

Abstract
SRC is a major regulator of many signaling pathways and contributes to cancer development. However, development of a selective SRC inhibitor has been challenging, and FDA-approved SRC inhibitors, dasatinib and bosutinib, are multitargeted kinase inhibitors. Here, we describe our efforts to develop a selective SRC covalent inhibitor by targeting cysteine 277 on the P-loop of SRC. Using a promiscuous covalent kinase inhibitor (CKI) SM1–71 as a starting point, we developed covalent inhibitor 15a, which discriminates SRC from other covalent targets of SM1–71 including TAK1 and FGFR1. As an irreversible covalent inhibitor, compound 15a exhibited sustained inhibition of SRC signaling both in vitro and in vivo. Moreover, 15a exhibited potent antiproliferative effects in nonsmall cell lung cancer cell lines harboring SRC activation, thus providing evidence that this approach may be promising for further drug development efforts.
Graphical Abstract

INTRODUCTION
The product of the c-SRC gene, the first discovered protooncogene, SRC kinase belongs to a family of 11 nonreceptor tyrosine kinases known as SRC family kinases (SFKs).1 SFKs have been suggested to play roles in various cancers, including breast, prostate, colon, head and neck, and lung.2,3 SRC is a central signaling hub that can be activated by many factors, including G protein-coupled receptors (GPCRs), adhesion receptors, cytokine receptors, and receptor tyrosine kinases (RTKs) such as EGFR, VEGFR, HER2, and PDGFR.4–6 As an effector of RTK signaling, SRC transduces survival signals to downstream effector pathways, such as PI3K/-AKT/mTOR and JAK/-STAT, to enhance diverse aspects of cancer development including proliferation, invasion, and differentiation.1,7–9 For example, SRC transfection potentiates EGF-induced oncogenesis;10 overexpression of SRC increases HER2–HER3 dimerization and enhances downstream signaling,11 and under hypoxic conditions, activated SRC can lead to upregulation of VEGFR and thus spur angiogenesis.12 SRC overexpression/activation has been implicated in conferring resistance to several inhibitors across different tumors.13 For example, elevated SRC expression confers resistance to an anti-HER2 agent, Trastuzumab, in breast cancer and to ALK inhibitors in nonsmall cell lung cancer (NSCLC).14–16 Due to its involvement in many cellular processes related to cancer development, SRC has long been considered a potential drug target in oncology.
Extensive medicinal chemistry efforts have yielded a number of small molecule SRC inhibitors. Several compounds representing a diverse range of chemical scaffolds have entered clinical trials with two compounds ultimately approved by the FDA: dasatinib (approved in 2006) and bosutinib (approved in 2012).17–25 However, both of these drugs are also very potent inhibitors of BCR-ABL, a known oncogenic driver of chronic myelogenous leukemia (CML), which indicates these agents have demonstrated clinical efficacy. It is, therefore, unclear whether SRC inhibition contributes to the drug response in CML, especially given that drugs such as imatinib and nilotinib that do not inhibit SRC are also efficacious in CML. Furthermore, dasatinib is known to inhibit >30 kinases, while bosutinib inhibits >45 kinases, making it impossible to use these compounds as selective mechanistic probes for SRC-dependent pharmacology.26–29 Similarly, most preclinical SRC inhibitors reported thus far lack selectivity and are not suitable for probing the biological functions of SRC. For example, although saracatinib (AZD0530) is a more selective SRC inhibitor than dasatinib, it also potently inhibits EGFR.30 To develop a SRC-selective small molecule inhibitor, we turned to covalent targeting whereby inhibitors containing a reactive warhead would be irreversibly bound to a unique cysteine on SRC.
Targeted covalent inhibitors (TCIs) are an emerging pharmacological modality that offers several potential advantages such as improved potency, selectivity, pharmacodynamics, and efficacy over conventional reversible inhibitors.31 The main liabilities of TCIs are their potential for indiscriminate reactivity due to the presence of the reactive warhead that could result in covalent adduct formation with unexpected targets and the potential for idiosyncratic toxicities. Despite these potential issues, TCIs have proven to be effective and safe in the clinic, leading to the FDA approval of six covalent kinase inhibitors for different indications, including acalabrutinib (BTK), afatinib (EGFR), ibrutinib (BTK), neratinib (HER2), osimertinib (EGFR), and dacomitinib (EGFR).32 Overall, TCIs represent a promising opportunity for the development of potent kinase inhibitors based on their ability to target specific cysteine residues while reducing reversible, off-target binding. A sequence alignment of the kinase family shows that SRC, along with eight other human protein kinases (Yes, Fgr, FGFR1–4, LIMK1, and TNK1), possess an equivalently positioned cysteine residue in the glycine-rich phosphate binding loop (P-loop) that can be targeted using TCIs. For example, we have previously described compound FIIN-2 that binds the P-loop cysteine of FGFRs (Cys477),33,34 while another study reported covalent targeting of SRC P-loop cysteine, Cys277, using the vinyl sulfonamide as the warhead.35
We recently reported that SM1–71, a promiscuous covalent kinase inhibitor (CKI), reacts with the P-loop cysteine of SRC (Cys277), suggesting that development of selective CKIs for SRC may be feasible.36–38 However, SRC is just one of many SM1–71 targets, as this compound reacts with cysteines located not only within the P-loop of a range of kinases but also at the DFG-1 position and within the activation loop as well. Here, we use SM1–71 as a starting point for a structure-guided medicinal chemistry campaign to engineer out covalent binding to cysteines at the DFG-1 position, activation loop, and the P-loop cysteine of FGFR1–4, FGR, LIMK1, and TNK1, while maintaining covalent SRC activity. Our efforts resulted in compound 15a, a SRC-directed CKI with an acrylamide warhead, that displayed efficient covalent target labeling and irreversible cellular inhibition of SRC signaling. Moreover, compound 15a demonstrated preferential cell killing of NSCLC cell lines harboring SRC activation, motivating further therapeutic development of covalent SRC inhibitors.
CHEMISTRY
The syntheses of compounds 5a–5e, 10a–10c, and 15a–15i are depicted in Scheme 1. 5-Bromo-2,4-dichloropyrimidine (1) was sequentially coupled with benzene-1,2-diamine and 4-(4-methylpiperazin-1-yl)aniline at the 4 and 2 positions of the pyrimidine, respectively, to generate the intermediate 3, which was subjected to a Suzuki coupling with an aryl or heteroaryl boronic acid or ester.
Scheme 1.

Syntheses of Compounds 5a–e, 10a–c, and 15a–i from Commercially Available 5-Substituted-2,4-Dichloropyrimidinea
aReagents and conditions: (a) benzene-1,2-diamine, DIEA, n-BuOH, reflux; (b) 4-(4-methylpiperazin-1-yl)aniline, TFA, s-BuOH, reflux; (c) aryl boronic acid or ester, Pd(dppf)Cl2·DCM, Et3N, dioxane/H2O; (d) acryloyl chloride, NaHCO3 aq., THF, ice bath; (e) different benzyl bromides, K2CO3, DMF, 50 °C; (f) different anilines, Et3N, DMF, ice bath; (g) benzene-1,2-diamine, DIEA, n-BuOH, rt.
Installation of the acrylamide on intermediate 4 with acryloyl chloride under basic aqueous conditions afforded final compounds 5a–e in overall moderate yield. The commercially available 2,4-dichloropyrimidin-5-ol (6) and 2,4-dichloropyrimidine-5-carbonyl chloride (11) were reacted with benzyl bromides and anilines, respectively, and the resulting intermediates 7 and 12 were converted to final compounds 10 and 15 using the same reaction route as for compound 5.
For the synthesis of compounds 17, we first established the amide group at the 5 position of the pyrimidine with the method used for intermediate 13a in Scheme 1. Then, the acrylamide was installed, leading to intermediate 16, which is stable under the acidic conditions used for the amination (Scheme 2). Scheme 3 shows the synthesis for compounds that bear different covalent warheads. First, a SN2 substitution reaction with meta-, ortho-, and para-benzenediamine was performed, followed by a second substitution at the 2 position of intermediate 18. Installation of an electrophile, such as chloroacetamide or vinyl sulfonamide, as the last step provided compounds 20a–20f.
Scheme 2.

Synthesis of Compounds 17a–f from 13aa
aReagents and conditions: (a) acryloyl chloride, NaHCO3 aq., THF, ice bath; (b) different anilines, TFA, sec-BuOH, reflux.
Scheme 3.

Synthesis of Compounds 20a–f from 12aa
aReagents and conditions: (a) different benzene diamines, DIEA, n-BuOH, rt; (b) 4-(4-methylpiperazin-1-yl)aniline, TFA, s-BuOH, reflux; (c) different electrophiles, NaHCO3 aq., THF, ice bath or Et3N, DCM, ice bath.
RESULTS
Prior to this work, we established that SM1–71 was a promiscuous CKI that labeled multiple cysteine sites, including at the DFG-1 position, at the activation loop, and on the P-loop.38 A chemoproteomics study of SM1–71 revealed that SRC is one of the modified proteins, and a trypsin digestion MS/MS experiment demonstrated covalent labeling of Cys277 on the P-loop. Additionally, the crystal structure of SM1–71 bound to SRC (PDB: 6ATE) revealed that anilinopyrimidine of SM1–71 engages the backbone of Met341 in the hinge region of SRC via two hydrogen bonds, thus bringing the acrylamide warhead in close proximity to Cys277 SRC. To better understand the binding mode and identify potential selectivity features, we performed covalent docking of SM1–71 into FGFR1 using Glide.39 FGFR1 was chosen for this analysis because our previous study revealed SM1–71-SRC binding and also showed that SM1–71 targeted FGFR1 via a P-loop cysteine.38 Our docking results suggested that SM1–71 interacts with SRC and FGFR1 in a very similar manner (Figure 1). Both the docking result of SM1–71 with FGFR1 and the previously solved crystal structure of SM1–71 with SRC (PDB: 6ATE) reveal that the hydrophobic pocket I is not occupied. Additionally, a cocrystal of SM1–71 bound to TAK1 (PDB: 5J7S), another one of its covalent targets, showed a smaller hydrophobic pocket I relative to SRC. We were, therefore, interested in investigating whether selectivity for SRC could be achieved by exploiting this pocket. Furthermore, an inspection of the gatekeeper residues revealed another difference: the apolar and bulkier valine of FGFR and methionine of TAK1 (Met104), versus SRC’s threonine, supporting the idea that developing a SRC-selective CKI may be feasible.
Figure 1.

(a) Superposition of the binding mode of SM1–71 in SRC with FGFR1 and TAK1 kinase domains. Red dashed lines indicate hinge hydrogen bonds. The spheres show the size of the gatekeeper residues: red for SRC, gold for FGFR1, and cyan for TAK1. The red, gold, and cyan ribbons represent SRC, FGFR1, and TAK1 kinases, respectively. DFG-1 Cys of TAK1 is shown in sphere representation with a carbon backbone in cyan. (b) Crystal structure of SM1–71 covalently bound to SRC kinase (PDB: 6ATE). SM1–71 covalently binds to P-loop Cys277. The structure alignment was done using Schrodinger Suite protein alignment tool with default settings, and the figures were generated with pymol.
Starting from SM1–71, we tailored the modifications at the C5 position on the anilinopyrimidine ring to fine-tune the selectivity for SRC. We used commercial Z’LYTE and LanthaScreen kinase assays from Invitrogen to measure all biochemical IC50’s reported in this work. As shown in Table 1, the first series of modifications involved the introduction of a monoaryl ring: thiophene (5a), furan (5b), and 6-methoxypyridine (5c) resulted in enhanced inhibitory activity against FGFR1, presumably as a consequence of improved hydrophobic interactions in the back-pocket. In contrast, the bulkiness of R1 groups on compounds 5a–5e significantly abrogate TAK1 inhibition, likely due to a steric clash with Met104. Compounds 5d and 5e that bear 4-acetophenone or 4-oxidibenzene start to lose activity against FGFR, albeit to a limited extent. Next, we asked whether extending the phenyl ring further into the pocket by adding an ether linkage would further improve the selectivity. We therefore generated compounds 10a, 10b, and 10c. Compounds 10a and 10b showed some improvement in selectivity, whereas 10c was the most advanced compound, which resulted in an IC50 > 1 μM on FGFR1, a 20-fold loss compared to SM1–71.
Table 1.
IC50 Values for Compounds 5a–e and 10a–c against SRC, FGFR1, and TAK1a
 | ||||
|---|---|---|---|---|
| Compound | R1 | SRC IC50 (nM) |
FGFR1 IC50 (nM) |
TAK1 IC50 (nM) |
| SM1–71 | Cl | 4 | 54 | 50 |
| 5a |  |
3 | 17 | 543 |
| 5b |  |
2 | 12 | 224 |
| 5c |  |
6 | 20 | 1890 |
| 5d |  |
6 | 72 | 1490 |
| 5e |  |
5 | 89 | 5460 |
| 10a | 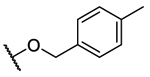 |
5 | 119 | _a |
| 10b |  |
3 | 373 | - |
| 10c |  |
6 | 1050 | - |
“-” means not applicable.
Next, we asked whether an amide bond, as a more polar functional group, at the C5 position influenced the activity on both SRC and FGFR1. Compared to compounds with a C–C (5a–5e) or C–O (10a–10c) bond, the SM1–71 derivatives we synthesized with an amide bond (15a–i) generally demonstrated significantly decreased activity on FGFR1 (with compounds 15b, 15d, 15e, 15h, and 15i showing activity above 10 μM) and a narrow structure–activity relationship (SAR) for SRC. For example, SRC tolerates a phenyl group with up to 3 substitutions such as methyl or chloride at the ortho or para position (15a, 15b, 15c, and 15d) but not at the meta position (15e, 15f, and 15g). Replacement of the methyl group on compound 15a with ethyl (15h) resulted in a 40-fold loss of activity on SRC, suggesting a steric clash. The trajectory of the amide-conjugated phenyl ring on compound 15i likely differs from ether-linked compound 10c, as a complete loss of activity on both SRC and FGFR1 was observed with 15i. Overall, in this series, compound 15a emerged as a lead for further optimization, given that it displayed an IC50 of 3 nM in a fixed time point assay against SRC, while offering a broad selectivity window (more than 103) versus FGFR1 (Table 2).
Table 2.
IC50 Values for Compounds 15a–i against SRC and FGFR1
 | |||
|---|---|---|---|
| Compound | R3 | SRC IC50 (nM) |
FGFR1 IC50 (nM) |
| 15a |  |
3 | 8340 |
| 15b |  |
9 | >10000 |
| 15c |  |
8 | >3330 |
| 15d |  |
14 | >10000 |
| 15e |  |
4010 | >10000 |
| 15f |  |
1010 | 6710 |
| 15g |  |
940 | 391 |
| 15h |  |
79 | >10000 |
| 15i |  |
>3330 | >10000 |
Given compound 15a’s superior potency and selectivity, we made further changes to the 15a scaffold, extending our optimization to parts of the molecule that mediate interactions with hydrophobic binding pocket II and the solvent-exposed region of SRC. The moiety that binds in the solvent-exposed region, such as piperazine, can be installed at the meta (17a) or para position (17b), whereas the ortho position (17c) results in loss of binding, likely due to interruption of binding to the kinase hinge interaction. A similar result was observed once a methoxy group was introduced at this position (17d and 17e). In contrast, a methyl group at the meta position is tolerated as shown by compound 17f, which displayed a single digit nanomolar IC50 on SRC (Table 3). These attempts to enhance binding by exploring interactions with hydrophobic binding pocket II and the solvent-exposed region on SRC resulted in no improvement over 15a as assessed by biochemical assays.
Table 3.
IC50 Values for Compounds 17a–f against SRC
 | |||||
|---|---|---|---|---|---|
| Compound | R4 | SRC IC50 (nM) |
Compound | R4 | SRC IC50 (nM) |
| 17a |  |
22 | 17d |  |
1210 |
| 17b |  |
5 | 17e |  |
3580 |
| 17c |  |
>10000 | 17f |  |
4 |
We next explored the position of the acrylamide warhead and demonstrated that the ortho position is optimal, as the meta or para position (20a and 20b) resulted in 10–15-fold loss of activity on SRC. Furthermore, the ortho position tolerates other short warheads such as chloroacetamide (20c) and vinyl sulfonamide (20d), but adding a bulkier methylene dimethylamine to the acrylamide likely caused a steric clash with the P-loop, compromising the IC50 of compound 20e (Table 4).
Table 4.
IC50 Values for Compounds 20a–e against SRC
 | ||
|---|---|---|
| Compound | R7 | SRC IC50 (nM) |
| 20a |  |
226 |
| 20b |  |
33 |
| 20c |  |
8 |
| 20d |  |
7 |
| 20e |  |
121 |
| 20f |  |
3 |
Collectively, our exploration around the hydrophobic pocket I provided a wide selectivity window over TAK1 and FGFR1, likely due to the different gatekeeper residues among these three kinases. The investigation of the solvent-exposed region and variation of the covalent warhead proved less fruitful in developing more potent and selective molecules. Thus, compound 15a remained our lead compound, and we focused on it for further detailed mechanistic validation and preliminary pharmacological characterization.
Compound 15a Selectively Modifies SRC Cys277.
To confirm the covalent bond formation of compound 15a with Cys277 on SRC, we first analyzed the protein by LC-MS after incubation with a 10-fold molar excess of 15a for 2 h at 37 °C and observed a mass shift consistent with 15a, indicating that 15a forms a single covalent adduct with SRC (Figure 2A). To confirm the site of modification, labeled protein was digested with trypsin, and peptides were analyzed by capillary electrophoresis-MS/MS. A database search revealed an exclusive modification of SRC by 15a at Cys277 (Figure 2B). Taken together, our mass spectrometry-based analysis showed that 15a forms a covalent adduct with SRC by reacting with a single cysteine (out of 9), Cys277, thus mirroring the reactivity of the parental, pan-CKI SM1–71.
Figure 2.

Src is exclusively labeled by 15a at cysteine 277. (A) Mass spectra (left) and zero-charge mass spectra (right) of SRC incubated (top) with DMSO or (bottom) with a 10-fold molar excess of 15a for 2 h at 37 °C. Unlabeled protein peaks are indicated with purple glyphs, and 15a modified protein peaks are highlighted with blue glyphs. (B) MS/MS spectrum of the SRC tryptic peptide 273LGQGC*FGEVWMGTWNGTTR287 modified with 15a at Cys 277 (indicated with “*” in the sequence). Ions of type y are indicated with red glyphs. Inhibitor modified ions are labeled with green glyphs.40
Kinase Profiling Elucidates SRC-Selective Inhibition by 15a.
To map the selectivity of our SRC lead CKI 15a, we performed KinomeScan profiling, which is an active site-directed binding assay for 468 kinases. The result represents the remaining kinase activity in the presence of the test compound compared to the DMSO control. As shown in Figure 3, while parental CKI SM1–71 hits more than 100 kinases at 1 μM, compound 15a, at the same concentration, is far more selective, inhibiting around 20 kinases (data shown in Tables S1 and S2; scores of >35%, as an indication of weak binding, are not shown). BTK, ABL, and most SFK family members such as HCK, BLK, LCK, and YES1 were the main off-targets as confirmed with low nanomolar biochemical IC50’s (Table S3). We tested HCK, one of the off-targets of 15a, in a cellular antiproliferation assay. 15a did not show antiproliferative activity on HCK-dependent Ba/F3 cells (IC50 > 2927nM; Table S4) despite its biochemical IC50 of ~3 nM. Unlike SRC, HCK lacks the P-loop cysteine, suggesting that 15a binds this kinase in a reversible manner and thus explains the disparity between cellular and biochemical data. Strikingly, MEK family kinases are not inhibited by compound 15a, although they are major targets of SM1–71.
Figure 3.

KinomeScan profiling for SM1–71 and 15a. (KinomeScan profiling for compound SM1–71 was previously published.36 Reprinted with permission from ref 36. Copyright 2017 Elsevier.) Compounds SM1–71 and 15a were screened at 1 μM against a panel of 468 human kinases. The results for the binding interactions are reported as “% Ctrl”, where larger circles indicate stronger hits. The top hits of 15a are SRC, YES1, ABL1, HCK, LCK, LYN, BTK, BLK, and GAK, shown as blue circles in the kinome tree. (See the biochemical IC50’s in Table S3.) The selectivity score was defined as the ratio of the number of kinases inhibited to a specified percentage versus the total number of kinases. For this experiment, specified percent inhibition was set at 35%, resulting in S(35) values of 0.43 for SM1–71 and 0.07 for compound 15a.
Growth Inhibitory Screen Reveals Potent Antiproliferative Effects in NSCLC Cells.
Next, we sought to evaluate the antiproliferative potency of 15a using a growth inhibition assay. We chose cell lines where SRC kinase activity is known to contribute to growth and proliferation.41–43 These included MDA-MB-231 (triple negative breast cancer cell line) and H1975 and HCC827 (two nonsmall cell lung cancer lines, NSCLC). We compared the growth inhibitory potency of 15a with that of its reversible (noncovalent) analog, 20f, as well as AZD0530 (saracatinib), a reversible (noncovalent) SRC inhibitor developed by AstraZeneca.44 Briefly, cells were treated with varying concentrations of the compounds for a period of 72 h, and cell viability was measured using the CellTiter-Glo assay. In order to account for confounding effects arising from varying doubling times across different cell lines, we implemented the recently described growth rate (GR) corrected calculations.40,45 This analysis results in GR50 (similar to IC50) and GRmax (similar to Emax) values, where a GRmax value that lies within 0 and 1 implies partial growth arrest, a value of 0 indicates complete growth arrest, and a negative value implies cytotoxic effects (Data S1). Our results showed that 15a induced strong growth inhibitory effects across all three cell lines with GR50 values of 0.3, 0.5, and 0.3 μM, respectively (Figure 4A, Table S5). Furthermore, the compound also led to cytotoxic effects in HCC827 (GRmax = −0.6) and MDA-MB-231 (GRmax = −0.4) cells (Table S5). The reversible analog 20f and AZD0530 also induced growth inhibition in these three cell lines but showed a weaker potency compared to 15a (Figure 4B). These results suggest that covalent and selective SRC targeting in these cell lines can achieve potent inhibition of growth and proliferation.
Figure 4.

Evaluating biological potency of SRC inhibitors in lung and breast cancer cell lines. (A) Comparing growth inhibitory potency of 15a in H1975 (nonsmall cell lung cancer, NSCLC), HCC827 (NSCLC), and MDA-MB-231 (triple negative breast cancer, TNBC) cell lines. (B) Comparing growth inhibitory potency of 15a, 20f, and AZD0530 across cell lines. Growth inhibition was carried out by treating cells with different concentrations of 15a, 20f, or AZD0530 for 72 h, followed by analysis using the CellTiter-Glo reagent. Dose–response curves and the corresponding GR values were calculated using the online tool GR calculator (http://www.grcalculator.org/grcalculator/). Both the curves and the GR50 values are representative of two independent experiments, each carried out in technical triplicate.
Cellular Target Engagement and Signaling Inhibition by 15a.
Having demonstrated binding of 15a to SRC in biochemical assays, we next investigated whether 15a could covalently bind SRC in cells. To assess cellular binding potency, we conducted a cellular target engagement analysis in two NSCLC cell lines, H1975 and HCC827, that showed strong growth inhibition by 15a (Figure 4A). We further chose these cell lines because SRC is known to promote tumor progression in NSCLC.43,46 H1975 and HCC827 cells were treated with 1 μM of 15a or 20f or DMSO for 2 h, followed by drug removal, cellular extraction, and lysis. The obtained lysates were then incubated with 1 μM of TL13–68 (Figure S2),37 a biotin-tagged version of SM1–71 that competes with 15a for covalent SRC binding. Western blotting was used to evaluate cellular target engagement. Our results showed that SRC was covalently modified by 15a in both cell lines, as indicated by the lack of SRC binding to TL13–68 and pulldown by streptavidin beads (Figure 5A). The reversible analog 20f resulted in weaker SRC inhibitory effects, presumably due to displacement by TL13–68 (Figure 5A). We further analyzed whether 15a can bind other cysteine-containing kinases such as MEK1/2 and ERK1/2, which have a DFG-1 cysteine. Our results showed 15a did not engage MEK1/2 or ERK1/2 in cells. In addition, we also inspected whether TL13–86 can compete with 15a for binding to YES1, a kinase sharing a similar P-loop cysteine. Although the YES1 expression level is low in both cells, a rescue of YES1 pulldown from the TL13–68 probe was detected, indicating YES1 is a potential covalent target for 15a.
Figure 5.

Target engagement and inhibition of SRC in vitro and in vivo. (A) Target engagement in H1975 and HCC827 cells treated with 1 μM of 15a or 20f for 2 h followed by overnight incubation with 1 μM TL13–68 (biotin compound), pulldown by streptavidin beads, and analysis using Western blotting. (B) Signaling inhibition in H1975 and HCC827 cells treated with 1 μM of 15a or 20f for 2 h followed by analysis using Western blotting. (C) Cellular target engagement and (D) signaling inhibition in H1975 cells treated with 1 μM of 15a, 20f, AZD0530, or DMSO for 2 h, followed by drug removal and replacement with drug-free medium. Cells were collected and lysed 0, 2, and 4 h postdrug washout and analyzed using Western blotting. For evaluating SRC binding (C), lysates were incubated with 1 μM of TL13–68 (biotin compound), followed by pulldown by streptavidin beads and visualization using Western blotting. Blots shown in (A–D) are representative of one of two biological replicates carried out. (Both blots for each experiment are shown in Figure S1.) (E, F) Pharmacodynamic analysis of (E) inhibition of p-SRCY416 signaling and (F) inhibition of SRC binding in spleen samples. Briefly, C57B6 mice (n = 2/condition) were treated with 5 mg/kg of 15a or vehicle three times and sacrificed 2 or 4 h following the last dose. Spleen samples collected were ground up, lysed, and analyzed using Western blotting.
Next, we sought to evaluate whether 15a could lead to inhibition of SRC signaling in cells. Briefly, H1975 and HCC827 cells were treated with 1 μM of 15a or 20f or DMSO for 2 h, followed by extraction and lysis. Western blotting was used to measure the inhibition of SRC phosphorylation. Our results showed that, consistent with SRC binding, 15a inhibited p-SRCY416 signaling in both H1975 and HCC827 cells (Figure 5B). Furthermore, 20f was also capable of inhibiting p-SRCY416 signaling in cells (Figure 5B). Together, our results indicated that 15a is capable of inducing potent SRC binding and inhibition of SRC signaling in cells.
Washout-Assays Reveal Superior Target Engagement by Covalent Inhibitor.
To assess the duration of inhibition of SRC in cells and further distinguish between covalent and noncovalent modes of binding, we conducted target engagement and signaling analyses under drug-washout conditions. H1975 cells were treated with 1 μM of 15a, 20f, AZD0530, or DMSO for 2 h, followed by drug washout and replacement of drug-containing medium with fresh drug-free medium. Cells were subsequently collected and lysed at 0, 2, and 4 h postwashout. For the cellular target engagement assay, the resulting lysate was incubated with 1 μM of TL13–68 (biotin compound) prior to visualization using Western blotting. For signaling analysis, lysates were directly processed to visualize Western blotting, without incubation with the biotin compound. Our results showed that 15a was able to inhibit SRC up to 4 h postwashout, unlike the reversible compounds, 20f and AZD0530 (Figure 5C). Similarly, 15a induced potent and sustained inhibition of p-SRCY416 signaling up to 4 h postwashout, unlike AZD0530 (Figure 5D). Despite showing weak target engagement, 20f led to moderate inhibition of p-SRCY416 signaling (Figure 5D). Together, these results indicate that covalent inhibition of SRC by 15a leads to potent and sustained target engagement, compared to reversible SRC compounds.
Pharmacodynamic Analysis in Mice Spleen Tissue Demonstrates in Vivo Target Engagement.
Next, we sought to evaluate target engagement and signaling analysis in vivo. We conducted a PK study for our lead compound 15a. We dosed 15a at 5 mg/kg IP in 5% DMSO/95% D5W in B6 mice. The compound exhibited a short half-life and high exposure (T1/2 = 1.29 h, AUC = 12 746.25 min·ng/mL). Since compound 15a has a short half-life, we decided to treat mice with three doses to ensure full target engagement. We conducted pharmacodynamic analysis in C57B6 mice (n = 2 per condition) using spleen tissue samples collected 2 and 4 h after administering the last dose of compound 15a. A total of 3 doses (5 mg/kg) was administered to the mice prior to sacrificing and collecting tissue samples. Spleen tissue samples were ground up and lysed, and Western blotting was used to measure target engagement and signaling changes. Our results showed that 15a led to inhibition of p-SRCY416 at 2 and 4 h postdosing, compared to the vehicle controls (Figure 5E). Similarly, 15a also demonstrated SRC binding and inhibition at both 2 and 4 h postdosing compared to the vehicle controls (Figure 5F). These results suggest that 15a is able to inhibit SRC for an extended duration, likely due to its ability to covalently bind the target.
DISCUSSION AND CONCLUSION
Promiscuous kinase inhibitors, or drugs with polypharmacology, have proven beneficial in treating cancers (e.g., sunitinib, sorafenib, and dasatinib), and they have also been useful as pharmacological probes to assess dependence on different signaling pathways.47–50 In the present study, we used our recently characterized pan-kinase covalent inhibitor SM1–71 as a chemical starting point for designing a selective, covalent SRC inhibitor. SM1–71 bears a 2,4-diaminopyrimidine, a prevalent scaffold among kinase inhibitors, and the presence of a chlorine at the C5 position allows binding to many kinases regardless of the gatekeeper residue. SM1–71 primarily reacts with cysteines that are positioned before the DFG motif (e.g., TAK1, MEK, ERK) or on the P-loop (e.g., SRC, FGFR1–4) through rotation of the acrylamide warhead.38 In designing 15a, we explored tuning the trajectory of the covalent warhead and the differences between the hydrophobic pockets of various SM1–71 targets. By inspecting the gatekeeper residues of TAK1, SRC, and FGFR1, we reasoned that both SRC and FGFR1 would tolerate a bulkier phenyl group at the C5 position of the pyrimidine but not TAK1, which has a larger methionine gatekeeper. Compound 15a, with a phenyl group linked through an amide bond at the C5 position, preserves potent binding to SRC while abolishing activity against TAK1 and FGFR1. We propose that the selectivity between FGFR1 and SRC is due to a hydrogen bond interaction between the amide and the threonine gatekeeper on SRC, which is disrupted by the valine on FGFR, a more hydrophobic residue. Due to the high sequence, and likely structure, similarity, other SRC family kinases (HCK, BLK, LCK, and YES1) have been indicated as potential off-targets for 15a on the basis of a low nanomolar biochemical IC50 value. YES1 and FGR are the only proteins in this family that have a P-loop cysteine and may potentially be covalent targets, while other kinases in this group are likely biochemically inhibited through noncovalent, reversible binding. Further optimization to remove these off-targets is the subject of a future medicinal chemistry effort.
Irreversible SRC inhibition by compound 15a led to sustained signaling inhibition, target engagement, and more potent antiproliferative effects across cancer cell lines. Although 15a showed a half-life of 5.2 min in mouse liver microsomes (data not shown), the PD study demonstrated an extended duration of SRC signaling blockade. This suggests that the irreversible SRC inhibitor 15a could have therapeutic potential and potential differentiation from noncovalent SRC inhibitors.
MATERIALS AND METHODS
Chemistry.
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on a 500 MHz Bruker A500, and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5 μM) using a gradient of 15–95% methanol in water or acetonitrile in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) or 35 min (45 min run time) at a flow rate of 20 mL/min. The purity of all the final compounds is ≥95%, and it is measured by analytical reverse-HPLC using a gradient of 15–85% acetonitrile in water containing TFA over 2.5 min at a flow rate of 2 mL/min.
N1-(5-Bromo-2-chloropyrimidin-4-yl)benzene-1,2-diamine (2).
To the solution of 5-bromo-2,4-dichloropyrimidine (1.0 g, 4.39 mmol) and benzene-1,2-diamine (474 mg, 4.39 mmol) in n-BuOH (15 mL) was added DIEA (1.13 g, 8.78 mmol) at room temperature. The reaction mixture was heated up to 100 °C for 2 h. The solvent was evaporated under vacuum, and the residue was purified with flash chromatography to yield the product (1.10 g, 3.68 mmol, 84%). (M +H)+ calculated 298.96; found 298.70, 300.70.
N4-(2-Aminophenyl)-5-bromo-N2-(4-(4-methylpiperazin-1-yl)-phenyl)pyrimidine-2,4-diamine (3).
To the solution of intermediate 2 (550 mg, 1.84 mmol) and 4-(4-methylpiperazin-1-yl)aniline (352 mg, 1.84 mmol) in s-BuOH (5 mL) was added TFA (314 mg, 2.76 mmol) at room temperature. The reaction mixture was heated up to 100 °C for 2 h. The mixture was cooled to room temperature and concentrated to get the residue. The residue was purified with flash chromatography to obtain the product (760 mg, 1.67 mmol, 91%). (M + H)+ calculated 454.13; found 454.19, 456.19.
N4-(2-Aminophenyl)-N2-(4-(4-methylpiperazin-1-yl)phenyl)-5-(thiophen-2-yl)pyrimidine-2,4-diamine (4a).
To the solution of intermediate 3 (100 mg, 0.22 mmol), thiophen-2-ylboronic acid (42 mg, 0.33 mmol), and K3PO4 (279 mg, 1.32 mmol) in 1,4-dioxane/H2O (2 mL/0.5 mL) was added Pd(dppf)Cl2·DCM (18 mg, 0.022 mmol) at room temperature under a nitrogen atmosphere. The reaction mixture was heated up to 80 °C for 3 h. The mixture was extracted with ethyl acetate, and the organic layer was concentrated to get the residue. The residue was purified with chromatography to yield the product (40 mg, 0.087 mmol, 40%). (M + H)+ calculated 458.20; found 457.88.
N-(2-((2-((4-(4-Methylpiperazin-1-yl)phenyl)amino)-5-(thiophen-2-yl)pyrimidin-4-yl)amino)phenyl)acrylamide (5a).
To the solution of intermediate 4a (40 mg, 0.087 mmol) in NaHCO3 (sat. aq., 1 mL) and THF (1 mL) was added acryloyl chloride (8.7 mg, 0.096 mmol) slowly under an ice bath. The reaction was extracted with ethyl acetate after all the intermediate 4a was consumed. The organic layer was concentrated to get residue. The residue was purified by HPLC to afford the title compound (9.1 mg, 0.014 mmol, 36%). 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 1H), 9.80 (s, 2H), 8.53 (s, 1H), 8.03 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.69 (dd, J = 4.9, 1.4 Hz, 1H), 7.40 (d, J = 8.5 Hz, 2H), 7.38–7.31 (m, 2H), 7.30–7.25 (m, 1H), 7.23–7.17 (m, 2H), 6.87 (d, J = 8.6 Hz, 2H), 6.41 (dd, J = 17.0, 10.2 Hz, 1H),6.15 (dd, J = 17.0, 2.0 Hz, 1H), 5.78 (dd, J = 10.1, 2.0 Hz, 1H), 3.75 (d, J = 13.2 Hz, 2H), 3.53 (d, J = 12.1 Hz, 2H), 3.18 (s, 2H), 2.96–2.84 (m, 5H). (M + H)+ calculated 512.22; found 511.88.
N-(2-((5-(Furan-3-yl)-2-((4-(4-methylpiperazin-1-yl)phenyl)-amino)pyrimidin-4-yl)amino)phenyl)acrylamide (5b).
Compound 5b was synthesized via the same route as compound 5a. 1H NMR (500 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.94 (s, 1H), 9.81 (s, 1H),8.57 (s, 1H), 8.02 (s, 1H), 7.94 (s, 1H), 7.85 (t, J = 1.7 Hz, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.44 (dd, J = 7.8, 1.7 Hz, 1H), 7.39–7.31 (m, 3H), 7.29 (td, J = 7.6, 1.6 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 1.8 Hz, 1H), 6.45 (dd, J = 16.9, 10.2 Hz, 1H), 6.20 (dd, J = 17.0, 2.0 Hz, 1H), 5.79 (dd, J = 10.1, 2.0 Hz, 1H), 3.74 (d, J = 13.1 Hz, 2H),3.53 (d, J = 12.2 Hz, 2H), 3.17 (s, 2H), 2.91 (d, J = 11.5 Hz, 2H),2.88 (s, 3H). (M + H)+ calculated 496.24; found 496.38.
N-(2-((5-(6-Methoxypyridin-3-yl)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)pyrimidin-4-yl)amino)phenyl)acrylamide (5c).
Compound 5c was synthesized via the same route as compound 5a. 1H NMR (500 MHz, DMSO-d6) δ 10.04 (s, 1H), 9.96 (s, 1H), 9.72 (s, 1H), 8.62 (s, 1H), 8.13 (d, J = 2.4 Hz, 1H), 7.86 (s, 1H), 7.73–7.66 (m, 2H), 7.35 (dd, J = 8.0, 1.7 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.25 (td, J = 7.7, 1.7 Hz, 1H), 7.20 (td, J = 7.6, 1.6 Hz, 1H), 6.85 (d, J =8.6 Hz, 1H), 6.81 (d, J = 8.6 Hz, 2H), 6.33 (dd, J = 17.0, 10.2 Hz, 1H), 6.00 (dd, J = 17.0, 2.0 Hz, 1H), 5.70 (dd, J = 10.1, 2.0 Hz, 1H),3.85 (s, 3H), 3.68 (d, J = 13.3 Hz, 2H), 3.46 (d, J = 12.2 Hz, 2H),3.10 (d, J = 10.9 Hz, 2H), 2.84 (d, J = 12.2 Hz, 2H), 2.80 (s, 3H). (M + H)+ calculated 537.26; found 537.39.
N-(2-((5-(4-Acetylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)-amino)pyrimidin-4-yl)amino)phenyl)acrylamide (5d).
Compound 5d was synthesized via the same route as compound 5a. 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.85 (s, 1H) 9.82 (s, 1H),8.50 (s, 1H), 8.05 (d, J = 8.2 Hz, 2H), 8.01 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.63–7.58 (m, 2H), 7.44–7.36 (m, 3H), 7.35–7.30 (m, 1H), 7.29–7.21 (m, 1H), 6.87 (d, J = 8.6 Hz, 2H), 6.40 (dd, J = 16.9,10.2 Hz, 1H), 6.01 (dd, J = 17.1, 2.0 Hz, 1H), 5.75 (dd, J = 10.1, 2.0 Hz, 1H), 3.75 (d, J = 13.2 Hz, 2H), 3.54 (d, J = 12.1 Hz, 2H), 3.17 (d, J = 10.7 Hz, 2H), 2.92 (d, J = 12.4 Hz, 2H), 2.88 (s, 3H), 2.64 (s, 3H). (M + H)+ calculated 548.27; found 548.49.
N-(2-((2-((4-(4-methylpiperazin-1-yl)phenyl)amino)-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (5e).
Compound 5e was synthesized via the same route as compound 5a. 1H NMR (500 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.77 (s, 2H), 8.41 (s, 1H), 7.86 (s, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.39–7.31 (m, 6H),7.30–7.23 (m, 2H), 7.22–7.15 (m, 1H), 7.15–7.09 (m, 1H), 7.07–7.00 (m, 4H), 6.81 (d, J = 8.5 Hz, 2H), 6.32 (dd, J = 17.0, 10.2 Hz, 1H), 6.01 (dd, J = 17.0, 2.0 Hz, 1H), 5.65 (dd, J = 10.1, 2.0 Hz, 1H),3.68 (d, J = 12.9 Hz, 2H), 3.46 (d, J = 12.2 Hz, 2H), 3.17–3.03 (m, 2H), 2.89–2.77 (m, 5H). (M + H)+ calculated 598.29; found 598.59.
2,4-Dichloro-5-((4-methylbenzyl)oxy)pyrimidine (7a).
To the solution of 2,4-dichloropyrimidin-5-ol (100 mg, 0.61 mmol), NaI (10 mg, 0.061 mmol), and K2CO3 (210 mg, 1.52 mmol) in acetone (3 mL) was added 4-methyl benzylbromide (169 mg, 0.91 mmol) at room temperature. All the starting material was consumed overnight. The mixture was evaporated under vacuum, and the residue was purified with flash chromatography to yield the product (150 mg, 0.56 mmol, 91%). (M + H)+ calculated 269.02; found 269.17.
N1-(2-Chloro-5-((4-methylbenzyl)oxy)pyrimidin-4-yl)benzene-1,2-diamine (8a).
To the solution of intermediate 7a (150 mg, 0.56 mmol) and benzene-1,2-diamine (60 mg, 0.56 mmol) in n-BuOH (3 mL) was added DIEA (145 mg, 1.12 mmol) at room temperature. The mixture was then heated up to 110 °C for 2 h. The mixture was cooled to room temperature and concentrated under vacuum. The residue was purified with flash chromatography to yield the product (170 mg, 0.50 mmol, 89%). (M + H)+ calculated 341.11; found 341.27.
N4-(2-Aminophenyl)-5-((4-methylbenzyl)oxy)-N2-(4-(4-methylpiperazin-1-yl)phenyl)pyrimidine-2,4-diamine (9a).
To the solution of intermediate 8a (170 mg, 0.50 mmol) and 4-(4-methylpiperazin-1-yl)aniline (96 mg, 0.50 mmol) in s-BuOH (3 mL) was added TFA (85 mg, 0.75 mmol) at room temperature. The mixture was then heated up to 90 °C for 2 h. The mixture was evaporated under vacuum, and the residue was purified with flash chromatography to yield the product (70 mg, 0.14 mmol, 28%). (M + H)+ calculated 469.27; found 469.48.
N-(2-((5-((4-Methylbenzyl)oxy)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)pyrimidin-4-yl)amino)phenyl)acrylamide (10a).
To the solution of intermediate 9a (70 mg, 0.14 mmol) in NaHCO3 (sat. aq., 2 mL) and THF (2 mL) was added acryloyl chloride (14 mg, 0.15 mmol) under an ice bath. The reaction was extracted with ethyl acetate after all 9a was consumed. The organic layer was concentrated under vacuum, and the residue was purified by HPLC to obtain the title compound (34 mg, 0.052 mmol, 37%). 1H NMR (500 MHz, DMSO-d6) δ 10.24 (s, 1H), 9.81 (s, 2H), 9.24 (s, 1H), 7.88–7.66 (m, 1H), 7.39–7.32 (m, 2H), 7.31–7.24 (m, 2H), 7.24–7.19 (m, 1H),7.18–7.12 (m, 3H), 7.06 (s, 1H), 6.96 (s, 1H), 6.85–6.77 (m, 2H),6.42 (dd, J = 17.0, 10.2 Hz, 1H), 6.16 (dd, J = 17.0, 1.9 Hz, 1H), 5.74 (dd, J = 10.2, 1.9 Hz, 1H), 5.05 (s, 2H), 3.73–3.62 (m, 2H), 3.51–3.39 (m, 2H), 3.15–3.04 (m, 2H), 2.88–2.76 (m, 5H), 2.26 (s, 3H). (M + H)+ calculated 550.29; found 550.29.
N-(2-((5-((2-Chloro-6-methylbenzyl)oxy)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)acrylamide (10b).
Compound 10b was synthesized via the same route as compound 10a. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H),9.96 (s, 1H), 9.77 (s, 1H), 9.17 (s, 1H), 7.82 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.33–7.25 (m, 5H), 7.25 (d, J = 7.8 Hz, 1H), 7.23–7.14 (m, 2H), 6.84 (d, J = 8.7 Hz, 2H), 6.28 (dd, J = 17.0, 10.2 Hz, 1H), 5.81 (dd, J = 16.9, 1.9 Hz, 1H), 5.60 (dd, J = 10.1, 1.9 Hz, 1H), 5.17 (s, 2H), 3.68 (d, J = 13.1 Hz, 2H), 3.46 (d, J = 12.2 Hz, 2H), 3.10 (s, 2H), 2.84 (d, J = 12.8 Hz, 2H), 2.80 (s, 3H), 2.34 (s, 3H). (M + H)+ calculated 584.25; found 584.89.
N-(2-((2-((4-(4-Methylpiperazin-1-yl)phenyl)amino)-5-((4-phenoxybenzyl)oxy)pyrimidin-4-yl)amino)phenyl)acrylamide (10c).
Compound 10c was synthesized via the same route as compound 10a. 1H NMR (500 MHz, DMSO-d6) δ 10.24 (s, 1H),9.88 (s, 2H), 9.33 (s, 1H), 7.79–7.73 (m, 2H), 7.51–7.46 (m, 2H),7.40–7.31 (m, 3H), 7.28 (ddd, J = 11.2, 6.3, 2.4 Hz, 3H), 7.22 (td, J =7.6, 1.6 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 7.02–6.93 (m, 4H), 6.86–6.79 (m, 2H), 6.41 (dd, J = 16.9, 10.2 Hz, 1H), 6.12 (dd, J = 17.0, 1.9 Hz, 1H), 5.68 (dd, J = 10.1, 2.0 Hz, 1H), 5.08 (s, 2H), 3.71–3.64 (m, 2H), 3.45 (d, J = 12.0 Hz, 2H), 3.09 (s, 2H), 2.85 (d, J = 12.5 Hz, 2H), 2.80 (s, 3H). (M + H)+ calculated 628.30; found 628.50.
2,4-Dichloro-N-(2-chloro-6-methylphenyl)pyrimidine-5-carboxamide (12a).
To the solution of 2-chloro-6-methylaniline (67 mg, 0.47 mmol) in THF (1 mL) was added 2,4-dichloropyrimidine-5-carbonyl chloride (100 mg, 0.47 mmol) at room temperature. The starting material was nearly consumed overnight. The mixture was evaporated under vacuum, and the residue was purified with flash chromatography to yield the product (120 mg, 0.38 mmol, 81%). (M + H)+ calculated 315.97; found 316.07.
4-((2-Aminophenyl)amino)-2-chloro-N-(2-chloro-6-methylphenyl)pyrimidine-5-carboxamide (13a).
To the solution of intermediate 12a (120 mg, 0.38 mmol) and benzene-1,2-diamine (41 mg, 0.38 mmol) in n-BuOH (2 mL) was added DIEA at room temperature. The mixture was then heated up to 110 °C for 2 h. The mixture was cooled to room temperature and concentrated under vacuum. The residue was purified with flash chromatography to yield the product (147 mg, 0.38 mmol, quant.). (M + H)+ calculated 388.07; found 388.27.
4-((2-Aminophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (14a).
To the solution of intermediate 13a (147 mg, 0.38 mmol) and 4-(4-methylpiperazin-1-yl)aniline (73 mg, 0.38 mmol) in s-BuOH (2 mL) was added TFA (65 mg, 0.57 mmol) at room temperature. The mixture was then heated up to 90 °C for 2 h. The mixture was cooled down to room temperature and concentrated under vacuum. The residue was purified with flash chromatography to yield the product (120 mg, 0.22 mmol, 58%). (M + H)+ calculated 543.23; found 543.29.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15a).
To the solution of intermediate 14a (20 mg, 0.037 mmol) in NaHCO3 (sat. aq., 1 mL) and THF (1 mL) was added acryloyl chloride (4 mg, 0.044 mmol) under an ice bath. The reaction was extracted with ethyl acetate after all intermediate 14a was consumed. The organic layer was concentrated, and the residue was purified with HPLC to obtain the title compound (3.6 mg, 0.0050 mmol, 14%). 1H NMR (500 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.83 (s, 1H), 9.69 (s, 3H), 8.82 (s, 1H), 8.04 (s, 1H), 7.46 (d, J = 8.3 Hz, 2H), 7.36–7.26 (m, 2H), 7.26–7.16 (m, 3H), 7.09 (t, J = 7.5 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 6.27 (dd, J = 17.1, 10.3 Hz, 1H), 6.06 (dd, J = 17.1, 2.0 Hz, 1H), 5.56 (dd, J = 10.2, 2.0 Hz, 1H), 3.68 (d, J = 13.1 Hz, 2H), 3.46 (d, J = 12.2 Hz, 2H), 3.11 (d, J = 11.2 Hz, 2H),2.82–2.77 (m, 5H), 2.16 (s, 3H). (M + H)+ calculated 597.24; found 597.39.
4-((2-Acrylamidophenyl)amino)-N-(2,4-dichloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15b).
Compound 15b was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.69 (s, 1H),9.85 (s, 1H), 9.70 (s, 1H), 9.65 (s, 2H), 8.82 (s, 1H), 8.00 (s, 1H),7.51 (dd, J = 9.8, 2.4 Hz, 1H), 7.45 (s, 2H), 7.37 (dd, J = 11.0, 2.4 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 6.28 (dd, J = 17.1, 10.3 Hz, 1H),6.06 (dd, J = 17.1, 2.0 Hz, 1H), 5.57 (dd, J = 10.2, 2.0 Hz, 1H), 3.68 (d, J = 13.2 Hz, 2H), 3.46 (d, J = 12.0 Hz, 2H), 3.11 (d, J = 12.8 Hz, 2H), 2.85 (d, J = 12.6 Hz, 2H), 2.80 (s, 3H), 2.16 (s, 3H). (M + H)+ calculated 611.26; found 610.79.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-4,6-dimethylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15c).
Compound 15c was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H),9.75 (s, 1H), 9.69 (s, 3H), 8.82 (s, 1H), 8.00 (s, 1H), 7.48–7.41 (m, 2H), 7.29 (d, J = 7.9 Hz, 1H), 7.23–7.13 (m, 2H), 7.09 (t, J = 7.7 Hz, 1H), 7.04 (d, J = 11.2 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 6.28 (dd, J =17.1, 10.2 Hz, 1H), 6.06 (dd, J = 17.1, 1.9 Hz, 1H), 5.57 (dd, J = 10.2, 2.0 Hz, 1H), 3.68 (d, J = 13.3 Hz, 2H), 3.46 (d, J = 12.0 Hz, 2H),3.11 (d, J = 10.8 Hz, 2H), 2.88–2.78 (m, 5H), 2.23 (d, J = 3.3 Hz, 3H), 2.11 (s, 3H). (M + H)+ calculated 631.20; found 630.70.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-4-cyano-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15d).
Compound 15d was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.61 (s, 1H), 10.05 (s, 1H), 9.70 (s, 1H), 9.68 (s, 2H), 8.82 (s, 1H), 7.96 (s, 2H), 7.76 (s, 1H), 7.48–7.43 (m, 2H), 7.29 (d, J = 7.9 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 6.82 (d, J = 8.1 Hz, 2H),6.27 (dd, J = 17.1, 10.3 Hz, 1H), 6.05 (dd, J = 17.1, 2.0 Hz, 1H), 5.57 (dd, J = 10.2, 2.0 Hz, 1H), 3.68 (d, J = 13.2 Hz, 2H), 3.46 (d, J = 12.0 Hz, 2H), 3.11 (q, J = 11.0 Hz, 2H), 2.88–2.78 (m, 5H), 2.21 (s, 3H). (M + H)+ calculated 622.24; found 621.89.
4-((2-Acrylamidophenyl)amino)-N-(3-chloro-5-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15e).
Compound 15e was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.86 (s, 1H),10.08 (s, 1H), 9.78 (s, 1H), 9.68 (s, 2H), 8.75 (s, 1H), 8.12 (s, 1H),7.68–7.62 (m, 1H), 7.47–7.43 (m, 2H), 7.39 (s, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.08 (t, J = 7.7 Hz, 1H), 6.94 (s, 1H), 6.86 (d, J = 8.5 Hz, 2H), 6.44 (dd, J = 17.1, 10.3 Hz, 1H), 6.18 (dd, J = 17.1, 2.0 Hz, 1H), 5.68 (dd, J = 10.2, 2.0 Hz, 1H), 3.70 (d, J = 13.0 Hz, 2H), 3.47 (d, J = 12.0 Hz, 2H), 3.10 (t, J = 10.9 Hz, 2H),2.86 (d, J = 12.7 Hz, 2H), 2.81 (s, 3H), 2.24 (s, 3H). (M + H)+ calculated 597.24; found 596.89.
4-((2-Acrylamidophenyl)amino)-N-(4,5-dimethoxy-2-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15f).
Compound 15f was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.92 (s, 1H), 9.73 (s, 1H), 9.69 (s, 1H), 9.60 (s, 1H), 9.56 (s, 1H), 8.76 (s, 1H), 8.02 (s, 1H), 7.47 (s, 2H), 7.32 (d, J = 7.9 Hz, 1H), 7.19 (t, J =7.5 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.86–6.80 (m, 3H), 6.78 (s, 1H), 6.34 (dd, J = 17.1, 10.3 Hz, 1H), 6.10 (dd, J = 17.1, 2.0 Hz, 1H),5.59 (dd, J = 10.3, 2.0 Hz, 1H), 3.69 (s, 3H), 3.67 (s, 2H), 3.65 (s, 3H), 3.46 (d, J = 12.1 Hz, 2H), 3.11 (d, J = 10.6 Hz, 2H), 2.85 (d, J =12.6 Hz, 2H), 2.80 (d, J = 2.9 Hz, 3H), 2.08 (s, 3H). (M + H)+ calculated 623.30; found 622.99.
4-((2-Acrylamidophenyl)amino)-N-(2,6-dichloro-3,5-dimethoxyphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15g).
Compound 15g was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.59 (s, 1H), 10.01 (s, 1H), 9.67 (s, 3H), 8.81 (s, 1H), 7.93 (s, 1H), 7.44 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 7.9 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 6.88 (s, 1H), 6.83–6.78 (m, 2H), 6.27 (dd, J =17.1, 10.2 Hz, 1H), 6.05 (dd, J = 17.1, 1.9 Hz, 1H), 5.55 (dd, J = 10.2, 2.0 Hz, 1H), 3.89 (s, 6H), 3.68 (d, J = 13.2 Hz, 2H), 3.46 (d, J = 12.1 Hz, 2H), 3.16–3.05 (m, 2H), 2.84 (d, J = 12.3 Hz, 2H), 2.80 (s, 3H). (M + H)+ calculated 677.21; found 676.80.
4-((2-Acrylamidophenyl)amino)-N-(2-ethylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (15h).
Compound 15h was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.87 (s, 1H),9.73 (s, 1H), 9.72 (s, 1H), 9.70 (s, 1H), 9.61 (s, 1H), 8.76 (s, 1H),8.05 (s, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.27–7.21 (m, 1H), 7.23–7.12 (m, 4H), 7.08 (t, J = 7.7 Hz, 1H), 6.84 (d, J = 8.6 Hz, 2H), 6.31 (dd, J = 17.1, 10.3 Hz, 1H), 6.08 (dd, J = 17.1, 2.0 Hz, 1H), 5.59 (dd, J = 10.2, 2.0 Hz, 1H), 3.69 (d, J = 13.2 Hz, 2H),3.46 (d, J = 12.0 Hz, 2H), 3.11 (d, J = 11.1 Hz, 2H), 2.85 (d, J = 12.6 Hz, 2H), 2.80 (d, J = 2.9 Hz, 3H), 2.55 (q, J = 7.5 Hz, 2H), 1.07 (t, J = 7.5 Hz, 3H). (M + H)+ calculated 577.30; found 576.89.
4-((2-Acrylamidophenyl)amino)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)-N-(4-phenoxyphenyl)pyrimidine-5-carboxamide (15i).
Compound 15i was synthesized via the same route as compound 15a. 1H NMR (500 MHz, DMSO-d6) δ 10.89 (s, 1H),10.12 (s, 1H), 9.81 (s, 1H), 9.61 (brs, 2H), 8.82 (s, 1H), 7.78–7.65 (m, 2H), 7.62–7.49 (m, 2H), 7.47–7.36 (m, 3H), 7.27 (t, J = 7.8 Hz, 1H), 7.21–7.08 (m, 2H), 7.08–6.96 (m, 4H), 6.91 (d, J = 8.4 Hz, 2H), 6.48 (dd, J = 17.1, 10.2 Hz, 1H), 6.22 (dd, J = 17.2, 2.0 Hz, 1H),5.77–5.71 (m, 1H), 3.80–3.73 (m, 2H), 3.59–3.51 (m, 2H), 3.25–3.12 (m, 2H), 2.96–2.85 (m, 5H). (M + H)+ calculated 641.29; found 641.50.
4-((2-Acrylamidophenyl)amino)-2-chloro-N-(2-chloro-6-methylphenyl)pyrimidine-5-carboxamide (16).
To the solution of intermediate 13a (54.4 mg, 0.15 mmol) in NaHCO3 (aq. sat., 1 mL) and THF (1 mL) was added acryloyl chloride (12 μL, 0.15 mmol) under an ice bath. After 5 min, the intermediate was consumed, and the mixture was extracted with ethyl acetate. The organic layer was concentrated under vacuum, and the residue was purified with flash chromatography to yield the title product (66 mg, 0.15 mmol, quant.). (M + H)+ calculated 442.08; found 441.78.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((3-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (17a).
To the solution of intermediate 16 (20 mg, 0.045 mol) and 3-(4-methylpiperazin-1-yl)aniline (10 mg, 0.054 mmol) in s-BuOH (1 mL) was added TFA (10 μL, 0.068 mmol) at room temperature. The mixture was then heated up to 90 °C for 30 min. The mixture was cooled down to room temperature, and the crude residue was purified by HPLC to provide the title compound (23 mg,0.032 mmol, 72%). 1H NMR (500 MHz, DMSO-d6) δ 10.73 (s, 1H),9.88 (s, 1H), 9.72 (s, 1H), 9.67 (s, 1H), 9.61 (s, 1H), 8.86 (s, 1H),8.00 (s, 1H), 7.36–7.28 (m, 2H), 7.25–7.12 (m, 5H), 7.09 (td, J =7.6, 1.6 Hz, 1H), 7.04 (t, J = 8.1 Hz, 1H), 6.60 (d, J = 8.9 Hz, 1H),6.29 (dd, J = 17.1, 10.3 Hz, 1H), 6.07 (dd, J = 17.1, 2.0 Hz, 1H), 5.57 (dd, J = 10.2, 2.0 Hz, 1H), 3.55 (d, J = 13.3 Hz, 2H), 3.39 (d, J = 11.9 Hz, 2H), 3.06–3.00 (m, 2H), 2.84–2.76 (m, 5H), 2.16 (s, 3H). (M +H)+ calculated 597.24; found 597.29.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((2-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (17b).
Compound 17b was synthesized via the same route as compound 17a. 1H NMR (500 MHz, DMSO-d6) δ 10.74 (s, 1H),9.93 (s, 1H), 9.74 (s, 2H), 8.86 (s, 1H), 8.53 (s, 1H), 7.91 (dt, J = 7.9, 2.3 Hz, 2H), 7.34 (dd, J = 7.5, 2.0 Hz, 1H), 7.29 (dd, J = 7.8, 1.7 Hz, 1H), 7.26–7.18 (m, 2H), 7.17 (s, 1H), 7.15–7.09 (m, 1H), 7.07 (s, 1H), 7.01 (td, J = 7.6, 1.7 Hz, 1H), 6.99–6.92 (m, 1H), 6.36–6.21 (m, 1H), 6.06 (dd, J = 17.1, 1.9 Hz, 1H), 5.57 (dd, J = 10.2, 2.0 Hz, 1H), 3.44 (d, J = 12.6 Hz, 2H), 3.23 (s, 2H), 3.13–3.04 (m, 2H), 2.98–2.87 (m, 2H), 2.82 (s, 3H), 2.16 (s, 3H). (M + H)+ calculated 597.24; found 596.89.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-(4-methylpiperazin-1-yl)piperidin-1-yl)phenyl)amino)-pyrimidine-5-carboxamide (17c).
Compound 17c was synthesized via the same route as compound 17a. 1H NMR (500 MHz, DMSO-d6) δ 10.83 (s, 1H), 9.95 (s, 1H), 9.85 (s, 2H), 9.79 (s, 1H), 8.90 (s, 1H), 8.06 (s, 1H), 7.55 (s, 2H), 7.45–7.36 (m, 2H), 7.32–7.24 (m, 3H), 7.19 (t, J = 7.6 Hz, 1H), 7.02 (s, 2H), 6.36 (dd, J = 17.1, 10.3 Hz, 1H), 6.14 (dd, J = 17.1, 1.9 Hz, 1H), 5.64 (dd, J = 10.2, 2.0 Hz, 1H), 3.74 (d, J = 12.1 Hz, 2H), 3.55 (s, 4H), 3.15 (s, 4H), 2.83 (s, 5H), 2.24 (s, 3H), 2.13–2.06 (m, 2H), 1.78–1.72 (m, 2H). (M + H)+ calculated 680.31; found 680.39.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (17d).
Compound 17d was synthesized via the same route as compound 17a. 1H NMR (500 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.91 (s, 1H), 9.75 (s, 2H), 9.69 (s, 1H), 8.77 (s, 2H), 7.95 (s, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.33 (dd, J = 7.4, 2.1 Hz, 1H), 7.25–7.16 (m, 3H), 7.08 (s, 1H), 7.03 (t, J = 7.3 Hz, 1H), 6.66 (d, J = 2.5 Hz, 1H), 6.47–6.41 (m, 1H), 6.25 (dd, J = 17.1, 10.3 Hz, 1H), 6.05 (dd, J = 17.1, 1.9 Hz, 1H), 5.55 (dd, J = 10.2, 2.0 Hz, 1H), 3.82 (d, J = 13.2 Hz, 2H), 3.74 (s, 3H), 3.49 (d, J = 12.0 Hz, 2H), 3.12 (d, J =10.6 Hz, 2H), 2.90 (t, J = 12.4 Hz, 2H), 2.82 (d, J = 2.7 Hz, 3H), 2.15 (s, 3H). (M + H)+ calculated 627.25; found 627.49.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((2-methoxy-4-morpholinophenyl)amino)pyrimidine-5-carboxamide (17e).
Compound 17b was synthesized via the same route as compound 17a. 1H NMR (500 MHz, DMSO-d6) δ 11.04 (s, 1H),10.10 (s, 1H), 9.83 (s, 1H), 9.05 (s, 1H), 8.81 (s, 1H), 7.99 (s, 1H),7.41 (dd, J = 7.3, 2.2 Hz, 1H), 7.34–7.24 (m, 3H), 7.18 (s, 2H), 6.69 (d, J = 2.5 Hz, 1H), 6.47 (s, 1H), 6.33 (dd, J = 17.1, 10.2 Hz, 1H),6.13 (dd, J = 17.0, 2.0 Hz, 1H), 5.64 (dd, J = 10.1, 2.0 Hz, 1H), 3.81 (s, 3H), 3.77 (t, J = 4.8 Hz, 4H), 3.17 (t, J = 4.8 Hz, 4H), 2.23 (s, 3H). (M + H)+ calculated 614.22; found 614.39.
4-((2-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((3-methyl-4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (17f).
Compound 17f was synthesized via the same route as compound 17a. 1H NMR (500 MHz, DMSO-d6) δ 10.77 (s, 1H), 9.85 (s, 1H), 9.69 (s, 1H), 9.66 (s, 1H), 9.61 (s, 1H), 8.84 (s, 1H), 8.01 (s, 1H), 7.46 (d, J = 2.6 Hz, 1H), 7.37–7.29 (m, 3H), 7.26–7.14 (m, 3H), 7.09 (t, J = 7.6 Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H),6.28 (dd, J = 17.1, 10.2 Hz, 1H), 6.06 (dd, J = 17.1, 2.0 Hz, 1H), 5.56 (dd, J = 10.3, 2.0 Hz, 1H), 3.43 (d, J = 11.7 Hz, 2H), 3.19–3.11 (m, 2H), 3.08 (d, J = 13.6 Hz, 2H), 2.88–2.80 (m, 5H), 2.16 (s, 3H),2.11 (s, 3H). (M + H)+ calculated 611.26; found 611.39.
4-((3-Aminophenyl)amino)-2-chloro-N-(2-chloro-6-methylphenyl)pyrimidine-5-carboxamide (18a).
To the solution of intermediate 12a (28 mg, 0.089 mmol) and bezene-1,3-diamine (9.6 mg, 0.089 mmol) in n-BuOH (2 mL) was added DIEA (31 μL, 0.18 mmol) at room temperature. The mixture was then heated up to 90 °C for 1 h. The crude residue was purified with flash chromatography to yield the product (34 mg, 0.089 mmol, quant.). (M + H)+ calculated 388.07; found 388.20.
4-((3-Aminophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (19a).
To the solution of intermediate 18a (34 mg, 0.089 mmol) and 4-(4-methylpiperazin-1-yl)aniline (19 mg, 0.098 mmol) in s-BuOH (1 mL) was added TFA (10 μL, 0.13 mmol) at room temperature. The mixture was heated up to 90 °C for 2 h. The mixture was cooled down to room temperature and was evaporated under vacuum to yield the crude product without further purification. (M + H)+ calculated 543.23; found 543.42.
4-((3-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (20a).
To the solution of intermediate 19a (crude, 0.089 mmol) in NaHCO3 (sat. aq., 1 mL) and THF (1 mL) was added acryloyl chloride (9 mg, 0.098 mmol) under an ice bath. The reaction was extracted with ethyl acetate after all 19a was consumed. The crude residue was purified by HPLC to provide the title compound as a white solid (21 mg, 0.034 mmol, 38% over 2 steps). 1H NMR (500 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.16 (s, 1H), 10.04 (s, 1H), 9.72 (s, 2H), 8.95 (s, 1H), 7.75 (s, 1H), 7.70–7.40 (s, 1H) 7.59 (d, J = 8.4 Hz, 2H), 7.43 (dd, J = 7.7, 1.9 Hz, 2H), 7.35–7.26 (m, 3H), 6.91 (s, 2H), 6.44 (dd, J = 16.9, 10.1 Hz, 1H), 6.26 (dd, J = 17.0, 2.0 Hz, 1H),5.77 (dd, J = 10.1, 2.0 Hz, 1H), 3.75–3.70 (m, 2H), 3.53 (d, J = 12.1 Hz, 2H), 3.23–3.14 (m, 2H), 2.94–2.86 (m, 5H), 2.28 (s, 3H). (M +H)+ calculated 597.24; found 597.39.
4-((4-Acrylamidophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (20b).
Compound 20b was synthesized via the same route as compound 20a. 1H NMR (500 MHz, DMSO-d6) δ 11.09 (s, 1H),10.17 (s, 1H), 10.02 (s, 1H), 9.74 (s, 2H), 8.93 (s, 1H), 7.73–7.49 (m, 6H), 7.42 (dd, J = 7.7, 1.8 Hz, 1H), 7.35–7.25 (m, 2H), 6.97 (d, J = 8.7 Hz, 2H), 6.45 (dd, J = 16.9, 10.1 Hz, 1H), 6.27 (dd, J = 16.9, 2.1 Hz, 1H), 5.77 (dd, J = 10.0, 2.1 Hz, 1H), 3.80 (d, J = 13.3 Hz, 2H), 3.57–3.51 (m, 2H), 3.20 (d, J = 10.4 Hz, 2H), 2.95 (t, J = 12.5 Hz, 2H), 2.89 (s, 3H), 2.27 (s, 3H). (M + H)+ calculated 597.24; found 597.29.
N-(2-Chloro-6-methylphenyl)-4-((2-(2-chloroacetamido)phenyl)-amino)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidine-5-carboxamide (20c).
4-((2-Aminophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)-pyrimidine-5-carboxamide (18 mg, 0.040 mmol) was dissolved in THF (1 mL) and sat. aqueous NaHCO3 (1 mL). Chloroacetyl chloride dissolved in THF was slowly titrated at 0 °C until the starting aniline was consumed. Water was added followed by extraction with EtOAc. Combined extracts were washed with brine, dried over Na2SO4, concentrated, and purified by HPLC to provide the title compound as a white solid (4.4 mg, 0.006 mmol, 15%). 1H NMR (500 MHz, DMSO-d6) δ 11.02 (s, 1H), 9.99 (s, 1H), 9.90 (s, 1H),9.77 (s, 2H), 8.93 (s, 1H), 8.15 (s, 1H), 7.55–7.51 (m, 2H), 7.42 (dd, J = 7.7, 1.8 Hz, 1H), 7.35–7.25 (m, 4H), 7.16 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 8.5 Hz, 2H), 4.15 (s, 2H), 3.81–3.74 (m, 2H), 3.54 (d, J = 12.1 Hz, 2H), 3.19 (s, 2H), 2.97–2.86 (m, 5H), 2.27 (s, 3H). (M + H)+ calculated 619.20; found 619.19.
N-(2-Chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)-4-((2-(vinylsulfonamido)phenyl)amino)pyrimidine-5-carboxamide (20d).
4-((2-Aminophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)-pyrimidine-5-carboxamide (23 mg, 0.042 mmol) was dissolved in MeCN (1 mL) and DMF (1 mL) and treated with Et3N (23 μL, 0.17 mmol). Then, 2-chloroethanesulfonyl chloride (9.2 mg, 0.056 mmol) in a small amount of MeCN was added at 0 °C. After stirring for 30 min, water was added, followed by extraction with CHCl3/IPA (3:1). Combined extracts were washed with brine, concentrated, and purified by HPLC to provide the title compound as a white solid (3.5 mg, 0.0055 mmol, 13%). 1H NMR (500 MHz, DMSO-d6) δ11.15 (s, 1H), 9.98 (s, 1H), 9.84 (s, 1H), 9.71 (s, 1H), 9.42 (s, 1H),8.93 (s, 1H), 8.24 (s, 1H), 7.52 (s, 2H), 7.43 (dd, J = 7.9, 1.7 Hz, 1H), 7.35–7.26 (m, 3H), 7.24 (dd, J = 8.0, 1.6 Hz, 1H), 7.11 (t, J =7.4 Hz, 1H), 6.93 (d, J = 8.5 Hz, 2H), 6.77 (dd, J = 16.5, 9.9 Hz, 1H),5.88 (d, J = 2.1 Hz, 1H), 5.86 (d, J = 4.8 Hz, 1H), 3.77 (d, J = 13.4 Hz, 2H), 3.53 (d, J = 12.2 Hz, 2H), 3.21–3.10 (m, 2H), 2.98–2.89 (m, 2H), 2.87 (s, 3H), 2.30 (s, 3H). (M + H)+ calculated 633.20; found 633.20.
(E)-N-(2-Chloro-6-methylphenyl)-4-((2-(4-(dimethylamino)but-2-enamido)phenyl)amino)-2-((4-(4-methylpiperazin-1-yl)phenyl)-amino)pyrimidine-5-carboxamide (20e).
4-((2-Aminophenyl)-amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)pyrimidine-5-carboxamide (23 mg, 0.042 mmol) was dissolved in THF (1 mL) and sat. aqueous NaHCO3 (1 mL). Then, 4-bromocrotonyl chloride in THF was slowly titrated until the starting aniline was consumed. Water was added, followed by extraction with CHCl3/IPA (3:1). Combined extracts were washed with brine, dried over Na2SO4, and concentrated. The residue was then dissolved in 1 mL of MeCN and 1 mL of DMF. At 50 °C, dimethylamine solution (100 μL, 2.0 M in THF) was added. After stirring for 30 min, the mixture was diluted with water, followed by extraction with CHCl3/IPA (3:1). Combined extracts were washed with brine, dried over Na2SO4, concentrated, and purified by HPLC to provide the title compound as a white solid (2.0 mg, 2.6 μmol, 6% over 2 steps). 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.98 (s, 1H), 9.96 (s, 1H), 9.94 (s, 1H), 9.70 (s, 1H), 8.90 (s, 1H), 8.20 (s, 1H), 7.55 (s, 2H), 7.42 (dd, J = 7.6, 2.0 Hz, 1H), 7.34–7.25 (m, 4H), 7.15 (t, J =7.7 Hz, 1H), 6.93 (d, J = 8.6 Hz, 2H), 6.67–6.57 (m, 1H), 6.40 (d, J = 15.3 Hz, 1H), 3.82 (d, J = 7.0 Hz, 2H), 3.76 (d, J = 12.9 Hz, 2H),3.55–3.52 (m, 2H), 3.18 (s, 2H), 2.98–2.89 (m, 2H), 2.88 (s, 3H),2.66 (s, 6H), 2.24 (s, 3H). (M + H)+ calculated 654.30; found 654.40.
N-(2-Chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)-phenyl)amino)-4-((2-propionamidophenyl)amino)pyrimidine-5-carboxamide (20f).
4-((2-Aminophenyl)amino)-N-(2-chloro-6-methylphenyl)-2-((4-(4-methylpiperazin-1-yl)phenyl)amino)-pyrimidine-5-carboxamide was dissolved in THF (1 mL) and sat. aqueous NaHCO3 (1 mL). Then, propionyl chloride in THF was slowly titrated until the starting aniline was consumed. Water was added, followed by extraction with EtOAc. Combined extracts were washed with brine, dried over Na2SO4, concentrated, and purified by HPLC to provide the title compound as a white solid (13 mg, 0.021 mmol, 19%). 1H NMR (500 MHz, DMSO-d6) δ 10.92 (s, 1H), 9.86 (s, 1H), 9.64 (s, 2H), 9.39 (s, 1H), 8.84 (s, 1H), 8.30–8.00 (s, 1H),7.48 (d, J = 8.2 Hz, 2H), 7.34 (dd, J = 7.6, 1.8 Hz, 1H), 7.27–7.11 (m, 4H), 7.03 (t, J = 7.6 Hz, 1H), 6.86 (d, J = 8.4 Hz, 2H), 3.70 (d, J = 13.2 Hz, 2H), 3.47 (d, J = 12.1 Hz, 2H), 3.12 (d, J = 11.2 Hz, 2H), 2.89–2.79 (m, 5H), 2.18 (s, 3H), 2.14 (q, J = 7.6 Hz, 2H), 0.87 (t, J =7.6 Hz, 3H). (M + H)+ calculated 599.26; found 598.89.
Cell Culture.
In this study, three cell lines were used including H1975 and HCC827 (nonsmall cell lung cancer, NSCLC) and MDAMB-231 (triple negative breast cancer, TNBC) cells. The cell lines were grown, maintained, and authenticated according to the methodology previously described.51
Growth Inhibition Assay.
For the growth inhibition assay, cells were plated in 384-well plates (3764, Corning) (2000 cells/well) using the Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific). The next day, cells were treated with varying doses of 15a, 20f, AZD0530 (saracatinib), or DMSO (0.2%) using the D300 digital dispenser (HP-D300 Digital Dispenser). The treatment period lasted 72 h, followed by the addition of 25 μL/well of CellTiter-Glo reagent (G7572, Promega). Cell viability was measured using the Synergy H1 microplate reader (Biotek), and analysis was carried out as previously described.51 The GR values were calculated using the online GR calculator (http://www.grcalculator.org/grtutorial/Home.html) according to the methodology recently described.40,45
Western Blotting.
For cellular target engagement and signaling analyses, H1975 and HCC827 NSCLC cells were treated with 1 μM of 15a, 20f, or DMSO for 2 h, followed by rinsing cells twice with cold PBS, extracting, and lysing with cold M-PER Mammalian Protein Extraction Reagent (78505, Thermo Fisher Scientific) substituted with 1× HALT protease and phosphatase inhibitor (78446, Thermo Fisher Scientific). In the case of the washout assay, drug-containing medium was replaced with drug-free medium and cells were lysed at 0, 2, or 4 h postwashout. To prepare samples for signaling analysis, a small volume of the lysate obtained was combined with loading buffer [5% v/v 2-mercaptoethanol (Sigma-Aldrich) + 4× Laemmli Sample Buffer (161–0747 Bio-Rad)]. To prepare samples for cellular target engagement, lysates were incubated with 1 μM TL13–68 with constant rocking at 4 °C overnight, followed by pulldown using streptavidin beads (20353, Thermo Fisher Scientific). Kinases pulled down on beads were then rigorously washed (5 times) using cold M-PER lysis buffer. Excess liquid was removed from the beads after washes and diluted using loading buffer. For both signaling analysis and target engagement, samples were assessed using Western blotting according to the methodology previously described.38,51 Kinases of interest were detected using the following antibodies from Cell Signaling Technology: phospho-SRCY416 (6943S, rabbit), SRC (2123S, rabbit), YES1 (65890S, rabbit), MEK1/2 (9126S, rabbit), and ERK1/2 (4696S, mouse). The β-actin antibody was purchased from Santa Cruz (sc-47778, mouse). Signaling and target engagement experiments were repeated twice, and blots from each independent experiment are presented in Figure S2.
Pharmacodynamic Analysis.
All mouse procedures were reviewed and approved by the Dana-Farber Animal Care and Use Committee (Protocol number: 16–015) and carried out in the pathogen-free animal facilities in Dana-Farber Cancer Institute (Boston, MA). Adult C57B6 mice were purchased from Jackson Laboratory (#000058, Bar Harbor, ME) and dosed by vehicle (5% DMSO, 95% D5W) or 15a (5 MPK) for 3 times every 12 h via intraperitoneal injection. Mouse spleens were harvested 2 or 4 h after the last dose.
Spleen tissue harvested from mice were lysed and homogenized using the M-PER Mammalian Protein Extraction Reagent (78505, Thermo Fisher Scientific) substituted with 1× HALT protease and phosphatase inhibitor (78446, Thermo Fisher Scientific). Sample preparation for both signaling and target engagement analyses was performed similar to the above cellular assays. Target inhibition was assessed using Western blotting according to the methodology described in Rao et al.38,51 Antibodies used in the study included phospho-SRCY416 (6943S, rabbit) and SRC (2123S, rabbit) from Cell Signaling Technologies and β-actin antibody from Santa Cruz (sc-47778, mouse).
Molecular Docking.
Docking of SM1–71 was performed with the Glide covalent docking function (version 2019 release 1). It was docked into the FGFR1 cocrystal structure (PDB code: 6MZW), and the Cys488 at the P-loop was chosen as the reactive residue. Hinge hydrogen bonds shown in Figure 1 were used as a constraint to ensure the proper binding mode. Default values were used for other parameters.
Mass Spectrometry.
Src protein (10 μg) was treated with DMSO or a 10-fold molar excess of DGY-06–116 for 2 h at 37 °C and analyzed by LC-MS using an HPLC (Shimadzu, Marlborough, MA) interfaced to an LTQ ion trap mass spectrometer (ThermoFisher Scientific, San Jose, CA). Protein (5 μg) was injected onto a reversed phase column (5 cm POROS 50R2, Applied Biosystems, Foster City, CA), desalted for 4 min (100% A), and gradient eluted (0–100% B in 1 min; A = 0.2 M acetic acid in water; B = 0.2 M acetic acid in acetonitrile) into the mass spectrometer (spray voltage = 4.8 kV). The mass spectrometer was programmed to acquire profile mass spectra (m/z 300–2000). Raw data was deconvoluted using MagTran version1.03b2.38 To determine which amino acid was modified by the inhibitor, labeled protein was first reduced (10 mM dithiothreitol for 30 min at 56 °C), alkylated (22.5 mM iodoacetamide for 30 min at room temperature and protected from light), and digested with trypsin. Peptides were then desalted by C18, dried by vacuum centrifugation, reconstituted in 50% MeCN, 1% formic acid, and 100 mM ammonium acetate, and analyzed by CE-MS using a ZipChip autosampler and CE system (908 Devices, Boston, MA) interfaced to a QE-HF mass spectrometer (Thermofisher Scientific). The mass spectrometer was operated in data-dependent mode, and the 5 most abundant ions in each MS scan (m/z 300–2000, resolution = 60K, target = 3 × 106, max fill time = 50 ms) were subjected to MS/MS (collision energy = 35%, target = 1 × 105, max fill time = 100 ms). Dynamic exclusion was enabled with a repeat count of 1 and an exclusion duration of 6 s. Raw data were converted to .mgf using the multiplierz toolset39 and searched using Mascot 2.6.1 against a forward reversed human refseq database (NCBI). Search parameters specified a precursor mass tolerance of 50 ppm, a product ion tolerance of 25 mmu, fixed carbamidomethylation of cysteine, and variable oxidation of methionine as well as variable DGY-06–116 modification of cysteine. Inhibitor related fragment ions were assigned as described.40
Supplementary Material
ACKNOWLEDGMENTS
We thank Jim Sun at the NMR facility of Dana-Farber cancer institute for his assistance on 1H NMR and 13C NMR data collection. Milka Kostic is greatly acknowledged for the editing and proof reading. This work was supported by Welch I-1829, ACS RSG-18-039-01-DMC, and a Career Enhancement Grant through NIH P50CA07090720 to K.D.W.
ABBREVIATIONS USED
- TCIs
targeted covalent inhibitors
- NSCLC
nonsmall cell lung cancer
- SFKs
SRC family kinases
- GPCRs
G protein coupled receptors
- RTKs
receptor tyrosine kinases
- CKI
covalent kinase inhibitor
- GR
growth rate
- DIEA
N,N-diisopropylethylamine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, hexa-fluorophosphate azabenzotriazole tetramethyl uronium
- Et3N
triethylamine
- DMF
dimethylformamide
- n-BuOH
n-butanol
- s-BuOH
s-butanol
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01502.
Western blot analysis characterizing target engagement and signaling inhibition in cells for compounds AZD0530, 15a, and 20f (Figure S1); structure of TL13–68 (Figure S2); KinomeScan profiling data for compound SM1–71 (Table S1); KinomeScan profiling data for compound 15a (Table S2); IC50’s of top off-targets for compound 15a (Table S3); compound 15a in a SRC-TEL and HCK-BCR Ba/F3 cell-based assay (Table S4); GR50 and GRmax values from a growth inhibitory screen across multiple cancer cell lines for compounds AZD0530, 15a, and 20f (Table S5) (PDF) Molecular formula strings (CSV)
Growth inhibtion assay for compounds AZD0530, 15a, and 20f (XLSX)
The authors declare the following competing financial interest(s): N.S.G. is a founder, SAB, and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives/has received funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield, and Sanofi. A.J.A. has consulted for Oncorus, Inc. and Merck & Co., Inc. and has research funding from Mirati Therapeutics and Deerfield. G.D., N.J.H., S.R., D.G., T.Z., K.D.W., and N.S.G. are inventors on SRC covalent inhibitor patents.
Contributor Information
Guangyan Du, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Cancer Biology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Suman Rao, Department of Biological Chemistry and Molecular Pharmacology and Laboratory of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Cancer Biology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Deepak Gurbani, Departments of Biochemistry and Radiation Oncology, The University of Texas Southwestern Medical Center at Dallas, Dallas, Texas 75390, United States.
Jianwei Che, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Cancer Biology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Annan Yang, Department of Medical Oncology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Jarrod A. Marto, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States
Andrew J. Aguirre, Department of Medical Oncology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States
Peter K. Sorger, Laboratory of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, United States
Kenneth D. Westover, Departments of Biochemistry and Radiation Oncology, The University of Texas Southwestern Medical Center at Dallas, Dallas, Texas 75390, United States.
Nathanael S. Gray, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States; Department of Cancer Biology, Dana Farber Cancer Institute, Boston, Massachusetts 02215, United States.
REFERENCES
- (1).Sen B; Johnson FM Regulation of SRC family kinases in human cancers. J. Signal Transduction 2011, 2011, 865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wheeler DL; Iida M; Dunn EF The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Irby RB; Yeatman TJ Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636. [DOI] [PubMed] [Google Scholar]
- (4).Thomas SM; Brugge JS Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol 1997, 13, 513–609. [DOI] [PubMed] [Google Scholar]
- (5).Finn R Targeting Src in breast cancer. Annals of Oncology 2008, 19, 1379–1386. [DOI] [PubMed] [Google Scholar]
- (6).Dehm SM; Bonham K SRC gene expression in human cancer: the role of transcriptional activation. Biochem. Cell Biol 2004, 82, 263–274. [DOI] [PubMed] [Google Scholar]
- (7).Lu Y; Yu Q; Liu JH; Zhang J; Wang H; Koul D; McMurray JS; Fang X; Yung WA; Siminovitch KA; et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem 2003, 278, 40057–40066. [DOI] [PubMed] [Google Scholar]
- (8).Cantley LC The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [DOI] [PubMed] [Google Scholar]
- (9).Lai SY; Johnson FM Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 67–78. [DOI] [PubMed] [Google Scholar]
- (10).Maa M-C; Leu T-H; Trandel BJ; Chang J-H; Parsons SJ A protein that is highly related to GTPase-activating protein-associated p62 complexes with phospholipase C gamma. Mol. Cell. Biol 1994, 14, 5466–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ishizawar R; Miyake T; Parsons S c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene 2007, 26, 3503. [DOI] [PubMed] [Google Scholar]
- (12).López-Ocejo O; Viloria-Petit A; Bequet-Romero M; Mukhopadhyay D; Rak J; Kerbel RS Oncogenes and tumor angiogenesis: the HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611. [DOI] [PubMed] [Google Scholar]
- (13).Zhang S; Yu D Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol. Sci 2012, 33, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Boland JM; Erdogan S; Vasmatzis G; Yang P; Tillmans LS; Johnson MRE; Wang X; Peterson LM; Halling KC; Oliveira AM; et al. Anaplastic lymphoma kinase immunoreactivity correlates with ALK gene rearrangement and transcriptional up-regulation in non–small cell lung carcinomas. Hum. Pathol 2009, 40, 1152–1158. [DOI] [PubMed] [Google Scholar]
- (15).Muthuswamy SK Trastuzumab resistance: all roads lead to SRC. Nat. Med 2011, 17, 416. [DOI] [PubMed] [Google Scholar]
- (16).Yoshida R; Sasaki T; Minami Y; Hibino Y; Okumura S; Sado M; Miyokawa N; Hayashi S; Kitada M; Ohsaki Y Activation of Src signaling mediates acquired resistance to ALK inhibition in lung cancer. Int. J. Oncol 2017, 51, 1533–1540. [DOI] [PubMed] [Google Scholar]
- (17).Das J; Chen P; Norris D; Padmanabha R; Lin J; Moquin RV; Shen Z; Cook LS; Doweyko AM; Pitt S; et al. 2-Aminothiazole as a novel kinase inhibitor template. Structure– activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl] amino)]-1, 3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J. Med. Chem 2006, 49, 6819–6832. [DOI] [PubMed] [Google Scholar]
- (18).Boschelli DH; Ye F; Wang YD; Dutia M; Johnson SL; Wu B; Miller K; Powell DW; Yaczko D; Young M; et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J. Med. Chem 2001, 44, 3965–3977. [DOI] [PubMed] [Google Scholar]
- (19).Puls LN; Eadens M; Messersmith W Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brandvold KR; Steffey ME; Fox CC; Soellner MB Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol 2012, 7, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liang X; Liu X; Wang B; Zou F; Wang A; Qi S; Chen C; Zhao Z; Wang W; Qi Z; et al. Discovery of 2-((3-Amino-4-methylphenyl) amino)-N-(2-methyl-5-(3-(trifluoromethyl) benzamido) phenyl)-4-(methylamino) pyrimidine-5-carboxamide (CHMFLABL-053) as a Potent, Selective, and Orally Available BCR-ABL/SRC/p38 Kinase Inhibitor for Chronic Myeloid Leukemia. J. Med. Chem 2016, 59, 1984–2004. [DOI] [PubMed] [Google Scholar]
- (22).Zhang C-H; Zheng M-W; Li Y-P; Lin X-D; Huang M; Zhong L; Li G-B; Zhang R-J; Lin W-T; Jiao Y; et al. Design, synthesis, and structure–activity relationship studies of 3-(Phenylethynyl)-1 H-pyrazolo [3, 4-d] pyrimidin-4-amine derivatives as a new class of Src inhibitors with potent activities in models of triple negative breast cancer. J. Med. Chem 2015, 58, 3957–3974. [DOI] [PubMed] [Google Scholar]
- (23).Fraser C; Dawson JC; Dowling R; Houston DR; Weiss JT; Munro AF; Muir M; Harrington L; Webster SP; Frame MC; et al. Rapid discovery and structure–activity relationships of pyrazolopyrimidines that potently suppress breast cancer cell growth via SRC kinase inhibition with exceptional selectivity over ABL kinase. J. Med. Chem 2016, 59, 4697–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tintori C; Fallacara AL; Radi M; Zamperini C; Dreassi E; Crespan E; Maga G; Schenone S; Musumeci F; Brullo C; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo [3, 4-d] pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem 2015, 58, 347–361. [DOI] [PubMed] [Google Scholar]
- (25).Huang W-S; Zhu X; Wang Y; Azam M; Wen D; Sundaramoorthi R; Thomas RM; Liu S; Banda G; Lentini SP; et al. 9-(Arenethenyl) purines as dual Src/Abl kinase inhibitors targeting the inactive conformation: design, synthesis, and biological evaluation. J. Med. Chem 2009, 52, 4743–4756. [DOI] [PubMed] [Google Scholar]
- (26).Kitagawa D; Yokota K; Gouda M; Narumi Y; Ohmoto H; Nishiwaki E; Akita K; Kirii Y Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes to cells 2013, 18, 110–122. [DOI] [PubMed] [Google Scholar]
- (27).Rix U; Hantschel O; Dürnberger G; Rix LLR; Planyavsky M; Fernbach NV; Kaupe I; Bennett KL; Valent P; Colinge J; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [DOI] [PubMed] [Google Scholar]
- (28).Remsing Rix LL; Rix U; Colinge J; Hantschel O; Bennett KL; Stranzl T; Muller A; Baumgartner C; Valent P; Augustin M; Till JH; Superti-Furga G Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 2009, 23, 477. [DOI] [PubMed] [Google Scholar]
- (29).Keller-von Amsberg G; Koschmieder S Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. OncoTargets Ther. 2013, 6, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hennequin LF; Allen J; Breed J; Curwen J; Fennell M; Green TP; Lambert-van der Brempt C; Morgentin R; Norman RA; Olivier A; et al. N-(5-Chloro-1, 3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl) ethoxy]-5-(tetrahydro-2 H-pyran-4-yloxy) quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem 2006, 49, 6465–6488. [DOI] [PubMed] [Google Scholar]
- (31).Ferguson FM; Gray NS Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 2018, 17, 353. [DOI] [PubMed] [Google Scholar]
- (32).Roskoski R Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res 2019, 144, 19. [DOI] [PubMed] [Google Scholar]
- (33).Liu Q; Sabnis Y; Zhao Z; Zhang T; Buhrlage SJ; Jones LH; Gray NS Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tan L; Wang J; Tanizaki J; Huang Z; Aref AR; Rusan M; Zhu S-J; Zhang Y; Ercan D; Liao RG; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E4869–E4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kwarcinski FE; Fox CC; Steffey ME; Soellner MB Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol 2012, 7, 1910–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tan L; Gurbani D; Weisberg EL; Hunter JC; Li L; Jones DS; Ficarro SB; Mowafy S; Tam C-P; Rao S; et al. Structure-guided development of covalent TAK1 inhibitors. Bioorg. Med. Chem 2017, 25, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tan L; Gurbani D; Weisberg EL; Jones DS; Rao S; Singer WD; Bernard FM; Mowafy S; Jenney A; Du G; et al. Studies of TAK1-centered polypharmacology with novel covalent TAK1 inhibitors. Bioorg. Med. Chem 2017, 25, 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rao S; Gurbani D; Du G; Everley RA; Browne CM; Chaikuad A; Tan L; Schröder M; Gondi S; Ficarro SB; et al. Leveraging compound promiscuity to identify targetable cysteines within the kinome. Cell chemical biology 2019, 26, 818–829.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhu K; Borrelli KW; Greenwood JR; Day T; Abel R; Farid RS; Harder E Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J. Chem. Inf. Model 2014, 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- (40).Clark NA; Hafner M; Kouril M; Williams EH; Muhlich JL; Pilarczyk M; Niepel M; Sorger PK; Medvedovic M GRcalculator: an online tool for calculating and mining dose– response data. BMC Cancer 2017, 17, 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sánchez-Bailón MP; Calcabrini A; Gómez-Domínguez D; Morte B; Martín-Forero E; Goómez-López G; Molinari A; Wagner K-U; Martín-Pérez J Src kinases catalytic activity regulates proliferation, migration and invasiveness of MDA-MB-231 breast cancer cells. Cell. Signalling 2012, 24, 1276–1286. [DOI] [PubMed] [Google Scholar]
- (42).Formisano L; D’Amato V; Servetto A; Brillante S; Raimondo L; Di Mauro C; Marciano R; Orsini RC; Cosconati S; Randazzo A; et al. Src inhibitors act through different mechanisms in Non-Small Cell Lung Cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Giaccone G; Zucali P Src as a potential therapeutic target in non-small-cell lung cancer. Annals of Oncology 2008, 19, 1219–1223. [DOI] [PubMed] [Google Scholar]
- (44).Green TP; Fennell M; Whittaker R; Curwen J; Jacobs V; Allen J; Logie A; Hargreaves J; Hickinson DM; Wilkinson RW; et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol. Oncol 2009, 3, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hafner M; Niepel M; Chung M; Sorger PK Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang J; Kalyankrishna S; Wislez M; Thilaganathan N; Saigal B; Wei W; Ma L; Wistuba II; Johnson FM; Kurie JM SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol 2007, 170, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ficarro SB; Browne CM; Card JD; Alexander WM; Zhang T; Park E; McNally R; Dhe-Paganon S; Seo H-S; Lamberto I; et al. Leveraging gas-phase fragmentation pathways for improved identification and selective detection of targets modified by covalent probes. Anal. Chem 2016, 88, 12248–12254. [DOI] [PubMed] [Google Scholar]
- (48).Papaetis GS; Syrigos KN Sunitinib. BioDrugs 2009, 23, 377–389. [DOI] [PubMed] [Google Scholar]
- (49).Wilhelm S; Carter C; Lynch M; Lowinger T; Dumas J; Smith RA; Schwartz B; Simantov R; Kelley S Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery 2006, 5, 835. [DOI] [PubMed] [Google Scholar]
- (50).Lee SJ; Wang JY Exploiting the promiscuity of imatinib. J. Biol 2009, 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Rao S; Du G; Hafner M; Subramanian K; Sorger PK; Gray NS A multi-targeted probe-based strategy to identify signaling vulnerabilities in cancers. J. Biol. Chem 2019, 294, 8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.