activated protein kinase and decreases luteinizing hormone secretion in LbetaT2 gonadotropes. Mol. Endocrinol 22(3), 760–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL, Hoyt KR, 2007. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett 411(2), 98–103. [DOI] [PubMed] [Google Scholar]
- Mendez-Armenta M, Rios C, 2007. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol 23(3), 350–358. [DOI] [PubMed] [Google Scholar]
- Meng X, Chu G, Yang Z, Qiu P, Hu Y, Chen X, Peng W, Ye C, He FF, Zhang C, 2016. Metformin protects neurons against oxygen-glucose deprivation/reoxygenation -induced injury by down-regulating MAD2B. Cell. Physiol. Biochem 40(3–4), 477–485. [DOI] [PubMed] [Google Scholar]
- Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H, 2001. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 20(21), 6028–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M, 1997. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg 99(4), 263–265. [DOI] [PubMed] [Google Scholar]
- Ota K, Nakamura J, Li W, Kozakae M, Watarai A, Nakamura N, Yasuda Y, Nakashima E, Naruse K, Watabe K, Kato K, Oiso Y, Hamada Y, 2007. Metformin prevents methylglyoxal-induced apoptosis of mouse Schwann cells. Biochem. Biophys. Res. Commun 357(1), 270–275. [DOI] [PubMed] [Google Scholar]
- Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S, 2014. Neuroprotective effect of metformin in mptp-induced Parkinson’s disease in mice. Neuroscience 277, 747–754. [DOI] [PubMed] [Google Scholar]
- Peng Q, Bakulski KM, Nan B, Park SK, 2017. Cadmium and Alzheimer’s disease mortality in U.S. adults: Updated evidence with a urinary biomarker and extended follow-up time. Environ. Res 157, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Liu Y, Chen L, Zhu Y, Xiao X, Wang D, Zhu Y, 2018. Nobiletin prevents cadmium-induced neuronal apoptosis by inhibiting reactive oxygen species and modulating JNK/ERK1/2 and Akt/mTOR networks in rats. Neurol. Res 40(3), 211–220. [DOI] [PubMed] [Google Scholar]
- Rigon AP, Cordova FM, Oliveira CS, Posser T, Costa AP, Silva IG, Santos DA, Rossi FM, Rocha JB, Leal RB, 2008. Neurotoxicity of cadmium on immature hippocampus and a neuroprotective role for p38 MAPK. Neurotoxicology 29(4), 727–734. [DOI] [PubMed] [Google Scholar]
- Rotermund C, Machetanz G, Fitzgerald JC, 2018. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol 9, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satarug S, Nazar S, 2010. Cadmium in food and human health: technologies for environmental restoration and rehabilitation January 15–17, 2010, Phitsanulok, Thailand. Toxicol. Lett 198(1), 2–6. [DOI] [PubMed] [Google Scholar]
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C, 2010. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci 123(Pt 15), 2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shati AA, 2019. Resveratrol protects against cadmium chloride-induced hippocampal neurotoxicity by inhibiting ER stress and GAAD 153 and activating sirtuin 1/AMPK/Akt. Environ. Toxicol 34(12), 1340–1353. [DOI] [PubMed] [Google Scholar]
- Singh J, Olle B, Suhail H, Felicella MM, Giri S, 2016. Metformin-induced mitochondrial function and ABCD2 up-regulation in X-linked adrenoleukodystrophy involves AMP-activated protein kinase. J. Neurochem 138(1), 86–100. [DOI] [PubMed] [Google Scholar]
- Tayara K, Espinosa-Oliva AM, Garcia-Dominguez I, Ismaiel AA, Boza-Serrano A, Deierborg T, Machado A, Herrera AJ, Venero JL, de Pablos RM, 2018. Divergent effects of metformin on an inflammatory model of Parkinson’s disease. Front. Cell. Neurosci 12, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I, 2018. Metformin as an Anticancer Agent. Trends Pharmacol. Sci 39(10), 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P, 2011. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 25(1), 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Du Y, 2013. Cadmium and its neurotoxic effects. Oxid. Med. Cell. Longev 2013, 898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J, 2001. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29(3), 629–643. [DOI] [PubMed] [Google Scholar]
- Williams BB, Li D, Wegrzynowicz M, Vadodaria BK, Anderson JG, Kwakye GF, Aschner M, Erikson KM, Bowman AB, 2010. Disease-toxicant screen reveals a neuroprotective interaction between Huntington’s disease and manganese exposure. J. Neurochem 112(1), 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Gu C, Gu H, Hu H, Han Y, Li Q, 2011. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma 58(6), 482–490. [DOI] [PubMed] [Google Scholar]
- Xu C, Wang X, Gu C, Zhang H, Zhang R, Dong X, Liu C, Hu X, Ji X, Huang S, Chen L, 2017. Celastrol ameliorates Cd-induced neuronal apoptosis by targeting NOX2-derived ROS-dependent PP5-JNK signaling pathway. J. Neurochem 141(1), 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang X, Zhu Y, Dong X, Liu C, Zhang H, Liu L, Huang S, Chen L, 2016. Rapamycin ameliorates cadmium-induced activation of MAPK pathway and neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A. Neuropharmacology 105, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yang S, Qian SY, Hong JS, Kadiiska MB, Tennant RW, Waalkes MP, Liu J, 2007. Cadmium-induced toxicity in rat primary mid-brain neuroglia cultures: role of oxidative stress from microglia. Toxicol. Sci 98(2), 488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YT, Yeh H, Su SH, Lin JS, Lee KJ, Shyu HW, Chen ZF, Huang SY, Su SJ, 2014. Phenethyl isothiocyanate induces DNA damage-associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations. Free Radic. Biol. Med 74, 1–13. [DOI] [PubMed] [Google Scholar]
- Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA, 2004. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem 279(46), 47898–47905. [DOI] [PubMed] [Google Scholar]
- Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, Wu YQ, Lin SY, Lin SC, 2016. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 24(4), 521–522. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang N, Zhang H, Liu C, Dong X, Wang X, Zhu Y, Xu C, Liu L, Yang S, Huang S, Chen L, 2017. Celastrol prevents cadmium-induced neuronal cell death by blocking reactive oxygen species-mediated mammalian target of rapamycin pathway. Br. J. Pharmacol 174(1), 82–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zeng Z, Gaur U, Fang J, Peng T, Li S, Zheng W, 2019. Metformin protects PC12 cells and hippocampal neurons from H2 O 2 -induced oxidative damage through activation of AMPK pathway. J. Cell Physiol doi: 10.1002/jcp.28337. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y, Jin H, He Y, Gu Q, Xu X, 2012. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 61(1), 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zheng Y, Zhang H, Sun H, 2016. Targeting cancer cell metabolism: The combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am. J. Transl. Res 8(11), 4812–4821. [PMC free article] [PubMed] [Google Scholar]
- Zhu XC, Jiang T, Zhang QQ, Cao L, Tan MS, Wang HF, Ding ZZ, Tan L, Yu JT, 2015. Chronic metformin preconditioning provides neuroprotection via suppression of NF-κB-mediated inflammatory pathway in rats with permanent cerebral ischemia. Mol. Neurobiol 52(1), 375–385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Untruncated images of blots in Fig. 1E. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 2. Untruncated images of blots in Fig. 2A. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 3. Untruncated images of blots in Fig. 3A and D. PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-c-Jun or Ad-GFP (as control), respectively, were pretreated with/without SP600125 (20 μM) for 1 h and then metformin (1 mM) for 24 h, or pretreated with/without metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h (for Western blotting). Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 4. Untruncated images of blots in Fig.4A and C. PC12 cells, SH-SY5Y cells and primary neurons, or PC12 cells infected with Ad-PP5 or Ad-GFP (as control), respectively, were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and/or 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 5. Untruncated images of blots in Fig. 5A. PC12 cells, SH-SY5Y cells and primary neurons were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 and 20 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 6. Untruncated images of blots in Fig. 5C. PC12 cells and primary neurons were pretreated with/without AICAR (2 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 7. Untruncated images of blots in Fig. 6A and D.PC12 cells, infected with Ad-dn-AMPK?, Ad-AMPK?-ca or Ad-GFP (as control), were pretreated with/without metformin (1 mM) for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 8. Untruncated images of blots in Fig. 7E. PC12 cells and primary neurons were pretreated with/without NAC (5 mM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.
Supplementary Fig. 9. Untruncated images of blots in Fig. 8C. PC12 cells and primary neurons were pretreated with/without Mito-TEMPO (10 μM) for 1 h and then metformin for 24 h, followed by exposure to Cd (10 μM) for 4 h. Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control.