

Abstract
Excessive glucocorticoid (GC) production in adipose tissue promotes the development of visceral obesity and metabolic syndrome (MS). 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) is critical for controlling intracellular GC production, and this process is tightly regulated by hexose-6-phosphate dehydrogenase (H6PDH). To better understand the integrated molecular physiological effects of adipose H6PDH, we created a tissue-specific knockout of the H6PDH gene mouse model in adipocytes (adipocyte-specific conditional knockout of H6PDH (H6PDHAcKO) mice). H6PDHAcKO mice exhibited almost complete absence of H6PDH expression and decreased intra-adipose corticosterone production with a reduction in 11β-HSD1 activity in adipose tissue. These mice also had decreased abdominal fat mass, which was paralleled by decreased adipose lipogenic acetyl-CoA carboxylase (ACC) and ATP-citrate lyase (ACL) gene expression and reduction in their transcription factor C/EBPα mRNA levels. Moreover, H6PDHAcKO mice also had reduced fasting blood glucose levels, increased glucose tolerance, and increased insulin sensitivity. In addition, plasma free fatty acid (FFA) levels were decreased with a concomitant decrease in the expression of lipase adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) in adipose tissue. These results indicate that inactivation of adipocyte H6PDH expression is sufficient to cause intra-adipose GC inactivation that leads to a favorable pattern of metabolic phenotypes. These data suggest that H6PDHAcKO mice may provide a good model for studying the potential contributions of fat-specific H6PDH inhibition to improve the metabolic phenotype in vivo. Our study suggests that suppression or inactivation of H6PDH expression in adipocytes could be an effective intervention for treating obesity and diabetes.
Introduction
Obesity is a major public health threat that dramatically increases the risk of developing metabolic syndrome (MS) and type 2 diabetes (T2DM) [1–3]. Glucocorticoid (GC) plays a critical role in regulating both energy metabolism and glucose homeostasis. It also contributes to the pathogenesis of MS [4]. Patients with excessive GCs (Cushing’s syndrome) develop MS (e.g. visceral fat deposition, lipid abnormalities, insulin resistance, and hypertension) [5,6]. GCs promote adipocyte proliferation and differentiation [7], but evidence also indicates that GCs can induce the expression of hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL), two key enzymes for adipose release of free fatty acid (FFA) that is linked to hyperlipidemia and systemic insulin resistance [8,9]. However, circulating cortisol levels are not elevated in the vast majority of patients with obesity and MS. Indeed, many adverse phenotypes of obesity and MS can be induced by enhancing local GC levels within adipose tissue by nicotinamide adenine dinucleotide phosphate (NADPH)-dependent 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), an intracellular endoplasmic reticulum (ER) lumen-resident enzyme. 11β-HSD1 converts inactive cortisone (in humans), 11-dehydrocorticosterone (11-DHC) into cortisol/corticosterone (in rodents) and thus enhances intracellular GC activity. 11β-HSD1 is largely expressed in metabolic tissues such as liver and adipose; 11β-HSD1 also regulates glucose homeostasis, adipose differentiation and fat metabolism through increased local tissue GC production. In humans that are obese and have MS, adipocyte 11β-HSD1 expression is increased [10]. Transgenic mice, overexpressing 11β-HSD1 in adipose, recapitulate MS phenotypes [11]. Additionally, 11β-HSD1 KO mice are resistant to obesity and MS [12]. In addition, pharmacologic inhibition of endogenous 11β-HSD1 has been shown to reduce weight gain and improve lipid metabolic profile and insulin sensitivity in obese animal models [13,14]. These results support the hypothesis that 11β-HSD1 plays a pathogenic role in the development of obesity and insulin resistance in adipose tissue through elevated active GC levels. These elevated active GC levels may promote adipogenesis and increase the lipid profile through enhancing lipogenesis and lipolysis in fat tissue.
At the molecular levels, 11β-HSD1 activation of GCs requires hexose-6-phosphate dehydrogenase (H6PDH) to provide its cofactor NADPH [15,16]. H6PDH is an ER-luminal enzyme that utilizes glucose 6-phosphate (G6P), provided by the G6P transporter (G6PT), to generate NADPH from NADH in the ER that can be used by NADPH-dependent 11β-HSD1 reductase activity. Thus, H6PDH is required for 11β-HSD1 amplification of intracellular GC activation [17,18]. Transgenic overexpression of H6PDH in adipose tissue increases 11β-HSD1-driven intra-adipose GC production and exhibits many typical features of abdominal obesity and MS [19]. Similarly, in patients with MS and T2DM, as well as in obese animals, adipose cortisol production is increased with elevated local H6PDH mRNA levels [20–23]. These studies highlight the central role of elevated adipose H6PDH in the pathogenesis of MS and obesity.
In contrast, mice with inactivation of H6PDH have reduced 11β-HSD1 amplification of active GCs in the metabolic tissues of liver, adipose, and muscle. In addition, the mice also have reduced fasting glucose levels and gonadal fat mass, although this did not alter their metabolic phenotype in a non-fasting state [24]. In contrast, inhibition of adipocyte 11β-HSD1 improves visceral obesity and MS associated with a reduction in adipose H6PDH in animal obesity models [25]. Moreover, patients with target gene mutations in H6PDH have impaired cortisol production [26,27]. Taken together, these studies suggest that adipose tissue H6PDH is able to couple the regulation of cellular GC metabolism and energy homeostasis relative to MS and visceral obesity. However, the contribution of H6PDH in adipocytes to the development of MS could not be elucidated using conventional global knockout mouse models. In addition, the potential contribution of selective H6PDH inhibitors or lack of H6PDH specifically in any specific tissue including adipose tissue to pre-receptor inactivation of fat GCs remains unknown. Importantly, the phenotypic consequences of reduction in fat-specific GC action caused by inactivation of H6PDH specifically in adipose tissue has not been explored.
To test the hypothesis that decreasing the H6PDH expression or activity exclusively in adipose tissue would reduce 11β-HSD1-driven GCs production and that this reduction would lead to a favorable metabolic phenotype, we generated a tissue-specific animal model of adipocyte-specific H6PDH deletion under the control of the murine adipocyte-specific adiponectin promoter (adipocyte-specific conditional knockout of H6PDH (H6PDHAcKO) mice). Here, we show that H6PDHAcKO mice have decreased intra-adipose-specific GC production and exhibited a favorable metabolic phenotype of abdominal fat distribution and insulin sensitivity associated with improved fat lipid homeostasis.
Methods
Generation of adipose specific knockout mice of H6PDH
Tissue-specific deletion of H6PDH in murine fat cells were achieved by breeding a floxed H6PDH allele mice with an adipocyte-specific expression Cre line. Briefly, conditional targeted H6PDH ES clones (EM: 07643) were obtained from the European Conditional Mouse Mutagenesis Program. ES cells were then microinjected into C57blk/6 blastocysts to generate chimeric mice. Germline transmitted chimeric mice were bred to a Flp recombinase line to obtain a Neomycin cassette-free H6PDH floxed line. The Neo-free H6PDH floxed mice were further crossed with an adipocyte-specific Cre (adipoQ-Cre mouse line, JAX mice 010803) BAC transgenic line to generate H6PDHAcKO mice.
Animal study and biochemical assays
H6PDHAcKO mice and their floxed allele controls (H6PDHfl/fl, lacking the Cre gene) were housed in a room maintained on a 12:12-h light–dark cycle with free access to water and standard laboratory chow. All animal experiments and procedures were approved by the Institutional Animal Care and Use Committee of Charles R. Drew University. All animal experiments were performed at Charles R. Drew University. Body weight, food intake, blood glucose levels and blood pressure (BP) were determined weekly. BP was measured by non-invasive tail-cuff plethysmography in mice using a Visitech 2000 automated device (Visitech Systems, Apex, NC). Animals were killed via 5% isoflurane inhalation. Blood samples were collected between 09:00 and 10:00, and adipose tissues were weighed, dissected, and frozen rapidly in liquid nitrogen and stored at −80°C for metabolic assays. Blood samples were analyzed for the level of plasma corticosterone, insulin, and FFA using commercially available kits (Abcam, Cambridge, MA, U.S.A.).
Intraperitoneal glucose and insulin tolerance tests
For glucose tolerance tests (GTTs), H6PDHAcKO mice and floxed/floxed controls (Flox controls) were fasted overnight (16 h) at 14 weeks age and then given an intraperitoneal injection of glucose (2 g/kg, body weight) [25]. Blood samples were collected from tail tips before, and at different times after, the glucose load. For the insulin tolerance test (ITT), mice were fasted for 8 h, and blood samples were collected at indicated time points following insulin injection (0.75 U/kg, ip; Humulin) for glucose measurement [9].
Histological analysis
At the end of the experiments, the epididymal fat pad from both the control and H6PDHAcKO mice were collected and fixed in 10% formalin, embedded in paraffin, and sectioned. Adipose morphometry was visualized by Hematoxylin and Eosin staining (H&E) and three representative images per section were taken from each mouse (three mice/group). Adipocyte size in adipose tissue was analyzed using the NIH ImageJ software as described previously [19].
Microsomal H6PDH activity assays in adipose tissue
The adipose microsomal pellet was obtained by centrifugation of the supernatant for 1 h at 100 000×g. The adipose microsomal H6PDH activity was measured as previously reported [19]. Briefly, protein (100 μg) from adipose microsomes were incubated with 0.5–5 mM G-6-P, 1–5 mM NADP, and 100 mM glycine buffer solution at 22°C for 0–10 min. H6PDH activities were calculated and expressed as micromoles of NADPH production per minute per milligram of protein using absorbance at 340 nm by spectrophotometry [28].
Adipose tissue 11β-HSD1 activity and corticosterone assays
Adipose 11β-HSD1 activity was measured by the addition of 1 mM NADPH and 250 nM 11-DHC with [3H]11-DHC as the tracer to microsomes in a KRB solution at 37°C for 1 h as previously described [25]. Steroids were extracted with ethyl acetate and separated by a thin layer chromatography system. Radioactivity for each fraction was determined by scintillation counting and enzyme activity was determined. Corticosterone concentrations were measured in adipose tissue as previously described [29]. Briefly, epididymal fat pads were weighed and homogenized in PBS buffer (PH 7.4) by a tissue solubilizer. Steroids in adipose tissue were extracted with ethyl acetate and fat corticosterone levels were measured using a corticosterone EIA kit [30,31].
RNA extraction and gene expression analysis
Total RNA was extracted from mouse tissues using TRIzol reagent (Thermo Fisher Scientific Inc. Waltham, MA) and the first-strand cDNA was synthesized from 2.0 μg mRNA using High-Capacity RNA-to-cDNA™ Kit (Thermo Fisher Scientific Inc. Waltham, MA). Real-time PCRprimers for mouse H6PDH (F: 5′-TGGCTACGGGTTGTTTTT GAA-3′; R: 5′-TATACACGGTACATCTCCTCTTCCT-3′), 11β-HSD1 (F: 5′-CCTTGGCC-TCATAGACACAGA AA C-3′; R: 5′-GGAGTCAAAGGCGATTTGTCAT-3′), ACC (F: 5′-TGT AAATCTGGCTGCATCCATTAT-3′; R: 5′-TGGTAGACTGCCCGTGTGA-3′), ATP-citrate lyase (ACL) (F: 5′ -ATGCCAAGACCATCCTCTCACT-3′; R: 5′-TCTCACAATGCCCTTGAAGGT-3′), HSL (F: 5′-GGGCAAAGAAGGATCGAAGAA-3′; R: 5′ GCGTAAATC CATGCTGTGTGA-3′), ATGL (F: 5′-TCGTGGATGTTGGTGGAGCT-3′; R: 5′-TGTGGCCTCATTCCTCCTA-3′) , C/EBPα (F: 5′-TGGACAAG AACAGCAACGAGTAC-3′; R: 5′-CGGTCATTGTCACTGGTCAACT-3′), and C/EBPβ (R:5′-CTGCGGGGTTGTTGATGT-3′; R: 5′-ATGCTCGAAACGGAAA GGT-3′) were designed by Primer express software 2.0 (Thermo Fisher Scientific Inc. Waltham, MA). Quantitative Real-time PCR analysis was performed with SYBER green kit following manufacturer’s protocol in the ABI StepOne Plus System (Thermo Fisher Scientific Inc. Waltham, MA). Threshold cycle (Ct) readings for each of the unknown samples were then used to calculate the amount of target genes. The expression for target genes was normalized to the 18S rRNA values.
Western immunoblotting analysis
Adipose tissues were homogenized and the protein concentration was determined by Bradford assay as previously described [20]. The proteins were separated on 4–12% acrylamide SDS/PAGE gels (Bio-Rad, Hercules, CA) to evaluate the expression of H6PDH (Cat# sc-67394, Santa Cruz, Dallas, Texas), 11β-HSDl (Cat# sc-20175, Santa Cruz, Dallas, Texas), ACC (Cat# C83B10, Cell Signaling, Danvers, MA), ACL (Cat# 4332, Cell Signaling, Danvers, MA), pSer79 ACC (Cat# D7D11, Cell Signaling, Danvers, MA), pSer455 ACL (Cat# 4331, Cell Signaling, Danvers, MA), total HSL (Cat# 4107S, Cell Signaling, Danvers, MA), phospho-Ser660 HSL(Cat# 4126S Cell Signaling, Danvers, MA), ATGL (Cat# 2138S, Cell Signaling, Danvers, MA), and β-actin (Cat# 3700S, Cell Signaling, Danvers, MA). Adipose proteins were transferred to nitrocellulose membranes and incubated with the appropriate primary and secondary antibodies. Protein bands were visualized using ECL (Thermo Fisher Scientific Inc. Waltham, MA) and quantified by Eagle Eye II Quantitation System (Stratagene, CA, U.S.A.).
Statistical analysis
All values are expressed as the mean ± SEM. Data were analyzed using an unpaired Student’s t-test. To compare multiple groups, we conducted one-way ANOVA at first. When ANOVA revealed significant differences, then group comparisons were performed using Newman–Keul’s post hoc test. A P-value <0.05 was considered statistically significant.
Results
H6PDH gene expression is impaired in adipose tissue of H6PDHAcKO mice
To evaluate the expression levels of H6PDH in adipose tissue, both H6PDH mRNA and protein expression were measured by RT-PCR and Western blot analysis. Figure 1A shows that H6PDH mRNA expression was reduced by 71% in epididymal fat and 74.1% in subcutaneous fat of the H6PDHAcKO mice, compared with flox controls (P<0.01). Western blot analysis further demonstrated that H6PDH protein expression was extremely low in epididymal fat as well as in the subcutaneous fat of H6PDHAcKO mice, compared with flox controls (Figure 1B–D). Consistent with these observations, both epididymal and subcutaneous fat microsomal H6PDH activity, as determined by NADPH production, was barely detectable in the H6PDHAcKO mice compared with controls (Figure 1E). In contrast, endogenous H6PDH mRNA levels in the liver (Figure 1F) and skeletal muscle, as well as in kidney, were not different between H6PDHAcKO mice and their age-matched controls. Similarly, no significant differences in H6PDH protein expression were observed between two genotypes (data not shown). These results demonstrate that our conditional KO of H6PDH deletes H6PDH expression effectively in adipocytes, but leaves H6PDH expression unaffected in other tissues.
Figure 1. H6PDH mRNA and protein expression in adipose and other tissues of H6PDHAcKO and control mice.
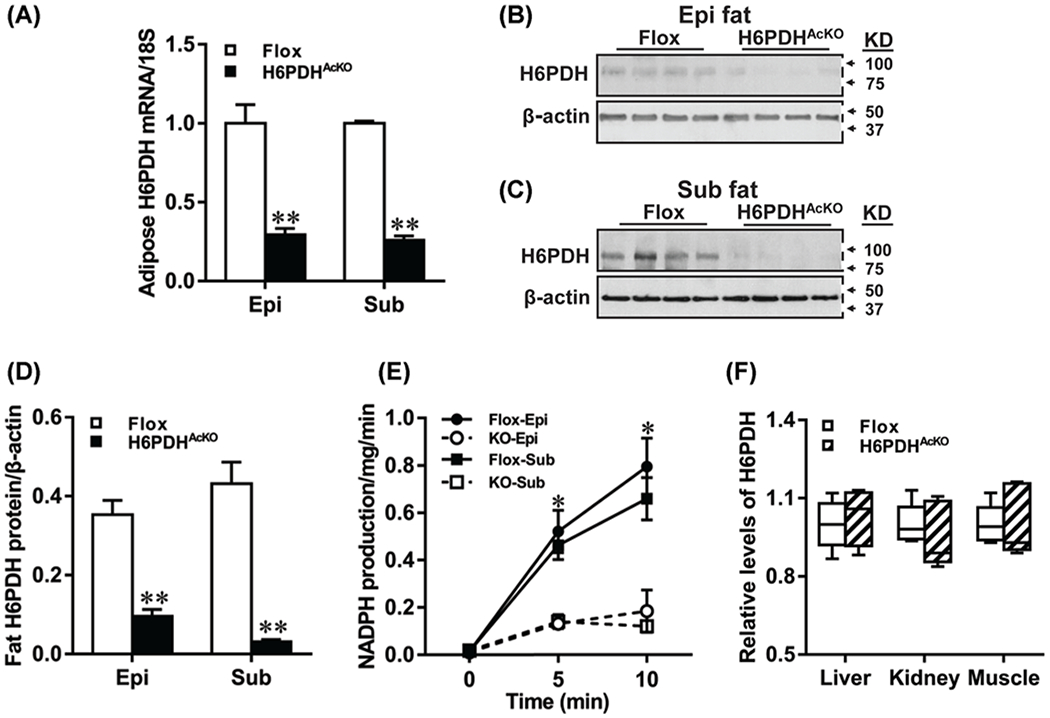
(A) Quantitative real-time PCR analysis exhibiting the mRNA expression of H6PDH in epididymal (Epi) and subcutaneous (Sub) fat of H6PDHAcKO mice and flox controls were normalized to 18S. (B–D) Western blotting images for H6PDH in Epi fat (B) and Sub fat (C) of the two genotypes and the quantification was relative to the amount of β-actin with NIH ImageJ (D). (E) H6PDH activity was measured in the adipose tissues using 2 mM G6P as substrate with the presence of NADP. (F) mRNA expression in liver, kidney and muscle of H6PDHAcKO mice and control littermates. Data are means ± SE of eight mice/group. *P<0.05 and **P<0.01 vs. Flox control mice.
Metabolic phenotype characterization of H6PDHAcKO mice
To determine the physiological effects of adipose H6PDH deletion, we then assessed the differences in body weight, food intake, and white adipose fat pad mass between H6PDHAcKO and their age-matched controls at 18 weeks old. As shown in Figure 2A, adipose tissue for H6PDHAcKO and flox control mice showed similar changes in weight gain. Similarly, there was no significant difference in food intake between the H6PDHAcKO mice and flox controls (data not shown). In addition, no significance was observed in the value of mean systolic blood pressure between H6PDHAcKO mice (102 ± 16 mmHg) and flox controls (108 ± 18 mmHg). In contrast, adipose fat pad weight was significantly reduced (by 53%) in epididymal fat, 34% in subcutaneous fat, and 62% in mesenteric fat pad (P<0.05) in H6PDHAcKO mice compared with controls (Figure 2B). Adipocyte size for H6PDHAcKO mice was decreased by 20% than controls (P<0.001, Figure 2C,D). H6PDHAcKO mice also showed decreased epididymal fat pad mass (0.33 ± 0.12 g; n=7) compared with controls (0.59 ± 0.14 g; n=7, P<0.01) in fasting conditions. These data indicate that H6PDHAcKO mice exhibited a favorable abdominal fat distribution to matched controls.
Figure 2. Body weight and fat pad mass of adipose-specific H6PDH knockout mice.
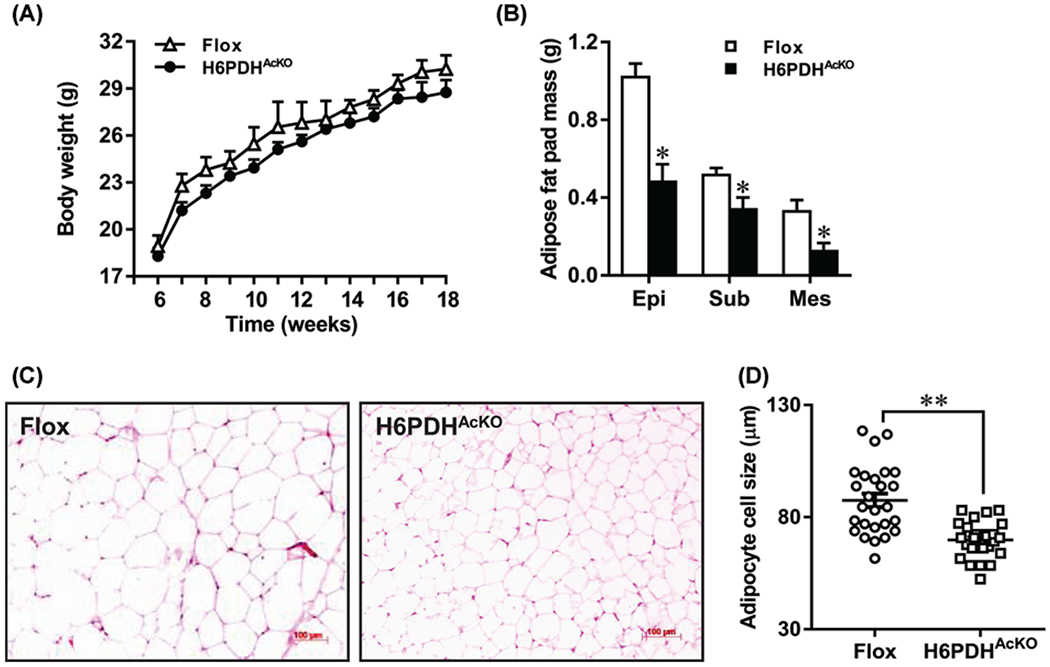
(A) Unchanged body weight gain of H6PDHAcKO mice on a chow diet (n=8–10 mice/group). (B) Weight of fat mass of H6PDHAcKO mice in fed state. Epi, epididymal fat; Sub, subcutaneous fat; Mes, mesenteric fat. Mice (n=8–10/group) were killed at the age of 18 weeks. (C,D) Average adipocytes size of epididymal fats on histological slides was measured with NIH ImageJ. *P<0.05 and **P<0.01 vs. Flox controls.
Decreased adipose lipogenesis in H6PDHAcKO mice
To determine if the reduced fat content in H6PDHAcKO adipocytes occurred in conjunction with changes in lipogenesis, we analyzed the expression of lipid synthase ACC and ACL, the major markers of lipid content in adipose tissue. Real-time RT-PCR revealed that epididymal fat ACC and ACL mRNA levels were decreased (61 and 65%, respectively) in H6PDHAcKO mice compared with controls (Figure 3A). Western blot analysis also confirmed that epididymal fat ACC and ACL protein levels were decreased with a reduction in phosphorylated levels of pSer79 ACC and pSer455 ACL in H6PDHAcKO mice (Figure 3C,E). Similarly, lower ACC and ACL mRNA levels (Figure 3B) with decreased ACC, pSer79 ACC, ACL, and pSer455 ACL protein expression were observed in subcutaneous fat of H6PDHAcKO mice (Figure 3D,F). These data indicate that reduction in adipose lipogenesis may contribute to abdominal fat loss observed in H6PDHAcKO mice.
Figure 3. Alterations of lipogenic gene mRNA and protein expression in epididymal fat (EF) and subcutaneous fat (SF) of H6PDHAcKO (∎) or Flox controls (◻).

(A,B) Relative mRNA expression levels of ACC and ACL in EF (A) and SF (B). (C–F) The expression and quantification of ACC, ACL, pSer79 ACC, and pSer455 ACL protein levels in EF (C,E) and SF (D,F) of H6PDHAcKO and Non-KO controls. Data are means ± SE of 6–8 mice/group. *P<0.05 and **P<0.01 vs. controls.
H6PDHAcKO mice had impaired adipose lipolysis and reduced plasma FFA levels
To study if adipose H6PDH inactivation could influence lipolysis, we next measured the plasma FFA levels and the expression of HSL and ATGL, two key lipolytic enzymes in adipose tissue. Figure 4A shows that H6PDHAcKO mice had significantly decreased plasma FFA levels compared with that of flox controls under both fasting and fed states (P<0.01 and P<0.05, respectively). Consistent with decreased FFA levels, epididymal fat mRNA levels of HSL were decreased (50 and 36%, in fasting and fed states) in H6PDHAcKO mice compared with that of controls (Figure 4B). This was confirmed by the results of Western blot analysis which demonstrated that the epididymal fat total for HSL protein level was decreased with strongly reduced phosphorylated levels of Ser660 HSL in H6PDHAcKO mice compared with controls (Figure 4C,D). Similarly, lower ATGL mRNA and protein levels were observed in epididymal fat from H6PDHAcKO mice compared with controls under both fasting and fed conditions (Figure 4B–D). These results indicate that the reduction in adipose lipolysis may contribute to decreased circulating FFA levels.
Figure 4. Adipose lipolytic enzymes mRNA and protein expression of H6PDHAcKO and Flox controls in fasted and fed condition.

(A) Serum FFA concentrations in H6PDHAcKO and Non-KO controls under both fasted and fed states. (B) Relatively mRNA expression changes of HSL and ATGL in Epi fat of H6PDHAcKO mice in both fasting and fed states. 18s rRNA was used as the internal normalizer. (C,D) Western blot analysis showed the alterations of ATGL, total and Ser660 phosphorylation (p) of HSL in Epi fat of H6PDHAcKO and their respective controls mice in fasted (C) and fed (D) states. Data are means ± SE of eight mice/group. *P<0.05 and **P<0.01 vs. Non-KO control mice.
Adipose H6PDHAcKO mice showed decreased fasting glucose levels and increased tolerance to glucose and insulin
To further explore the impact of adipose H6PDH deletions on glucose homeostasis and insulin sensitivity, we assessed glucose handling and insulin sensitivity in H6PDHAcKO mice and flox controls by performing glucose and insulin tolerance tests. As shown in Figure 5A, the fasting glucose levels were decreased in H6PDHAcKO mice (71 ± 12 mg/dl) compared with controls (102 ± 11 mg/dl; P=0.002). In parallel with fasting hypoglycemia, H6PDHAcKO mice showed decreased glucose levels during GTT. In addition, the area under the curve (AUC) levels of glucose tolerance in H6PDHAcKO mice were reduced compared with controls (Figure 5B). Moreover, decreased plasma insulin levels were detected in H6PDHAcKO mice in comparison with controls during GTT (Figure 5C). This is supported by the results of the ITT which showed that H6PDHAcKO mice had improved glucose disposal in response to insulin (Figure 5D). Consistent with these observations, fasting plasma insulin levels were decreased in H6PDHAcKO mice compared with that of controls (P<0.01; Figure 5E). These data indicate that the H6PDHAcKO mice exhibited an improved glucose handing with increased insulin sensitivity.
Figure 5. Glucose metabolism in H6PDHAcKO and Flox mice.

(A) GTT of H6PDHAcKO and control mice aged 14 weeks. (B) The glucose AUC during GTT. (C) Insulin concentrations at 0, 30, and 120 min during the GTT. (D) ITT in knockout and control mice. ITT was performed on mice after an 8-h fast, and 0.75 U/kg insulin was used. (E) Plasma insulin level in H6PDHAcKO and Flox mice. Data are means ± SE of eight mice/group. *P<0.05 and **P<0.01 vs. Flox controls.
H6PDHAcKO mice exhibit decreased endogenous 11β-HSD1 and corticosterone levels in adipose tissue
Because H6PDH is required for 11β-HSD1 activity, we next examined the impact of adipose H6PDH deletion on 11β-HSD1 expression and GC metabolism in adipose tissue. 11β-HSD1 mRNA level was reduced by 59% in epididymal fat (P<0.01) and 45% in subcutaneous fat (P<0.01) of H6PDHAcKO mice compared with their controls (Figure 6A). Western blot analysis indicated that epididymal and subcutaneous adipose 11β-HSD1 protein expression was substantially down-regulated in H6PDHAcKO mice compared with controls (Figure 6B–D). Likewise, 11β-HSD1 reductase activity was decreased by 2.4-fold in epididymal fat and 1.85-fold in subcutaneous fat (P<0.01; Figure 6E) in H6PDHAcKO mice compared with controls. In addition, real-time RT-PCR analysis showed that H6PDHAcKO mice also markedly reduced C/EBPa mRNA levels in epididymal fat (40%; P<0.01) and subcutaneous fat (37%; Figure 6F), a key transcription factor for 11β-HSD1. However, H6PDHAcKO mice maintained C/EBPα mRNA expression in adipose tissue. Moreover, there was a positive correlation between adipose 11β-HSD1 and C/EBPa mRNA expression in H6PDHAcKO mice and controls (R2 = 0.8752, P<0.001). In support of our findings, adipose corticosterone levels were significantly decreased by 2.3-fold in epididymal fat and 4.1-fold in subcutaneous fat in H6PDHAcKO mice (Figure 6G; P<0.001). In spite of this, H6PDHAcKO mice did not have altered plasma corticosterone levels compared with their controls (Figure 6H).
Figure 6. 11β-HSD1 and corticosterone levels in white adipose tissue of H6PDHAcKO mice and non-KO controls.

(A) mRNA expression of 11 β-HSD1 in epididymal (Epi) and subcutaneous (Sub) of H6PDHAcKO mice and Flox controls were normalized to 18S. (B–D) Western blot analysis of the protein expression of 11β-HSD1 in Epi fat (B) and Sub fat (C) between the two groups and the quantification was determined relative to the amount of β-actin (D). (E) Adipose 11β-HSD1 reductase activity was measured in the adipose microsomes using 11-DHC as substrate in the presence of NADPH. (F) mRNA expression of C/EBPα and C/EBPβ in Epi fat (EF) and Sub fat (SF) were determined by real-time PCR and normalized to 18S. (G,H) Adipose (G) and plasma (H) corticosterone level in H6PDHAcKO and Flox mice. Data are means ± SE of 7–8 mice/group. *P<0.05 and **P<0.01 vs. Flox controls.
Discussion
We have established a mouse model for exclusive adipocyte-specific knockdown of H6PDH under the control of the murine adiponectin promoter (H6PDHAcKO). We confirmed that the expression of H6PDH in H6PDHAcKO mice was diminished in adipocytes, but unaffected in other tissues. We demonstrated that H6PDHAcKO mice had reduced expression and activity of 11β-HSD1 and exhibited a favorable metabolic phenotype. We observed that corticosterone levels were decreased in the adipose tissue of H6PDHAcKO mice that was associated with a reduction in adipose 11β-HSD1 activity. These findings indicate that the conversion of corticosterone from 11-DHC is impaired in response to inactivation of H6PDH expression in the adipose tissue of H6PDHAcKO mice. Since 11β-HSD1 activity is dependent on the production of H6PDH-driven NADPH in target tissues, reduced adipose H6PDH expression decreased ER luminal NADPH availability to 11β-HSD1-driven GC production in adipose tissue. These data suggest that H6PDHAcKO mice effectively decrease adipose GC production through the reduction in the GC-amplifying effects of endogenous 11β-HSD1 in adipose tissue. This interaction is supported by recent studies reporting that intracellular GC reactivation is impaired in the metabolic tissues of adipose, liver and muscle in H6PDH knockout mice [24,32,33]. However, H6PDHAcKO mice did not have altered endogenous H6PDH expression in non-adipocyte tissues for liver, muscle, kidney, and brain. Thus, reduced adipose H6PDH expression can account for the phenotype observed in H6PDHAcKOmice, independent of non-adipocyte H6PDH expression.
Importantly, we observed that H6PDHAcKO mice exhibited fasting hypoglycemia, decreased insulin levels, and decreased plasma FFA levels suggesting that H6PDHAcKO mice exhibit improved insulin sensitivity and decreased fatty acid flux. The major difference between H6PDHAcKO and global H6PDH knockout mice is that H6PDHAcKO mice showed both improved glucose and insulin tolerance in response to decreased adipose GCs production while maintaining normal corticosterone levels, whereas global H6PDH knockout mice had normal glucose tolerance and insulin sensitivity with increased plasma GC levels [24,32,34]. Similarly, 11β-HSD1 KO mice showed no changes in these glucose metabolic parameters and plasma FFA levels [35,36], but elevated plasma GC levels [36,37]. Different circulating GC levels might contribute to the difference observed in glucose tolerance and systemic insulin sensitivity between H6PDHAcKO and global H6PDH knockout mice. Our data indicate that adipose-specific reduction in H6PDH is sufficient to account for the fasting hypoglycemia and insulin-sensitive phenotype, as observed in H6PDHAcKO mice, independent of circulating and non-adipocyte GC effects.
In adipocytes, GCs promote fat lipolysis by stimulation of both HSL and ATGL, two key enzymes for the hydrolysis of triacylglycerol as well as for the release of FFA into the circulation that leads to hyperlipidemia linked to systemic insulin resistance [38,39]. Increased adipose 11β-HSD1 and H6PDH have been shown to be important mediators for enhancing the contribution of GCs to elevated plasma FFA levels and insulin resistance [19]. In contrast, pharmacological inhibition or target knockout of 11β-HSD1 what shown to reduce lipemia and improve insulin sensitivity in rodents as well as in humans [40,41]. In accord with this concept, H6PDHAcKO mice exhibited lower levels of both HSL and ATGL expression in response to reduced local GC production in the abdominal adipose tissue and decreased levels of plasma FFA, suggesting an impaired lipolytic activity in adipose tissue in H6PDHAcKO mice. This could explain the decreased plasma FFA levels and may thus provide an additional mechanism for maintaining the phenotype of glucose homeostasis and insulin sensitivity observed in H6PDHAcKO mice. Our findings are supported by a recent study reporting that global H6PDH knockout mice showed decreased fat HSL mRNA levels [24]. In contrast, adipose-specific activation of H6PDH induced adipose lipase and elevated plasma FFA levels. This resulted in fasting hyperglycemia and glucose intolerance in response to elevated GC production in adipose tissue [19]. These findings support the notion that the reduction in fat lipolysis most likely occurs via reduced intra-adipose GC production-induced fat lipase expression.
In addition, H6PDHAcKO mice develop a lean fat metabolic phenotype, which is similar to global H6PDH knockout mice that also show reduced gonadal fat weight in the fed state. The difference is that H6PDHAcKO mice pronounced decreased epididymal fat mass in both fed and fasted states, while global H6PDH knockout mice did not have altered fasted gonadal fat mass [24]. Moreover, fat tissue histology revealed that H6PDHAcKO mice had small adipose cell size that differs from the normal fat cell size of H6PDH knockout mice [24]. These data could explain the difference in phenotype of reduced abdominal fat-depot mass between mice who lack global and adipose-specific H6PDH. More importantly, we observed that H6PDHAcKO mice have less intra-abdominal mesenteric fat and epididymal fat mass than seen in subcutaneous fat. However, in H6PDHAcKO mice, this did not affect blood pressure, despite the relationship between adipocytes and blood pressure [42,43]. Similarly, 11β-HSD1 KO mice had normal blood pressure but did not change mesenteric fat weight [35,44]; this is different from what is seen in H6PDHAcKO mice.
In fact, intra-abdominal fat accumulation is thought to be a leading cause of adverse metabolic consequence relative to the development of visceral obesity and MS [45,46]. Clinical studies have revealed that patients with obesity and MS have increased H6PDH expression and elevated local activation of cortisol from cortisone with induction of 11β-HSD1 in visceral fat [21,22]. Additionally, the key lipogenic transcription factor, mRNA encoding C/EBPα, is decreased with concomitantly reduced lipogenic enzymes ACC and ACL with reduction in 11β-HSD1 in adipose tissue of H6PDHAcKO mice. Decreased C/EBPα could attenuate the progress of lipogenesis and thus contribute to reduced abdominal fat mass. This interpretation is supported by studies reporting that enhanced adipose H6PDH increased fat lipogenesis and induced visceral fat accumulation with elevated C/EBPα, which itself is activated by GCs [11,19,47]. Our present data are consistent with a recent study reporting that inhibition of adipose 11β-HSD1 coupled with suppression of H6PDH, improved the phenotype of visceral obesity [25]. Moreover, C/EBPα is also a key transcriptional activator of 11β-HSD1 in adipose and liver [48,49]. Adipose H6PDH knockdown-mediated reduction in C/EBPα may thus represent an additional mechanism to promote adipose 11β-HSD1 reduction that is linked to decreased lipogenesis and fat loss in H6PDHAcKO mice. Additionally, adipocyte 11β-HSD1 can also be stimulated by insulin [50,51] and H6PDHAcKO mice had decreased insulin levels in response to the favorable metabolic phenotype with reduction of adipose 11β-HSD1 activity and gene expression observed in H6PDHAcKO mice. These findings imply that H6PDHAcKO mice-mediated induction of hypoinsulinemia may contribute to reduced adipose 11β-HSD1 activity. This interpretation is supported by earlier observations that insulin directly increases 11β-HSD1 production, though did not affect H6PDH in 3T3-L1 adipocytes [51]. Decreased insulin level itself may thus provide another possible mechanism to account for reduced adipose 11β-HSD1 production linked to the phenotype of H6PDHAcKO mice. In addition, reduced fasting plasma insulin levels are associated with increased insulin sensitivity and decreased plasma FFA levels in H6PDHAcKO mice. This interpretation is supported by earlier observations that reduction in plasma FFA levels contribute to low basal insulin levels [52,53].
One of the major strength of our study is the use of adipose-specific conditional KO. This enabled us to delineate the crucial contribution of H6PDH in adipocyte from other tissues (including liver or muscle) related to adipose function and glucose homeostasis in rodents. However, considering H6PDHAKO mice are conditional (not inducible) knockout mice, compensatory changes that develop in knockout mice cannot be ruled out, even in the adipose-specific KO mice, which could have contributed to the current results. Importantly, it is unanswered yet whether the absence of adipose H6PDH could prevent or reduce the adverse effects of high-calorie food. This represents a caveat of the current study, as we did not assess if these mice are protected against the metabolic changes that occur after exposure to high-fat diet in mice.
In summary, the present study demonstrates that reduction in adipose-specific H6PDH leads to reduced fat cell-specific production of GCs, and decreased adipose tissue lipolysis with resultant decreased plasma FFA level. This is known to be responsible for both global insulin sensitivity and glucose homeostasis. In addition, the mice exhibited a favorable metabolic phenotype. These findings also imply that inhibition of H6PDH in adipose tissue may represent a novel and potential therapeutic approach to prevent or delay the progress of MS and obesity.
Clinical perspectives.
H6PDH is able to couple the regulation of cellular GC activity and plays an important role in the etiology of MS and obesity. However, the functional consequences of selective H6PDH inhibition, or lack in adipose tissue, remains unclear.
In the present study, we established an adipocyte-specific knockdown mouse model of H6PDH. This specific model showed decreased adipose-specific GC production and exhibited a favorable metabolic phenotype of insulin sensitivity and abdominal fat distribution with reduced abdominal adipose mass independent of circulating and nonadipocyte GC effects.
Adipose-specific inhibition of H6PDH may have potential as a novel and effective pharmaco-therapeutic approach to target visceral fat GC action for prevent or delay the progress of MS and obesity.
Acknowledgments
We acknowledge Robin Faria (Director, CTSI Grants Submission Unit, UCLA Clinical and Translational Science Institute (CTSI)) for editing our manuscript.
Funding
This work was supported by the NIH [grant numbers SC1DK104821, S21 MD000103 (to Y.j.L.)]; the LSI [grant number F00333003007 (to Y.j.L.)]; the Accelerating Excellence in Translational Science (AXIS) [grant number #U54MD007598 (to T.C.F.)]; the Diversity-Promoting Institution Drug Abuse Research Program (DIDARP) [grant number R24DA017298 (to T.C.F.)]; the California Tobacco-Related Disease Research Program (TRDRP) [grant number 251P003 (to T.C.F.)]; the Project of National Natural Science Foundation of China [grant numbers 8147102, 8127086 (to L.L.)]; the Shanghai Leading Talent [grant number SLJ15055 (to L.L.)]; and the UCLA CTSI [grant number UL1TR001881 (to R.F.)].
Abbreviations
- ACC
acetyl-CoA carboxylase
- ACL
ATP-citrate lyase
- ATGL
adipose triglyceride lipase
- C/EBPs
CCAAT-enhancer-binding proteins
- ER
endoplasmic reticulum
- FFA
free fatty acid
- GC
glucocorticoid
- G6P
glucose 6-phosphate
- GTT
glucose tolerance test
- HSL
hormone-sensitive lipase
- H6PDH
hexose-6-phosphate dehydrogenase
- H6PDHAcKO
adipocyte-specific conditional knockout of H6PDH
- ITT
insulin tolerance test
- MS
metabolic syndrome
- NADPH
nicotinamide adenine dinucleotide phosphate
- 11-DHC
11-dehydrocorticosterone
- 11β-HSD1
11β-hydroxysteroid dehydrogenase type 1.
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Despres JP and Lemieux I (2006) Abdominal obesity and metabolic syndrome. Nature 444, 881–887, 10.1038/nature05488 [DOI] [PubMed] [Google Scholar]
- 2.Despres JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E et al. (2008) Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol 28,1039–1049, 10.1161/ATVBAHA.107.159228 [DOI] [PubMed] [Google Scholar]
- 3.Shimabukuro M (2009) Cardiac adiposity and global cardiometabolic risk: new concept and clinical implication. Circ. J 73, 27–34, 10.1253/circj.CJ-08-1012 [DOI] [PubMed] [Google Scholar]
- 4.Vegiopoulos A and Herzig S (2007) Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol 275, 43–61, 10.1016/j.mce.2007.05.015 [DOI] [PubMed] [Google Scholar]
- 5.Abraham SB, Rubino D, Sinaii N, Ramsey S and Nieman LK (2013) Cortisol, obesity, and the metabolic syndrome: a cross-sectional study of obese subjects and review of the literature. Obesity (Silver Spring) 21, E105–E117, 10.1002/oby.20083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A and Nieman LK (2009) Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J. Clin. Endocrinol. Metab 94, 3857–3864, 10.1210/jc.2008-2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cristancho AG and Lazar MA (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol 12, 722–734, 10.1038/nrm3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH and Sul HS (2004) Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem 279, 47066–47075, 10.1074/jbc.M403855200 [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Nakagawa Y, Liu L, Wang W, Ren X, Anghel A et al. (2011) Tissue-specific dysregulation of hexose-6-phosphate dehydrogenase and glucose-6-phosphate transporter production in db/db mice as a model of type 2 diabetes. Diabetologia 54, 440–450, 10.1007/s00125-010-1956-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alberti L, Girola A, Gilardini L, Conti A, Cattaldo S, Micheletto G et al. (2007) Type 2 diabetes and metabolic syndrome are associated with increased expression of 11beta-hydroxysteroid dehydrogenase 1 in obese subjects. Int. J. Obes 31,1826–1831, 10.1038/sj.ijo.0803677 [DOI] [PubMed] [Google Scholar]
- 11.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR et al. (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294, 2166–2170, 10.1126/science.1066285 [DOI] [PubMed] [Google Scholar]
- 12.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P et al. (1997) 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A 94, 14924–14929, 10.1073/pnas.94.26.14924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh H, Jeong KH, Han HY, Son HJ, Kim SS, Lee HJ et al. (2015) A potent and selective 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, SKI2852, ameliorates metabolic syndrome in diabetic mice models. Eur. J. Pharmacol 768,139–148, 10.1016/j.ejphar.2015.10.042 [DOI] [PubMed] [Google Scholar]
- 14.Anagnostis P, Katsiki N, Adamidou F, Athyros VG, Karagiannis A, Kita M et al. (2013) 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism 62, 21–33, 10.1016/j.metabol.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 15.Odermatt A, Arnold P, Stauffer A, Frey BM and Frey FJ (1999) The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J. Biol. Chem 274, 28762–28770, 10.1074/jbc.274.40.28762 [DOI] [PubMed] [Google Scholar]
- 16.Atanasov AG, Nashev LG, Schweizer RA, Frick C and Odermatt A (2004) Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett. 571,129–133, 10.1016/j.febslet.2004.06.065 [DOI] [PubMed] [Google Scholar]
- 17.Hewitt KN, Walker EA and Stewart PM (2005) Minireview: hexose-6-phosphate dehydrogenase and redox control of 11{beta}-hydroxysteroid dehydrogenase type 1 activity. Endocrinology 146, 2539–2543, 10.1210/en.2005-0117 [DOI] [PubMed] [Google Scholar]
- 18.McCormick KL, Wang X and Mick GJ (2006) Evidence that the 11 beta-hydroxysteroid dehydrogenase (11 beta-HSD1) is regulated by pentose pathway flux. Studies in rat adipocytes and microsomes. J. Biol. Chem 281, 341–347 [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Liu L, Du H, Nagaoka Y, Fan W, Lutfy K et al. (2014) Transgenic overexpression of hexose-6-phosphate dehydrogenase in adipose tissue causes local glucocorticoid amplification and lipolysis in male mice. Am. J. Physiol. Endocrinol. Metab 306, E543–E551, 10.1152/ajpendo.00491.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan C, Yang H, Wang Y, Dong Y, Yu F, Wu Y et al. (2016) Increased glycogen synthase kinase-3beta and hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to glucocorticoid-induced mouse visceral adiposity. Int. J. Obes 40,1233–1241, 10.1038/ijo.2016.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uckaya G, Karadurmus N, Kutlu O, Corakci A, Kizildag S, Ural AU et al. (2008) Adipose tissue 11-beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase gene expressions are increased in patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract 82, S135–S140, 10.1016/j.diabres.2008.09.022 [DOI] [PubMed] [Google Scholar]
- 22.Torrecilla E, Fernandez-Vazquez G, Vicent D, Sanchez-Franco F, Barabash A, Cabrerizo L et al. (2012) Liver upregulation of genes involved in cortisol production and action is associated with metabolic syndrome in morbidly obese patients. Obes. Surg 22, 478–486, 10.1007/s11695-011-0524-9 [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Garcia MA, San-Millan JL and Escobar-Morreale HF (2012) The R453Q and D151A polymorphisms of hexose-6-phosphate dehydrogenase gene (H6PD) influence the polycystic ovary syndrome (PCOS) and obesity. Gene 497, 38–44, 10.1016/j.gene.2012.01.047 [DOI] [PubMed] [Google Scholar]
- 24.Bujalska IJ, Hewitt KN, Hauton D, Lavery GG, Tomlinson JW, Walker EA et al. (2008) Lack of hexose-6-phosphate dehydrogenase impairs lipid mobilization from mouse adipose tissue. Endocrinology 149, 2584–2591, 10.1210/en.2007-1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Nakagawa Y, Wang Y, Liu L, Du H, Wang W et al. (2008) Reduction of hepatic glucocorticoid receptor and hexose-6-phosphate dehydrogenase expression ameliorates diet-induced obesity and insulin resistance in mice. J. Mol. Endocrinol 41, 53–64, 10.1677/JME-08-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zajkowska A, Rydzewska M, Wojtkielewicz K, Pomaski J, Romer T and Bossowski A (2017) A follow-up history of young man with apparent cortisone reductase deficiency (ACRD) - several years after diagnosis. Pediatr. Endocrinol. Diabetes Metab 23, 42–48, 10.18544/PEDM-23.01.0073 [DOI] [PubMed] [Google Scholar]
- 27.Lavery GG, Idkowiak J, Sherlock M, Bujalska I, Ride JP, Saqib K et al. (2013) Novel H6PDH mutations in two girls with premature adrenarche: ‘apparent’ and ‘true’ CRD can be differentiated by urinary steroid profiling. Eur. J. Endocrinol 168, K19–K26, 10.1530/EJE-12-0628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du H, Liu L, Wang Y, Nakagawa Y, Lyzlov A, Lutfy K et al. (2013) Specific reduction of G6PT may contribute to downregulation of hepatic 11beta-HSD1 in diabetic mice. J. Mol. Endocrinol 50, 167–178, 10.1530/JME-12-0223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Yan C, Liu L, Wang W, Du H, Fan W et al. (2015) 11beta-Hydroxysteroid dehydrogenase type 1 shRNA ameliorates glucocorticoid-induced insulin resistance and lipolysis in mouse abdominal adipose tissue. Am. J. Physiol. Endocrinol. Metab 308, E84–E95, 10.1152/ajpendo.00205.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ronquist-Nii Y and Edlund PO (2005) Determination of corticosteroids in tissue samples by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal 37, 341–350, 10.1016/j.jpba.2004.10.044 [DOI] [PubMed] [Google Scholar]
- 31.Szymczak J, Milewicz A, Thijssen JH, Blankenstein MA and Daroszewski J (1998) Concentration of sex steroids in adipose tissue after menopause. Steroids 63, 319–321, 10.1016/S0039-128X(98)00019-1 [DOI] [PubMed] [Google Scholar]
- 32.Lavery GG, Hauton D, Hewitt KN, Brice SM, Sherlock M, Walker EA et al. (2007) Hypoglycemia with enhanced hepatic glycogen synthesis in recombinant mice lacking hexose-6-phosphate dehydrogenase. Endocrinology 148, 6100–6106, 10.1210/en.2007-0963 [DOI] [PubMed] [Google Scholar]
- 33.Lavery GG, Walker EA, Turan N, Rogoff D, Ryder JW, Shelton JM et al. (2008) Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J. Biol. Chem 283, 8453–8461, 10.1074/jbc.M710067200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogoff D, Ryder JW, Black K, Yan Z, Burgess SC, McMillan DR et al. (2007) Abnormalities of glucose homeostasis and the hypothalamic-pituitary-adrenal axis in mice lacking hexose-6-phosphate dehydrogenase. Endocrinology 148, 5072–5080, 10.1210/en.2007-0593 [DOI] [PubMed] [Google Scholar]
- 35.Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ et al. (2014) 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. U.S.A 111, E2482–E2491, 10.1073/pnas.1323681111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P et al. (1997) 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A 94, 14924–14929, 10.1073/pnas.94.26.14924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR and Holmes MC (2001) Intracellular regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11β-HSD-1-deficient mice. Endocrinology 142, 114–120, 10.1210/endo.142.1.7887 [DOI] [PubMed] [Google Scholar]
- 38.Campbell JE, Peckett AJ, D’Souza AM, Hawke TJ and Riddell MC (2011) Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol 300, C198–C209, 10.1152/ajpcell.00045.2010 [DOI] [PubMed] [Google Scholar]
- 39.Xu C, He J, Jiang H, Zu L, Zhai W, Pu S et al. (2009) Direct effect of glucocorticoids on lipolysis in adipocytes. Mol. Endocrinol 23,1161–1170, 10.1210/me.2008-0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhat BG, Younis H, Herrera J, Palacio K, Pascual B, Hur G et al. (2008) Antisense inhibition of 11betahydroxysteroid dehydrogenase type 1 improves diabetes in a novel cortisone-induced diabetic KK mouse model. Biochem. Biophys. Res. Commun 365, 740–745, 10.1016/j.bbrc.2007.11.032 [DOI] [PubMed] [Google Scholar]
- 41.Hughes KA, Webster SP and Walker BR (2008) 11-Beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity. ExpertOpin. Investig. Drugs 17, 481–496, 10.1517/13543784.17.4.481 [DOI] [PubMed] [Google Scholar]
- 42.Huang Cao ZF, Stoffel E and Cohen P (2017) Role of perivascular adipose tissue in vascular physiology and pathology. Hypertension 69, 770–777, 10.1161/HYPERTENSIONAHA.116.08451 [DOI] [PubMed] [Google Scholar]
- 43.Shu J, Matarese A and Santulli G (2019) Diabetes, body fat, skeletal muscle, and hypertension: the ominous chiasmus? J. Clin. Hypertens 21, 239–242, 10.1111/jch.13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C et al. (2004) Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53, 931–938, 10.2337/diabetes.53.4.931 [DOI] [PubMed] [Google Scholar]
- 45.Catalano KJ, Stefanovski D and Bergman RN (2010) Critical role of the mesenteric depot versus other intra-abdominal adipose depots in the development of insulin resistance in young rats. Diabetes 59,1416–1423, 10.2337/db08-0675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu KH, Chu WC, To KW, Ko FW, Ng SS, Ngai JC et al. (2014) Mesenteric fat thickness is associated with increased risk of obstructive sleep apnoea. Respirology 19, 92–97, 10.1111/resp.12164 [DOI] [PubMed] [Google Scholar]
- 47.Liu L, Wang Y, Wang J, Dong Y, Chang S, Liu X et al. (2018) Enhanced hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to diet-induced visceral adiposity. Int. J. Obes 42,1999–2011, 10.1038/s41366-018-0041-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams LJ, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V et al. (2000) C/EBP regulates hepatic transcription of 11beta-hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J. Biol. Chem 275, 30232–30239, 10.1074/jbc.M001286200 [DOI] [PubMed] [Google Scholar]
- 49.Yang Z, Guo C, Zhu P, Li W, Myatt L and Sun K (2007) Role of glucocorticoid receptor and CCAAT/enhancer-binding protein alpha in the feed-forward induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J. Endocrinol 195, 241–253, 10.1677/J0E-07-0303 [DOI] [PubMed] [Google Scholar]
- 50.Handoko K, Yang K, Strutt B, Khalil W and Killinger D (2000) Insulin attenuates the stimulatory effects of tumor necrosis factor alpha on 11beta-hydroxysteroid dehydrogenase 1 in human adipose stromal cells. J. Steroid Biochem. Mol. Biol 72,163–168, 10.1016/S0960-0760(00)00029-7 [DOI] [PubMed] [Google Scholar]
- 51.Balachandran A, Guan H, Sellan M, van Uum S and Yang K (2008) Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 149, 4069–4079, 10.1210/en.2008-0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fulcher GR, Walker M, Catalano C, Agius L and Alberti KGMM (1992) Metabolic effects of suppression of nonesterified fatty acid levels with acipimox in obese NIDDM subjects. Diabetes 41, 1400–1408, 10.2337/diab.41.11.1400 [DOI] [PubMed] [Google Scholar]
- 53.Fery F, Plat L, Melot C and Balasse EO (1996) Role of fat-derived substrates in the regulation of gluconeogenesis during fasting. Am. J. Physiol 270, E822–E830, 10.1152/ajpendo.1996.270.5.E822 [DOI] [PubMed] [Google Scholar]