

Abstract
The precuneus (PreC; Brodmann area 7), a key hub within the default mode network (DMN) displays amyloid and tau-containing neurofibrillary tangle (NFT) pathology during the onset of Alzheimer’s disease (AD). PreC layer III projection neurons contain lysosomal hydrolase cathepsin D (CatD), 14)a marker of neurons vulnerable to NFT pathology. Here we applied single population laser capture microdissection coupled with custom-designed microarray profiling to determine the genetic signature of PreC CatD-positive-layer III neurons accrued from postmortem tissue obtained from the Rush Religious Orders Study (RROS) cases with a premortem clinical diagnosis of no cognitive impairment (NCI), mild cognitive impairment (MCI) and AD. Expression profiling revealed significant differential expression of key transcripts in MCI and AD compared to NCI that underlie signaling defects, including dysregulation of genes within the endosomal-lysosomal and autophagy pathways, cytoskeletal elements, AD-related genes, ionotropic and metabotropic glutamate receptors, cholinergic enzyme and receptors, markers of monoamine neurotransmission as well as steroid-related transcripts. Pervasive defects in both MCI and AD were found in select transcripts within these key gene ontology categories, underscoring the vulnerability of these corticocortical neurons during the onset and progression of dementia. Select PreC dysregulated genes detected via custom-designed microarray analysis were validated using qPCR. In summary, expression profiling of CatD positive PreC layer III neurons revealed significant dysregulation of a mosaic of genes in MCI and AD that were not previously appreciated in terms of their indication of systems-wide signaling defects in a key hub of the DMN.
Keywords: Alzheimer’s disease, expression profiling, laser capture microdissection, mild cognitive impairment, microarray, neurons, precuneus, RRID
Introduction
The precuneus [PreC; Brodmann area 7 (BA7)], a key component of the default mode network (DMN), which plays a role in episodic memory retrieval (Wagner et al., 2005), displays high metabolic activity during conscious rest and selectively deactivates during non-self-directed cognitive tasks in the healthy human brain (Buckner et al., 2008; Raichle et al., 2001; Sperling et al., 2010 ). By contrast, the PreC is dysregulated in aging (Andrews-Hanna et al., 2007), inactivates during cognitive tasks and is compromised during the early stages of Alzheimer’s disease (AD) (Herholz et al,. 2002; Matsuda., 2007; Rombouts et al., 2005). Pittsburgh Compound B ([3H]PiB) amyloid imaging demonstrates the DMN is prone to amyloid-beta peptide (Aβ) deposition in the earliest preclinical stages of AD (Mintun et al., 2006; Sheline et al., 2010; Sperling et al., 2009). The PreC also displays a greater degree of atrophy in early compared to late-onset AD (Frisoni et al., 2005; Ishii et al., 2005; Karas et al., 2007), a reduction in synapse number in AD but not in mild cognitive impairment (MCI) (Scheff et al., 2013) and a relationship between synaptic failure, functional and structural disconnection, and amyloid deposition prior to the onset of dementia (Drzezga et al., 2011; Yan et al., 2013). Despite extensive amyloid PiB binding, neurofibrillary tangle (NFT) bearing neurons are relatively sparse in the PreC compared to other vulnerable hubs of the DMN (Nelson et al., 2009; Perez et al., 2015). Currently, molecular factors associated with PreC neuron dysfunction during the onset of AD remain under investigated.
Dysregulation of the endosomal-lysosomal (EL) system is an early event in the pathogenesis of neocortical (Cataldo et al., 1995), hippocampal, and cholinergic basal forebrain neurons in prodromal AD (Ginsberg et al., 2000, 2010, 2011), which precedes amyloid and tau pathology (Cataldo et al., 1997). Numerous studies indicate the EL system plays a key role in amyloid-β precursor protein (APP) processing and Aβ generation in AD (Pasternak et al., 2004; Nixon, 2007, 2017). Vulnerable neurons display an increase in the size and number of lysosomes containing cathepsin D (CatD) (Cataldo et al., 1997), a ubiquitous and abundant lysosomal hydrolase. Transcriptomic and protein based assays demonstrate upregulation of CatD mRNA and protein levels in hippocampal and neocortical layer III and V pyramidal neurons in AD, including NFT-pyramidal neurons (Ginsberg et al., 2000; Cataldo et al., 1996), indicating an increase in the activation of the EL system in neurons prone to tau pathology (Ginsberg et al., 2010; Cataldo et al., 1995). CatD mediates programed cell death (Deiss et al., 1996) and carriers of the T-allele of rs17571 located on the CatD gene show an increased risk for AD (Riemenschneider et al., 2006). Genetic ablation of CatD markedly potentiates tau-induced neurotoxicity, indicating a role for this encoded protein in neuroprotection (Khurana et al., 2010). In the present study, we investigated the genetic signature of PreC CatD-positive layer III neurons in tissue obtained from participants in the Rush Religious Orders Study (RROS) that died with a premortem clinical diagnosis of no cognitive impairment (NCI), MCI, or AD. These findings will aid in understanding the mechanisms underlying cortical cellular neurodegeneration and potential resilience to disease within the DMN memory and executive function circuit.
Materials and Methods
Subjects
A total of 31 cases with an antemortem clinical diagnosis of NCI (n=11, 8F/3M, 86.17 ± 4.95 years), MCI (n=11, 9F/2M, 87.36 ± 4.58 years), and AD (n=9, 6F/3M, 88.48± 5.36 years) from the RROS (Mufson et al., 1999; Bennett et al., 2005, 2006) were examined (Table 1). Each participant agreed to an annual detailed premortem clinical evaluation, brain donation at the time of death and a postmortem neuropathological evaluation. Written informed consent for research and autopsy was obtained from family or guardians of each subject participating in the study. The Human Investigations Committee of Rush University Medical Center approved the study.
Table 1.
Clinical and Neuropathological Case Characteristics
| NCI (N=11) | MCI (N=11) | AD (N=9) | Overall p-value | Pair-wise comparisons | ||
|---|---|---|---|---|---|---|
| Age (years) at death | Mean ± SD | 86.17 ± 4.95 | 87.36 ± 4.58 | 88.48 ± 5.36 | 0.59 | ----- |
| Number of males (%) | 3 (27) | 18 (33) | 3 (33) | 0.74 | ----- | |
| Years of education | Mean ± SD | 16.91 ± 3.65 | 18.36 ± 2.80 | 18.22 ± 4.02 | 0.57 | ----- |
| Number with ApoE ε4 allele (%) | 0 (0) | 5 (45) | 2 (22) | 0.04 | ----- | |
| MMSE | Mean ± SD | 28.00 ± 1.55 | 26.91 ± 2.91 | 20.33 ± 2.43 | <0.001 | NCI, MCI > AD |
| GCS | Mean ± SD | −0.16 ± 0.25 | −0.41 ± 0.44 | −1.27 ± 0.68 | <0.001 | NCI, MCI > AD |
| Episodic memory z-score | Mean ± SD | 0.23 ± 0.45 | −0.33 ± 0.65 | −1.68 ± 1.15 | <0.001 | NCI, MCI > AD |
| Semantic memory z-score | Mean ± SD | −0.42 ± 0.65 | −0.37 ± 0.71 | −0.74 ± 0.77 | 0.46 | ----- |
| Working memory z-score | Mean ± SD | −0.18 ± 0.49 | −0.19 ± 0.69 | −0.58 ± 0.59 | 0.25 | ----- |
| Perceptual speed z-score | Mean ± SD | −0.71 ± 0.66 | −0.84 ± 0.59 | −2.40 ± 0.72 | <0.001 | NCI, MCI > AD |
| Visuospatial z-score | Mean ± SD | −0.41 ± 0.53 | −0.80 ± 0.63 | −1.12 ± 0.89 | 0.08 | ---- |
| Post-mortem interval (hours) | Mean ± SD | 5.38 ± 2.11 | 5.44 ± 2.18 | 4.14 ± 1.43 | 0.27 | ----- |
| Brain weight (g) | Mean ± SD | 1166.91 ± 86.22 | 1154.27 ± 141.18 | 1150.56 ± 88.26 | 0.83 | ----- |
| Distribution of Braak scores | 0 | 0 | 0 | 0 | 0.76 | ----- |
| I/II | 2 | 2 | 1 | |||
| III/IV | 8 | 5 | 6 | |||
| V/VI | 1 | 4 | 2 | |||
| NIA Reagan (likelihood of AD) | No AD | 0 | 0 | 0 | 0.17 | ----- |
| Low | 6 | 3 | 1 | |||
| Intermediate | 5 | 5 | 6 | |||
| High | 0 | 3 | 2 | |||
| CERAD | No AD | 2 | 2 | 0 | 0.10 | ----- |
| Possible | 6 | 6 | 6 | |||
| Probable | 3 | 0 | 0 | |||
| Definite | 0 | 3 | 3 | |||
Clinical and Neuropathological Evaluation
Clinical, demographic and neuropathological case details are reported in Table 1. Clinical evaluation and criteria for diagnosis of AD and MCI have been published previously (Mufson et al., 1999; Bennett et al., 2005; Davis et al., 1999; Perez et al., 2012; Schmitt et al., 2012). Briefly, following review of clinical data and participant examination, clinical diagnoses were made by a board-certified neurologist with expertise in gerontology. A neurologist reviewed medical history, medications, findings from neurologic examinations, cognitive performance scores and the neuropsychologist’s opinion of cognitive impairment and the presence of dementia. Each subject was evaluated in their home, emphasizing clinically relevant findings. AD diagnosis of dementia followed the recommendations of the joint working group of the National Institute of Neurological and Communicative Disorders and the Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA) (McKhann et al., 1984). Clinical classification of MCI are compatible with those used by others in the field to describe persons who are not cognitively normal, but do not meet accepted criteria for dementia (Rubin et al., 1989; Petersen et al., 1995; Ebly et al., 1995; Devanand et al., 1997; Albert et al., 1991; Flicker et al., 1991). MCI was defined as persons rated as impaired on neuropsychological testing by a neuropsychologist but not found to have dementia by the examining neurologist. Average time from the last clinical evaluation to death was ~8 months.
Among the MCI cases, five are amnestic (45.5%) and six are non-amnestic (54.5%) (Schmitt et al., 2000). The Mini-mental status exam (MMSE) and a neuropsychological battery was also available within two years prior to death, from which a global cognitive z-score (GCS) comprised of 19 individual tests was computed, as was an episodic memory z-score (Albert et al., 1991) based on seven tests (East Boston Memory immediate and delayed recall, Logical Memory immediate and delayed story recall, CERAD Word List Memory immediate and delayed recall, and CERAD Word List Recognition). At a consensus conference a neurologist and neuropsychologist reviewed all clinical data, medical records, interviews with family members and assigned a final clinical diagnosis for each subject. Postmortem neuropathological diagnosis was performed as reported previously (Mufson et al., 1999; Bennett et al., 2005; Perez et al., 2012; Schmitt et al., 2012), which included NIA-Reagan criteria (Newell et al., 1999), recommendations of the CERAD (Mirra, 1997), Braak staging of NFTs, amyloid deposition scores (Braak & Braak, 1991) and TDP-43 reactivity (Josephs et al., 2016). Cases with other pathologies (e.g., cerebral amyloid angiopathy, dementia with Lewy bodies, hippocampal sclerosis, Parkinson’s disease, and large strokes) were excluded from the study. None of the subjects examined were treated with anticholinesterase inhibitors.
Brain samples
PreC tissue was dissected using fiduciary landmarks and fixed at autopsy in 4% paraformaldehyde (pH 7.4) for at least 5 days, cryoprotected in a solution containing 30% glycerol, 30% ethylene glycol, in 0.1 M phosphate buffer, cut in parallel series at 40 μm on a freezing sliding microtome and stored in the same cryoprotectant at −20 °C until processing (Perez et al., 2012; Mufson et al., 1997). Cortical slabs from the opposite hemisphere containing the PreC were snap frozen and kept at −80 ° C.
Immunohistochemistry
Floating PreC sections were washed in phosphate buffer and Tris-buffered saline (TBS) before a 20 min incubation in 0.1 M sodium metaperiodate (Sigma-Aldrich, St. Louis, MO) in TBS to inactivate endogenous peroxidase activity. Tissue sections were blocked in TBS containing 0.25% Triton-X 100 and 3% goat serum for 1 h. Sections were incubated with a mouse monoclonal to human CatD primary antibody generated from recombinant human Cathepsin D (21-412 aa) and purified using protein G (Lifespan Biosciences, Seattle, WA, Cat# LS-B2931 RRID:AB_1931140; 1:200 dilution). Western blotting detected a single band for CatD and did not cross react with other protein targets (Lifespan Biosciences). Sections were incubated overnight at room temperature in 0.25% Triton X-100, 1% goat serum TBS solution in a humidified chamber. Sections were washed in TBS containing 1% goat serum prior to a 1 h incubation with the biotinylated goat anti-mouse secondary antibody (1:200 dilution; Vector Laboratories Cat# PK-7100, RRID:AB_2336827). Following TBS washes, sections were incubated using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) for 1 h, rinsed in 0.2 M sodium acetate, 1.0 M imidazole buffer (pH 7.4) and developed in acetate-imidazole buffer containing 0.05% 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich) and 1 gram nickel ammonium sulfate. The reaction was terminated in acetate-imidazole buffer. Sections were mounted on glass slides, dried overnight and dehydrated in 100% ethanol. Immunohistochemical controls consisted of the elimination of the primary antibody resulting in a lack of immunoreactivity. To avoid differences in staining across cases, we immunostained and developed all the cases at the same time, which controls for batch to batch differences in antibodies and reagents. CatD positive neurons appeared dark blue-black in all cases examined (Fig. 1a–c).
Fig. 1.
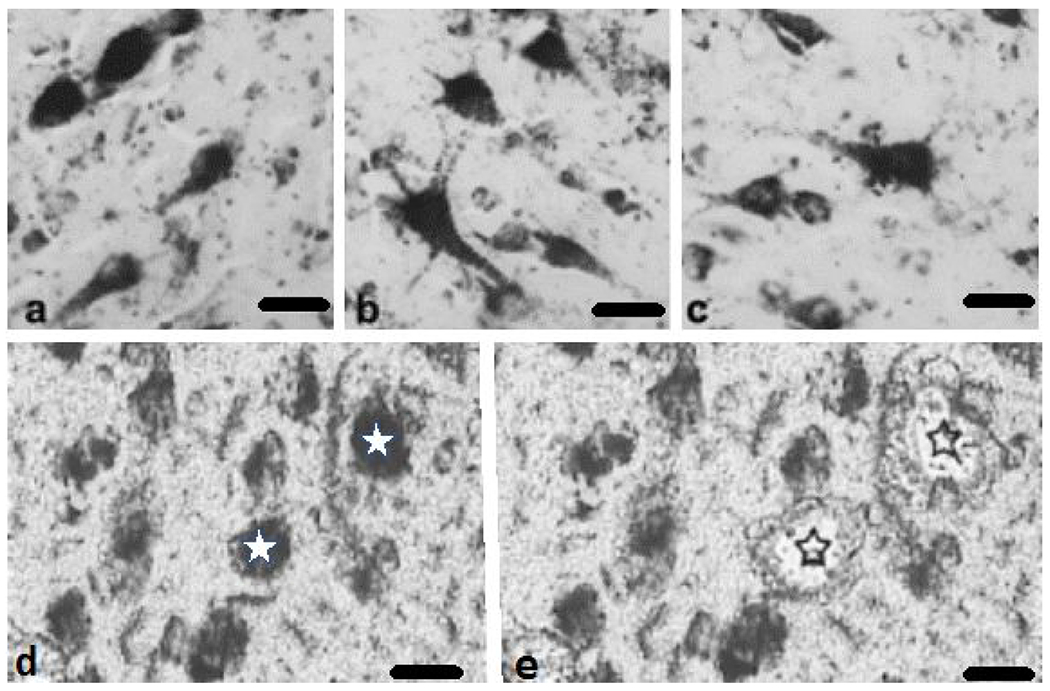
Photomicrographs showing PreC CatD-positive layer III neurons in NCI (a), MCI (b), and AD (c). CatD immunolabeled neurons pre (d, white stars) and post (e, black outlines stars) microdissection from a non-cognitively impaired 84-year old female. Distorted appearance of the images (d and e) is due to the fact that the pictures were taken from uncover slipped sections with the aid of a Zeiss PALM III LCM.. Scale bars = 25 μm
Laser capture microdissection and Terminal Continuation (TC) RNA amplification
The procedure for laser capture microdissection (LCM) and TC RNA amplification combined with custom-designed microarray technology has been described previously (Che & Ginsberg, 2004; Alldred et al., 2008, 2009; Ginsberg, 2008). Briefly individual PreC CatD-immunopositive layer III pyramidal neurons were microdissected (Fig. 1d–e) using a Zeiss PALM LCM III instrument (Zeiss, Oberkochen, Germany). Thirty PreC CatD-positive layer III neurons were captured per reaction for population cell analysis (Alldred et al., 2008; Ginsberg & Che, 2005). A total of two reactions were performed per human brain. TC RNA amplification protocol is available at http://cdr.rfmh.org/pages/ginsberglabpage.html. Total RNA from captured cells was extracted using Trizol reagent (ThermoFisher Scientific, Waltham, MA) according to manufacturer’s instruction. RNAs were first reverse transcribed into cDNA. After RNase H digestion and re-annealing of T7-TC primer to generate cDNAs with double-stranded regions at the primer interfaces, single stranded cDNA was purified using Ambion Ultra 0.5 mL centrifugal filters (UFC 503024, EMD Millipore, Billerica, MA). Hybridization probes were synthesized by in vitro transcription using 33P incorporation in 40 mM Tris (pH 7.5), 6 mM MgCl2, 10 mM NaCl, 2 mM spermidine, 10 mM DTT, 125uM ATP, GTP and CTP, 2.5 μM of cold UTP, 20 U of RNase inhibitor, 2 KU of T7 RNA polymerase (TH950K, Lucigen, Sommerset, WI), and 60 μCi of 33P -UTP (Perkin-Elmer, Boston, MA) (Alldred et al., 2008; Ginsberg, 2008). The reaction was performed at 37 °C for 4 h. Radiolabeled TC RNA probes were hybridized to custom-designed cDNA arrays without further purification.
Custom-designed cDNA array platforms and hybridization
Array platforms consisted of 1 μg of linearized cDNA purified from plasmid preparations adhered to high-density nitrocellulose membrane (Hybond XL, GE Healthcare, Piscataway, NJ). Each cDNA and/or expressed sequence-tagged (EST) cDNA was verified by sequence analysis and restriction digestion. Human and some mouse clones were employed on the custom-designed array. Approximately 576 cDNAs/ESTs were utilized on the current array platform. Although the majority of genes are represented by one transcript on the array platform, several genes have representation at 3’ and 5’ regions. Following overnight hybridization at 42 °C, arrays were washed sequentially in 2X SSC/0.1% SDS, 1X SSC/0.1% SDS and 0.5X SSC/0.1% SDS for 15 min each at 37 °C and exposed to a phosphor screen for 24 hours and developed using a phosphor imager (GE Healthcare, Chicago, IL). All array phosphor images were adjusted to the same brightness and contrast levels for data acquisition and analysis.
Quantitative polymerase chain reaction (qPCR)
RNA was extracted from 75 mg of frozen dissected PreC tissue from NCI (n=9), MCI (n=9) and AD (n=8) cases. Total RNA was purified on a column with DNase I treatment using a PureLink RNA Mini Kit (12183018A, ThermoFisher Scientific) according to manufacturer’s instructions. Total RNA was quantitated using Infinite M200 PRO (Tecan, Männedorf, Switzerland). qPCR was carried out in triplicate on a real-time qPCR cycler Applied Biosystem QuantStudio 6 Flex (ThermoFisher Scientific) in 96 well optical plates with coverfilm using TaqMan Universal Master Mix II with no UNG (ThermoFisher Scientific) and TaqMan gene expression assays. Genes analyzed via qPCR and the respective specific primers are as follows: galanin receptor 1 (GALR1; Hs00175668_m1), BRCA1 associated protein-1 (BAP1; Hs01109276_g1), RNA binding motif protein 3 (RBM3; Hs00943160_g1), RAB11A (Hs00366449_g1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Hs02758991_g1), HOMER1 (Hs01029333_m1), G-protein gamma 2 (GNG2; Hs00828232_m1), metabotropic glutamate receptor 1 (GRM1; Hs00168250_m1), glucose transporter 5 (SLC2a5; Hs01086390_m1), tissue inhibitor of metalloproteinase 1 (TIMP1; Hs01092512_g1), ribosomal protein S6 kinase, 70kDa, polypeptide 1 (RPS6KB1; Hs00356367_m1) and choline acetyltransferase (CHAT; Hs00758143_m1 ) (Table 2) (ThermoFisher Scientific). qPCR conditions were as follows: one cycle at 95° C for 10 min and 40 cycles of 95 °C for 15 sec and 60 °C for 1 min.
Table 2.
qPCR TaqMan probes
| Gene | Assay ID |
|---|---|
| GALR1 | Hs00175668_m1 |
| BAP1 | Hs01109276_g1 |
| RBM3 | 00943160_g1 |
| RAB11A | Hs00366449_g1 |
| HOMER1 | 01029333_m1 |
| GNG2 | Hs00828232_m1 |
| GRM1 | Hs00168250_m1 |
| SLC2A5 | 01086390_m1 |
| TIMP1 | Hs01092512_g1 |
| RPS6KB1 | Hs00356367_m1 |
| GAPDH | Hs02758991_g1 |
| CHAT | Hs00758143_m1 |
Statistical analysis
Clinical and demographic characteristics (e.g., age, educational level, GCS, MMSE, and PMI) were compared using a one-way analysis of variance (ANOVA) with Bonferroni-type correction for pairwise comparisons (Ginsberg et al., 2006a, 2006b; Counts et al., 2007). Gender and ApoE allele status were assessed across clinical conditions using a Fisher’s exact test. Neuropathologic classifications (NIA-Reagan, CERAD, and Braak scoring) were compared among clinical diagnostic groups by the Kruskal-Wallis test (Ginsberg et al., 2006a, 2006b). Hybridization signal intensity was determined utilizing ImageQuant software (GE Healthcare, Chicago, IL). Statistical procedures for custom-designed microarray analysis have been described in detail (Ginsberg et al., 2006a; Ginsberg, 2008). Briefly, expression of TC amplified RNA bound to each linearized cDNA (approximately 576 cDNAs/ESTs on the array) minus background was expressed as a ratio of the total hybridization signal intensity of the array (a global normalization approach). Global normalization effectively minimizes variation due to differences in the specific activity of the synthesized probe and the absolute quantity of probe (Ginsberg, 2008; Eberwine & Crino, 2001). These data do not allow the absolute quantitation of mRNA levels. However, an expression profile of relative changes in mRNA levels was generated. Relative changes in total hybridization signal intensity and percentage of cDNA clones above negative control were analyzed as described previously (Ginsberg et al., 2006a; Counts et al., 2007; SAS Institute Inc, 2004). Since multiple cells were measured in each subject, between-subject versus within-subject (between-cell) variation in gene expression was analyzed by variance component analysis and intraclass correlation coefficients (Ginsberg et al., 2006b). False discovery rate (FDR) was used to correct for multiple comparisons among the correlations. The FDR was applied to each domain separately so that the significance level adjustment was based on 23 comparisons. Real time PCR used GAPDH as a reference gene. Relative mRNA expression levels of target genes were calculated via 2−ΔΔCT method (Schmittgen & Livak, 2008). ANOVA on Ranks was used to analyze the significant difference of gene expression of qPCR. For microarray analysis, level of statistical significance was set at p≤0.01, p values between 0.01 and 0.05 were considered as a trend level (t) (Ginsberg et al., 2010, 2019; Alldred et al., 2018). For other analyses, the level of statistical significance was set at p < 0.05.
Results
Case demographics
Clinical groups did not differ by age (p=0.59), sex (p=0.74), education level (p=0.57), postmortem interval (PMI, p=0.27), or brain weight (p=0.83) (Table 1). There were significantly more cases with an ApoE ε4 genotype in MCI (45 %) and AD (28.6%) (p=0.04) compared to NCI (0%). MMSE (p<0.001), GCS (p<0.001), and episodic memory z-score (p<0.001) were significantly lower in AD compared to MCI and NCI. No significant differences were found for the Semantic Memory z-score (p=0.46), Working Memory z-score (p=0.25), and Visuospatial z-score (p=0.08) in AD compared to MCI and NCI. Significant differences for these cognitive variables were not found between NCI and MCI (Table 1). Neuropathological examination revealed that 89% of AD, 82% of MCI and 82% of NCI cases were Braak stage III-V. NIA-Reagan criteria revealed that 45% of NCI, 73% of MCI, and 89% of AD cases were classified as intermediate to high likelihood of AD. CERAD diagnosis revealed that 27% of NCI, 27% of MCI, and 33% of AD cases received a diagnosis of probable or definite AD. Among the three clinical diagnostic groups no differences were found in Braak scores (p=0.76), NIA-Reagan (p=0.17) or CERAD diagnosis (p=0.1) (Table 1). We divided the 11 NCI cases into those with low Braak scores (I-III; n=5) and high Braak score (IV-V; n=6) and found that only 3 genes (0.52%) {USP8 (p=0.01), AGER (p=0.02t) and PPP2CA (p=0.04t)} were significantly different (n=1) or trend-level different (n=2) between these NCI cases, suggesting that Braak score did not influence results in the NCI group. Interestingly, only 10 of 576 genes (1.73%) {EIF2AK2 (p=0.001), GNG2 (p=0.004), ATG4D (p=0.006), RPS6KB1 (p=0.01), MSR1 (p=0.02t), GABRA6 (p=0.03t), RAP1B (p=0.03t), MAPT2N6D (p=0.05t), TUBB2C (p=0.05t), and MAPT1N6P (p=0.05t)} were significant (n=4) or trend-level different (n=6) between non-amnestic and amnestic MCI subjects. Data for TDP 43 was available for a subset of the cases (n=23) examined. Of these cases TDP-43 accumulation was found in 4 NCI (stage 1, amygdala), 2 MCI (stage 1, amygdala) and 4 AD (stage 2-3, amygdala, entorhinal cortex, and subiculum) cases according to the recent schema for TDP 43 pathology (Josephs et al., 2016). Quantitative analysis did not reveal differences in average intensity levels of CatD immunostaining among NCI (71.1 ± 12.2), MCI (72.9 ± 12.7) and AD (68.3 ± 10.9) (p=0.69) cases.
Differential expression in PreC CatD-positive layer III neurons between MCI and NCI
Custom-designed microarray analysis revealed upregulation of 10 genes and downregulation of 9 genes out of the 576 transcripts belonging to 11 different gene oncology categories (GOCs) in PreC CatD-positive layer III neurons in MCI compared to NCI (Table 3 and Fig. 2a). Comparing MCI and NCI, we found transcript upregulation for truncated tau 6D isoform with E2 and E3 (MAPT2N6D; p=0.003), truncated tau 6P isoform with only E2 (MAPT1N6P; p=0.01), B-cell CLL/lymphoma 2 (BCL2; p=0.01; Fig. 3a), dynein light chain LC8-type 1 (DNCLC1; p=0.01), RAB4A, (p=0.003; Fig. 4a), beclin 1, (BECN1; p=0.03t; Fig. 4a), glutamate receptor NMDA R2C (GRIN2C; p=0.003), dipeptidyl-peptidase 6 (DPP6; p=0.02t), dipeptidyl-peptidase 10 (DPP10; p=0.004) and aminopeptidase puromycin sensitive (PSA; p=0.008). A comparison between MCI and NCI revealed transcript downregulation for islet amyloid polypeptide (IAPP; p=0.005), nestin (NES; p=0.005), neurofilament heavy polypeptide (NEFH; p=0.008), EPH receptor A2, epithelial cell protein tyrosine kinase (EPHA2; p=0.04t), glutamate receptor ionotropic kainate 5 (GRIK5; p=0.03t), cocaine- and amphetamine-regulated transcript (CART; p=0.04t), gamma-aminobutyric acid (GABA) A receptor, alpha 1 (GABRA1; p=0.01), calcium channel, voltage-dependent, N type, alpha 1B subunit (CACNA1B; p=0.01), and macrophage scavenger receptor 1 (MSR1; p=0.04t).
Table 3.
Differentially expressed transcripts in PreC CatD-positive layer III neurons in MCI compared to NCI
| Gene | Gene Ontology Category Group | Gene Full Name | p | FDR | MCI |
|---|---|---|---|---|---|
| MAPT2N6D | AD | truncated tau 6D isoform with E2 and E3 | 0.003 | 0.305 | ↑ |
| IAPP | AD | islet amyloid polypeptide alias amylin | 0.005 | 0.305 | ↓ |
| MAPT1N6P | AD | truncated tau 6P isoform with only E2 | 0.01 | 0.452 | ↑ |
| BCL2 | CD | B-cell CLL/lymphoma 2 | 0.01 | 0.452 | ↑ |
| NES | CYT | nestin | 0.005 | 0.305 | ↓ |
| NEFH | CYT | neurofilament, heavy polypeptide | 0.008 | 0.452 | ↓ |
| DNCLC1 | CYT | dynein light chain LC8-type 1 | 0.01 | 0.452 | ↑ |
| RAB4A | EALS | RAB4A, member RAS oncogene family | 0.003 | 0.305 | ↑ |
| BECN1 | EALS | beclin 1, autophagy related | 0.03t | 0.468 | ↑ |
| EPHA2 | PP/K | EPH receptor A2; epithelial cell protein tyrosine kinase | 0.04t | 0.594 | ↓ |
| GR1K5 | GLUR | glutamate receptor ionotropic kainate 5 | 0.03t | 0.468 | ↓ |
| GR1N2C | GLUR | glutamate receptor, ionotropic NMDA 2C | 0.003 | 0.305 | ↑ |
| CART | MONO | cocaine- and amphetamine-regulated transcript | 0.04t | 0.594 | ↓ |
| GABRA1 | GABA | gamma-aminobutyric acid (GABA) A receptor, alpha 1 | 0.01 | 0.452 | ↓ |
| CACNA1B | CH | calcium channel, voltage-dependent, N type, alpha 1B subunit | 0.01 | 0.452 | ↓ |
| DPP6 | CH | dipeptidyl-peptidase 6 | 0.02t | 0.468 | ↑ |
| DPP10 | CH | dipeptidyl-peptidase 10 | 0.004 | 0.305 | ↑ |
| MSR1 | GLIA | macrophage scavenger receptor 1 | 0.04t | 0.594 | ↓ |
| PSA | PEP | aminopeptidase puromycin sensitive | 0.008 | 0.452 | ↑ |
: trend level change.
Fig. 2.
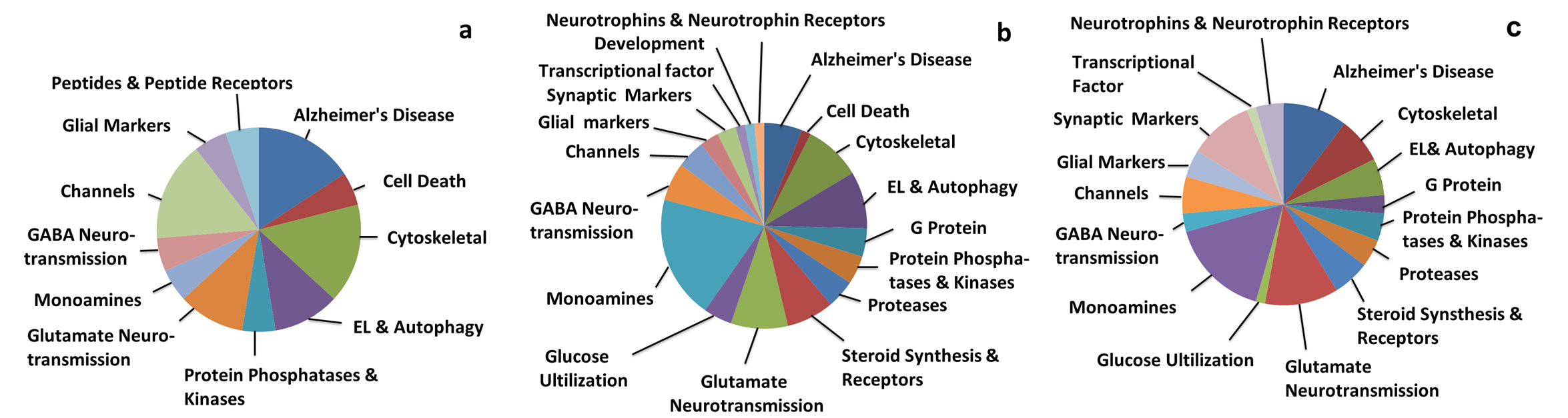
Pie charts showing gene ontology categories (GOCs) of differentially expressed genes in PreC CatD-positive layer III neurons across clinical groups, a: GOCs of differentially expressed genes in PreC CatD-positive layer III neurons of MCI compared to NCI; b: GOCs of differentially expressed genes in PreC CatD-positive layer III neurons in AD compared to NCI; c: GOCs of differentially expressed genes in PreC CatD-positive layer III neurons in AD compared to MCI.
Fig. 3.
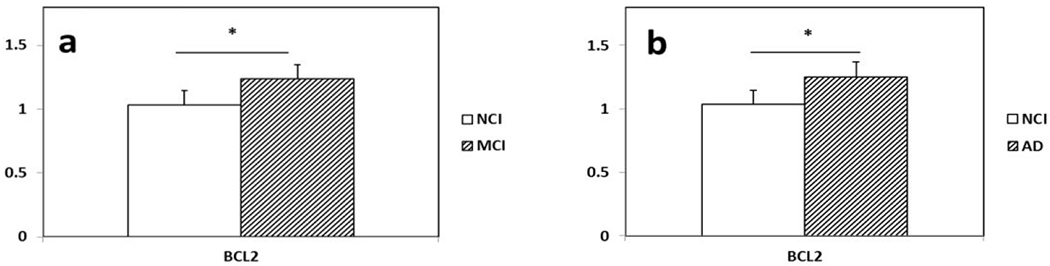
Histograms showing upregulation of BCL2 in PreC CatD-positive layer III neurons in MCI (a) and AD (b) compared to NCI.
Fig. 4.
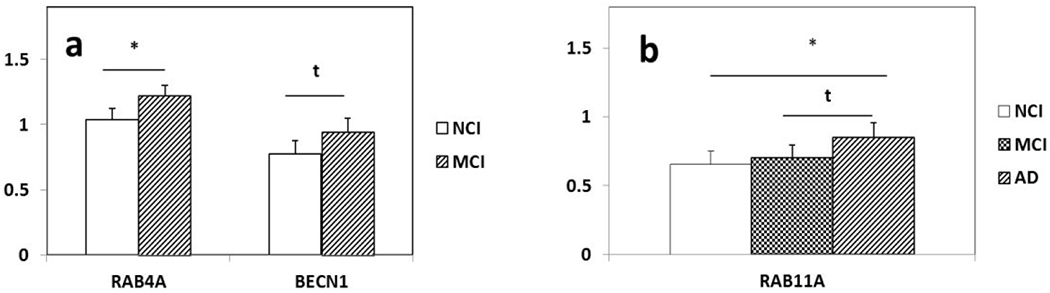
Differentially expressed endosomal lysosomal (EL) and autophagy transcripts in PreC CatD-positive layer III neurons in MCI and AD. a: Significant upregulation of RAB4A (p=0.003) and upregulation trend of BECN1 (p=0.03t) in MCI compared to NCI. b: Upregulation of RAB11A in AD compared to NCI (p=0.009) and upregulation trend of RAB11a (p=0.04t) in AD compared to MCI. Key: t-trend; 0.01<P<0.05; *: P⩽0.01.
Differential expression in PreC CatD positive layer III neurons in AD compared to NCI
Custom-designed microarray analysis revealed upregulation of 49 genes and downregulation of 18 genes in PreC CatD-positive layer III neurons in AD compared to NCI (Table 4) that belonged to 18 different GOCs (Table 4, Fig 2b). Comparison between AD and NCI revealed upregulation of BCL2 (p=0.01; Fig 3b), 3 EL and autophagy related genes {RAB5A(p=0.01), RAB11A (p=0.009;Fig. 4b) and cathepsin C (CTSC; p=0.05t}, upregulation of monoamine neurotransmission genes {choline acetyltransferase (CHAT; p=0.04t; Fig. 5), butyrylcholinesterase (BCHE; p=0.02t; Fig. 5), nicotinic cholinergic receptor alpha 2 (CHRNA2; p=0.03t; Fig. 5), alpha 4 (CHRNA4; p=0.02t; Fig 5), and alpha 5 (CHRNA5; p=0.02t), serotonin transporter (SERT) solute carrier family 6 member 4 (SLC6A4; p=0.003), noradrenaline transporter (NET) solute carrier family 6 member 2 (SLC6A2; p=0.01), dopamine receptor D2 (DRD2; p=0.02t) and D4 (DRD4; p=0.04t)} and 5 glutamate receptor subunits (kainate 3 (GRIK3; p=0.003), NMDAR 2B (GRIN2B; p=0.03t), metabotropic glutamate receptor subunits GBM1 (p=0.004; Fig. 6), metabotropic 2 (GRM2; p=0.02t; Fig. 6), and metabotropic 5 (GRM5; p=0.009; Fig. 6)}. A comparison between AD and NCI revealed upregulation of 6 cytoskeletal elements including DNCLC1 (p=0.002), neurofilament light polypeptide (NEFL; p=0.0002; Fig. 7), neurofilament medium polypeptide (NEFM; p=0.002; Fig. 7), β-actin (ACTB; p=0.002; Fig. 7), microtubule-associated protein 2 (MAP2; p=0.008; Fig. 7), and clathrin heavy polypeptide (CLTC; p=0.04t), 4 AD-related transcripts {amyloid A4 (SAA4; p=0.01), nicastrin (NCSTN; p=0.01), high density lipoprotein binding protein (HDLBP; p=0.02t), serum amyloid P component (APCS; p=0.03t)}, and select steroid-related genes {hydroxysteroid (17-beta) dehydrogenase 1(HSD17B1; p=0.0004; Fig. 8), nuclear receptor subfamily 5 group A member 1 (NR5A1 also known as SF1; p=0.002; Fig. 8), and nuclear receptor subfamily 4 group A member 2 (NR4A2 also known as NURR1; p=0.04t)}.
Table 4.
Differentially expressed transcripts in PreC CatD-positive layer III neurons in AD compared to NCI
| Gene | Gene Oncology Category Group | Gene Full name | p | FDR | AD |
|---|---|---|---|---|---|
| SAA4 | AD | serum amyloid A4 | 0.01 | 0.211 | ↑ |
| NCSTN | AD | nicastrin | 0.01 | 0.211 | ↑ |
| HDLBP | AD | high density lipoprotein binding protein | 0.02t | 0.351 | ↑ |
| APCS | AD | amyloid P component, serum | 0.03t | 0.351 | ↑ |
| BCL2 | CD | B-cell CLL/lymphoma 2 | 0.01 | 0.211 | ↑ |
| NEFL | CYT | neurofilament, light polypeptide | 0.0002 | 0.035 | ↑ |
| NEFM | CYT | neurofilament, medium polypeptide | 0.002 | 0.073 | ↑ |
| ACTB | CYT | β-actin | 0.002 | 0.073 | ↑ |
| DNCLC1 | CYT | dynein light chain LC8-type 1 | 0.002 | 0.096 | ↑ |
| MAP2 | CYT | microtubule-associated protein 2 | 0.008 | 0.211 | ↑ |
| CLTC | CYT | clathrin, heavy polypeptide (He) | 0.04t | 0.357 | ↑ |
| RAB11A | EALS | RAB11 A, member RAS oncogene family | 0.009 | 0.211 | ↑ |
| LAMP1 | EALS | lysosomal-associated membrane protein 1 | 0.008 | 0.211 | ↓ |
| ATG10 | EALS | autophagy related 10 | 0.002 | 0.073 | ↓ |
| CTSC | EALS | cathepsin C | 0.046t | 0.357 | ↑ |
| RAB5A | EALS | RAB5A, member RAS oncogene family | 0.01 | 0.211 | ↑ |
| PPT1 | EALS | palmitoyl-protein thioesterase 1 | 0.02t | 0.351 | ↓ |
| GNG2 | GP | guanine nucleotide binding protein (G protein), gamma 2 | 0.0008 | 0.073 | ↓ |
| RGS9 | GP | regulator of G-protein signaling 9 | 0.04t | 0.357 | ↑ |
| RGS13 | GP | regulator of G-protein signaling 13 | 0.04t | 0.357 | ↑ |
| PPP3CC | PP/K | protein phosphatase 3, catalytic subunit, gamma isoform | 0.0003 | 0.052 | ↑ |
| RPS6KB1 | PP/K | ribosomal protein S6 kinase, 70kDa, polypeptide 1 | 0.01 | 0.211 | ↓ |
| PRKCE | PP/K | protein kinase C, epsilon | 0.02t | 0.211 | ↑ |
| TIMP1 | PROT | tissue inhibitor of metalloproteinase 1 | 0.009 | 0.211 | ↑ |
| GLRX | PROT | glutaredoxin (thioltransferase) | 0.04t | 0.351 | ↑ |
| CAST | PROT | calpastatin | 0.04t | 0.357 | ↑ |
| HSD17B1 | ST | hydroxysteroid (17-beta) dehydrogenase 1 | 0.0004 | 0.073 | ↑ |
| NR5A1/SF1 | ST | nuclear receptor subfamily 5, group A, member 1 | 0.002 | 0.073 | ↑ |
| NCOR2 | ST | nuclear receptor corepressor 2 | 0.01 | 0.211 | ↓ |
| NR4A2 | ST | nuclear receptor subfamily 4, group A, member 2 alias NURR1 | 0.04t | 0.357 | ↑ |
| NR3C1 | ST | nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | 0.046t | 0.357 | ↓ |
| GRIK3 | GLUR | glutamate receptor ionotropic kainate 3 | 0.003 | 0.096 | ↑ |
| GRM1 | GLUR | glutamate receptor, metabotropic 1 | 0.004 | 0.211 | ↑ |
| GRM2 | GLUR | glutamate receptor, metabotropic 2 | 0.02t | 0.211 | ↑ |
| GRM5 | GLUR | glutamate receptor, metabotropic 5 | 0.009 | 0.211 | ↑ |
| GRIN2B | GLUR | glutamate receptor, ionotropic NMDA 2B | 0.03t | 0.351 | ↑ |
| GRIA1 | GLUR | glutamate receptor ionotropic AMPA 1 | 0.045t | 0.357 | ↓ |
| SLC2A4 | GLUC | solute carrier family 2 (glucose transporter), member 4 | 0.02t | 0.211 | ↑ |
| SLC2A5 | GLUC | solute carrier family 2 (glucose transporter), member 5 | 0.002 | 0.073 | ↓ |
| SLC2A3 | GLUC | solute carrier family 2 (glucose transporter), member 3 | 0.001 | 0.073 | ↑ |
| SLC6A4/SERT | MONO | solute carrier family 6 (neurotransmitter transporter, serotonin), member 4, | 0.003 | 0.211 | ↑ |
| SLC6A2/NET | MONO | solute carrier family 6 (neurotransmitter transporter, noradrenalin), member 2 | 0.01 | 0.211 | ↑ |
| CHRNB4 | MONO | cholinergic receptor, nicotinic, beta 4 | 0.03t | 0.351 | ↓ |
| DRD3 | MONO | dopamine receptor D3 | 0.03t | 0.351 | ↓ |
| CART | MONO | cocaine- and amphetamine-regulated transcript | 0.03t | 0.351 | ↓ |
| DRD4 | MONO | dopamine receptor D4 | 0.04t | 0.351 | ↑ |
| ADRA2A | MONO | adrenergic receptor, alpha 2a | 0.04t | 0.351 | ↓ |
| CHAT | MONO | choline acetyltransferase | 0.04t | 0.351 | ↑ |
| CHRNA2 | MONO | cholinergic receptor, nicotinic, alpha2 | 0.03t | 0.351 | ↑ |
| BCHE | MONO | butyrylcholinesterase | 0.02t | 0.211 | ↑ |
| CHRNA5 | MONO | cholinergic receptor, nicotinic, alpha5 | 0.02t | 0.211 | ↑ |
| DRD2 | MONO | dopamine receptor D2 | 0.02t | 0.211 | ↑ |
| CHRNA4 | MONO | cholinergic receptor, nicotinic, alpha4 | 0.02t | 0.211 | ↑ |
| GABRA1 | GABA | gamma-aminobutyric acid (GABA) A receptor, alpha 1 | 0.02t | 0.211 | ↓ |
| GABRG3 | GABA | gamma-aminobutyric acid (GABA) A receptor, gamma 3 | 0.03t | 0.351 | ↑ |
| GABRA4 | GABA | gamma-aminobutyric acid (GABA) A receptor, alpha 4 | 0.006 | 0.211 | ↑ |
| GABBR1 | GABA | gamma-aminobutyric acid (GABA) B receptor, 1 | 0.02t | 0.211 | ↑ |
| KCNIP1 | CH | Kv channel interacting protein 1 | 0.03t | 0.351 | ↓ |
| KCNA2 | CH | potassium voltage-gated channel, shaker-related subfamily, member 2 | 0.02t | 0.211 | ↓ |
| CACNA1B | CH | calcium channel, voltage-dependent, N type, alpha 1B subunit | 0.005 | 0.211 | ↓ |
| MSR1 | GLIA | macrophage scavenger receptor 1 | 3.40E-06 | 0.035 | ↓ |
| CSF1 | GLIA | colony stimulating factor 1 (macrophage) | 0.02t | 0.351 | ↑ |
| HOMER1 | SYN | homer homolog 1 | 0.03t | 0.351 | ↑ |
| SNCB | SYN | synuclein, beta | 0.049t | 0.357 | ↑ |
| ERCC1 | TF | excision repair cross-complementing rodent repair deficiency, complementation group 1 | 0.0002 | 0.035 | ↑ |
| DAB1 | DV | disabled 1 | 0.04t | 0.351 | ↑ |
| IGF2R | NT | insulin-like growth factor 2 receptor | 0.04t | 0.357 | ↑ |
: trend level change
Fig. 5.
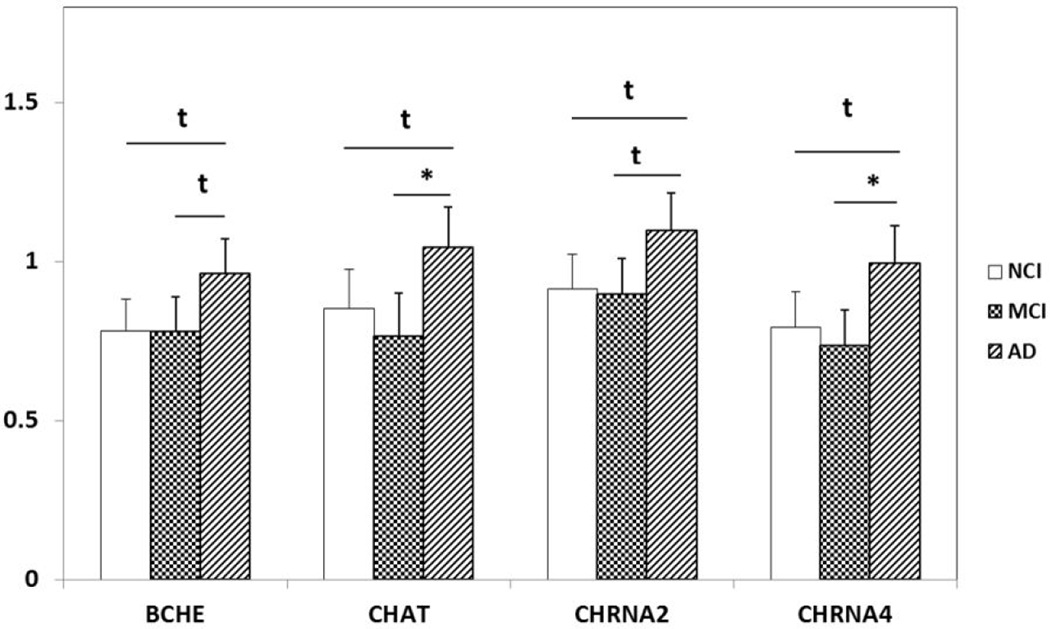
Differential expression of cholinergic enzymes (BCHE and CHAT) and nicotinic acetylcholine receptors (CHRNA2 and CHRNA4) in PreC CatD-positive layer III neurons in AD compared to NCI and MCI. Upregulation trend of BCHE (AD>NCI, p=0.2t; AD>MCI, p=0.02t) and CHRNA2 (AD>NCI, p=0.03t; AD>MCI, p=0.02t) in AD compared to NCI and MCI. Upregulation trend of CHAT (AD>NCI, p=0.04t) and CHRNA4 (AD>NCI, p=0.02t) in AD compared to NCI and significant upregulation of CHAT(AD>MCI, p=0.005) and CHRNA4 (AD>MCI, p=0.003) in AD compared to MCI. Key: t-trend: 0.01<p<0.05; *: p⩽0.01.
Fig. 6.
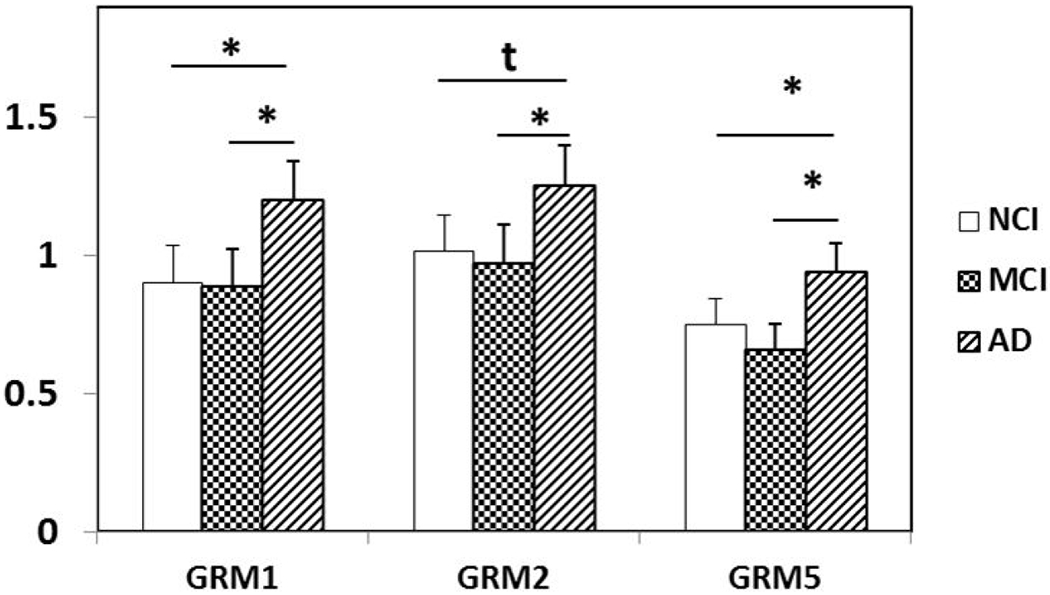
Differential expression of GRM1, GRM2, and GRM5 in PreC CatD-positive layer III neurons in AD compared to NCI and MCI. GRM1 (AD>NCI, p=0.004; AD>MCI, p=0.003) and GRM5 (AD>NCI, p=0.009; AD>MCI, p=0.01) are upregulated in AD compared to NCI and MCI. Upregulation trend of GRM2 (AD>NCI, p=0.02t) in AD compared to NCI and significant upregulation of GRM2 (AD>MCI, p=0.007) in AD compared to MCI. Key: t-trend: 0.01<p<0.05; *: p⩽0.01.
Fig. 7.
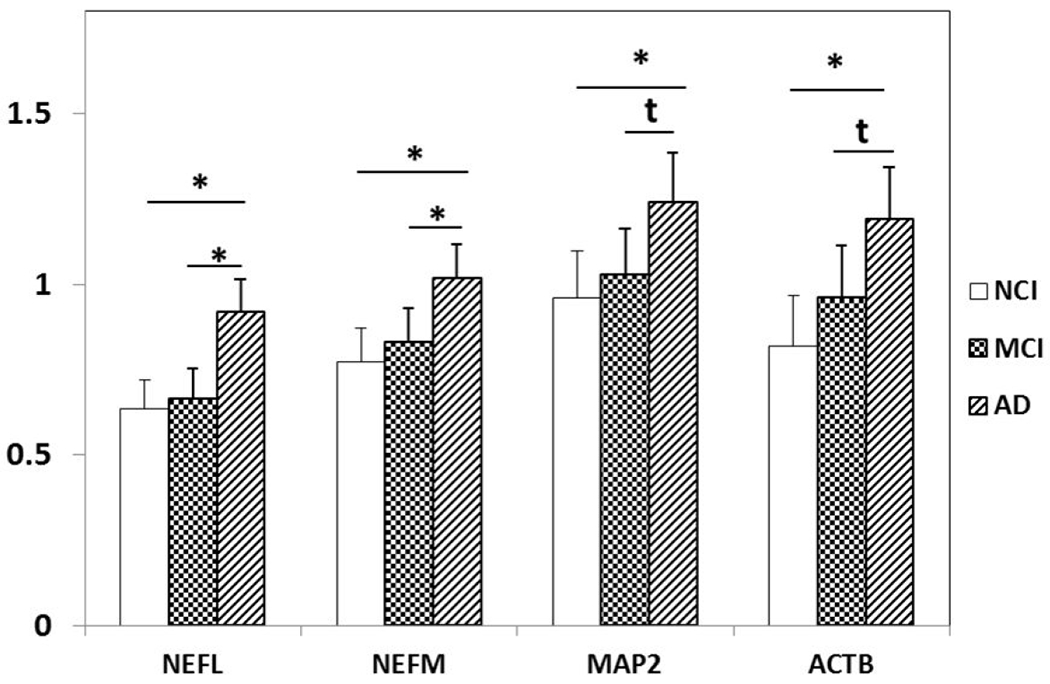
Upregulation of cytoskeletal elements NEFL NEFM, MAP2 and ACTB in PreC CatD-positive layer III neurons in AD compared to NCI and MCI. NEFL (p=0.0002), NEFM (p=0.002), MAP2 (p=0.008) and ACTB (p=0.002) are significantly upregulated in AD compared to NCI. Significant upregulation of NEFL (p=0.0008) and NEFM (p=0.01) and upregulation trend for MAP2 (p=0.04t) and ACTB (p=0.04t) in AD compared to MCI. Key: t-trend: 0.01<p<0.05, *: p≤0.01.
Fig 8.
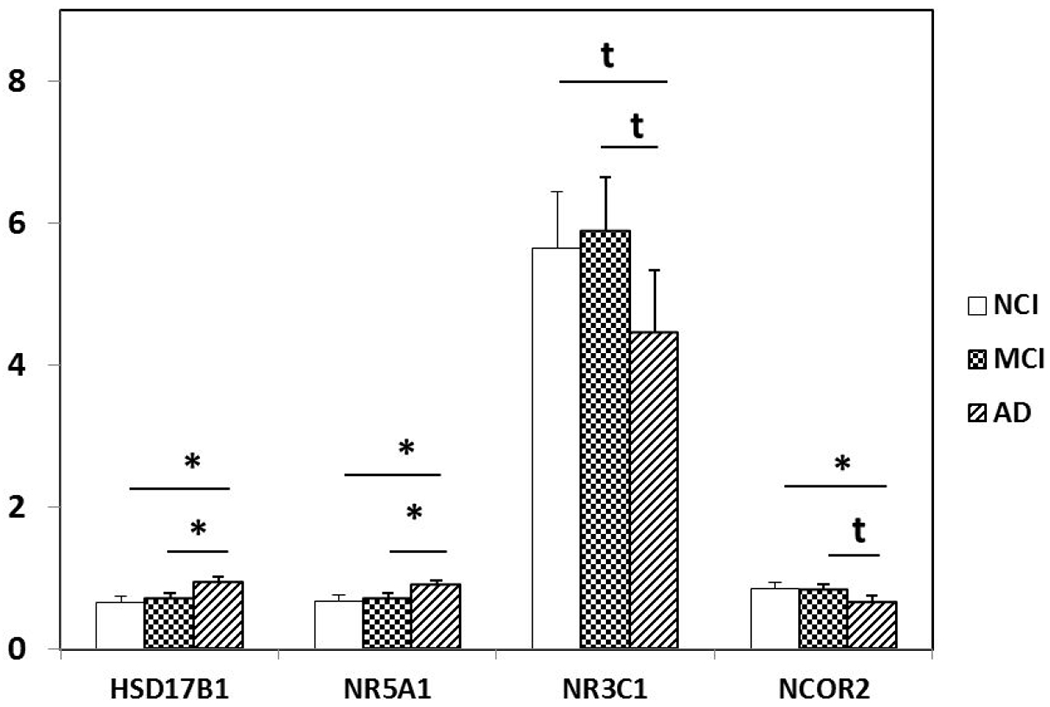
Differentially expressed HSD17B1, NR5A1, NR3C1 and NCOR2 in PreC CatD-positive layer III neurons in AD compared to NCI and MCI. HSD17B1 (AD>NCI, p=0.0004; AD>MCI, p=0.002) and NR5A1 (AD>NCI, p=0.002; AD>MCI, p=0.006) are significantly upregulated in PreC CatD-positive layer III neurons in AD compared to NCI and MCI. Downregulation trend for NR3C1 (AD<NCI, p=0.046t; AD<MCI, p=0.02t) in AD compared to NCI and MCI. Significant downregulation of NCOR2 (AD<NCI, p=0.01) in AD compared to NCI and downregulation trend of NCOR2 (AD<MCI, p=0.02t) in AD compared to MCI. Key: t-trend: 0.01<p<0.05, *: p≤0.01.
Further analysis of AD and NCI demonstrated downregulation of channel-related transcripts {potassium voltage-gated channel (Kv) shaker-related subfamily member 2 (KCNA2; p=0.02t), Kv channel interacting protein 1 (KCNIP1; p=0.03t), CACNA1B (p=0.005)}, EL and autophagy related genes {lysosomal-associated membrane protein 1 (LAMP1; p=0.008), autophagy related 10 (ATG10; p=0.002) and palmitoyl-protein thioesterase 1 (PPT1; p=0.02t)}, steroid receptor transcripts nuclear receptor corepressor 2 (NCOR2) (p=0.01; Fig. 8) and glucocorticoid receptor nuclear receptor subfamily 3 group C member 1 (NR3C1; p=0.05t; Fig. 8) as well as monoamine neurotransmission genes {nicotinic beta cholinergic receptor 4 (CHRNB4; p=0.03t), dopamine receptor D3 (DRD3; p=0.03t), adrenergic receptor alpha 2A (ADRA2A; p=0.04t), and CART (p=0.03t)}.
Differential expression in PreC CatD-positive layer III neurons in AD compared to MCI
Custom-designed microarray analysis revealed upregulation of 54 genes and downregulation of 14 genes in PreC CatD-positive layer III neurons, which belonged to 16 different GOCs (Table 5, Fig. 2c). Comparing AD and MCI revealed upregulation of 2 EL and autophagy pathway transcripts RAB11A (p=0.04t; Fig 4b) and myosin VB (MYO5B; p=0.006), upregulation of 7 cholinergic neurotransmission transcripts {CHAT (p=0.005; Fig. 5), BCHE (p=0.02t; Fig. 5), muscarinic cholinergic receptors CHRM1 (p=0.03t) and CHRM3 (p=0.01), nicotinic cholinergic receptors CHRNA1 (p=0.05t), CHRNA2 (p=0.02t; Fig. 5), and CHRNA4 (p=0.003; Fig 5)}, 3 dopamine receptors {DRD2 (p=0.002) and DRD4 (p=0.02t), SLC6A4 (p=0.0005)} and serotonin receptor 3A (HTR3A; p=0.03t). This comparison also revealed upregulation of 8 glutamate neurotransmission transcripts {ionotropic glutamate receptors kainate 2 (GRIK2; p=0.006) and NMDAR 2D (GRIN2D; p=0.003) and metabotropic glutamate receptors GRM1 (p=0.003; Fig. 6), GRM2 (p=0.007; Fig. 6), GRM5 (p=0.01; Fig. 6), GRM7 (p=0.03t), and GRM8 (p=0.02t), and glial high-affinity glutamate transporter EAAT1 (solute carrier family 1 member3; SLC1A3; p=0.03t)}, upregulation of 5 cytoskeletal elements {NEFL (p=0.0008; Fig. 7), ACTB (p=0.04t; Fig. 7), NEFM (p=0.01; Fig. 7), alpha tubulin 1 (TUBA1; p=0.05t) and MAP2 (p=0.04t; Fig. 7)}. Comparing AD and MCI further demonstrated upregulation of 7 AD related transcripts {APP (p=0.003), HDLBP (p=0.0008), SAA4 (p=0.004), beta-2-microglobulin (B2M; p=0.01), low density lipoprotein receptor-related protein 1 (LRP1; p=0.02t), NCSTN (p=0.03t) and epiphycan (EPYC) (p=0.04t)}, and 2 steroid-related genes HSD17B1 (p=0.002; Fig. 8) and NR5A1(SF1; p=0.006; Fig. 8).
Table 5.
Differentially expressed transcripts in PreC CatD-positive layer III neurons in AD compared to MCI
| Gene | Gene Oncology Category Group | Gene Full name | p | FDR | AD |
|---|---|---|---|---|---|
| HDLBP | AD | high density lipoprotein binding protein | 0.0008 | 0.122 | ↑ |
| APP | AD | amyloid beta (A4) precursor protein | 0.003 | 0.137 | ↑ |
| SAA4 | AD | serum amyloid A4 | 0.004 | 0.137 | ↑ |
| B2M | AD | beta-2-microglobulin | 0.01 | 0.181 | ↑ |
| LRP1 | AD | low density lipoprotein receptor-related protein 1 alias A2MR | 0.02t | 0.230 | ↑ |
| NCSTN | AD | nicastrin | 0.03t | 0.233 | ↑ |
| EPYC/ | AD | epiphycan | 0.04t | 0.233 | ↑ |
| NEFL | CYT | neurofilament, light polypeptide | 0.0008 | 0.069 | ↑ |
| ACTB | CYT | β-actin | 0.04t | 0.233 | ↑ |
| NEFM | CYT | neurofilament, medium polypeptide | 0.01 | 0.181 | ↑ |
| TUBA1 | CYT | tubulin, alpha 1 | 0.0499t | 0.283 | ↑ |
| MAP2 | CYT | microtubule-associated protein 2 | 0.04t | 0.233 | ↑ |
| MYO5B | EALS | myosin VB | 0.006 | 0.137 | ↑ |
| RAB11A | EALS | RAB11 A, member RAS oncogene family | 0.04t | 0.233 | ↑ |
| BECN1 | EALS | beclin 1, autophagy related | 0.002 | 0.122 | ↓ |
| LAMP1 | EALS | lysosomal-associated membrane protein 1 | 0.005 | 0.137 | ↓ |
| RGS9 | GP | regulator of G-protein signaling 9 | 0.03t | 0.233 | ↑ |
| GNG2 | GP | guanine nucleotide binding protein (G protein), gamma 2 | 0.01 | 0.181 | ↓ |
| PPP3CC | PP/K | protein phosphatase 3, catalytic subunit, gamma isoform | 0.009 | 0.181 | ↑ |
| RPS6KB1 | PP/K | ribosomal protein S6 kinase, 70kDa, polypeptide 1 | 0.01 | 0.181 | ↓ |
| PPP2CA | PP/K | protein phosphatase 2 (formerly 2A), catalytic subunit, alpha isoform | 0.02t | 0.230 | ↓ |
| SOD1 | PROT | superoxide dismutase 1 | 0.02t | 0.181 | ↑ |
| SOD2 | PROT | superoxide dismutase 2 | 0.02t | 0.181 | ↑ |
| TIMP1 | PROT | tissue inhibitor of metalloproteinase 1 | 0.04t | 0.233 | ↑ |
| HSD17B1 | ST | hydroxysteroid (17-beta) dehydrogenase 1 | 0.002 | 0.122 | ↑ |
| NR5A1/SF1 | ST | nuclear receptor subfamily 5, group A, member 1 | 0.006 | 0.137 | ↑ |
| NR3C1 | ST | nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | 0.02t | 0.181 | ↓ |
| NCOR2 | ST | nuclear receptor corepressor 2 | 0.02t | 0.230 | ↓ |
| GRM1 | GLUR | glutamate receptor, metabotropic 1 | 0.003 | 0.122 | ↑ |
| GRM2 | GLUR | glutamate receptor, metabotropic 2 | 0.007 | 0.149 | ↑ |
| GRM5 | GLUR | glutamate receptor, metabotropic 5 | 0.01 | 0.181 | ↑ |
| GRM7 | GLUR | glutamate receptor, metabotropic 7 | 0.03t | 0.233 | ↑ |
| GRM8 | GLUR | glutamate receptor, metabotropic 8 | 0.02t | 0.181 | ↑ |
| GRIK2 | GLUR | glutamate receptor ionotropic kainate 2 | 0.006 | 0.137 | ↑ |
| GRIN2D | GLUR | glutamate receptor, ionotropic NMDA 2D | 0.003 | 0.122 | ↑ |
| SLC1A3/EAAT1 | GLUR | solute carrier family 1 (glial high-affinity glutamate transporter), member 3 | 0.03t | 0.233 | ↑ |
| SLC2A5 | GLUC | solute carrier family 2 (facilitated glucose transporter), member 5 | 0.004 | 0.137 | ↓ |
| BCHE | MONO | butyrylcholinesterase | 0.02t | 0.230 | ↑ |
| CHAT | MONO | choline acetyltransferase | 0.005 | 0.137 | ↑ |
| SLC6A4/SERT | MONO | solute carrier family 6 (neurotransmitter transporter, serotonin), member 4, | 0.0005 | 0.069 | ↑ |
| CHRM1 | MONO | cholinergic receptor, muscarinic 1 | 0.03t | 0.233 | ↑ |
| CHRM3 | MONO | cholinergic receptor, muscarinic 3 | 0.01 | 0.181 | ↑ |
| CHRNA1 | MONO | cholinergic receptor, nicotinic, alphal | 0.046t | 0.283 | ↑ |
| CHRNA2 | MONO | cholinergic receptor, nicotinic, alpha2 | 0.02t | 0.181 | ↑ |
| CHRNA4 | MONO | cholinergic receptor, nicotinic, alpha4 | 0.003 | 0.137 | ↑ |
| DRD2 | MONO | dopamine receptor D2 | 0.002 | 0.1221 | ↑ |
| DRD4 | MONO | dopamine receptor D4 | 0.02t | 0.181 | ↑ |
| HTR3A | MONO | 5-hydroxytryptamine (serotonin) receptor 3A | 0.03t | 0.233 | ↑ |
| SLC6A11/GAT4 | GABA | solute carrier family 6 (neurotransmitter transporter, GABA), member 11 | 0.03t | 0.233 | ↑ |
| SLC6A13/GAT3 | GABA | solute carrier family 6 (neurotransmitter transporter, GABA), member 13 | 0.04t | 0.283 | ↑ |
| KCNA2 | CH | potassium voltage-gated channel, shaker-related subfamily, member 2 | 0.049t | 0.283 | ↓ |
| KCNA6 | CH | potassium voltage-gated channel, shaker-related subfamily, member 6 | 0.04t | 0.233 | ↓ |
| DPP6 | CH | dipeptidyl-peptidase 6 | 0.03t | 0.233 | ↓ |
| DPP10 | CH | dipeptidyl-peptidase 10 | 0.02t | 0.181 | ↓ |
| HSP90AA1 | GLIA | heat shock protein 90kDa alpha (cytosolic), class A member 1 | 0.02t | 0.181 | ↑ |
| DNAJA1 | GLIA | DnaJ (Hsp40) homolog, subfamily A, member 1 alias HDJ2 | 0.03t | 0.233 | ↑ |
| MSR1 | GLIA | macrophage scavenger receptor 1 | 0.0001 | 0.052 | ↓ |
| ERBB2IP | SYN | erbb2 interacting protein densin-180 | 0.006 | 0.137 | ↑ |
| DLG3 | SYN | discs, large homolog 3 synapse-associated protein 102 | 0.008 | 0.181 | ↑ |
| HOMER1 | SYN | homer homolog 1 | 0.01 | 0.181 | ↑ |
| SNCAIP | SYN | synuclein, alpha interacting protein (synphilin) | 0.01 | 0.181 | ↑ |
| SAP47 | SYN | synapse-associated protein 47kD | 0.02t | 0.181 | ↑ |
| SNCG | SYN | synuclein, gamma | 0.04t | 0.283 | ↑ |
| STX7 | SYN | syntaxin 7 | 0.02t | 0.181 | ↓ |
| ERCC1 | TF | excision repair cross-complementing rodent repair deficiency, complementation group 1 | 0.0003 | 0.052 | ↑ |
| NGFB | NT | nerve growth factor β | 0.03t | 0.233 | ↑ |
| NRP1 | NT | neuropilin 1 | 0.03t | 0.233 | ↑ |
| IGF2R | NT | insulin-like growth factor 2 receptor | 0.003 | 0.122 | ↑ |
: trend level change.
A comparison between AD and MCI revealed downregulation of 4 channel-related transcripts {KCNA2 (p=0.05t), Kv shaker-related subfamily, member 6 (KCNA6; p=0.04t), DPP6 (p=0.03t), and DPP10 (p=0.02t)}, the EL gene LAMP1 (p=0.005), autophagy gene BECN1 (p=0.002), and steroid-related genes glucocorticoid receptors NR3C1 (p=0.02t; Fig. 8) and NCOR2 (p=0.02t; Fig. 8).
Select temporal transcript dysregulation during the progression of AD
Among the 19 differentially expressed genes found between NCI and MCI, significant upregulation of BCL2 and DNCLC1 transcripts occurred in MCI, ultimately resulting in significant upregulation in AD, demonstrating a clear progression. Similarly, significant downregulation of CACNA1B was found in MCI and AD. There was a trend for downregulation of MSR1 in MCI, which reached significance in AD. Of the differentially expressed genes found in AD compared to NCI and MCI, 20 showed either significant or trend level upregulation, whereas 5 genes displayed significant or trend level downregulation in AD compared to NCI and MCI. From NCI to MCI, expression levels of SAA4, NEFL, PPP3CC, HSD17B1, NR5A1, GRM1, GRM5, LAMP1, and SLC2A5 were stable. Expression levels of SAA4, NEFL, PPP3CC, HSD17B1, NR5A1, GRM1, and GRM5 displayed significant upregulation in AD compared to NCI and MCI. Concomitantly, LAMP1 and SLC2A5 expression levels were significantly downregulated in AD compared to NCI and MCI. Expression levels of RGS9, BCHE, CHRNA2, NR3C1, and KCNA2 were unchanged from NCI to MCI. In contrast, expression levels of RGS9, BCHE, and CHRNA2 displayed trend level upregulation whereas expression levels of NR3C1 and KCNA2 were downregulated in AD compared to NCI and MCI. ACTB, MAP2, RAB11A and TIMP1 expression levels were similar between NCI and MCI. In contrast, ACTB, MAP2, RAB11A and TIMP1 levels were significantly upregulated in AD compared to NCI and displayed trend level upregulation in AD compared to MCI. NCOR2 expression was significantly downregulated in AD compared to NCI. There was a trend level downregulation of NCOR2 in AD compared to MCI. Moreover, transcript levels of HDLBP, GRM2, CHAT, CHRNA4, DRD2, and HOMER1 displayed significant upregulation in AD compared to MCI and trend level upregulation in AD compared to NCI. These ‘step up’ and ‘step down’ expression levels between NCI, MCI, and AD indicate we were able to discriminate temporal events of select changes for many targets during the progression of dementia within this vulnerable PreC CatD-positive layer III cell type.
qPCR validation
Since it was not practical to validate every gene, we initially chose three stably expressed genes (GALR1, BAP1, and RBM3) from the custom-designed microarray analysis for validation as positive controls. qPCR revealed changes in expression of GALR1 (p=0.6), BAP1 (p=0.4), and RBM3 (p=0.2) similar to expression direction derived from the microarray analysis of PreC CatD-positive layer III neurons showing that no transcript differences between NCI, MCI, and AD (Table 6, Fig. 10). qPCR was also used to evaluate expression levels of RAB11A, HOMER1, GNG2, GRM1, SLC2A5, TIMP1, and RPS6KB1, which were differentially expressed in PreC neurons in AD compared to NCI and MCI. qPCR showed significant upregulation for RAB11A (p=0.01) and HOMER1 (p=0.04) in AD compared to NCI (Table 6, Fig 9) similar to the single population microarray data. qPCR analysis found no changes in the expression of RAB11A (p=0.4) and HOMER1 (p=0.63) in AD versus MCI compared to the upregulation derived from the single population microarray analysis. qPCR product expression revealed stable levels for GNG2 (p=0.6), SLC2A5 (p=0.6), and RPS6KB1 (p=0.2), which did not follow the downregulation seen in the single cell microarray analysis among NCI, MCI, and AD groups. Expression of qPCR products for GRM1 (p=0.5) and TIMP1 (p=0.5) were stable in PreC among NCI, MCI, and AD, which is different from the upregulation of GRM1 and TIMP1 seen in AD compared to NCI and MCI derived from the microarray data (Table 6, Fig. 9). CHAT expression level was below the limit of qPCR detection, so no comparison to the single cell microarray analysis was possible.
Table 6.
Quantitative PCR validation of select mRNA expression of PreC transcripts
|
Gene |
NCI (Mean±SD) (N=9) | MCI (Mean±SD) (N=9) | AD (Mean±SD) (N=8) |
p |
Multiple comparison |
|---|---|---|---|---|---|
| GALR1 | 0.26±0.137 | 0.40±0.334 | 0.22±0.065 | 0.644 | unchanged |
| BAP1 | 0.636±0.101 | 0.823±0.334 | 0.709±0.192 | 0.427 | unchanged |
| RBM3 | 0.207±0.154 | 0.169±0.155 | 0.324±0.233 | 0.202 | unchanged |
| RAB11A | 0.308±0.103 | 0.380±0.046 | 0.423±0.047 | 0.016 | AD>NCI |
| HOMER1 | 0.092±0.060 | 0.138±0.060 | 0.181±0.085 | 0.045 | AD>NCI |
| GNG2 | 0.247±0.208 | 0.196±0.0.178 | 0.242±0.069 | 0.623 | unchanged |
| GRM1 | 0.249±0.226 | 0.212±0.158 | 0.314±0.141 | 0.514 | unchanged |
| SLC2A5 | 0.996±1.237 | 0.813±0.440 | 1.240±1.537 | 0.572 | unchanged |
| TIMP1 | 1.823±1.874 | 2.531±2.125 | 1.586±1.358 | 0.451 | unchanged |
| RPS6KB1 | 0.168±0.066 | 0.240±0.098 | 0.215±0.062 | 0.159 | unchanged |
| CHAT | 0 | 0 | 0 |
Fig. 9.
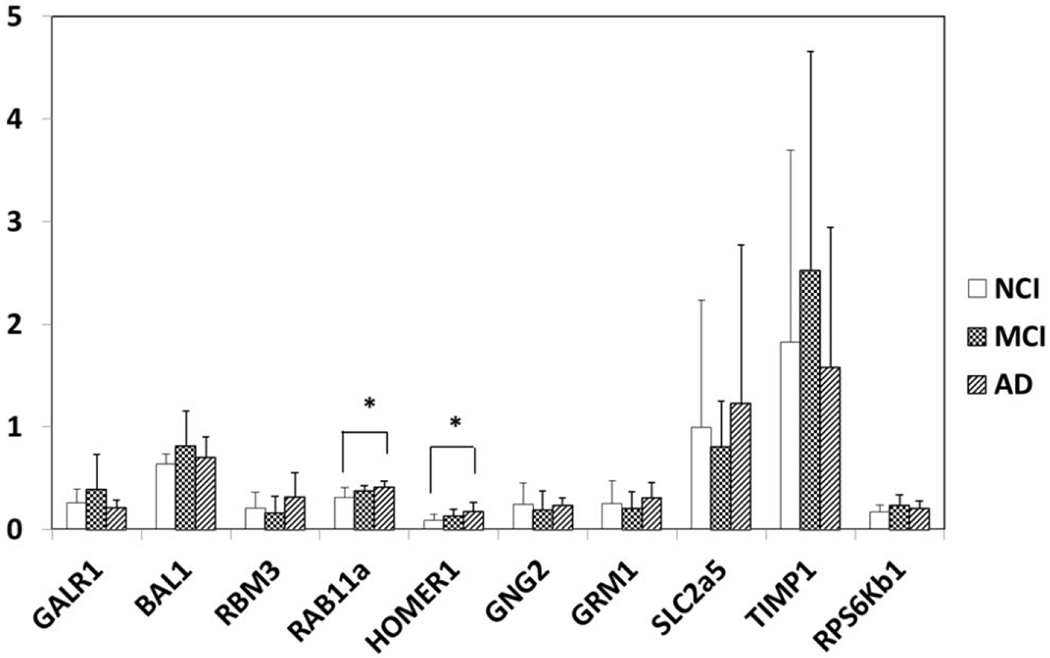
qPCR of select genes in the PreC of NCI, MCI and AD. Upregulation of RAB11A (p=0.02) and HOMER1 (p=0.045) in AD compared to NCI and unchanged mRNA levels of GALR1 (p=0.64), BAP1 (p=0.43) and RBM3 (p=0.2) validating microarray observations. GNG2 (p=0.6), GRM1 (p=0.5), SLC2A5 (p=0.6), TIMP1 (p=0.5) and RPS6KB1 (p=0.2) are unchanged among NCI, MCI and AD. *: p<0.05.
Discussion
A custom-designed array platform of 576 genes was used to define gene expression profiles of individual PreC CatD-positive layer III neurons combined with qPCR validation of select transcripts. Because literally dozens of genes displayed dysregulation in MCI and AD, we highlighted those transcripts either upregulated or downregulated relevant towards understanding mechanisms underlying the vulnerability of this cortical cell type. For example, results showed significant upregulation of BCL2 in MCI and AD compared to NCI. BCL2 inhibits apoptosis and protects neurons against cell death (Yang & Korsmeyer, 1996). Although Western blotting (Kitamura et al., 1998) and immunostaining for BCL2 immunostaining (Satou et al., 1995) revealed increased protein in hippocampus, entorhinal and temporal cortex, respectively in AD compared to normal controls, reduced BCL2 staining was found in neurons positive for the tau epitopes AT8 and PHF-1 (Satou et al., 1995). By contrast, we reported immunoblotting data showing an increase in BCL2 protein in the PreC in AD compared to NCI (Perez et al., 2015). These observations together with our current findings of a significant upregulation of BCL2 in MCI and AD suggest that PreC CatD-positive layer III neurons are exhibiting a cellular plasticity response related to cell survival during the progression of AD that may be a valuable marker for therapeutic development.
Transcript analysis revealed selective upregulation of several EL and autophagy genes including RAB4A, RAB11A, and BECN1. RAB4A regulates membrane trafficking and is associated with early endosomes and is involved in cell sorting and recycling (Mohrmann et al., 2002; van der Sluijs et al., 1992). Upregulation of RAB4A protein and mRNA correlates with measurements of cognitive decline in individuals with MCI and AD (Ginsberg et al., 2011). RAB11A is associated with recycling endosomes and regulating vesicular trafficking (Ullrich et al., 1996). RAB11 is involved in the recycling of β-secretase to the plasma membrane and affects Aβ production (Udayar et al., 2013). Although the present study found upregulation of BECN1 in MCI compared to NCI in PreC CatD-positive layer III neurons, others report no change of BECN1 in CA1 pyramidal neurons in AD (Bordi et al., 2016) or downregulation of BECN1 protein levels in midfrontal cortex of moderate to severe AD patients (Pickford et al., 2008). Together, these findings suggest cell- and region-specificity of BECN1 gene and encoded protein regulation during the progression of dementia.
The current findings revealed a trend toward an upregulation of several cholinergic transcripts (BCHE, CHAT, CHRNA2 and CHRNA4) in PreC layer III Cat D positive neurons in AD compared to NCI. CHAT and CHRNA4 displayed significant upregulation, whereas BCHE and CHRNA2 show trend level upregulation in AD compared to MCI. However, qPCR and Nanonstring Counter (data not shown) findings failed to detect CHAT mRNA in the PreC. Although several studies report cortical CHAT protein derived from the cholinergic neurons of the nucleus basalis of Meynert (nbM) (Mufson et al., 2007), most gene expression studies have failed to detect CHAT mRNA in the cortex (Butcher et al., 1992; Ibáñez et al., 1991; Oh et al., 1992), which would explain the lack of expression in the present study. On the other hand, BCHE, a serine hydrolase that is widely distributed throughout the central nervous system and also catalyzes the hydrolysis of acetylcholine, is localized to cortical neurons and is associated with NFTs and senile plaques in the AD cortex (Wright et al., 1993; Moran et al., 1994; Gomez-Ramos & Moran, 1997), supporting the current transcript findings of this cholinoceptive marker in PreC neurons. Select nicotinic acetylcholine receptor subtypes were also found to be reduced in the AD cerebral cortex (Whitehouse et al., 1986). Prior studies revealed a significant reduction in [3H]nicotine binding sites in the putamen and nbM and a reduction of [3H]QNB binding in the nbM and hippocampus in AD (Shimohama et al., 1986). In cortex, protein activity of CHRNA4 is reduced while mRNA level remains stable in AD, suggesting a disconnect between transcription and translation of CHRNA4 in AD (Nordberg, 2001). High-affinity nicotine binding showed a decrease in α4/β2 nicotinic receptors in hippocampus and temporal neocortex in AD (Perry et al., 1995). The present data showing an increase PreC neuronal BCHE, CHRNA2, and CHRNA4 transcripts support their involvement in the vulnerability of these corticocortical projection neurons.
A significant upregulation of metabotropic glutamate receptors GRM1 and GRM5 and trend level upregulation for GRM2 was found in AD compared to NCI. In addition, we found significant upregulation of the metabotropic glutamate receptors GRM1, GRM2 and GRM5 in AD compared to MCI. These data suggest progressive dysregulation of several presynaptic and postsynaptic metabotropic glutamate receptors in PreC neurons. On the other hand, GRM1 was downregulated in AD frontal cortex, which correlated with the progression of AD (Albasanz et al., 2005). Activation of GRM1 accelerates the processing of APP into non-amyloidogenic products by α-secretase in vitro (Lee et al., 1995) and also initiates PI3 kinase activity preventing neuronal apoptosis (Rong et al., 2003). Similar to the PreC, GRM2 is upregulated in the AD hippocampus and colocalizes with NFTs (Lee et al., 2009), suggesting a role in the cellular pathogenesis of this disease. Taken together, these finding suggest an interplay between a putative neuroprotective action related to GRM1 and GRM5 upregulation versus upregulation of GRM2, which may lead to PreC neuronal dysfunction early in the onset of AD.
In terms of regulating cytoskeletal elements, we observed upregulation of NEFL, NEFM, MAP2, and ACTB in AD compared to NCI. Significant upregulation of NEFL and NEFM and trend for upregulation of MAP2 and ACTB were found in AD compared to MCI. Western blotting studies demonstrated hyperphosphorylation of NEFH and NEFM and a significant increase in levels of NEFH, NEFM and NEFL in AD neocortex (Wang et al., 2001). O-GlcNAcylation and phosphorylation of NEFM regulate each other in cultured neuroblastoma cells and decreased O-GlcNAcylation and increased phosphorylation of NEFM were reported in the medial temporal cortex in AD (Deng et al., 2008), suggesting a role for these transcripts in AD neuropathology.
The present study also showed differential regulation of several steroid-related markers including significant upregulation of HSD17B1 and NR5A1 and trend towards downregulation for NR3C1 in AD compared MCI and NCI. We also observed a significant downregulation of NCOR2 in AD compared to NCI and trend level downregulation of NCOR2 between AD and MCI. HSD17B1, the enzyme catalyzing conversion of estrone to estradiol, was upregulated in the PreC similar to that seen in the prefrontal cortex in AD (Luchetti et al., 2011). In Down syndrome, a genetic model of AD, women with homozygous alleles at rs676387 (intron 4), rs605059 (exon 6), and rs598126 (exon 4) of HSD17B1 have an increased risk of dementia (Lee et al., 2012). Set-association analysis revealed that a rare haplotype in the 5’ regulatory region of the HSD17B1 gene was a risk factor for sporadic AD (de Quervain et al., 2004). NR5A1 (SF1) is a steroidogenic tissue-specific nuclear receptor that regulates nearly all genes of the glycolytic pathway. Knockdown of NR5A1 by small interfering RNA reduces the production of ATP, nicotinamide adenine dinucleotide phosphate, and downregulates genes involved in glucose metabolism (Baba et al., 2014). NR3C1, a glucocorticoid receptor that regulates glucocorticoid-responsive expression is related to cognitive impairment and neurodegeneration in rodents (Montkowski et al., 1995; Murphy et al., 2002). NCOR2 (also known as SMART) is a nuclear receptor co-repressor that mediates transcriptional silencing of target genes that regulate mitochondrial oxidative metabolism and aging processes (Reilly et al., 2010). Interestingly, a repressor complex containing NCOR2 and the epigenetic marker HDAC1 regulates Notch signaling transduction pathways (Kao et al., 1998). NCOR2 forms a stable complex with HDAC3 and functions as an activating cofactor of this epigenetic factor. In contrast, NCOR2 does not directly activate class II HDACs (Guenther et al., 2001; Fischle et al., 2002). Class II HDACs regulate transcription by bridging the NCOR1/NCOR2-HDAC3 complex (Fischle et al., 2002). Together, these findings suggest that transcripts related to steroid production and regulation of their receptors are involved in gene transcription and epigenetic modulation related to the cellular pathogenesis of PreC neurons in AD.
In select cases, there was a disconnect between the findings at the single population level in the custom-designed microarray analysis and that derived from regional PreC findings determined by qPCR. This discrepancy is not uncommon due to the admixed cell types within regional tissue samples required for qPCR analysis. Similar to our previous human postmortem nbM and hippocampal microarray and qPCR analyses (Ginsberg et al., 2000, 2006a, 2006b, 2010; Counts et al., 2007, 2008, 2009), we were able to validate several transcripts, often those with moderate to high expression levels, providing clear evidence of validation. Here, we chose 5 upregulated genes (RAB11A, HOMER1, GRM1, CHAT and TIMP1) and 3 downregulated genes (GNG2, SLC2A5 and RPS6KB1) for qPCR validation that were related to AD in comparison to NCI and MCI. These 8 genes belong to 7 gene ontology classes with relative higher expression levels and small p-values (p≤0.01) in at least one comparison (e.g., AD vs NCI and AD vs MCI). qPCR confirmed upregulation of RAB11A and HOMER1 in AD compared to NCI verifying observations from the microarray experiments. qPCR detected unchanged levels of GRM1, TIMP1, GNG2, SLC2A5, and RPS6KB1 in PreC among NCI, MCI and AD, which did not match our array findings. Further, the discrepancy between the present single population microarray and our inability to perform qPCR validation related to CHAT upregulation may be related to the fact that this gene is not highly expressed in cortical neurons, which can be overwhelmed by the complexity of gene expressions related to multiple cell types located within the cortical layers of the PreC. An alternative approach for validation of single population microarray findings is in situ hybridization (Ginsberg et al., 2000), which can be applied to a population of individual neurons within a well-defined cortical lamina similar to the LCM-based microarray approach used in the current study. However, in situ hybridization carries its own limitations and pitfalls, especially in postmortem human brain tissue. Due to a lack of available tissue samples for in situ hybridization, we selected the more conventional qPCR approach to validate custom-designed microarray results.
A caveat of this and other postmortem human brain tissue studies is the role that end of life agonal state exerts upon RNA expression levels in the brain. The effects of agonal conditions on nucleic acids are well documented and an average PMI of 3–4 h is considered optimal for gene expression analysis (Blair et al., 2016). Current PMIs were ~5 h which are appropriate for gene expression studies. Another consideration is that this investigation examined PreC CatD-immunopositive neurons irrespective of NFT status. Since we did not differentiate accrued from NFT-positive or negative neurons, as done in prior studies (Tiernan et al., 2016, 2018), we were unable to attribute the effect of tau pathology on specific expression profiles during the progression of dementia. This neuronal distinction will be investigated upon access to additional precuneal sections and the maximization of double immunolabeling for LCM experiments. Another limitation is the relatively small number of APOE ε4 carriers, particularly homozygous individuals, which likely affect the associations described here. Future studies consisting of a greater balance of APOE ε4 carriers and non-carriers are needed. Moreover, the individuals in the present investigation were from a community-based group of highly educated retired clergy who were provided excellent health care and nutrition (Bennett et al., 2012; Mufson et al., 2012). Volunteer subjects may introduce bias by decreasing pathology but this, in part, is mitigated by the high follow-up and autopsy rates of the RROS cohort (Bennett et al., 2005). Strengths of the study include uniform premortem clinical and postmortem pathological evaluation and that final pathologic classification was performed without knowledge of the clinical evaluation.
In summary, PreC CatD-positive layer III neurons display significant dysregulation of anti-apoptotic, EL and autophagy genes in MCI and AD indicating early involvement of cell survival and cellular trafficking systems in the pathogenesis of these neurons. Upregulation of type I metabotropic glutamate receptor (GRM1 and GRM5), type II metabotropic glutamate receptor (GRM2) in AD compared to NCI and MCI also suggest a role in neuronal pathogenesis. Upregulation of mRNA for the cholinoceptive marker BCHE and acetylcholine receptors CHRNA2 and CHRNA4 reveal dysregulation of cholinergic-related genes in the pathogenesis of PreC layer III neurons in AD. In addition, upregulation of cytoskeletal element transcripts (NEFL, NEFM, MAP2, and ACTB) also indicate participation in the pathogenesis of AD. Differential expression of steroid-related marker and receptors implicate involvement in gene transcription and epigenetic regulation in the cellular pathogenesis of PreC neurons in AD. Together, the present findings suggest that PreC CatD-positive layer III neurons undergo a diverse dysregulation of multiple transcripts affecting different functional pathways during the onset of AD.
Precuneus (PreC) layer III projection neurons, which contain lysosomal hydrolase cathepsin D (CatD) are dysfunctional in Alzheimer’s disease (AD). Expression profiling of CatD positive PreC layer III neurons revealed significant dysregulation of a mosaic of genes in tissue from subjects with mild cognitive impairment and AD that was not previously appreciated in terms of their indication of systems-wide signaling defects in a key hub of the default mode memory network.
Acknowledgements:
We are indebted to the nuns, priests, and lay brothers who participated in the Rush Religious Orders Study and to the members of the Rush ADC.
Funding: This study was supported by grants PO1 AG014449, RO1 AG043375, RO1 AG010161, P01 AG017617, and RO1 AG042146 from the National Institute on Aging, Barrow Neurological Institute Barrow and Beyond and the Fine Foundation.
Footnotes
Conflict of Interest: Authors have no conflict of interest to declare.
Data Sharing and Data Accessibility: The data that support the findings of this study are available on request from the corresponding author. These data are not publicly available due to privacy or ethical restrictions.
References
- Albasanz JL, Dalfo E, Ferrer I, & Martin M (2005). Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol Dis, 20 (3), 685–693. DOI: 10.1016/j.nbd.2005.05.001 [DOI] [PubMed] [Google Scholar]
- Albert M, Smith LA, Scherr PA, Taylor JO, Evans DA, & Funkenstein HH (1991). Use of brief cognitive tests to identify individuals in the community with clinically diagnosed Alzheimer’s disease. Int J Neurosci, 57 (3-4), 167–178. DOI: 10.3109/00207459109150691 [DOI] [PubMed] [Google Scholar]
- Alldred MJ, Che S, & Ginsberg SD (2008). Terminal Continuation (TC) RNA amplification enables expression profiling using minute RNA input obtained from mouse brain. Int J Mol Sci, 9 (11), 2091–2104. DOI: 10.3390/ijms9112091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Che S, & Ginsberg SD (2009). Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods, 177 (2), 381–385. DOI: 10.1016/j.jneumeth.2008.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Chao HM, Lee SH, Beilin J, Powers BE, Petkova E, … Ginsberg SD. (2018). CA1 pyramidal neuron gene expression mosaics in the Ts65Dn murine model of Down syndrome and Alzheimer’s disease following maternal choline supplementation. Hippocampus, 28 (4), 251–268. DOI: 10.1002/hipo.22832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, & Buckner RL (2007). Disruption of large-scale brain systems in advanced aging. Neuron, 56 (5), 924–935. DOI: 10.1016/j.neuron.2007.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Otake H, Sato T, Miyabayashi K, Shishido Y, Wang CY … Morohashi K. (2014). Glycolytic genes are targets of the nuclear receptor Ad4BP/SF-1. Nat Commun, 5, 3634–3646. DOI: 10.1038/ncomms4634 [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, & Wilson RS (2005) Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology, 64 (5), 834–841. DOI: 10.1212/01.WNL.0000152982.47274.9E [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah R , & Wilson RS (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology, 66 (12), 1837–1844. DOI: 10.1212/01.wnl.0000219668.47116.e6 [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Boyle PA, Buchman AS, & Schneider JA (2012). Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol, 72 (4), 599–609. DOI: 10.1002/ana.23654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair JA, Wang C, Hernandez D, Siedlak SL, Rodgers MS, Achar RK, …Lee HG. (2016). Individual Case Analysis of Postmortem Interval Time on Brain Tissue Preservation. PLoS One, 11 (3),e0151615 DOI: 10.1371/journal.pone.0151615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, … Nixon RA. (2016). Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy, 12 (12),2467–2483. DOI: 10.1080/15548627.2016.1239003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, & Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 82 (4), 239–259. DOI: 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, & Schacter DL (2008). The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci, 1124, 1–38. DOI: 10.1196/annals.1440.011 [DOI] [PubMed] [Google Scholar]
- Butcher LL, Oh JD, Woolf NJ, Edwards RH, & Roghani A (1992). Organization of central cholinergic neurons revealed by combined in situ hybridization histochemistry and choline-O-acetyltransferase immunocytochemistry. Neurochem Int, 21 (3), 429–445. DOI: 10.1016/0197-0186(92)90195-w [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, … Nixon RA. (1995). Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron, 14 (3), 671–680. DOI: 10.1016/0896-6273(95)90324-0 [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, & Nixon RA (1996). Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci, 16 (1), 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, & Nixon RA (1997). Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci, 17 (16),6142–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che S, & Ginsberg SD (2004). Amplification of RNA transcripts using terminal continuation. Lab Invest, 84 (1), 131–137. DOI: 10.1038/labinvest.3700005 [DOI] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, & Mufson EJ (2007). Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol, 64 (12), 1771–1776. DOI: 10.1001/archneur.64.12.1771 [DOI] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ginsberg SD, & Mufson EJ (2008). Galanin hyperinnervation upregulates choline acetyltransferase expression in cholinergic basal forebrain neurons in Alzheimer’s disease. Neurodegener Dis, 5, 228–331. DOI: 10.1159/000113710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ginsberg SD, & Mufson EJ (2009). Galanin fiber hyperinnervation preserves neuroprotective gene expression in cholinergic basal forebrain neurons in Alzheimer’s disease. J Alzheimers Dis, 18, 885–896. DOI: 10.3233/JAD-2009-1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DG, Schmitt FA, Wekstein DR, & Markesbery WR (1999). Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol, 58 (4), 376–388. DOI: 10.1097/00005072-199904000-00008 [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, … Papassotiropoulos A. (2004). Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum Mol Genet, 13 (1), 47–52. DOI: 10.1093/hmg/ddg361 [DOI] [PubMed] [Google Scholar]
- Deiss LP, Galinka H, Berissi H, Cohen O, & Kimchi A (1996). Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J, 15 (15), 3861–3870. [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Li B, Liu F, Iqbal K, Grundke-Iqbal I, Brandt R, & Gong CX (2008). Regulation between O-GlcNAcylation and phosphorylation of neurofilament-M and their dysregulation in Alzheimer disease. FASEB J, 22 (1), 138–145. DOI: 10.1096/fj.07-8309com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanand DP, Folz M, Gorlyn M, Moeller JR, & Stern Y (1997). Questionable dementia: clinical course and predictors of outcome. J Am Geriatr Soc, 45 (3),321–328. DOI: 10.1111/j.1532-5415.1997.tb00947.x [DOI] [PubMed] [Google Scholar]
- Drzezga A, Becker JA, Van Dijk KR, Sreenivasan A, Talukdar T, Sullivan C, … Sperling RA. (2011). Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain, 134 (Pt 6), 1635–1646. DOI: 10.1093/brain/awr066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, & Crino P (2001). Analysis of mRNA populations from single live and fixed cells of the central nervous system. Curr Protoc Neurosci, Chapter 5: Unit 5.3. DOI: 10.1002/0471142301.ns0503s00 [DOI] [PubMed] [Google Scholar]
- Ebly EM, Hogan DB, & Parhad IM (1995). Cognitive impairment in the nondemented elderly. Results from the Canadian Study of Health and Aging. Arch Neurol, 52 (6), 612–619. DOI: 10.1001/archneur.1995.00540300086018 [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, & Verdin E (2002). Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell, 9 (1), 45–57. DOI: 10.1016/s1097-2765(01)00429-4 [DOI] [PubMed] [Google Scholar]
- Flicker C, Ferris SH, & Reisberg B (1991). Mild cognitive impairment in the elderly: predictors of dementia. Neurology, 41 (7), 1006–1009. DOI: 10.1212/wnl.41.7.1006 [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Testa C, Sabattoli F, Beltramello A, Soininen H, & Laakso MP (2005). Structural correlates of early and late onset Alzheimer’s disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry, 76 (1), 112–114. DOI: 10.1136/jnnp.2003.029876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, & Trojanowski JQ (2000). Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann Neurol, 48 (1), 77–87. [PubMed] [Google Scholar]
- Ginsberg SD, & Che S (2005). Expression profile analysis within the human hippocampus: comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol, 487(1), 107–118. DOI: 10.1002/cne.20535 [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, & Mufson EJ (2006a) Single cell gene expression profiling in Alzheimer’s disease. NeuroRx,3 (3), 302–318. DOI: 10.1016/j.nurx.2006.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, & Mufson EJ. (2006b). Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Neurochem, 96 (5), 1401–1408. DOI: 10.1111/j.1471-4159.2005.03641.x [DOI] [PubMed] [Google Scholar]
- Ginsberg SD (2008). Transcriptional profiling of small samples in the central nervous system. Methods Mol Biol, 439, 147–158. DOI: 10.1007/978-1-59745-188-8_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, … Che S. (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry, 68 (10), 885–893. DOI: 10.1016/j.biopsych.2010.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, & Che S (2011). Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Chem Neuroanat, 42 (2),102–110. DOI: 10.1016/j.jchemneu.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Malek-Ahmadi MH, Alldred MJ, Che S, Elarova I, Chen Y, … Mufson EJ. (2019). Selective decline of neurotrophin and neurotrophin receptor genes within CA1 pyramidal neurons and hippocampus proper: Correlation with cognitive performance and neuropathology in mild cognitive impairment and Alzheimer’s disease. Hippocampus, 29 (5), 422–439. DOI: 10.1002/hipo.22802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Ramos P, & Morán MA (1997). Ultrastructural localization of butyrylcholinesterase in senile plaques in the brains of aged and Alzheimer disease patients. Mol Chem Neuropathol, 30 (3), 161–173. DOI: 10.1007/bf02815095 [DOI] [PubMed] [Google Scholar]
- Guenther MG, Barak O, & Lazar MA (2001). The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol, 21 (18), 6091–6101. DOI: 10.1128/mcb.21.18.6091-6101.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz K, Salmon E, Perani D, Baron JC, Holthoff V, Frölich L, … Heiss W. (2002). Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage, 17 (1), 302–316. DOI: 10.1006/nimg.2002.1208 [DOI] [PubMed] [Google Scholar]
- Ibáñez CF, Ernfors P, & Persson H (1991). Developmental and regional expression of choline acetyltransferase mRNA in the rat central nervous system. J Neurosci Res, 29 (2),163–171. DOI: 10.1002/jnr.490290205 [DOI] [PubMed] [Google Scholar]
- Ishii K, Kawachi T, Sasaki H, Kono AK, Fukuda T, Kojima Y, & Mori E (2005). Voxel-based morphometric comparison between early- and late-onset mild Alzheimer’s disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol, 26 (2), 333–340. [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, … Dickson DW. (2016). Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol, 131(4), 571–585. DOI: 10.1007/s00401-016-1537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, … Kadesch T. (1998). A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev, 12 (15), 2269–2277. DOI: 10.1101/gad.12.15.2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas G, Scheltens P, Rombouts S, van Schijndel R, Klein M, Jones B, … Barkhof F. (2007). Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology, 49 (12),967–976. DOI: 10.1007/s00234-007-0269-2 [DOI] [PubMed] [Google Scholar]
- Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, … Feany MB (2010). Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet, 6 (7), e1001026 DOI: 10.1371/journal.pgen.1001026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, … Taniguchi T. (1998). Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Research, 780 (2),260–269. DOI: 10.1016/s0006-8993(97)01202-x [DOI] [PubMed] [Google Scholar]
- Lee HG, Zhu X, Casadesus G, Pallas M, Camins A, O’Neill MJ, … Smith MA. (2009). The effect of mGluR2 activation on signal transduction pathways and neuronal cell survival. Brain Res, 1249, 244–250. DOI: 10.1016/j.brainres.2008.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gurney S, Pang D, Temkin A, Park N, Janicki SC, … Schupf N. (2012). Polymorphisms in HSD17B1: Early Onset and Increased Risk of Alzheimer’s Disease in Women with Down Syndrome. Curr Gerontol Geriatr Res, 2012, 361218 DOI: 10.1155/2012/361218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RK, Wurtman RJ, Cox AJ, & Nitsch RM (1995). Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci U S A, 92 (17), 8083–8087. DOI: 10.1073/pnas.92.17.8083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti S, Bossers K, Van de Bilt S, Agrapart V, Morales RR, Frajese GV, & Swaab DF (2011). Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol Aging, 32 (11), 1964–1976. DOI: 10.1016/j.neurobiolaging.2009.12.014 [DOI] [PubMed] [Google Scholar]
- Matsuda H (2007). The role of neuroimaging in mild cognitive impairment. Neuropathology, 27: 570–577. DOI: 10.1111/j.1440-1789.2007.00794.x [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, & Stadlan EM (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology, 34 (7), 939–944. DOI: 10.1212/wnl.34.7.939 [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, … Morris JC. (2006). [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology, 67 (3), 446–452. DOI: 10.1212/01.wnl.0000228230.26044.a4 [DOI] [PubMed] [Google Scholar]
- Mirra SS (1997). The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging, 18 (4 Suppl), S91–94. DOI: 10.1016/s0197-4580(97)00058-4 [DOI] [PubMed] [Google Scholar]
- Mohrmann K, Gerez L, Oorschot V, Klumperman J, & van der Sluijs P (2002). Rab4 function in membrane recycling from early endosomes depends on a membrane to cytoplasm cycle. J Biol Chem, 277 (35), 32029–32035. DOI: 10.1074/jbc.M203064200 [DOI] [PubMed] [Google Scholar]
- Montkowski A, Barden N, Wotjak C, Stec I, Ganster J, Meaney M, … Holsboer, (1995). Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J Neuroendocrinol, 7 (11), 841–845. DOI: 10.1111/j.1365-2826.1995.tb00724.x [DOI] [PubMed] [Google Scholar]
- Morán MA, Mufson EJ, & Gomez-Ramos P (1994). Cholinesterases colocalize with sites of neurofibrillary degeneration in aged and Alzheimer’s brains. Acta Neuropathol, 87 (3), 284–292. DOI: 10.1007/bf00296744 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, & Saragovi HU (1997). Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp Neurol, 146 (1), 91–103. DOI: 10.1006/exnr.1997.6504 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, & Kordower JH (1999). Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol, 158 (2), 469–490. DOI: 10.1006/exnr.1999.7086 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Fahnestock M, & Ginsberg SD (2007). Cholinotrophic molecular substrates of mild cognitive impairment in the elderly. Curr Alzheimer Res, 4 (4), 340–350. DOI: 10.2174/156720507781788855 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, … Scheff SW. (2012). Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol, 123 (1), 13–30. DOI: 10.1007/s00401-011-0884-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EK, Spencer RL, Sipe KJ, & Herman JP (2002). Decrements in nuclear glucocorticoid receptor (GR) protein levels and DNA binding in aged rat hippocampus. Endocrinology, 143 (4), 1362–1370. DOI: 10.1210/endo.143.4.8740 [DOI] [PubMed] [Google Scholar]
- Nelson PT, Abner EL, Scheff SW, Schmitt FA, Kryscio RJ, Jicha GA, … Markesbery WR. (2009). Alzheimer’s-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett, 450 (3), 336–339. DOI: 10.1016/j.neulet.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell KL, Hyman BT, Growdon JH, & Hedley-Whyte ET (1999). Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol, 58 (11), 1147–1155. DOI: 10.1097/00005072-199911000-00004 [DOI] [PubMed] [Google Scholar]
- Nixon RA (2007). Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci, 120 (Pt 23), 4081–4091. DOI: 10.1242/jcs.019265 [DOI] [PubMed] [Google Scholar]
- Nixon RA (2017). Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J, 31, 2729–2743. DOI: 10.1096/fj.201700359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A (2001). Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry, 49 (3), 200–210. DOI: 10.1016/s0006-3223(00)01125-2 [DOI] [PubMed] [Google Scholar]
- Oh JD, Woolf NJ, Roghani A, Edwards RH, & Butcher LL (1992). Cholinergic neurons in the rat central nervous system demonstrated by in situ hybridization of choline acetyltransferase mRNA. Neuroscience, 47 (4), 807–822. DOI: 10.1016/0306-4522(92)90031-v [DOI] [PubMed] [Google Scholar]
- Pasternak SH, Callahan JW, & Mahuran DJ (2004). The role of the endosomal/lysosomal system in amyloid-beta production and the pathophysiology of Alzheimer’s disease: reexamining the spatial paradox from a lysosomal perspective. J Alzheimers Dis, 6 (1), 53–65. DOI: 10.3233/jad-2004-6107 [DOI] [PubMed] [Google Scholar]
- Perez SE, Getova DP, He B, Counts SE, Geula C, Desire L, … Mufson EJ. (2012). Rac1b increases with progressive tau pathology within cholinergic nucleus basalis neurons in Alzheimer’s disease. Am J Pathol, 180 (2), 526–540. DOI: 10.1016/j.ajpath.2011.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, He B, Nadeem M, Wuu J, Scheff SW, Abrahamson EE, … Mufson EJ. (2015). Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer’s disease. Biol Psychiatry, 77 (8), 693–703. DOI: 10.1016/j.biopsych.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, … Perry RH. (1995). Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience, 64 (2), 385–395. DOI: 10.1016/0306-4522(94)00410-7 [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Ivnik RJ, Tangalos EG, Schaid DJ, Thibodeau SN, … Kurland LT. (1995). Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. JAMA, 273 (16), 1274–1278. [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, … Wyss-Coray T. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest, 118 (6), 2190–2199. DOI: 10.1172/JCI33585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, & Shulman GL (2001). A default mode of brain function. Proc Natl Acad Sci U S A, 98 (2), 676–682. DOI: 10.1073/pnas.98.2.676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Bhargava P, Liu S, Gangl MR, Gorgun C, Nofsinger RR, … Lee CH. (2010). Nuclear receptor corepressor SMRT regulates mitochondrial oxidative metabolism and mediates aging-related metabolic deterioration. Cell Metab, 12 (6), 643–653. DOI: 10.1016/j.cmet.2010.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemenschneider M, Blennow K, Wagenpfeil S, Andreasen N, Prince JA, Laws SM, … Kurz A. (2006). The cathepsin D rs17571 polymorphism: effects on CSF tau concentrations in Alzheimer disease. Hum Mutat, 27 (6), 532–537. DOI: 10.1002/humu.20326 [DOI] [PubMed] [Google Scholar]
- Rombouts SA, Barkhof F, Goekoop R, Stam CJ, & Scheltens P (2005). Altered resting state networks in mild cognitive impairment and mild Alzheimer’s disease: an fMRI study. Hum Brain Mapp, 26 (4), 231–239. DOI: 10.1002/hbm.20160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, … Ye K. (2003). PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci, 6 (11), 1153–1161. DOI: 10.1038/nn1134 [DOI] [PubMed] [Google Scholar]
- Rubin EH, Morris JC, Grant EA, & Vendegna T (1989). Very mild senile dementia of the Alzheimer type. I. Clinical assessment. Arch Neurol, 46 (4), 379–382. [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT 9.1 User’s Guide. (2004). SAS Publishing; Cary, NC. [Google Scholar]
- Satou T, Cummingsa BJ, & Cotmana CW (1995). Immunoreactivity for Bcl-2 protein within neurons in the Alzheimer’s disease brain increases with disease severity. Brain Res, 697 (1–2), 35–43. DOI: 10.1016/0006-8993(95)00748-f [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, & Mufson EJ (2013). Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J Alzheimers Dis, 35 (3), 599–609. DOI: 10.3233/JAD-122353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, & Markesbery WR (2000). “preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology, 55 (3), 370–376. DOI: 10.1212/wnl.55.3.370 [DOI] [PubMed] [Google Scholar]
- Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, … Kryscio RJ. (2012). University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res, 9 (6), 724–733 DOI: 10.2174/156720512801322591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, & Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc, 3 (6), 1101–1108. DOI: 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, & Mintun MA (2010). Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry, 67 (6), 584–587. DOI: 10.1016/j.biopsych.2009.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Taniguchi T, Fujiwara M, & Kameyama, (1986). M. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J Neurochem, 46(1), 288–293. DOI: 10.1111/j.1471-4159.1986.tb12960.x [DOI] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M , … Johnson KA. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron, 63 (2), 178–188. DOI: 10.1016/j.neuron.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, … Johnson KA. (2010). Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med, 12 (1), 27–43. DOI: 10.1007/s12017-009-8109-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiernan CT, Ginsberg SD, Guillozet-Bongaarts AL, Ward SM, He B, Kanaan NM, … Counts SE. (2016). Protein homeostasis gene dysregulation in pretangle-bearing nucleus basalis neurons during the progression of Alzheimer’s disease. Neurobiol Aging, 42, 80–90. DOI: 10.1016/j.neurobiolaging.2016.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiernan CT, Ginsberg SD, He B, Ward SM, Guillozet-Bongaarts AL, Kanaan NM, … Counts SE. (2018). Pretangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer’s disease. Neurobiol Dis, 117, 125–136. DOI: 10.1016/j.nbd.2018.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayar V, Buggia-Prevot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, … Rajendran L. (2013). A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep, 5 (6), 1536–1551. DOI: 10.1016/j.celrep.2013.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbe S, Zerial M, & Parton RG (1996). Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol, 135 (4), 913–924. DOI: 10.1083/jcb.135.4.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hull M, Webster P, Mâle P, Goud B, & Mellman I (1992). The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell, 70 (5), 729–740. DOI: 10.1016/0092-8674(92)90307-x [DOI] [PubMed] [Google Scholar]
- Wagner AD, Shannon BJ, Kahn I, & Buckner RL (2005). Parietal lobe contributions to episodic memory retrieval. Trends Cogn Sci, 9 (9), 445–453. DOI: 10.1016/j.tics.2005.07.001 [DOI] [PubMed] [Google Scholar]
- Wang J, Tung YC, Wang Y, Li XT, Iqbal K, & Grundke-Iqbal I (2001). Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett, 507 (1), 81–87. DOI: 10.1016/s0014-5793(01)02944-1 [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Martino AM, Antuono PG, Lowenstein PR, Coyle JT, Price DL, & Kellar KJ (1986). Nicotinic acetylcholine binding sites in Alzheimer’s disease. Brain Res, 371(1), 146–151. DOI: 10.1016/0006-8993(86)90819-x [DOI] [PubMed] [Google Scholar]
- Wright CI, Geula C, & Mesulam MM (1993). Neurological cholinesterases in the normal brain and in Alzheimer’s disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol, 34 (3), 373–384. DOI: 10.1002/ana.410340312 [DOI] [PubMed] [Google Scholar]
- Yan H, Zhang Y, Chen H, Wang Y, & Liu Y (2013). Altered effective connectivity of the default mode network in resting-state amnestic type mild cognitive impairment. J Int Neuropsychol Soc, 19 (4), 400–409. DOI: 10.1017/S1355617712001580 [DOI] [PubMed] [Google Scholar]
- Yang E, & Korsmeyer SJ (1996). Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood, 88 (2), 386–401. [PubMed] [Google Scholar]