

Abstract
The phenomenon of chemical or conformational exchange in NMR spectroscopy has enabled detailed characterization of time-dependent aspects of biomolecular function, including folding, molecular recognition, allostery, and catalysis, on time scales from μs-s. Importantly, NMR methods based on a variety of spin relaxation parameters have been developed that provide quantitative information on interconversion kinetics, thermodynamic properties, and structural features of molecular states populated to a fraction of a percent at equilibrium and otherwise unobservable by other NMR approaches. The ongoing development of more sophisticated experimental techniques and the necessity to apply these methods to larger and more complex molecular systems engenders a corresponding need for theoretical advances describing such techniques and facilitating data analysis in applications. The present Review surveys current aspects of the theory of chemical exchange, as utilized in ZZ-exchange; Hahn and Carr-Purcell-Meiboom-Gill (CPMG) spin-echo; and R1ρ, chemical exchange saturation transfer (CEST), and dark state saturation transfer (DEST) spin-locking experiments. The Review emphasizes theoretical results for kinetic topologies with more than two interconverting states, both to obtain compact analytical forms suitable for data analysis and to establish conditions for distinguishability between alternative kinetic schemes.
Keywords: Bloch-McConnell, Dynamics, Kinetics, NMR spectroscopy, Spin relaxation
1. Introduction
Knowledge about the dynamics of proteins and nucleic acids can assist in understanding and modulating their functions. A significant part of research in structural biology is devoted to exploration of the conformations of biomolecules. Each conformation corresponds to a specific energetic state; the equilibrium populations of various conformations depend on their conformational energies. “Conformational dynamics” describes the nature and distribution of various conformations of a given biomolecule. Equilibrium between populations is established and maintained by kinetic processes, which correspond to paths between the individual conformational states. For two conformations 1 and 2 with populations p1 and p2, the corresponding rate constants for conversion between the state satisfy p2/p1 = k12/k21. The sum of all populations is unity and does not contain any temporal information; the magnitudes of the rate constants is not defined by the energetic states of the conformational ensembles. The sum k21 + k12 contains temporal information and is defined by the energy barriers along the path connecting the two states. This kinetic information adds the dimension of time to the description of a biomolecule (Ban, Smith, de Groot, Griesinger, & Lee, 2017), which would define the term “dynamics” in a narrower sense. This simple example can be extended to arbitrary numbers of conformational or chemical states.
Dynamic processes in biomolecules occur over a wide range of time scales. This review focuses on NMR methods to study kinetic processes in the μs-s range, which include major conformational changes, molecular recognition, folding and allosteric events. Such phenomenal are generally classified as “chemical exchange” or “conformational exchange” in NMR spectroscopy. Chemical exchange in the μs-ms range is characterized by CPMG and R1ρ relaxation dispersion and related methods, such as CEST and DEST. Slower chemical exchange in the 102-103 ms range, limited by longitudinal relaxation, is characterized by ZZ-exchange or EXSY methods (Farrow, Zhang, Forman-Kay, & Kay, 1995; Furukawa, Konuma, Yanaka, & Sugase, 2016; Montelione & Wagner, 1989; A. G. Palmer, 2014). Faster processes in the ps-ns range mostly can be attributed to more local intramolecular motions. A number of other NMR techniques are used to study these processes, including determination of order parameters, correlation times, residual dipolar couplings, chemical shifts, and cross-correlated relaxation (Charlier, Cousin, & Ferrage, 2016; Kovermann, Rogne, & Wolf-Watz, 2016; A. G. Palmer, 2004; Salmon & Blackledge, 2015). The kinetics of processes too slow for application of the above methods can be studied with real-time NMR spectroscopy (Meier & Ernst, 1979; Rennella & Brutscher, 2013; Wagner et al., 1985).
The kinetics of protein folding occurs over a large range of timescales and protein folding therefore is an ideal problem for application of NMR spectroscopy (Meinhold & Wright, 2011; Zhuravleva & Korzhnev, 2017). Unfolded states of stably folded proteins are closely related to the ensembles of states populated by intrinsically disordered proteins, for which specialized NMR techniques have been developed (Brutscher et al., 2015; Dyson & Wright, 2004; Salvi, Abyzov, & Blackledge, 2017). NMR is an ensemble method uniquely providing multiple types of data that can be highly resolved in many different customized ways. Temperature, pressure, and solution conditions can be changed as physical parameters to alter the energetic landscapes of proteins to study folding kinetics (Roche, Royer, & Roumestand, 2017; Tugarinov, Libich, Meyer, Roche, & Clore, 2015). NMR spectroscopy is usually thought of an equilibrium method (Ban et al., 2017); however, it can be combined with transient methods in order to study protein folding intermediates. Rapid mixing (Fazelinia, Xu, Cheng, & Roder, 2014; Manoharan, Furtig, Jaschke, & Schwalbe, 2009; Roder, Elove, & Englander, 1988; Udgaonkar & Baldwin, 1988), light activation (Nash et al., 2011; Rubinstenn et al., 1999), and most recently pressure-jump (Charlier, Courtney, Alderson, Anfinrud, & Bax, 2018; Furtig et al., 2007; Kremer et al., 2011) methods, classical physical chemistry techniques, have been adapted to NMR spectroscopy. Relaxation dispersion NMR has been applied to study folding intermediates of SH3 domains (Roche et al., 2017; Tollinger, Skrynnikov, Mulder, Forman-Kay, & Kay, 2001; Tugarinov et al., 2015), PBX-homeodomain (Farber, Slager, & Mittermaier, 2012), villin headpiece (Grey et al., 2006; Tang, Grey, McKnight, Palmer, & Raleigh, 2006), the FF domain from HYPA/FBP11 (Korzhnev, Religa, Lundstrom, Fersht, & Kay, 2007), and apo-myoglobin (Dyson & Wright, 2017; Meinhold & Wright, 2011). While amyloid disease and misfolding is mostly studied by other techniques, including solid-state NMR (Chiti & Dobson, 2006), examples for systems that have been studied by solution NMR relaxation dispersion include human ADA2h (Pustovalova, Kukic, Vendruscolo, & Korzhnev, 2015), beta2-microglobulin (Franco, Gil-Caballero, Ayala, Favier, & Brutscher, 2017), amyloid β protofibril interactions (Fawzi, Libich, Ying, Tugarinov, & Clore, 2014), and transthyretin misfolding(Lim, Dyson, Kelly, & Wright, 2013).
For intramolecular μs-ms dynamics, circumscribed structural features causing the exchange process can be frequently identified. These include as examples disulfide bond isomerization in basic pancreatic trypsin inhibitor (Grey, Wang, & Palmer, 2003) and altered coordination of a Zn ion by cysteine sulfides (Deshmukh, Tugarinov, Appella, & Clore, 2018). For FGFR kinase 2, a major drug target, the ability of gain-of function cancer mutant K659E to modulate μs-ms dynamics of the DFG motif and surrounding residues has been revealed by CPMG relaxation dispersion (Chen et al., 2017). Differences in flexibility between S. aureus wild-type DHFR and the methicillin-resistant S1 DHFR mutant were identified by linebroadening and CPMG relaxation dispersion (Sahasrabudhe et al., 2017). The ability of an external allosteric agent to influence internal dynamic processes was demonstrated for imidazole glycerol phosphatase synthase (Lisi et al., 2016) and for dihydrofolate reductase (Oyen et al., 2017).
Due to extended search interactions between protein and ligands, intermolecular processes can occur on intermediate or slow timescales. Examples the initial formation of an X-dimer in the E-Cadherin dimerization process (Y. Li et al., 2013), GB1(A34F) dimerization (Jee, Ishima, & Gronenborn, 2008), formation of a complex between the intrinsically disordered protein MKK4 and MAPK p38α (Delaforge et al., 2018), profilin self-assembly (Rennella, Sekhar, & Kay, 2017), and amyloid β protofibril interactions (Fawzi et al., 2014).
Chemical exchange methods pioneered in applications to proteins increasingly are being extended to investigations of kinetic processes in nucleic acids(Xue et al., 2015). The bases of the experimental methods are unchanged, but the different spin systems and topologies must be considered in optimization of these methods for nucleic acids. For example, rapid imino hydrogen exchange with solvent in nucleic acids makes the imino 1H and 15N spins in nucleic aids less suitable than the protein 1HN and 15N amide spins in proteins as probes of chemical exchange and RNA completely lacks methyl groups, which also are powerful probes in proteins. Recent applications include a series of relaxation dispersion studies of base-pair rearrangement in DNA by Al-Hashimi and coworkers, and detailed characterization of the fluoride riboswitch by Zhang and coworkers (Kimsey et al., 2018; Szymanski, Kimsey, & Al-Hashimi, 2017; Zhao, Guffy, Williams, & Zhang, 2017).
Two-site exchange is the paradigmatic example of the effects of kinetic processes on NMR phenomena and this simple system allows derivation of exact or nearly exact theoretical descriptions. As consequence of both the theory and methods available, most experimental investigations of chemical exchange have utilized the 2-site model to interpret experimental data. Beyond the 2-site assumption, the majority of established N-site processes is based on 3-site linear kinetic schemes. Examples have been established in the area of protein folding (Farber et al., 2012; Grey et al., 2006; Tang et al., 2006; Tugarinov et al., 2015) as well as intermolecular processes (Rennella et al., 2017). Kinetically more complex models have been rarely studied by relaxation dispersion to date. Deshmukh and coworkers have suggested a 4-site star scheme for binding kinetics in HIV-1 protease - Gag interactions (Deshmukh, Tugarinov, Louis, & Clore, 2017) and a triangular scheme for the coordination of a zinc ion by thioester groups in NCp7 (Deshmukh, Ghirlando, & Clore, 2014). A quadratic scheme has been used for the folding of a FynSH3 triple mutant with the chaperonin GroEL (Libich, Tugarinov, & Clore, 2015). A recent study on Watson-Crick mismatches in nucleic acids features R1ρ data that was conclusively described a triangular scheme, but not a 4-site star scheme (Kimsey et al., 2018). Relatively few research groups have generated the majority of examples for N-site exchange probed by relaxation dispersion. This suggests that the main reason for the limited number of examples are practical difficulties to record or process the data, rather than rarity of interesting biological processes. The possibility of N-site (N > 2) exchange is being raised in some studies, warranting further research once appropriate methods are available (Jee et al., 2008). Intramolecular dynamic processes as well as an intramolecular strand-swapping have been identified in separate classical cadherin subtypes (Y. Li et al., 2013; Miloushev et al., 2008). A combination of both phenomena might occur in some cadherin proteins, yielding an N-site exchange situation. Four-site exchange processes have been suggested for unfolding of carbonic anhydrase (Uversky & Ptitsyn, 1996). A theoretical study exploring the possibility to model synthetic data with a 4-site scheme exposed the computational challenges associated with numerical approaches to efficiently fit data, but also highlighted how coupled allostery could give rise to N-site exchange (P. Li, Martins, & Rosen, 2011). One candidate for this type of N-site exchange is the cSH2 domain in phospholipase Cγ1, for which a dynamic allosteric network involving a combination of faster and slower exchange processes was associated with the variable binding of an intramolecular proline-rich, long flexible linker, or to an externally supplied peptide ligand (Koss, Bunney, et al., 2018). A number of exchange processes of different timescales is also qualitatively observed in an NMR study of the β-adrenergic receptor, illustrating that N-site exchange is very frequent but still not easily quantified (Solt et al., 2017).
Evaluation of complex exchange phenomena in large proteins is facilitated by increasingly powerful experimental methods that can be applied to protein side chains. In addition to methyl groups (Gill & Palmer, 2011; Korzhnev, Kloiber, Kanelis, Tugarinov, & Kay, 2004; Skrynnikov, Mulder, Hon, Dahlquist, & Kay, 2001; Tugarinov, Sprangers, & Kay, 2004; Weininger et al., 2013), chemical exchange can be probed in a number of other side chains (Mackenzie & Hansen, 2018; Weininger, Brath, Modig, Teilum, & Akke, 2014). The extraction of structural features from minor conformations using relaxation dispersion experiments is tightly coupled to the ability to extract structural information from chemical shifts (Robustelli, Stafford, & Palmer, 2012), which renders dispersion experiments for nuclei other than 15N or 1H particularly relevant (Gopalan, Hansen, & Vallurupalli, 2018; Hansen, Vallurupalli, Lundstrom, Neudecker, & Kay, 2008; Korzhnev, Religa, Banachewicz, Fersht, & Kay, 2010). Relaxation dispersion experiments can also yield RDCs (Hansen, Vallurupalli, & Kay, 2008b; Igumenova, Brath, Akke, & Palmer, 2007), CSAs (Vallurupalli, Hansen, & Kay, 2008a), or information about side chain conformations (Baldwin, Hansen, Vallurupalli, & Kay, 2009; Hansen, Neudecker, Vallurupalli, Mulder, & Kay, 2010), which also can be used in structure calculations (Gopalan et al., 2018; Vallurupalli, Hansen, & Kay, 2008b).
This review begins by outlining theoretical approaches for characterizing chemical exchange phenomena, with an emphasis on extensions from 2 to N-site kinetic topologies. General approximations have recently been obtained for N-site exchange in both CPMG and R1ρ experiments that are valid for most experimentally accessible time scales (Koss, Rance, & Palmer, 2017, 2018). These results are used to examine conditions under which various N-site kinetic schemes can be distinguished by CPMG or R1ρ experiments. The review concludes by discussing key aspects of the main experimental methods applied to proteins and nucleic acids, but is not intended as a comprehensive reference to the myriad of experimental techniques available. Other reviews of chemical exchange cover, among other aspects, the theoretical basis (Farber & Mittermaier, 2015; A. G. Palmer, 2014, 2016; Vallurupalli, Sekhar, Yuwen, & Kay, 2017; Xue et al., 2015), the history (A. G. Palmer, 2014, 2016; Sauerwein & Hansen, 2015; Vallurupalli et al., 2017), applications (Farber & Mittermaier, 2015; Lisi & Loria, 2016; Osawa, Takeuchi, Ueda, Nishida, & Shimada, 2012; A. G. Palmer, 2016; Sauerwein & Hansen, 2015), practical considerations (Gopalan et al., 2018; Ishima, 2014; Kempf & Loria, 2004; Walinda, Morimoto, & Sugase, 2018a, 2018b; Xue et al., 2015), comprehensive overview of existing experiments (Sauerwein & Hansen, 2015; Vallurupalli et al., 2017; Walinda et al., 2018b), and special topics, including CEST (Vallurupalli et al., 2017), R1ρ (Francesca Massi & Peng, 2018; A. G. Palmer & Massi, 2006), high-power relaxation dispersion (Ban et al., 2017), applications to nucleic acids (Xue et al., 2015), and structure determination from relaxation dispersion(Gopalan et al., 2018).
2. Theory
The effects of chemical or conformational exchange kinetic process on NMR observables are described by the stochastic Liouville equation:
| (1) |
In Eq. 1, , N is the number of interconverting sites or species (containing the spin of interest), is a M × 1 column vector containing the time-dependent amplitudes of the M basis operators Bm for the Liouville space for the nth species, the elements of are defined by for m = 1,…,M, σn(t) is the density operator for the nth state, and is defined in similar fashion using the equilibrium density operator. The matrix is block diagonal with elements , in which Ln is the M × M matrix for coherent evolution for the nth state (containing chemical shifts, scalar couplings, and radiofrequency fields, for example), the matrix is block diagonal with elements , in which Rn is the M × M matrix for relaxation the nth state from processes other than chemical exchange (dipole-dipole, chemical shift anisotropy, and quadrupole relaxation, for example). The exchange matrix , in which IM is an M × M identity matrix and K is an N × N kinetic matrix with elements Kij = kji, kji is the (pseudo-) first-order rate constant for transition from state j to state i for i ≠ j and
| (2) |
The second term on the right hand side of Eq. 1 ensures that relaxation processes return the spin system to equilibrium or steady state at long times. Equation 1 is valid if the system is at chemical equilibrium and the transitions exchange coherences or populations between states, but do not change the coherences or populations themselves.
Equation 1 has a formal solution given by:
| (3) |
and clearly this expression can be evaluated numerically for any number of sites and basis operators. The amplitude of the mth basis operator in the nth state is obtained as
| (4) |
in which [0,…,δjk,…,0]V is an V × 1 row vector. As noted by Allerhand and Thiele, the kinetic matrix K can be symmetrized by a matrix S with elements , in which pn is the population of the nth site, which can simplify analytic or numerical calculations (Allerhand & Thiele, 1966).
Many NMR experiments are designed so that the signal of interest s(T), proportional to a particular basis operator or function of basis operators, has simple functional dependence on a relaxation delay, T. for example, if the signal decay for s(T)∝<Bmn>(T) is monoexponential, then relaxation rate constant for the mth basis operator constant is defined as:
| (5) |
This equation also can be used to define an effective (T-dependent) relaxation rate constant for a multi-exponential decay process. Analytic expressions for relaxation rate constants Rnm′ generally have been obtained either by calculation of the evolution of the average magnetization (and then using Eq. 5) or by calculation of the eigenvalues of (Abergel & Palmer, 2004). Exact results are available only for cases in which the product MN is small (≤ 4, but more often 2) or in the limits of very fast or very slow kinetic processes. Approximate expressions applicable to all kinetic rate scales and topologies have been obtained for both the average magnetization and eigenvalue approaches. Analytical (if MN ≤ 4) or numerical eigenvalues (for any MN) can be obtained from the roots of the characteristic equation (Koss et al., 2017; Koss, Rance, et al., 2018):
| (6) |
Approximate roots of Eq. 6 frequently provide simplified expressions. The nth Newton-Raphson and Halley (linearized Laguerre) approximations are, respectively:
| (7) |
| (8) |
with λ0 = 0. If the variation in Rn is modest, the effects of are given to first order by:
| (9) |
in which λn0 is calculated using Eqs. 6, 7, or 8 assuming . A sufficient condition for accuracy of approximations of either type is that that the equilibrium population of one state is much larger than for the other N −1 states. The dominant state is normally denoted as state 1. If site 1 is not dominant, then another sufficient condition is that kinetic exchange amongst the states 2, …, N is fast. The above expressions frequently yield compact algebraic expressions that are also sufficiently accurate for analysis of experimental data; the present Review will emphasize results obtained using the first-order Newton-Raphson and Halley approaches, but both more accurate, but more complex (Koss et al., 2017; Koss, Rance, et al., 2018), and less accurate, but simpler (Trott & Palmer, 2004), expressions have been described. The recently developed geometric approximation approach, in which numerical solutions to Eq. 3 are pre-calculated and stored as a library of response surfaces, is an efficient alternative to “on-the-fly” numerical calculations and to the theoretical approximations that the focus of the present Review (Chao & Byrd, 2016, 2017).
2.1. Free Precession
Free precession of transverse magnetization provides a simple example of the application of the above theoretical description. In this situation, M = 1, , Ln = iΩn, Ωn is the resonance offset of a nuclear spin in the nth state, Rn = −R2n, and R2n is the transverse relaxation rate constant for a nuclear spin in the nth state. For N = 2 states, Eq. 1 becomes:
| (10) |
This equation is the Bloch-McConnell equation for transverse magnetization undergoing 2-site jumps. In this case, the two eigenvalues of the relaxation rate matrix can be obtained exactly. The desired smaller of the two relaxation rate constants is given by the negative of the real part of the eigenvalue:
| (11) |
in which pn is the equilibrium population of the nth site, Δωij =Ωi−Ωj, ΔRn1=R2n−R21, and kex = k12 + k21. In this case, the first-order Newton-Raphson estimate of the eigenvalue yield the estimated rate constant
| (12) |
in which the second line results from the first-order treatment of ΔR21. The exact and first-order approximate values of R2 are compared in Figure 1. The Halley approximation is extremely accurate for a 2-state system and results are not shown for clarity. First-order approximations are accurate when exchange is relatively slow (kex/|Δω21| < 1) or relatively fast (kex/|Δω21| >> 1); higher order approximations are required for high accuracy in the intermediate exchange regime (kex/|Δω21| ~ 1). Allerhand and Theile defined C-type or R-type exchange depending upon whether the effects of differences in resonance frequencies or differences in relaxation rate constants, respectively, between sites have a dominant effect. In either limiting case, Δω21 → ∞ or ΔR21 → ∞, R2 → R21 + p2kex = k12.
Figure 1.
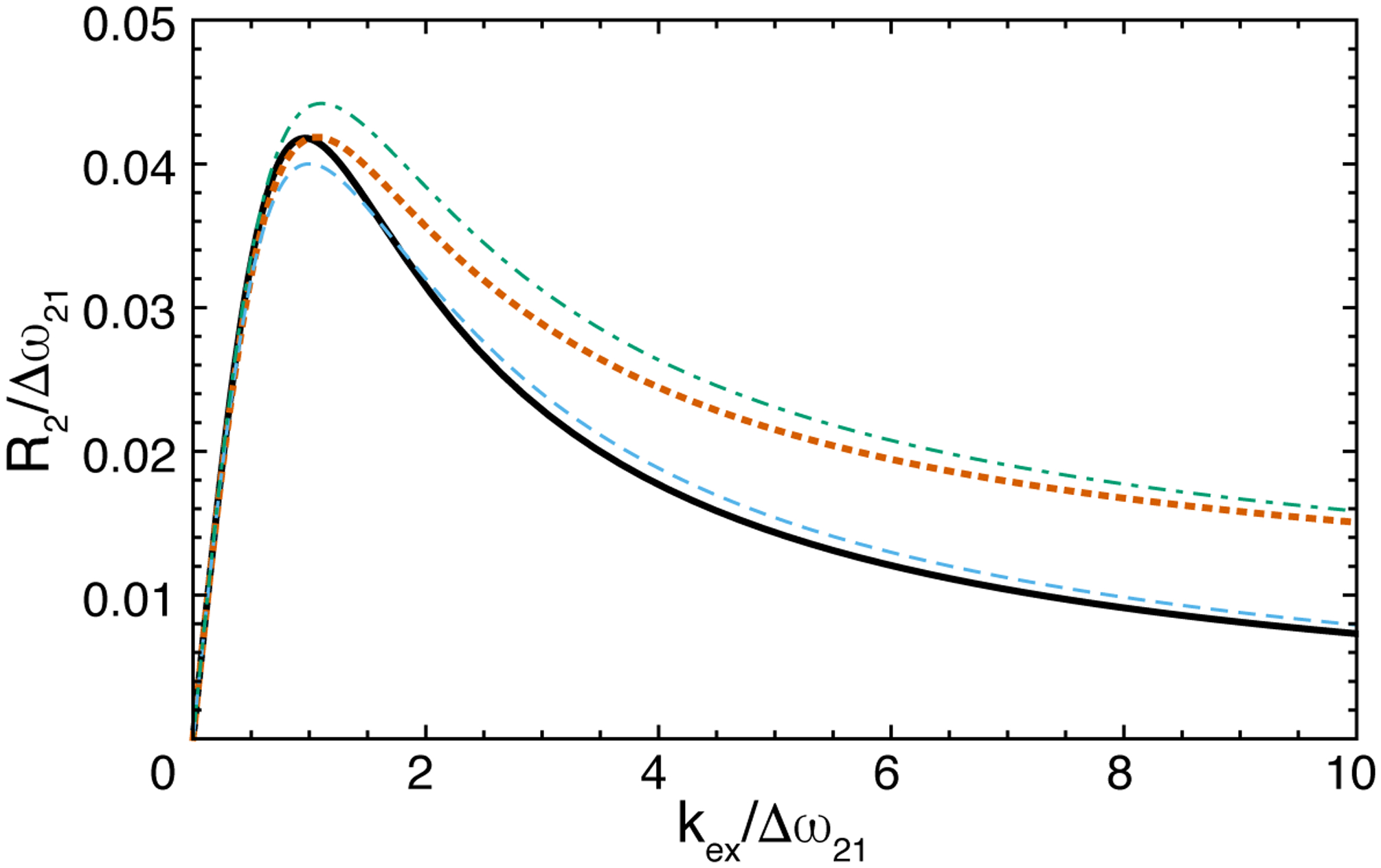
Free-precession transverse relaxation rate constant for 2-site chemical exchange with p2 = 0.08 and R21 = 0. R2 obtained from (black, solid) exact eigenvalue and (blue, dashed) first-order approximation assuming ΔR21 = 0. R2 obrtained from (orange, dotted) exact eigenvalue and (green, dash-dotted) first-order approximation ΔR21/Δω21 = 0.1.
2.2. Carr-Purcell-Meiboom-Gill Relaxation
The CPMG experiment consists of m repetitions of the pulse sequence element:
| (13) |
applied to transverse magnetization. The effect of ideal 180° pulses is to invert the signs of Ωi.
Thus, evolution of magnetization through the CPMG sequence becomes:
| (14) |
in which the asterisk indicates complex conjugation, , and other variables are defined as for the case of free-precession. For 2-site exchange, the Carver-Richards equation gives the relaxation rate constant as (Carver & Richards, 1969):
| (15) |
in which,
The Carver-Richards equation for 2-site exchange of single-quantum coherence has been extended to multiple-quantum relaxation (Korzhnev et al., 2004).
In order to provide additional insight for N-site exchange, Koss and coworkers have shown that M(t) obeys a coarse-grained differential equation (Koss, Rance, et al., 2018):
| (16) |
with an average Liouvillian matrix H(τcp) defined as:
| (17) |
The matrix logarithm is determined to high accuracy by Padé approximation. For example, four such approximations are:
| (18) |
in which Z(τcp)=exp{2Λ*τcp}exp{2Λτcp} and the subscripts in the expressions Hjk(τcp) give the highest order power in the numerator and denominator polynomials in Z(τcp). The CPMG relaxation rate constant Rcpmg(τcp) then is obtained from the largest (least negative) eigenvalue of the average Liouvillian H(τcp) / (4τcp), because the eigenvalues are unaffected by the similarity transformation in Eq. 16.
The Cayley-Hamilton theorem allows an arbitrary Padé approximation Hjk(τcp) and its inverse to be expressed as:
| (19) |
in which the coefficients, an, bn, cn, and ε depend on {TrZ(τcp), … TrZN−1(τcp)} and |Z(τcp)|. The determinant has the simple form:
| (20) |
so the main computational effort is determining the traces {TrZ(τcp), … TrZN−1(τcp)} The coefficients an depend on bn, cn, and ε, but not on the exact functional form of these parameters. Once the representation using bn, cn, and ε has been transformed to the representation using an, then the resulting expressions can be used with any set of bn, cn, and ε. Consequently, the use of higher-order Padé approximations is not computationally prohibitive. In addition, the inverse H−1(τcp), needed for application of the eigenvalue approximations, is obtained simply by replacing ε by 1/ε and exchanging bn and cn.
Application of these equations for 3-site exchange are presented in the Appendix. Low-order Padé approximations frequently yield compact analytical expressions. For example, using H11(τcp) for 3-site exchange,
| (21) |
in which Z(τcp) is calculated using and is assumed to be symmetrized (vide supra).
If chemical exchange is slow, (kij + kji < |Δωij|/10), then Koss and coworkers showed that H10(τcp) is given by (Koss, Rance, et al., 2018):
| (22) |
For N = 2,
| (23) |
A further simplification of this equation is obtained by applying another similarity transformation:
| (24) |
which yields:
| (25) |
This equation illustrates an analogy between the CPMG experiment and the chemical exchange formalism presented above, in which
| (26) |
The relaxation rate constant obtained from the above equation is:
| (27) |
in which the second line is the first-order approximation of the eigenvalue. For 3-sites in a linear topology B-A-C, in which the population p1 >> p2, p3 (1, 2, and 3 denote sites A, B, and C), the first-order approximation to the relaxation rate constant is:
| (28) |
in which B0 = (k12+k13)(k21+ΔR21)(k31+ΔR31) and B1 = (k21+ΔR21)(k31+ΔR31) + (k12+k13) (k21+k31+ΔR21+ΔR31). Expressions for other kinetic schemes have been presented by Koss and co-workers (Koss, Rance, et al., 2018). The above equations demonstrate the main effect of R-type relaxation for the CPMG experiment: ΔR21 becomes important only if k21 is not >> | ΔR21|. Essentially, the difference in relaxation rates must be large enough that relaxation happens efficiently within the lifetime of sites 2,…, N.
The approximations obtained above for 2-site exchange using the exact eigenvalue of H33(τcp) and a second-order approximation to the eigenvalue of the slow exchange Liouvillian (Eq. 8) are compared to the Carver-Richards equation in Figure 2. The H33(τcp) approximation is extremely accurate over all time scales, while the slow exchange result is accurate when kex < |ΔωAB|/10.
Figure 2.
CPMG relaxation dispersion curves for 2-site chemical exchange for p2 = 0.05, and R22 = R21 = 0. (a) Δω21 /kex = 5: (black solid lower curve) Carver-Richards equation, (blue, dotted) exact eigenvalue of H33(τcp), (orange, dashed), second-order slow exchange equation. Δω21 /kex = 10: (black, solid, upper curve) Carver-Richards equation, (blue, dash-dotted) exact eigenvalue of H33(τcp) , (reddish-purple, dash-dot-dotted), second-order slow exchange equation. (b) Δω21 /kex = 2: (black, solid, lower curve) Carver-Richards equation, (blue, dotted) exact eigenvalue of H33(τcp) ; Δω21 /kex = 1: (black, solid, middle curve) Carver-Richards equation, (orange, dashed) exact eigenvalue of H33(τcp) ; Δω21 /kex = 0.2: (black, solid, lower curve) Carver-Richards equation, (reddish-purple, dash-dotted) exact eigenvalue of H33(τcp) ; curves for Δω21 /kex = 0.2 have been multiplied by 10 for clarify.
The above approximations for 3-site triangular exchange are illustrated in the left panel of Figure 3. In the given example, kex > |ΔωAB|/10 for all sites and the slow exchange approximations for H10 (Eq. 22) are not expected to be accurate. Approximations based on the exact matrix exponential (Eq. 17) are more appropriate, even though the second-order approximation based on Eq. 21 also gives a satisfactory fit in this case. As shown in the Figure, Padé approximations of increasing accuracy can greatly enhance accuracy, provided that the eigenvalue approximation also is sufficiently accurate. The right panel of Fig. 3 illustrates CPMG dispersion curves for 4-site exchange with various kinetic schemes. In most cases shown here, a second-order eigenvalue approximation is sufficient to fit the exact solution; however, for the star scheme, a second-order (Halley’s method) eigenvalue approximation was required to fit data at large τcp; these more complex approximations are recommended if data are available at very large τcp and if a wide parameter space is being considered (for validity of the single eigenvalue approximation vide infra). The examples of 4-site exchange examples in Fig. 3 also illustrate that the CPMG curve is influenced less by exchange processes between minor sites at large values of τcp, while those processes become more influential at smaller τcp (Koss et al, 2018).
Figure 3.
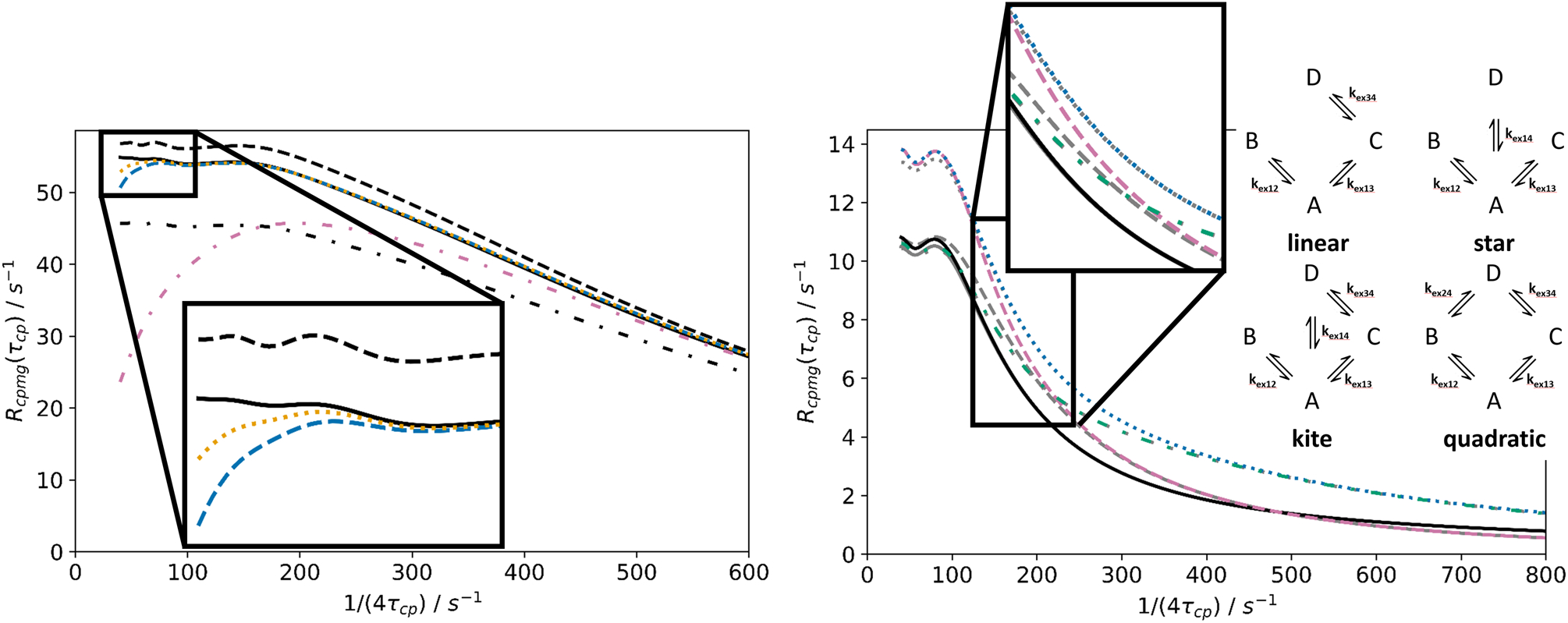
Approximations for N-site kinetic schemes in CPMG experiments. Left: 3-site triangular exchange (see Fig. 6 for the respective R1ρ figure). (Solid, black) Exact solution; (not black) Hnn(τcp) approximations, eigenvalue approximated to second order (Halley’s method), with (reddish-purple, dashed-dotted) n = 1, (blue, dashed) n = 2; (orange, dotted) n = 3; (black, not solid) slow exchange approximations from matrix exponential series truncation, with eigenvalue approximation of order k, with (dashed-dotted) k = 1; (dashed) k = 2 (Halley’s method). Parameters (in analogy to parameters in Fig. 1, Koss et. al. 2018): k12 + k21 = 200 s−1; k13 + k31 = 300 s−1; k23 + k32 = 600 s−1; ΔωAB = 1800 s−1; ΔωAC = −2600 s−1; pA = 0.85; pB = 0.08; pC = 0.07. Right: CPMG curves can help to differentiate between various 4-site schemes. A schematic illustration of 4-site schemes is shown within the figure. Exact solutions (not grey), Hnn(τcp) approximations with n = 2 (grey) for CPMG curves for a variety of four-site schemes: kite (blue, dotted), star (reddish-purple, dashed), quadratic (green, dashed-dotted) and linear (black, solid). Eigenvalue approximations to first order almost overlap with the exact solutions, except for the star scheme; however, a second-order eigenvalue approximation (Halley’s formula) gives a perfect overlap in this case. Parameters: k12 + k21 = 200 s−1; k13 + k31 = 20 s−1; k14 + k41 = 70 s−1; k24 + k42 = 400 s−1; k34 + k43 = 150 s−1; ΔωAB = 1000 s−1; ΔωAC = −2900 s−1; ΔωAD = 1100 s–1; pA = 0.88; pB = 0.05; pC = 0.03; pD = 0.04.
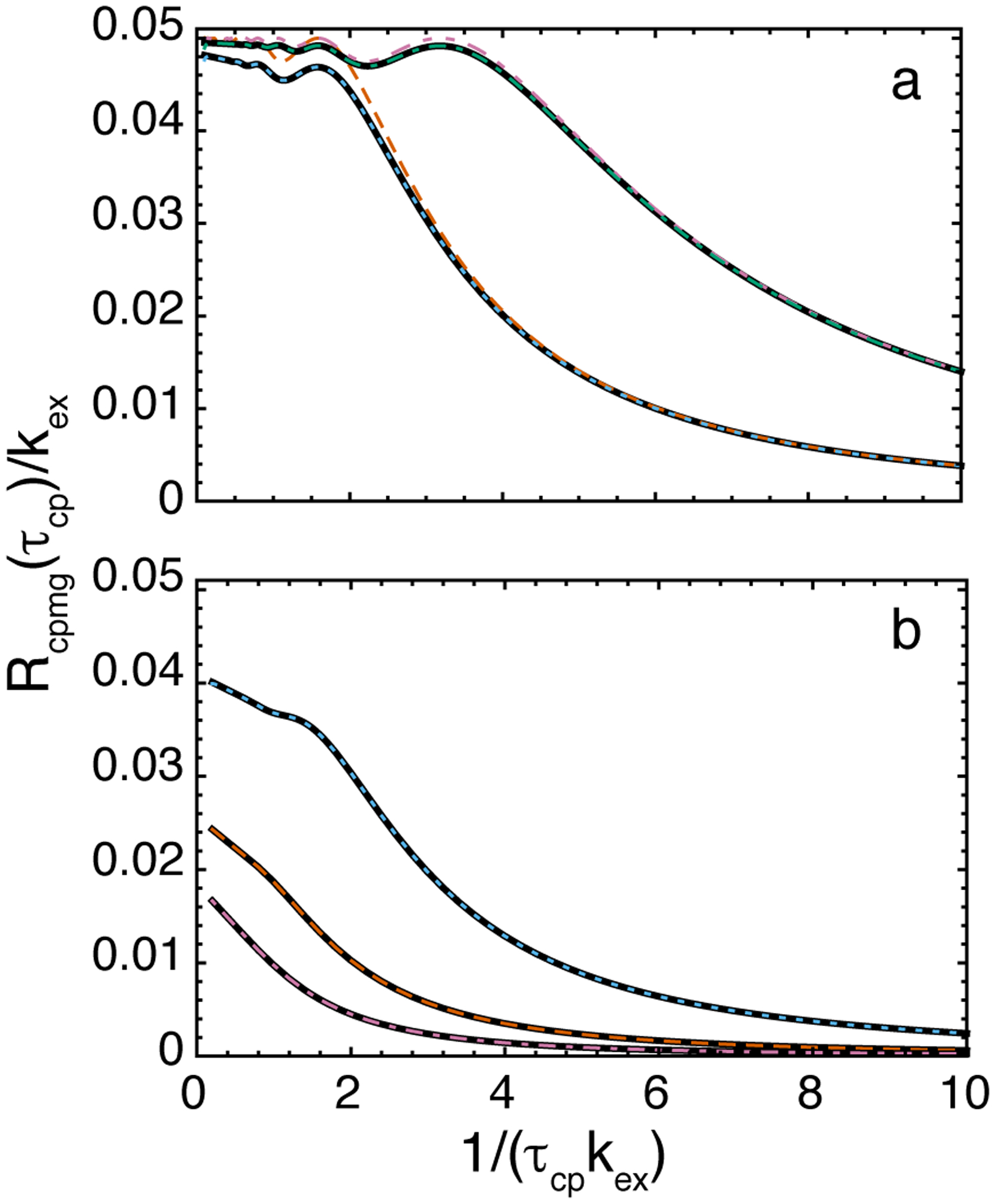
Relaxation rate constants defined as the least negative eigenvalue of H(τcp) or by Eq. 5 differ because if T = 4mτcp is not sufficiently long, then contributions to the magnetization decay from other eigenvalues will not have been damped to zero. The effective relaxation rate constant obtained form Eq. 5 will then contains a T-dependent contribution from the residual multi-exponential components of the magnetization decay. This situation arises in particular if exchange is slow and the population of site 1 is not dominant. Baldwin has derived an expression for 2-site CPMG relaxation based on Eq. 5 that provides a correction the Carver-Richards equation, which is itself the negative of the largest (least negative) eigenvalue of H(τcp) (Baldwin, 2014). A more formal (without reduction to explicit algebraic form) derivation of the effective relaxation rate constant derived from Eq. 5 for the N-site CPMG experiment is given in the Appendix.
Both the results of Baldwin and those presented in the Appendix (Eq. A14 and A15) show that the difference between the two estimates of the relaxation rate constant depends upon 1/T = 1/(4mτcp). Thus these results support the use of constant relaxation time CPMG experimental methods. This correction to the Carver-Richards equation also accounts for different behavior in the slow exchange limit of Eq. 27 (for ΔR21 = 1) and the expression obtained by Tollinger and coworkers (Tollinger et al., 2001):
Figure 4 compares the Carver-Richards equation (Eq. 15) with various expressions that estimate Rcpmg(τcp) based on more than a single eigenvalue. “Exact” Rcpmg(τcp) rate constants were defined by a monoexponential fit to simulated multiexponential decays. Equation 29 reproduces the exact results best at extremely slow exchange (Fig. 4, left panel); for slow or intermediate exchange, the Carver-Richards equation and Eq 27 are more appropriate. Baldwin’s result for 2-site schemes, and equivalently the correction for N-site exchange (Eq. A14), give the effective Rcpmg(τcp) rates for experiments with constant T and analyzed as in Eq. 5. These Rcpmg(τcp) values fall between the results for Eq. 15 and 29. For exchange which is not too slow (Fig. 4, right panel), simpler approximations for this correction factor to Eq. 15 can be used (Eqs. A16, A17).
Figure 4.
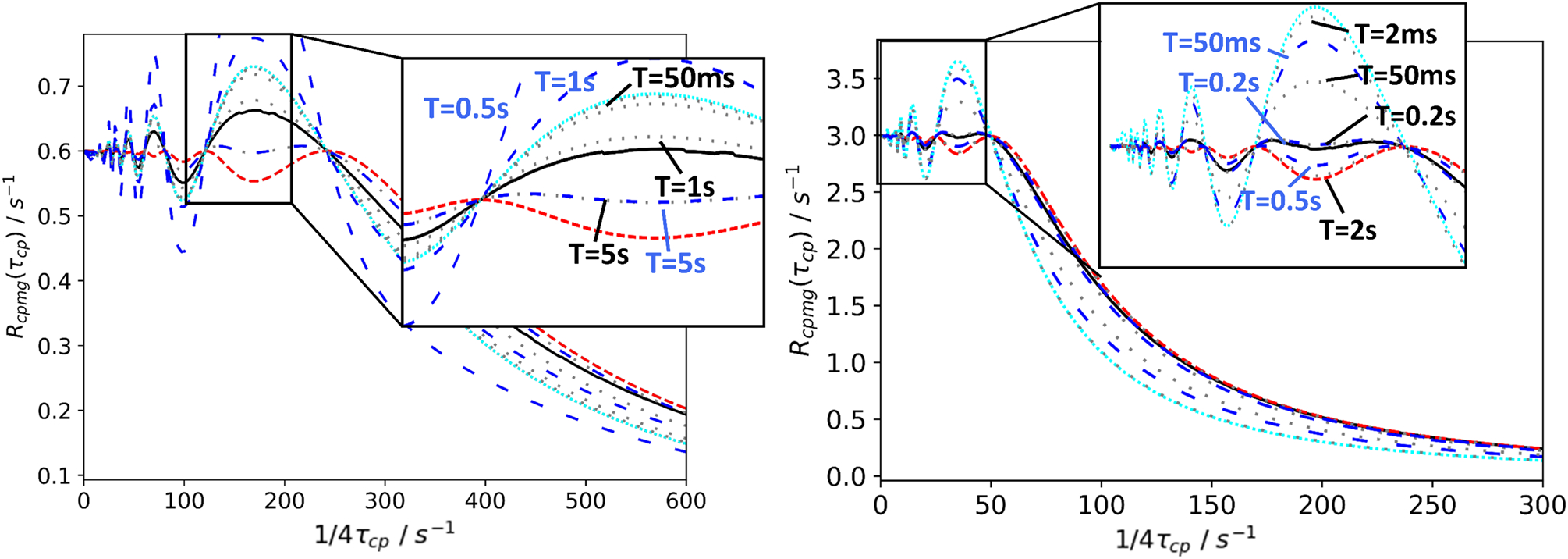
Exact solutions two-site CPMG experiments for Rcpmg obtained from more than one eigenvalue, and/or at constant T. (Solid black) Exact solution, calculated from multiexponential decays from two eigenvalues of Hnn(τcp). Rcpmg was extracted from monoexponential fits to the decays, by evenly sampling the curve along the magnetization axis; (red, dense-dashed) Carver- Richards equation / single eigenvalue exact solution; (cyan, dense-dotted) Eq. 29 (Tollinger et al., 2001) for extremely slow exchange; (gray, sparse-dotted) exact solutions for various constant T, based on Eq. A14, equivalent to Baldwin’s expressions (Baldwin, 2014); (blue, sparse-dashed) approximations for constant T expressions (Eq. A16). Left panel: T = {0.05 s, 0.2 s, 0.5s, 1 s, 5s}; labels in figure. Parameters, equivalent to Tollinger et al.: k12 + k21 =2 s−1; ΔωAB = 6 ppm for 15N at 800 MHz (1H frequency); pA = 0.7; pB = 0.3. Right panel: (dotted) T = {0.002 s, 0.05 s, 0.2 s, 0.5 s, 2 s}; labels in figure. Parameters: k12 + k21 = 30 s−1; ΔωAB/(2π) = 100 Hz; pA = 0.9; pB = 0.1.
2.3. Rotating-Frame Relaxation
The above formalism is particularly powerful for spin-locking experiments, including R1ρ, CEST, and DEST, because the dimensions of the evolution matrix are 3N × 3N and even for 2-site exchange exact analytical results cannot be obtained. For N-sites,
| (29) |
in which the spin-locking field is assumed to have x-phase and amplitude ω1 and resonance offsets are relative to the frequency of the spin-locking radiofrequency field, ωrf. The relaxation rate constant R1ρ is the relaxation rate constant for magnetization spin-locked along the effective field (R2ρ relaxation of magnetization orthogonal to the effective field has been discussed by Garwood and co-workers (Mangia, Traaseth, Veglia, Garwood, & Michaeli, 2010; Michaeli, Sorce, Idiyatullin, Ugurbil, & Garwood, 2004; Traaseth et al., 2012)). The R1ρ relaxation rate constant is obtained as the largest (least negative) eigenvalue of the evolution matrix, as discussed above. For N = 2 sites and assuming R1 = R2 and p1 >> p2, the first-order approximation to the eigenvalue gives (Trott & Palmer, 2002):
| (30) |
in which and are the population-average relaxation rate constants in the absence of exchange , is the population-average resonance offset, , and . The second line is obtained if p1 is sufficiently large that ωA2 ≈ ωB2. This equation demonstrates the main advantage of spin-locking experiments compared with lineshape analysis and CPMG experiments: the relaxation rate constant depends on the resonance offset in the B site (site 2), even if the population of that site is so low that the resonance signal is not directly observable. In contrast, as shown by the Carver-Richards equation (Eq. 15), the CPMG experiment depends only on Δω212 and hence provides information on the magnitude of the resonance frequency difference between sites, but not on the absolute sign (vide supra).
The above equation has been extended both to higher order of approximation and to include differences in relaxation rates R21 and R22 between sites (differences between R11 and R12 are generally small enough to be ignored). In the former case, the second-order result is (Miloushev & Palmer, 2005):
| (31) |
The latter case has been treated to first order by Baldwin and Kay (Baldwin & Kay, 2013); the resulting equation is lengthy and not reproduced herein. The expression obtained from Eq. 8:
| (32) |
is more compact, but less accurate. This expression is easily obtained by noting that:
| (33) |
in which Diag[ ] is a diagonal matrix and performing the matrix manipulations term by term. The first term in the brackets of Eq. 32 corrects for the different effective fields for spins in the two states, which makes a contribution even if ΔR21 = 0, and the second term corrects for ΔR21 ≠ 0. If ΔR21 = 0, this equation reduces to Eq. 30 if p1 >> p2 or exchange is in the fast limit. As for the CPMG experiment, ΔR21 becomes important only if k21 is not >> | ΔR21|.
The behavior of the above expressions for R1ρ relaxation for 2-site exchange are illustrated in Figure 5. In contrast to the CPMG experiment, in which only τcp can be varied, R1ρ relaxation is altered by changing both ω1 and ωrf. Even in relatively fast exchange, the asymmetry in the profile of R1ρ relaxation rate constants for values of ωrf upfield or downfield of the observable resonance is significant and allows determination of Ω2 (Bothe, Stein, & Al-Hashimi, 2014; Kimsey, Petzold, Sathyamoorthy, Stein, & Al-Hashimi, 2015).
Figure 5.
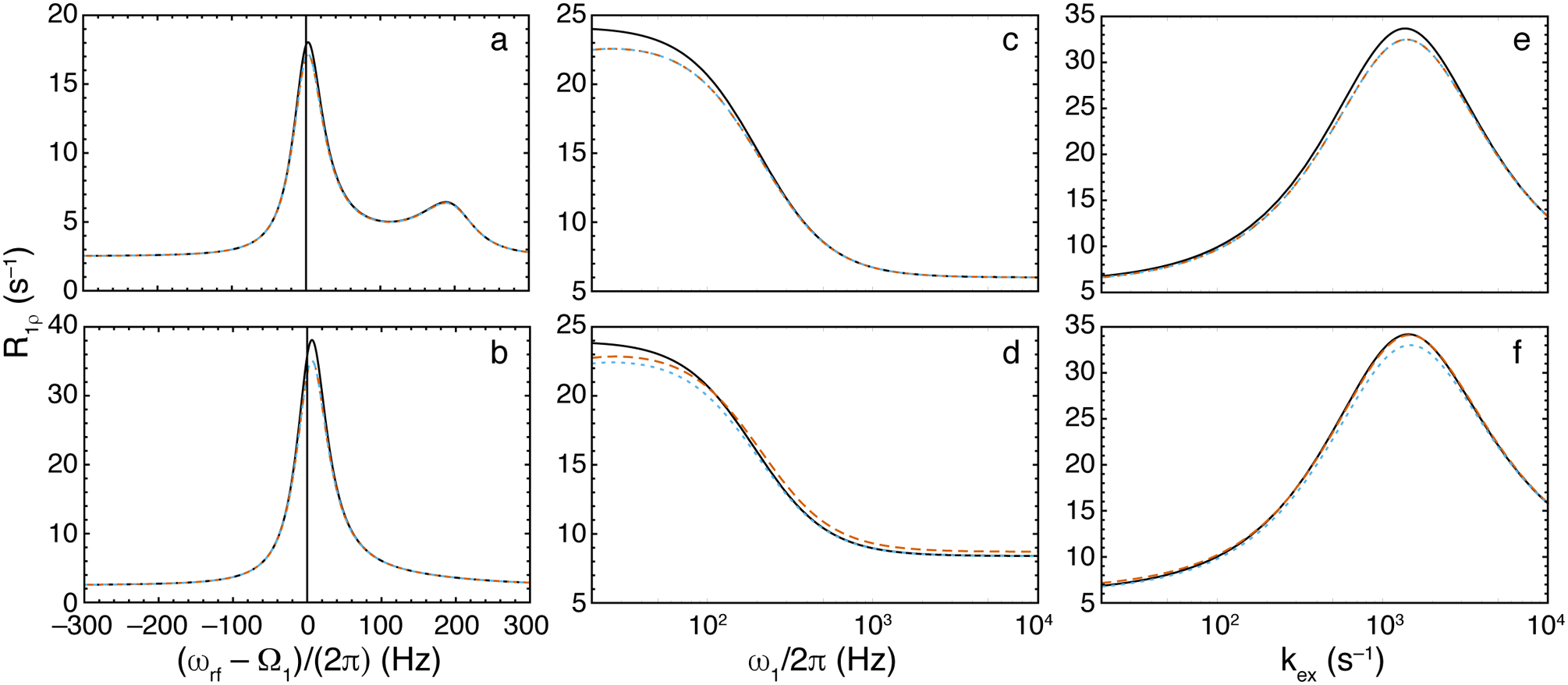
R1ρ relaxation dispersion curves for 2-site chemical exchange. (black, solid) Numerical eigenvalue, (orange, dashed), Trott-Palmer expression, Eq. 30, (blue, dotted), Eq. 32. Parameters common to all calculations are ω21/(2π) = 200 Hz, p2 = 0.05, and R11 = R12 = 2.5 s−1. Other parameters are (a) kex = 250 s−1, ω1/(2π) = 25 Hz, R21 = R22 = 6 s−1; (b) as in (a) except kex = 1200; (c) ωrf/(2π) = 0 Hz, kex = 400 s−1, R21 = R22 = 6 s−1; (d) as in (c) except R22 = 60 s−1; (e) ωrf/(2π) = 0 Hz, ω1/(2π) = 100 Hz, R21 = R22 = 6 s−1; and (f) as in (e), except R22 = 60 s−1.
Koss and coworkers have illustrated results for 3- and 4-site kinetic schemes (Koss et al., 2017). The simplest N-site cases (N > 2) are linear topologies, because the 3N × 3N characteristic equations can be reduced to 3 × 3 matrix polynomials of leading order N. Using this approach, the linear B-A-C scheme has a first-order approximation:
| (34) |
in which . The above expression does not include the contribution from relaxation other than chemical exchange. The effects of relaxation can be treated by incorporating the relaxation matrix Rn into , approximated as , or calculated to first order as in Eq. 9.
Higher-order Newton-Raphson approximations based on the full 3N × 3N matrices L+K+R give results of arbitrary accuracy, which is useful when reduction to N × N matrices schemes is difficult. The examples shown in Figure 6 demonstrate that for complex kinetic schemes (triangular, kite), a second-order Halley approximation for the eigenvalue is applicable to the practically relevant parameter space, and in many cases superior to the approximations of the Woodbury type reported previously (Koss et al., 2017).
Figure 6.
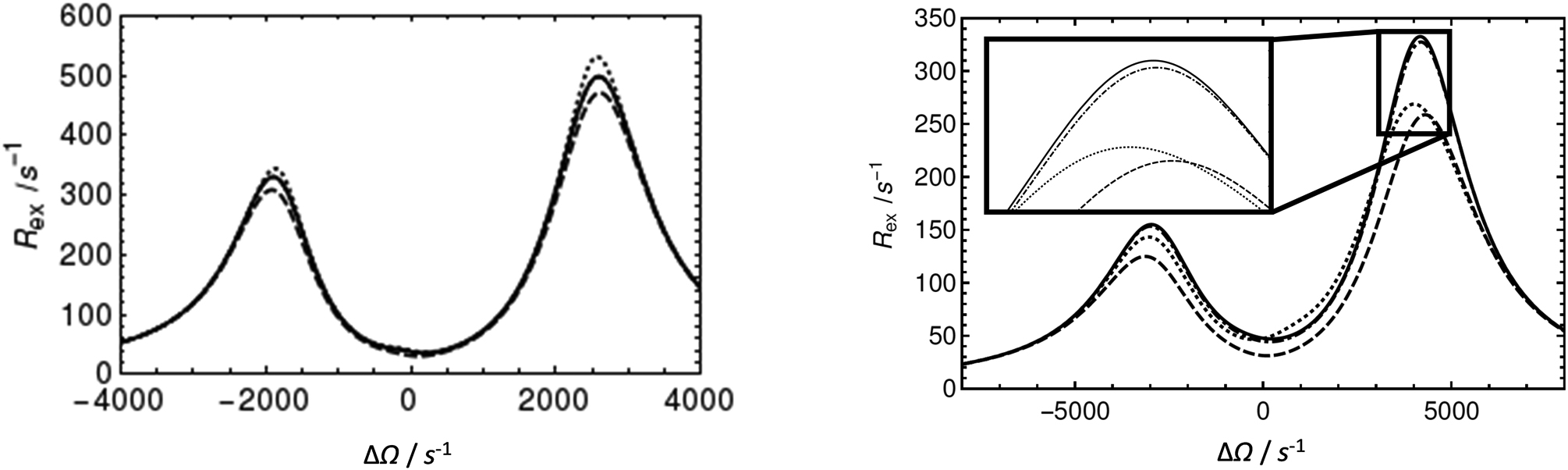
Approximations for N-site kinetic schemes for the Rex contribution in R1ρ experiments. (Solid) Numerical calculation of Rex=−λ/sin2θ from the least negative real eigenvalue of the 12 × 12 evolution matrix, (dashed) calculation from the first order approximation from Eq. 11 in Koss et al., 2017, (dotted) calculation from the Woodbury approximation from Eq. 50 in Koss et al., 2017, (dashed-dotted) calculation from the new second-order eigenvalue (Halley) approximation from Eq. 27 in this paper and Eq. 11 in Koss et al, 2017. This (dashed-dotted) approximation overlaps almost perfectly with the numerical solution. Left: Approximations for the triangular scheme (see Fig. 3 for the respective CPMG figure). k12 + k21 = 200 s−1; k13 + k31 = 300 s−1; k23 + k32 = 600 s−1; ΔωAB = 1800 s−1; ΔωAC = −2600 s−1; pA = 0.85; pB = 0.08; pC = 0.07. Right: Approximations for the 4-site kite scheme. The insets exemplify regions in which the results of the calculations differ. Parameters used for all calculations were used for all calculations were ω1 = 1250 s−1; k12 + k21 = 140 s−1, k13 + k31 = 350 s−1, k34 + k43 = 700 s−1 and k14 + k41 = 350 s−1, ΩB − ΩA = −850 s−1, ΩC − ΩA = 2550 s−1, and ΩD − ΩA = −4250 s−1. pA = 0.79, pB = 0.08, pC = 0.06, pD = 0.07.
3. Experimental Techniques and Examples
A large number of variations of NMR experimental methods for investigation of chemical exchange in proteins and nucleic acids have been reported. Many of these experiments have been developed to account for particular isotopic labeling patterns or spin topologies. The following sections address issues that arise in application of the above theoretical results when analyzing experimental data, rather than discussing particular pulse sequences; for the latter, see recent reviews (Francesca Massi & Peng, 2018; A. G. Palmer, 2014; A. G. Palmer, 3rd, Grey, & Wang, 2005; Sauerwein & Hansen, 2015; Vallurupalli et al., 2017; Walinda et al., 2018b). Practical experimental guidelines have been discussed in other reviews (Ishima, 2014; Kempf & Loria, 2004; Francesca Massi & Peng, 2018; Walinda et al., 2018a, 2018b; Xue et al., 2015).
3.1. ZZ-exchange
If chemical exchange is slow enough and populations of minor sites large enough that the resonances for minor sites are observable, then chemical exchange rate constants are conveniently obtained using ZZ-exchange experiments. These experiments are the analogues to the 1H NOESY experiments, but have the advantage of lacking numerous confounding NOE cross peaks. In proteins, for example, 15N ZZ-exchange experiments are devoid of 15N-15N NOE cross-peaks owing to the low magnetogyric ratio of the 15N nucleus and the long distance between 15N atoms. ZZ-exchange experiment for uniformly 13C labeled molecules may contain 13C-13C NOE cross peaks, usually between directly bonded atoms. Pulse sequences have been described in which the chemical exchange occurs between longitudinal magnetization (Farrow et al., 1995; Hwang & Kay, 2005; H. Wang et al., 2002), longitudinal two-spin order (Montelione & Wagner, 1989; Sprangers, Gribun, Hwang, Houry, & Kay, 2005), and single transition operators (Y. Li & Palmer, 2009), and in which detection of the signal utilizes HSQC, TROSY-detection (Hwang & Kay, 2005; Sahu, Clore, & Iwahara, 2007; Sprangers et al., 2005), and TROSY-selection (Y. Li & Palmer, 2009) methods.
For 2-site exchange,
| (35) |
The above equation is written for z-magnetization, but as noted ZZ-exchange experiments can be based on other components of the density operator, such as longitudinal two-spin order. The integrated solution to this equation is:
| (36) |
in which the eigenvalues of the evolution matrix are:
| (37) |
and the mixing coefficients are given by:
| (38) |
In a conventional two-dimensional ZZ-exchange experiment, t1 frequency labeling occurs prior to the mixing time t, resulting in two auto-peaks and two cross-peaks for each exchanging spin. The integrated auto- and cross-peak amplitudes resulting from this experiment are given by Ijk(t) = ajk(t) Mzk(0).
Direct global fitting of the time dependence of the two auto- and cross-peak intensities (measured as peak heights) is prone to systematic errors from the effects of differential relaxation during parts of the experiment other than the relation delay, t, particularly the t2 acquisition period. Miloushev and coworkers proposed a simplified analysis (Miloushev et al., 2008):
| (39) |
that is insensitive to such effects. In addition, this equation is independent of moderate differences between R11 and R12, does not require iterative curve-fitting, and is easily used in a global analysis of multiple spins subject to the same exchange process, because the right-hand-side depends only on the exchange rate constants. In order to obtain k12 and k21 individually, the equilibrium constant Keq = k12/k21 must be known independently or measured from integrated peak volumes using the HSQC0 approach of Markley and coworkers (Hu, Westler, & Markley, 2011). In an alternative method, Kloiber and coworkers noted that if a second experiment is recorded in which the mixing period is moved prior to the t1 period, then only auto-peaks are observed with amplitudes given by Ij(t) = [ajj(t) + ajk(t)]Mzk(0). A global analysis of the time dependence of I11(t) and I22(t), I1(t), and I2(t) allows determination of k12, k21, R11, and R12.(Kloiber, Spitzer, Grutsch, Kreutz, & Tollinger, 2011) This approach also is robust against variations in relaxation during the t2 acquisition period. For time delays from ~0 to ~0.5/(k12+k21), simulations, such as those shown in Figure 7, suggest that the two methods have similar precisions (although the second approach requires acquiring two spectra at each mixing time). Which approach is optimal in a given situation then depends on whether both I12(t) and I21(t) cross-peaks are resolved in the ZZ-exchange spectra for a suitable number of spins, which is required for the convenient global analysis offered by the approach of Miloushev and coworkers.
Figure 7.
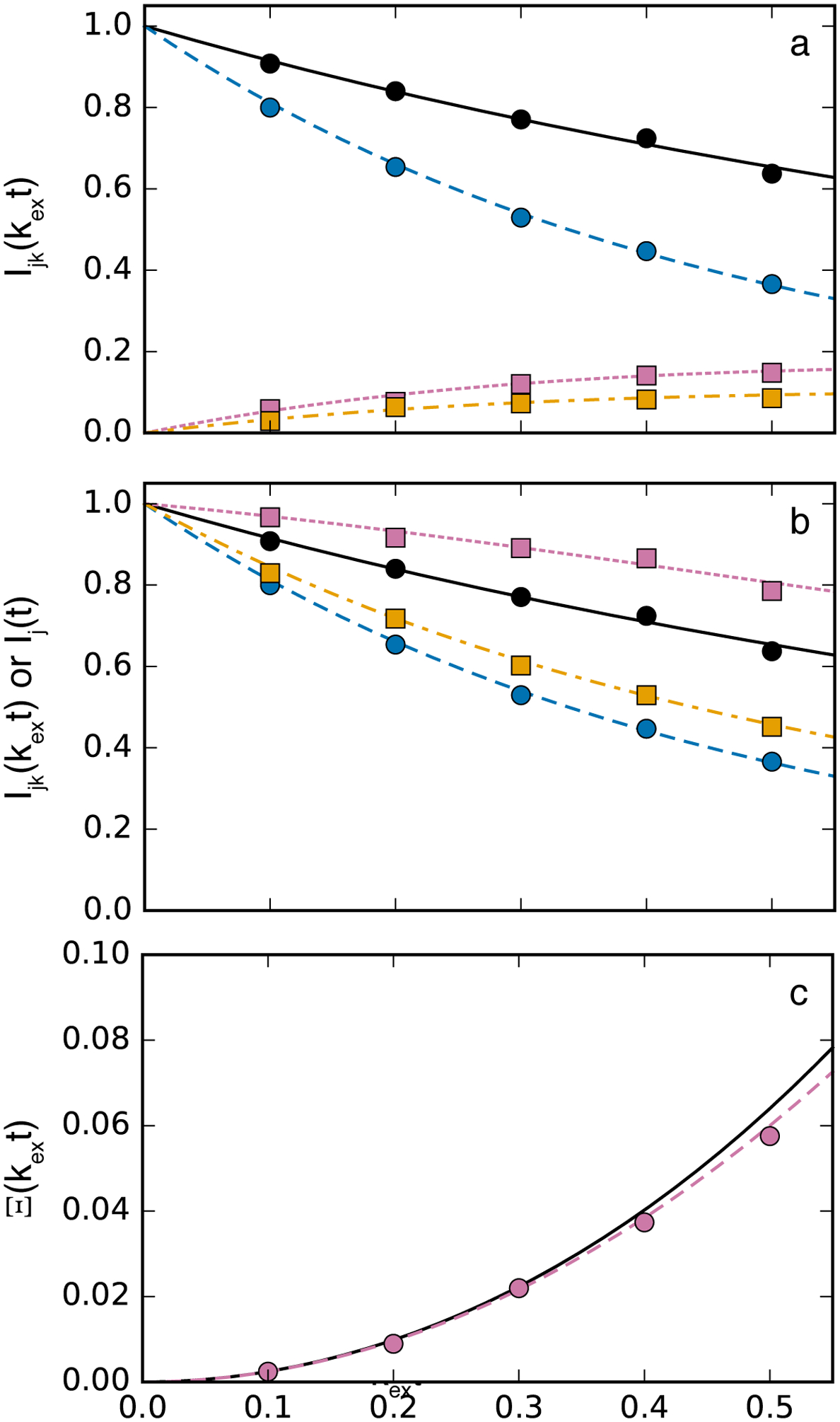
ZZ-exchange relaxation. (top) Normalized amplitudes (black, solid) I11(t), (blue, dashed), I22(t), (reddish-purple, dotted) I12(t), and (orange, dash-dotted) I21(t). (middle) Normalized amplitudes (black, solid) I11(t), (blue, dashed), I22(t), (reddish-purple, dotted) I1(t), and (orange, dash-dotted) I2(t). (bottom) Ξ(T) with (black, solid) calculated from best fit values from the analysis of the middle figure and (reddish-purple, dotted) calculated from Eq. 39. Circles, with same color coding as lines, show one of the simulated data sets and lines are calculated from best fit values. Simulations used R11/kex = 0.5, R12/kex = 1.5, p1 = 0.6, p2 = 0.4. Five hundred simulations were performed assuming a standard deviation for amplitudes of 0.01.
A recent example of the use of the 1H-15N ZZ-exchange experiment investigated folding kinetics of autonomously folding protein domains derived from the ribosomal protein L9 using high-pressure NMR spectroscopy (Zhang et al., 2016). Figure 8 shows results for three residues in the C-terminal domain of the I98A mutant of L9. As in the original work, data for individual peak intensities shown in Fig. 8a–c were fit simultaneously using Eq. 38 to give individual kinetic rate constants for each residue. Figure 8d shows the global analysis of data for the three residues using Eq. 39 to obtain k12k21. The simplicity of the global analysis is evident. However, as shown, only data at shorter mixing times should be used in the global analysis; at long values of T, the denominator in Eq. 39 approaches 0 and uncertainties in Ξ(T) are amplified.
Figure 8.
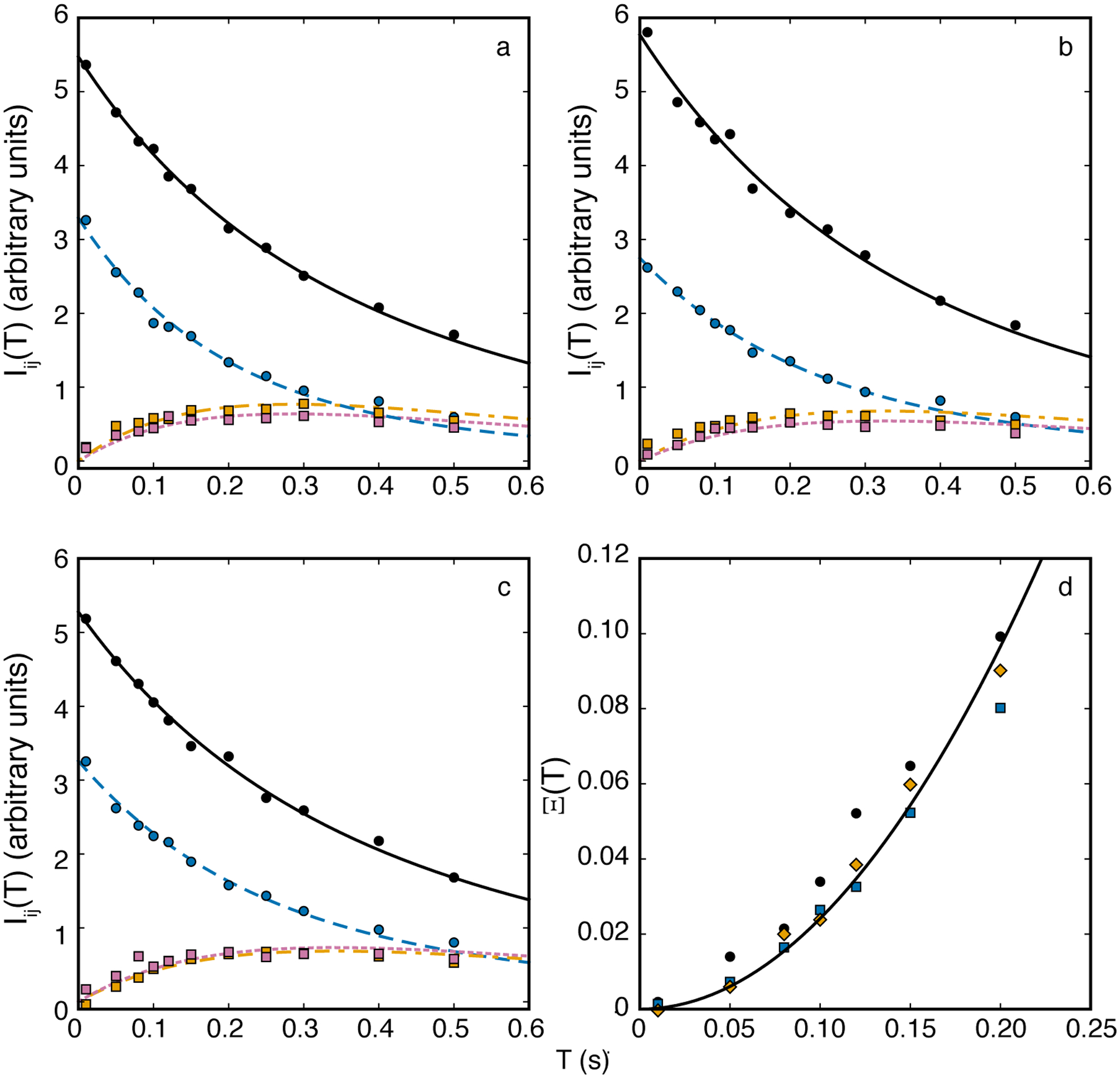
ZZ-exchange spectroscopy for the C-terminal domain of the I98A mutant of L9 protein for residues (a) 70, (b) 71, and (c) 72 at a static pressure of 300 bar. Solid lines in each panel are fits of the auto-peaks for (black, solid, circles) folded and (blue, dashed, circles) unfolded states and two cross peaks for (reddish-purple, dotted, squares) folded → unfolded and (orange, dashed, squares) unfolded → folded states using Eq. 38. (d) Global fit of Ξ(T) for residues (black) 70, (blue) 71, and (orange) 72 fit to Eq. 39. The fitted value of k12k21 = 2.08 ± 0.16 in (d) agrees well with the average value 2.16 ± 0.23 obtained from the fits in (a-c). Data were obtained from Zhang et al (Zhang et al., 2016).
3.2. Hahn-Echo, CPMG and R1ρ Experiments
Hahn-echo relaxation experiments measure the transverse relaxation rate constant from a spectrum recorded with one or two spin-echo periods, in which the total time T is as long as feasible (consistent with sensitivity) to mimic the free-precession relaxation rate constant. In the simplest approach, a second spectrum is recorded in which the spin-echo periods are omitted. The relaxation rate constant is obtained from the ratio of signal intensities in the two experiments, I(T)/I(0), using Eq. 5. As shown in Figure 9, numerical simulations for a single spin echo, compared with the exact free-precession R2 (given by the real part of Eq. 11) show that the Hahn-echo experiment closely approximates the free-precession relaxation rate constant if exchange is fast (kex/Δω21 > 4) or T is sufficiently long (T greater than ~10/Δω21). Differential relaxation, in which R22 ≠ R21 improves agreement between the exact eigenvalue and the Hahn-echo result (not shown).
Figure 9.
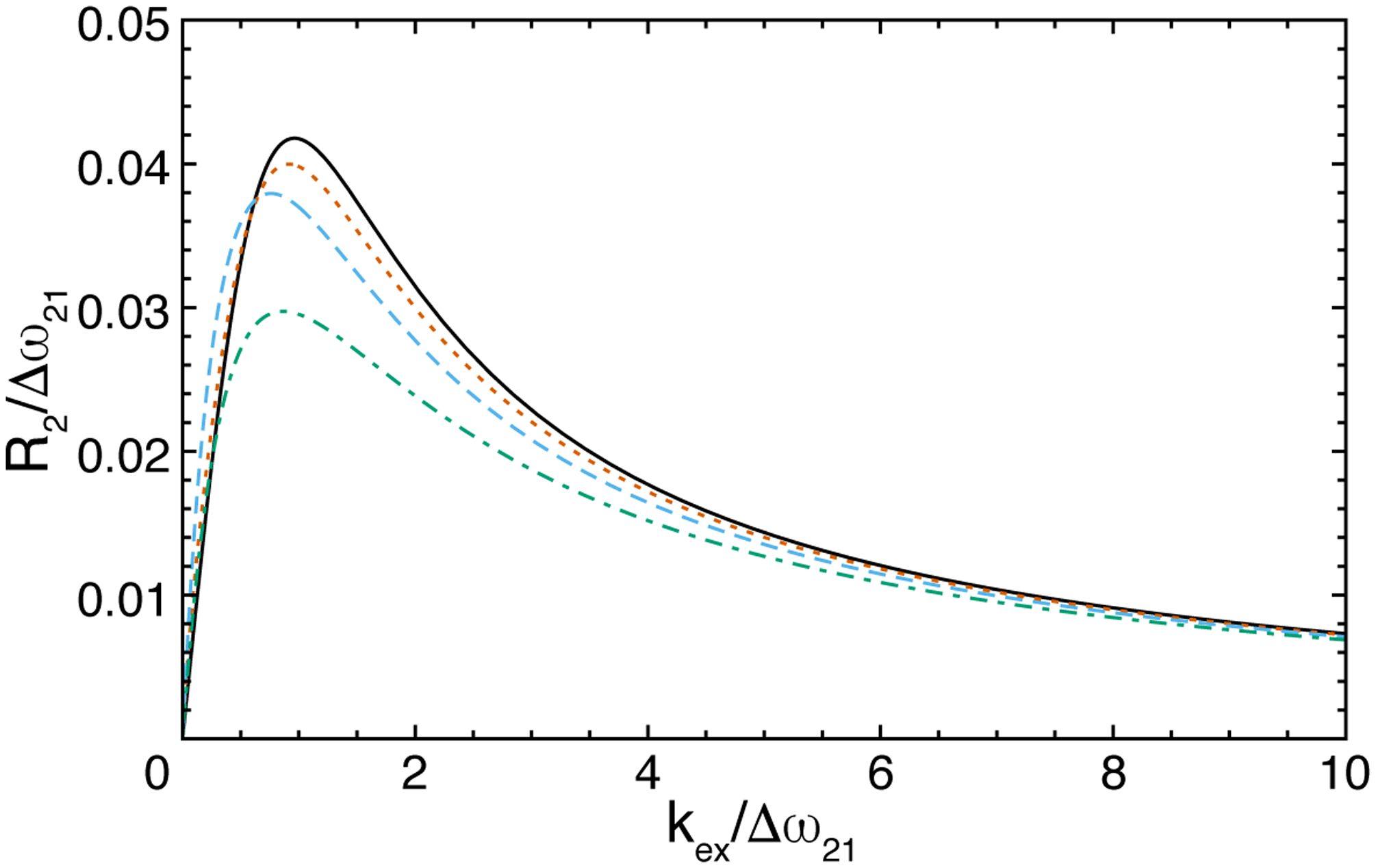
Hahn-echo transverse relaxation rate constant for 2-site chemical exchange with p2 = 0.08 and R21 = R22 = 0. (black, solid) Exact R2 given as the real part of eigenvalue for free-precession (Eq. 11) and Hahn-echo for single spin-echo with total time (blue, dashed) t = 25/Δω21, (orange, dotted) t = 10/Δω21, and (green, dash-dotted) t = 5/Δω21.
A number of Hahn-echo experimental methods have been described in the literature. Examples include the 1H-15N TROSY Hahn-echo (C. Wang, Rance, & Palmer, 2003), 13Cα Hahn-echo (O’Connell et al., 2009), and the 13CH3 methyl zero-quantum TROSY Hahn-echo experiments (Gill & Palmer, 2011; Ollerenshaw, Tugarinov, Skrynnikov, & Kay, 2005; Tugarinov et al., 2004). In particular, the 13CH3 methyl zero-quantum TROSY Hahn-echo experiment recently has been applied to characterization of chemical exchange in the DNA-repair enzyme AlkB (Gill & Palmer, 2011) and in Gα proteins (Toyama et al., 2017).
The CPMG experiment depends on acquisition of a relaxation dispersion curve in which the relaxation rate constants for spins of interest are measured as functions of τcp. In coupled spin systems, the evolution of scalar coupling interactions also varies as τcp is changed. This results in deleterious τcp-dependent averaging of relaxation rate constants of in-phase and anti-phase magnetization. The first solution to this difficulty was the relaxation-compensated CPMG experiment, in which in-phase and anti-phase magnetization components are interchanged halfway through the CPMG pulse train in order to exactly average the in-phase and anti-phase relaxation rate constants, rendering the average independent of τcp (Loria, Rance, & Palmer, 1999a). Since the original description of the relaxation-compensated 15N CPMG experiment, numerous variants have been developed for applications to a variety of spin systems in both proteins and nucleic acids (Sauerwein & Hansen, 2015; Walinda et al., 2018b). The second solution to this difficulty employs spin-decoupling methods to prevent evolution of the scalar coupling. Decoupling must be carefully executed to avoid interfering with proper refocusing of the CPMG spin echoes, but has the advantage of avoiding averaging between in-phase and anti-phase magnetization (Hansen, Vallurupalli, & Kay, 2008a) TROSY(Loria, Rance, & Palmer, 1999b) and BEST-TROSY (Franco et al., 2017) versions of this experiment that improve sensitivity also have been reported.
Only chemical exchange on a time scale covered by the τcp range of the CPMG experiment can be detected. For slow exchange, τcp therefore has to be large. For large τcp, oscillations in the CPMG curve can be observed for slow exchange. Tollinger et al. have found that the oscillations can be described by a functional form depending on sinc(Δω21τcp) for very slow exchange (Eq 2 in (Tollinger et al., 2001)). Recently, Koss and coworkers derived analytical expressions for slow N-site exchange (N ≥ 2) that include sinc2(Δωijτcp) terms (Eqs. 27 and 28 for example) (Koss, Rance, et al., 2018). These expressions approximate the dominant single eigenvalue of the average magnetization evolution matrix; higher-order eigenvalue eigenvalue approximations yield results which are very similar to those obtained from the Carver-Richards equation (for N = 2). The maxima for sinc(Δω21τcp) and sinc2(Δω21τcp), are identical, at τcp = nπ/|Δω21|; the integral for both functions (0 < τcp < ∞) is π/2. The sinc(Δω21τcp) functional dependence describes oscillations in the dispersion profile better for very slow exchange and very large minor populations (Tollinger et al., 2001); in these special cases, exchange cannot be described using the dominant single eigenvalue. For slow-to-intermediate exchange or smaller minor site populations, the sinc2(Δω21τcp) functional dependence gives more accurate results. Analysis of the oscillatory region might be useful to distinguish kinetic schemes: For two-site exchange, sinc2(Δω21τcp) (or sinc(Δω21τcp)) depends on Δω21τcp and directly describes the oscillations, while N-site (N > 2) exchange gives more complex oscillatory patterns. For kinetic schemes with increasing complexity, approximations based on a single dominant eigenvalue become more reliable because the single-eigenvalue model only become invalid for large τcp when the number of sites connected to the major site is 1 or 2, in combination with exclusively very slow exchange, including very slow exchange between minor sites. CPMG data at large τcp can be obtained be obtained either by increasing T without changing the number of CPMG cycles, which reduces sensitivity, or by reducing the number of CPMG cycles during a given T. Relaxation-compensated CPMG experiments, however, require a minimum number of 2 cycles. CPMG experiments using 1H continuous wave decoupling during the relaxation element can be used to overcome this problem by maintaining in-phase magnetization throughout the relaxation delay (Hansen, Vallurupalli, et al., 2008a).
A drawback of the CPMG experiment is that only the magnitude and not the sign of Δωn1 is obtained from analysis of CPMG relaxation dispersion curves. This disadvantage of the CPMG experiment, compared to the R1ρ experiment, is offset by the number of available experimental methods and relative ease of CPMG methods. For 2-site exchange, Kay and coworkers have shown that differences in F1 resonance frequencies between HSQC and HMQC experiments are indicative of the sign of chemical shift difference between states, provided exchange is not too fast (Skrynnikov, Dahlquist, & Kay, 2002). Thus, this technique is an important complement to 15N and 13C CPMG experiments.
As described above, the main strength of R1ρ measurements over other approaches for characterizing chemical exchange when ZZ-exchange measurements are not feasible, compared with Hahn-echo and CPMG methods, is the sensitivity of the R1ρ relaxation rate constant to the resonance frequencies of otherwise unobservable sparsely populated states. In many cases, experimental methods for R1ρ measurements are similar to those used for CPMG relaxation dispersion studies, except that the CPMG pulse train is replaced by a spin-locking sequence, which in the simplest case is continuous-wave radiofrequency field, but might be phase-modulated or applied as an adiabatic sweep. Particular experimental methods differ in how the desired spin magnetization is aligned with the effective field in the rotating frame and in how the effects of scalar coupling interactions are treated. As for other experiments, TROSY and non-TROSY versions of many sequences have been reported (Francesca Massi & Peng, 2018). More recently, R1ρ measurements for methyl groups (Weininger et al., 2013) or aromatic side chains (Weininger et al., 2014) have come into focus, as well as experiments to characterize slow dynamics in nucleic acids (Xue et al., 2015; Zhao, Hansen, & Zhang, 2014).
3.3. CEST and DEST
As originally described by Allerhand and Thiele, the limiting effect of chemical exchange on spin relaxation can be described as C-type or R-type, depending on whether the differences in chemical shifts or relaxation rates between states dominates (Allerhand & Thiele, 1966). These two cases have been exemplified in modern CEST (Bouvignies & Kay, 2012; Vallurupalli, Bouvignies, & Kay, 2012) and DEST (Fawzi, Ying, Ghirlando, Torchia, & Clore, 2011) experiments, respectively. Both experiments record changes in resonance signal intensities as functions of the resonance offset and amplitude of a spin-locking radiofrequency field. As such, both CEST and DEST experiments can be considered as variants of the general R1ρ experiment and are described by the same theory as described above for R1ρ relaxation (A. G. Palmer, 2014; Zhao et al., 2014). Methods for improving the time efficiency of CEST experiments have been developed that use multi-site excitation with DANTE (Yuwen, Kay, & Bouvignies, 2018) or appropriately designed shaped pulses (Leninger, Marsiglia, Jerschow, & Traaseth, 2018). For 1H-CEST experiments, a longitudinal relaxation optimized sequence has been developed (Yuwen & Kay, 2017).
More specifically, CEST or DEST experiment measures I(T)/I(0) for values of the resonance frequency of the radiofrequency field, ωrf, stepped through the spectral region of interest, and usually repeated for at least two values of ω1. As a consequence of the above considerations, the observed intensity signal in the CEST or DEST experiments is:
| (40) |
in which T is the length of the irradiation period. In contrast to conventional R1ρ experiments, the spin magnetization is not rotated to the tilted reference frame prior T and back to the rotating after T. Thus, in Eq. 40, one factor of cosθ results from projection of the initial longitudinal magnetization onto the tilted reference frame (this factor rapidly approaches unity for weak radiofrequency fields off-resonance with respect to the population-average chemical shift) and a second factor of cosθ results from projection of the magnetization locked along the direction of the effective field during T back onto the z-axis after the end of the irradiation period T (transverse magnetization after the irradiation period is dephased by a z-filter gradient pulse). This analysis does not consider the contribution near resonance from evolution of magnetization components orthogonal to the tilted z-axis; for long irradiation times used in CEST and DEST experiments, these components dephase owing to R2ρ relaxation. An alternative, equally valid, approach treats CEST and DEST experiments as modifications of the classic saturation transfer method introduced by Hoffman and Forsén (Forsén & Hoffman, 1963). In this approach, data analysis requires curve-fitting by numerical integration of the Bloch-McConnell equations, for example Eq. 1 with parameters defined in Eq. 29, rather than direct application of the analytical expression Eq. 40.
In support of the above interpretation, Figure 10 illustrates the approach of the evolving magnetization towards the state described by Eq. 40. Large oscillations arising from magnetization components orthogonal to the effective field are observed when the spin-locking field is on-resonance with the major state (Fig. 10b). The magnetization decays toward zero through a combination of R1ρ and R2ρ processes, while B1 inhomogeneity accelerates the damping process. Off-resonance from the major state, the contribution of orthogonal magnetization is reduced (Fig. 10a and c). Importantly, the effects of orthogonal magnetization are reduced on-resonance with the minor state (Figure 10d). In the example shown, T ≥ 0.25 s assures that Eq. 40 is applicable. The effects of B1 inhomogeneity are reduced off-resonance from the major state, when resonance offset dominates ω1, but become more significant as ω1 is increased. Thus, R2ρ relaxation contributes to damping of orthogonal magnetization components in the “transient regime” (unless B1 inhomogeneity or ω1 is large), but once these components decay to zero at times T commonly employed in practice, Eq. 40 is accurate description of the CEST profile.
Figure 10.
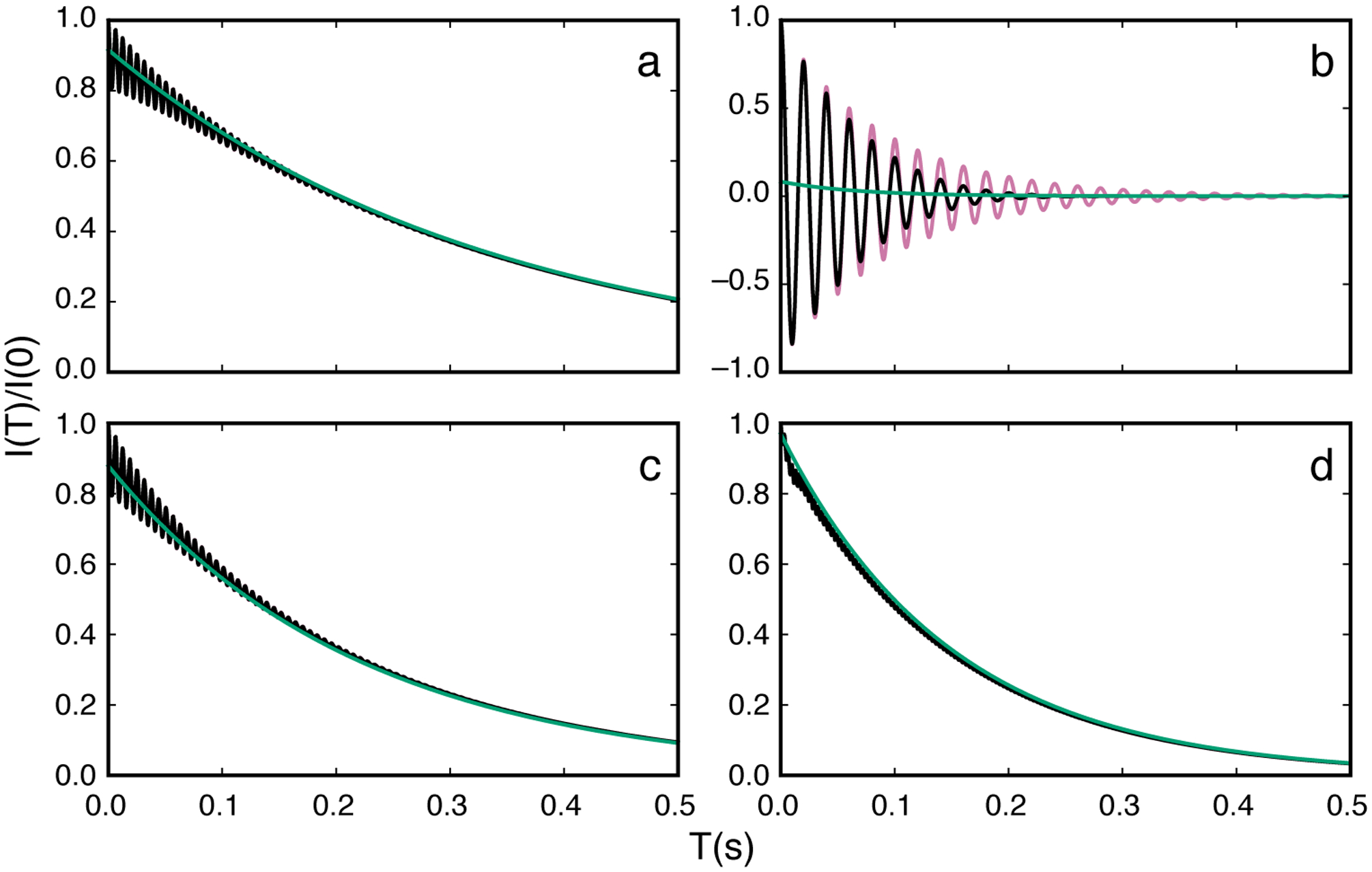
Time evolution of magnetization in CEST experiments. Parameters used for calculations were Δω21/2π = 200 Hz, ω1/2π = 50 Hz, kex = 100 s−1, R11 = R12 = 2.0 s−1, R21 = R22 = 20 s−1, and T = 0.3 s. (reddish-purple or gray, oscillating) Numerical integration of the Bloch-McConnell equations for the nominal value of ω1, (black, oscillating) numerical integration of the Bloch-McConnell equations for a Gaussian distribution of spin-lock field strengths centered on the nominal value of ω1 with a standard deviation of 3%, and (green or light gray) Eq. 40 using a numerical eigenvalue of L+K+R. Panels show decay curves for ωrf/2π = (a) −100 Hz, (b) 0 Hz (on-resonance with major state), (c) 100 Hz, and (d) 200 Hz (on-resonance with minor state).
Equation 40 assumes that either spin-spin interactions are decoupled, or the applied radiofrequency field has amplitude ω1 >> πJ, in which J is the scalar coupling constant. If these conditions are not satisfied, then each multiplet component in the irradiated spin system experiences a different resonance offset, and hence different relaxation decay profile. This situation has been investigated for R1ρ relaxation by Igumenova and Palmer and for CEST by Zhou and Yang (Igumenova et al., 2007; Zhou & Yang, 2014). In the simplest case of an IS spin system in which the S spin is irradiated,
| (41) |
In these expressions “α” refers to the S spin magnetization for which the coupling partner is in the α spin state (and similarly for “β”). The offset frequencies used to calculate the tilt angle and effective fields becomes Ωi ± πJ, depending on the sign of the scalar coupling and whether the α or β spin states were being considered. The resulting CEST profiles then depend on whether coupled or decoupled in-phase magnetization is detected, as in an HSQC or HMQC experiment, or a single multiplet component is detected, as in a TROSY-selected experiment. Note that decoupling or spin-state selection may be infeasible for homonuclear scalar coupling interactions. Simulations of these cases are shown in Figure 11. In addition, as discussed by Igumenova and Palmer, in the spin-locked tilted frames of reference, cross-relaxation between these magnetization components can occur (Igumenova & Palmer, 2006). The cross relaxation rate constant is dominated by dipole-dipole interactions with remote spins. Thus, cross-relaxation is reduced in molecules in which remote sites are highly deuterated.
Figure 11.
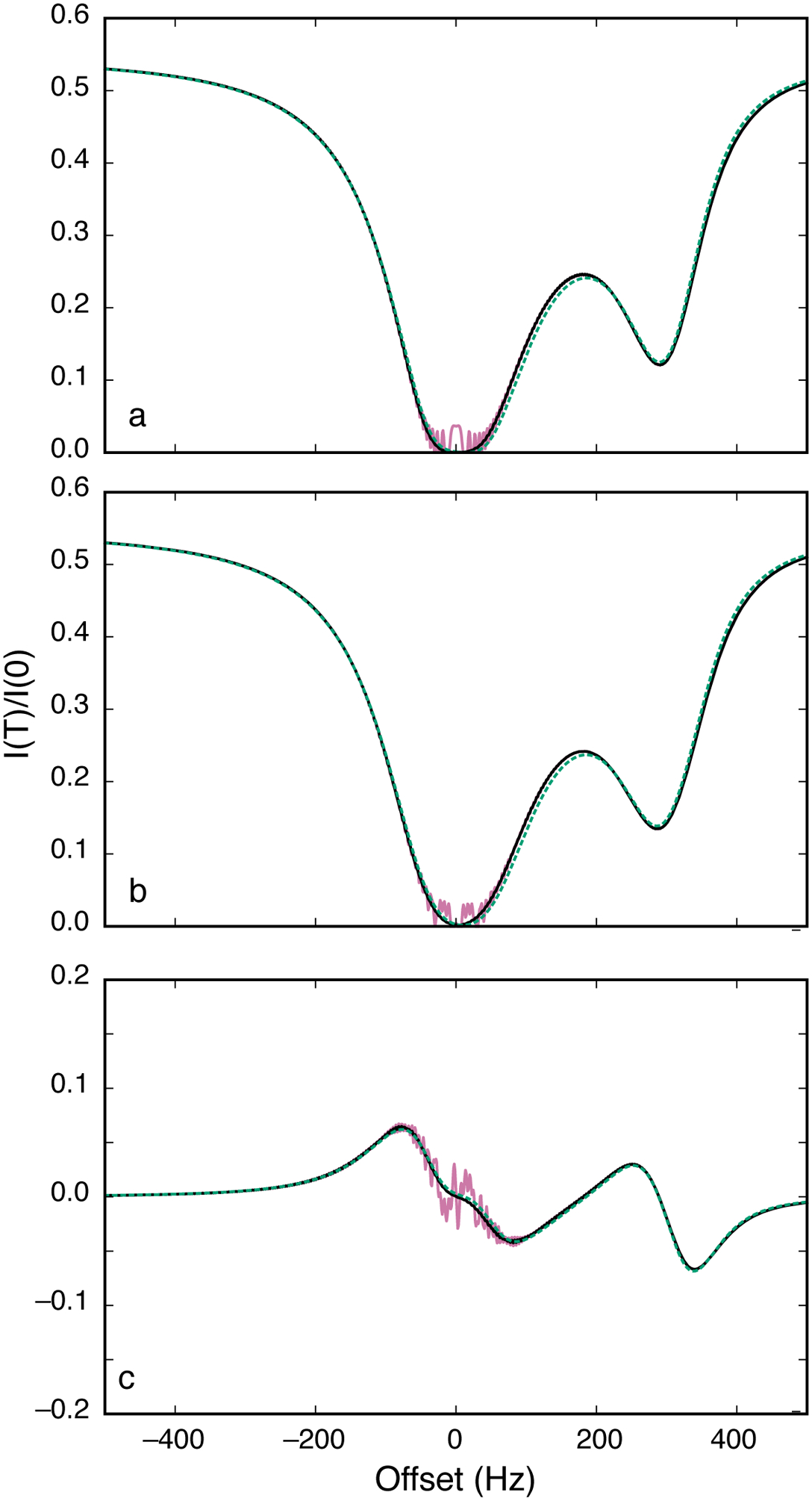
CEST profiles. Parameters used for calculations were Δω21/2π = 300 Hz, ω1/2π = 50 Hz, kex = 100 s−1, R11 = R12 = 2.0 s−1, R21 = R22 = 20 s−1, T = 0.3 s, and J = 33 Hz. (reddish-purple or grey, solid) Numerical integration of the Bloch-McConnell equations for the nominal value of ω1, (black, solid) numerical integration of the Bloch-McConnell equations for a Gaussian distribution of spin-lock field strengths centered on the nominal value of ω1 with a standard deviation of 3%, and (green, dotted) Eq. 40 using a numerical eigenvalue of L+K+R. (a) Results are calculated (a) detection of total longitudinal magnetization with decoupling of scalar coupling interactions during T (using Eq. 40) (b) detection of total longitudinal magnetization with no decoupling of scalar coupling interaction during T (using Eq. 42) and (c) detection of longitudinal two-spin order without decoupling of scalar coupling constants (using Eq. 42).
The conceptual simplicity of the CEST experiment can be complicated further by effects other than chemical exchange. As a prominent example, conventional 1H CEST experiments contain contributions from 1H-1H dipole-dipole cross relaxation as well as the effects of the desired chemical exchange phenomena. Yuwen and Kay have described an elegant approach to suppress the artifacts from 1H-1H dipole-dipole cross relaxation for amide and methyl groups that makes use of the difference between R1ρα and R1ρβ (Yuwen, Sekhar, & Kay, 2017). When the amplitude of the 1H radiofrequency field used for CEST irradiation satisfies ω1 < πJXH, then 1H components of the density operator in which the attached heteronuclear spin is in the α or β state are differentially affected by exchange during T, as shown by Eq. 41. As a result, longitudinal two-spin order, 2IzSz, is created during T. Subsequently, this two-spin order, rather than z-magnetization as in conventional CEST experiments, is selected and detected. The resulting spectra yield CEST profiles free of dipole-dipole artifacts, because dipole-dipole cross relaxation cannot generate two-spin order. CSA-dipole cross-correlated relaxation does lead to build-up of two-spin order, and can be eliminated by a more complex irradiation scheme, as shown by Yuwen and Kay, but appear to be negligible in practice. An example CEST profile arising from this approach is shown in Fig. 11c.
Equation 32 or the expression obtained by Baldwin and Kay both are accurate for the CEST experiment, provided that ΔR21 is not large (Baldwin & Kay, 2013). However, the DEST experiment relies on ΔR21 >> 0, as for a protein or other molecule interacting with a very large particle, such as a fibril. Equation 32, which is first-order ΔR21, is inaccurate in this limit. The expression obtained by Baldwin and Kay remains accurate, as does the simple result obtained from perturbation theory:
| (42) |
in which the second line is obtained as ΔR21 → ∞. In this limit, nearly instantaneous relaxation of spins in site 2 prevents back transfer of magnetization from site 2 to site 1; essentially, the two sites become non-secular in the sense of relaxation theory (vide supra). This result is identical to the the result obtained for C-type exchange if Δω21 → ∞. In addition, if ΔR21 >> Δω21, then Eq. 41 becomes insensitive to C-type exchange and relaxation dispersion arises from R-type relaxation only. Thus, if ΔR21 > 0, chemical exchange can be characterized by R1ρ relaxation dispersion even if Δω21 = 0 (Yuwen, Brady, & Kay, 2018). Examples of DEST profiles calculated using the above expressions or by numerical integration of the Bloch-McConnell equations (Eq. 1 with parameters as defined in Eq. 29) are shown in Figure 12. The R1ρ-based approach is seen to be highly accurate in describing the DEST experiment as well as the CEST experiment.
Figure 12.
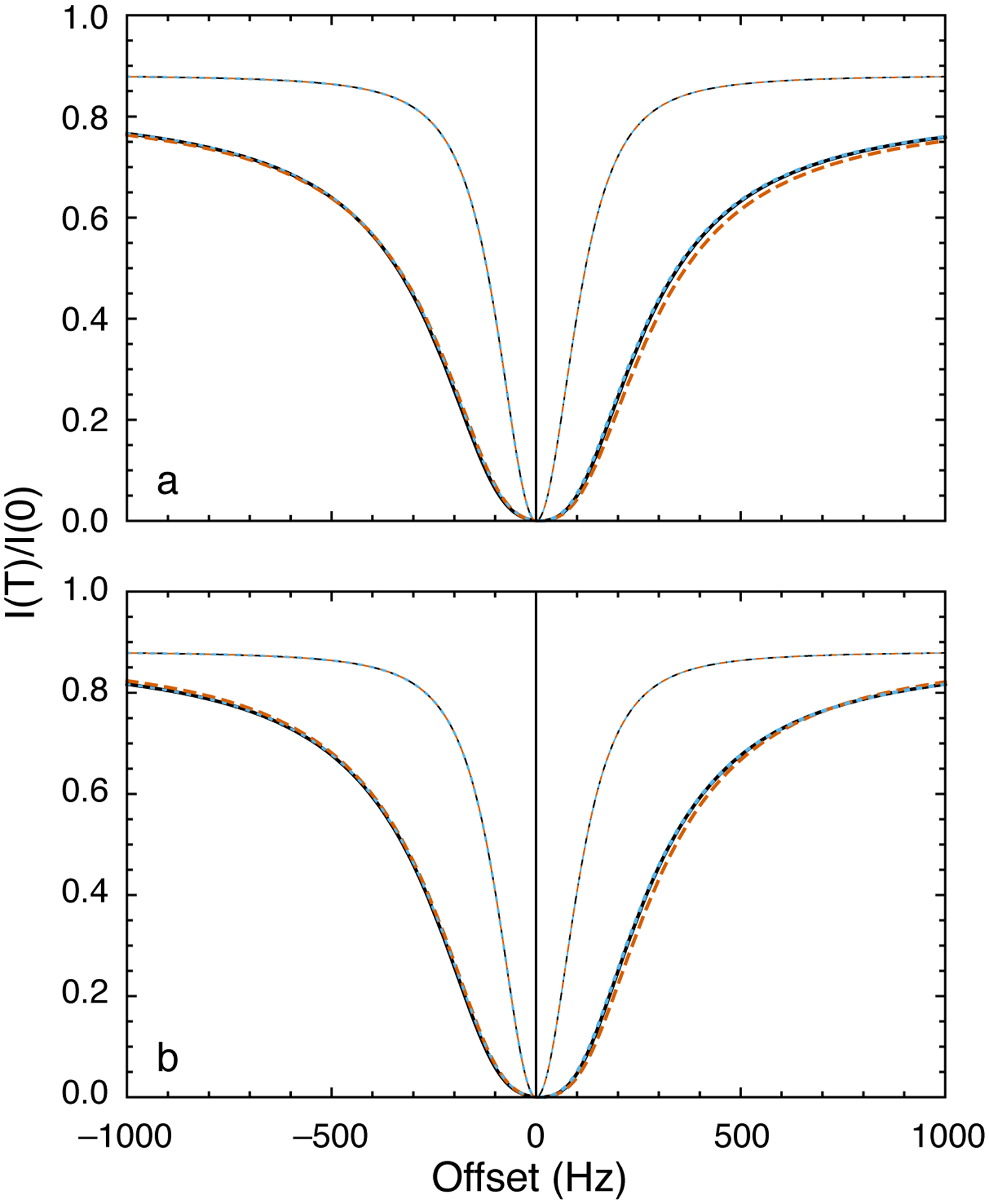
DEST profiles. (a) Parameters used for calculations were Δω21/2π = 200 Hz, ω1/2π = 200 Hz, kex = 1200 s−1, R11 = R12 = 2.5 s−1, R21 = 6 s−1, and ΔR21 = 20,000 s−1. Thick lines show calculations for p2 = 0.05 and thin lines show calculations for p2 → 0. (black, solid) Numerical integration of the Bloch-McConnell equations, (blue, dotted) Baldwin-Kay equation,(Baldwin & Kay, 2013) and (orange, dashed) Eqs. 40 and 42. (b) Results are calculated with ΔR21 = 200,000 s−1 and other parameters as in (a). Lines are drawn as in (a) except that the dashed orange line is the limit of Eq. 42 as as ΔR21 → ∞.
Zhang and coworkers have reported a very complete comparison of ZZ-exchange, R1ρ, and CEST methods applied to the fluoride riboswitch (Zhao et al., 2014). The CEST data were originally analyzed by numerical integration of the Bloch-McConnell equations. The 13C CEST data reported for the C8 position of guanine 8 of the riboswitch are shown in Figure 13a. The solid lines show the fit to Eqs. 40 and 42 to highlight the facility of the above interpretation of the CEST experiment. The data analysis using Eqs. 40 and 42 (or the combination of Eqs. 40 and 32) is more efficient than integration of the Bloch-McConnell equations. Of course, R1ρ can be calculated from the CEST data by inversion of Eq. 40 and a graph of the results obtained by this approach are shown in Figure 13b. The R1ρ data obtained independently also are shown in Fig. 13c for comparison. The overall profiles are similar, but quantitative differences arise because the initial magnetization in the R1ρ experiment was selected as the magnetization of the dominant state, as opposed to the CEST experiment, in which the initial magnetization was proportional to the equilibrium magnetization (Xue et al., 2015; Zhao et al., 2014).
Figure 13.
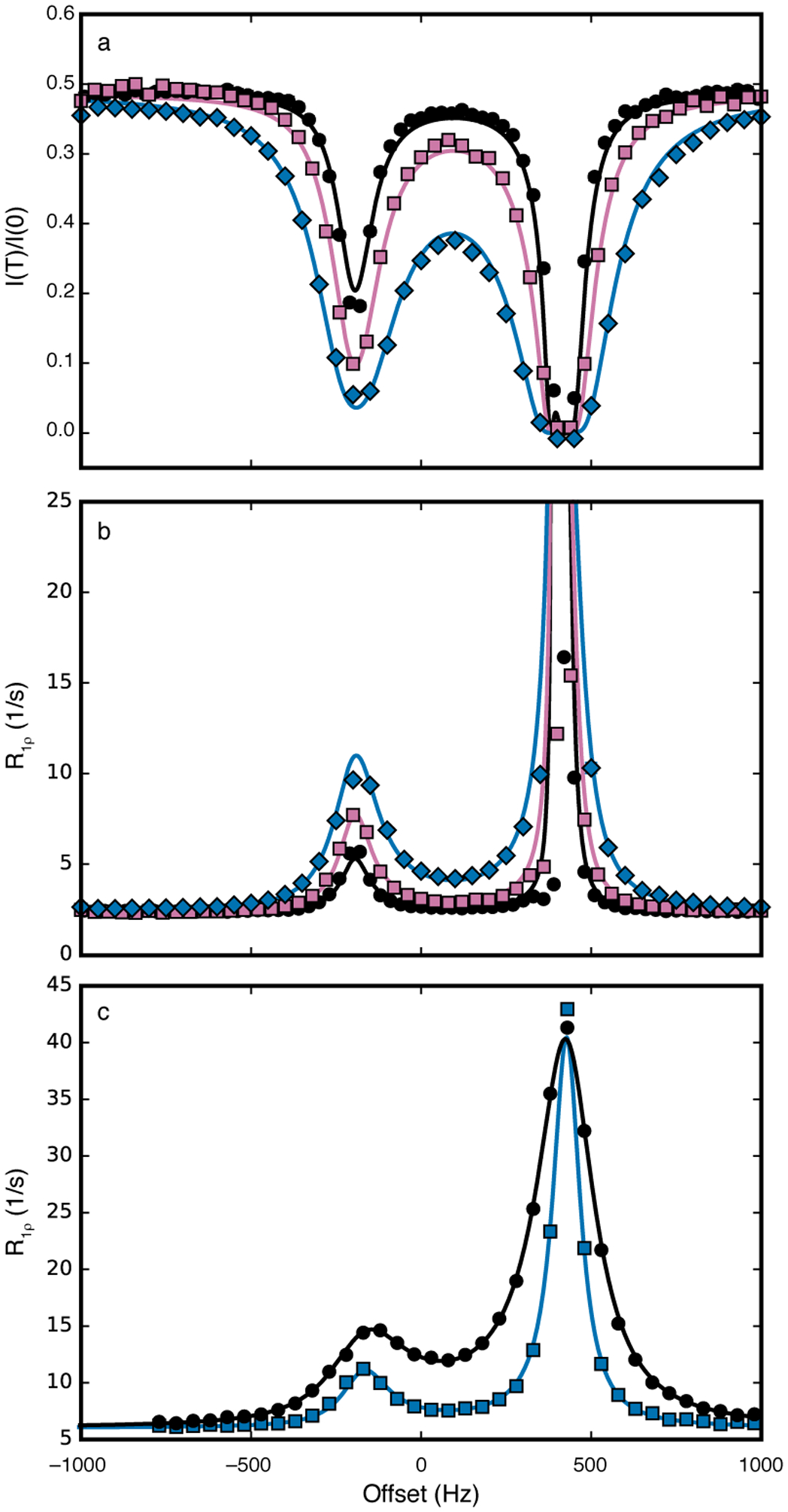
13C CEST profiles and R1ρ dispersion for the C-8 position of Guanine 8 in the fluoride riboswitch. (a) Data were recorded for ω1/2π = (black, circles) 17.7 Hz, (reddish-purple, squares) 27.9 Hz, and (blue, diamonds) 48.2 Hz. Solid lines represent fits to Eqs 40 and 42. (b) CEST data shown in (a) were converted to R1ρ values using Eq. 40. Solid lines are calculated from Eq. 42 using fitted parameters from (a). Color scheme as in (a). (c) R1ρ relaxation dispersion measured using ω1/2π = (blue, squares) 48.2 Hz and (black, circles) 102.4 Hz. Solid lines are fit with Eq. 42. Data were obtained from Zhao, et al. (Zhao et al., 2014).
3.4. Determination of Rex from relaxation dispersion experiments
In many situations, differentiating between the effects on relaxation rates or linewidths from chemical exchange, Rex, and other relaxation mechanisms, such as dipole-dipole and CSA interactions, R0, is of interest. Thus, a given relaxation rate constant, R, might be partitioned as:
| (43) |
in which ω = ωe for R1ρ -type experiments and 1/(4τcp) for CPMG-type experiments. Thus, relaxation experiments generally do not directly measure the exchange contribution to relaxation, Rex, because dipole-dipole and CSA relaxation processes, R1j and R2j, also contribute to spin relaxation through R0 (vide supra). Because R or R(ω → 0) usually can be approximated in a relatively straightforward manner, as in a Hahn-echo experiment, R0 or R(ω → ∞) must be measured separately to obtain Rex. Information on R0 or R(ω → ∞) also can usefully constrain fitting algorithms for relaxation dispersion data (Grey et al., 2006; O’Connell et al., 2009).
One way to estimate R0 involves extrapolation of relaxation data that have been recorded at different field strengths to B0 = 0 (Millet, Loria, Kroenke, Pons, & Palmer, 2000). For example, the static magnetic field dependence of Hahn-echo relaxation rate constants, provided CSA values are available, allows separation of chemical exchange, dipole-dipole, and CSA contributions to relaxation (O’Connell et al., 2009; Phan, Boyd, & Campbell, 1996; Toyama et al., 2017). A second approach estimates the desired R0 from a relaxation rate constant, such as a cross-correlation relaxation rate constant, that is insensitive to chemical exchange effects but has a similar dependence on other relaxation mechanisms (Kroenke, Loria, Lee, Rance, & Palmer, 1998; F. Massi, Wang, & Palmer, 2006).
Chemical exchange effects also can be suppressed in a reference relaxation dispersion experiment performed very high spin-lock power in R1ρ experiments or very small τcp in CPMG experiments to obtain R(ω → ∞). The realization that modern cryogenically cooled probeheads can withstand 6.4 kHz 15N-spin lock fields with durations of 20 ms has led to the development of high-power relaxation dispersion experiments, which facilitate determination of R20 (Ban et al., 2017). Thus, the spin-lock based HEROINE experiment can be used as a reference for constant-time relaxation-compensated CPMG experiments (Ban et al., 2013). An analogous approach has been reported for multi-quantum Hahn-Echo experiments (Toyama, Osawa, Yokogawa, & Shimada, 2016). A related approach, suited as a reference experiment for in-phase magnetization relaxation dispersion experiments, has been developed previously by Hansen et al. to obtain exchange-free values R2 from a linear combination of relaxation rate constants for a specific set of longitudinal and spin-locked 1H-15N coherences (Hansen et al., 2007). R20 also can be determined from CPMG by setting τcp to be sufficiently small. However, when the duty cycle (ratio of 180° pulsing time to τcp) is more than 1/10, as rough approximation, evolution during the pulses may become relevant and complicates data analysis. Reddy et al. have demonstrated in the “extreme CPMG” experiment that Rcpmg(τcp) estimates can be obtained at a CPMG frequency of up to 1/(τcp) = 6.4 kHz for 15N nuclei, which is essentially equivalent to τcp → 0 (Reddy et al., 2018); an intensive cooling regime is required to protect sample and probe from overheating in this limit. Such a “windowless” extreme-CPMG experiment is equivalent to an R1ρ experiment with phase jumps (due to the x,x,y,−y phase cycle). Simulations show that relaxation rates are, however, not dramatically changed in comparison to a phase cycle with x-phases only (equivalent to a continuous wave spin-lock). “Extreme-CPMG” experiments can cover a broad chemical shift range, while R1ρ experiments have the advantage of avoiding complicated evolution of magnetization during 180° pulses, either due to pulse imperfections or phase changes from pulse to pulse.
3.5. Choosing appropriate relaxation dispersion experiments
The complexity of the kinetic schemes and the large number of parameters for N-site kinetic models might suggest that available data would be insufficient to characterize 4-site models like the kite or the quadratic scheme. However, CPMG and R1ρ/CEST experiments yield an enormous amount of data. First, the static magnetic field B0 can be varied in all of these experiments (and indeed is essential even for 2-site exchange) (Millet et al., 2000). In addition, these experiments typically simultaneously record data for a number of spins with different chemical shifts. For CPMG experiments, theoretical calculations illustrated in Figure 14 suggest that various 2- and 3-site models often can be distinguished as long as data are recorded at two different B0 fields, and as long as data for a sufficient number of spins (preferably > 15) are analyzed (Koss, Rance, et al., 2018). For R1ρ/CEST experiments, data should be recorded preferably at 2 different B0 fields and at two ω1 fields. In an R1ρ experiment, theoretical calculations suggest that a combination of spins with different shifts, in the given example five, can be sufficient to drastically enhance distinguishability between two- and three-site (triangular/BAC) models for a wide range of parameters (Figure 15). Interestingly, a combination of a small and large ω1 fields generally performs well, but in particular also for cases in which exchange between sites A and B and between sites A and C is slow, but between minor sites B and C is fast. Based on these simulations, complicated N-site schemes may be distinguishable, for example by the combination of the following experiments, all at two different B0 fields: CEST experiments for at least 3 spins, and an R1ρ experiment at a higher ω1 field (to complement the CEST data); additional CPMG data can also be useful in many cases.
Figure 14:
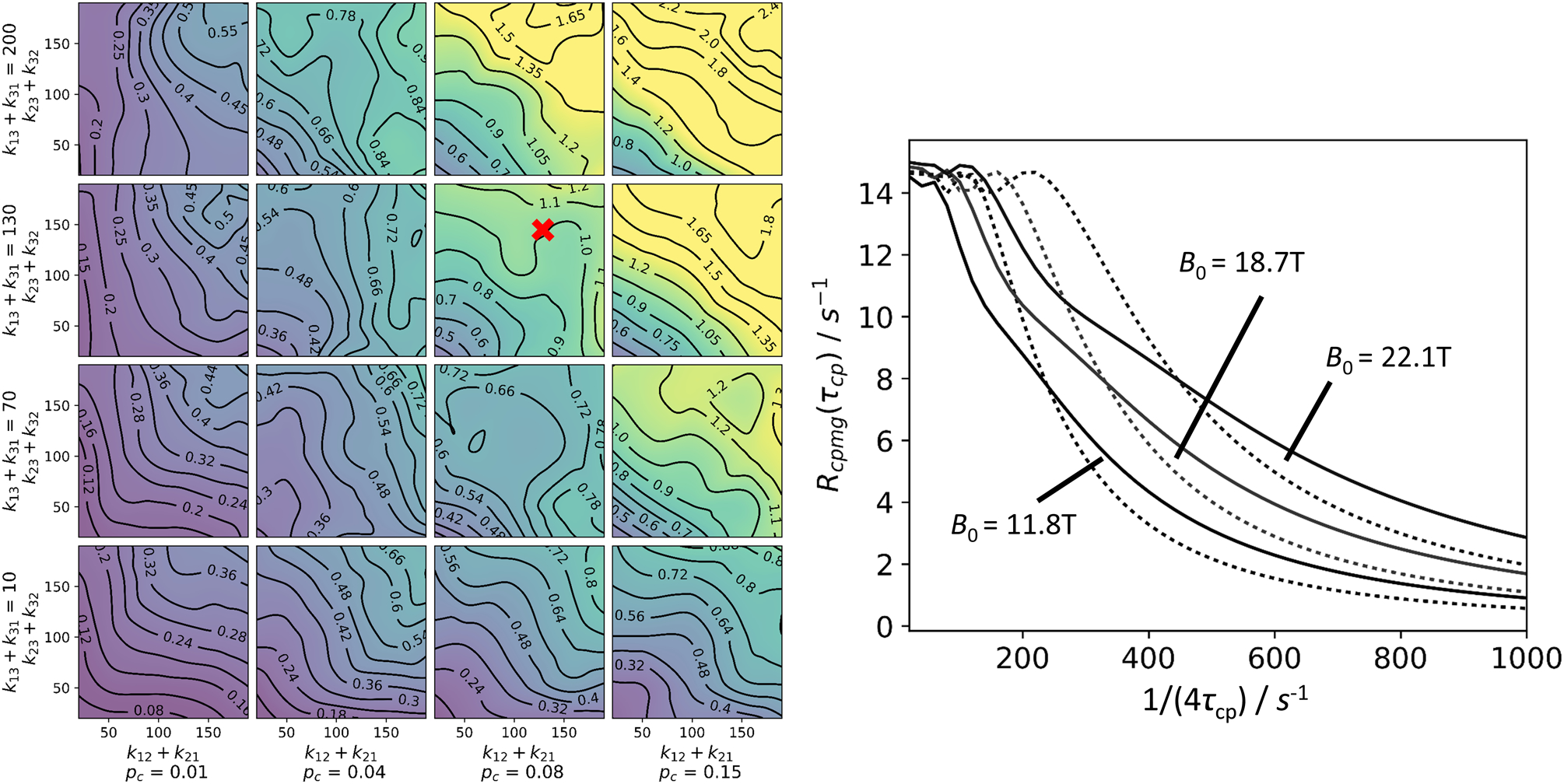
Distinguishability of the triangular kinetic scheme from the two-state model for CPMG experiments. Left: General conditions for distinguishability between triangular and linear schemes. In each of the 16 subpanels, 25 points with different values of k12/k23 were analyzed by globally fitting synthetic three-state curves to a two-state model, with a 1/(4τcp) range 20–1000 s−1. For each point, 20 RMS values, comparing the synthetic and the fitted data set, were obtained. For the global fitting procedure, a set of 3 × 32 CPMG curves was used, with 32 randomized ΔωAB (700–2000 s−1 at 11.7 T) and ΔωAC(±700–2000 s−1), and with 3 magnetic fields (11.7 T, 18.7 T, 22.2 T). The median, which was obtained from the 20 RMS values, is shown. pb was set to 0.04. To aid in convergence, some variables were constrained between upper and lower bounds during fitting: pb between 0.005 and 0.25; k12 + k21 between 10 s−1 and 400 s−1; ΔωAB between 450 s−1 and 2500 s−1. Right: Example plot for fits of synthetic triangular data to a two-state model (slow exchange approximations from truncation of the matrix exponential series, with eigenvalue approximation of 2nd order, Halley’s method), point of interest labeled in left panel. (solid) Synthetic triangular scheme data; (dotted) best fits. In this global fit, 32 randomized spin sets (see left panel) were used; plots are shown for a single spin set, ΔωAB (1892 s−1) and ΔωAC(−722 s−1), at three magnetic fields, as indicated in the figure. Other parameters: k12 + k21 = 100 s−1; k13 + k31 = 130 s−1; k23 + k32 = 150 s−1; pA = 0.88; pB = 0.04; pC = 0.08. The RMS deviation in this example is 1.38 s−1for the 1/(4τcp) region 20–300 s−1.
Figure 15.
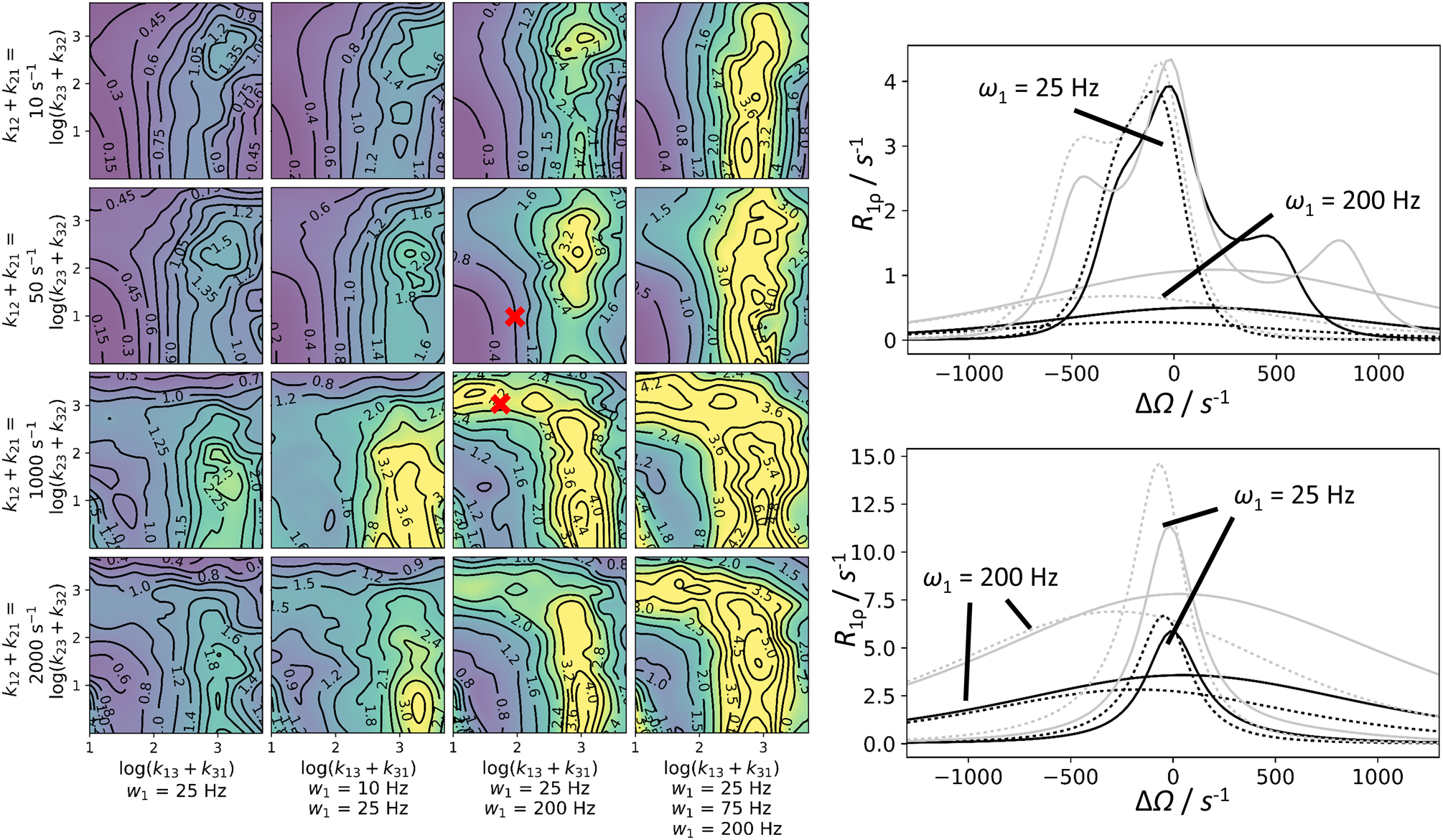
Distinguishability of the 3-state linear and triangular kinetic scheme from the two-state model in R1ρ experiments. Left panel: General conditions for distinguishability. Each subpanel has been created from 90 data points; each corresponds to the median of 20 separately calculated RMS values. For each RMS value, five sets of spins with randomly generated chemical shifts ΔωAB (200–800 s−1 at 11.7 T) and ΔωAC (±200–800 s−1) were included. In addition, two B0 fields (11.7 T, 18.7 T) and various combinations of one to three ω1 fields (see figure) were used. This yielded 10 to 30 numerically generated R1ρ curves for three sites. These curves were globally fit to a two-site model (ΔΩ range: −1300 to 1300 s−1). The RMS for each of the synthetic and fitted curve combinations was calculated. The results are shown in the contour plot. The points from which the sample fits in the right panels were generated are marked. Multiple parameters are shown in the plot, other parameters are: pB = 0.05; pC = 0.02. To aid in convergence, some variables were constrained between upper and lower bounds during fitting: pB between 0.005 and 0.23; k12 + k21 between 5 s−1 and 7000 s−1; ΔωAB between 100 s−1 and 1200 s−1. Right panel: Example curves corresponding to various kinetic situations marked in the right panel (top: k12 + k21 = 50 s−1; k13 + k31 = 100 s−1; k23 + k32 = 10 s−1; ω1/(2π) = 25 and 200 Hz; bottom: k12 + k21 = 1000 s−1; k13 + k31 = 50 s−1; k23 + k32= 1000 s−1; ω1/(2π) = 25 and 200 Hz). Numerical three-state solutions (solid lines) are shown alongside the best two-state fits (dotted lines). The fits result from global fits of 20 curves: five spins (in plot only shown: ΔωAB = 536 s−1; ΔωAC = −312 s−1); B0 = 11.7 T, black; B0 = 18.7 T, grey; ω1/(2π) = 50 Hz and 200 Hz, as indicated in the figure. The RMS deviation for the given examples are 0.74 s−1 (top) and 3.48 s−1 (bottom).
Conclusion
The 2-site model in which a molecule or system of molecules interconverts reversibly between two states at chemical equilibrium is powerful in descriptions of chemical exchange in NMR spectroscopy (and other areas of biophysics) because it is analytically tractable, allowing both qualitative insight and facile data analysis. More complex biological systems are becoming accessible to NMR spectroscopy, for example due to techniques focusing on the side chains of large proteins, or embedment of membrane proteins into nanodiscs. In those systems, multiple states may interconvert on multiple timescales. The complexity of the resulting Bloch-McConnell or stochastic Liouville equations obscures insight and complicates data analysis. The examples surveyed in this review illustrate theoretical advances leading to improved experimental methods, formulations facilitating global analysis of experimental data for improved power, and analytical approximations generalizing the simple 2-site model to arbitrary kinetic schemes.
Acknowledgments.
We thank Catherine Royer (Rensselaer Polytechnic Institute) and Qi Zhang (Univ. North Carolina, Chapel Hill) for providing the data used to prepare Figures 8 and 13, respectively. We thank Mark Rance (Univ. Cincinnati) for fruitful collaboration and helpful discussions. Support from National Institutes of Health grant R01 GM059273 to A.G.P. is acknowledged gratefully.
Abbreviations
- CEST
Chemical exchange saturation transfer
- CPMG
Carr-Purcell-Meiboom-Gill
- DEST
Dark state exchange saturation transfer
Appendix
A.1. Cayley-Hamilton theorem
The Cayley-Hamilton theorem states that a matrix A satisfies its own characteristic equation. Thus, for a 2 ×2 matrix,
| (A1) |
in which 0 is the zero matrix. Any matrix equation containing powers of A > 1 can be reduced by repeated use of the above expression. For example,
| (A2) |
which has the form of Eq. 19:
| (A3) |
with
| (A4) |
The equation
| (A5) |
is formally solved for a0 and a1 by diagonalizing Z(τcp) (this matrix is Hermitian and consequently has real eigenvalues) to obtain the algebraic equations:
| (A6) |
in which Λn is an eigenvalue of Z(τcp). The solution to this equation gives:
| (A7) |
in which the relationships between the eigenvalues, the trace and determinant have been used. This expression can be used with any Padé approximation to the Liouvillian for 2-site exchange. Using the above example,
| (A8) |
As noted in the text, this approach allows the inverse matrix to be easily obtained as:
| (A9) |
and the lowest-order approximation to the relaxation rate constant is given by:
| (A10) |
For 3-site exchange, the evolution matrices have dimensions 3 ×3 and the Cayley-Hamilton theorem is:
| (A11) |
The same approach as outlined for 2-site exchange, but more complicated algebraically, allows the Hamiltonian H11(tcp) to be expressed using an = dn/D with:
| (A12) |
A.2. Effective relaxation rate constant in CPMG experiments
A formal solution for N-state CPMG obtained from Eq. 14 is (with detection of the major species):
| (A13) |
in which <δ1n| is a 1 × N vector with values δ1n for n = {1,..,N}; S is diagonal with ;|uj> and {λj} are the eigenvectors and eigenvalues of (symmetrized) evolution matrix , in which |uj> is the jth column of U and < uj| is the the jth row of U−1; |ψj> and Λj are the eigenvectors and eigenvalues of the symmetrized average Liouvillian H(τcp), in which <ψj| is the complex transpose of ; and H(τcp) is defined in Eq. 17.
From Eq. 5, an effective relaxation rate constant is defined as:
| (A14) |
in which
| (A15) |
and . On the last line of the above equations, Λ1 is the least-negative eigenvalue of H(τcp), which is approximated as described in Section 2.2, and Rcorr gives the difference between the eigenvalue estimate of the relaxation rate constant and the effective relaxation rate constant obtained from Eq. 5.
The matrix H(τcp) is Hermitian, so the eigenvalues Λj are real and the factors exp[(Λj−Λ1)4mτcp] are pure exponential decays. The eigenvalues λi are complex and the factors exp[(λi−λk)τcp] are oscillatory. These contribute significantly when τcp is long (or exchange is very slow). If T = 4nτcp is sufficiently long, as is usually the case if significant magnetization decay occurs during T, then |(Λ2 − Λ1)|4mτcp >> 1, exp[(Λj−Λ1)4mτcp]→0 and the j > 1 terms vanish:
| (A16) |
In addition, if the correction is small, then the logarithm can be expanded to first-order:
| (A17) |
The correction term Rcorr vanishes as T = 4nτcp becomes large. The correction term also will vanish as pn→0 for n > 1, or exchange becomes fast.
The eigenvalues and eigenvectors of are obtained at low computational cost by standard methods. If the approximations of Eqs. A16 or A17 are appropriate, the eigenvector ψ1 can be estimated efficiently by inverse iteration:
| (A18) |
with z(0) = [0 … 1]T and the approximate largest (least negative) eigenvalue of H(τcp), Λ10, as described in Section 2.2, Numerical calculations suggest that ψ1(2) or ψ1(3) are sufficiently accurate. Note that for N = 2, the eigenvalue Λ1 and the eigenvector ψ1 can be calculated exactly, leading to the result reported by Baldwin (Baldwin, 2014). Additional considerations arising from detection when exchange occurs on the intermediate time scale also have been discussed by Baldwin.
References
- Abergel D, & Palmer AG (2004). Approximate solutions of the Bloch-McConnell equations for two-site chemical exchange. Chemphyschem, 5, 787–793. doi: 10.1002/cphc.200301051 [DOI] [PubMed] [Google Scholar]
- Allerhand A, & Thiele E (1966). Analysis of Carr—Purcell Spin-Echo NMR Experiments on Multiple-Spin Systems. II. The Effect of Chemical Exchange. J Chem Phys, 45, 902–916. doi: 10.1063/1.1727703 [DOI] [Google Scholar]
- Baldwin AJ (2014). An exact solution for R2,eff in CPMG experiments in the case of two site chemical exchange. J Magn Reson, 244, 114–124. doi: 10.1016/j.jmr.2014.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AJ, Hansen DF, Vallurupalli P, & Kay LE (2009). Measurement of methyl axis orientations in invisible, excited states of proteins by relaxation dispersion NMR spectroscopy. J Am Chem Soc, 131, 11939–11948. doi: 10.1021/ja903896p [DOI] [PubMed] [Google Scholar]
- Baldwin AJ, & Kay LE (2013). An R1ρ expression for a spin in chemical exchange between two sites with unequal transverse relaxation rates. J Biomol NMR, 55, 211–218. doi: 10.1007/s10858-012-9694-6 [DOI] [PubMed] [Google Scholar]
- Ban D, Mazur A, Carneiro MG, Sabo TM, Giller K, Koharudin LM, Becker S, Gronenborn AM, Griesinger C, & Lee D (2013). Enhanced accuracy of kinetic information from CT-CPMG experiments by transverse rotating-frame spectroscopy. J Biomol NMR, 57, 73–82. doi: 10.1007/s10858-013-9769-z [DOI] [PubMed] [Google Scholar]
- Ban D, Smith CA, de Groot BL, Griesinger C, & Lee D (2017). Recent advances in measuring the kinetics of biomolecules by NMR relaxation dispersion spectroscopy. Arch Biochem Biophys, 628, 81–91. doi: 10.1016/j.abb.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Bothe JR, Stein ZW, & Al-Hashimi HM (2014). Evaluating the uncertainty in exchange parameters determined from off-resonance R1ρ relaxation dispersion for systems in fast exchange. J Magn Reson, 244, 18–29. doi: 10.1016/j.jmr.2014.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvignies G, & Kay LE (2012). Measurement of proton chemical shifts in invisible states of slowly exchanging protein systems by chemical exchange saturation transfer. J Phys Chem B, 116, 14311–14317. doi: 10.1021/jp311109u [DOI] [PubMed] [Google Scholar]
- Brutscher B, Felli IC, Gil-Caballero S, Hosek T, Kummerle R, Piai A, Pierattelli R, & Solyom Z (2015). NMR Methods for the Study of Instrinsically Disordered Proteins Structure, Dynamics, and Interactions: General Overview and Practical Guidelines. Adv Exp Med Biol, 870, 49–122. doi: 10.1007/978-3-319-20164-1_3 [DOI] [PubMed] [Google Scholar]
- Carver JP, & Richards RE (1969). A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J Magn Reson, 6, 89–105. doi: 10.1016/0022-2364(72)90090-X [DOI] [Google Scholar]
- Chao FA, & Byrd RA (2016). Geometric Approximation: A new computational approach to characterize protein dynamics from NMR adiabatic relaxation dispersion experiments. J Am Chem Soc, 138, 7337–7345. doi: 10.1021/jacs.6b02786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao FA, & Byrd RA (2017). Application of geometric approximation to the CPMG experiment: Two- and three-site exchange. J Magn Reson, 277, 8–14. doi: 10.1016/j.jmr.2017.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Courtney JM, Alderson TR, Anfinrud P, & Bax A (2018). Monitoring 15N chemical shifts during protein folding by pressure-jump NMR. J Am Chem Soc, 140, 8096–8099. doi: 10.1021/jacs.8b04833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Cousin SF, & Ferrage F (2016). Protein dynamics from nuclear magnetic relaxation. Chem Soc Rev, 45, 2410–2422. doi: 10.1039/c5cs00832h [DOI] [PubMed] [Google Scholar]
- Chen H, Marsiglia WM, Cho MK, Huang Z, Deng J, Blais SP, Gai W, Bhattacharya S, Neubert TA, Traaseth NJ, & Mohammadi M (2017). Elucidation of a four-site allosteric network in fibroblast growth factor receptor tyrosine kinases. Elife, 6. doi: 10.7554/eLife.21137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, & Dobson CM (2006). Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem, 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- Delaforge E, Kragelj J, Tengo L, Palencia A, Milles S, Bouvignies G, Salvi N, Blackledge M, & Jensen MR (2018). Deciphering the dynamic interaction profile of an intrinsically disordered protein by NMR exchange spectroscopy. J Am Chem Soc, 140, 1148–1158. doi: 10.1021/jacs.7b12407 [DOI] [PubMed] [Google Scholar]
- Deshmukh L, Ghirlando R, & Clore GM (2014). Investigation of the structure and dynamics of the capsid-spacer peptide 1-nucleocapsid fragment of the HIV-1 gag polyprotein by solution NMR spectroscopy. Angew Chem Int Ed Engl, 53, 1025–1028. doi: 10.1002/anie.201309127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh L, Tugarinov V, Appella DH, & Clore GM (2018). Targeting a dark excited state of HIV-1 nucleocapsid by antiretroviral thioesters revealed by NMR spectroscopy. Angew Chem Int Ed Engl, 57, 2687–2691. doi: 10.1002/anie.201713172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh L, Tugarinov V, Louis JM, & Clore GM (2017). Binding kinetics and substrate selectivity in HIV-1 protease-Gag interactions probed at atomic resolution by chemical exchange NMR. Proc Natl Acad Sci U S A, 114, E9855–E9862. doi: 10.1073/pnas.1716098114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, & Wright PE (2004). Unfolded proteins and protein folding studied by NMR. Chem Rev, 104(8), 3607–3622. doi: 10.1021/cr030403s [DOI] [PubMed] [Google Scholar]
- Dyson HJ, & Wright PE (2017). How does your protein fold? Elucidating the apomyoglobin folding pathway. Acc Chem Res, 50, 105–111. doi: 10.1021/acs.accounts.6b00511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber PJ, & Mittermaier A (2015). Relaxation dispersion NMR spectroscopy for the study of protein allostery. Biophys Rev, 7, 191–200. doi: 10.1007/s12551-015-0166-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber PJ, Slager J, & Mittermaier AK (2012). Local folding and misfolding in the PBX homeodomain from a three-state analysis of CPMG relaxation dispersion NMR data. J Phys Chem B, 116, 10317–10329. doi: 10.1021/jp306127m [DOI] [PubMed] [Google Scholar]
- Farrow NA, Zhang O, Forman-Kay JD, & Kay LE (1995). Comparison of the backbone dynamics of a folded and an unfolded SH3 domain existing in equilibrium in aqueous buffer. Biochemistry, 34, 868–878. doi: 10.1021/bi00003a021 [DOI] [PubMed] [Google Scholar]
- Fawzi NL, Libich DS, Ying J, Tugarinov V, & Clore GM (2014). Characterizing methyl-bearing side chain contacts and dynamics mediating amyloid beta protofibril interactions using 13C(methyl)-DEST and lifetime line broadening. Angew Chem Int Ed Engl, 53, 10345–10349. doi: 10.1002/anie.201405180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi NL, Ying J, Ghirlando R, Torchia DA, & Clore GM (2011). Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR. Nature, 480, 268–272. doi: 10.1038/nature10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazelinia H, Xu M, Cheng H, & Roder H (2014). Ultrafast hydrogen exchange reveals specific structural events during the initial stages of folding of cytochrome c. J Am Chem Soc, 136, 733–740. doi: 10.1021/ja410437d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsén S, & Hoffman RA (1963). Study of moderately rapid chemical exchange reactions by means of nuclear magnetic double resonance. J Chem Phys, 39, 2892–2901. doi: 10.1063/1.1734121 [DOI] [Google Scholar]
- Franco R, Gil-Caballero S, Ayala I, Favier A, & Brutscher B (2017). Probing conformational exchange dynamics in a short-lived protein folding intermediate by real-time relaxation-dispersion NMR. J Am Chem Soc, 139, 1065–1068. doi: 10.1021/jacs.6b12089 [DOI] [PubMed] [Google Scholar]
- Furtig B, Buck J, Manoharan V, Bermel W, Jaschke A, Wenter P, Pitsch S, & Schwalbe H (2007). Time-resolved NMR studies of RNA folding. Biopolymers, 86, 360–383. doi: 10.1002/bip.20761 [DOI] [PubMed] [Google Scholar]
- Furukawa A, Konuma T, Yanaka S, & Sugase K (2016). Quantitative analysis of protein-ligand interactions by NMR. Prog Nucl Magn Reson Spectrosc, 96, 47–57. doi: 10.1016/j.pnmrs.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Gill ML, & Palmer AG (2011). Multiplet-filtered and gradient-selected zero-quantum TROSY experiments for 13C1H3 methyl groups in proteins. J Biomol NMR, 51, 245–251. doi: 10.1007/s10858-011-9533-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan AB, Hansen DF, & Vallurupalli P (2018). CPMG experiments for protein minor conformer structure determination. Methods Mol Biol, 1688, 223–242. doi: 10.1007/978-1-4939-7386-6_11 [DOI] [PubMed] [Google Scholar]
- Grey MJ, Tang Y, Alexov E, McKnight CJ, Raleigh DP, & Palmer AG (2006). Characterizing a partially folded intermediate of the villin headpiece domain under non denaturing conditions: contribution of His41 to the pH-dependent stability of the N-terminal subdomain. J Mol Biol, 355, 1078–1094. doi: 10.1016/j.jmb.2005.11.001 [DOI] [PubMed] [Google Scholar]
- Grey MJ, Wang C, & Palmer AG (2003). Disulfide bond isomerization in basic pancreatic trypsin inhibitor: Multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J Am Chem Soc, 125, 14324–14335. doi: 10.1021/ja0367389 [DOI] [PubMed] [Google Scholar]
- Hansen DF, Neudecker P, Vallurupalli P, Mulder FA, & Kay LE (2010). Determination of Leu side-chain conformations in excited protein states by NMR relaxation dispersion. J Am Chem Soc, 132, 42–43. doi: 10.1021/ja909294n [DOI] [PubMed] [Google Scholar]
- Hansen DF, Vallurupalli P, & Kay LE (2008a). An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J Phys Chem B, 112, 5898–5904. doi: 10.1021/jp074793o [DOI] [PubMed] [Google Scholar]
- Hansen DF, Vallurupalli P, & Kay LE (2008b). Quantifying two-bond 1HN-13CO and one-bond 1Hα−13Cα dipolar couplings of invisible protein states by spin-state selective relaxation dispersion NMR spectroscopy. J Am Chem Soc, 130, 8397–8405. doi: 10.1021/ja801005n [DOI] [PubMed] [Google Scholar]
- Hansen DF, Vallurupalli P, Lundstrom P, Neudecker P, & Kay LE (2008). Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: how well can we do? J Am Chem Soc, 130, 2667–2675. doi: 10.1021/ja078337p [DOI] [PubMed] [Google Scholar]
- Hansen DF, Yang D, Feng H, Zhou Z, Wiesner S, Bai Y, & Kay LE (2007). An exchange-free measure of 15N transverse relaxation: an NMR spectroscopy application to the study of a folding intermediate with pervasive chemical exchange. J Am Chem Soc, 129, 11468–11479. doi: 10.1021/ja072717t [DOI] [PubMed] [Google Scholar]
- Hu K, Westler WM, & Markley JL (2011). Simultaneous quantification and identification of individual chemicals in metabolite mixtures by two-dimensional extrapolated time-zero 1H-13C HSQC (HSQC0). J Am Chem Soc, 133, 1662–1665. doi: 10.1021/ja1095304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, & Kay LE (2005). Solution structure and dynamics of integral membrane proteins by NMR: a case study involving the enzyme PagP. Methods Enzymol, 394, 335–350. doi: 10.1016/S0076-6879(05)94013-5 [DOI] [PubMed] [Google Scholar]
- Igumenova TI, Brath U, Akke M, & Palmer AG (2007). Characterization of chemical exchange using residual dipolar coupling. J Am Chem Soc, 129, 13396–13397. doi: 10.1021/ja0761636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igumenova TI, & Palmer AG (2006). Off-resonance TROSY-selected R1ρ experiment with improved sensitivity for medium- and high-molecular-weight proteins. J Am Chem Soc, 128, 8110–8111. doi: 10.1021/ja061692f [DOI] [PubMed] [Google Scholar]
- Ishima R (2014). CPMG relaxation dispersion. Methods Mol Biol, 1084, 29–49. doi: 10.1007/978-1-62703-658-0_2 [DOI] [PubMed] [Google Scholar]
- Jee J, Ishima R, & Gronenborn AM (2008). Characterization of specific protein association by 15N CPMG relaxation dispersion NMR: the GB1(A34F) monomer-dimer equilibrium. J Phys Chem B, 112, 6008–6012. doi: 10.1021/jp076094h [DOI] [PubMed] [Google Scholar]
- Kempf JG, & Loria JP (2004). Measurement of Intermediate Exchange Phenomena. Methods Mol Biol, 278, 185–232. doi: 10.1385/1-59259-809-9:185 [DOI] [PubMed] [Google Scholar]
- Kimsey IJ, Petzold K, Sathyamoorthy B, Stein ZW, & Al-Hashimi HM (2015). Visualizing transient Watson-Crick-like mispairs in DNA and RNA duplexes. Nature, 519, 315–320. doi: 10.1038/nature14227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimsey IJ, Szymanski ES, Zahurancik WJ, Shakya A, Xue Y, Chu CC, Sathyamoorthy B, Suo Z, & Al-Hashimi HM (2018). Dynamic basis for dG*dT misincorporation via tautomerization and ionization. Nature, 554, 195–201. doi: 10.1038/nature25487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloiber K, Spitzer R, Grutsch S, Kreutz C, & Tollinger M (2011). Longitudinal exchange: an alternative strategy towards quantification of dynamics parameters in ZZ exchange spectroscopy. J Biomol NMR, 51, 123–129. doi: 10.1007/s10858-011-9547-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, & Kay LE (2004). Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J Am Chem Soc, 126, 3964–3973. doi: 10.1021/ja039587i [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, & Kay LE (2010). A transient and low-populated protein-folding intermediate at atomic resolution. Science, 329, 1312–1316. doi: 10.1126/science.1191723 [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Lundstrom P, Fersht AR, & Kay LE (2007). The folding pathway of an FF domain: characterization of an on-pathway intermediate state under folding conditions by 15N, 13Cα and 13C-methyl relaxation dispersion and 1H/2H-exchange NMR spectroscopy. J Mol Biol, 372, 497–512. doi: 10.1016/j.jmb.2007.06.012 [DOI] [PubMed] [Google Scholar]
- Koss H, Bunney TD, Esposito D, Martins M, Katan M, & Driscoll PC (2018). Dynamic allostery in PLCgamma1 and its modulation by a cancer mutation revealed by MD simulation and NMR. Biophys J, 115, 31–45. doi: 10.1016/j.bpj.2018.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss H, Rance M, & Palmer AG (2017). General expressions for R1ρ relaxation for N-site chemical exchange and the special case of linear chains. J Magn Reson, 274, 36–45. doi: 10.1016/j.jmr.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss H, Rance M, & Palmer AG (2018). General expressions for Carr-Purcell-Meiboom-Gill relaxation dispersion for N-Site chemical exchange. Biochemistry, 57, 4753–4763. doi: 10.1021/acs.biochem.8b00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovermann M, Rogne P, & Wolf-Watz M (2016). Protein dynamics and function from solution state NMR spectroscopy. Q Rev Biophys, 49, e6. doi: 10.1017/S0033583516000019 [DOI] [PubMed] [Google Scholar]
- Kremer W, Arnold M, Munte CE, Hartl R, Erlach MB, Koehler J, Meier A, & Kalbitzer HR (2011). Pulsed pressure perturbations, an extra dimension in NMR spectroscopy of proteins. J Am Chem Soc, 133, 13646–13651. doi: 10.1021/ja2050698 [DOI] [PubMed] [Google Scholar]
- Kroenke CD, Loria JP, Lee LK, Rance M, & Palmer AG (1998). Longitudinal and transverse1H−15N dipolar/15N chemical shift anisotropy relaxation interference: Unambiguous determination of rotational diffusion tensors and chemical exchange effects in biological macromolecules. J Am Chem Soc, 120, 7905–7915. doi: 10.1021/ja980832l [DOI] [Google Scholar]
- Leninger M, Marsiglia WM, Jerschow A, & Traaseth NJ (2018). Multiple frequency saturation pulses reduce CEST acquisition time for quantifying conformational exchange in biomolecules. J Biomol NMR, 71, 19–30. doi: 10.1007/s10858-018-0186-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Martins IR, & Rosen MK (2011). The feasibility of parameterizing four-state equilibria using relaxation dispersion measurements. J Biomol NMR, 51, 57–70. doi: 10.1007/s10858-011-9541-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Altorelli NL, Bahna F, Honig B, Shapiro L, & Palmer AG (2013). Mechanism of E-cadherin dimerization probed by NMR relaxation dispersion. Proc Natl Acad Sci U S A, 110, 16462–16467. doi: 10.1073/pnas.1314303110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, & Palmer AG (2009). TROSY-selected ZZ-exchange experiment for characterizing slow chemical exchange in large proteins. J Biomol NMR, 45, 357–360. doi: 10.1007/s10858-009-9385-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libich DS, Tugarinov V, & Clore GM (2015). Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc Natl Acad Sci U S A, 112, 8817–8823. doi: 10.1073/pnas.1510083112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Dyson HJ, Kelly JW, & Wright PE (2013). Localized structural fluctuations promote amyloidogenic conformations in transthyretin. J Mol Biol, 425, 977–988. doi: 10.1016/j.jmb.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, & Loria JP (2016). Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function. Prog Nucl Magn Reson Spectrosc , 92-93, 1–17. doi: 10.1016/j.pnmrs.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, Manley GA, Hendrickson H, Rivalta I, Batista VS, & Loria JP (2016). Dissecting dynamic allosteric pathways using chemically related small-molecule activators. Structure, 24, 1155–1166. doi: 10.1016/j.str.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria JP, Rance M, & Palmer AG (1999a). A relaxation-compensated Carr−Purcell−Meiboom−Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc, 121, 2331–2332. doi: 10.1021/ja983961a [DOI] [Google Scholar]
- Loria JP, Rance M, & Palmer AG (1999b). A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR, 15, 151–155. doi: 10.1023/A:1008355631073 [DOI] [PubMed] [Google Scholar]
- Mackenzie HW, & Hansen DF (2018). Arginine side-chain hydrogen exchange: Quantifying arginine side-chain interactions in solution. Chemphyschem. doi: 10.1002/cphc.201800598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangia S, Traaseth NJ, Veglia G, Garwood M, & Michaeli S (2010). Probing slow protein dynamics by adiabatic R1ρ and R2ρ NMR experiments. J Am Chem Soc, 132, 9979–9981. doi: 10.1021/ja1038787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoharan V, Furtig B, Jaschke A, & Schwalbe H (2009). Metal-induced folding of Diels-Alderase ribozymes studied by static and time-resolved NMR spectroscopy. J Am Chem Soc, 131, 6261–6270. doi: 10.1021/ja900244x [DOI] [PubMed] [Google Scholar]
- Massi F, & Peng JW (2018). Characterizing Protein Dynamics with NMR R1ρ Relaxation Experiments. Methods Mol Biol, 1688, 205–221. doi: 10.1007/978-1-4939-7386-6_10 [DOI] [PubMed] [Google Scholar]
- Massi F, Wang C, & Palmer AG (2006). Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry, 45, 10787–10794. doi: 10.1021/bi060764c [DOI] [PubMed] [Google Scholar]
- Meier BH, & Ernst RR (1979). Elucidation of chemical exchange networks by two-dimensional NMR spectroscopy: the heptamethylbenzenonium ion. J Am Chem Soc, 101, 6441–6442. doi: 10.1021/ja00515a053 [DOI] [Google Scholar]
- Meinhold DW, & Wright PE (2011). Measurement of protein unfolding/refolding kinetics and structural characterization of hidden intermediates by NMR relaxation dispersion. Proc Natl Acad Sci U S A, 108, 9078–9083. doi: 10.1073/pnas.1105682108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli S, Sorce DJ, Idiyatullin D, Ugurbil K, & Garwood M (2004). Transverse relaxation in the rotating frame induced by chemical exchange. J Magn Reson, 169, 293–299. doi: 10.1016/j.jmr.2004.05.010 [DOI] [PubMed] [Google Scholar]
- Millet O, Loria JP, Kroenke CD, Pons M, & Palmer AG (2000). The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc, 122, 2867–2877. doi:DOI 10.1021/ja993511y [DOI] [Google Scholar]
- Miloushev VZ, Bahna F, Ciatto C, Ahlsen G, Honig B, Shapiro L, & Palmer AG (2008). Dynamic properties of a type II cadherin adhesive domain: implications for the mechanism of strand-swapping of classical cadherins. Structure, 16, 1195–1205. doi: 10.1016/j.str.2008.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloushev VZ, & Palmer AG (2005). R1ρ relaxation for two-site chemical exchange: general approximations and some exact solutions. J Magn Reson, 177, 221–227. doi: 10.1016/j.jmr.2005.07.023 [DOI] [PubMed] [Google Scholar]
- Montelione GT, & Wagner G (1989). 2D Chemical exchange NMR spectroscopy by proton-detected heteronuclear correlation. J Am Chem Soc, 111, 3096–3098. doi: 10.1021/ja00190a072 [DOI] [Google Scholar]
- Nash AI, McNulty R, Shillito ME, Swartz TE, Bogomolni RA, Luecke H, & Gardner KH (2011). Structural basis of photosensitivity in a bacterial light-oxygen-voltage/helix-turn-helix (LOV-HTH) DNA-binding protein. Proc Natl Acad Sci U S A, 108, 9449–9454. doi: 10.1073/pnas.1100262108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell NE, Grey MJ, Tang Y, Kosuri P, Miloushev VZ, Raleigh DP, & Palmer AG (2009). Partially folded equilibrium intermediate of the villin headpiece HP67 defined by 13C relaxation dispersion. J Biomol NMR, 45, 85–98. doi: 10.1007/s10858-009-9340-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollerenshaw JE, Tugarinov V, Skrynnikov NR, & Kay LE (2005). Comparison of 13CH3, 13CH2D, and 13CHD2 methyl labeling strategies in proteins. J Biomol NMR, 33, 25–41. doi: 10.1007/s10858-005-2614-2 [DOI] [PubMed] [Google Scholar]
- Osawa M, Takeuchi K, Ueda T, Nishida N, & Shimada I (2012). Functional dynamics of proteins revealed by solution NMR. Curr Opin Struct Biol, 22, 660–669. doi: 10.1016/j.sbi.2012.08.007 [DOI] [PubMed] [Google Scholar]
- Oyen D, Fenwick RB, Aoto PC, Stanfield RL, Wilson IA, Dyson HJ, & Wright PE (2017). Defining the structural basis for allosteric product release from E. coli dihydrofolate reductase using NMR relaxation dispersion. J Am Chem Soc, 139, 11233–11240. doi: 10.1021/jacs.7b05958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG (2004). NMR characterization of the dynamics of biomacromolecules. Chem Rev, 104, 3623–3640. doi: 10.1021/cr030413t [DOI] [PubMed] [Google Scholar]
- Palmer AG (2014). Chemical exchange in biomacromolecules: past, present, and future. J Magn Reson, 241, 3–17. doi: 10.1016/j.jmr.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG (2016). A dynamic look backward and forward. J Magn Reson, 266, 73–80. doi: 10.1016/j.jmr.2016.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG, Grey MJ, & Wang C (2005). Solution NMR spin relaxation methods for characterizing chemical exchange in high-molecular-weight systems. Methods Enzymol, 394, 430–465. doi: 10.1016/S0076-6879(05)94018-4 [DOI] [PubMed] [Google Scholar]
- Palmer AG, & Massi F (2006). Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev, 106, 1700–1719. doi: 10.1021/cr0404287 [DOI] [PubMed] [Google Scholar]
- Phan IQ, Boyd J, & Campbell ID (1996). Dynamic studies of a fibronectin type I module pair at three frequencies: Anisotropic modelling and direct determination of conformational exchange. J Biomol NMR, 8, 369–378. doi: 10.1007/BF00228140 [DOI] [PubMed] [Google Scholar]
- Pustovalova Y, Kukic P, Vendruscolo M, & Korzhnev DM (2015). Probing the residual structure of the low populated denatured state of ADA2h under folding conditions by relaxation dispersion nuclear magnetic resonance spectroscopy. Biochemistry, 54, 4611–4622. doi: 10.1021/acs.biochem.5b00345 [DOI] [PubMed] [Google Scholar]
- Reddy JG, Pratihar S, Ban D, Frischkorn S, Becker S, Griesinger C, & Lee D (2018). Simultaneous determination of fast and slow dynamics in molecules using extreme CPMG relaxation dispersion experiments. J Biomol NMR, 70, 1–9. doi: 10.1007/s10858-017-0155-0 [DOI] [PubMed] [Google Scholar]
- Rennella E, & Brutscher B (2013). Fast real-time NMR methods for characterizing short-lived molecular states. Chemphyschem, 14, 3059–3070. doi: 10.1002/cphc.201300339 [DOI] [PubMed] [Google Scholar]
- Rennella E, Sekhar A, & Kay LE (2017). Self-assembly of human profilin-1 detected by Carr-Purcell-Meiboom-Gill nuclear magnetic resonance (CPMG NMR) spectroscopy. Biochemistry, 56, 692–703. doi: 10.1021/acs.biochem.6b01263 [DOI] [PubMed] [Google Scholar]
- Robustelli P, Stafford KA, & Palmer AG (2012). Interpreting protein structural dynamics from NMR chemical shifts. J Am Chem Soc, 134, 6365–6374. doi: 10.1021/ja300265w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J, Royer CA, & Roumestand C (2017). Monitoring protein folding through high pressure NMR spectroscopy. Prog Nucl Magn Reson Spectrosc, 102–103, 15–31. doi: 10.1016/j.pnmrs.2017.05.003 [DOI] [PubMed] [Google Scholar]
- Roder H, Elove GA, & Englander SW (1988). Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature, 335, 700–704. doi: 10.1038/335700a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstenn G, Vuister GW, Zwanenburg N, Hellingwerf KJ, Boelens R, & Kaptein R (1999). NMR experiments for the study of photointermediates: application to the photoactive yellow protein. J Magn Reson, 137, 443–447. doi: 10.1006/jmre.1999.1705 [DOI] [PubMed] [Google Scholar]
- Sahasrabudhe P, Shanmugasundaram V, Flanagan M, Borzilleri KA, Heaslet H, Rane A, McColl A, Subashi T, Karam G, Sarver R, Harris M, Chrunyk BA, Subramanyam C, Magee TV, Fahnoe K, Lacey B, Putz H, Miller JR, Cho J, Palmer AG, & Withka JM (2017). Driving drug discovery with biophysical information: : Application to Staphylococcus aureus dihydrofolate reductase (DHFR) In Zartler D. H. a. E. R. (Ed.), Applied Biophysics for Drug Discovery (pp. 241–261): John Wiley & Sons Ltd. [Google Scholar]
- Sahu D, Clore GM, & Iwahara J (2007). TRO SY-based z-exchange spectroscopy: application to the determination of the activation energy for intermolecular protein translocation between specific sites on different DNA molecules. J Am Chem Soc, 129, 13232–13237. doi: 10.1021/ja074604f [DOI] [PubMed] [Google Scholar]
- Salmon L, & Blackledge M (2015). Investigating protein conformational energy landscapes and atomic resolution dynamics from NMR dipolar couplings: a review. Rep Prog Phys, 78, 126601. doi: 10.1088/0034-4885/78/12/126601 [DOI] [PubMed] [Google Scholar]
- Salvi N, Abyzov A, & Blackledge M (2017). Atomic resolution conformational dynamics of intrinsically disordered proteins from NMR spin relaxation. Prog Nucl Magn Reson Spectrosc, 102–103, 43–60. doi: 10.1016/j.pnmrs.2017.06.001 [DOI] [PubMed] [Google Scholar]
- Sauerwein A, & Hansen DF (2015). Relaxation dispersion NMR spectroscopy Protein NMR, Biological Magnetic Resonance (Vol. 32, pp. 75–132): Springer. [Google Scholar]
- Skrynnikov NR, Dahlquist FW, & Kay LE (2002). Reconstructing NMR spectra of “invisible” excited protein states using HSQC and HMQC experiments. J Am Chem Soc, 124, 12352–12360. doi: 10.1021/ja0207089 [DOI] [PubMed] [Google Scholar]
- Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, & Kay LE (2001). Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J Am Chem Soc, 123, 4556–4566. doi: 10.1021/ja004179p [DOI] [PubMed] [Google Scholar]
- Solt AS, Bostock MJ, Shrestha B, Kumar P, Warne T, Tate CG, & Nietlispach D (2017). Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat Commun, 8, 1795. doi: 10.1038/s41467-017-02008-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Gribun A, Hwang PM, Houry WA, & Kay LE (2005). Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc Natl Acad Sci U S A, 102, 16678–16683. doi: 10.1073/pnas.0507370102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski ES, Kimsey IJ, & Al-Hashimi HM (2017). Direct NMR evidence that transient tautomeric and anionic states in dG.dT form Watson-Crick-like base pairs. J Am Chem Soc, 139, 4326–4329. doi: 10.1021/jacs.7b01156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Grey MJ, McKnight J, Palmer AG, & Raleigh DP (2006). Multistate folding of the villin headpiece domain. J Mol Biol, 355, 1066–1077. doi: 10.1016/j.jmb.2005.10.066 [DOI] [PubMed] [Google Scholar]
- Tollinger M, Skrynnikov NR, Mulder FAA, Forman-Kay JD, & Kay LE (2001). Slow Dynamics in Folded and Unfolded States of an SH3 Domain. J Am Chem Soc, 123, 11341–11352. doi: 10.1021/ja011300z [DOI] [PubMed] [Google Scholar]
- Toyama Y, Kano H, Mase Y, Yokogawa M, Osawa M, & Shimada I (2017). Dynamic regulation of GDP binding to G proteins revealed by magnetic field-dependent NMR relaxation analyses. Nat Commun, 8, 14523. doi: 10.1038/ncomms14523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama Y, Osawa M, Yokogawa M, & Shimada I (2016). NMR method for characterizing microsecond-to-millisecond chemical exchanges utilizing differential multiple-quantum relaxation in high molecular weight proteins. J Am Chem Soc, 138, 2302–2311. doi: 10.1021/jacs.5b12954 [DOI] [PubMed] [Google Scholar]
- Traaseth NJ, Chao FA, Masterson LR, Mangia S, Garwood M, Michaeli S, Seelig B, & Veglia G (2012). Heteronuclear Adiabatic Relaxation Dispersion (HARD) for quantitative analysis of conformational dynamics in proteins. J Magn Reson, 219, 75–82. doi: 10.1016/j.jmr.2012.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, & Palmer AG (2002). R1ρ relaxation outside of the fast-exchange limit. J Magn Reson, 154, 157–160. doi: 10.1006/jmre.2001.2466 [DOI] [PubMed] [Google Scholar]
- Trott O, & Palmer AG (2004). Theoretical study of R1ρ rotating-frame and R2 free-precession relaxation in the presence of n-site chemical exchange. J Magn Reson, 170, 104–112. doi: 10.1016/j.jmr.2004.06.005 [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Libich DS, Meyer V, Roche J, & Clore GM (2015). The energetics of a three-state protein folding system probed by high-pressure relaxation dispersion NMR spectroscopy. Angew Chem Int Ed Engl, 54, 11157–11161. doi: 10.1002/anie.201505416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Sprangers R, & Kay LE (2004). Line narrowing in methyl-TROSY using zero-quantum 1H-13C NMR spectroscopy. J Am Chem Soc, 126, 4921–4925. doi: 10.1021/ja039732s [DOI] [PubMed] [Google Scholar]
- Udgaonkar JB, & Baldwin RL (1988). NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature, 335, 694–699. doi: 10.1038/335694a0 [DOI] [PubMed] [Google Scholar]
- Uversky VN, & Ptitsyn OB (1996). Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J Mol Biol, 255, 215–228. doi: 10.1006/jmbi.1996.0018 [DOI] [PubMed] [Google Scholar]
- Vallurupalli P, Bouvignies G, & Kay LE (2012). Studying “invisible” excited protein states in slow exchange with a major state conformation. J Am Chem Soc, 134, 8148–8161. doi: 10.1021/ja3001419 [DOI] [PubMed] [Google Scholar]
- Vallurupalli P, Hansen DF, & Kay LE (2008a). Probing structure in invisible protein states with anisotropic NMR chemical shifts. J Am Chem Soc, 130, 2734–2735. doi: 10.1021/ja710817g [DOI] [PubMed] [Google Scholar]
- Vallurupalli P, Hansen DF, & Kay LE (2008b). Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci U S A, 105, 11766–11771. doi: 10.1073/pnas.0804221105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallurupalli P, Sekhar A, Yuwen T, & Kay LE (2017). Probing conformational dynamics in biomolecules via chemical exchange saturation transfer: a primer. J Biomol NMR, 67, 243–271. doi: 10.1007/s10858-017-0099-4 [DOI] [PubMed] [Google Scholar]
- Wagner G, Bodenhausen G, Mueller N, Rance M, Sørensen OW, Ernst RR, & Wuethrich K (1985). Exchange of two-spin order in nuclear magnetic resonance: separation of exchange and cross-relaxation processes. J Am Chem Soc, 107, 6440–6446. doi: 10.1021/ja00309a002 [DOI] [Google Scholar]
- Walinda E, Morimoto D, & Sugase K (2018a). Overview of relaxation dispersion NMR spectroscopy to study protein dynamics and protein-ligand interactions. Curr Protoc Protein Sci, 92, e57. doi: 10.1002/cpps.57 [DOI] [PubMed] [Google Scholar]
- Walinda E, Morimoto D, & Sugase K (2018b). Resolving biomolecular motion and interactions by R2 and R1ρ relaxation dispersion NMR. Methods, 148, 28–38. doi: 10.1016/j.ymeth.2018.04.026 [DOI] [PubMed] [Google Scholar]
- Wang C, Rance M, & Palmer AG (2003). Mapping chemical exchange in proteins with MW > 50 kD. J Am Chem Soc, 125, 8968–8969. doi: 10.1021/ja035139z [DOI] [PubMed] [Google Scholar]
- Wang H, He Y, Kroenke CD, Kodukula S, Storch J, Palmer AG, & Stark RE (2002). Titration and exchange studies of liver fatty acid-binding protein with 13C-labeled long-chain fatty acids. Biochemistry, 41, 5453–5461. doi: 10.1021/bi011914g [DOI] [PubMed] [Google Scholar]
- Weininger U, Blissing AT, Hennig J, Ahlner A, Liu Z, Vogel HJ, Akke M, & Lundstrom P (2013). Protein conformational exchange measured by 1H R1ρ relaxation dispersion of methyl groups. J Biomol NMR, 57, 47–55. doi: 10.1007/s10858-013-9764-4 [DOI] [PubMed] [Google Scholar]
- Weininger U, Brath U, Modig K, Teilum K, & Akke M (2014). Off-resonance rotating-frame relaxation dispersion experiment for 13C in aromatic side chains using L-optimized TROSY-selection. J Biomol NMR, 59, 23–29. doi: 10.1007/s10858-014-9826-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Kellogg D, Kimsey IJ, Sathyamoorthy B, Stein ZW, McBrairty M, & Al-Hashimi HM (2015). Characterizing RNA excited states using NMR relaxation dispersion. Methods Enzymol, 558, 39–73. doi: 10.1016/bs.mie.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuwen T, Brady JP, & Kay LE (2018). Probing conformational exchange in weakly interacting, slowly exchanging protein systems via off-resonance R1ρ experiments: Application to studies of protein phase separation. J Am Chem Soc, 140, 2115–2126. doi: 10.1021/jacs.7b09576 [DOI] [PubMed] [Google Scholar]
- Yuwen T, & Kay LE (2017). Longitudinal relaxation optimized amide 1H-CEST experiments for studying slow chemical exchange processes in fully protonated proteins. J Biomol NMR, 67, 295–307. doi: 10.1007/s10858-017-0104-y [DOI] [PubMed] [Google Scholar]
- Yuwen T, Kay LE, & Bouvignies G (2018). Dramatic decrease in CEST measurement times using multi-site excitation. Chemphyschem, 19, 1707–1710. doi: 10.1002/cphc.201800249 [DOI] [PubMed] [Google Scholar]
- Yuwen T, Sekhar A, & Kay LE (2017). Separating dipolar and chemical exchange magnetization transfer processes in 1H-CEST. Angew Chem Int Ed Engl, 56, 6122–6125. doi: 10.1002/anie.201610759 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kitazawa S, Peran I, Stenzoski N, McCallum SA, Raleigh DP, & Royer CA (2016). High pressure ZZ-exchange NMR reveals key features of protein folding transition states. J Am Chem Soc, 138, 15260–15266. doi: 10.1021/jacs.6b09887 [DOI] [PubMed] [Google Scholar]
- Zhao B, Guffy SL, Williams B, & Zhang Q (2017). An excited state underlies gene regulation of a transcriptional riboswitch. Nat Chem Biol, 13, 968–974. doi: 10.1038/nchembio.2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Hansen AL, & Zhang Q (2014). Characterizing slow chemical exchange in nucleic acids by carbon CEST and low spin-lock field R1ρ NMR spectroscopy. J Am Chem Soc, 136, 20–23. doi: 10.1021/ja409835y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, & Yang D (2014). Effects of J couplings and unobservable minor states on kinetics parameters extracted from CEST data. J Magn Reson, 249, 118–125. doi: 10.1016/j.jmr.2014.10.010 [DOI] [PubMed] [Google Scholar]
- Zhuravleva A, & Korzhnev DM (2017). Protein folding by NMR. Prog Nucl Magn Reson Spectrosc, 100, 52–77. doi: 10.1016/j.pnmrs.2016.10.002 [DOI] [PubMed] [Google Scholar]