

Approximately 15% of chronic lymphocytic leukemia/small lymphocytic lymphomas (CLL/SLL) transform into aggressive lymphomas, often diffuse large B cell lymphomas (DLBCL), known as Richter Syndrome (RS) [1]. RS shows poor prognosis with a median survival of 2.5–8 months [2]. Recent studies demonstrated that RS is driven by two major, mutually-exclusive genetic pathways. The main pathway involves inactivation of CDKN2A/B and loss of TP53, along with MYC amplification or translocation, and 13q14 loss in a subset of cases [3]. The clonal relationship between CLL/SLL and the aggressive lymphoma is a critical factor influencing treatments. On the basis of direct IGHV gene sequencing, studies have found that 80% of RS cases featured clonal relationships between both lymphoma populations [4]. The clonally unrelated cases had a much longer median survival (~5years) [4], despite more complex genomic alterations seen in de novo DLBCL compared to RS [5]. Therefore, the clonal relationship affects therapeutic choices and risk/benefit calculus regarding the role of autologous or allogeneic stem cell transplantation (alloSCT) consolidation.
Herein, we describe a 49year-old male with CD5+ CLL/SLL associated with CD10+ high grade B cell lymphoma (HGBCL). We provide unequivocal molecular evidence of their clonal relationship despite stark differences in immunophenotype. Furthermore, we postulate that the CLL/SLL and HGBCL likely arose from a common neoplastic progenitor, in a branched pattern of disease evolution.
The patient presented in June 2006 with low-volume lymphadenopathy, mild splenomegaly, and lymphocytosis (WBC 17.0K/mcL). He was diagnosed with CLL/SLL and placed on observation. In November 2008, the patient presented with bulky lymphadenopathy and WBC ~40K/mcL, prompting treatment with six cycles of fludarabine, cyclophosphamide, and rituximab with good response. An end-of-treatment computed tomography (CT) scan showed resolution of lymphadenopathy with low level persistent CLL/SLL in peripheral blood (PB) (~6–7% of WBC by flow cytometry (FC)). In December 2013, the patient developed diffuse, palpable lymphadenopathy, splenomegaly, and PB absolute lymphocytosis (64.8K/mcL). Bone marrow biopsy (BMBx) in January 2014 showed marrow with extensive involvement by CLL/SLL. Fluorescence in situ hybridization (FISH) studies showed 13q and 17p deletions. In February 2014, the patient showed disease progression (WBC 115.4 K/mcL) and started on ibrutinib. Given the patien’s young age and 17p alteration, alloSCT was discussed, but the patient declined.
In August 2017, the patient was admitted with pancytopenia and febrile neutropenia. Prednisone and intravenous immunoglobulin were given for suspected autoimmune hemolytic anemia. PET/CT revealed diffuse FDG uptake throughout the marrow, splenomegaly, and diffuse lymphadenopathy. Brain MRI showed no leptomeningeal disease or intracranial abnormality, but demonstrated marrow abnormality in the left mandibular ramus. A BMBx demonstrated extensive involvement by HGBCL, coexisting with CLL/SLL. Cytogenetic and molecular studies were performed and are described below (Figure 1 and Table 1).
Figure 1.
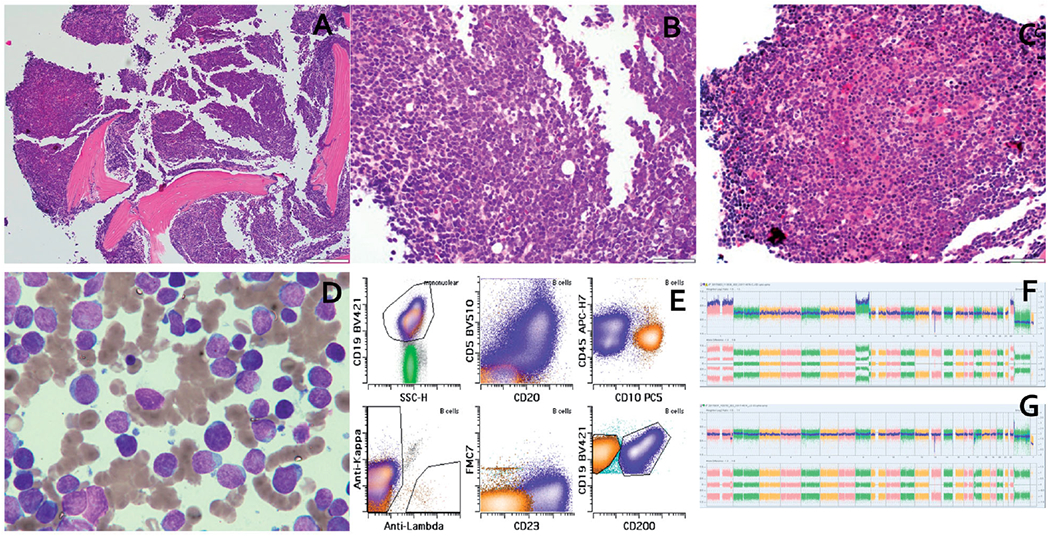
(a) Low power view showing hypercellular marrow. (b) High power view showing “blastoid” morphology in high grade B-cell lymphoma (HGBCL). (c) High power view showing CLL/SLL. (d) Aspirate smear showing dimorphic population of small lymphocytes with mature chromatin and large lymphoid cells with dispersed chromatin and prominent nucleoli. (e) Flow cytometric plots showing immunophenotype of HGBCL (orange) and CLL/SLL (blue). (f) SNP array findings in HGBCL showing homozygous deletion at 13q14.2 (including RB1 and SETDB2) and CN-LOH of 17p terminal to 17p11.2 (including TP53). (g) SNP array findings in CLL/SLL showing a 13q14.2 hemizygous deletion for RB1 and homozygous deletion for SETDB2 and loss of 17p13.1 to 17p12 (including TP53).
Table 1.
Comparison of genetic alterations and Variant Allele Frequencies (VAF) between the CD5+ CLL/SLL and CD10+ High Grade B-Cell Lymphoma.
| Gene | Type | Alteration | Location | Sorted CD5+ CLL cells | Sorted CD10+ B-Cell Lymphoma |
|---|---|---|---|---|---|
| Mutations | |||||
| SF3B1 | Missense mutation | p.K666E | Exon 14 | 49.8% | 48.8% |
| TP53 | Missense mutation | p.M237I | Exon 7 | 8.6% | 99.3% |
| UBR5 | Missense mutation | p.Q1822E | Exon 39 | 19.5% | 62.4% |
| TP53 | Missense mutation | p.G244C | Exon 7 | 29.9% | − |
| TP53 | Frameshift mutation | p.L257Gfs*6 | Exon 7 | 5.3% | − |
| TP53 | Missense mutation | p.E271K | Exon 8 | 35.7% | − |
| CIITA | Missense mutation | p.S1095I | Exon 18 | 22.1% | − |
| ERG | Missense mutation | p.R354Q | Exon 10 | 21.6% | − |
| KMT2D | Frameshift mutation | p.G2806Cfs*43 | Exon 34 | 16.8% | − |
| KMT2D | Frameshift mutation | p.P2938Tfs*4 | Exon 34 | − | 88.5% |
| KDM5C | Missense mutation | p.R1166L | Exon 23 | − | 97.3% |
| Copy number alterations | |||||
| SETDB2 | Whole gene | Deletion | 13q14.2 | + | + |
| TP53 | Whole gene | Loss | 17p13.1 | + | − |
| ALOX12B | Whole gene | Loss | 17p13.1 | + | − |
| RB1 | Intragenic | Deletion | Exons 24-25 | + | − |
| RB1 | Intragenic | Deletion | Exons 18-27 | − | + |
| TP53 | Whole gene | Copy Neutral-Loss of Heterozygosity | 17p terminal to 17p11.2 | − | + |
| MYC | Whole gene | Gain | 8q24.21 | − | + |
| Other chromosomal level alterations | |||||
| IGH/MYC | Translocation | Translocation | t(8;14)(q24;q32 | − | + |
The patient then underwent 4 cycles of dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; rituximab (R) was omitted because of CD20 negativity in HGBCL). After cycle 2, lumbar puncture exhibited equivocal evidence of leptomeningeal disease, prompting addition of intra-thecal methotrexate. Despite an initial disappearance of circulating HGBCL, after cycle 4 he developed marked lymphocytosis and cytopenias, consistent with primary refractory disease. A subsequent BMBx demonstrated persistent disease: 70% HGBCL and 30% CLL/SLL. In November 2017, the patient was started on IVAC (etoposide, ifosfamide, cytarabine, methotrexate) with plan for alloSCT consolidation. The patien’s course after two cycles was complicated by neutropenic fever, thrombocytopenia, E. Coli bacteremia, respiratory failure with multiple viral infections, bacterial pneumonia, sepsis and subarachnoid hemorrhage, resulting in delay in his planned alloSCT. Although he achieved remission briefly, in March 2018, the patient was diagnosed with recurrent HGBCL and was started on venetoclax, subsequently pembrolizumab without response. The patient remained refractory to therapies with disease progression and severe cytopenias, requiring daily transfusion. In April 2018, the patient died.
Pathologic examination of the August 2017 BMBx revealed 90% involvement by HGBCL coexisting with 10% CLL/SLL (Figure 1(a–d)). Immunophenotypes of the two populations by immunohistochemical (IHC) staining and FC (Figure 1(e)) demonstrated CLL/SLL: CD20+, PAX5+, CD5+, CD10−, CD23+, LEF1+, CD200+, FMC7−, and negative for BCL2, BCL6 and CMYC; HGBCL: CD20−, PAX5+, CD5−, CD10+, CD23−, LEF1−, CD200−, FMC7−, CD34−, TdT−, CMYC+ and negative for BCL2 and BCL6. The Ki67 was ~90% in the HGBCL and ~10% in CLL/SLL. FISH studies using probes flanking TP53, MYC, IGH were performed on FC-sorted cells from bone marrow aspirates (200–300 cells counted), and revealed loss of TP53 in 85% of CLL/SLL cells, but no loss of TP53 in the HGBCL cells. There was absence of MYC abnormalities in the CLL/SLL cells, but MYC rearrangement in 98% of the HGBCL cells, confirmed as t(8;14)/MYC-IGH by karyotype and IGH/MYC dual fusion probes (Figure 2(a–d) and Table 1).
Figure 2.
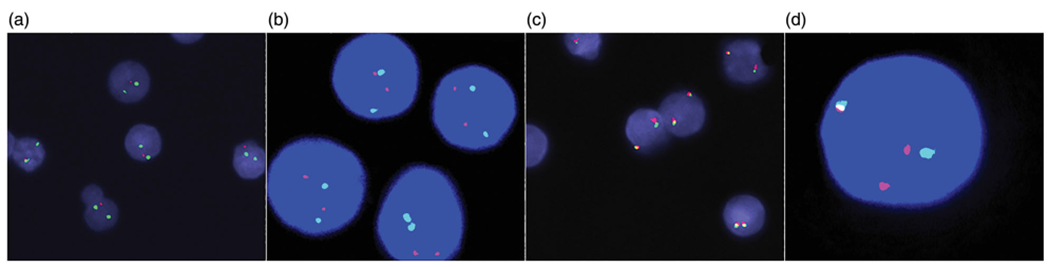
FISH findings on flow-sorted cells. (a) Loss of TP53 in 85% cells in CLL/SLL component. (b) No loss of TP53 in HGBCL component. (c) No MYC rearrangement in CLL/SLL component. (d) MYC rearrangement in 98% of cells in HGBCL component. (a–b) TP53 (17p13) probe (Red) and CEP17 (chr. 17 centromere) probe (Green). (c–d) MYC (8q24) break-apart probes.
SNP array-based DNA copy number analysis was performed using Affymetrix CytoScan™ HD Assay. SNP array on FC-sorted cells revealed alterations of chromosomes 13 and 17 in both lymphoma populations. The CLL/SLL population harbored 13q14.2 deletion (hemizygous deletion of RB1 involving the 3′ end of the gene only and homozygous deletion of the entire SETDB2 gene), as well as loss of 17p13.1 to 17p12, encompassing TP53. The HGBCL exhibited homozygous loss of 13q14.2 (also involving the 3′ end of the RB1 gene and the entire SETDB2 gene), and copy neutral-loss of heterozygosity (CN-LOH) of 17p terminal to 17p11.2, including TP53 (Figure 1(f–g) and Table 1).
Somatic mutation profiling was performed using a targeted, 400 gene panel NGS-based assay relevant to hematologic malignancies [6], on FC-sorted cells, with germline variant filtering using patien’s saliva. Both shared and unique mutations can be seen (Table 1). The unique mutations in each lymphoma were confirmed to be absent even at low allele frequencies in the other sample based on manual review.
IGH clonal rearrangement studies were performed using a NGS-based assay, Lymphotrack® (Invivoscribe, Inc., San Diego, CA) on FC-sorted populations and unsorted marrow samples. Clonal calling and analyses were performed as described previously [7]. All samples exhibited a single, identical clonal sequence with unmutated (0.0%) IGHV mutation status, and V-J segment usage of V3-30-J4.
CLL/SLL is characterized by CD5 expression, which is generally retained in most cases of RS [8,9]. A profound shift in immunophenotype during transformation is uncommon. CD10 expression can be seen in follicular lymphoma, DLBCL, Burkitt lymphoma, and some HGBCL, but is exceedingly rare in RS [10,11]. Adjacent lymphomas with profoundly different immunophenotypic profiles can raise the possibility of collision tumors. Nevertheless, we provided unequivocal evidence of clonally-related CD5+ CLL/SLL and CD10+ HGBCL in the same patient, including shared mutations in SF3B1, TP53, UBR5; shared alterations in chr.13q14 and 17p; and identical clonal IGH rearrangements. Demonstration of the clonal relationship in this patient has important prognostic and therapeutic implications, as RS cases have been shown to have inferior survival rate compared to de novo DLBCL. R-EPOCH would have been potentially curative therapy for a de novo aggressive lymphoma . In contrast, R-CHOP or R-EPOCH would have suboptimal efficacy for RS. In RS cases, consolidation with autologous or alloSCT would be preferable to achieve durable remission [12]. Unfortunately, this patien’s HGBCL recurred before he proceeded with alloSCT.
Large-scale genomic studies have characterized RS as genetically distinct from CLL and DLBCL, with intermediate genomic complexity [3,5]. The HGBCL in our patient harbored a genetic signature consistent with one of the two major pathways implicated in RS [5], including MYC-IGH rearrangement, loss of 13q14 with RB1 disruption, and deleterious TP53 mutations. By immunohistochemical stains, the HGBCL showed the expected finding of high expression of C-MYC. C-MYC protein, a global transcription factor regulating an estimated 10–15% of all human genes, is essential for normal early B-cell development and maintenance of B-cell identity. However, it is repressed in germinal center dark zone to limit the numbers of cell division for normal antigen affinity-dependent maturation. During lymphomagenesis, upregulation of C-MYC protein expression, as a result of translocation or gene amplification, results in a loss of regulation of a variety of cellular pathways involved in cell cycle and growth, metabolism, biosynthesis, and mitochondrial function. It also allows the B cells to circumvent affinity-dependent maturation, increasing the chances of further oncogenic events [13]. The CLL/SLL in our patient exhibited TP53 alterations, which are found more frequently in CLL/SLL with aggressive disease courses [4,5]. Therefore, somatic mutation profiling and cytogenetics studies provided further insight into underlying disease biology.
RS transformation from CLL/SLL most commonly occurs in a linear pattern, through the CLL/SLL clone’s accumulation of additional genetic alterations [3]. Alternatively, CLL/SLL and aggressive B-cell lymphoma can rarely arise in a branched pattern of disease evolution, in which a postulated early neoplastic progenitor gives rise to two tumors. They would share some genetic abnormalities, but also harbor unique alterations through independent evolution, similar to the observation in our case. Despite the efficacy of Bruton’s tyrosine kinase (BTK) inhibitors such as ibrutinib, phosphoinositide 3-kinase inhibitors, and BCL2 inhibitors in treating CLL, a subset of patients still showed refractory disease, occasionally with the development of aggressive B-cell lymphomas [14]. Several mechanisms to BTK inhibitors resistance and progression of disease under selective pressure from therapy are known, including expansion of subclones and acquisition of additional driver mutations [15,16]. Interestingly, our patient was started on ibrutinib therapy approximately 3.5 years prior to the diagnosis of HGBCL. Although difficult to ascertain, ibrutinib might have provided selective pressure facilitating disease evolution. In the milieu of TP53 alterations and SF3B1 mutation, conferring genetic instability and chemotherapeutic insensitivity, respectively, these two related lymphomas progressed in an aggressive fashion.
In summary, we describe a 49year-old male with strong molecular evidence of clonally -related CD5+ CLL/ SLL associated with CD10+ high grade B cell lymphoma (HGBCL) after ibrutinib treatment. The similarities and differences in genetic alterations between the two suggest the presence of a common neoplastic progenitor with a branched pattern of disease evolution, rather than a linear progression of conventional Richter Syndrome (RS).
Acknowledgements
We also want to acknowledge the members of the MSKCC diagnostic molecular laboratory, flow cytometry laboratory, and immunohistochemical stain laboratory for their support in performing the relevant assays.
Funding
This study was supported by the Comprehensive Cancer Center Core Grant [P30 CA008748] at Memorial Sloan-Kettering Cancer Center (MSKCC) from the National Institutes of Health, USA.
Disclosure statement
Dr. Arcila has served as a consultant and received honoraria from Invivoscribe, Inc. Dr. Ho has received honoraria from Invivoscribe, Inc. Dr. Roshal has served as a consultant for BD Biosciences, Agios, Celgene, as well as contract research funding for Agios, Roche, BMS, and Bayer. Dr. Dogan has received compensation from Roche, Novartis, Celgene, Seattle Genetics, and Corvus Pharmaceuticals for consulting/ advisory activities. Other authors declare no relevant conflict of interest.
Footnotes
Data availability statement
The complete data supporting the findings in this case report are available from the corresponding author upon reasonable request.
References
- [1].Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2017. (Steven H Swerdlow MEC, MD, PhD; Lee Harris Nancy, MD; Jaffe Elaine S., MD; Pileri Stefani A., MD, PhD; Stein Harald, MD; Thiele Jurgen, MD, PhD, editor. World Health Organization Classification of Tumours; ). [Google Scholar]
- [2].Tsimberidou AM, Keating MJ. Richter syndrome: biology, incidence, and therapeutic strategies. Cancer. 2005;103(2): 216–228. [DOI] [PubMed] [Google Scholar]
- [3].Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–3401. [DOI] [PubMed] [Google Scholar]
- [5].Chigrinova E, Rinaldi A, Kwee I, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673–2682. [DOI] [PubMed] [Google Scholar]
- [6].Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based Next-Generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arcila ME, Yu W, Syed M, et al. Establishment of Immunoglobulin Heavy (IGH) Chain clonality testing by Next-Generation sequencing for routine characterization of B-Cell and plasma cell neoplasms. J Mol Diagn. 2019;21(2):330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Matolcsy A, Chadburn A, Knowles DM. De novo CD5-positive and Richter’s syndrome-associated diffuse large B cell lymphomas are genotypically distinct. Am J Pathol. 1995;147(1): 207–216. [PMC free article] [PubMed] [Google Scholar]
- [9].Miyamura K, Osada H, Yamauchi T, et al. Single clonal origin of neoplastic B-cells with different immunoglobulin light chains in a patient with Richter’s syndrome. Cancer. 1990; 66(1):140–144. [DOI] [PubMed] [Google Scholar]
- [10].Kroft SH, Dawson DB, McKenna RW. Large cell lymphoma transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma. A flow cytometric analysis of seven cases. Am J Clin Pathol. 2001;115(3):385–395. [DOI] [PubMed] [Google Scholar]
- [11].Woroniecka R, Rymkiewicz G, Grygalewicz B, et al. Cytogenetic and flow cytometry evaluation of Richter syndrome reveals MYC, CDKN2A, IGH alterations with loss of CD52, CD62L and increase of CD71 antigen expression as the most frequent recurrent abnormalities. Am J Clin Pathol. 2015;143(1):25–35. [DOI] [PubMed] [Google Scholar]
- [12].Kharfan-Dabaja MA, Kumar A, Hamadani M, et al. Clinical practice recommendations for use of allogeneic hematopoietic cell transplantation in chronic lymphocytic leukemia on behalf of the guidelines committee of the american society for blood and marrow transplantation. Biol Blood Marrow Tr. 2016;22(12):2117–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nguyen L, Papenhausen P, Shao H. The role of c-MYC in B-cell lymphomas: Diagnostic and molecular aspects. Genes (Basel). 2017;8(4):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Delgado J, Bea S, Valdes-Mas R, et al. Linear and branching progression of chronic lymphocytic leukemia (CLL) under PI3K inhibition (PI3Ki). Blood. 2017;130:3000. [Google Scholar]
- [15].Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7(1): 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]