

Abstract
Cervical nerve root injury induces a host of inflammatory mediators in the spinal cord that initiate and maintain neuronal hyperexcitability and pain. Secretory phospholipase A2 (sPLA2) is an enzyme that has been implicated as a mediator of pain onset and maintenance in inflammation and neural injury. Although sPLA2 modulates nociception and excitatory neuronal signaling in vitro, its effects on neuronal activity and central sensitization early after painful nerve root injury are unknown. This study investigated whether inhibiting spinal sPLA2 at the time of nerve root compression modulates the pain, dorsal horn hyperexcitability, and spinal genes involved in glutamate signaling, nociception, and inflammation that are seen early after injury. Rats underwent a painful C7 nerve root compression injury with immediate intrathecal administration of the sPLA2 inhibitor thioetheramide-PC (TEA-PC). Additional groups underwent either injury alone or a sham surgery. One day after injury, behavioral sensitivity, spinal neuronal excitability, and spinal cord gene expression for glutamate receptors (mGluR5, NR1) and transporters (GLT1, EAAC1), the neuropeptide substance P, and pro-inflammatory cytokines (TNFα, IL1α, IL1β) were assessed. Treatment with the sPLA2 inhibitor prevented mechanical allodynia, attenuated neuronal hyperexcitability in the spinal dorsal horn, restored the proportion of spinal neurons classified as wide dynamic range, and reduced genes for mGluR5, substance P, IL1α, and IL1β to sham levels. These findings indicate spinal regulation of central sensitization after painful neuropathy and suggest that spinal sPLA2 is implicated in those early spinal mechanisms of neuronal, perhaps via glutamate signaling, neurotransmitters, and/or inflammatory cascades.
Keywords: radiculopathy, injury, neuronal hyperexcitability, sPLA2, inflammation, pain
INTRODUCTION
The cervical nerve root is a common source of painful neuropathy resulting from neck trauma and/or disc disease [1]. Nerve root compression induces a host of inflammatory mediators in the spinal cord that initiate and maintain neuronal hyperexcitability and pain [2].. Spinal glutamate signaling has a role in synaptic plasticity and is altered after painful nerve root compression in the rat, with increased expression of the metabotropic glutamate receptor mGluR5 and decreased astrocytic glutamate transporter GLT-1 expression [3;4]. Although spinal hyperexcitability has been reported at later times after nerve root injury when pain persists and is attenuated by pain-relieving neuromodulatory drugs [4], it is unknown if spinal hyperexcitability is established early after painful injury paralleling when behavioral sensitivity, spinal cytokine upregulation, and spinal microglial proliferation are all evident [2;5].
The enzyme phospholipase A2 (PLA2) hydrolyzes the sn-2 position of membrane glycerophospholipids, which catalyzes the release of arachidonic acid from cell membrane phospholipids and initiates the synthesis of the inflammatory agents [6]. Secretory PLA2 (sPLA2) has been implicated as an early mediator of pain from inflammation and neural injury and is upregulated in the spinal cord as early as 4 hours after spinal cord injury in the rat [6]. Injecting sPLA2 in the rat induces mechanical allodynia and activates spinal astrocytes and microglia within hours and increases TNFα expression [7;8]; its inhibition prevents hyperalgesia and attenuates inflammatory responses [9]. Beyond inducing inflammation, sPLA2 potentiates excitatory glutamate signaling and is cytotoxic in neuronal culture [6;10]. Although sPLA2 is implicated in neuronal excitotoxicity and excitatory signaling in vitro [10], the mechanisms through which sPLA2 modulates spinal neuronal activity after painful root injury are unknown.
Since sPLA2 modulates behavioral sensitivity [7;9], inflammatory cascades implicated in central sensitization [6;7;9;11], and neuronal excitability [9;10], this study investigated whether inhibiting spinal sPLA2 at the time of a painful root injury can prevent pain and central sensitization that occur early after injury. The effects of sPLA2 inhibition on mechanical hyperalgesia, dorsal horn neuron excitability, and spinal gene expression were evaluated one day after \ root compression.
METHODS
All experimental procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and carried out under the guidelines of the International Association for the Study of Pain [12]. Male Holtzman rats (306–462g) were housed under USDA- and AAALAC-compliant conditions.
Compression of the right C7 nerve root was performed with rats under inhalation isoflurane anesthesia (Fig. 1A) [4;13]. A C6/C7 hemilaminectomy were performed on the right side. The C7 dorsal nerve root was exposed and compressed with a 10-gf microvascular clip (World Precision Instruments; Sarasota, FL, USA) to apply a nerve root compression (NRC) injury for 15-mins [13]. Immediately after compression, the sPLA2 inhibitor thioetheramide-PC (0.25mg/mL; TEA-PC; Cayman Chemical Co., Ann Abor, MI, USA) in phosphate-buffered saline (PBS; 40–60μL) was administered by lumbar puncture (NRC+sPLA2 inh; n=11) at a dose known to reduce pain [11;14]. Additional groups of rats underwent compression only (NRC; n=11) or a sham surgery (sham; n=10) where the nerve root was exposed but not compressed.
Fig. 1. Schematic of the experimental protocol.
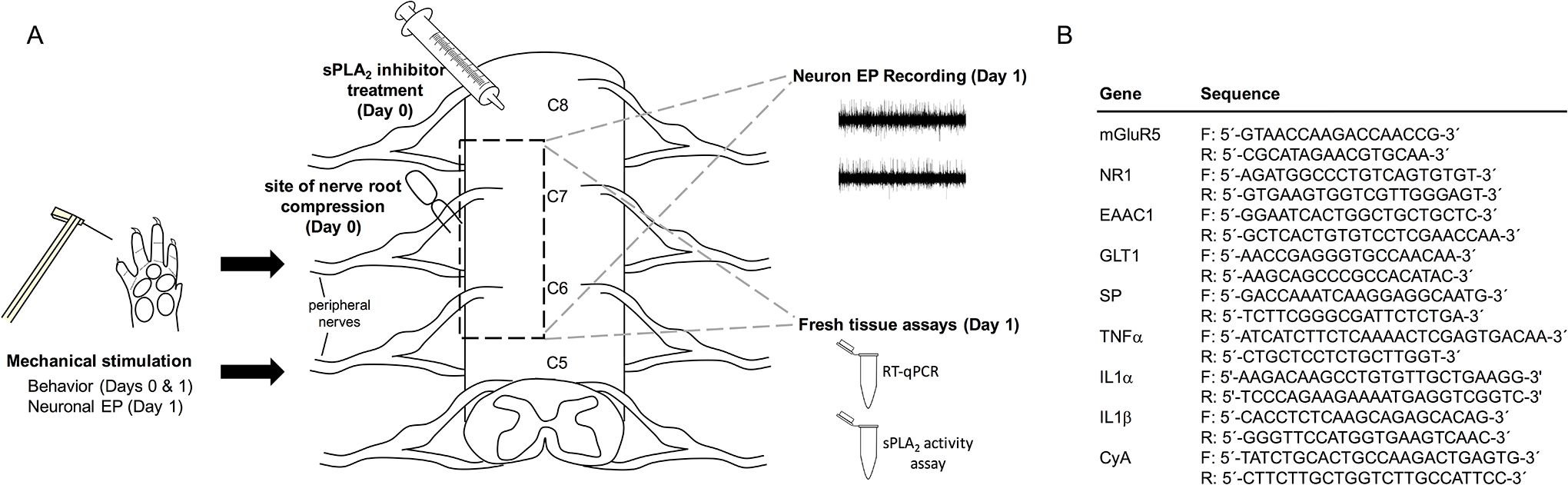
(A) Rats underwent a right C7 nerve root compression immediately followed by intrathecal treatment with an sPLA2 inhibitor, no treatment, or a sham surgery. Behavioral testing was performed on the ipsilateral forepaw on days 0 and 1 and electrophysiology recording of the ipsilateral C6 or C7 spinal cord during mechanical stimulation of the ipsilateral forepaw was performed on day 1. C6 spinal cord tissue was used for either RT-qPCR or sPLA2 activity analysis. (B) Genes for glutamate receptors and transporters, substance P (SP) and pro-inflammatory cytokines were assayed using published primer sequences [2;15–18].
Mechanical hyperalgesia was measured in the right forepaw both before (baseline; day 0) and one day after surgery (Fig. 1A). The right forepaw was stimulated with von Frey filaments of increasing strengths (1.4g-26g) as previously described [13] to obtain the lowest strength filament to elicit a response (paw withdrawal threshold; PWT). A repeated-measures ANOVA with post-hoc Tukey HSD test compared PWTs between groups over time.
In a subset of rats (NRC n=3; NRC+sPLA2 inh n=4; sham n=3), extracellular electrophysiological recordings were acquired in the dorsal horn of the cervical spinal cord on day 1, as previously described [4] (Fig. 1A). Electrophysiology recordings were also obtained from a naive rat. Rats were anesthetized intraperitoneally (i.p.) with sodium pentobarbital (45mg/kg) and given supplementary doses (5–10mg/kg, i.p.) as needed [4]. The C6/C7 spinal cord was exposed and a glass-insulated tungsten electrode (FHC; Bowdoin, ME, USA) was lowered into the ipsilateral dorsal horn to record evoked extracellular potentials using Spike2 software (CED; Cambridge, UK) [4].
Mechanoreceptive neurons in the spinal laminae were identified by brushing the plantar surface of the forepaw with four von Frey filaments (1.4g,4g,10g,26g) [4]. Voltage recordings were spike-sorted using Spike2 (CED) to isolate and count spikes evoked by each stimulus for individual neurons [4]. Neuron phenotypes were classified by their evoked responses [4]: wide dynamic range (WDR) neurons responded in a graded manner to increasing stimuli, low-threshold mechanoreceptive (LTM) neurons responded more to non-noxious (1.4g,4g) filaments, and nociceptive specific (NS) neurons responded only to noxious (10g,26g) filaments. Evoked responses for each stimulus were compared between groups using separate ANOVAs with a post hoc Tukey’s HSD test. Differences in the proportion of neuron phenotypes were assessed between groups through Pearson χ2 tests.
In a subset of rats (NRC n=6; NRC+sPLA2 inh n=6; sham n=6), the ipsilateral C6 spinal cord was harvested to evaluate the effects of sPLA2 inhibitor treatment on genes involved in neuronal signaling and inflammation (Fig. 1A). Spinal levels of genes for glutamate receptors (mGluR5, NR1) and transporters (GLT1, EAAC1), substance P, and pro-inflammatory cytokines (TNFα, IL1α, IL1β) (Fig. 1B) were quantified using quantitative reverse transcription polymerase chain reaction (RT-qPCR) methods [2;15–18]. Samples were run in duplicate and the relative target gene expression was analyzed using the comparative ΔΔCt method [2]. Gene expression was normalized to the housekeeping gene cyclophilin-A (CyA) and calculated as fold-change over normal (n=1). mRNA levels for each gene were compared between groups using ANOVAs with a post hoc Tukey’s HSD test.
Inhibitor effectiveness was evaluated in spinal tissue at day 1 using a commercially-available sPLA2 Assay Kit (Cayman Chemical; Ann Arbor, MI, USA) [8] (Fig. 1A). Ipsilateral C6 spinal cord was harvested (NRC n=5; NRC+sPLA2 inh n=5; sham n=4) and sPLA2 activity was measured as per assay instructions. sPLA2 activity was also measured in un-operated rats (n=2). Activity rates were compared between groups using an ANOVA with a post hoc Tukey’s HSD test.
RESULTS
Blocking spinal sPLA2 immediately after NRC prevents the development of mechanical hyperalgesia that is evident at day 1 (Fig. 2). PWT after an NRC is significantly lower than its baseline (p<0.001) (Fig. 2). In contrast, the PWT with sPLA2 inhibitor treatment remains at baseline (p=0.86) and is significantly higher than NRC alone (p=0.002) (Fig. 2). Although the PWT after NRC is significantly lower than sham on day 1 (p<0.004), there is no difference between sham and NRC+sPLA2 inh (p=1) at that time (Fig. 2).
Fig. 2. Behavioral sensitivity.
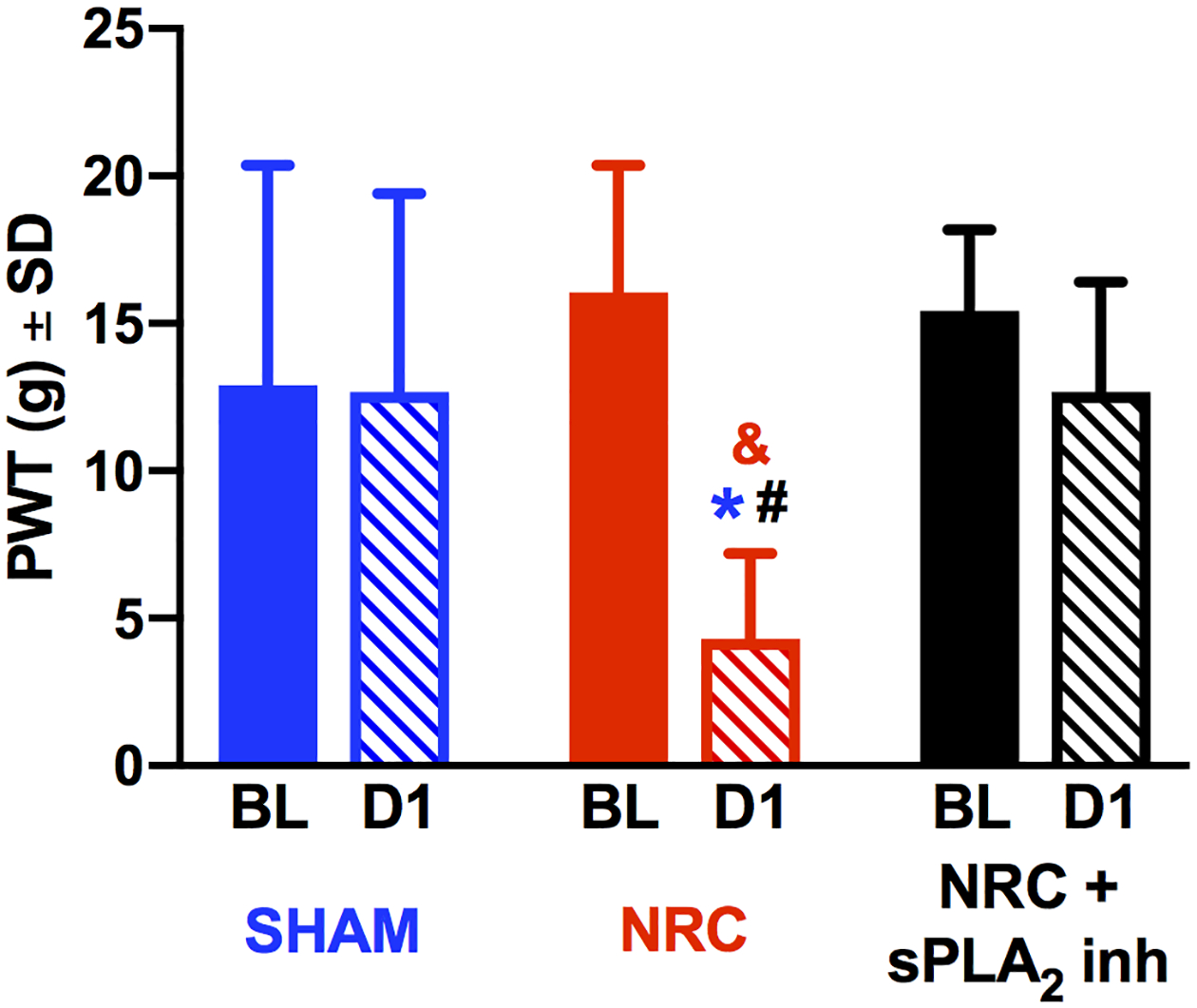
Treatment with sPLA2 inhibitor (NRC+sPLA2 inh) prevents the reduction in PWT at day 1 (D1) that is evident after a root compression (NRC) (***p<0.003). At day 1 after NRC, PWTs decrease from baseline (BL) (**p<0.001) and are lower than sham levels at that day (*p<0.004). PWTs for sham and NRC+sPLA2 inh are not different from each other.
sPLA2 inhibitor treatment prevents the spinal neuronal hyperexcitability that is observed at day 1 after NRC (Figs. 3A & 3B). Neuron activity was recorded from a total of 107 neurons (NRC n=20; NRC+sPLA2 inh n=37; sham n=34; normal n=16) at 533±238μm below the pial surface. At day 1 after NRC, the number of evoked action potentials is significantly higher than that in a normal rat (p<0.04) for all filaments, and over the number of spikes in sham rats for all filaments except 1.4g (p<0.02) (Fig. 3B). However, sPLA2 inhibition maintains neuron firing for all stimuli at sham and normal levels (Fig. 3B), with significant reductions compared to NRC for all filaments except 1.4g (p<0.04). sPLA2 inhibition also prevents the increase in neurons classified as WDR in the dorsal horn that is produced by NRC (Fig. 3C). In the NRC+sPLA2 inh group, 43% of neurons is classified as WDR, which is significantly less (p<0.02) than after NRC (78%) (Fig. 3C). The phenotypic distribution for the NRC+sPLA2 inh group is not different (p>0.74) than that in normal or sham (Fig. 3C).
Fig. 3. Extracellular spike activity and neuronal classifications in the spinal dorsal horn.
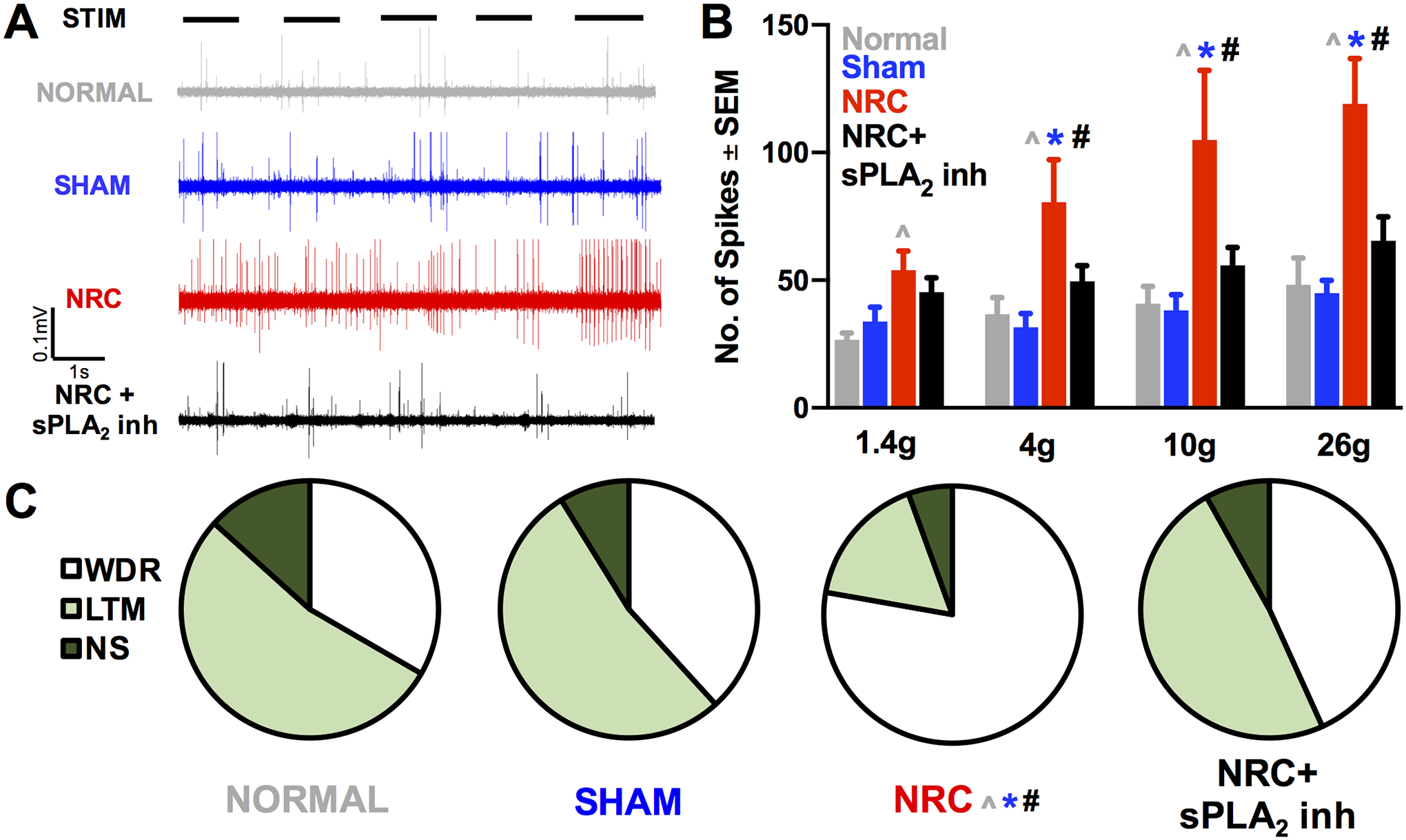
(A) Representative recordings during noxious 26g filament stimulus (STIM) showing a reduction in evoked activity with sPLA2 inhibitor compared to NRC. (B) Evoked activity after NRC is increased over normal levels for all stimuli (*p<0.04) and over sham levels for all filaments except the nonnoxious 1.4g filament (**p<0.02). sPLA2 inhibitor treatment (NRC+sPLA2 inh) prevents that increase compared to NRC in neuron firing for all stimuli except the 1.4g filament (***p<0.04). (C) There is a shift in the distribution of neuron phenotypes (WDR, LTM, NS) after NRC compared to normal (*p<0.02), sham (**p<0.02), and NRC+sPLA2 inh (***p<0.03) with no differences between any other groups.
Spinal sPLA2 inhibition reduces (p<0.05) spinal genes for mGluR5, substance P, and IL1 that are upregulated early after NRC (Figs. 4A–4C). Root compression significantly increases mGluR5 levels (p<0.03) and decreases glutamate transporter (GLT1, EAAC1) genes (p<0.04) compared to sham (Fig. 4A). However, NR1 is unchanged with injury (p=0.88) (Fig. 4A). sPLA2 inhibition does not reverse the decrease (p<0.01) in glutamate transporters that is evident after compression (Fig. 4A). A painful compression also induces elevated substance P (p<0.05) and TNFα and IL1α (p<0.02) over sham (Figs. 4B & 4C). Although sPLA2 inhibition also decreases TNFα levels that are elevated after NRC (p<0.05), that reduction is not significant (p=0.07) (Fig. 4C). sPLA2 activity significantly increases after NRC compared to sham (p<0.0004) (Fig. 4D), and this increase is prevented by sPLA2 inhibitor treatment (p<0.003) (Fig. 4D).
Fig. 4. Assays of mRNA and sPLA2 activity in C6 ipsilateral spinal cord tissue.
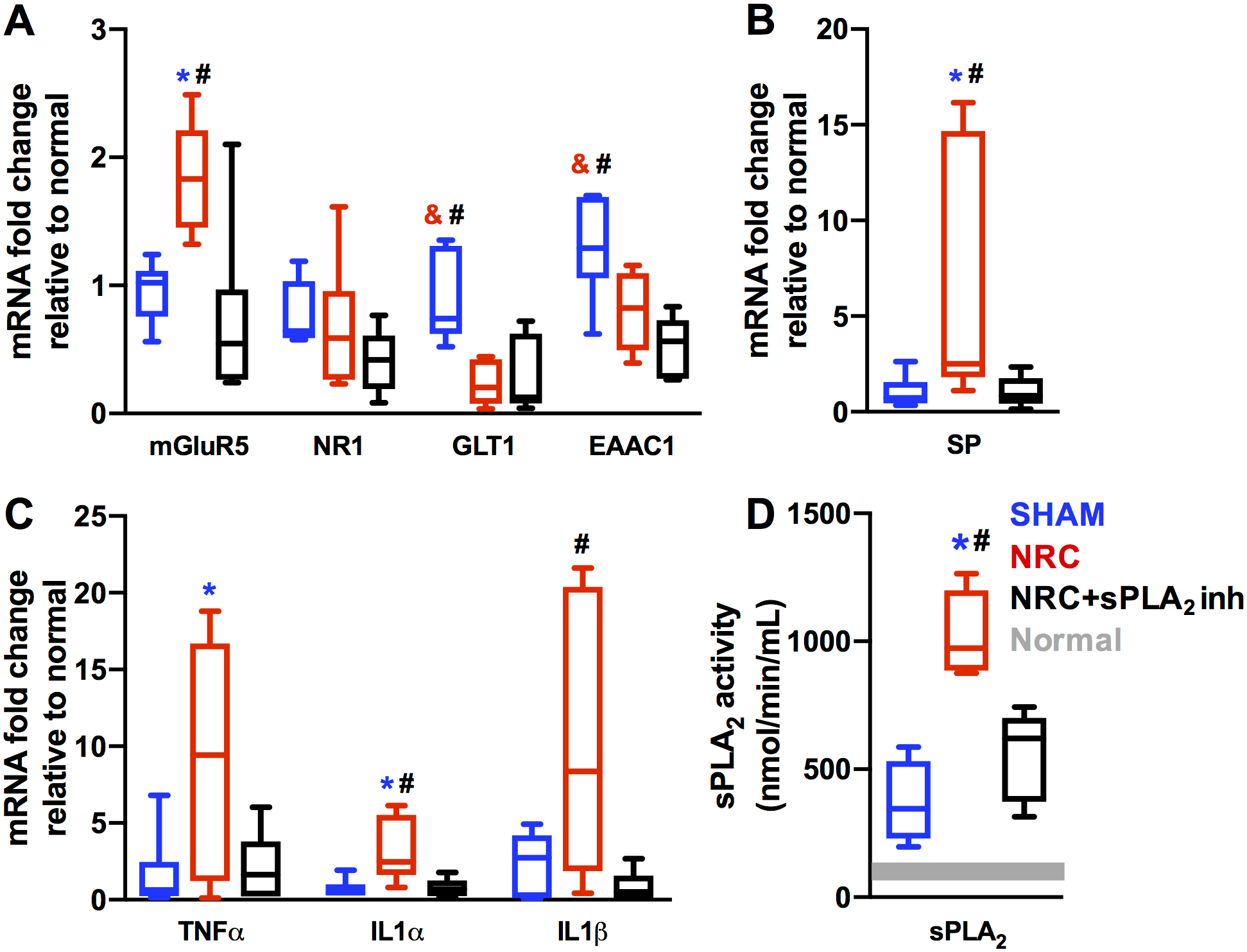
(A) mGluR5 mRNA is elevated after NRC relative to sham (*p<0.03), and treatment with sPLA2 inhibitor (NRC+sPLA2 inh) prevents this increase (**p<0.01). NR1 mRNA levels are not different in any group. Both GLT1 and EAAC1 mRNA levels are reduced after NRC relative to sham (***p<0.04). Inhibitor treatment does not prevent this decrease as NRC+sPLA2 inh mRNA levels are lower than sham (**p<0.01) and not different from NRC for both GLT1 and EAAC1. (B) Substance P (SP) mRNA is elevated after NRC relative to both sham (*p<0.05) and NRC+sPLA2 inh (**p<0.05). (C) TNFα mRNA is elevated after NRC relative to sham (*p<0.02). IL1α mRNA is elevated after NRC relative to both sham (*p<0.01) and NRC+sPLA2 inh (**p<0.02). IL1β mRNA is elevated after NRC relative to NRC+sPLA2 inh (**p<0.03), but not sham. (D) Spinal sPLA2 activity is significantly elevated after NRC relative to sham (*p<0.0004) and treatment with sPLA2 inhibitor (NRC+sPLA2 inh) prevents this increase (**p<0.003). sPLA2 activity levels in sham and NRC+sPLA2 inh are not different. Grey bar represents sPLA2 activity in normal tissue.
DISCUSSION
A single dose of the anti-inflammatory sPLA2 inhibitor given at the time of a painful nerve root compression suppresses inflammatory responses (Fig. 4C), interrupts glutamate signaling regulation (Fig. 4A), and prevents elevated spinal neuron activity (Fig. 3B). sPLA2 inhibition also maintains substance P at sham levels (Fig. 4B), accompanying a similar suppression of spinal hyperexcitability (Fig. 3). Elevated spinal substance P mRNA after painful injury supports its role as a regulator in the development of central sensitization through its activation of post-synaptic neurokinin receptors [19]. Increased pro-inflammatory cytokines (Fig. 4C) can modulate neuronal activity by activating their receptors in the spinal cord or promoting the release of nociceptive mediators or excitatory glutamate from glial cells [20], which may be responsible for preventing increases in mGluR5 genes (Fig. 4A). Since both increased cytokine mRNA and neuronal hyperexcitability are attenuated by sPLA2 inhibition immediately after injury (Figs. 3B & 4C), findings support its role in neuroinflammatory processes that facilitate spinal hyperexcitability with neuropathic pain [11].
The novel finding of dorsal horn neuronal excitability at an early time after a painful transient root injury parallels the quick development of pain in this model [5]. Central sensitization contributes to pain onset after neuropathic injury via the early functional alterations of neuromodulators [21] like substance P and mGluR5 which are upregulated at day 1 (Figs. 4A & 4B) and also at later times after this injury when spinal neuron hyperexcitability and phenotypic shifts are also observed [4;22]. sPLA2 inhibition may prevent the neuron phenotypic switch via neuropeptide-mediated mechanisms. Substance P release from low threshold Aβ fibers after injury enables those non-nociceptive fibers to signal pain [23]; if decreased spinal substance P mRNA is taken as a proxy for decreased substance P release by Aβ afferents, sPLA2 inhibition may also prevent hyperexcitability of post-synaptic spinal neurons.
sPLA2 potentiates glutamatergic synaptic function and enhances glutamate-induced excitotoxicity [10]; its downstream cleavage product arachidonic acid inhibits synaptic glutamate uptake [14]. Excess synaptic glutamate resulting from increased activation of glutamate receptors and reduced transporter expression enhances nociceptive synaptic transmission [21]. sPLA2 inhibition reduces spinal mGluR5 levels (Fig. 4A), but does not restore glutamate transporter genes to sham levels (Fig. 4A), suggesting sPLA2 may mediate pain and hyperexcitability through mechanisms other than spinal glutamate uptake.
This is the first study demonstrating that a single intrathecal dose of sPLA2 inhibitor given immediately after a neuropathic injury is sufficient to prevent the pain and spinal cellular respsonses that develop otherwise by day 1. Intracerebroventricular injection of the sPLA2 inhibitor 12-epi-scalaradial immediately after carrageenan injection prevents mechanical hyperalgesia, suggesting that blocking the sPLA2 pathway directly in the central nervous system after an insult may also relieve pain [24]. Pain and central sensitivity suppression with sPLA2 inhibition in this study (Figs. 3 & 4) suggest the specific blockade of sPLA2 may be a potential therapeutic approach for painful neuropathy that is upstream of the targets of traditional treatments like non-steroidal anti-inflammatory drugs.
Acknowledgements:
Special thanks to Prabesh Ghimire for providing training on RT-qPCR techniques. This study was supported by the Catherine Sharpe Foundation, the NIH (NS199892l) and fellowship support from an NIH/NINDS T32 (NS043126).
Source of Funding: This study was supported by the Catherine Sharpe Foundation, the NIH (NS199892l) and fellowship support from an NIH/NINDS T32 (NS043126).
Footnotes
Conflict of Interest Statement: None declared.
REFERENCES
- 1.Woods BI, Hilibrand AS. Cervical radiculopathy: Epidemiology, etiology, diagnosis, and treatment. J. Spinal Disord. Tech 2015; 28 (5): E251–E259. [DOI] [PubMed] [Google Scholar]
- 2.Rothman SM, Huang Z, Lee KE, Weisshaar CL, Winkelstein BA. Cytokine mRNA Expression in Painful Radiculopathy. J. Pain 2009; 10 (1): 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson KJ, Guarino BB, Winkelstein BA. Transient nerve root compression load and duration differentially mediate behavioral sensitivity and associated spinal astrocyte activation and mGluR5 expression. Neuroscience 2012; 209: 187–195. [DOI] [PubMed] [Google Scholar]
- 4.Nicholson KJ, Gilliland TM, Winkelstein BA. Upregulation of GLT-1 by treatment with ceftriaxone alleviates radicular pain by reducing spinal astrocyte activation and neuronal hyperexcitability. J. Neurosci. Res 2014; 92 (1): 116–129. [DOI] [PubMed] [Google Scholar]
- 5.Rothman SM, Winkelstein BA. Cytokine antagonism reduces pain and modulates spinal astrocytic reactivity after cervical nerve root compression. Ann. Biomed. Eng 2010; 38 (8): 2563–2576. [DOI] [PubMed] [Google Scholar]
- 6.Titsworth WL, Cheng X, Yan K, Deng L, Burckardt KA, Pendleton C, et al. Differential Expression of sPLA2 Following Spinal Cord Injury and a Functional Role for sPLA2-IIA in Mediating Oligodendrocyte Death. Glia 2009; 57: 1521–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chacur M, Milligan ED, Sloan EM, Wieseler-frank J, Barrientos RM, Martin D, et al. Snake venom phospholipase A2s (Asp49 and Lys49) induce mechanical allodynia upon peri-sciatic administration : involvement of spinal cord glia, proinflammatory cytokines and nitric oxide. Pain 2004; 108: 180–191. [DOI] [PubMed] [Google Scholar]
- 8.Liu N, Zhang YP, Titsworth WL, Jiang X, Han S, Lu P, et al. A Novel Role of Phospholipase A2 in Mediating Spinal Cord Secondary Injury. Ann. Neurol 2006; 59: 606–619. [DOI] [PubMed] [Google Scholar]
- 9.Svensson CI, Lucas KK, Powell HC, Dennis EA, Yaksh TL. Spinal phospholipase A2 in inflammatory hyperalgesia: role of the small, secretory phospholipase A1. Neuroscience 2005; 133: 543–553. [DOI] [PubMed] [Google Scholar]
- 10.Decoster MA, Lambeau G, Lazdunski M, Bazan NG. Secreted Phospholipase A2 Potentiates Glutamate-Induced Calcium Increase and Cell Death in Primary Neuronal Cultures. J. Neurosci. Res 2002; 67: 634–645. [DOI] [PubMed] [Google Scholar]
- 11.Morioka N, Takeda K, Kumagai K, Hanada T, Ikoma K. Interleukin-1b-induced substance P release from rat cultured primary afferent neurons driven by two phospholipase A2 enzymes: secretory type IIA and cytosolic type IV. J. Neurochem 2002; 80: 989–997. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann M Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983; 16 (2): 109–110. [DOI] [PubMed] [Google Scholar]
- 13.Kartha S, Weisshaar CL, Philips H, Winkelstein BA. Pre-treatment with meloxicam prevents the spinal inflammation and oxidative stress in DRG neurons that accompany painful cervical radiculopathy. Neuroscience 2018; 388: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung B, Wang S, Zhou B, Lim G, Yang L, Zeng Q, et al. Altered spinal arachidonic acid turnover after peripheral nerve injury regulates regional glutamate concentration and neuropathic pain behaviors in rats. Pain 2007; 131 (1–2): 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labombarda F, Florencia M, José M, Federico A, Nicola D, Laura S. Neuropathic pain and temporal expression of preprodynorphin, protein kinase C and N-methyl- d -aspartate receptor subunits after spinal cord injury. Neurosci. Lett 2008; 447: 115–119. [DOI] [PubMed] [Google Scholar]
- 16.Doi T, Ueda Y, Takaki M, Willmore LJ. Differential molecular regulation of glutamate in kindling resistant rats. Brain Res. 2011; 1375: 1–6. [DOI] [PubMed] [Google Scholar]
- 17.Mantilla CB, Bailey JP, Zhan W, Sieck GC. Phrenic motoneuron expression of serotonergic and glutamatergic receptors following upper cervical spinal cord injury. Exp. Neurol 2012; 234 (1): 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui X, Li L, Hu Y, Ren S, Zhang M, Li W-B. Sulbactam Plays Neuronal Protective Effect Against Brain Ischemia via Upregulating GLT1 in Rats. Mol Neurobiol 2015; 51: 1322–1333. [DOI] [PubMed] [Google Scholar]
- 19.Khasabov SG, Malecha P, Noack J, Tabakov J, Giesler GJ, Simone DA. Hyperalgesia and sensitization of dorsal horn neurons following activation of NK-1 receptors in the rostral ventromedial medulla. J. Neurophysiol 2017; 118 (5): 2727–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015; 96 (PA): 70–82. [DOI] [PubMed] [Google Scholar]
- 21.Bleakman D, Alt A, Nisenbaum ES. Glutamate receptors and pain. Semin. Cell Dev. Biol 2006; 17: 592–604. [DOI] [PubMed] [Google Scholar]
- 22.Hubbard RD, Chen Z, Winkelstein BA. Transient cervical nerve root compression modulates pain: load thresholds for allodynia and sustained changes in spinal neuropeptide expression. J. Biomech 2008; 41 (3): 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devor M Ectopic discharge in AB afferents as a source of neuropathic pain. Exp. brain Res 2009; 196: 115–128. [DOI] [PubMed] [Google Scholar]
- 24.Yeo JF, Ong WY, Ling SF, Farooqui AA. Intracerebroventricular injection of phospholipases A 2 inhibitors modulates allodynia after facial carrageenan injection in mice. Pain 2004; 112 (1–2): 148–155. [DOI] [PubMed] [Google Scholar]