

Abstract
Antibody-dependent cell-mediated cytotoxicity (ADCC) is an effector function of immunoglobulins (IgGs) involved in the killing of target cells by a cytotoxic effector cell. Recognition of IgG by Fc receptors expressed on natural killer cells, mostly FcγIII receptors (FcγRIII), underpins the ADCC mechanism, thus motivating investigations of these interactions. In this paper, we describe the combination of hydrogen deuterium exchange (HDX) and fast photochemical oxidation of proteins (FPOP) coupled with mass spectrometry (MS) to study the interactions between the human IgG1/FcγRIII complex. Using these orthogonal approaches, we identified critical peptide regions and residues involved in the recognition of IgG1 by FcγRIII. The footprinting results are consistent with the previously published crystal structure of the IgG1 Fc/FcγRIII complex. Additionally, our FPOP results reveal the conformational changes of the Fab domain upon Fc domain binding to FcγRIII. These data demonstrate the value of footprinting as part of a comprehensive toolbox for identifying the changes in the higher order structure of therapeutic antibodies in solution.
Graphical Abstract
Introduction
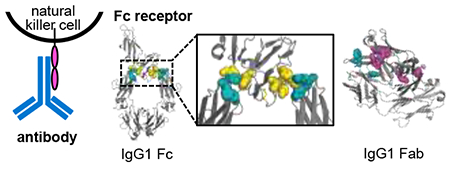
Immunoglobulins (IgGs) have become essential therapeutic tools for management of malignant, infectious, and autoimmune diseases.1–4 An effector function of an IgG, mostly IgG1, is to promote killing of target cells (including tumor cells) by cytotoxic effector cells.5–7 This process is termed antibody-dependent cell-mediated cytotoxicity (ADCC).8,9 ADCC starts with an antibody binding to a target cell and is triggered through the interaction of target-bound antibodies with certain Fc receptors on the effector cell surface (Figure 1). The typical ADCC mechanism involves recruitment of natural killer (NK) cells by the antibodies.10,11 One of the Fc receptors expressed by NK cells is the FcγIII receptor (FcγRIII).12 Once FcγRIII binds to the Fc region of IgG, the NK cell releases proteases, known as granzymes, to cause lysis of the target cell.13–15
Figure 1.

A. Cartoon representation of antibody-dependent cell-mediated cytotoxicity (ADCC), and B. the crystal structure of the complex of FcγIII receptor and IgG1 Fc. In A. the surface antigen (in yellow) on the target cells interacts with the antibody (in blue) that recognizes the antigen. The Fc receptor (magenta) on the surface of the natural killer (NK) cells can then interact with the antibody. In the crystal structure of the complex (B) (PDB: 1E4K), the FcγIII receptor and IgG1 Fc are also displayed in magenta and blue, respectively. The disulfide bond that links two heavy chains is shown in yellow. The glycosylated sites on FcγIII receptor and IgG1 are shown in spheres.
Investigators have achieved a better understanding of how ADCC activity is modulated, particularly for IgG1 antibodies, by using X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy.16,17 Thus far, crystal structures of FcγRIII extracellular domain and the FcγRIII/IgG1 Fc complex provide the principal structural means to understand the recognition in ADCC (Figure 1). Additionally, mutagenesis studies have demonstrated that human IgG1 in the ADCC process highly relies on the glycosylation of its Fc portion and on the polymorphism of the Fcγ receptors.18–22 There are gaps, however, in the current understanding regarding structure-function relationships associated with ADCC.
Mass spectrometry (MS) based approaches have become effective in probing protein higher order structure (HOS)23–30 Here, we combined hydrogen deuterium exchange (HDX) and fast photochemical oxidation of proteins (FPOP) with mass spectrometry to investigate the interaction between human IgG1 and FcγRIII. HDX reports changes in H-bonding and solvent accessibility in two or more states of a protein by monitoring the replacement of protein backbone amide hydrogens with deuterium. On the other hand, FPOP reports the changes in the solvent accessibility of side chains by following their differential modifications by reacting the protein with reactive radicals (e.g., OH and carbene radicals). Although there are various approaches to OH radical labeling, the approach we employed here, FPOP, was first established by Hambly and Gross in 2005.31 FPOP has been applied to study protein/ligand interactions,32–34 protein hidden conformations,35,36 and amyloid aggregation.37
In this paper, we mapped the binding interfaces and conformational changes of IgG1 and FcγRIII interaction in solution by using HDX and FPOP coupled with MS. HDX-MS determines the binding interface at the peptide level, and FPOP further identified some of the critical binding residues. The outcomes compare favorably with predictions from the solid-state crystal structure of the FcγRIII/IgG1 Fc complex. FPOP results also demonstrate that the binding of FcγRIII to the Fc region causes conformational changes in the Fab domain. Based on our results, we propose a putative mechanism for FcγRIII binding to IgG1. The results also show that FPOP can follow conformational changes hidden from other structural biology techniques including HDX.
Materials and Methods
Materials and sample preparation
Human FcγRIII protein was purchased from Sino Biological Inc. (Beijing, China). Anti-hCD20-hIgG1 antibody was purchased from InvivoGen (San Diego, CA). The UniProt accession number of IgG1 is P01857, and the UniProt accession of human FcγRIII protein is P08637. D2O was purchased from Cambridge Isotopic Laboratories (Andover, MA). Trypsin and chymotrypsin were purchased from Promega (Madison, WI). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
The protein samples of FcγIII receptor (FcγRIII) and IgG1 were dissolved in water and then buffer exchanged into 1 X PBS. For the bound state of FcγRIII, a molar ratio of 1:2 was used for FcγRIII:IgG1. For the bound state of IgG1, the molar ratio of IgG1:FcγRIII was 1:2. A molar ratio of 1:2 was used to ensure the completion of the binding. The solution of protein complex was incubated at 25 °C for 1 h before conducting the footprinting experiments.
HDX experiments
For each protein state that was studied in HDX, a stock solution of 30 µM such protein and 60 µM binding partner was prepared. A solution of 8 M guanidine chloride and 200 mM TCEP was adjusted to pH 2.5 and was kept on ice for quenching the HDX.38 Additionally, a saturated solution of fungal XIII was prepared in water with 1% formic acid (FA) and was held on ice for off-line protease digestion.
To initiate HDX, 2 µL of protein stock solution was diluted into 16 µL of D2O in 1 X PBS buffer. The extents of HDX were measured at various time intervals (10 s, 30 s, 60 s, 360 s, 900 s, 3600 s, and 14400 s) on ice. A quench solution of 16 µL was added to stop the HDX reaction, and then 16 µL of fungal protease XIII solution was immediately added to initiate off-line digestion. The mixture solution was kept on ice for 3 min and then immediately submitted to a custom-packed pepsin column (2 mm × 20 mm) for on-line digestion at a flow rate of 100 µL/min. The resulting peptides were further captured on a C8 cartridge (2.1 mm × 15 mm, Agilent, Santa Clara, CA) at a flow rate of 100 μL/min of H2O with 0.1% trifluoracetic acid (TFA). Digestion and desalting were carried out in an ice-water bath for 3 min. The resulting peptides were then separated by liquid chromatography (LC) consisting of a reversed-phase C18 column (2.1 mm × 5 cm, 3 µm Hypersil Gold, Thermo Fisher Scientific, Waltham, MA) at a flow rate of 100 µL/min by using a linear gradient of 4% to 40% acetonitrile with 0.1% FA in 4 min, the total length of the gradient is 9.5 min. To minimize back exchange, the valves, both trap and analytical columns, and tubing for protein digestion and separation were submerged in an ice-water bath. MS analysis of the peptides was conducted on an LTQ-FT mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a positive-ion electrospray ionization source. All the experiments were conducted in duplicate.
HDX data analysis
Prior to conducting HDX, experiments in the absence of D2O were performed to map the peptides generated from the digestion of each protein, generating a list of peptides that were to be followed during HDX-MS data acquisition. Mass spectra of product-ions were collected in a data-dependent mode, in which the six most abundant ions in each scan were selected for MS/MS analysis. The resulting MS/MS files from three parallel runs were then converted to mzXML files using MM File Conversion and submitted to MassMatrix (version 2.4.2) for peptide identification.39 Additionally, this search was carried out against a reversed sequence to discard ambiguous identifications. Data analysis of the HDX experiments was performed on HDExaminer (version 1.1.0, Sierra Analytics, Inc., Modesto, CA), and the deuterium uptake for each peptide was manually inspected.
FPOP experiments
Two stock solutions were prepared, one for interrogating the IgG1 and another for interrogating FcγRIII. The concentration of the protein being interrogated was 200 µM and that of the binding protein was 400 µM in 1X PBS. A stock solution of 40 µM YGGFL was prepared and used as a reporter peptide for the FPOP time-dependent experiments. Additionally, stock solutions of 0.5 mM, 5 mM, 25 mM, and 100 mM histidine, as scavenger, were prepared in 1X PBS buffer.
The FPOP time-dependent experiments were performed at four different scavenger conditions: 0.10, 1.0, 5.0, and 20 mM histidine to afford time-dependent measurements. Solutions were prepared starting with a 50-µL sample solution consisting of 2 µL protein solution (200 µM protein of interest and 400 µM of binding partner), 10 µL reporter peptide, 10 µL histidine stock solution, and 23 µL 1X PBS buffer solution. Hydrogen peroxide solution (5 µL of 300 mM H2O2) was added to the sample just prior to infusing the solution into the tubing for laser irradiation (FPOP experimental procedures were described in detail previously32,33). The final concentration of the protein of interest was 8.0 μM whereas that of the binding partner was 16.0 μM.
The power of the KrF excimer laser (GAM Laser Inc., Orlando, FL, USA) was adjusted to 24 mJ/pulse, and its pulse frequency was set to 7.5 Hz. The width of the laser beam at the intersection with the tubing was 2.4 mm. The flow rate was adjusted to 25 µL/min to ensure a 25% exclusion volume to minimize repeated •OH exposure (double hits). Samples were collected in Eppendorf tubes containing 10 mM catalase and 20 mM Met to exhaust the left-over H2O2. Additionally, two replicate control samples were prepared and handled in the same manner except no laser-irradiation was used.
Proteolysis for peptide analysis
A 10-μL aliquot of each FPOP sample was fully dried and re-suspended in 50 μL of ammonium bicarbonate buffer (50 mM, pH 7.8) and 1 μL of Protease MAX surfactant (Promega, WI, USA). The solution was heated for 10 min at 80 °C for denaturation, followed by reduction with dithiothreitol (5 μL of 300 mM solution) for 30 min at 56 °C. The solution was further cooled to room temperature and alkylated with iodoacetamide (2 μL of 100 mM solution) in dark for 30 min. The sample was digested overnight with trypsin or chymotrypsin at 37 °C with a protease-to-protein ratio of 1:20 (w/w). The digestion was terminated by acidifying the sample with 1% FA. In the LC-MS/MS analysis, peptide fragments were separated on a Thermo C18 column (75 μm × 25 cm, 3 μm, 100 Å) by using a Nano UltiMate 3000 Rapid Separation system (Dionex Co., Sunnyvale, CA) and analyzed with a Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
Identification and quantification of FPOP modification
Product-ion spectra obtained with the orbitrap mass spectrometer were searched for peptide identification using Byonic™ software (Protein Metrics, San Carlos, CA). For the FPOP data analysis, the false discovery rate was 1%. Modification sites on the peptide were assigned based on product-ion spectra (MS/MS data). The assignments were further validated by manual inspection of their accurate mass and product-ion spectra. Modification fractions for certain peptides were calculated with Byologic™ software (Protein Metrics, San Carlos, CA) and double-checked with Thermo Xcalibur (Thermo Fisher Scientific, Bremen, Germany).
Results and discussion
Hydrogen/Deuterium Exchange.
We began this study by using HDX-MS to locate interaction interfaces on FcγRIII upon binding to IgG1 (HDX plots of four representative peptides of FcγRIII are shown in Figure 2A, and HDX plots of all the peptides of FcγRIII are provided in the SI). We obtained a sequence coverage of ~75 % for FcγRIII by using pepsin and fungal digestion (Figure S1). As we do not include deglycosylation in our HDX workflow, we are not able to cover the regions that are glycosylated in the HDX experiments.
Figure 2.
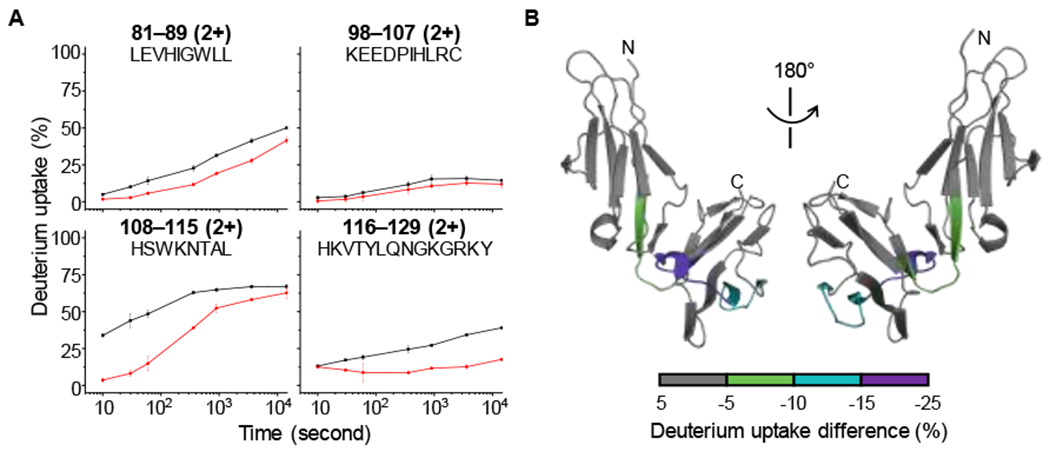
(A) HDX kinetic plots of representative peptides of FcγRIII in the unbound (black) and bound (red) states. The peptide, its charge state, and its amino acid sequence are labeled on each panel. (B) Differential deuterium uptake percentage of FcγRIII mapped onto the crystal structure. The color scale representing the difference between bound and unbound is shown in the bottom of the panel. Mirrored views are provided for clarity.
Comparison between the apo and holo states (Figure 2A) establishes that the peptides 81-89, 108-115, and 116-129 show significant decrease and peptide 98-107 smaller decrease in deuterium uptake. By mapping the differential deuterium uptake onto the crystal structure of FcγRIII (Figure 2B), we find that the protected regions are loops connecting two β sheets, an observation that is consistent with the crystal structure of the FcγRIII/IgG1 complex in which those loops are at the interaction interface (Figure 1). No additional significant conformational changes occur as indicated by nearly identical HDX over the remainder of FcγRIII in both states.
We then expanded our HDX inquiry to probe the interaction interfaces of IgG1 upon binding to FcγRIII. Sequence coverage percentage obtained was ~94% and ~96% for the heavy chain and light chain of IgG1, respectively (Figure S4). Deuterium uptake extents of IgG1 heavy chain (HC) and light chain (LC) (Figure S5) are consistent with the crystal structures of IgG1 Fc region and Fab region. (A summary of the deuterium uptake kinetic plots of the peptides of IgG1 heavy chain and light chain is provided in Figure S6 and S7, respectively.) Examination of these HDX kinetic plots shows that differences in the deuterium uptake occur for regions represented by heavy chain peptides 239-245, 246-256, and 286-303 (Figure 3A). Peptide 239-245 of IgG1 shows ~10% difference between the unbound and bound states at the first three time points (10 s, 30 s, and 60 s), whereas a difference of ~50 % in deuterium uptake is observed for peptide 246-256 at the last three incubation time points (900 s, 3600 s, and 14400 s). The difference in HDX extents between the unbound and bound states of peptide 286-303 is ~20 % and found in the first four time points (10 s, 30 s, 60 s, and 360 s). There are no significant deuterium update differences were observed for any of the light chain peptides. By mapping protected regions on the crystal structure of IgG1 Fc (Figure 3B), we find that our HDX data are consistent with the available crystal structure of the FcγRIII/IgG1 Fc complex. Furthermore, no conformational change of the IgG1 Fab region is detected upon Fc region binding to FcγRIII based on our HDX results.
Figure 3.
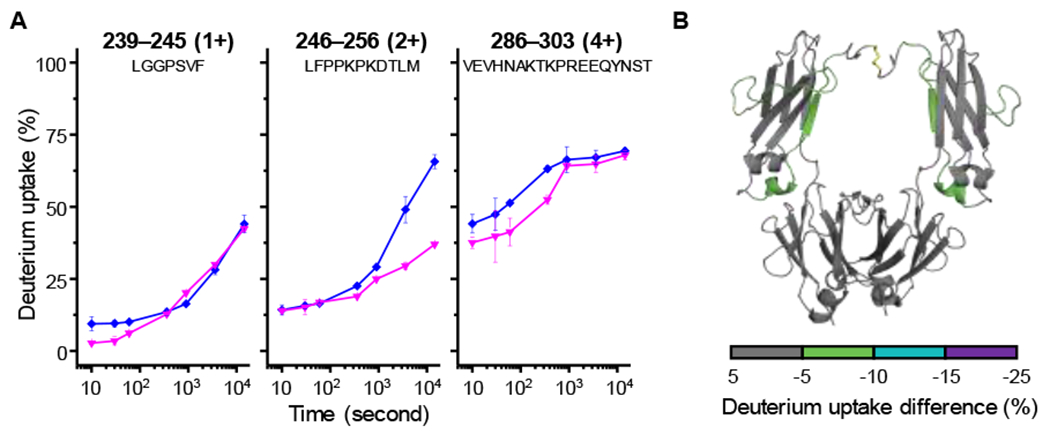
(A) HDX plots of representative peptides of IgG1 in the unbound (blue) and bound (magenta) states. The peptide, charge state, and the amino acid sequence are indicated in each panel. (B) Differential deuterium uptake of IgG1 mapped onto the crystal structure of IgG1 Fc. The color scale used to indicate percentage differences is given at the bottom of the panel. Mirrored views of the protein are provided for clarity.
FPOP.
We also examined the interaction between human IgG1 and FcγRIII by using FPOP coupled with proteomic analysis by MS. Here, we adopted a reporter peptide approach to obtain quantitative measurements of reactive residues.40 This methodology, developed in our laboratory, involves including a reporter peptide in the solution submitted to FPOP. In this scheme, the protein and the reporter peptide are modified by FPOP together under the same conditions. Incorporation of the reporter resolves discrepancies in the radical dosage by normalizing the outcome independent of changes (e.g., adventitious scavengers) in one sample and not another. Moreover, its modification extent is also a marker for the time of footprinting (i.e., the greater the extent of modification, the longer the exposure time). By varying the scavenger concentration (i.e., the radical lifetime), the reporter peptide approach permits a normalized, time-dependent measurement of the modification on the protein.
In the FPOP experiments of FcγRIII, we achieved ~97% coverage of FcγRIII by using a combination of trypsin and chymotrypsin (Figure S9). All the measurements conducted here do not involve deglycosylation, and as a result, we only sampled the peptides that are non-glycosylated. We identified and quantified 22 residues of FcγRIII that undergo oxidative modification (Figure S11). To demonstrate time-dependent measurements, we plotted the fraction of leu-enkephalin modified at each scavenger (histidine) concentration versus the corresponding fraction modified for each peptide of the protein (here FcγRIII) on the y-axis. The reporter-fraction modified reports •OH lifetime (i.e., longer times give more modification), and the plots show the time-dependent FcγRIII modification, affording a similar output to that of HDX kinetics data for peptides except the times are orders of magnitude shorter.
For comparing the modification of the two states of FcγRIII and IgG1, we used the Student’s t-test on both states (bound and unbound) for each peptide and residue to determine if the two sets of data are significantly different from each other. We applied a two-sample, two-tailed t-test assuming unequal variances (the resulting P-values for each peptide and residue of FcγRIII and IgG1 are listed in Tables S1–S6). Those peptides and residues with a P value that is smaller than 0.050 are considered as critical peptide and residue involved in the interaction between FcγRIII and IgG1. Five residues that show a difference in modification extent between unbound and bound states of FcγRIII (Figure 4A) are I85, W87, H116, K117, and H131; all show more extensive modification for the unbound state.
Figure 4.
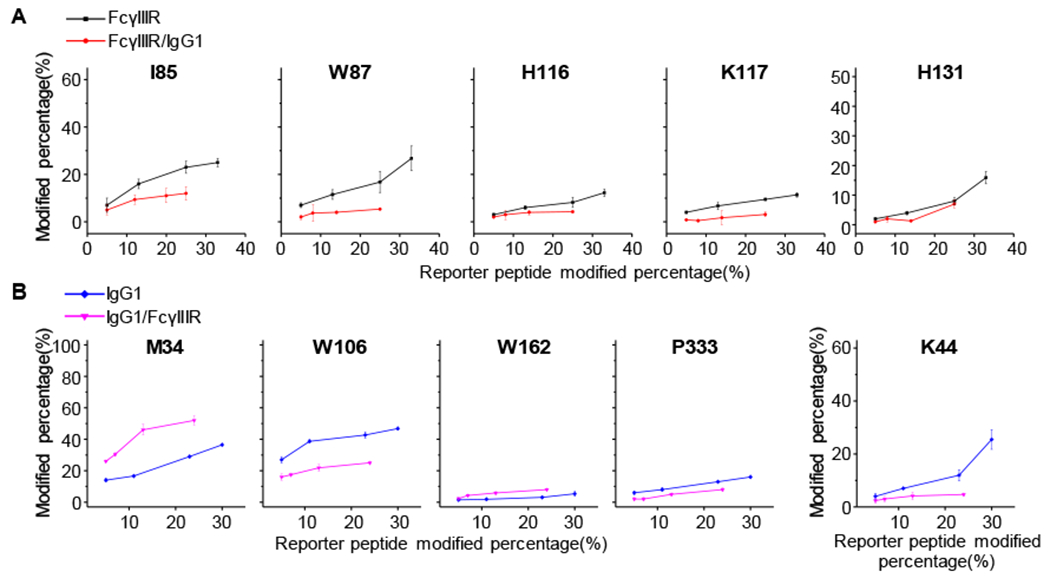
(A) FPOP kinetic plots of representative residues of FcγRIII. The unbound and bound states of FcγIII receptor are shown in black and red, respectively. (B) FPOP kinetic plots of representative residues of IgG1 heavy chain (left) and light chain (right). The unbound and bound states of IgG1 are shown in blue and magenta, respectively. In each FPOP kinetic plot, the FPOP modified percentage is normalized by the modified percentage of reported peptide under each scavenging condition.
Considering the conformational changes of IgG1 upon binding to FcγRIII as determined by FPOP, we obtained coverages of ~97% and ~98% for IgG1 heavy and light chains, respectively (Figure S10). A total of 51 residues of IgG1 heavy chain and 24 residues of IgG1 light chain are modified by FPOP under these conditions (Figure S12 and S13); Figure 4B summarizes FPOP representative kinetic plots of residue M34, W106, W162, and P333 on the heavy chain and K44 on the light chain. Nine residues of IgG1 heavy chain and one residue of IgG1 light chain display FPOP modification differences upon IgG1 binding to FcγRIII. Four residues (W106, P242, H272, and P333) on the IgG1 heavy chain and one residue (K44) on the IgG1 light chain exhibit protection upon binding. In contrast, five residues (M34, W36, W47, F154, and W162) in the heavy chain show increased exposure upon binding according to the FPOP measurements. It is interesting that all the exposed residues are in the Fab portion of the molecule.
Mapping the critical residues identified by FPOP on the crystal structure of the FcγRIII /IgG1 Fc complex (Figure 5A) and the IgG1 Fab region (Figure 5B) affords a better understanding of how recognition of IgG1 by FcγRIII affects the IgG1 structure. The differential footprinting of FcγRIII indicates that residues I85, W87, H116, K117, and H131/H132 are involved in the interaction of FcγRIII with IgG1 (Figure 5A). These critical residues of FcγRIII are consistent with the reported crystal structure of the FcγRIII /IgG1 complex. Key binding residues P242, H272, and P333 of IgG1 identified by FPOP are also consistent with the crystal structure of the FcγRIII/IgG1 Fc complex. Furthermore, we find that two residues (HC W106 and LC K44) show protection and five residues (HC M34, W36, W47, F154, and W162) undergo exposure in the Fab domain of IgG1. Currently, there is no available crystal structure for the complex of FcγRIII with the full-length IgG1. Our data provide direct evidence that the binding of IgG1 Fc to FcγRIII affects the conformations of the IgG1 Fab region, and this conclusion cannot be made from the X-ray crystal structure.
Figure 5.
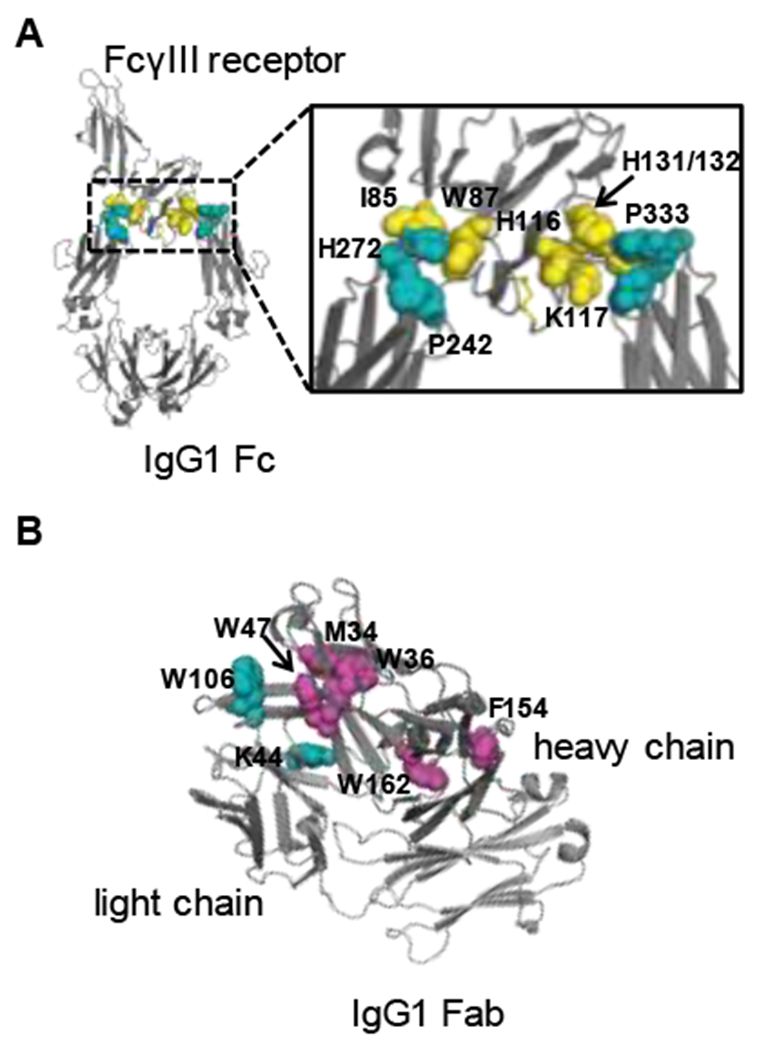
(A) Critical binding residues of FcγIII receptor and IgG1 identified by FPOP mapped on the crystal structure of FcγIII receptor/IgG1 Fc complex (PDB: 1E4K). The critical residues in FcγIII receptor are shown in yellow, and the critical residues in IgG1 Fc are shown in cyan. (B) Critical residues of IgG1 identified by FPOP mapped on the crystal structure of IgG1/Fab (PDB: 4KAQ). The residues that display protection in FPOP are shown in cyan, and magenta is used to represent the residues that display exposure in FPOP.
Our findings also reveal the structural basis, for the first time by MS-based footprinting, that the Fab domain may have an impact on the recognition of FcγRIII by IgG1. In our measurements, certain residues in the Fab domain remote from binding site become more exposed. One explanation is that Fab and Fc domains are in close contact conformationally in its “resting” state, even forming non-covalent interactions. Upon interaction with FcγRIII, Fab is released from the interaction with Fc and become more exposed. This indicates that Fab domain has an impact on the binding of the Fc domain to the FcγRIII. It is consistent with previous surface plasmon resonance assay and affinity studies,41 where even with the same Fc domain, only a subset of IgG1s are capable of eliciting ADCC activity. We were not able to observe, however, significant protection in the Fab regions according to both HDX and FPOP results, indicating that the Fab domain of IgG1 is not directly interacting with the FcγRIII molecule that binds to the Fc domain of IgG1. This finding is different compared to the two-pronged binding mechanism of IgG1 to the FcRn proposed by Rand and his co-workers.42 Both of these studies, however, indicate that binding to Fc receptors is not an isolated interaction that occurs only in the Fc region; rather, it affects the conformation of the entire mAb.
The critical binding regions as determined on the peptide level by HDX and FPOP are in remarkable agreement (Figure 6). (Figure S14 and S15 summarize the FPOP modification of peptides of FcγRIII and IgG1, respectively.) Specifically, FPOP shows that for FcγRIII, regions 73-87 and 112-140 are involved in the interaction with IgG1, in agreement with the HDX data of FcγRIII. The critical binding regions of IgG1 Fc region as characterized by FPOP are peptides 227-252, 256-279, and 331-338. The findings of IgG1 Fc region by FPOP are also consistent with the HDX measurements.
Figure 6.
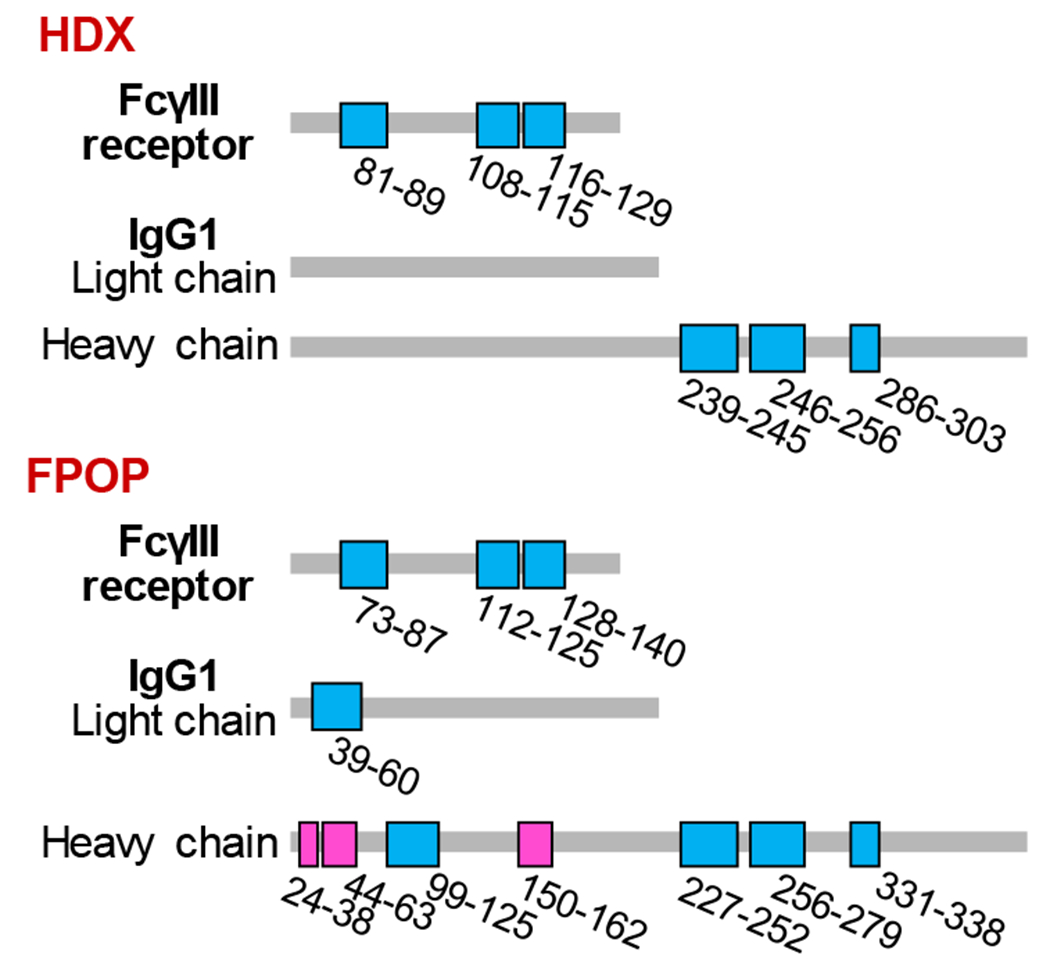
Binding interfaces and conformational changes determined by HDX (top) and (FPOP) mapped on the linear sequence of FcγIII receptor and IgG1. The regions that show protection upon binding in both HDX and FPOP are shown in blue. Magenta is used to represent the regions that show exposure in FPOP results. See text for details.
Furthermore, peptides 39-60 from the IgG1 light chain and 99-125 from the heavy chain are protected upon binding, according to FPOP, indicating a remote conformational decrease in solvent accessibility in these regions. We also note that regions 24-63 and 150-162 in the IgG1 Fab region undergo increasing exposure upon binding, indicating another remote conformational change. The binding affinity between the Fc receptor and Fc measured by the ultracentrifugation experiments is 0.4 µM.17 Based on the binding affinity and our experimental condition, the relative fraction of the target protein that is in complex with binding partner is 92% and 95% for the HDX and FPOP experiments, respectively. Therefore, any differences between the HDX and FPOP experiments should not be attributed to the binding fraction difference in these two types of measurements.
Our findings demonstrate the high sensitivity and capability of FPOP to probe side chain interactions (i.e., protein higher order structure (HOS)). HDX-MS is incapable of capturing these hidden conformational changes, as HDX is insensitive to fast dynamics. Moreover, the results afford a more comprehensive understanding of the recognition of IgG1 by FcγRIII and highlight the sensitivity of FPOP for detecting fast structural changes owing to the short half-life of hydroxyl radicals and higher coverage compared to HDX.
Conclusions
Interaction between human IgG1 and FcγRIII can be interrogated with high spatial resolution by using complementary footprinting and structural mapping (i.e., HDX-MS and FPOP). The finding that FcγRIII binding affects the conformation of the Fab domain in IgG1 sheds light onto the underying mechanism of ADCC activity elicited by certain IgG1s. Those “hidden” motions in structure and/or dynamics are invisible to HDX and presumably other slow footprinting methods. The combined use of two approaches not only serves to categorize and interpret changes in footprinting, but also adds confidence to characterize hidden conformational changes where traditional biophysical methods are challenged. This integrated approach can be applied to optimize the design of protein therapeutics and more generally shows great utility for characterizing protein and protein complexes.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge support for this research from the NIH, NIGMS (P41GM103422) and Eli Lilly and Company (Lilly Research Award Program).
Footnotes
Associated Content: Supporting Information
Sequence coverage and heat maps of FcγRIII and IgG1 in HDX experiments; summary of deuterium uptake plots of representative peptides identified in FcγRIII unbound and bound states; summary of deuterium uptake plots of representative peptides identified for IgG1 unbound and bound states; sequence coverage of FcγRIII and IgG1 in FPOP experiments; a summary of FPOP kinetic plots of residues identified in FcγRIII and IgG1; and LC chromatograms and product-ion (MS/MS) spectra of critical residues of FcγRIII and IgG1 identified by FPOP; and tabulated Student’s t to compare bound and unbound states. This material is available free of charge via the Internet.
References
- 1.Scott AM, Wolchok JD, Old LJ (2012) Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287. [DOI] [PubMed] [Google Scholar]
- 2.Peipp M, Dechant M, Valerius T (2008) Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr. Opin. Immunol 20, 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tobinai K, Klein C, Oya N, Fingerle-Rowson G (2017) A Review of obinutuzumab (GA101), a novel type II anti-CD20 monoclonal antibody, for the treatment of patients with B-cell malignancies. Adv. Ther 34, 324–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ (2010) CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin. Hematol 47, 107–114. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Mathieu M, Brezski RJ (2018) IgG Fc engineering to modulate antibody effector functions. Protein Cell. 9, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, Vielmetter J, Carmichael DF, Hayes RJ, Dahiyat BI (2006) Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A 103, 4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jegaskanda S, Reading PC, Kent SJ (2014) Influenza-specific antibody-dependent cellular cytotoxicity: toward a universal influenza vaccine. J. Immunol 193, 469–475. [DOI] [PubMed] [Google Scholar]
- 8.Vanderven HA, Jegaskanda S, Wheatley AK, Kent SJ (2017) Antibody-dependent cellular cytotoxicity and influenza virus. Curr. Opin. Virol 22, 89–96. [DOI] [PubMed] [Google Scholar]
- 9.Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inogés S, Melero I, Berraondo P (2017) Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol 95, 347–355. [DOI] [PubMed] [Google Scholar]
- 10.Feugier P (2015) A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Future Oncol. 11, 1327–1342. [DOI] [PubMed] [Google Scholar]
- 11.Kellner C, Otte A, Cappuzzello E, Klausz K, Peipp M (2017) Modulating Cytotoxic Effector Functions by Fc Engineering to Improve Cancer Therapy. Transfus. Med. Hemother 44, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nimmerjahn F, Gordan S, Lux A (2015) FcγR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 36, 325–336. [DOI] [PubMed] [Google Scholar]
- 13.Adams GP, Weiner LM (2005) Monoclonal antibody therapy of cancer. Nat. Biotechnol 23, 1147–1157. [DOI] [PubMed] [Google Scholar]
- 14.Carter P (2001) Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 1, 118–129. [DOI] [PubMed] [Google Scholar]
- 15.Carter PJ (2006) Potent antibody therapeutics by design. Nat. Rev. Immunol 6, 343–357. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Boesen CC, Radaev S, Brooks AG, Fridman WH, Sautes-Fridman C, Sun PD Crystal structure of the extracellular domain of a human FcγRIII. (2000) Immunity 13, 387–395. [DOI] [PubMed] [Google Scholar]
- 17.Sondermann P, Huber R, Oosthuizen V, Jacob U (2000) The 3.2-Å crystal structure of the human IgG1 Fc fragment-FcγRIII complex. Nature 406, 267–273. [DOI] [PubMed] [Google Scholar]
- 18.Hayes JM, Wormald MR, Rudd PM, Davey GP (2016) Fc gamma receptors: glycobiology and therapeutic prospects. J. Inflamm. Res 9, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, Umaña P, Benz J (2011) Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. U.S.A 108, 12669–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayes JM, Frostell A, Cosgrave EF, Struwe WB, Potter O, Davey GP, Karlsson R, Anneren C, Rudd PM (2014) Fc gamma receptor glycosylation modulates the binding of IgG glycoforms: a requirement for stable antibody interactions. J. Proteome. Res 13, 5471–5485. [DOI] [PubMed] [Google Scholar]
- 21.Jiang N, Chen W, Jothikumar P, Patel JM, Shashidharamurthy R, Selvaraj P, Zhu C (2016) Effects of anchor structure and glycosylation of Fc gamma receptor III on ligand binding affinity. Mol Biol Cell. 27, 3449–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiyoshi M, Caaveiro JMM, Tada M, Tamura H, Tanaka T, Terao Y, Morante K, Harazono A, Hashii N, Shibata H, Kuroda D, Nagatoishi S, Oe S, Ide T, Tsumoto K, Ishii-Watabe A (2018) Assessing the Heterogeneity of the Fc-Glycan of a Therapeutic Antibody Using an engineered FcγReceptor IIIa-Immobilized Column. Sci. Rep 8, 3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu GH, and Chance MR (2007) Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev 107, 3514–3543. [DOI] [PubMed] [Google Scholar]
- 24.Li KS, Shi L, Gross ML (2018) Mass spectrometry-based fast photochemical oxidation of proteins (FPOP) for higher order structure characterization. Acc. Chem. Res 51, 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Cui WD, and Gross ML (2014) Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. Febs. Lett 588, 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konermann L, Vahidi S, Sowole MA (2014) Mass Spectrometry Methods for Studying Structure and Dynamics of Biological Macromolecules, Anal. Chem 86, 213–232. [DOI] [PubMed] [Google Scholar]
- 27.Wang LW, and Chance MR (2011) Structural Mass Spectrometry of Proteins Using Hydroxyl Radical Based Protein Footprinting, Anal Chem 83, 7234–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konermann L, Pan JX, Liu YH (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chemical Society reviews 40, 1224–1234. [DOI] [PubMed] [Google Scholar]
- 29.Marcsisin SR, Engen JR (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal. Bioanal. Chem 397, 967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takamoto K, Chance MR (2006) Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct 35, 251–276. [DOI] [PubMed] [Google Scholar]
- 31.Hambly DE, Gross ML (2005) Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom 16, 2057–2063. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY-C, Julien SH, Lin Z, Moore K, Mueller L, Noriega C, Sejwal P, Sheppard P, Stevens B, Chen G, Tymiak AA, Gross ML, Schneeweis LA (2017) Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal. Chem 89, 2250–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li KS, Chen G, Mo J, Huang RY-C, Deyanova EG, Beno BR, O’Neil SR, Tymiak AA, Gross ML (2017) Orthogonal mass spectrometry-based footprinting for epitope mapping and structural characterization: The IL-6 Receptor upon binding of protein therapeutics. Anal. Chem 89, 7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Y, Chen G, Wei H, Huang RY-C, Mo J, Rempel DL, Tymiak AA, Gross ML (2014) Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. J. Am. Soc. Mass Spectrom 25, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hart KM, Ho CM, Dutta S, Gross ML, Bowman GR (2016) Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat. Commun 7, 12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gau B, Garai K, Frieden C, Gross ML (2011) Mass Spectrometry-Based Protein Footprinting Characterizes the Structures of Oligomeric Apolipoprotein E2, E3, and E4. Biochemistry 50, 8117–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li KS, Rempel DL, Gross ML (2016)Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J. Am. Chem. Soc 138, 12090–12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen E, Salinas ND, Huang Y, Ntumngia F, Plasencia MD, Gross ML, Adams JH, Tolia NH (2016) Broadly neutralizing epitopes in the Plasmodium vivax vaccine candidate Duffy Binding Protein. Proc. Natl. Acad. Sci. U.S.A 113, 6277–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu H, Freitas MA (2009) MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 9, 1548–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niu B, Mackness BC, Rempel DL, Zhang H, Cui W, Matthews CR, Zitzewitz JA, Gross ML (2017) Incorporation of a reporter peptide in FPOP compensates for adventitious scavengers and permits time-dependent measurements. J. Am. Soc. Mass Spectrom 28, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T (2011) Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab. Dispos 39, 1469–1477. [DOI] [PubMed] [Google Scholar]
- 42.Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD (2017) A two-pronged binding mechanism of IgG to the neonatal Fc receptor controls complex stability and IgG serum half-life. Mol. Cell Proteomics 16, 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.