

Abstract
Restoration of kidney tubular epithelium following sublethal injury sequentially involves partial epithelial–mesenchymal transition (pEMT), proliferation, and further redifferentiation into specialized tubule epithelial cells (TECs). Because the immunosuppressant cyclosporine-A produces pEMT in TECs and inhibits the peptidyl-prolyl isomerase (PPIase) activity of cyclophilin (Cyp) proteins, we hypothesized that cyclophilins could regulate TEC phenotype. Here we demonstrate that in cultured TECs, CypA silencing triggers loss of epithelial features and enhances transforming growth factor β (TGFβ)-induced EMT in association with upregulation of epithelial repressors Slug and Snail. This pro-epithelial action of CypA relies on its PPIase activity. By contrast, CypB emerges as an epithelial repressor, because CypB silencing promotes epithelial differentiation, prevents TGFβ-induced EMT, and induces tubular structures in 3D cultures. In addition, in the kidneys of CypB knockout mice subjected to unilateral ureteral obstruction, inflammatory and pro-fibrotic events were attenuated. CypB silencing/knockout leads to Slug, but not Snail, downregulation. CypB support of Slug expression depends on its endoplasmic reticulum location, where it interacts with calreticulin, a calcium-buffering chaperone related to Slug expression. As CypB silencing reduces ionomycin-induced calcium release and Slug upregulation, we suggest that Slug expression may rely on CypB modulation of calreticulin-dependent calcium signaling. In conclusion, this work uncovers new roles for CypA and CypB in modulating TEC plasticity and identifies CypB as a druggable target potentially relevant in promoting kidney repair.
Keywords: cyclophilins, epithelial phenotype, Slug, TGFβ, UUO, fibrosis
Graphical abstract

Introduction
Restoration of the renal tubular epithelium following sublethal kidney injury relies on the complex interplay of dedifferentiation of tubule epithelial cells (TECs), proliferation, and further redifferentiation into functionally specialized TECs (Chang-Panesso and Humphreys, 2017). Failure of these processes can lead to pathological conditions such as fibrosis or carcinogenesis. Within the kidney, injured TECs play a major role in fibrosis by releasing inflammatory cytokines and pro-fibrotic factors, in particular transforming growth factor β (TGFβ) (Liu et al., 2018). TGFβ drives transient loss of epithelial features and acquisition of mesenchymal features in TECs, the so-called partial epithelial–mesenchymal transition (pEMT). EMT-activating transcription factors Snail, Slug, zinc finger E-box-binding homeobox 1 and 2 (ZEB1/2), and Twist1/2 negatively regulate epithelial markers, including E-cadherin expression (Moreno-Bueno et al., 2006), which is a major hallmark of EMT. Unraveling the mechanisms modulating these epithelial repressors could provide therapeutic tools to mitigate kidney fibrosis after renal damage.
Cyclosporine-A (CsA) is a potent immunosuppressant whose clinical use is limited by its nephrotoxicity, including tubular atrophy and tubule–interstitial fibrosis (Bennett et al., 1996). In vitro, CsA induces loss of epithelial phenotype of TECs by inducing Snail expression, TGFβ release, and pro-inflammatory cytokine production (Slattery et al., 2005; González-Guerrero et al., 2017). CsA binds to cyclophilins, a family of ubiquitous highly conserved peptidyl-prolyl cis-trans isomerases (PPIase), inhibiting their enzymatic activity. cyclophilins play prominent roles as chaperones by mediating protein trafficking, protein–protein interactions, and assembly of macromolecular complexes (Wang and Heitman, 2005). Cyclophilin A (CypA) and cyclophilin B (CypB) are the best studied members of the family. CypA is cytosolic, whereas CypB has an N-terminal signal peptide targeting it to the endoplasmic reticulum (ER) (Wang and Heitman, 2005). CypB also contains a C-terminal ER-retention sequence (VEKPFAIAKE) directing CypB to ER subcompartments related to calcium signaling, where it colocalizes with the calcium-buffering chaperone calreticulin (CRT) (Arber et al., 1992). In addition, CypB interacts with the calcium signal-modulating cyclophilin ligand (CAML), potentially regulating intracellular calcium release (Bram and Crabtree, 1994). CypB is secreted in the presence of CsA (Price et al., 1994), and both CypA and CypB are released in response to inflammatory stimuli acting as pro-inflammatory mediators via their potent chemotactic effects on various immune cells (Bukrinsky, 2015). In the kidney, CypB interacts with kidney androgen-regulated protein (KAP), a protein exclusively expressed in proximal TECs that protects against CsA toxicity (Tornavaca et al., 2011).
Considering the loss of epithelial phenotype triggered by CsA and its inhibitory action on cyclophilins, we hypothesized that these proteins could play a key role in the maintenance of the epithelial phenotype and that, consequently, their inhibition would trigger the development of a pEMT and, eventually, kidney fibrosis.
Results
CypA and CypB differentially affect epithelial phenotype of TECs
To investigate the involvement of cyclophilins in the regulation of the epithelial phenotype, we silenced CypA and CypB in human kidney-2 (HK-2) cells (Figure 1A) by short hairpin RNA (shRNA) and analyzed epithelial markers. Control cells reached confluency at Day 2 postseeding, progressively increasing the levels of E-cadherin, zonula occludens-1 (ZO-1), occludin, and keratin (Figure 1B), and the activity of proximal tubule brush border alkaline phosphatase (AP) (Figure 1C) within days in culture. AP activity was markedly enhanced in CypB-silenced cells and reduced in cyclophilin-silenced cells, with the greatest differences between control and cyclophilin-silenced cells observed at Day 5 (Figure 1C). At this time point, CypB silencing upregulated E-cadherin and occludin greatly and ZO-1 and keratin to a less extent at both protein (Figure 1D) and mRNA levels (Figure 1E). CypB silencing also intensified plasma membrane (PM) E-cadherin staining (Figure 1F), suggesting a gain in functionality, and increased proliferation indices (Figure 1G). On the other hand, CypA silencing reduced occludin, ZO-1, and keratin levels (Figure 1C). This downregulation correlated with reduced transepithelial electric resistance (TEER) (Figure 1H) and increased fluorescein isothiocyanate (FITC)-dextran permeability (Figure 1I).
Figure 1.

CypA and CypB differentially affect epithelial phenotype of TECs (HK-2 cells). (A) Western blot analysis (WB) showing CypA and CypB expression after cyclophilin silencing. (B) Protein levels of E-cadherin, ZO-1, occludin, and keratin in control cells cultured for different days. (C) AP activity after 2, 5, and 10 days of culture. (D and E) WB (D) and quantitative polymerase chain reaction (qPCR; E) showing expression levels of the indicated epithelial markers in CypA- and CypB-silenced cells after Day 5 of culture. (F) Immunofluorescence staining of E-cadherin (green) in cells cultured for 5 days. Calnexin (red) is used to stain ER. Scale bar, 25 μm. (G) Cell proliferation as measured by CFSE labeling followed by flow cytometry. Values indicate the ratio FInd/MFI, where MFI is the median fluorescence intensity of all viable cells at collection and FInd is the peak fluorescence intensity of the viable non-divided cells. (H and I) To assess monolayer integrity, TEER (H) and FITC-labeled dextran permeability (I) were measured in cells cultured for 5 days. (J) The mRNA levels of the indicated transcriptional repressors were analyzed by qPCR. (K and L) Protein levels of Slug and Snail in control cells after 2, 5, and 10 days of culture (K) or in cyclophilin-silenced cells at Day 5 of culture (L). To simplify the graphs in C, only the statistically significant differences within each group are shown. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
We next explored whether these phenotypic changes correlated with altered levels of epithelial repressors. Snail mRNA levels were slightly upregulated in both CypA and CypB-silenced cells, whereas the expression of Slug was strongly increased in CypA-silenced cells but almost undetectable after CypB silencing (Figure 1J). By contrast, Twist1 and Zeb1 levels were unaffected in either case (Figure 1J). At the protein level, Slug and Snail levels progressively decreased from Day 2 to Day 10 (Figure 1K), correlating with the increase in epithelial markers. At Day 5 postseeding, Slug protein levels were increased in CypA-silenced cells compared to control and were almost undetectable after CypB silencing (Figure 1L). Snail protein levels were increased further than their mRNAs in both CypA and CypB knockdown conditions, suggesting additional post-transcriptional regulation (Figure 1L).
The effects of cyclophilin silencing on E-cadherin, occludin, Slug, and Snail were also observed in the human telomerase reverse transcriptase (hTERT) immortalized human renal proximal tubular epithelial cell line (RPTEC/TERT1) (Supplementary Figure S1A and B). In RPTEC/TERT1, E-cadherin upregulation was less dramatic at the protein level, likely due to its higher basal expression compared to HK-2 cells (Supplementary Figure S1C).
CypA and CypB oppositely modulate TGFβ-induced phenotypic changes
Considering cyclophilin modulation of TEC phenotype, we next aimed to investigate the effects of cyclophilin silencing on TGFβ-induced EMT. TGFβ treatment induced a time-dependent decrease in E-cadherin and occludin as well as an increase in fibronectin, without affecting CypA or CypB levels (Figure 2A). These molecular changes were consistent with a morphological switch from a cobblestone-like monolayer to formation of cell aggregates containing a combination of poorly interconnected rounded cells and spindle-shaped cells with filopodia (Figure 2C, shCon), and to an increase in cell-to-substrate adhesiveness (shCon data of Figure 2D). CypB silencing partially prevented TGFβ-induced EMT by maintaining E-cadherin and occludin levels closer to control and by preventing fibronectin expression (Figure 2B), TGFβ-induced morphological changes (Figure 2C), and the increase in cell-to-substrate adhesiveness (Figure 2D). By contrast, CypA silencing enhanced TGFβ-induced EMT by further decreasing E-cadherin and occludin and increasing fibronectin levels. Similar results were observed in RPTEC cells (Supplementary Figure S2).
Figure 2.
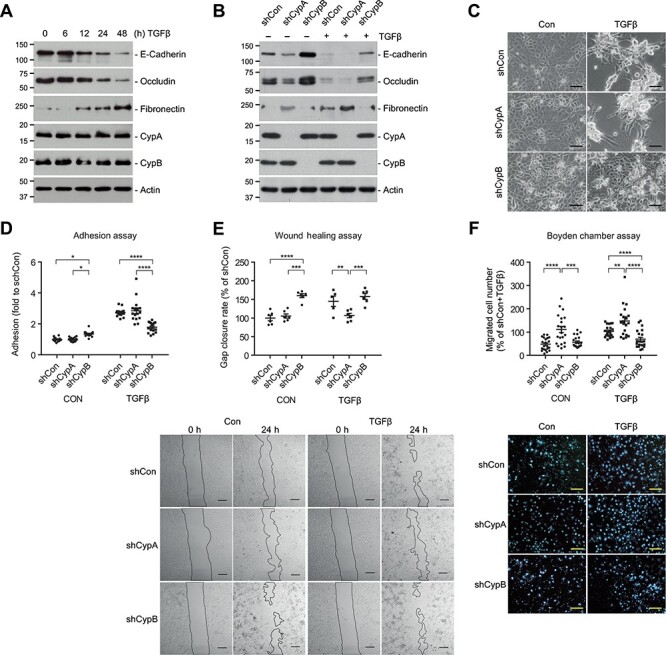
CypA and CypB oppositely modulate TGFβ-induced phenotypic changes. (A) HK-2 cells were treated with 1.5 ng/ml TGFβ for different times and the expression levels of the indicated proteins were analyzed by WB. (B–D) CypA- and CypB-silenced HK-2 cells were treated with TGFβ for 24 h and analyzed as above (B), or treated for 48 h and visualized under a light microscope (C; scale bar, 100 μm) or assayed for cell adhesion (D). (E) Wound healing assay of CypA- and CypB-silenced HK-2 cells treated or not with TGFβ. The rate of gap closure was measured. Scale bar, 250 μm. (F) HK-2 cells were seeded on Matrigel™-coated transwells and treated with TGFβ for 24 h. The number of cells that migrated through the transwell was monitored. Scale bar, 100 μm. Values are expressed as fold or percentage of controls. Only statistically significant differences within control or TGFβ-treated groups are shown. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Epithelial cells either migrate collectively as multicellular sheets maintaining their intercellular junctions, or undergo a pEMT and migrate as individual mesenchymal-like cells, although these features combine in variable degrees (Campbell and Casanova, 2016). We analyzed collective cell migration by means of a wound healing assay and individual migration by a Boyden chamber assay. Our results show that in scratch assays and in basal conditions, CypB-silenced cells exhibited faster gap closure than control or CypA-silenced cells (Figure 2E). TGFβ-stimulated control cells migrated at rates similar to untreated CypB-silenced cells, whereas CypA silencing left cells unresponsive to TGFβ’s pro-migratory effects. In contrast, in Boyden chamber assays, CypA-silenced cells showed higher rates of cell migration in both basal and TGFβ conditions, whereas CypB silencing partially prevented TGFβ-induced individual cell migration (Figure 2F).
Cyclophilin modulation of TGFβ-induced phenotypic effects coincides with Slug and SMAD6/7 expression patterns but not SMAD2/3 activation
We next explored whether cyclophilin modulation of TGFβ-induced EMT correlated with Slug and Snail levels and the activation of SMAD2/3, which canonically transduce TGFβ signals. TGFβ treatment of HK-2 cells induced a time-dependent expression of Slug and Snail that was preceded by SMAD2/3 activation (Figure 3A). Both CypA and CypB silencing enhanced TGFβ-induced Snail expression. CypA silencing also potentiated TGFβ-induced Slug expression, whereas CypB silencing prevented Slug induction by TGFβ (Figure 3B). These effects were also observed at the mRNA level (Figure 3B right, bottom). Cyclophilin modulation of TGFβ-induced Slug and Snail levels occurred without changes in SMAD4 levels or TGFβ-induced SMAD2/3 phosphorylation (Figure 3B) and nuclear translocation (Figure 3C). Treatment with the proteasome inhibitor MG132 inhibited Slug degradation, thereby increasing Slug protein levels (Figure 3D). This increase was diminished in CypB-silenced cells, confirming that CypB silencing reduced de novo Slug expression. Simultaneous treatment with TGFβ and MG132 did not increase Slug levels above those observed with MG132 alone. By contrast, CypB silencing downregulated Slug levels more effectively when TGFβ and MG132 were added together compared to MG132 alone. Because TGFβ limits its own signaling by initiating an autoinhibitory feedback loop (Yan and Chen, 2011), these results suggest that CypB silencing might overactivate TGFβ-induced termination signaling.
Figure 3.

Cyclophilin modulation of TGFβ-induced phenotypic effects coincides with Slug and I-SMADs expression patterns but not SMAD2/3 activation. (A) HK-2 cells were treated with 1.5 ng/ml TGFβ for the indicated times and the expression levels of Slug, Snail, and total and phosphorylated SMAD3 and SMAD2 were analyzed by WB. (B) CypA- and CypB-silenced HK-2 cells were treated with TGFβ for 4 or 24 h. Protein levels were analyzed by WB (left) and quantified for Slug/actin and Snail/actin ratios, expressed as percentage of that of control shRNA cells exposed to TGFβ for 4 h (right, upper), and the mRNA levels of Slug and Snail were analyzed by qPCR after 24 h of TGFβ treatment (right, bottom). (C) Control and CypB-silenced cells were treated with TGFβ for 24 h and Slug levels were analyzed by WB. When indicated, 5 μM of the proteasome inhibitor MG132 was added to cells for the last 16 h of TGFβ treatment. (D) Nuclear translocation of SMAD3 and SMAD2 (green) after treatment with TGFβ or vehicle for 4 h. Nuclei were stained with Hoechst (blue). Scale bar, 25 μm. (E) The mRNA levels of of the indicated genes were analyzed by qPCR after treatment with TGFβ for 24 h. Unless otherwise indicated values are expressed as fold to controls. Only statistically significant differences within control or TGFβ-treated groups are shown. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
We analyzed inhibitory SMADs (I-SMADs) SMAD6/7 and Ski-like (SKIL) which mediate a TGFβ autoregulatory feedback loop (Yan and Chen, 2011). In addition to upregulation of these genes by TGFβ, we observed that CypB silencing further increased the levels of SMAD6/7 and SKIL in basal and TGFβ-treated cells, whereas CypA silencing slightly reduced them (Figure 3E). We also analyzed the levels of bone morphogenic protein (BMPs) 2 and 7, which counteract TGFβ-induced EMT in a SMAD7-dependent way (Yang et al., 2009). BMP2 mRNA was found to be upregulated in CypB-silenced cells (Figure 3E), whereas we were unable to detect BMP7 in HK-2 cells (data not shown).
Disruption of the PPIase activity of CypA and CypB induces Slug expression, whereas mislocalization of CypB out of the ER abrogates Slug expression
To further investigate the mechanisms underlying Slug and Snail modulation by cyclophilins, we reintroduced either wild-type (wt) or mutant CypA and CypB lacking PPIase activity (ΔPPI) or CypB’s ER-directing signal peptide (ΔER), in the corresponding silenced cells. Our results show that all constructs were correctly expressed (Figure 4A, B, and E) and that wt-CypB and ΔPPI-CypB were properly located in the ER (Figure 4F) and secreted to the extracellular medium (Figure 4E, CM blot), whereas ΔER-CypB was not.
Figure 4.

CypA PPIase activity is required to downregulate Snail and Slug, whereas ER location of CypB is mandatory to sustain Slug expression. (A) Scheme summarizing the mutations introduced in CypA and CypB expression vectors to generate HA-tagged shRNA-resistant wt, PPIase-defective (ΔPPI), and signal peptide-defective (ΔER) cyclophilins. (B) The expression levels of wt-CypA and ΔPPI-CypA forms reintroduced into CypA-silenced cells were determined by WB. (C and D) Slug and Snail levels in CypA-silenced cells carrying wt-CypA or ΔPPI-CypA, either untreated (C) or treated with TGFβ for 4 h (D). (E) The expression levels of wt-CypB and mutant forms reintroduced into CypB-silenced cells were analyzed by WB in cell extracts and CM. (F) Immunofluorescence staining showing that wt-CypB and ΔPPI-CypB colocalized with the ER marker calnexin (CNX), whereas ΔER-CypB did not. Scale bar, 25 μm (left) and 90 μm (right, magnifications). (G and H) WB of Slug and Snail levels after reintroduction of wt-CypB, ΔPPI-CypB, or ΔER-CypB, either in untreated cells (G) or in cells treated with TGFβ for 4 h (H). In all cases, Ø corresponds to the empty expression vector. In D and H, the statistically significant differences with shCon+ Ø are not shown. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Reintroduction of wt-CypA in CypA-silenced cells downregulated Slug and Snail to the levels found in control cells, whereas reintroduction of ΔPPI-CypA failed to do so (Figure 4C). These results were also observed in the presence of TGFβ (Figure 4D). On the other hand, reintroduction of wt-CypB, but not of ΔER-CypB, rescued Slug expression in CypB-silenced cells (Figure 4G). Remarkably, ΔPPI-CypB increased Slug levels even further than those achieved by wt-CypB (Figure 4G). As before, these effects were reproduced in the presence of TGFβ (Figure 4H).
Slug modulation by CypB is independent of the CD147 receptor and extracellular CypB
Extracellular CypA and CypB are important mediators of intercellular communications, both in physiological and pathological processes (Yurchenko et al., 2010; Bukrinsky, 2015). Accordingly, we explored whether cyclophilins could regulate Slug and Snail in an autocrine/paracrine manner. Analysis of conditioned media (CM) showed that CypB and CypA were progressively secreted into the extracellular milieu and that their secretion was unaffected by TGFβ treatment (Figure 5A). Pretreatment with brefeldin-A (BFA), a secretion inhibitor that impedes ER–Golgi protein transport, blocked both basal and CsA-induced CypB secretion but had no effect on that of CypA (Figure 5B). This, together with the lack of CsA effect on CypA release, confirms that CypA and CypB secretion involves different mechanisms. BFA prevented basal and TGFβ-induced Slug expression (Figure 5C) but, unlike CypB silencing, also reduced Snail expression and SMAD2/3 activation. We next silenced CD147, the only known receptor for extracellular CypB and CypA (Yurchenko et al., 2010). CD147 silencing increased rather than decreased Slug and Snail levels, which was reminiscent of CypA silencing (Figure 5D). Finally, exogenously added recombinant human CypB (rCypB) was unable to restore Slug or downregulate Snail to levels found in non-silenced cells (Figure 5E). ERK1/2 phosphorylation was used as a control for CypB activity (Yurchenko et al., 2001). Taken together these results argue against an autocrine loop in Slug modulation by CypB but hint that CypA–CD147 interaction could be key in Slug and Snail regulation.
Figure 5.

Slug modulation by CypB is independent of the CD147 receptor and extracellular CypB. (A) HK-2 cells were treated with 1.5 ng/ml TGFβ for the indicated times, and the presence of CypA and CypB in the CM was analyzed by WB. Culture medium was used as a negative control. (B) HK-2 cells were treated with 1 μM BFA for 30 min before addition of either 1.5 ng/ml TGFβ or 0.5 μM CsA for 4 h and CM were analyzed as above. (C) HK-2 cells were treated with 1 μM BFA for 30 min before addition of either 1.5 ng/ml TGFβ or not for 4 h. The levels of Slug, Snail, and total and phosphorylated SMAD3 and SMAD2 were analyzed by WB. (D) HK-2 cells were stably silenced for CD147 and treated or not with TGFβ for 4 h before WB for the indicated protein levels. (E) Control and CypB-silenced cells were treated with increasing doses of rCypB for 4 h and the levels of Slug, Snail, and total and phosphorylated ERK1/2 were analyzed by WB.
CypB reduces ionomycin- and thapsigargin-induced calcium release and prevents ionomycin-induced Slug expression
CypB and CypA participate in ER calcium regulation in lymphocytes (Bram and Crabtree, 1994) and platelets (Rosado et al., 2010), respectively. Since Slug expression is modulated by calcium (Hayashida et al., 2006), we investigated whether Slug regulation by cyclophilins involved calcium signaling. Control and cyclophilin-silenced cells were loaded with a fluorescent Ca2+ indicator and subsequently treated with the calcium ionophore ionomycin or the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) inhibitor thapsigargin. Ionomycin induced a stronger Ca2+ increase that rapidly peaked and gradually declined to a steady-state above baseline (Figure 6A), whereas thapsigargin triggered a weaker and more flattened response (Figure 6B). CypB silencing reduced the calcium rise elicited by both compounds. Interestingly, Slug expression was either transiently increased or reduced after ionomycin (Figure 6C) or thapsigargin (Figure 6D) treatment, respectively. These striking differences may be attributable to the distinct intensities and kinetics in calcium release of each compound. Ionomycin was unable to induce Slug expression in the absence of CypB (Figure 6E) or when CypB was not localized within the ER (Figure 6F). By contrast, CypB PPIase activity was unnecessary for ionomycin-induced Slug expression. Conversely, the reduction in Slug levels after thapsigargin treatment was not inhibited but instead increased in CypB-silenced cells (Supplementary Figure S3), indicating that both CypB silencing and thapsigargin effects are not mutually exclusive.
Figure 6.

Effect of CypB silencing on ionomycin and thapsigargin-induced cytosolic calcium increase and Slug expression in HK-2 cells. (A) Left: cytosolic calcium increase in ionomycin-treated cyclophilin-silenced cells, expressed as the ratio between maximal fluo-4 fluorescence achieved after the addition of 1 μM ionomycin (Fmax) and the fluorescence before treatment (F0). Dotted line indicates fluorescent value before treatment. Right: cytosolic calcium kinetics in two representative cells. The arrow points the time when ionomycin was added. (B) Same as A after addition of 5 μM thapsigargin. Data represent mean ± SEM of 5–10 replicates per group. *corresponds to significant statistical difference (P < 0.05) respect to shCon group. (C and D) HK-2 cells were treated with 1 μM ionomycin (C) or 5 μM thapsigargin (D) for the indicated times and Slug expression was analyzed by WB. (E and F) Slug levels were analyzed after 4-h treatment with 1 μM ionomycin in CypA- and CypB-silenced cells (E) or in CypB-silenced cells carrying wt or mutated CypB forms (F). (G) HA-tagged wt-CypB, ΔPPI-CypB, and ΔER-CypB were immunoprecipitated with an anti-HA affinity matrix, followed by immunoblotting with CRT and HA. (H) Immunofluorescence staining of CRT (ER luminal and cytosolic; in red) and CNX (ER transmembrane; in green). Scale bar, 25 μm (left) and 90 μm (right, magnifications).
The ER calcium-buffering chaperone calreticulin (CRT) has been shown to be involved in Slug upregulation and EMT promotion (Hayashida et al., 2006). Coimmunoprecipitation (coIP) assays with reintroduced wt or mutant CypB showed that CRT interacted with wt-CypB and to a higher extent with ΔPPI-CypB, but not with ΔER-CypB (Figure 6G). We next explored whether CypB could be modulating CRT by altering its subcellular localization. Figure 6H shows that CRT localization was not affected by either CypA or CypB silencing.
CypB and CypA differentially affect epithelial phenotype of TECs in 3D cultures
Three-dimensional (3D) cultures increase cell differentiation and partially recapitulate tissue-specific morphology (Secker et al., 2018). To validate two-dimensional (2D) results, we performed 3D ‘sandwich’ experiments where control and cyclophilin-silenced cells were embedded in between two layers of Matrigel™ and allowed to differentiate for 14 days (Figure 7A). Our results show that HK-2 cells formed a multilayered cell sheet-like structure with higher cell density and multicellular protrusions in the edges of the cell sheet. Interestingly, the sheets of CypB-silenced cells formed longer and more branched multicellular protrusions than sheets of control or CypA-silenced cells, resembling tubular-like structures (Figure 7B). In addition, and as in 2D, 3D-cultured CypB-silenced cells exhibited higher levels of epithelial markers and lower levels of Slug than control or CypA-silenced cells (Figure 7C and D).
Figure 7.

CypA and CypB differentially affect epithelial phenotype of TECs in 3D cultures. (A) Control or CypA- or CypB-silenced HK-2 cells were embedded between two layers of Matrigel™ and allowed to differentiate for 14 days. (B) Bright-field images showing HK-2 cells after 14 days. Scale bar, 500 μm (for 2.5× magnification) and 125 μm (for 10× magnification). (C and D) WB (C) and qPCR (D) of cell extracts obtained from HK-2 cells embedded in Matrigel™. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
CypB deletion ameliorates inflammation and early events of renal fibrosis after unilateral ureteral obstruction (UUO)
To expand on our in vitro results, we investigated the contribution of CypB in the development of fibrosis using global CypB knockout (KO) mice and wt littermates subjected to UUO (Figure 8A). To study early events in renal fibrosis, mice underwent ureteral ligation of the left kidney for 1 week and were compared with contralateral (CL) non-ligated right kidneys (Figure 8B). In non-obstructed kidneys, there were no apparent histological differences between wt and CypB KO mice. By contrast, kidneys from CypB KO mice were partially protected from the UUO-induced tubular distension and inflammation (Figure 8C). In these conditions, no overt fibrosis was observed using Masson’s trichrome staining (data not shown), likely due to the short-term obstruction. We next analyzed the expression of relevant pro-fibrotic and pEMT-related markers at the mRNA level. In wt kidneys, UUO did not have significant effects on CypB, BMP2, SMAD7, and SMAD6 mRNA levels, but significantly reduced CypA, E-cadherin, CD147, and BMP7 whereas increased fibronectin, collagen-Ia, matrix metalloproteinase 9 (MMP-9), TGFβ, Snail, Slug, and BMP6 levels. Genetic deletion of CypB significantly prevented the decrease of CypA, E-cadherin, CD147, and BMP7 and the increase of fibronectin, MMP9, and Slug produced by UUO. CypB knockdown further increased the UUO-induced expression of Snail and BMP-6 levels. Finally, there were no significant differences in collagen-Ia, TGFβ, or BMP2 levels between obstructed kidneys of wt and CypB KO mice. SMAD6/7 and Slug were the only genes that were upregulated or downregulated, respectively, in non-ligated kidneys of CypB KO mice. Regarding pro-inflammatory cytokines, UUO-induced tumor necrosis factor α (TNFα), macrophage chemoattracting protein 1 (MCP1), and the pan-macrophage marker CD68 were all significantly reduced in CypB KO mice (Figure 8E). To unravel whether cytokine reduction underlies a downregulation of pro-inflammatory signaling in TECs, we analyzed the activity of nuclear factor kappa B (NFκB) in HK-2 cells. Results in Figure 8F show that NFκB promoter activity was strongly increased after CypA silencing and considerably decreased in CypB-silenced cells.
Figure 8.

CypB depletion ameliorates inflammation and early events of renal fibrosis after UUO. (A) WB showing CypB levels in kidneys from CypB KO (CypB−/−) mice and control wt littermates. (B) Experimental scheme: CypB KO mice and control littermates were subjected to UUO as a model of renal fibrosis. Mice were sacrificed 7 days after obstruction and total RNA was extracted from the right CL kidneys and left obstructed kidneys (UUO). (C) Left: hematoxylin and eosin (H&E) and mouse macrophage marker F4/80 staining of kidney sections. Scale bar, 100 μm. Right: semi-quantitative immunohistochemistry analysis for tubular dilatation and inflammation; each parameter was assessed independently by two pathologists and was scored on a scale of 0–4, ranging from none (0), minimum (1), mild (2), moderate (3), to severe (4). Mann–Whitney U test was used to detect differences. (D and E) The mRNA levels of the indicated genes were detected by qPCR. Dots represent individual values for either CL or UUO kidneys, with eight mice in each group. (F) The NFκB promoter activity in HK-2 cells transfected with a control empty plasmid pCMV-HA or the p65 subunit of NFκB was analyzed with a luciferase reporter assay. Only statistically significant differences within empty vector or p65-transfected groups are shown. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Discussion
This paper shows that CypA and CypB are opposite regulators of TEC plasticity since silencing of CypA hampered, whereas silencing of CypB promoted the epithelial phenotype of kidney cells (HK-2 cells). Accordingly, we postulate that CypA favors kidney epithelial differentiation, whereas CypB acts as an epithelial repressor. We also propose that, based on our in vivo results in a model of fibrosis caused by UUO, CypB may participate in the development of kidney fibrosis and inflammation in the early stages.
CypA silencing results in loss of epithelial characteristics, including downregulation of tight junction proteins, decreased TEER and increased dextran permeability, reduced AP activity, and increased invasion and fibronectin expression. Although some of these effects were previously reported in CypA-silenced human primary TECs (Pallet et al., 2008), we now describe for the first time that CypB silencing promotes epithelial differentiation and prevents TGFβ-induced EMT-like phenotypic changes. Those included downregulation of epithelial markers, altered cell morphology, and increased cell-to-substrate adhesion and invasion. There are, however, some similarities between CypB silencing and TGFβ in promoting collective cell migration and cell-to-substrate adhesion that might involve different mechanisms of action. Faster collective migration of CypB-silenced cells would result from upregulation of adherens and tight junction proteins. This ought to facilitate transmission of the traction force between leader and follower cells and between follower cells, as described (Campbell and Casanova, 2016; Friedl and Mayor, 2017). Conversely, TGFβ treatment acts by enhancing the migratory phenotype of leader cells at the edge of the gap, accelerating wound closure. The small increase in cell-to-substrate adhesion in CypB-silenced cells may result from upregulation of hemi-desmosomal protein keratin-8, as previously described (Liu et al., 2008). The stronger TGFβ pro-adhesive effect may relate to acquisition of a rear–front polarity, which, in turn, is inhibited by CypB silencing.
Cells cultured in 3D more accurately reproduce in vivo behavior (Secker et al., 2018). In a 3D environment, HK-2 cells formed multicellular protrusions, which resembled the collective sprouting underlying the branching morphogenesis of the kidney (Gray et al., 2010; Friedl and Mayor, 2017). The growing sprout moves collectively with a row of front–rear polarized leader cells forming a terminal end bud, coupled to lumen-enclosing apicobasal polarized follower cells by adherens, desmosomal, and tight junctions (Shamir and Ewald, 2015; Huebner et al., 2016). In 3D-cultured HK-2, extension of multicellular protrusions was much longer and branched in CypB-silenced cells. We hypothesize that, while the EMT-like processes in the tip of sprouting strands could be occurring in a similar way in all cell lines, the higher proliferation and collective migration rates of follower cells with silenced CypB could boost sprout extension.
The strikingly opposite actions of CypB and CypA on HK-2 epithelial phenotype revealed differential regulation of the epithelial repressors Snail and Slug. In HK-2 cells, CypA acts as a repressor of Slug and Snail, whereas CypB represses Snail but is mandatory for Slug expression. This raises the question of whether Slug and Snail function in redundant, specific, or even antagonistic manners. Interestingly, there are examples of both cooperativity and antagonism depending on the context (Nieto, 2002; Moreno-Bueno et al., 2006). Snail upregulation in CypB-silenced cells could also involve a compensatory mechanism to counterbalance the decrease in Slug levels, as previously described (Ganesan et al., 2016). Regardless, our results indicate that, at least in CypB-silenced cells, Slug deficiency, rather than augmented Snail levels, determines cell phenotype. This predominant role of Slug is particularly relevant because, so far, Snail has been considered as sufficient and necessary to induce EMT and fibrosis in the mouse kidney (Grande et al., 2015).
Our results indicate that cyclophilin-mediated regulation of Slug and Snail might be exerted through I-SMADs. Because I-SMADs expression is, in turn, regulated by BMPs, upregulation of BMP2 in CypB-silenced HK-2 cells, as well as BMP6/7 in CypB KO mice further supports this concept. Curiously, BMP2 upregulation was not observed in CypB KO mice. As BMPs have both redundant and isoform-specific functions (Yuasa and Fukuda, 2008), we hypothesize that BMP2-mediated CypB-silencing effects in HK-2 cells would be carried out by BMP6 or BMP7 in kidneys of CypB KO mice.
In this work we also demonstrate that CypA PPIase activity is necessary to repress Slug and Snail expression. Interestingly, while reintroducing ΔPPI-CypA reproduced CypA-silencing effects on Slug and Snail, reintroduction of ΔPPI-CypB did not mimic CypB silencing. In contrast, ΔPPI-CypB further increased Slug levels above those observed with wt-CypB, suggesting that mutation of CypB PPIase activity may represent a gain of function over CypB silencing. Thus, while CypA and CypB exert almost opposite actions in regulating epithelial phenotype of TECs, mutation to block either of their PPIase activities resulted in Slug upregulation. As CsA inhibits the PPIase activities of both CypA and CypB, our results suggest that CsA pro-fibrotic action could be mediated, at least in part, by Slug upregulation consequent to the inhibition of CypA and CypB activity.
Accumulating evidence points to an important role for extracellular CypA and CypB in mediating intercellular communications through receptor-mediated autocrine and paracrine signaling pathways (Bukrinsky, 2015). However, no evidence was found to support an extracellular role of CypB in modulating Slug expression in HK-2 cells because: (i) no differences in CypB secretion were observed after TGFβ treatment; (ii) silencing of CD147, the only known receptor for extracellular cyclophilins, did not prevent but rather increased Slug expression; and (iii) addition of recombinant CypB in the extracellular milieu of CypB-silenced cells was unable to restore Slug levels. Because CD147 silencing upregulated Slug and Snail levels as in CypA-silenced cells, we hypothesize that interaction between extracellular CypA and CD147 could account for Slug and Snail repression in differentiated TECs.
Slug modulation by CypB might involve calcium signaling because CypB silencing prevents ionomycin-induced calcium release and Slug expression. Although the SERCA inhibitor thapsigargin also increased calcium release, it reduced Slug levels. This disparity reflects the fact that calcium signals differing in amplitude, spatial, and temporal properties trigger different transcriptional responses (Carrasco et al., 2004). Ionomycin catalyzes calcium transport not only across the PM but also out of intracellular calcium stores (Kao et al., 2010). Because both ionomycin and thapsigargin commonly target the ER calcium store, reduced calcium release in CypB-silenced cells could reflect lower ER calcium levels or buffering capacity. This was further supported by CypB interaction with the calcium-buffering chaperone CRT. CypB–CRT interaction only occurred when CypB was located in the ER, and was stronger when CypB lacked its PPIase activity. Because this interaction pattern paralleled that of Slug expression, we speculate that CRT–CypB interaction might be crucial for Slug expression. In agreement with this, CRT has been previously involved in Slug upregulation and EMT induction (Hayashida et al., 2006). Although CypB mislocalization to the cytosol prevented ionomycin-induced Slug expression, CypB modulation of calcium signaling could involve other cellular compartments apart from the ER. Recent publications highlight the importance of ER–PM contacts in calcium signaling (Wang et al., 2018). Interestingly, CypB associates with the calcium channel, transient receptor potential cation channel subfamily V member 6 (TRPV6), regulating calcium uptake (Stumpf et al., 2008). TRPV6 is part of the store-operated calcium entry (SOCE) mechanism and translocates to the PM via Orai1 (Raphaël et al., 2014). CypB, through its chaperone activity, could be involved in the formation of multiprotein complexes containing TRPV6–Orai1 in the ER–PM junctions, thereby modulating Ca2+ influx and downstream Slug expression.
In vivo, we observed that kidneys of CypB KO mice exhibited downregulated and upregulated Slug and SMAD6/7 levels, respectively, largely recapitulating the results observed in cultured TECs. In addition, kidneys from CypB KO mice were protected from the UUO-induced tubular distension and inflammation after 1-week UUO. No overt fibrosis was observed at this time point but the upregulation of pro-fibrotic factors indicated that early events of renal fibrosis were taking place. Considering our in vitro results showing a pro-epithelial role of CypA, the reduction of CypA levels seen in the UUO model might also be considered to possibly exert a pro-fibrotic action. As CypB knockdown not only partially prevents UUO-induced fibronectin, MMP9 and Slug upregulation but also CypA and E-cadherin downregulation, we postulate that CypB may participate in kidney fibrosis. In addition, CypB also mediates UUO-induced inflammation because CypB knockdown reduced the expression of pro-inflammatory markers. Moreover, our results showing reduced activity of the pro-inflammatory transcription factor NFκB in CypB-silenced HK-2 cells support a central role of TEC’s in the development of interstitial inflammation (Liu et al., 2018).
Altogether, our results demonstrate key roles for CypA and CypB in kidney epithelial cell plasticity and have, moreover, pinpointed Slug as a key regulator of EMT. We suggest a role of CypB at the crossroads between the inflammatory and the fibrogenic processes by regulating the inflammatory response of TECs.
Materials and methods
Materials
The source of all materials used in this work is described in Supplementary Table S1.
Cell culture
Human kidney proximal tubule cells HK-2 (ATCC) and RPTEC/TERT1 (ATCC), which retain morphological and functional attributes of normal adult human proximal tubular epithelium (Ryan et al., 1994; Wieser et al., 2008), were cultured as indicated in Supplementary material. Unless otherwise indicated, cells were seeded at 2.25 × 104 cells/cm2 and experiments were performed at Day 5 postseeding. Before treatment, cells were starved in medium A supplemented with 0.1% fetal bovine serum (FBS) for 16 h.
Gene silencing and rescue experiments
CypA, CypB, and CD147 silencing and cyclophilin reintroduction were performed using lentiviral particles as described in Supplementary material. All shRNA sequences are provided in Supplementary Table S2. Briefly, for shRNA rescue experiments, we first generated a shRNA-resistant wt-CypB by introducing silent mutations into the shRNA-targeted sequence to prevent RNA-induced silencing complex (RISC)-mediated degradation. ShRNA against CypA was directed to the 3′UTR, and thus reintroduction of wt-CypA did not require further modification. Within these constructs, we mutated critical residues to inactivate PPIase activity (ΔPPI) of CypA (c.161_162delinsGC; p.Arg55Ala) and CypB (c.259_260delinsGC; p.Arg62Ala) or to eliminate CypB ER-directing signal peptide (ΔER) (c.4_75del; p.2_25del). All constructs were stably transduced into cyclophilin-silenced cells using lentiviral particles as for shRNA infection.
Immunoblotting, immunocytochemistry, and coIP
A detailed description of immunoblotting, immunocytochemistry, and coIP protocols is provided in Supplementary material. For WB of extracellular cyclophilins, CM were collected and centrifuged 5 min at 1500× g. Supernatants were mixed with 5× sample buffer, and 50 μl of the resulting mix was resolved via gel electrophoresis. A list of all antibodies used in this work is shown in Supplementary Table S3.
RNA extraction and qPCR
RNA extraction and qPCR were performed as indicated in Supplementary material. All probes used in this work are listed in Supplementary Table S4. Analysis of relative gene expression was performed using the 2–ΔΔCt method normalizing to TATA-binding protein (TBP) expression.
Cell proliferation
Cells were trypsinized, washed with phosphate buffered saline (PBS), and incubated with carboxyfluorescein diacetate succinimidyl ester (CFSE; Sigma-Aldrich) at 2.5 μM final concentration for 10 min at 37°C in a cell culture incubator. An aliquot of cells was left unlabeled to set background fluorescence. CFSE was then quenched by washing cells twice with complete medium, and a portion of cells was taken to measure fluorescence at the beginning of the experiment. The rest of labeled cells were plated and incubated at 37°C for 5 days. Fluorescence was measured with a flow cytometer (Becton Dickinson) and analyzed with Cell Quest software (Becton Dickinson).
TEER and FITC-dextran permeability
Cells were seeded on 24-well transwell plates with 0.4-μm pore polyester membrane inserts (Sigma-Aldrich) and measurements were performed 5 days later. TEER was measured using an epithelial voltmeter (Millipore) with STX100C electrode (World Precision Instruments) according to manufacturer’s instructions. For permeability assays, 40 kDa FITC-dextran (Sigma-Aldrich) in a final concentration of 100 μg/ml was added to the apical compartment of the cells. Three hours later, samples were collected from the basolateral compartment and absorbance (λex/λem = 485/528 nm) was measured using a microplate reader (Molecular Devices).
AP activity
AP activity was analyzed as described in Supplementary material.
Cell adhesion assay
Cells were trypsinized and washed twice with culture medium to completely eliminate trypsin. Cells were counted and 2 × 104 cells/well were seeded onto two duplicated 96-well plates and allowed to adhere at 37°C for 30 min. After the incubation period, unattached cells from one of the plates were removed by washing twice with PBS. The respective amounts of remaining attached cells (from the washed plate) and total of cells (from the unwashed plate) were determined using the tetrazolium salt (XTT) assay. Results were expressed as the ratio of XTT values between washed and unwashed plate.
Wound migration assay
For wound migration assay, 7.5 × 104 cells were seeded on each of the two compartments (growth area: 0.22 cm2/compartment) of silicone culture inserts (Ibidi GmbH), grown for 5 days, and starved for 16 h. At the onset of the experiment, the silicone insert was removed leaving a gap free of cells of 500 ± 100 μm width (according to manufacturer). Cells were gently washed with serum-free media to remove cell debris and treated as indicated. Digital images were obtained with an Olympus CellR microscope and area measurements were performed using ImageJ software. The rate of wound closure was measured by subtracting the area of the wound at 24 h from the area at 0 h.
Transwell migration assay
For transwell migration assays, polycarbonate cell culture inserts with 8-μm pore size and 0.47 cm2 growth area (ThermoFischer) were used. The inserts were coated with 50 μl growth factor reduced (GFR)-Matrigel™ matrix (BD Biosciences) diluted 10 times in serum-free media and incubated at 37°C for 1.5 h. A total of 200 μl serum-free media containing 5 × 104 serum-starved HK-2 cells were placed in the top well chamber, whereas the bottom chamber was filled with 500 μl complete medium enriched with up to 10% FBS. Media in the top and bottom chambers were supplemented with vehicle or TGFβ. After 24 h of treatment, cells were washed in PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Cell nuclei were then stained with Hoechst (ThermoFischer). Cells from the top surface of the membrane were gently removed with a cotton swab. Membranes were then cut out from the inserts with a scalpel blade and mounted on microscope slides using antifade mountant (ThermoFischer), with the lower surface facing up. Nuclei were visualized with an Olympus IX71 fluorescence microscope and counted using Image J.
Cytosolic calcium measurement
Attached HK-2 cells were loaded with 5 μM fluo-4 (ThermoFischer) in control buffer (containing 140 mM NaCl, 3.6 mM KCl, 1.2 mM MgSO4, 1 mM CaCl2, 20 mM HEPES, pH 7.4) for 1 h at 37°C. Time-dependent changes in cell fluorescence (Ex/Em 488/528 nm) secondary to the addition of ionomycin (1 μM) or thapsigargin (5 μM) were monitored every 5 sec using a multimode microplate fluorometer (Molecular Devices).
3D cultures
Three-dimensional cell cultures were performed in 24-well plates as indicated in (Secker et al., 2018), with the following modifications: each well was first coated with 210 μl Matrigel™ (BD Biosciences) and allowed to polymerize at 37°C for 1 h. Thereafter, 0.15 × 106 HK-2 cells/cm2 were seeded onto the Matrigel™ layer and left to attach for 16 h at 37°C. Subsequently, medium was removed and cells were overlaid with 150 μl/well Matrigel™. After polymerization of the new layer of Matrigel™, 400 μl/well medium was added and cultures were maintained for 14 days at 37°C, with medium replacement every 48 h. Images were obtained using a Leica DM IRBE inverted microscope.
For protein or RNA isolation from cells embedded in Matrigel™, cell cultures were washed with cold PBS followed by addition of 500 μl 0.5 mM EDTA/PBS per well and incubated on ice for 15 min. Cell/Matrigel™ mixture was dislodged from the dish with a p1000 pipet with the tip cutoff and transferred to a microcentrifuge tube. Cells were then pelleted by centrifugation and resuspended in either radioimmunoprecipitation assay (RIPA) buffer or Trizol for protein or RNA extraction, respectively.
UUO procedure
UUO was performed as previously described (Kie et al., 2008). Briefly, mice were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg). Through a 2-cm midline abdominal incision, the left kidney was exposed by retraction of the intestines using a self-retaining microdissection retractor and the left ureter was carefully dissected from surrounding tissue. The ureter was then doubly ligated at the midpoint between the kidney and the bladder using sterile 6-0 silk suture. The surgical incision was then closed and the mouse was allowed to recover from anesthesia; postoperative analgesia (buprenorphine, 0.1 mg/kg, SQ) was administered. Sham-operated control mice underwent a similar surgical procedure without ligation of the ureter.
NFκB activity
NFκB promoter activity was measured as indicated in Supplementary material.
Statistics
All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software). In all charts, dots represent individual values from at least three independent experiments and the horizontal and vertical bars represent the mean and SEM, respectively. Statistical differences were determined by one-way analysis of variance (ANOVA) (Figures 1D,1F–H, 1J, 4C,4D, 4G, 4H, 6A, 6B, and7D) or two-way ANOVA (Figures 1I,2D–F,3B,3E, 8C–F) followed by Tukey’s multiple comparison test.
Study approval
Animal studies were conducted as approved by the Institutional Animal Care and Use Committee of Mayo Clinic and in accordance with National Institutes of Health guidelines.
Funding
E.S. and M.D. were supported by the generous contribution of Asdent Patients Association. This work was supported in part by grants from Ministerio de Ciencia e Innovación (SAF201459945-R and SAF201789989-R to A.M.), the Fundación Senefro (SEN2019 to A.M.), Instituto de Salud CarlosIII (PIE13/00027), and Red de Investigación Renal REDinREN (12/0021/0013). K.A.N. is supported by National Institutes of Health (NIH) DK 47060. A.M. group holds the Quality Mention from the Generalitat de Catalunya (2017 SGR).
Conflict of interest: none declared.
Author contributions: E.S. and A.M. conceived and designed the research studies. E.S., M.D., A.R., A.J.C., K.A.N., M.T.S., and J.H.G. conducted the experiments. R.J.B. provided the CypB KO mice. E.S., A.M., R.J.B., and D.B. analyzed the data. E.S., A.M., D.B., K.A.N., and R.J.B. draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
References
- Arber, S., Krause, K.H., and Caroni, P. (1992). S-Cyclophilin is retained intracellularly via a unique COOH-terminal sequence and colocalizes with the calcium storage protein calreticulin. J. Cell. Biol. 116, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, W.M., DeMattos, A., Meyer, M.M., et al. (1996). Chronic cyclosporine nephropathy: the Achilles’ heel of immunosuppressive therapy. Kidney Int. 50, 1089–1100. [DOI] [PubMed] [Google Scholar]
- Bram, R.J., and Crabtree, G.R. (1994). Calcium signalling in T cells stimulated by a cyclophilin B-binding protein. Nature 371, 355–358. [DOI] [PubMed] [Google Scholar]
- Bukrinsky, M. (2015). Extracellular cyclophilins in health and disease. Biochim. Biophys. Acta 1850, 2087–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, K., and Casanova, J. (2016). A common framework for EMT and collective cell migration. Development 143, 4291–4300. [DOI] [PubMed] [Google Scholar]
- Carrasco, M.A., Jaimovich, E., Kemmerling, U., et al. (2004). Signal transduction and gene expression regulated by calcium release from internal stores in excitable cells. Biol. Res. 37, 701–712. [DOI] [PubMed] [Google Scholar]
- Chang-Panesso, M., and Humphreys, B.D. (2017). Cellular plasticity in kidney injury and repair. Nat. Rev. Nephrol. 13, 39–46. [DOI] [PubMed] [Google Scholar]
- Friedl, P., and Mayor, R. (2017). Tuning collective cell migration by cell–cell junction regulation. Cold Spring Harb. Perspect. Biol. 9, pii: a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan, R., Mallets, E., and Gomez-Cambronero, J. (2016). The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 10, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Guerrero, C., Cannata-Ortiz, P., Guerri, C., et al. (2017). TLR4-mediated inflammation is a key pathogenic event leading to kidney damage and fibrosis in cyclosporine nephrotoxicity. Arch. Toxicol. 91, 1925–1939. [DOI] [PubMed] [Google Scholar]
- Grande, M.T., Sánchez-Laorden, B., López-Blau, C., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. [DOI] [PubMed] [Google Scholar]
- Gray, R.S., Cheung, K.J., and Ewald, A.J. (2010). Cellular mechanisms regulating epithelial morphogenesis and cancer invasion. Curr. Opin. Cell Biol. 22, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, Y., Urata, Y., Muroi, E., et al. (2006). Calreticulin represses E-cadherin gene expression in Madin-Darby canine kidney cells via Slug. J. Biol. Chem. 281, 32469–32484. [DOI] [PubMed] [Google Scholar]
- Huebner, R.J., Neumann, N.M., and Ewald, A.J. (2016). Mammary epithelial tubes elongate through MAPK-dependent coordination of cell migration. J. Cell Sci. 129, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, J.P.Y., Li, G., and Auston, D.A. (2010). Practical aspects of measuring intracellular calcium signals with fluorescent indicators. Methods Cell Biol.99, 113–152. [DOI] [PubMed] [Google Scholar]
- Kie, J.-H., Kapturczak, M.H., Traylor, A., et al. (2008). Heme oxygenase-1 deficiency promotes epithelial–mesenchymal transition and renal fibrosis. J. Am. Soc. Nephrol. 19, 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B.C., Tang, T.T., Lv, L.L., et al. (2018). Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 93, 568–579. [DOI] [PubMed] [Google Scholar]
- Liu, F., Chen, Z., Wang, J., et al. (2008). Overexpression of cell surface cytokeratin 8 in multidrug-resistant MCF-7/MX cells enhances cell adhesion to the extracellular matrix. Neoplasia 10, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno, G., Cubillo, E., Sarrió, D., et al. (2006). Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial–mesenchymal transition. Cancer Res. 66, 9543–9556. [DOI] [PubMed] [Google Scholar]
- Nieto, M.A. (2002). The Snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3, 155–166. [DOI] [PubMed] [Google Scholar]
- Pallet, N., Bouvier, N., Bendjallabah, A., et al. (2008). Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am. J. Transplant. 8, 2283–2296. [DOI] [PubMed] [Google Scholar]
- Price, E.R., Jin, M., Lim, D., et al. (1994). Cyclophilin B trafficking through the secretory pathway is altered by binding of cyclosporin A. Proc. Natl Acad. Sci. USA 91, 3931–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphaël, M., Lehen'kyi, V., Vandenberghe, M., et al. (2014). TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl Acad. Sci. USA 111, E3870–E3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado, J.A., José, A.P., Pedro, C.R., et al. (2010). SERCA2b activity is regulated by cyclophilins in human platelets. Arter. Thromb. Vasc. Biol. 30, 419–425. [DOI] [PubMed] [Google Scholar]
- Ryan, M.J., Johnson, G., Kirk, J., et al. (1994). HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 45, 48–57. [DOI] [PubMed] [Google Scholar]
- Secker, P.F., Luks, L., Schlichenmaier, N., et al. (2018). RPTEC/TERT1 cells form highly differentiated tubules when cultured in a 3D matrix. ALTEX 35, 223–234. [DOI] [PubMed] [Google Scholar]
- Shamir, E.R., and Ewald, A.J. (2015). Adhesion in mammary development: novel roles for E-cadherin in individual and collective cell migration. Curr. Top. Dev. Biol. 353–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery, C., Campbell, E., McMorrow, T., et al. (2005). Cyclosporine A-induced renal fibrosis: a role for epithelial–mesenchymal transition. Am. J. Pathol. 167, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, T., Zhang, Q., Hirnet, D., et al. (2008). The human TRPV6 channel protein is associated with cyclophilin B in human placenta. J. Biol. Chem. 283, 18086–18098. [DOI] [PubMed] [Google Scholar]
- Tornavaca, O., Eduard, S., Gloria, P., et al. (2011). KAP degradation by calpain is associated with CK2 phosphorylation and provides a novel mechanism for cyclosporine A-induced proximal tubule injury. PLoS One 6, e25746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P., and Heitman, J. (2005). The cyclophilins. Genome Biol. 6, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q.C., Wang, X., Tang, T.S., et al. (2018). EB1 traps STIM1 and regulates local store-operated Ca2+ entry. J. Cell Biol. 217, 1895–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser, M., Stadler, G., Jennings, P., et al. (2008). hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am. J. Physiol. Physiol. 295, F1365–F1375. [DOI] [PubMed] [Google Scholar]
- Yan, X., and Chen, Y.G. (2011). Smad7: not only a regulator, but also a cross-talk mediator of TGF-β signalling. Biochem. J. 434, 1–10. [DOI] [PubMed] [Google Scholar]
- Yang, Y.L., Liu, Y.S., Chuang, L.Y., et al. (2009). Bone morphogenetic protein-2 antagonizes renal interstitial fibrosis by promoting catabolism of type I transforming growth factor-β receptors. Endocrinology 150, 727–740. [DOI] [PubMed] [Google Scholar]
- Yuasa, S., and Fukuda, K. (2008). Multiple roles for BMP signaling in cardiac development. Drug Discov. Today Ther. Strateg. 5, 209–214. [Google Scholar]
- Yurchenko, V., Constant, S., Eisenmesser, E., et al. (2010). Cyclophilin–CD147 interactions: a new target for anti-inflammatory therapeutics. Clin. Exp. Immunol. 160, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko, V., O'Connor, M., Dai, W.W., et al. (2001). CD147 is a signaling receptor for cyclophilin B. Biochem. Biophys. Res. Commun. 288, 786–788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.