

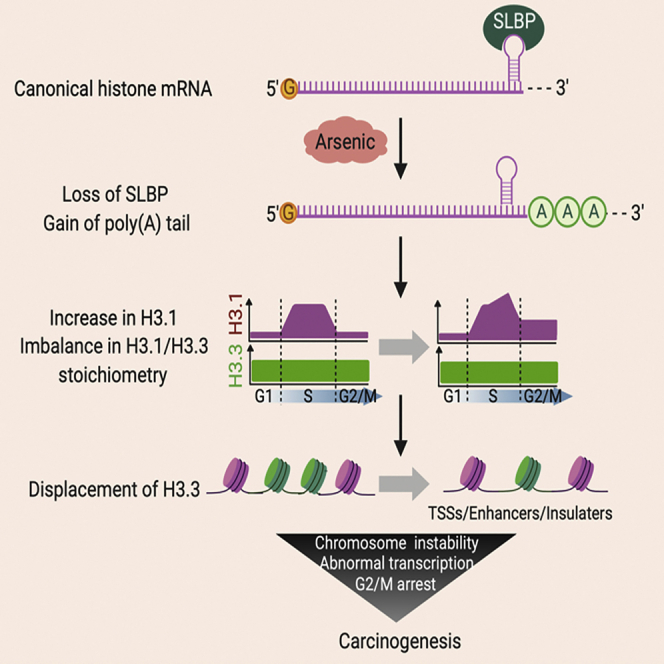
Summary
Replication-dependent canonical histone messenger RNAs (mRNAs) do not terminate with a poly(A) tail at the 3′ end. We previously demonstrated that exposure to arsenic, an environmental carcinogen, induces polyadenylation of canonical histone H3.1 mRNA, causing transformation of human cells in vitro. Here we report that polyadenylation of H3.1 mRNA increases H3.1 protein, resulting in displacement of histone variant H3.3 at active promoters, enhancers, and insulator regions, leading to transcriptional deregulation, G2/M cell-cycle arrest, chromosome aneuploidy, and aberrations. In support of these observations, knocking down the expression of H3.3 induced cell transformation, whereas ectopic expression of H3.3 attenuated arsenic-induced cell transformation. Notably, arsenic exposure also resulted in displacement of H3.3 from active promoters, enhancers, and insulator regions. These data suggest that H3.3 displacement might be central to carcinogenesis caused by polyadenylation of H3.1 mRNA upon arsenic exposure. Our findings illustrate the importance of proper histone stoichiometry in maintaining genome integrity.
Subject Areas: Toxicology, Cell Biology, Omics
Graphical Abstract
Highlights
-
•
Arsenic induces polyadenylation of canonical histone H3.1 mRNA in vivo
-
•
Polyadenylation of H3.1 mRNA promotes tumor formation in nude mice
-
•
The variant H3.3 is displaced from regulatory elements by polyadenylated H3.1 mRNA
-
•
Polyadenylated H3.1 mRNA causes abnormal transcription and chromosomal instability
Toxicology; Cell Biology; Omics
Introduction
Histone genes are highly conserved through evolution. Although replication-dependent histones, referred to as canonical, constitute the majority of histone genes, various replication-independent histone variants have evolved. Each variant retains distinct locus-specific properties and is unique in terms of gene and protein sequences (Buschbeck and Hake, 2017). Canonical histone genes (e.g., histone H3.1) lack introns and are the only genes found in multicellular organisms whose messenger RNAs (mRNAs) lack a poly(A) tail at their 3′ end (Marzluff et al., 2008). Instead, canonical histone mRNAs terminate with a stem-loop structure, which is comprised of a highly conserved 26-nucleotide sequence that binds an important factor, stem-loop binding protein (SLBP). SLBP is required for canonical histone mRNA processing and translation (Whitfield et al., 2000).
Canonical histone mRNAs are rapidly degraded at the end of S phase, since they exhibit less stability because of the absence of a poly(A) tail (Harris et al., 1991; Whitfield et al., 2000). The absence of the poly(A) tail is vital for maintaining the steady state levels of canonical histone mRNAs during S phase. Disruption of pre-mRNA processing by the loss of SLBP results in polyadenylation of canonical histone mRNAs, increasing their stability and elevating the level of H3.1 protein (Lanzotti et al., 2002; Sullivan et al., 2001). This could disrupt histone stoichiometry and induce genome instability. Deletion and mutation of SLBP have been found to induce polyadenylation of canonical histone mRNAs in Drosophila, causing failure of chromosome condensation that blocks normal mitosis process, resulting in zygotic lethality (Henikoff and Smith, 2015; Lanzotti et al., 2002; Sullivan et al., 2001, 2009).
We previously demonstrated that the 3′ end of canonical histone H3.1 mRNA can be polyadenylated following exposure to arsenic (Brocato et al., 2014). Arsenic causes human lung, bladder, and skin cancers (Mendez et al., 2017) and is linked to cancers of the kidney, liver, and prostate (Roh et al., 2017; Smith et al., 2018; Wang et al., 2014). Overexpression of polyadenylated H3.1 mRNA enhanced anchorage-independent growth of human bronchial epithelial BEAS-2B cells, demonstrating that polyadenylation of H3.1 mRNA induces cell transformation (Brocato et al., 2015).
Polyadenylated canonical histone H3.1 mRNA could induce genomic instability and cell transformation by disrupting the balance between canonical and variant histones. Deposition of canonical histone H3.1 is coupled with DNA replication, and its expression is largely limited to the S phase of the cell cycle, whereas histone variant H3.3 is expressed throughout the cell cycle independent of DNA replication (Brown et al., 1985; Green et al., 2005; Tagami et al., 2004; Wu et al., 1982; Wunsch and Lough, 1987). Polyadenylated canonical H3.1 mRNAs following arsenic exposure were increased during S phase and were also presented outside of S phase (Brocato et al., 2014). It is conceivable that accumulated or mistimed expression of H3.1 during the cell cycle affects H3.3 assembly, causing significant impacts on chromatin landscape and genome integrity, because H3.3 has unique missions in chromatin that are distinct from canonical H3 (Buschbeck and Hake, 2017; Szenker et al., 2011).
This study investigates the mechanisms that underlie the tumorigenic effects of polyadenylated H3.1 mRNA. We find that polyadenylated H3.1 mRNA displaces the variant H3.3 from important gene regulatory elements such as active promoters, enhancers, and insulators. Reducing H3.3 expression, which mimics H3.3 depletion by H3.1 polyadenylation, induces cell transformation, whereas overexpression of H3.3 rescues cell transformation induced by arsenic. Arsenic exposure also results in displacement of H3.3 from active promoters, enhancers, and insulators. H3.3 displacement appears to be central to the tumorigenic responses caused by polyadenylated H3.1 mRNA following arsenic exposure. Our findings add important insights not only into genomic instability induced by an imbalance in histone stoichiometry but also into the oncogenic role of H3.3.
Results
Polyadenylation of Canonical Histone H3.1 mRNA Enhances Tumor Formation in Nude Mice
We previously demonstrated that arsenic induces polyadenylation of canonical histone H3.1 mRNA, by depletion of SLBP in cell culture (Brocato et al., 2014). To study the impact of arsenic-induced polyadenylation of canonical histone H3.1 mRNA on carcinogenesis, we analyzed the effect of H3.1 mRNA polyadenylation on cell proliferation, anchorage-independent growth, and tumor formation in vivo. An H3.1poly(A)-expressing plasmid was constructed that contained a poly(A) signal immediately downstream of an H3.1 cDNA fragment to express polyadenylated histone H3.1 mRNA (Figure 1A). Because there were no antibodies against H3 that distinguish the different types of histone H3, human bronchial epithelial cells (BEAS-2B) were stably transfected with FLAG-tagged H3.1poly(A) plasmid (FH3.1poly(A)) (Figure 1A). Overexpression of both total and polyadenylated H3.1 mRNA was confirmed using RT-qPCR (Figure 1B). Total and polyadenylated mRNA was measured using cDNAs synthesized with random or oligo (dT) primers, respectively (Figure 1B). Total H3 protein levels were increased by 2.67-fold compared with control (lanes 1 and 2 in Figure 1C left panel; 1.90 + 2.03/1.47 = 2.67 versus 1/1 = 1), which is comparable to about the two-fold increase induced by arsenic exposure (1.44/0.78 = 1.85 in Figure 1C right panel and Brocato et al. (2014)). Thus, results using the FH3.1poly(A) plasmid mimic arsenic-induced H3.1 polyadenylation.
Figure 1.

Polyadenylation of Canonical Histone H3.1 mRNA Enhances Tumor Formation in Nude Mice
(A) An H3.1 cDNA fragment was inserted into the multiple clone sites (MCSs) of pcDNA-FLAG vector, which contains a poly(A) signal in the downstream of MCSs and generates polyadenylated mRNAs.
(B) pcDNA-FLAG-Empty (EV) or FH3.1poly(A) vector was stably transfected into BEAS-2B cells. The amount of total and polyadenylated H3.1 mRNA was measured by RT-qPCR using cDNAs synthesized with random or oligo (dT) primers, respectively. mRNA levels for H3.1 were normalized to Actin. The data shown are the mean ± S.D. (n = 3).
(C) Western blot shows exogenous H3.1 protein generated by transfection of FH3.1poly(A)(left) and the increase in H3 protein following arsenic treatment (right).
(D) Cell growth rate was measured by MTT. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗∗p < 0.01.
(E) Soft agar assays. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗∗p < 0.01.
(F–H) A total of five million transformed or control cell clones were injected into nude mice. After 5 months postinjection, the mice were sacrificed. Tumor number (F), volume (G), and weight (H) were calculated. Tumor diameters were measured with calipers, and the tumor volume was calculated. Results represent the mean ± S.D. (n = 18). Student's t test was applied for statistical significance: ∗p < 0.05.
The rate of cell proliferation was increased in BEAS-2B cells transfected with FH3.1poly(A) plasmid compared with empty vector (Figure 1D). Consistent with previous findings (Brocato et al., 2015), BEAS-2B/FH3.1poly(A) cells displayed enhanced anchorage-independent growth compared with BEAS-2B cells transfected with or without empty vector (Figure 1E). To evaluate the tumorigenic response of polyadenylated H3.1 mRNA in vivo, soft agar clones formed by control and FH3.1poly(A)-expressing BEAS-2B cells were collected and injected into nude mice to assess tumor formation. Juxtaposing the two groups, mice injected with FH3.1poly(A) cells exhibited 50% tumor formation compared with 11% in the control group (Figure 1F). Moreover, xenograft of FH3.1poly(A) cells in mice displayed significantly higher volume and weight than the control (Figures 1G and 1H). Taken together, these data suggest that polyadenylation of H3.1 mRNA promotes tumor cell growth in nude mice.
Insertion of a Stem-Loop Sequence before the Poly(A) Signal Attenuates Cell Transformation Induced by Polyadenylation of H3.1 mRNA
To examine whether acquisition of a poly(A) tail is the determining factor for cell transformation induced by transfection of FH3.1poly(A) plasmid, a plasmid H3.1Loop that contained a stem-loop sequence in front of the poly(A) signal was constructed (Figure 2A). Following binding of SLBP and other 3′ pre-mRNA processing factors to the stem-loop sequence, a majority of transcriptions were terminated immediately downstream of the sequence, before the RNA polymerase could transcribe the poly(A) signal. Thus, the presence of the stem-loop sequence generates mRNAs with less poly(A) tails (Figure 2A). To rule out the potential effects resulting from FLAG tag (Figure 1), we prepared H3.1poly(A) and H3.1Loop plasmids lacking FLAG (Figure 2A). RT-qPCR demonstrated that transfection with the plasmid-containing H3.1Loop sequence significantly reduced the amount of polyadenylated H3.1 mRNA compared with cells transfected with H3.1poly(A) vector (Figure 2B). Western blot revealed lower levels of H3 protein in cells transfected with H3.1Loop compared with H3.1poly(A)(Figure 2C). The difference seemed not to have resulted from the variation of inserted DNA copy numbers, because digital droplet PCR (ddPCR) experiments showed even more DNA copy numbers of ectopic H3.1Loop DNA in H3.1Loop-transfected cells than the copy numbers of ectopic H3.1poly(A) DNA in H3.1poly(A)-transfected cells (Figure 2D). RNA stability assays exhibited a dramatic decrease of ectopic H3.1Loop mRNA as compared with ectopic H3.1poly(A) mRNA in the cells treated with actinomycin D (Figure 2E), supporting the idea that the reduction of H3 protein in H3.1Loop-transfected cells was due to the instability and degradation of unpolyadenylated histone mRNAs. Taken together, any differential results obtained from using H3.1poly(A) and H3.1Loop are likely attributable to the effects of mRNA polyadenylation.
Figure 2.

Insertion of a Stem-Loop Sequence before the Poly(A) Signal Attenuates Cell Transformation Induced by Polyadenylation of H3.1 mRNA
(A) An H3.1 cDNA fragment was inserted into the MCSs of pcDNA vector, which contains a poly(A) signal in the downstream of MCSs. H3.1Loop plasmid: a stem-loop sequence was inserted before the poly(A) signal, whose transcription terminates near the stem-loop sequence, generating H3.1 mRNAs mostly without poly(A) tail. pcDNA-Empty (EV), H3.1poly(A), or H3.1Loop vector was stably transfected into BEAS-2B cells.
(B) RT-qPCR results using oligo (dT) primers for reverse transcription to capture polyadenylated H3.1 mRNAs. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗p < 0.01.
(C) Western blot analysis. The band intensities were quantified using ImageJ software. The controls in lane 1 were used as references (set to 1).
(D) Digital droplet PCR (ddPCR). Total DNA was extracted from BEAS-2B cells stably expressing H3.1poly(A) or H3.1Loop plasmid. The copy number of exogenous H3.1 cDNA was analyzed by ddPCR with primers designed to amplify a region spanning T7 and H3.1 cDNA on the vector. The ribonuclease P/MRP 30 kDa subunit (RPP30) was used as an internal control. The data shown are the mean ± S.D. (n = 3).
(E) RNA stability assay for exogenous H3.1poly(A) mRNA and H3.1Loop mRNA. BEAS-2B cells stably expressing H3.1poly(A) or H3.1Loop were treated with 10 μg/mL actinomycin D for the indicated time. Total RNA was extracted, and the mRNA level was analyzed by qPCR with primers designed to amplify the exogenous H3.1 mRNA. The relative expression level was normalized by the reference gene GAPDH. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗∗p < 0.01.
(F) The cell growth rate was measured by MTT. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗p < 0.05; ∗∗p < 0.01.
(G) Soft agar assays. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗∗p < 0.01. See also Figure S1.
(H) Transwell invasion assays. The migrated cells were fixed, stained, and counted. Cell number was determined and averaged from three fields randomly selected from each sample. Bars indicate S.D. (n = 3). Student's t test was applied for statistical significance: ∗p < 0.05.
We next examined how transfection of H3.1poly(A) and H3.1Loop differentially affected cell growth, transformation, and invasion. Although H3.1poly(A)-transfected cells exhibited accelerated growth compared with controls, transfection with H3.1Loop reduced the rate of cell growth to levels in control (Figure 2F). Anchorage-independent growth of BEAS-2B cells was enhanced by the transfection of H3.1poly(A) compared with control (Figure 2G), consistent with the results seen after transfection of FLAG-tagged H3.1poly(A)(Figure 1E), suggesting that the tag did not cause cell transformation. Insertion of the Loop sequence upstream of the poly(A) signal attenuated anchorage-independent growth (Figure 2G). Similar results were obtained with human prostate epithelial PZ-HPV-7 cells and human embryonic kidney 293 cells (Figure S1), suggesting that the impact of H3.1 polyadenylation on cell transformation is not limited to BEAS-2B. Likewise, H3.1poly(A) cells exhibited a much higher rate of cell migration than H3.1Loop and control cells in Transwell invasion assays (Figure 2H). The above results demonstrate generality across multiple human cell types of the role of polyadenylation of H3.1 mRNA in cell transformation.
Overexpression of Polyadenylated H3.1 mRNA Displaces Histone Variant H3.3 from Critical Gene Regulatory Elements
Polyadenylation of H3.1 mRNA results in the increase of H3.1 protein (Figures 1C and 2C). Moreover, arsenic exposure induces polyadenylation of canonical histone H3.1 mRNA, resulting in the accumulation of H3.1 mRNA in mid-to-late S and M phase (Brocato et al., 2014). The expression of canonical histone H3.1 peaks during S phase, whereas variant H3.3 is expressed throughout the cell cycle. Thus, both an increase in H3.1 protein and the presence of H3.1 protein outside of S phase from polyadenylation may have an impact on the assembly of histone H3.3, which differs from H3.1 by only five amino acids. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) was utilized to assess the potential effects of polyadenylated H3.1 mRNA on H3.3 assembly. BEAS-2B cells stably expressing FLAG-tagged H3.3 were transfected with untagged H3.1poly(A) plasmid or empty vector. RT-qPCR results confirmed the overexpression of polyadenylated H3.1 mRNA (Figure S2A). Notably, the total cellular level of FLAG-H3.3 was not changed by ectopic expression of H3.1poly(A) (Figure S2B).
H3.3-containing nucleosomes (FLAG-tagged) were isolated by anti-FLAG affinity-gel purification followed by ChIP-seq to examine how genome-wide H3.3 deposition was influenced by ectopic expression of H3.1poly(A). Results revealed highly enriched H3.3 in the promoter regions of 2,000 of the most highly expressed genes in control cells (blue line, left panel in Figures 3A and S3). This was consistent with previous findings in another cell line (Jin et al., 2009). The deposition of H3.3, however, was greatly reduced in cells transfected with untagged-H3.1poly(A) (green line, left panel in Figures 3A and S3), suggesting that ectopic expression of polyadenylated H3.1 mRNA compromises the assembly of H3.3 in nucleosomes around active promoters. Similar results were seen for DNaseI hypersensitive sites (DHSs)—defined as enhancers (middle panel in Figures 3A and S3)—and CTCF binding sites—defined as insulators (right panel in Figures 3A and S3). The same results were obtained when the ChIP-seqs were repeated (not shown). Collectively, these results indicate that increased H3.1 protein following polyadenylation of H3.1 mRNA competes with H3.3 for deposition at promoter regions of active genes and other regulatory regions, thereby disrupting proper assembly of H3.3.
Figure 3.

Overexpression of Polyadenylated H3.1 mRNA Displaces Histone Variant H3.3 from Critical Gene Regulatory Elements
(A) Profile of FLAG-tagged H3.3-containg nucleosomes in FLAG-tagged cells across the transcription start sites (TSSs) for the top 2,000 highly expressed genes (active promoters) (left), DNase I hypersensitive sites (enhancers) (middle), and CTCF-binding sites (insulators) (right). H3.3-containing nucleosomes were isolated followed by ChIP-seq with FLAG antibody. Relative H3.3 levels were calculated by dividing average ChIP signal by average Input signal per 20 bps bin. See also Figures S2 and S3.
(B) MA plot showing differential H3.3 level in FH3.3-NT3.1 compared with FH3.3 on all genes.
(C) MA plot showing differential H3.3 level in FH3.3-NT3.1 compared with FH3.3 on the 2,000 highly expressed genes.
(D) Differential H3.3 level in FH3.3-NT3.1 compared with FH3.3 on all genes (light gray) and on the 2,000 most highly expressed genes (red). p value was calculated by unpaired two-tailed Student's t test.
(E) Differential H3.3 level in FH3.3-NT3.1 compared with FH3.3 on top (red), middle (light gray), and bottom (blue) 2,000 genes, ranked by H3.3 level in FH3.3 cells. p value was calculated by unpaired two-tailed Student's t test.
(F) Profile of endogenous H3.3-containg nucleosomes in untagged cells around the transcription start sites (TSSs) on the 2,000 most highly expressed genes (active promoters) (left), DNase I hypersensitive sites (enhancers) (middle), or CTCF-binding sites (insulators) (right). H3.3-containing nucleosomes were isolated followed by ChIP-seq with the H3.3-specific antibody.
(G) Profile of H3.3-containg nucleosomes in untagged cells around the transcription start sites (TSSs) on the top 2,000 genes (ChIP top 2k) (left), top 2,000 DNase I hypersensitive sites (Enhancers top 2k) (middle), and top 2,000 CTCF-binding sites (Insulators top 2k) (right), ranked by H3.3-ChIP signal level in NT-Empty cells. H3.3-containing nucleosomes were isolated followed by ChIP-seq with the H3.3-specific antibody.
(H) Profile of H3.3-containg nucleosomes in cells treated with or without arsenic (As) around the transcription start sites (TSSs) on the 2,000 most highly expressed genes (active promoters) (left), DNase I hypersensitive sites (enhancers) (middle), or CTCF-binding sites (insulators) (right). H3.3-containing nucleosomes were isolated followed by ChIP-seq with the H3.3-specific antibody.
The ChIP-seq profile for H3.3 at active promoters (left panel in Figure 3A) measures combined read counts (RPKM) of the top 2,000 highly expressed genes. It does not reflect the changes in each promoter. To explore how H3.3 level in the promoter of each of the top 2,000 highly expressed genes is changed by transfection of H3.1poly(A), the normalized read count (RPKM) around TSS (ranging from −150 bp to +50 bp) of every gene, including the top 2,000 highly expressed genes in FLAG-H3.3 (FH3.3) cells transfected either with untagged H3.1(NTH3.1) or empty vector, was calculated. We then compared the H3.3-binding differences in FH3.3-NTH3.1 cells versus that of the FH3.3-empty cells for each gene (Figure 3B). Although a large number of genes among the top 2,000 displayed increased levels of H3.3 after transfection of H3.1poly(A) (Figure 3C), the overall H3.3 level in the promoters of the top 2,000 genes in FH3.3-NTH3.1 cells demonstrated significant decreases compared with FH3.3-Empty cells, in the context of all genes (p < 2.4 × 10−11, Figure 3D). These results indicate that H3.3 assembly in promoters of most active genes is more likely to be compromised by polyadenylation of H3.1 mRNA.
To further understand the nature of H3.3 reduction in the promoter regions after transfection of H3.1poly(A), all genes were sorted based on H3.3 enrichment prior to transfection at TSS (from −150 bp to +50 bp) from strongest to weakest, and then the top 2,000, middle 2,000, and bottom 2,000 genes were chosen from the list. Next, the extent of H3.3 reduction induced by polyadenylation of H3.1 mRNA among these three sets of genes were compared. Although average H3.3 levels in the promoter regions of the middle 2,000 genes were not changed by H3.1poly(A) transfection, the levels for the top 2,000 genes were significantly decreased (Figure 3E). These data indicate that H3.3 in active promoters and/or in promoters with high level of H3.3 are preferentially displaced by the increased H3.1 protein caused by polyadenylation of the mRNA.
Insertion of a Stem-Loop Sequence before the Poly(A) Signal Attenuates H3.3 Displacement from Gene Regulatory Elements
ChIP-seq analysis of H3.3-containing nucleosomes (FLAG-tagged), purified using affinity-gel from FLAG-H3.3-expressing cells, demonstrated displacement of H3.3 (FLAG-tagged) by polyadenylated H3.1 mRNA from the active promoters, enhancers, and insulators (Figure 3A). To determine if endogenous H3.3 is displaced by polyadenylation of H3.1 mRNA, ChIP-seq with the H3.3-specific antibody was employed with BEAS-2B cells that express empty vector or H3.1poly(A). Endogenous H3.3-containing nucleosomes were enriched in the promoter regions of the 2,000 most highly expressed genes and in enhancers and insulator regions in control cells (blue lines in Figure 3F). The pattern was very similar to that seen for FLAG-H3.3 nucleosomes (Figure 3A) except that FLAG-H3.3 nucleosomes were depleted at the +1 position, whereas relatively high level of H3.3-ChIP signals (endogenous H3.3) were detected at the same site (compare left panels in Figures 3A and 3F). Importantly, the endogenous H3.3 was displaced from immediately upstream of the active TSSs, enhancers, and insulator regions in the cells transfected with H3.1poly(A) plasmid (green lines in Figure 3F). This was consistent with the results obtained with FLAG-affinity gel purification in cells expressing FLAG-H3.3.
To explore whether polyadenylation of the 3′ end was the defining factor for H3.3 displacement by H3.1 mRNA, we compared the level of H3.3 in the cells transfected with H3.1poly(A) or H3.1Loop in the top 2,000 promoters, enhancers, and insulators, ranked by H3.3 level in the empty vector-transfected cells. Whereas overexpression of H3.1poly(A) depleted H3.3 at these sites (green lines in Figure 3G), overexpressing H3.1Loop decreased neither the ChIP-seq signal for H3.3 at enhancers and insulators nor the signal to a lesser extent than H3.1poly(A) in the promoter regions (red lines in Figure 3G). We conclude that polyadenylation is the determining factor for H3.3 displacement from critical regulatory elements by ectopic expression of H3.1poly(A).
Arsenic Exposure Leads to Displacement of H3.3 from Gene Regulatory Elements
Arsenic exposure induces polyadenylation of H3.1 mRNA. To correlate H3.1 mRNA polyadenylation with arsenic effects, we compared the relevance of arsenic exposure and polyadenylation of H3.1 mRNA on H3.3 nucleosome assembly genome-wide by ChIP-seq with H3.3 antibody in the absence and presence of arsenic. H3.3 was enriched in active promoters, enhancers, and insulators in control (blue lines in Figure 3H). Ninety-six hours after the treatment with 1 μM of arsenic, H3.3 was almost entirely abolished from these sites (green lines in Figure 3H), a pattern that resembled H3.3 profiles following overexpression of H3.1poly(A). These results demonstrate the overlapping effects of arsenic exposure and H3.1 mRNA polyadenylation on H3.3 deposition. Given the importance of H3.3 in gene regulation, cell memory, and maintenance of genome integrity, these data suggest that the displacement of H3.3 at important genomic loci through polyadenylation of canonical H3.1 mRNA is a potentially significant contributor to arsenic-induced carcinogenesis.
Ectopic Expression of Polyadenylated H3.1 mRNA Deregulates Cancer-Associated Genes
Reduction of the occupancy of histone variant H3.3 in critical gene regulatory elements such as active promoters, enhancers, and insulator regions by overexpression of polyadenylated H3.1 mRNA (Figure 3) implicates that polyadenylation of H3.1 mRNA might alter gene expression. To investigate global changes in transcription induced by ectopic expression of polyadenylated H3.1 mRNA, RNA-Seqs using BEAS-2B cells stably transfected with empty vector, H3.1poly(A), or H3.1Loop were carried out. The heatmap and volcano plot showed differential expression of hundreds of genes in H3.1poly(A) cells as compared with H3.1Loop cells (Figures 4A and 4B), indicating that transcription profile of the cells overexpressing H3.1poly(A) differ from the cells overexpressing H3.1Loop. By comparing transcriptomes among H3.1poly(A)-transfected cells, H3.1Loop-transfected cells, and the empty vector control, we then identified 380 genes that were upregulated in H3.1poly(A)(log2FC > 0.263; adj. p < 0.1) and were either upregulated in H3.1Loop with a lower fold change (<0.9 in H3.1poly(A)) or not differentially expressed in H3.1Loop as compared with the empty vector control. Another 326 genes that were downregulated in H3.1poly(A)(log2FC < −0.263; adj. p < 0.1) and were either downregulated in H3.1Loop with a lower fold change (<0.9 in H3.1poly(A)) or not differentially expressed in H3.1Loop as compared with the empty vector control were also characterized. We identified a total of 706 genes that were likely regulated by polyadenylation of H3.1 mRNA as poly(A)-specific differentially expressed genes (DEGs). Next, Ingenuity pathway analysis (IPA) was performed on 706 poly(A)-specific DEGs to characterize cellular pathways associated with polyadenylation of H3.1 mRNA. Among the top five diseases and disorders associated with the DEGs, cancer was ranked number 1 (Figure 4C). Key cancer pathways that were analogous to arsenic-targeted cancers were identified (Figure 4D), including non-small-cell lung cancer, small-cell lung cancer, and pancreatic adenocarcinoma signaling (Kuo et al., 2017; Liu-Mares et al., 2013).
Figure 4.

Ectopic Expression of Polyadenylated H3.1 mRNA Deregulates Cancer-Associated Genes
(A) Heatmap showing z-scored gene expression levels (log2 of TPM+1) of differentially expressed genes in two replicates of H3.1Loop and two replicates of H3.1poly(A). Up- and downregulated genes, 362 and 227, respectively, were identified by comparing transcriptomes in H3.1poly(A)-transfected cells and in H3.1Loop-transfected cells, with fold-change cutoffs at 1.2 (log2FC > 0.263 or log2FC < −0.263) and adjusted p value cutoff of 0.1 (adj.p < 0.1).
(B) Volcano plot showing log2 of fold change (x axis) and –log10 of adj.p (y axis) of gene expression changes comparing H3.1poly(A) versus H3.1 Loop. Differentially expressed genes (|log2FC|>0.263 and adj.p < 0.1) were marked as red dots. The horizontal black dotted line represents adj.p cutoff of 0.1; the vertical black dotted line represents log2FC = 0.
(C–F) Ingenuity pathway analyses (IPA) using transcriptome data generated from RNA sequencing (RNA-Seq) of BEAS-2B cells stably transfected with untagged H3.1poly(A), H3.1Loop, or empty vector. See also Figure S4. (C) Illustrations of top diseases and disorders associated with 706 poly(A)-specific DEGs. (D) Illustration of cancer pathway analysis based on 706 poly(A)-specific DEGs. (E) Illustrations of top upstream regulators associated 706 poly(A)-specific DEGs. (F) Illustration of top canonical pathways based on 380 poly(A)-specific upregulated genes.
(G) Illustration of top networks associated with 380 poly(A)-specific upregulated genes.
Top upstream regulators of H3.1poly(A)-controlled genes included TP53, ADRB, TGFB1, and RB1 (Figure 4E). Moreover, top canonical pathway of H3.1poly(A)-regulated genes included G2/M DNA damage checkpoint regulation, death receptor signaling, ATM signaling, and p53 signaling (Figure 4F). High frequency of mutations in TP53 gene is common in lung cancers (Shajani-Yi et al., 2018). About 8% of lung cancers showed ATM mutations (Ding et al., 2008), suggesting that H3.1poly(A) may play a role in lung carcinogenesis through similar pathways initiated by TP53 or ATM mutations. In addition, top associated networks for 380 poly(A)-specific upregulated genes included RNA posttranscriptional modification, cell growth and proliferation, embryonic development, cell cycle, DNA replication, recombination, and repair (Figure 4G). Top associated networks for downregulated genes also included RNA posttranscriptional modification (Figure S4). Cancer was a top disease, and mitochondrial dysfunction was one of the top canonical pathways associated with H3.1poly(A) downregulated genes (Figure S4). These data suggest that ectopic expression of H3.1poly(A) can engender differential expression of cancer-related genes, thereby activating pathways involved in driving tumorigenic properties of human bronchial epithelial cells.
Polyadenylation of H3.1 mRNA Causes Mitotic Arrest and Genomic Instability
Polyadenylation of H3.1 mRNA disrupts H3.3 assembly at gene regulatory regions such as promoters, enhancers, and insulators (Figure 3). H3.3 is also localized at other critical genomic loci, including pericentric heterochromatin regions and telomeres (Szenker et al., 2011; Udugama et al., 2015). The deposition of H3.3 at these regions may also be disrupted by polyadenylation of H3.1 mRNA. Moreover, analysis of top networks for differentially expressed genes by polyadenylated H3.1 mRNA identified cell-cycle progression and DNA repair, among others, as top associated functions (Figure 4). Thus, polyadenylation of canonical histone H3.1 mRNAs may trigger the dysregulation of cell cycle control and induce chromosomal instability. To examine this possibility, flow cytometry was used to compare alterations in cell cycle between control cells transiently transfected with empty vector and cells transiently transfected with H3.1poly(A) or with H3.1Loop. Significantly less cells were found in G1 phase when transfected with H3.1poly(A) compared with control and H3.1Loop cells (Figures 5A and 5C). Furthermore, cells transfected with H3.1poly(A) experienced a greater G2/M arrest than control and H3.1Loop (Figures 5A and 5C). To further distinguish between G2 and M arrest, flow cytometry was employed to determine the cellular level of histone H3S10 phosphorylation, a mitosis marker. The phosphorylation of H3S10 increased two-fold following transfection of H3.1poly(A) plasmid compared with control (Figures 5B and 5C). H3.1Loop-transfected cells displayed phosphorylation of H3S10 at a level similar to empty vector (Figures 5B and 5C). These results indicate that ectopic expression of H3.1 poly(A) mRNA causes mitotic arrest and that polyadenylation of its 3’ end is critical.
Figure 5.

Polyadenylation of H3.1 mRNA Causes Mitotic Arrest and Genomic Instability
(A) Flow cytometry analysis of the cell cycle. BEAS-2B cells that transiently transfected with empty vector (EV), H3.1Loop, or H3.1poly(A) were stained with PI.
(B) Flow cytometry analysis of the cells stained with antibodies specially against phosphorylated H3S10 (H3S10p) to monitor M phase cells.
(C) The experiment shown in (A) and (B) was repeated three times, and statistical analysis on the data was performed. The plot was represented as the percentage of cells in each cell cycle phase, polyploid/aneuploid cells, or H3S10p positive cells. The data shown are the mean ± S.D. from experiments performed in triplicate. Student's t test was applied for statistical significance: ∗∗p < 0.01.
(D–G) Giemsa-stained spread chromosomes from BEAS-2B cells transfected with EV, H3.1poly(A), or H3.1Loop plasmid. (D) Metaphase spread from control cells showing normal chromosome spread (scale bar, 5 μm). (E) Metaphase spread from H3.1poly(A) cells showing abnormal chromosome spread (scale bar, 5 μm), including chromatid break (a), deletion (b), dicentric chromosome (c), triradial chromosome (d) etc. (F) Metaphase spread from H3.1Loop cells showing mostly normal chromosome spread (scale bar, 5 μm). (G) Quantification of frequency of chromosome aberration after BEAS-2B cells were transfected with EV, H3.1poly(A), or H3.1Loop plasmid. The plot was represented as the rate of aberrations per 60 chromosome spreads for each cell line. The table was represented as the ratio of different types of aberration per 60 chromosomes for each cell line.
Flow cytometry showed that the number of aneuploid cells increased in H3.1poly(A)-overexpressing cells by 3.4- and 2.8-fold when compared with the control (from 3.19% to 10.85%) and the H3.1Loop (from 3.84% to 10.85%), respectively (Figure 5C). These results indicate that polyadenylation of H3.1 mRNAs causes defective chromosome segregation. To study chromosomal aberrations induced by polyadenylated H3.1 mRNA, cytogenetic analysis with empty vector, H3.1poly(A), or H3.1Loop cells was carried out. Although empty vector-transfected cells displayed normal metaphase spread (Figures 5D and 5G), deletions, chromatid breaks, and dicentric and triradial chromosomes were observed in H3.1poly(A)-transfected cells (Figure 5E). Moreover, among 60 chromosome spreads tested, about 90% exhibited some aberrations in H3.1poly(A)-transfected cells (Figure 5G). In contrast, only 3% displayed deletion or chromatid break in H3.1Loop cells (Figure 5G). Collectively, these results show that polyadenylation of H3.1 mRNA induces mitotic arrest and genomic instability.
Ectopic Expression of H3.3 Attenuates Arsenic-Induced Anchorage-Independent Cell Growth
Polyadenylation of H3.1 mRNA induced by arsenic exposure compromised assembly of H3.3 at important genomic loci. Numerous previous findings have linked abnormal H3.3 to cancer initiation and development (Buschbeck and Hake, 2017; Jang et al., 2015; Shi et al., 2017; Soria et al., 2012; Szenker et al., 2011; Talbert and Henikoff, 2010). Moreover, H3.3-knockout mice were embryonically lethal and displayed severe chromosomal aberrations (Bush et al., 2013; Jang et al., 2015), suggesting that disruption of H3.3 assembly might be a significant contributor to arsenic carcinogenesis. H3.3 was knocked down to assess effects on anchorage-independent cell growth. Both H3.3 mRNA levels (Figure 6A) as well as protein (Figure 6B) were reduced by transfection of two different types of H3.3 siRNAs in BEAS-2B cells compared with control siRNA. Soft agar growth showed that knocking down histone H3.3 enhanced colony formation (Figure 6C). To address whether the effect of H3.3 knockdown and arsenic treatment was additive or synergistic in causing cell transformation, we performed soft agar assays with H3.3 siRNA and control siRNA cells in the presence and absence of arsenic. In the case of H3.3 siRNA 1, the effect of H3.3 knockdown plus arsenic equaled the individual effects combined, whereas with H3.3 siRNA 2, the effect was obviously greater than the individual responses combined (Figure S5). Given that H3.3 siRNA 1 alone increased colony formation in soft agar by more than two-fold versus H3.3 siRNA2 (Figure S5), if the effect of H3.3 knockdown and arsenic is additive or synergistic appeared to be depending on the extent of the individual effect of H3.3 knockdown.
Figure 6.

Ectopic Expression of H3.3 Attenuates Arsenic-Induced Anchorage-Independent Cell Growth
(A–C) Knockdown of H3.3 facilitates colony formation of BEAS-2B cells on soft agar. (A) RT-qPCR analysis of H3.3 mRNA levels in BEAS-2B cells after 48-h transfection with control (Ctrl) siRNA or with two distinct siRNAs for H3.3. (B) Western blot using indicated antibodies. (C) Soft agar assays. The cells were grown in soft agar for 6 weeks. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗∗p < 0.01. See also Figure S5.
(D and E) Overexpression of H3.3 rescues arsenic-induced cell transformation. BEAS-2B cells were transiently transfected with pcDNA-empty (EV) or with pcDNA-FLAG-H3.3 (FH3.3) in the presence or absence of 1 μM arsenic. (D) Western blot using indicated antibodies. (E) Soft agar assays. The data shown are the mean ± S.D. (n = 3). Student's t test was applied for statistical significance: ∗p < 0.05; ∗∗p < 0.01. See also Figure S6.
To further examine whether inhibition of H3.3 assembly was the underlying defect in arsenic-induced cell transformation, H3.3 was overexpressed (Figure 6D) to assess if this could have antagonized arsenic-induced soft agar growth. Anchorage-independent cell growth increased following treatment of BEAS-2B cells with arsenic for either 3 days at 1 μM or 1 week at 0.5 μM (Figure 6E). Overexpression of H3.3 attenuated arsenic-induced colony growth in soft agar (Figure 6E), suggesting that aberrant histone H3.3 assembly is a significant contributor to arsenic transformation.
Polyadenylation of H3.1 mRNA and the inhibition of H3.3 assembly were critical for arsenic-induced cell transformation. To further assess the importance of this pathway in arsenic carcinogenesis, SLBP was overexpressed to examine if this could prevent arsenic transformation. BEAS-2B cells were stably transfected with empty vector or SLBP-expressing plasmid. Western blot and RT-qPCR showed that the protein and mRNA levels of SLBP were increased by about three-fold in cells expressing SLBP compared with the empty vector (Figures S6A and S6B). Polyadenylated H3.1 mRNA increased after arsenic treatment in control cells, but arsenic-induced polyadenylation was significantly reduced in the cells that expressed SLBP (Figure S6C). Similarly, overexpression of SLBP inhibited arsenic-induced anchorage-independent growth (Figure S6D), indicating the essential role for SLBP depletion/H3.1 mRNA polyadenylation/disruption of H3.3 assembly in arsenic transformation.
Gain of Polyadenylated H3.1 mRNA by Arsenic Exposure In Vivo
To determine whether arsenic is able to reduce SLBP and alter canonical histone H3.1 mRNA processing in vivo, SLBP and polyadenylation of H3.1 mRNA were measured in male A/J mice exposed to arsenic (0, 100, and 200 μg/L) by oropharyngeal aspiration every other day for 1 week. Lung tissues were collected, and RT-qPCR was first employed to measure H3.1 polyadenylation and SLBP mRNA after arsenic exposure. RT-qPCR showed that polyadenylated H3.1 mRNA was elevated with increasing arsenic exposure (Figure 7A). A reduction in SLBP mRNA was observed most significantly in the 200 μg/L treatment group (Figure 7B). Western blot analysis further demonstrated a downward trend in SLBP protein levels with arsenic exposure (Figure 7C). SLBP is essential for processing of canonical histone pre-mRNA, thus loss of SLBP caused polyadenylation of canonical histone mRNAs in vivo. The notion that loss of SLBP and gain of polyadenylated H3.1 mRNA were detected in mice exposed to arsenic suggests a potential role for polyadenylation of canonical histone H3.1 mRNA in arsenic-induced toxicity and carcinogenesis in vivo.
Figure 7.

Gain of Polyadenylated H3.1 mRNA and Loss of SLBP by Arsenic Exposure In Vivo
(A–C) A/J mice were treated with NaAsO2 via oropharyngeal aspiration every other day for one week. Lung tissues were collected and mRNA or protein levels were analyzed by RT-qPCR (A and B) or Western blot (C). GAPDH and β-actin were used as a loading control for qPCR and Western blot, respectively. The data are shown for each individual mouse. Relative protein levels were calculated based on band intensity. Error bars represent S.D. (n = 8). Student's t test was applied for statistical significance: ∗p < 0.05.
Discussion
We have demonstrated that arsenic exposure induces polyadenylation of canonical histone H3.1 mRNA in vivo and that polyadenylated H3.1 mRNA causes chromosome instability and cell transformation by displacing histone H3.3 from critical genomic loci.
The polyadenylation of H3.1 mRNA increased cellular H3.1 protein, resulting in excess H3.1 not only during S phase but also at other cell cycle phases (Brocato et al., 2014). Although H3.1 was incorporated into the chromatin exclusively by the replication-dependent chromatin assembly factor 1 (CAF-1) complex, many histone chaperones such as HIRA, DAXX, and DEK have been identified as H3.3-specific histone chaperones for replication-independent H3.3 assembly (Burgess and Zhang, 2013; Buschbeck and Hake, 2017; Szenker et al., 2011). CAF-1 complex is composed of three subunits, i.e., p150, p60, and p48. Whereas p150 is specific for H3.1, p60 and p48 exist in HIRA complex as well (Tagami et al., 2004). Thus, excess H3.1 protein might increase the association of H3.1 with CAF-1, including histone chaperones p60 and p48, which may result in shortages of these two chaperones available for HIRA, decreasing the interaction between H3.3 and HIRA complex, thereby compromising H3.3 assembly. Another scenario would be that excess H3.1 protein during M phase may directly compete with H3.3 variant for deposition onto genomic loci with higher H3.3 turnover. This appears to be possible since the overexpression of canonical H3.1 increased replication-independent H3.1 assembly outside of S phase in flies (Sakai et al., 2009). Moreover, a study showed that altering the ratio of H3.1 to H3.3 by increasing H3.1 protein resulted in replacement of H3.3 with H3.1 in regulatory regions of skeletal muscle cells (Harada et al., 2015). Inhibition of H3.3 assembly did not appear to result from downregulation of H3.3-specific chaperones based on RNA-seq data (not shown). However, we cannot rule out the possibility that posttranslational modifications of H3.3 could be altered indirectly by H3.1 polyadenylation, contributing to altered assembly of H3.3 into nucleosomes. The reduction of H3.3 (FLAG-tagged) at these genomic loci was not due to the downregulation of FLAG-H3.3 expression, because (a) FLAG-tagged H3.3 protein levels were not decreased after transfection of H3.1poly(A) plasmid (Figure S2), and (b) FLAG-H3.3 levels were not decreased at all the genomic loci tested, but in fact, were slightly increased in the promoters of silenced genes (Figure 3E). In addition, although H3.3 displacement was more likely to occur in promoters of highly expressed genes as well as genes whose promoters were enriched with H3.3 (Figures 3D and 3E), a direct relationship between the extent of H3.3 loss and gene downregulation was not found (not shown). Therefore, the H3.3 reduction was not likely a secondary event following transcriptional downregulation by H3.1 polyadenylation.
Displacement of H3.3 from critical genomic loci would have significant impact on carcinogenesis. H3.3 depletion resulted in cell death and mitotic defects, leading to karyotypical abnormalities such as lagging chromosomes, aneuploidy, and polyploidy (Jang et al., 2015). H3.3-knockout mice exhibited early embryonic lethality (Bush et al., 2013; Jang et al., 2015). Moreover, recent studies have directly linked histone variant H3.3 to oncogenesis. Sequencing studies revealed a high frequency of mutations in H3.3 genes in several cancers including pediatric high-grade glioblastoma and bone tumors (Lowe et al., 2019; Schwartzentruber et al., 2012; Wu et al., 2012; Yuen and Knoepfler, 2013). Downregulation of H3.3 expression in adult glioblastoma disrupted chromatin organization and promoted cancer stem cell properties (Gallo et al., 2015). Besides mutation and transcriptional downregulation, disruption of the H3.3 assembly was also implicated in cancers (Elsasser et al., 2011; Heaphy et al., 2011; Jiao et al., 2011; Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012). For example, somatic mutations in the DAXX and ATRX, the histone chaperones for H3.3 deposition at telomeric and pericentromeric regions, were identified in pediatric glioblastoma tumors (Dyer et al., 2017; Mahmud and Liao, 2019; Newhart et al., 2013; Sarai et al., 2013). Forty-three percent of pancreatic neuroendocrine tumors (PanNETs) had mutations in genes that encode ATRX and DAXX (Heaphy et al., 2011). A DAXX missense mutation was observed in acute myeloid leukemia (Ding et al., 2012). Mutation of DEK significantly reduced H3.3 loading in human myeloid leukemia patients (Sawatsubashi et al., 2010; Soekarman et al., 1992). These studies indicated an association between disruption of H3.3 assembly and cancer. Arsenic-induced polyadenylation of canonical H3.1 mRNA led to inhibition of H3.3 assembly at critical genomic loci by disrupting the stoichiometry of replication-dependent and -independent histones, causing chromosomal instability. Interestingly, incorporation of H3.1/H3.2 has recently been shown to be reduced by metastasis inducers, triggering deposition of H3.3 into chromatin, which is essential for tumor progress (Gomes et al., 2019). These data suggest that histone variant H3.3 participates in different stages of cancer by differential mechanisms.
Appropriate histone stoichiometry is essential for maintaining genomic integrity. Histone stoichiometry could be altered by either decreased or increased protein levels for a particular histone gene. It has been relatively well known that decreases in histone protein level can induce genomic instability by facilitating chromatin accessibility (Gossett and Lieb, 2012; Han et al., 1987; Kim et al., 1988; Wyrick et al., 1999). The mechanisms that underlie genomic instability induced by increased amounts of histones have not been well studied (Meeks-Wagner and Hartwell, 1986; Singh et al., 2010). Our present study demonstrates that upregulation of canonical histone protein induces chromosome instability by displacing their specific replacement histones from critical genomic loci in mammals.
In Drosophila, SLBP depletion led to polyadenylation of all core histones (Lanzotti et al., 2002; Sullivan et al., 2001, 2009), implicating that mRNAs for other canonical histones besides H3.1 may also be polyadenylated by arsenic. In fact, RT-qPCR results showed that the levels of polyadenylated mRNAs for H2A, H2B, H3.2, and H4 were increased by arsenic (Figures S7A–S7E). The protein levels for H2A, H2B, total H3, and H4 were also upregulated by arsenic (Figures S8A–S8E). The increase in canonical histone proteins other than H3.1 may also affect chromatin assembly and structure. However, the impact of polyadenylation of canonical histone H3 mRNAs is likely to be the most significant. H3.3 seems to be the only histone variant that serves as a “replacement histone.” For example, over a lifespan of the mouse, H3.3 replaces canonical H3 and becomes the predominant H3 protein in nondividing cells. Although histone H2A has several variants, such as H2A.X, H2A.Z, and macroH2A, they are not likely serving as “replacement histones.” Based on the work from Marzluff's group, a subset of canonical histone genes was expressed independently of DNA replication as polyadenylated mRNAs and likely served as “replacement histones” for the H2A, H2B, and H4 in terminally differentiated cells (Lyons et al., 2016). The implication of these observations is that although mRNAs for all canonical histone genes were polyadenylated by arsenic treatment and produced more respective proteins, increased canonical H3 proteins, i.e., H3.1 and H3.2, may have a greater impact on chromatin structure because they can displace the “replacement histone” H3.3, which has distinct roles that differ from canonical H3.1 and H3.2.
A marked increase in polyadenylated mRNA for H3.1 in arsenic-transformed BEAS-2B clones, but not in chromium (VI)-transformed BEAS-2B, has been reported (Brocato et al., 2014). Depletion of SLBP and increase in polyadenylated H3.1 mRNA was also observed following nickel treatment, whereas polyadenylation of H3.1 mRNA was not evident in cadmium-exposed cells (Jordan et al., 2017). These data suggest that the polyadenylation of canonical histone mRNAs is targeted by certain metals. Because both arsenic and chromium (VI) induce oxidative stress, it is unlikely that oxidative stress caused polyadenylation of canonical histone mRNAs. SLBP depletion was a key factor for arsenic-induced polyadenylation of canonical histones, because the transcription of other genes involved in canonical histone mRNA processing, including U7 snRNP (LSM10), CPSF2, 3, XRN2, and ZNF473, was not changed by arsenic exposure (Brocato et al., 2014). Arsenic- and nickel-induced SLBP depletion was epigenetically regulated by its promoter and also by proteasomal degradation. It would be important to identify which histone-/DNA-modifying enzymes or protein kinases/phosphatases were involved in regulating arsenic-induced SLBP depletion (Djakbarova et al., 2016; Koseoglu et al., 2008; Zhang et al., 2014). Arsenic was able to release zinc from proteins that contain 3-cysteine or 4-cysteine zinc finger motifs, thereby changing protein structures and enzyme activities (Zhou et al., 2011), whereas nickel inhibits enzymatic activities of dioxygenases such as histone demethylase JMJD1A by replacing the ferrous iron in the catalytic centers (Chen et al., 2010). Therefore, it would be interesting to examine if the above-characterized enzymes contain zinc finger motif or require iron for their activity.
In summary, arsenic-induced polyadenylation of canonical histone H3.1 mRNA displaces H3.3 variant from promoters, enhancers, and insulator regions, resulting in transcriptional deregulation, cell-cycle arrest, genomic instability, and tumor formation in nude mice. Polyadenylation of canonical histone H3.1 and disruption of H3.3 assembly has been described as an important mechanism for arsenic-induced carcinogenesis. This finding also sheds light on the oncogenic role for H3.3. In addition, a perspective on genomic instability induced by alterations of histone stoichiometry has been uncovered. In the future, studies should address to what extent polyadenylation of canonical histones accounts for the effects of arsenic and by what mechanisms arsenic induces SLBP depletion and increased H3.1 displaces H3.3 at regulatory regions. The findings from these studies will provide a potential therapeutic and preventive target for arsenic-induced cancers.
Limitations of the Study
Although our study is relatively complete using cell culture models, it would be strengthened by showing that polyadenylation of H3.1 mRNAs causes cancer in an animal model by making a transgenic mouse that expresses polyadenylated H3.1 mRNAs.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Max Costa (max.costa@nyulangone.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
All sequencing data created during this study are available at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE135637.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the NYULMC Genome Technology Center, partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. The authors thank Drs. Sitharam Ramaswami, Thomas Des Marais and Gabriele Grunig for technical assistance and Suresh Cuddapah for helpful comments. The graphic abstract was created with BioRender.com. This work was supported by grants from the US National Institutes of Health: R01ES026138 (M.C. and C.J.), P30ES000260 (M.C. and Pilot Project Program to C.J.), R01ES029359 (M.C. and C.J.), R01ES030583 (C.J. and M.C.), R01ES022935 (M.C.), R35GM133712 (C.Z.), and K22CA204439 (C.Z.).
Author Contributions
C.J. and M.C. conceived the study. D.C. and Q.Y.C. performed most of the experiments and data analysis. Z.W. performed most of the computational analysis under supervision of C.Z.; Y.Z., T.K., W.T., J.L., F.W., L.F., X.Z., R.H., S.S., and H.S. performed experiments and/or data analysis; Q.Y.C., D.C., Z.W., C.Z., C.J., and M. C. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101518.
Contributor Information
Chongzhi Zang, Email: zang@virginia.edu.
Chunyuan Jin, Email: chunyuan.jin@nyulangone.org.
Max Costa, Email: max.costa@nyulangone.org.
Supplemental Information
References
- Brocato J., Chen D., Liu J., Fang L., Jin C., Costa M. A potential new mechanism of arsenic carcinogenesis: depletion of stem-loop binding protein and increase in polyadenylated canonical histone H3.1 mRNA. Biol. Trace Elem. Res. 2015;166:72–81. doi: 10.1007/s12011-015-0296-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocato J., Fang L., Chervona Y., Chen D., Kiok K., Sun H., Tseng H.C., Xu D., Shamy M., Jin C. Arsenic induces polyadenylation of canonical histone mRNA by down-regulating stem-loop-binding protein gene expression. J. Biol. Chem. 2014;289:31751–31764. doi: 10.1074/jbc.M114.591883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.T., Wellman S.E., Sittman D.B. Changes in the levels of three different classes of histone mRNA during murine erythroleukemia cell differentiation. Mol. Cell. Biol. 1985;5:2879–2886. doi: 10.1128/mcb.5.11.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess R.J., Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M., Hake S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017;18:299–314. doi: 10.1038/nrm.2016.166. [DOI] [PubMed] [Google Scholar]
- Bush K.M., Yuen B.T., Barrilleaux B.L., Riggs J.W., O'Geen H., Cotterman R.F., Knoepfler P.S. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin. 2013;6:7. doi: 10.1186/1756-8935-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Giri N.C., Zhang R., Yamane K., Zhang Y., Maroney M., Costa M. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem. 2010;285:7374–7383. doi: 10.1074/jbc.M109.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M.D., Cibulskis K., Sougnez C., Greulich H., Muzny D.M., Morgan M.B. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Ley T.J., Larson D.E., Miller C.A., Koboldt D.C., Welch J.S., Ritchey J.K., Young M.A., Lamprecht T., McLellan M.D. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakbarova U., Marzluff W.F., Koseoglu M.M. DDB1 and CUL4 associated factor 11 (DCAF11) mediates degradation of Stem-loop binding protein at the end of S phase. Cell Cycle. 2016;15:1986–1996. doi: 10.1080/15384101.2016.1191708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M.A., Qadeer Z.A., Valle-Garcia D., Bernstein E. ATRX and DAXX: mechanisms and mutations. Cold Spring Harb. Perspect. Med. 2017;7:a026567. doi: 10.1101/cshperspect.a026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S.J., Allis C.D., Lewis P.W. Cancer. New epigenetic drivers of cancers. Science. 2011;331:1145–1146. doi: 10.1126/science.1203280. [DOI] [PubMed] [Google Scholar]
- Gallo M., Coutinho F.J., Vanner R.J., Gayden T., Mack S.C., Murison A., Remke M., Li R., Takayama N., Desai K. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell. 2015;28:715–729. doi: 10.1016/j.ccell.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Gomes A.P., Ilter D., Low V., Rosenzweig A., Shen Z.J., Schild T., Rivas M.A., Er E.E., McNally D.R., Mutvei A.P. Dynamic incorporation of histone H3 variants into chromatin is essential for acquisition of aggressive traits and metastatic colonization. Cancer Cell. 2019;36:402–417 e413. doi: 10.1016/j.ccell.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett A.J., Lieb J.D. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet. 2012;8:e1002771. doi: 10.1371/journal.pgen.1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E.M., Antczak A.J., Bailey A.O., Franco A.A., Wu K.J., Yates J.R., 3rd, Kaufman P.D. Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol. 2005;15:2044–2049. doi: 10.1016/j.cub.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M., Chang M., Kim U.J., Grunstein M. Histone H2B repression causes cell-cycle-specific arrest in yeast: effects on chromosomal segregation, replication, and transcription. Cell. 1987;48:589–597. doi: 10.1016/0092-8674(87)90237-6. [DOI] [PubMed] [Google Scholar]
- Harada A., Maehara K., Sato Y., Konno D., Tachibana T., Kimura H., Ohkawa Y. Incorporation of histone H3.1 suppresses the lineage potential of skeletal muscle. Nucleic Acids Res. 2015;43:775–786. doi: 10.1093/nar/gku1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.E., Bohni R., Schneiderman M.H., Ramamurthy L., Schumperli D., Marzluff W.F. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol. Cell. Biol. 1991;11:2416–2424. doi: 10.1128/mcb.11.5.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy C.M., de Wilde R.F., Jiao Y., Klein A.P., Edil B.H., Shi C., Bettegowda C., Rodriguez F.J., Eberhart C.G., Hebbar S. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Smith M.M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015;7:a019364. doi: 10.1101/cshperspect.a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C.W., Shibata Y., Starmer J., Yee D., Magnuson T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015;29:1377–1392. doi: 10.1101/gad.264150.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Shi C., Edil B.H., de Wilde R.F., Klimstra D.S., Maitra A., Schulick R.D., Tang L.H., Wolfgang C.L., Choti M.A. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark 'nucleosome-free regions' of active promoters and other regulatory regions. Nat. Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A., Zhang X., Li J., Laulicht-Glick F., Sun H., Costa M. Nickel and cadmium-induced SLBP depletion: a potential pathway to metal mediated cellular transformation. PLoS One. 2017;12:e0173624. doi: 10.1371/journal.pone.0173624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U.J., Han M., Kayne P., Grunstein M. Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 1988;7:2211–2219. doi: 10.1002/j.1460-2075.1988.tb03060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseoglu M.M., Graves L.M., Marzluff W.F. Phosphorylation of threonine 61 by cyclin a/Cdk1 triggers degradation of stem-loop binding protein at the end of S phase. Mol. Cell. Biol. 2008;28:4469–4479. doi: 10.1128/MCB.01416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C.C., Moon K.A., Wang S.L., Silbergeld E., Navas-Acien A. The association of arsenic metabolism with cancer, cardiovascular disease, and diabetes: a systematic review of the epidemiological evidence. Environ. Health Perspect. 2017;125:087001. doi: 10.1289/EHP577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzotti D.J., Kaygun H., Yang X., Duronio R.J., Marzluff W.F. Developmental control of histone mRNA and dSLBP synthesis during Drosophila embryogenesis and the role of dSLBP in histone mRNA 3' end processing in vivo. Mol. Cell. Biol. 2002;22:2267–2282. doi: 10.1128/MCB.22.7.2267-2282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Mares W., Mackinnon J.A., Sherman R., Fleming L.E., Rocha-Lima C., Hu J.J., Lee D.J. Pancreatic cancer clusters and arsenic-contaminated drinking water wells in Florida. BMC Cancer. 2013;13:111. doi: 10.1186/1471-2407-13-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe B.R., Maxham L.A., Hamey J.J., Wilkins M.R., Partridge J.F. Histone H3 mutations: an updated view of their role in chromatin deregulation and cancer. Cancers (Basel) 2019;11:660. doi: 10.3390/cancers11050660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons S.M., Cunningham C.H., Welch J.D., Groh B., Guo A.Y., Wei B., Whitfield M.L., Xiong Y., Marzluff W.F. A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues. Nucleic Acids Res. 2016;44:9190–9205. doi: 10.1093/nar/gkw620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud I., Liao D. DAXX in cancer: phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019;47:7734–7752. doi: 10.1093/nar/gkz634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff W.F., Wagner E.J., Duronio R.J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 2008;9:843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks-Wagner D., Hartwell L.H. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell. 1986;44:43–52. doi: 10.1016/0092-8674(86)90483-6. [DOI] [PubMed] [Google Scholar]
- Mendez W.M., Jr., Eftim S., Cohen J., Warren I., Cowden J., Lee J.S., Sams R. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J. Expo. Sci. Environ. Epidemiol. 2017;27:235–243. doi: 10.1038/jes.2016.58. [DOI] [PubMed] [Google Scholar]
- Newhart A., Rafalska-Metcalf I.U., Yang T., Joo L.M., Powers S.L., Kossenkov A.V., Lopez-Jones M., Singer R.H., Showe L.C., Skordalakes E. Single cell analysis of RNA-mediated histone H3.3 recruitment to a cytomegalovirus promoter-regulated transcription site. J. Biol. Chem. 2013;288:19882–19899. doi: 10.1074/jbc.M113.473181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh T., Lynch C.F., Weyer P., Wang K., Kelly K.M., Ludewig G. Low-level arsenic exposure from drinking water is associated with prostate cancer in Iowa. Environ. Res. 2017;159:338–343. doi: 10.1016/j.envres.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A., Schwartz B.E., Goldstein S., Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr. Biol. 2009;19:1816–1820. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarai N., Nimura K., Tamura T., Kanno T., Patel M.C., Heightman T.D., Ura K., Ozato K. WHSC1 links transcription elongation to HIRA-mediated histone H3.3 deposition. EMBO J. 2013;32:2392–2406. doi: 10.1038/emboj.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatsubashi S., Murata T., Lim J., Fujiki R., Ito S., Suzuki E., Tanabe M., Zhao Y., Kimura S., Fujiyama S. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 2010;24:159–170. doi: 10.1101/gad.1857410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A., Tonjes M. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Shajani-Yi Z., de Abreu F.B., Peterson J.D., Tsongalis G.J. Frequency of somatic TP53 mutations in combination with known pathogenic mutations in colon adenocarcinoma, non-small cell lung carcinoma, and gliomas as identified by next-generation sequencing. Neoplasia. 2018;20:256–262. doi: 10.1016/j.neo.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L., Wen H., Shi X. The histone variant H3.3 in transcriptional regulation and human disease. J. Mol. Biol. 2017;429:1934–1945. doi: 10.1016/j.jmb.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.K., Liang D., Gajjalaiahvari U.R., Kabbaj M.H., Paik J., Gunjan A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle. 2010;9:4236–4244. doi: 10.4161/cc.9.20.13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.H., Marshall G., Roh T., Ferreccio C., Liaw J., Steinmaus C. Lung, bladder, and kidney cancer mortality 40 Years after arsenic exposure reduction. J. Natl. Cancer Inst. 2018;110:241–249. doi: 10.1093/jnci/djx201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soekarman D., von Lindern M., van der Plas D.C., Selleri L., Bartram C.R., Martiat P., Culligan D., Padua R.A., Hasper-Voogt K.P., Hagemeijer A. Dek-can rearrangement in translocation (6;9)(p23;q34) Leukemia. 1992;6:489–494. [PubMed] [Google Scholar]
- Soria G., Polo S.E., Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol. Cell. 2012;46:722–734. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Sturm D., Witt H., Hovestadt V., Khuong-Quang D.A., Jones D.T., Konermann C., Pfaff E., Tönjes M., Sill M., Bender S. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Sullivan E., Santiago C., Parker E.D., Dominski Z., Yang X., Lanzotti D.J., Ingledue T.C., Marzluff W.F., Duronio R.J. Drosophila stem loop binding protein coordinates accumulation of mature histone mRNA with cell cycle progression. Genes Dev. 2001;15:173–187. doi: 10.1101/gad.862801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K.D., Steiniger M., Marzluff W.F. A core complex of CPSF73, CPSF100, and Symplekin may form two different cleavage factors for processing of poly(A) and histone mRNAs. Mol. Cell. 2009;34:322–332. doi: 10.1016/j.molcel.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker E., Ray-Gallet D., Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- Talbert P.B., Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Udugama M., FT M.C., Chan F.L., Tang M.C., Pickett H.A., JD R.M., Mayne L., Collas P., Mann J.R., Wong L.H. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 2015;43:10227–10237. doi: 10.1093/nar/gkv847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xie Z., Lin Y., Zhang D. Association of inorganic arsenic exposure with type 2 diabetes mellitus: a meta-analysis. J. Epidemiol. Community Health. 2014;68:176–184. doi: 10.1136/jech-2013-203114. [DOI] [PubMed] [Google Scholar]
- Whitfield M.L., Zheng L.X., Baldwin A., Ohta T., Hurt M.M., Marzluff W.F. Stem-loop binding protein, the protein that binds the 3' end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell. Biol. 2000;20:4188–4198. doi: 10.1128/mcb.20.12.4188-4198.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.S., Tsai S., Bonner W.M. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell. 1982;31:367–374. doi: 10.1016/0092-8674(82)90130-1. [DOI] [PubMed] [Google Scholar]
- Wu G., Broniscer A., McEachron T.A., Lu C., Paugh B.S., Becksfort J., Qu C., Ding L., Huether R., Parker M. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunsch A.M., Lough J. Modulation of histone H3 variant synthesis during the myoblast-myotube transition of chicken myogenesis. Dev. Biol. 1987;119:94–99. doi: 10.1016/0012-1606(87)90210-7. [DOI] [PubMed] [Google Scholar]
- Wyrick J.J., Holstege F.C., Jennings E.G., Causton H.C., Shore D., Grunstein M., Lander E.S., Young R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–421. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- Yuen B.T., Knoepfler P.S. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Tan D., DeRose E.F., Perera L., Dominski Z., Marzluff W.F., Tong L., Hall T.M. Molecular mechanisms for the regulation of histone mRNA stem-loop-binding protein by phosphorylation. Proc. Natl. Acad. Sci. U S A. 2014;111:E2937–E2946. doi: 10.1073/pnas.1406381111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Sun X., Cooper K.L., Wang F., Liu K.J., Hudson L.G. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem. 2011;286:22855–22863. doi: 10.1074/jbc.M111.232926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data created during this study are available at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE135637.