

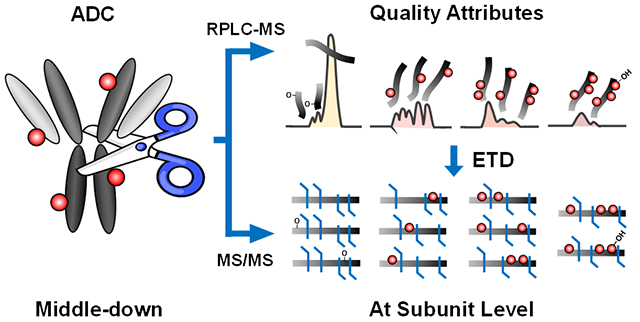
Abstract
Antibody-drug conjugates (ADCs) are designed to combine the target specificity of monoclonal antibodies and potent cytotoxin drugs to achieve better therapeutic outcome. Comprehensive evaluation of the quality attributes of ADCs is critical for drug development but remains challenging due to heterogeneity of the construct. Currently, peptide mapping with reversed-phase liquid chromatography (RPLC) coupled to mass spectrometry (MS) is the predominant approach to characterize ADCs. However, it is suboptimal for sequence characterization and quantification of ADCs because it lacks a comprehensive view of co-existing variants and suffers from varying ionization effects of drug-conjugated peptides compared to unconjugated counterparts. Here, we present the first middle-down RPLC-MS analysis of both cysteine (Adcetris®; BV) and lysine (Kadcyla®; T-DM1) conjugated ADCs at the subunit level (~25 kDa) with electron transfer dissociation (ETD). We successfully achieved high-resolution separation of subunit isomers arising from different drug conjugation and subsequently localized the conjugation sites. Moreover, we obtained a comprehensive overview of the micro-variants associated with each subunits and characterized them such as oxidized variants with different sites. Furthermore, we observed relatively high levels of conjugation near complementarity-determining regions (CDRs) from the heavy chain but no drug conjugation near CDRs of light chain (Lc) from lysine conjugated T-DM1. Based on the extracted ion chromatograms, we measured accurate average drug to antibody ratio (DAR) values and relative occupancy of drug-conjugated subunits. Overall, the middle-down MS approach enables the evaluation of multiple quality attributes including DAR, positional isomers, conjugation sites, occupancy, and micro-variants, which potentially open up a new avenue to characterize ADCs.
Graphical Abstract
INTRODUCTION
Over the past two decades, antibody drug conjugates (ADCs) have emerged as a novel class of bioconjugates consisting of cytotoxic agents and recombinant monoclonal antibodies (mAbs) interconnected by stable chemical linkers.1–3 The concept of ADCs exploits the specificity and stability of mAbs as well as the potency of the small molecule drugs against antigen-expressing targets, for instance, cancer cells.1, 4, 5 Although the idea of ADC being the “magic bullet” appears highly promising and attractive, the translation of ADCs into clinical practice and success has been enormously challenging. Currently, only 4 FDA-approved ADCs are on the market: brentuximab vedotin (BV; Adcetris®), ado-trastuzumab emtansine (T-DM1; Kadcyla®), inotuzumab ozogamicin (Besponsa®), and gemtuzumab ozogamicin (Mylotarg®) whereas more than 65 ADC candidates are under clinical evaluation.4
The majority of these ADCs on the market and under clinical trials are based on lysine or cysteine conjugation. Site specific conjugation technologies are promising but still under further development.6 Lysine conjugation occurs mainly on solvent accessible lysine residues (e.g., T-DM1), whereas cysteine conjugation takes place at the cysteine residues after the reduction of interchain disulfide bonds (e.g., BV).2 Consequently, both conjugation schemes manifest various positional isomers for a given drug-to-antibody ratio (DAR) species. The heterogeneity generated by the inherently random lysine or cysteine conjugation further complicates the analysis and characterization of ADCs, in addition to the high hydrophobicity introduced by the small molecule conjugation and the readily presence of post-translational modification (PTM) variants (e.g., amino acid clipping, oxidation, and glycosylation, etc.) on the mAbs. As a result, the efficacy, stability, and safety of the ADCs could be compromised.7 Therefore, a comprehensive characterization of the quality attributes is critically important during ADC development and quality control.
Among various analytical techniques used for analyzing ADCs, liquid chromatography coupled to mass spectrometry (LC-MS) remains one of the most versatile and powerful methods to obtain quality attributes.8, 9 In particular, peptide mapping with reversed phase liquid chromatography (RPLC)-MS, is a well-established and sensitive approach to identify and confirm amino acid sequences, variants, and modifications of ADCs.10 However, the bottom-up approach is prone to the introduction of artifacts due to prolonged sample preparation steps, and lacks of a comprehensive view on ADCs with different variants due to sequence variation, drug conjugation, and PTMs.11, 12 Moreover, because of the drastic differences in size, hydrophobicity, and protonation propensity, the attachment of small molecule drugs to peptides often results in strong retention on the column resin, poor recovery, and subdued ionization efficiency when compared to the unconjugated peptide counterparts. Hence, it makes the relative quantitation with the drug-conjugated and unconjugated peptides based solely on LC-MS peak intensity less reliable without full consideration of potential interferences.13
Alternatively, top-down14–17 and middle-down18, 19 approaches from the intact (~150 kDa) or subunits (~25 kDa) levels can provide a shorter sample preparation time, a more global and comprehensive view on the heterogeneity and variants,20, 21 and conceivably a more reliable quantitation (less dominant impact from drug-linker than the case of conjugated peptides).22, 23 Although mAbs have been extensively investigated in the past few years by top-down or middle-down MS,24–30 there are very limited studies on ADCs with these approaches.21 Recently, Dyachenko et al. successfully demonstrated native tandem MS (MS/MS) of a cysteine conjugated ADC, BV.21 In addition to profiling the overall heterogeneity, they were able to isolate individual DAR species and performed MS/MS to obtain information on drug localization.21 However, because of the large molecular weight of ADCs, MS/MS from an intact level results in limited sequence coverage and therefore remain challenging to precisely localize conjugation site to a single amino acid residue. Furthermore, positional isomers and low abundance micro-variants are difficult to separate and characterize from a top-down approach. Alternatively, subunits at a size around 25 kDa produced by enzymes such as IdeS appear to be more feasible and promising.31 Nevertheless, middle-down LC-MS methods with MS/MS to characterize ADCs, particularly to pinpoint conjugation sites of isomers and to characterize small variants, remain unexplored.
Here, we developed a middle-down RPLC-MS strategy with ETD to investigate both cysteine and lysine conjugated ADCs (i.e., BV and T-DM1) at the subunit levels. The middle-down method evaluates multiple analytical attributes including average DAR, positional isomers, conjugation sites, occupancy, and micro-variants. For BV, we explored both IdeS32 (FabRICATOR) and KGP33 (GingisKHAN) enzymes as well as partial (only inter-chain reduced) and full reduction of all disulfide bonds in order to maximize the fragmentation around the hinge region for confident assignment of the conjugation sites (Scheme 1). For T-DM1, IdeS digestion and full reduction was used to achieve high yields of subunits for RPLC separation and efficient fragmentation. Overall, the described middle-down RPLC-MS approach with ETD shows a great potential for examining multiple quality attributes of ADCs during product development and quality control.
Scheme 1.
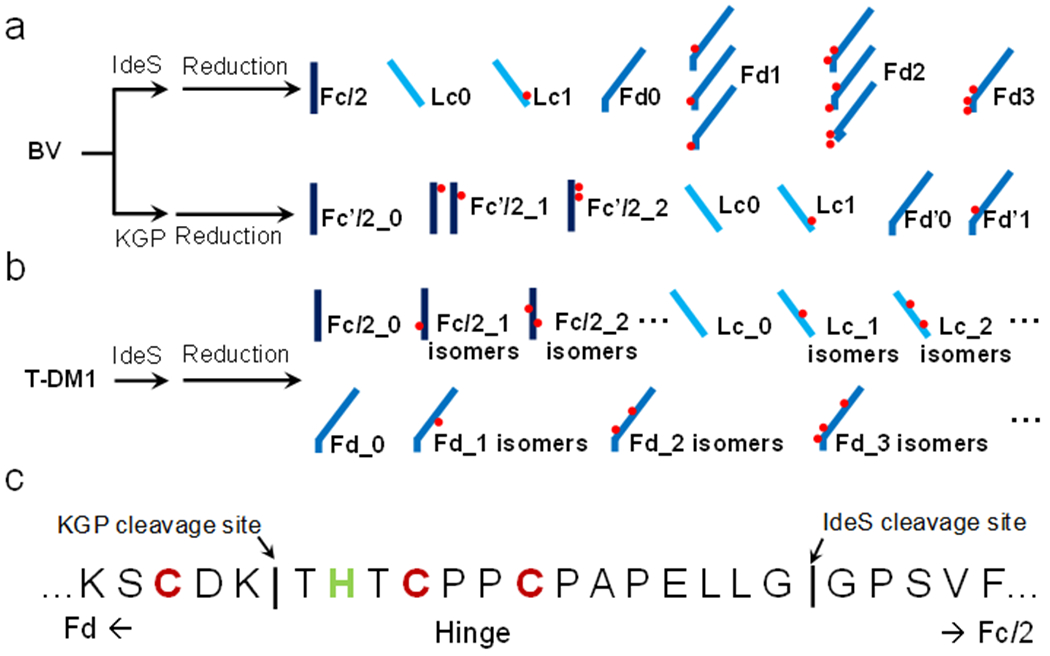
(a) Illustration of the subunits generated from BV (Adcetris) by IdeS and KGP enzymes. (b) Illustration of the subunits generated from T-DM1 (Kadcyla) by IdeS. (c) Cleavage sites of IdeS or KGP enzymes near the hinge region.
MATERIAL AND METHOD
Reagents.
HPLC grade water and isopropyl alcohol (IPA) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Formic acid (FA), 3,3,3-Trifluoropropionic acid (TFPA), 2-amino-2-(hydroxymethyl)-1,3-propanediol hydrochloride (Tris-HCl), dithiothreitol (DTT), tris(2-carboxyehtyl)phosphine hydrochloride (TCEP) and guanidine hydrochloride (HCl) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Sample preparation.
Brentuximab vedotin (BV; Adcetris®; Seattle Genetics), Ado-trastuzumab emtansine (T-DM1; Kadcyla®; Roche) were purchased and provided by AbbVie (North Chicago, IL, USA). FabRICATOR (IdeS), GingisKHAN (KGP), and GlyCINATOR (EndoS2) were purchased from Genovis (Sweden). IdeS and KGP proteolysis was carried out based on manufacturer’s recommendation. Additional details are provided in the Supporting Information.
LC-MS/(MS).
A Waters ACQUITY UPLC M-class system (Milford, MA, USA) was coupled to a maXis II ETD Q-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). 150 mm × 0.5 mm BIOshell A400 Protein C4 columns with 3.4 μm particle size (Sigma-Aldrich, St. Louis, MO, USA) was used for the reversed-phase separation. On column desalting was performed and bypassed the buffer salts into waste. For the mass spectrometer, the end plate offset and capillary voltage were optimized at 500 V and 4500 V, respectively. The nebulizer was set to 0.5 bar, and the dry gas flow rate was 4.0 L/min at 220 °C. Collision cell RF was set to 2000 Vpp, transfer time to 110 μs, and pre pulse storage to 10 μs for ion desolvation and transmission. The quadruple low mass was set to 500 m/z and the scan range was from 200-3500 m/z at 1Hz. ETD reagent was 3, 4-hexanedione (114 m/z negative ion). Precursor accumulation was ranging from 400 to 1000 ms, the reagent time ranging from 5-22 ms, and the reaction time ranging from 0-2 ms. Additional details are provided in the Supporting Information.
Data Analysis.
All data were processed and analyzed by Compass DataAnalysis 4.3 and MASH Suite Pro34. Extracted ion chromatograms were created based on top-5 most abundant charge state ions with an isolation window of 2 m/z for DAR calculation and 0.5 m/z for oxidation investigation. Chromatograms were smoothed for illustration. Area under curve was subsequently integrated for DAR calculation. Maximum Entropy was used for spectrum deconvolution with resolution set 80,000 for the high-resolution spectra. SNAP algorithm was used for peak picking, in which quality threshold was set to 0.4. Small molecule drugs and drug linkers were treated as variable PTMs and tentatively assigned to putative conjugation sites for sequence coverage comparison. We then manually validated all fragment ions in MASH Suite Pro and assigned conjugation sites based on the number of fragment ions as well as the presence of diagnostic ions (drug conjugated fragment ions).
RESULTS AND DISCUSSION
RPLC-MS/(MS) for the cysteine conjugated ADC
IdeS cleaves BV below the hinge region followed by a reduction step to produce subunits around 25 kDa (Scheme 1)35, 36. Sample treatment with IdeS, GlycINATOR, and a disulfide reduction step (partial or full) resulted in a total of seven separated subunits on a RPLC-MS run including Fc/2, Lc without drug (Lc0), Lc with 1 drug (Lc1), Fd without drug (Fd0), Fd with 1-3 drugs (Fd1-3, respectively) (Figure 1 and Figure S1). Among all the subunits, each of Fd1 and Fd2 contained two isomeric peaks, namely, Fd1a, Fd1b, and Fd2a, Fd2b. We assigned these putative identifications based on accurate masses of the subunits and evaluated the reproducibility and sample loading with replicates (Figure S2).
Figure 1.
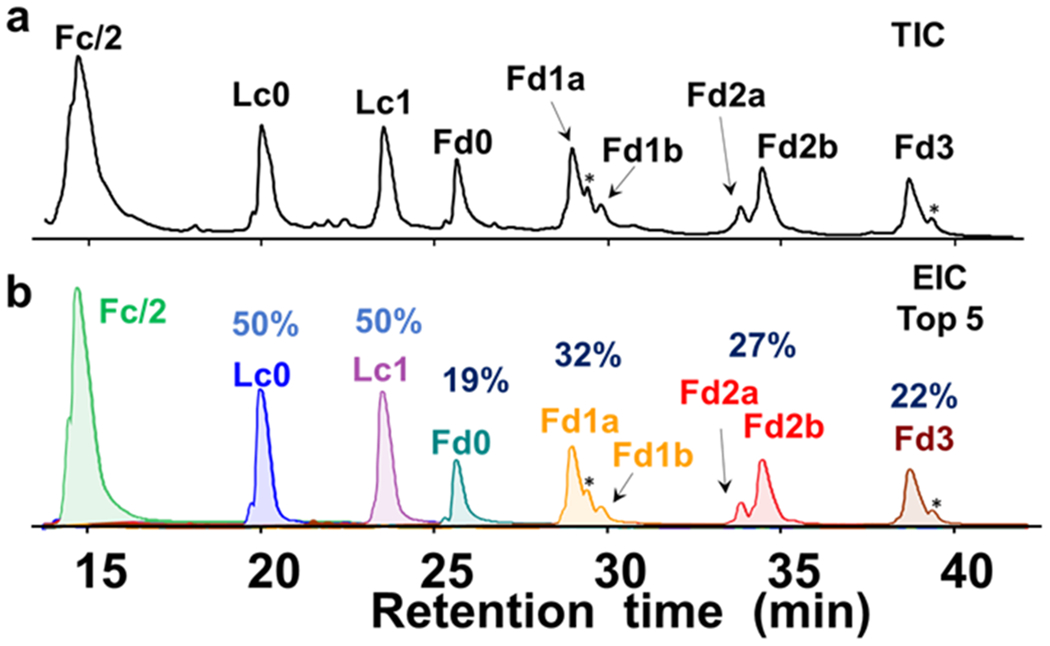
Online RPLC-MS of fully reduced BV digested by IdeS. (a) Total ion chromatogram (TIC) and (b) Extracted ion chromatograms of top-5 charge states of each subunits. Relative percentage of unconjugated and conjugated species for Lc and Fd were shown. * represents +18 Da species corresponding to the hydrolysis of one maleimide ring on the drug linkers.
While we observed all seven subunits from the partial and full reduction results, chromatogram with better separation and higher recovery of Fd2 and Fd3 were achieved when all disulfide bonds were reduced (Figure 1). Moreover, full reduction is preferred, because precise control of the partial reduction was difficult, which led to the observation of small and stronger retaining peaks corresponding to subunits with one more intra-chain disulfide bond reduced (Figure S1). Because of the small size of the drug with respect to the large subunits, we expect similar ionization efficiency among unconjugated and conjugated subunits based on previous studies.22, 37–40 By extracting the top-5 most abundant charge states and calculating the AUC of the EIC of each subunit from the fully reduced samples (Figure 1), we found 50% of Lc were conjugated with 1 drug and 32%, 27%, and 22% of Fd were conjugated with 1, 2, and 3 drugs respectively. Importantly, when fully reduced, the drug-linker portion of Fd2 and Fd3 subunits were more susceptible to fragmentation (loss of 761 Da) during ionization and ion transfer process in the mass spectrometer (Figure S3). Therefore, we have included the top-5 charge states of the Fd2 and Fd3 subunits with a loss of 761 Da (corresponding to the peak at 762 m/z) in addition to the intact Fd2 and Fd3 for the average DAR calculation. An average DAR of 4.0, which was in good agreement with the reference value using UV measurement, were obtained for BV based on the EIC (Table S1)41. Our data demonstrated that calculation of average DAR value using EIC of subunits was consistent with UV results.
The MS1 spectra of the middle-down approach also provided a comprehensive view of each subunit and its associated micro-variants. For example, the EIC of Fc/2 showed a minor peak eluting slightly earlier than the major peak (Figure 2a). The deconvoluted spectrum of peak (i) reveals the oxidized (+16 Da) and the unclipped C-terminal lysine (+128 Da) variants of Fc/2 (Figure 2b). Both of these two species are more hydrophilic, leading to an earlier elution than the main peak (ii). The deconvoluted spectrum of the main peak (ii) corresponds to Fc/2 with all intra-chain disulfide bonds reduced (27093.00 Da) and the variants with the core GlcNAc and fucose after deglycosylation (GlycINATOR), conforming the site of N-glycan attachment of Fc chain. Minor variants of water loss (−18 Da) and afucosylated species (−164 Da) were observed (Figure 2b). 42 We also observed a prevalent +18 Da variants of drug conjugated subunits (i.e., Lc1, Fd1, Fd2, and Fd3), likely due to the hydrolysis of succinimide-thioether ring43. The EIC of Fd3 shows the separation of the minor peak (ii) from main peak (i) (Figure 2c). Deconvoluted mass spectra reveal the later eluting peak (ii) corresponds to Fd3 with one succinimide ring being hydrolyzed on the drug linker moiety, with an additional mass shift of 18 Da (Figure 2d).
Figure 2.
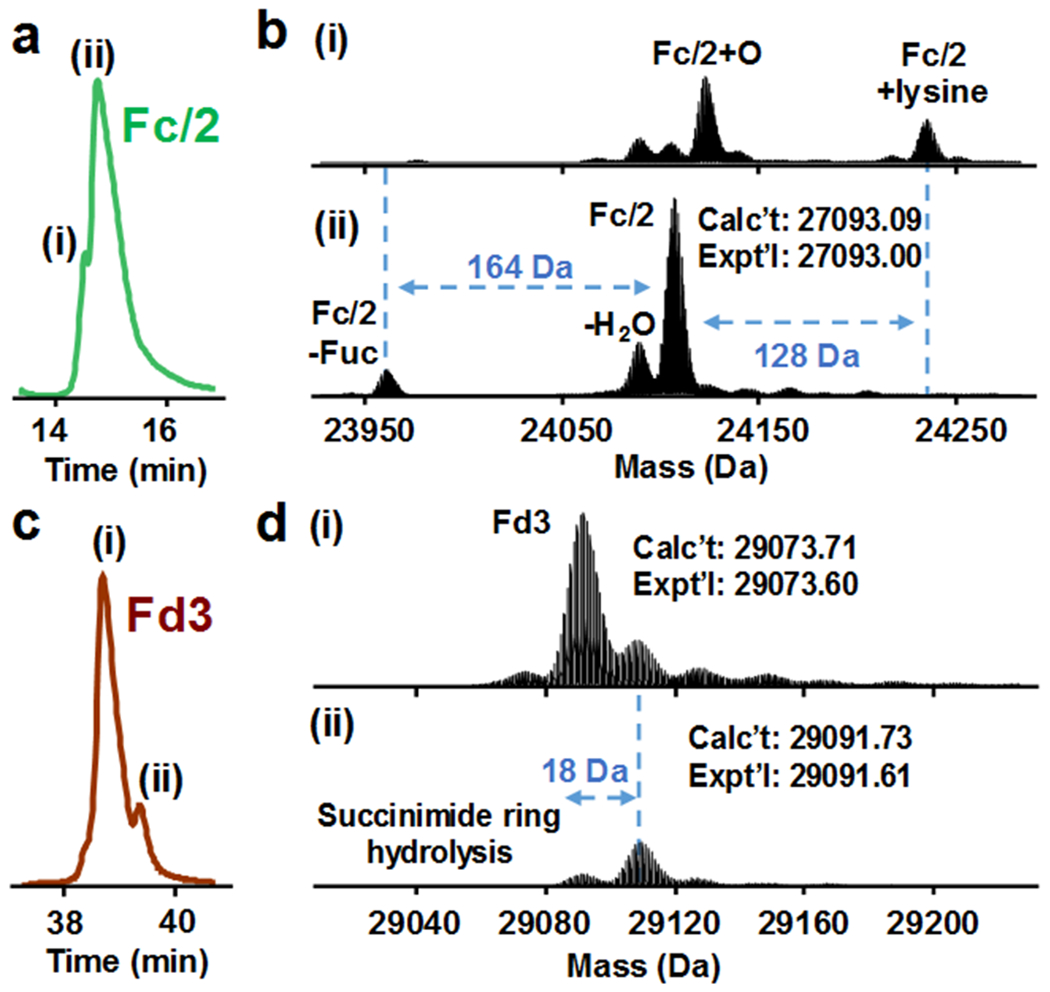
Micro-variants of BV subunits revealed by middle-down RPLC-MS. (a) Extracted ion chromatogram of Fc/2 showing two peaks (i) and (ii). (b) Deconvoluted spectra of peak (i) and (ii). (c) Extracted ion chromatogram of Fd3 showing two peaks (i) and (ii). (d) Deconvoluted spectra of peak (i) and (ii).
Next, we developed a middle-down approach with MS/MS aiming to characterize micro-variants and localize conjugation sites. When first applying collision-induced dissociation (CID) during MS/MS experiments, we found CID preferentially triggers the immolation of p-aminobenzyl alcohol fragmentation around the carbamate bonds of the drug-linker portion of BV, leading to predominantly drug fragments and no subunit backbone cleavages (Figure S4). Therefore, we subsequently applied electron-transfer dissociation (ETD) to all the subunits, which was known to provide rich sequence information with the preservation of labile bonds.44
Among all the observed micro-variants, oxidation in the Fc/2 subunit of BV is particularly interesting, as methionine or tryptophan oxidation on the Fc region can impact the binding of Fc receptors, stability, and half-life of the mAb.45, 46 By extracting the top-5 most abundant charge states of oxidized Fc/2 and Fc/2 with a narrower isolation window (0.1 m/z), we observed the separation of more hydrophilic oxidized Fc/2 from un-oxidized Fc/2 (Figure 3a). Two distinct peaks were observed from the EIC of oxidized Fc/2, indicating the potential existence of isomers and more than one oxidation sites. Subsequent ETD MS/MS comparison between the oxidized Fc/2 isomers (i and ii) and unoxidized Fc/2 (iii) confirmed our speculation (Figure 3b). Oxidized z●57 was detected with no observation of oxidized c57 from oxidized Fc/2 isomer i, whereas oxidized c57 was observed with no detection of oxidized z●57 from oxidized Fc/2 isomer ii. The finding suggested that there are at least two oxidation sites, one near the C-terminus (Met16 or Tyr41), and the other near N-terminus of Fc/2 (Tyr181 or Met192). The locations of the oxidation sites were in good agreement a previous mAb study in which forced degradation were performed to induce two oxidation sites.27 Overall, 11% of the Fc/2 were oxidized without force degradation; and a higher level of oxidation was found near the N-terminus (ii) than C-terminus (i) of Fc/2 based on the AUC of the EICs. Therefore, the developed middle-down method is capable to investigate oxidation at a relatively low level. It is worth noting that part of the oxidation was caused by elevated column temperature and long gradient time47. Although slightly lower level of oxidation could be achieved with shorter gradient and lower temperature, separation of positional isomers and accumulation of MS/MS scans would be compromised.
Figure 3.
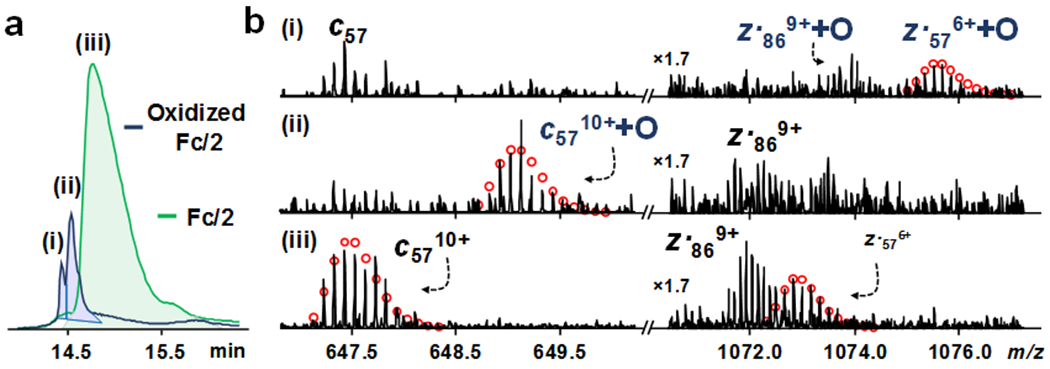
ETD LC-MS/MS characterization of oxidation of BV subunit Fc/2. (a) Extracted ion chromatogram of oxidized Fc/2 (i and ii isomers) and un-oxidized Fc/2 (iii) (b) ETD spectra validating the present of oxidized Fc/2 isomers. The intensities were normalized and 1071-1077 m/z was zoomed in 1.7 times. Theoretical ion distributions are indicated by the red dots.
For the analysis of different subunits, most subunits with or without conjugated drugs yielded a sequence coverage about 35% with ETD when all the intra-chain disulfide bonds were reduced (Figure 4a, S5 and S6). The N-glycosylation site on the Fc/2 was therefore confidently assigned with many fragment ions containing the core GlcNAc with fucose after deglycosylation (GlyCINATOR). When intra-chain disulfide bonds were not reduced, the overall sequence coverage was about 15% for most of the subunits (Figure 4b, S7 and S8). The seemingly low fragmentation efficiency and sequence coverage was caused by the presence of disulfide bonds, which led to nearly no cleavages of the residues within the disulfide bridges.48 However, as a result, more extensive ETD fragmentation were observed in the middle of the sequence outside of the disulfide bonds confinement, particularly in the middle of the sequence.
Figure 4.

ETD LC-MS/MS characterization of fully (a) and partially (b) reduced Fd1a isomer of BV digested by IdeS. (a) ETD Fragmentation maps of fully reduced Fd1a with (b) representative ETD fragment ion with drug (z●244+ and z●10811+) and without drug (c12512+). The red circles with a dash line indicate possible localization of the single conjugation site. (c) ETD Fragmentation maps of partially reduced Fd1a with (d) diagnostic ETD fragment ion with drug (z●24 4+) and without drug (z●15 2+ and z●16 2+), localizing the conjugation site to Cys220. The green circle with a solid line indicates confident localization of the conjugation site. Theoretical ion distributions are indicated by the red dots.
Although the fully reduced ETD spectra provided higher overall sequence coverage and confidently localize the drug conjugation sites in Lc1 and Fd3, it has yet to unambiguously localize the conjugation sites in Fd1a compared to the partially reduced ETD spectra. In the partially reduced ETD spectrum (Figure 4c), we were able to pinpoint the conjugation site of isomer Fd1a to the cysteine above the hinge region based on the detection of unconjugated (z●15 and z●16) and drug conjugated (z●24) diagnostic fragment ions from the partially reduced conditions (Figure 4d). In contrast, even though the fully reduced ETD spectrum provided high sequence coverage (Figure 4a and b), key diagnostic ions (e.g., z●15 and z●16) to preclude possible conjugation on hinge region cysteines (CPPC) were undetectable. Therefore, partial reduction may work better from a site localization perspective, because the disulfide linkages limit ETD cleavages to only certain regions, resulting in simpler spectra with higher signal to noise.
For Fd1b, Fd2a, and Fd2b, confident assignment of conjugation sites from both partially and fully reduced ETD spectra remained challenging. Although the fully reduced subunits bear more charges and therefore a high reaction cross section during ETD event, diagnostic ions around the hinge regions of Fd were still not detected, leaving ambiguous localization of conjugation sites (Figure S6). For the partially reduced subunits, the z●23 ion from Fd2a and Fd2b was only able to confirm that there are two drugs located near to the C-terminus hinge regions out of three possible conjugation cysteines. (Figure S8). We speculated that this is due to the lack of charged residues on the C-terminus (around the hinge region) of Fd and potential steric hindrance of the drug and proline residues blocking electron/radical transfer on the protein backbone.
In order to differentiate positional isomer and localize conjugation sites around the hinge region, we have further investigated the use of enzyme KGP to cleave BV above the hinge region into new subunits below 30 kDa (Scheme 1 and Figure S9). With KGP, the hinge region locates near the N-terminus of Fc’/2 (near C-terminus of Fd from IdeS). c-type fragment ions therefore includes a charge from N-terminal amine. The c6 ion (THTCPP|CPAP) between two potential conjugated cysteine residues were observed in the Fc’/2 as well as Fc’2 with 1 drug (Fc’2_1) during ETD experiments (Figure S10 and S11a). However, drug conjugated c6 ion were not detected neither in Fc’2_1 nor in Fc’2_2 (Figure S11). Therefore, drug conjugated isomers around the hinge region (CPPC) remain indistinguishable with our current middle down method (see extended discussion in supplementary information).
Overall, all the subunits of BV and some positional isomers were well-separated in the newly developed middle-down RPLC-MS/(MS) method. We obtained an average DAR value of 4.0 and assessed relative conjugation levels of each subunits for BV from AUC of EICs of top-5 most abundant charge states. From the MS1 spectra, micro-variants associated with the subunits were separated and revealed. Moreover, we examined the relatively low level of oxidation of Fc/2, showing the presence of more than one oxidation sites by using ETD. We further characterized partially and fully reduced BV subunits. Although the overall sequence coverage was higher when subunits were fully reduced, more extensive fragmentation around the middle and the C-terminus of drug conjugated Fd were obtained when subunits were partially reduced with intra-chain disulfide bonds intact. Nevertheless, our middle-down with IdeS and KGP as well as the current peptide mapping approaches were unable to differentiate the two cysteine residues around the hinge regions (THTCPPCPAP),23 possibly because of the lack of charged residues and the steric hindrance of the small molecule drug during fragmentation events.
RPLC-MS/(MS) for the lysine conjugated ADC
T-DM1 is an ADC that could have drug-linker attachment to any of the 88 Lysine residues and the N-terminal amines of Lc and Hc. Because of the complexity arising from all the possible conjugation sites, peptide mapping approach has been exclusively used to localize conjugation sites and to quantify their relative abundance. However, potential drawbacks include prolonged sample preparation, complicated inference of the whole ADC, and ionization suppression of drug conjugated peptides during quantification. Here, we developed and evaluated a RPLC middle-down approach with ETD, aiming to provide a more global view of the lysine-conjugated, localization of the abundant conjugation sites, and reliable assessment of relative abundance.
To facilitate MS/MS analysis, IdeS and strong reducing conditions were used to produce high yield of subunits with complete reduction of inter-chain and intra-chain disulfide bonds. From the TIC of T-DM1 (Figure 5a), we observed subunits with an elution order of Fc/2_0, Fc/2_1, Lc0, Lc1, Lc1, Fc/2_2, Fd0, Lc2, Fd1, Fd2, Fd3, along with some possible low abundance truncated species (Mr 20404.23 Da) at the beginning of the gradient. We next extracted the top-5 most abundant ions of each subunits, revealing the existence of multiple isomers of drug conjugated subunits in the EIC (Figure 5b, c, d). Only EIC traces of the corresponding subunits at the right elution time were shown due to the complexity of overlapping charge states of other subunits (Figure S12). In particular, in Fd1, multiple isomeric peaks were observed (Figure 5d). For Fc/2 and Lc, subunits with up to 2 drug conjugation were detected. Although the abundance was relatively low, Fd subunit with up to 3 drug conjugation was observed. By integrating AUC of the EIC of each subunit, we found that about 50% of the subunits were conjugated with small molecule drugs, and the average DAR of 3.5 can be therefore calculated (Table S2), in good correspondence to the reference value of 3.5.49
Figure 5.
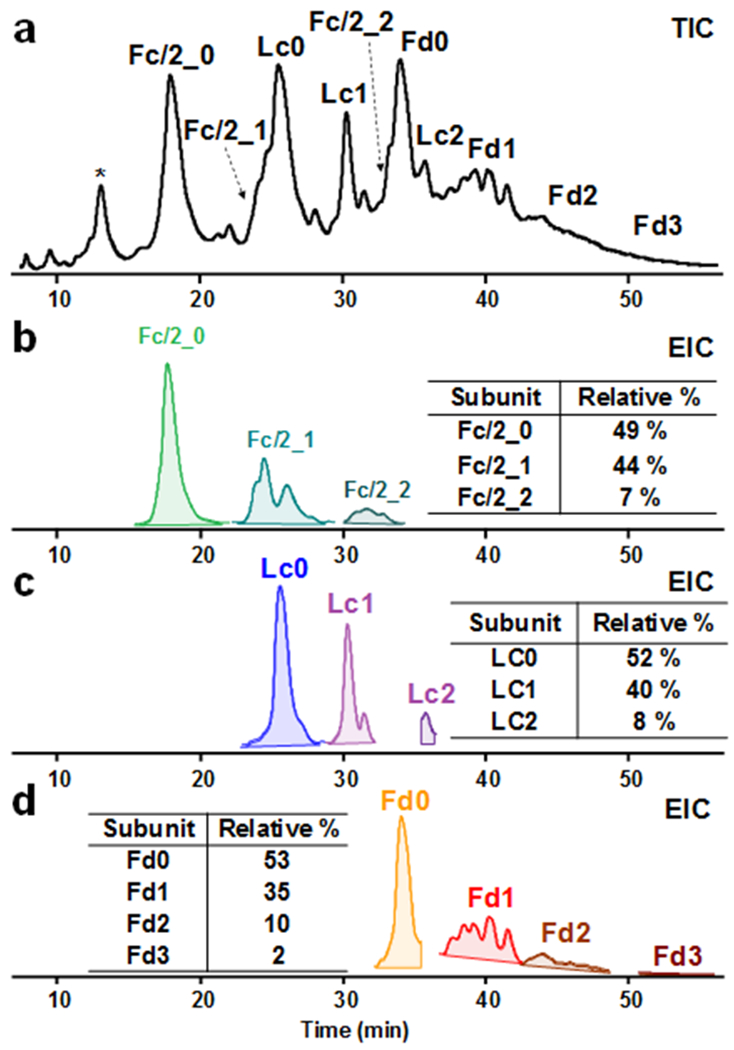
LC-MS analysis of subunits of T-DM1 digested by IdeS. (a) Total ion chromatogram (TIC).* corresponds to a possible truncation with a mass of 20404.23 Da. (b) Extracted ion chromatogram (EIC) of Fc/2 with 0, 1, and 2 drugs (Fc/2_0, Fc/2_1, and Fc/2_2). (c) EIC of Lc with 0, 1, and 2 drugs (Lc0, Lc1, and Lc2). (d) EIC of Fd with 0, 1, 2, and 3 drugs (Fd0, Fd1, Fd2, and Fd3). The inset tables show the relative percentage of each subunit based on the area under curve. Intensities are normalized in b, c, and d.
Next, we investigated conjugation sites and their relative abundance, particularly for Lc1 and Fd1. Lc and Fd subunits are important targets to study because they contain complementarity determining regions (CDRs) and drug conjugation on certain sites can potentially influence antigen binding. Furthermore, subunits with a single drug conjugation sites consisted of majority of drug conjugated species and more feasible for further investigation. We therefore applied ETD to localize conjugation sites of the relatively abundant Lc1 and Fd1.
We further optimized the gradient to improve separation particularly for Fd1 subunits. As shown in Figure 6a, 2 peaks were observed for Lc1 isomers, and 11 peaks were separated for Fd1 isomers from the EICs. For Lc1, although isomers were not readily resolved from the EIC, we examined the targeted ETD MS/MS spectra along the corresponding elution window. Interestingly, after examining the fragment ions, no drug-conjugated c-ions were observed. In fact, we found that the conjugation sites were all localized near the C-terminus, away from the CDRs regions of the Lc (Figure 6b). The close proximity of these potential conjugated sites (K169, K183, K188, K190, and K207) near the C-terminus explains the inefficiency of separating the Lc1 isomers (Figure 6a and Figure S13).
Figure 6.
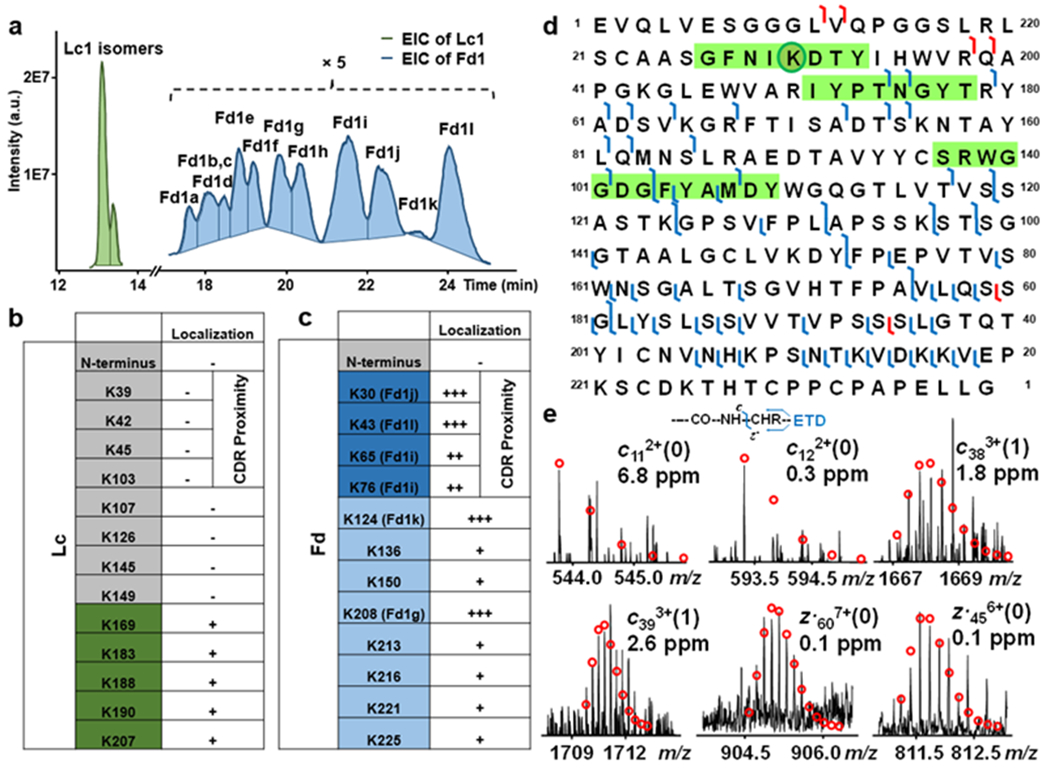
ETD LC-MS/MS characterization of T-DM1 Lc1 and Fd1 subunits digested by IdeS. (a) Extracted ion chromatograms (EICs) of Lc1 (green) and Fd1 (blue) (5x zoom-in). Summary of potential and confirmed conjugation sites of (b) Lc1 and (c) Fd1 species of T-DM1 digested by IdeS with ETD LC-MS/MS. +++: confidently localized conjugation sites; ++: highly possible conjugation sites; +: possible conjugation sites; and −: no conjugation observed. The darker blue shades indicates relatively higher abundance (d) Fragmentation map of Fdj localizing the conjugation sites of K30 within CDR1-VH. The highlighted regions in the sequence maps correspond to the CDR regions in the heavy chain. (e) Representative ETD fragment ions with one drug (c383+ and c393+) and without drug (c112+, c122+, z.396+ and z.115115+), corresponding to the red cleavages in d. Theoretical ion distributions are indicated by the red dots.
For Fd1, at least 11 isomers can be observed from the EIC (Figure 6a). We were able to unambiguously localize the conjugation sites of four of these isomers, and narrowed down the location of the conjugation sites for the others (Figure 6c, S14 and S15). We also found that Fd1 isomers with conjugation sites near the C-terminus tend to elute earlier than those isomers with conjugation sites near the N-terminus. Among these, high level of conjugation was found in the middle of CDR1-VH (K30) as well as near the other two CDR regions (CDR2-VH K43 and CDR3-VH K65/K76) (Figure 6c). Specifically, the conjugation site of K30 corresponds to Fd1j. Figure 6d shows the representative fragmentation map of Fdj from an online RPLC-ETD MS/MS analysis. Confident localization of the conjugation site to K30 was based on accurate detection of diagnostic fragment ions (Figure 6d). However, accurate quantification requires baseline separation of these isomers, which still remains challenging for those with drug conjugation sites in close proximity.
Our analysis demonstrated the potential of applying middle down approaches to characterize lysine-conjugated ADCs. Because of the complexity of lysine conjugation, analysis of subunits with more than one conjugated drug remains challenging. However, our data revealed that majority of the drug-containing subunits contains only one drug. Subunits with more than one drugs would likely to be a combinatorial arrangement of the conjugation sites detected on the subunits with one drug conjugated. Therefore, the analysis of subunits containing one drug should shed light on the location of conjugation sites and their relative abundance as demonstrated for Fd1 isomers from our data.
Although peptide mapping is prevalent and widely-used for T-DM1, various results of characterizing T-DM1 were reported. Recently, Chen et al. used a peptide mapping approach to characterize the conjugation sites and compared their relative abundance.50 They quantified over 35 conjugation sites with various abundance. Sang et al. also investigated the conjugation sites of T-DM1 in their more recent work, in which they were only able to localize and quantify less than a half of the conjugation sites found in Chen et al.’s work.13 However, instead of directly comparing the intensity of the unconjugated and the conjugated peptides, they performed serial dilution to create a calibration curve to account for the ionization and detection difference of each drug conjugated peptides. Therefore, other than the localization of conjugation sites, relative abundance of the same sites in these two works are vastly different. Our results revealed that for Lc1, nearly all drug conjugation is located far away from the CDRs (Lc K169, K183, K188, K190, and K207), which is consistent with the data Sang et al. observed. On the other hand, high level of conjugation was found on and around the CDR regions of the Fd subunit from the Hc, in particular, K65/K76 (Fd1i), K30 (Fd1j), and K43 (Fd1l), which provides an alternative perspective to the peptide mapping approach. All of these were in relatively high abundance compared to the other Fd1 isomers and are in close proximity with the CDRs of the heavy chain, which could be a potential risk impacting the specificity of antigen binding. Furthermore, by using the AUC of the EIC of each subunits, comparable average DAR value (3.5) to reference value (3.5) can be obtained. Therefore, we reason that the conjugation of a small molecule drug (1 kDa) to a relatively large size subunit (25 kDa) appears not to severely alter the ionization efficiency. Overall, although full dissection of all conjugation sites remains challenging, our results showed that middle-down analysis with ETD is a promising way to gain information about drug conjugation sites, and provides a better relative quantification considering the significant influence of small molecule drug on peptide ionization.
CONCLUSION
In this work, we present a comprehensive middle-down investigation on both cysteine (BV) and lysine (T-DM1) conjugated ADCs with ETD to probe multiple quality attributes. With this middle-down approach, we not only detected variants associated with each subunit from a comprehensive view, but also demonstrated that relatively low level of oxidation can be investigated and quantified. Because of the small size of the drug molecule and the similar molecular weight of each subunit, we were able to obtain accurate average DAR values (4.0 and 3.5 for BV and T-DM1 respectively) and relative percentage of drug-conjugated subunits from the EICs. In particular, we applied ETD on the separated positional isomers of drug-conjugated subunits from BV and T-DM1, demonstrating the feasibility of separating and localizing conjugation sites of positional isomers in addition to assessing their relative abundance. With the other emerging effective fragmentation techniques such as UVPD51 and activated ion ETD (AI-ETD),52 as well as further improvement on separation, middle-down MS characterization of ADCs will become more effective and powerful. Taken together, we have shown the middle-down methods with high-resolution MS allows for the determination of average DAR, positional isomers, conjugations sites, occupancies, and variants from the subunit levels, providing a useful and complementary tool for characterizing multiple analytical attributes of ADCs during the development of novel therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by AbbVie Inc. We acknowledge NIH grants, GM117058, GM125085, R01HL109810, R01HL096971 and high-end instrument grant S10OD018475 (to Y.G.). We would also like to thank Dr. Linjie Han and Dr. Shiyue Zhou for helpful discussion and critical reading of the manuscript.
Footnotes
Supporting Information Available:
This material is available free of charge via the Internet at http://pubs.acs.org. Additional information as noted in the text.
REFRENCES
- 1.Beck A; Goetsch L; Dumontet C; Corvaia N, Strategies and challenges for the next generation of antibody drug conjugates. Nature Reviews Drug Discovery 2017, 16 (5), 315–337. [DOI] [PubMed] [Google Scholar]
- 2.Ducry L; Stump B, Antibody-Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjugate Chemistry 2010, 21 (1), 5–13. [DOI] [PubMed] [Google Scholar]
- 3.Chari RVJ; Miller ML; Widdison WC, Antibody- Drug Conjugates: An Emerging Concept in Cancer Therapy. Angewandte Chemie-International Edition 2014, 53 (15), 3796–3827. [DOI] [PubMed] [Google Scholar]
- 4.Lambert JM; Berkenblit A, Antibody-Drug Conjugates for Cancer Treatment In Annual Review of Medicine, Vol 69, Caskey CT., Ed. 2018; Vol. 69, pp 191–207. [DOI] [PubMed] [Google Scholar]
- 5.Sievers EL; Senter PD, Antibody-Drug Conjugates in Cancer Therapy In Annual Review of Medicine, Vol 64, Caskey CT, Ed. 2013; Vol. 64, pp 15–29. [DOI] [PubMed] [Google Scholar]
- 6.Panowski S; Bhakta S; Raab H; Polakis P; Junutula JR, Site-specific antibody drug conjugates for cancer therapy. Mabs 2014, 6 (1), 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strop P; Liu SH; Dorywalska M; Delaria K; Dushin RG; Tran TT; Ho WH; Farias S; Casas MG; Abdiche Y; Zhou DH; Chandrasekaran R; Samain C; Loo C; Rossi A; Rickert M; Krimm S; Wong T; Chin SM; Yu J; Dilley J; Chaparro-Riggers J; Filzen GF; O’Donnell CJ; Wang F; Myers JS; Pons J; Shelton DL; Rajpal A, Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chemistry & Biology 2013, 20 (2), 161–167. [DOI] [PubMed] [Google Scholar]
- 8.Fekete S; Guillarme D; Sandra P; Sandra K, Chromatographic, Electrophoretic, and Mass Spectrometric Methods for the Analytical Characterization of Protein Biopharmaceuticals. Analytical Chemistry 2016, 88 (1), 480–507. [DOI] [PubMed] [Google Scholar]
- 9.Wagh A; Song HT; Zeng M; Tao L; Das TK, Challenges and new frontiers in analytical characterization of antibody-drug conjugates. Mabs 2018, 10 (2), 222–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouchahoir T; Schiel JE, Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal Bioanal Chem 2018, 410 (8), 2111–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren D; Pipes GD; Liu DJ; Shih LY; Nichols AC; Treuheit MJ; Brems DN; Bondarenko PV, An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal Biochem 2009, 392 (1), 12–21. [DOI] [PubMed] [Google Scholar]
- 12.Krokhin OV; Antonovici M; Ens W; Wilkins JA; Standing KG, Deamidation of -Asn-Gly-sequences during sample preparation for proteomics: Consequences for MALDI and HPLC-MALDI analysis. Analytical Chemistry 2006, 78 (18), 6645–6650. [DOI] [PubMed] [Google Scholar]
- 13.Sang H; Lu GY; Liu YZ; Hu QY; Xing WF; Cui DB; Zhou F; Zhang JW; Hao HP; Wang GJ; Ye H, Conjugation site analysis of antibody-drug-conjugates (ADCs) by signature ion fingerprinting and normalized area quantitation approach using nano-liquid chromatography coupled to high resolution mass spectrometry. Analytica Chimica Acta 2017, 955, 67–78. [DOI] [PubMed] [Google Scholar]
- 14.Chen BF; Brown KA; Lin ZQ; Ge Y, Top-Down Proteomics: Ready for Prime Time? Analytical Chemistry 2018, 90 (1), 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toby TK; Fornelli L; Kelleher NL, Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai WX; Tucholski TM; Gregorich ZR; Ge Y, Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev Proteomic 2016, 13 (8), 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregorich ZR; Ge Y, Top-down proteomics in health and disease: Challenges and opportunities. Proteomics 2014, 14 (10), 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristobal A; Marino F; Post H; van den Toorn HWP; Mohammed S; Heck AJR, Toward an Optimized Workflow for Middle-Down Proteomics. Analytical Chemistry 2017, 89 (6), 3318–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin YT; Wei LM; Cai WX; Lin ZQ; Wu ZJ; Peng Y; Kohmoto T; Moss RL; Ge Y, Complete Characterization of Cardiac Myosin Heavy Chain (223 kDa) Enabled by Size-Exclusion Chromatography and Middle-Down Mass Spectrometry. Analytical Chemistry 2017, 89 (9), 4922–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campuzano IDG; Netirojjanakul C; Nshanian M; Lippens JL; Kilgour DPA; Van Orden S; Loo JA, Native-MS Analysis of Monoclonal Antibody Conjugates by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Analytical Chemistry 2018, 90 (1), 745–751. [DOI] [PubMed] [Google Scholar]
- 21.Dyachenko A; Wang GB; Belov M; Makarov A; de Jong RN; van den Bremer ETJ; Parren PWHI; Heck AJR, Tandem Native Mass-Spectrometry on Antibody-Drug Conjugates and Submillion Da Antibody-Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Analytical Chemistry 2015, 87 (12), 6095–6102. [DOI] [PubMed] [Google Scholar]
- 22.Pesavento JJ; Mizzen CA; Kelleher NL, Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: Human histone H4. Analytical Chemistry 2006, 78 (13), 4271–4280. [DOI] [PubMed] [Google Scholar]
- 23.Janin-Bussat MC; Dillenbourg M; Corvaia N; Beck A; Klinguer-Hamour C, Characterization of antibody drug conjugate positional isomers at cysteine residues by peptide mapping LC-MS analysis. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences 2015, 981, 9–13. [DOI] [PubMed] [Google Scholar]
- 24.Mao Y; Valeja SG; Rouse JC; Hendrickson CL; Marshall AG, Top-Down Structural Analysis of an Intact Monoclonal Antibody by Electron Capture Dissociation-Fourier Transform Ion Cyclotron Resonance-Mass Spectrometry. Analytical Chemistry 2013, 85 (9), 4239–4246. [DOI] [PubMed] [Google Scholar]
- 25.Tsybin YO; Fornelli L; Stoermer C; Luebeck M; Parra J; Nallet S; Wurm FM; Hartmer R, Structural Analysis of Intact Monoclonal Antibodies by Electron Transfer Dissociation Mass Spectrometry. Analytical Chemistry 2011, 83 (23), 8919–8927. [DOI] [PubMed] [Google Scholar]
- 26.Fornelli L; Damoc E; Thomas PM; Kelleher NL; Aizikov K; Denisov E; Makarov A; Tsybin YO, Analysis of Intact Monoclonal Antibody IgG1 by Electron Transfer Dissociation Orbitrap FTMS. Mol Cell Proteomics 2012, 11 (12), 1758–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fornelli L; Ayoub D; Aizikov K; Beck A; Tsybin YO, Middle-Down Analysis of Monoclonal Antibodies with Electron Transfer Dissociation Orbitrap Fourier Transform Mass Spectrometry. Analytical Chemistry 2014, 86 (6), 3005–3012. [DOI] [PubMed] [Google Scholar]
- 28.Cotham VC; Brodbelt JS, Characterization of Therapeutic Monoclonal Antibodies at the Subunit-Level using Middle-Down 193 nm Ultraviolet Photodissociation. Analytical Chemistry 2016, 88 (7), 4004–4013. [DOI] [PubMed] [Google Scholar]
- 29.Fornelli L; Srzentic K; Huguet R; Mullen C; Sharma S; Zabrouskoy V; Fellers RT; Durbin KR; Compton PD; Kelleher NL, Accurate Sequence Analysis of a Monoclonal Antibody by Top-Down and Middle-Down Orbitrap Mass Spectrometry Applying Multiple Ion Activation Techniques. Analytical Chemistry 2018, 90 (14), 8421–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B; Lin Z; Alpert AJ; Fu C; Zhang Q; Pritts WA; Ge Y, Online Hydrophobic Interaction Chromatography–Mass Spectrometry for the Analysis of Intact Monoclonal Antibodies. Analytical Chemistry 2018, 90 (12), 7135–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner-Rousset E; Janin-Bussat MC; Colas O; Excoffier M; Ayoub D; Haeuw JF; Rilatt I; Perez M; Corvaia N; Beck A, Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. Mabs 2014, 6 (1), 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevreux G; Tilly N; Bihoreau N, Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal Biochem 2011, 415 (2), 212–214. [DOI] [PubMed] [Google Scholar]
- 33.Moelleken J; Endesfelder M; Gassner C; Lingke S; Tomaschek S; Tyshchuk O; Lorenz S; Reiff U; Molhoj M, GingisKHAN (TM) protease cleavage allows a high-throughput antibody to Fab conversion enabling direct functional assessment during lead identification of human monoclonal and bispecific IgG1 antibodies. Mabs 2017, 9 (7), 1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai WX; Guner H; Gregorich ZR; Chen AJ; Ayaz-Guner S; Peng Y; Valeja SG; Liu XW; Ge Y, MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol Cell Proteomics 2016, 15 (2), 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resemann A; Jabs W; Wiechmann A; Wagner E; Colas O; Evers W; Belau E; Vorwerg L; Evans C; Beck A In Full validation of therapeutic antibody sequences by middle-up mass measurements and middle-down protein sequencing, MAbs, Taylor & Francis: 2016; pp 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sjögren J; Olsson F; Beck A, Rapid and improved characterization of therapeutic antibodies and antibody related products using IdeS digestion and subunit analysis. Analyst 2016, 141 (11), 3114–3125. [DOI] [PubMed] [Google Scholar]
- 37.Ge Y; Rybakova IN; Xu QG; Moss RL, Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. P Natl Acad Sci USA 2009, 106 (31), 12658–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zabrouskov V; Ge Y; Schwartz J; Walker JW, Unraveling Molecular Complexity of Phosphorylated Human Cardiac Troponin I by Top Down Electron Capture Dissociation/Electron Transfer Dissociation Mass Spectrometry. Mol Cell Proteomics 2008, 7 (10), 1838–1849. [DOI] [PubMed] [Google Scholar]
- 39.Ayaz-Guner S; Zhang J; Li L; Walker JW; Ge Y, In Vivo Phosphorylation Site Mapping in Mouse Cardiac Troponin I by High Resolution Top-Down Electron Capture Dissociation Mass Spectrometry: Ser22/23 Are the Only Sites Basally Phosphorylated. Biochemistry-Us 2009, 48 (34), 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Y; Gregorich ZR; Valeja SG; Zhang H; Cai WX; Chen YC; Guner H; Chen AJ; Schwahn DJ; Hacker TA; Liu XW; Ge Y, Top-down Proteomics Reveals Concerted Reductions in Myofilament and Z-disc Protein Phosphorylation after Acute Myocardial Infarction. Mol Cell Proteomics 2014, 13 (10), 2752–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deslandes A In Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies, MAbs, Taylor & Francis: 2014; pp 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin YT; Lin ZQ; Xu QG; Fu CX; Zhang ZR; Zhang QY; Pritts WA; Ge Y, Comprehensive characterization of monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. Mabs 2019, 11 (1), 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tumey LN; Charati M; He T; Sousa E; Ma D; Han X; Clark T; Casavant J; Loganzo F; Barletta F, Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjugate chemistry 2014, 25 (10), 1871–1880. [DOI] [PubMed] [Google Scholar]
- 44.Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. P Natl Acad Sci USA 2004, 101 (26), 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang WR; Vlasak J; Li YS; Pristatsky P; Fang YL; Pittman T; Roman J; Wang Y; Prueksaritanont T; Ionescu R, Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol 2011, 48 (6-7), 860–866. [DOI] [PubMed] [Google Scholar]
- 46.Bertolotti-Ciarlet A; Wang WR; Lownes R; Pristatsky P; Fang YL; McKelvey T; Li YZ; Li YS; Drummond J; Prueksaritanont T; Vlasak J, Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fc gamma receptors. Mol Immunol 2009, 46 (8-9), 1878–1882. [DOI] [PubMed] [Google Scholar]
- 47.Zhang B; Jeong J; Burgess B; Jazayri M; Tang Y; Zhang YT, Development of a rapid RP-UHPLC–MS method for analysis of modifications in therapeutic monoclonal antibodies. Journal of Chromatography B 2016, 1032, 172–181. [DOI] [PubMed] [Google Scholar]
- 48.Rush MJP; Riley NM; Westphall MS; Coon JJ, Top-Down Characterization of Proteins with Intact Disulfide Bonds Using Activated-Ion Electron Transfer Dissociation. Analytical Chemistry 2018, 90 (15), 8946–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim MT; Chen Y; Marhoul J; Jacobson F, Statistical Modeling of the Drug Load Distribution on Trastuzumab Emtansine (Kadcyla), a Lysine-Linked Antibody Drug Conjugate. Bioconjugate Chemistry 2014, 25 (7), 1223–1232. [DOI] [PubMed] [Google Scholar]
- 50.Chen LX; Wang L; Shion H; Yu CF; Yu YQ; Zhu L; Li M; Chen WB; Gao K, In-depth structural characterization of Kadcyla (R) (ado-trastuzumab emtansine) and its biosimilar candidate. Mabs 2016, 8 (7), 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw JB; Li WZ; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS, Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J Am Chem Soc 2013, 135 (34), 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riley NM; Westphall MS; Coon JJ, Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Analytical Chemistry 2015, 87 (14), 7109–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.