

Abstract
Proximal tubular epithelial cells (PTEC) in the S1 segment of the kidney abundantly express sodium-glucose co-transporters (SGLT) that play a critical role in whole body glucose homeostasis. We recently reported suppression of RECK (Reversion Inducing Cysteine Rich Protein with Kazal Motifs), a membrane anchored endogenous MMP inhibitor and anti-fibrotic mediator, in the kidneys of db/db mice, a model of diabetic kidney disease (DKD), as well as in high glucose (HG) treated human kidney proximal tubule cells (HK–2). We further demonstrated that empagliflozin (EMPA), an SGLT2 inhibitor, reversed these effects. Little is known regarding the mechanisms underlying RECK suppression under hyperglycemic conditions, and its rescue by EMPA. Consistent with our previous studies, HG (25 mM) suppressed RECK expression in HK-2 cells. Further mechanistic investigations revealed that HG induced superoxide and hydrogen peroxide generation, oxidative stress-dependent TRAF3IP2 upregulation, NF-κB and p38 MAPK activation, inflammatory cytokine expression (IL-1β, IL-6, TNF-α, and MCP-1), miR-21 induction, MMP2 activation, and RECK suppression. Moreover, RECK gain-of-function inhibited HG-induced MMP2 activation and HK-2 cell migration. Similar to HG, advanced glycation end products (AGE) induced TRAF3IP2 and suppressed RECK, effects that were inhibited by EMPA. Importantly, EMPA treatment ameliorated all of these deleterious effects, and inhibited epithelial-to-mesenchymal transition (EMT) and HK-2 cell migration. Collectively, these findings indicate that hyperglycemia and associated AGE suppress RECK expression via oxidative stress/TRAF3IP2/NF-κB and p38 MAPK/miR-21 induction. Furthermore, these results suggest that interventions aimed at restoring RECK or inhibiting SGLT2 have the potential to treat kidney inflammatory response/fibrosis and nephropathy under chronic hyperglycemic conditions, such as DKD.
Keywords: SGLT2, Diabetic kidney disease, Fibrosis, Inflammation
1. Introduction
Persistent hyperglycemia, and the resultant chronic oxidative stress, has been shown to be the major causative factor in diabetic kidney disease (DKD), a disease which afflicts approximately 40% of individuals suffering from diabetes [1]. In this regard, DKD is associated with excessive activation of multiple pathways, each contributing to redox imbalance and oxidative stress [2–4]. In addition, DKD is characterized by increased accumulation of advanced glycation end products (AGEs) that result from non-enzymatic glycation of proteins and lipids that promote injury to glomeruli and proximal tubules [5]. Like hyperglycemia, AGEs also stimulate increased generation of reactive oxygen species (ROS), suggesting that oxidative stress is the principal contributing factor to kidney inflammation, injury and fibrosis [5].
Among the different cell types in the kidney, proximal tubular epithelial cells (PTEC) contribute to oxidative stress, inflammation and fibrosis that eventually progress to DKD [6,7]. PTEC abundantly express a pair of sodium-glucose co-transporters (SGLT) that function to help regulate whole body glucose homeostasis by coordinating the reabsorption of filtered glucose. They include SGLT2, which is localized to the brush border membranes of the S1 segment of proximal tubules (PT) and is the major glucose reabsorption pathway in the kidney and SGLT1, located predominantly in the S3 segment of PT and plays an important but lesser role in glucose reabsorption. Identification of SGLT2 and elucidation of its function ultimately led to development of several SGLT2 inhibitors that were designed to lower blood glucose (independent of insulin) by promoting glucose excretion through urine [8–10]. SGLT2 inhibitors were first approved to treat type 2 diabetes (T2D) and initially included canagliflozin, dapagliflozin, and empagli-flozin. Subsequently, these drugs have shown promise in ameliorating cardiovascular complications associated with T2D [11–16]. Indeed, recent data indicate that empagliflozin (EMPA) may reduce the risk of kidney events and slow progression of chronic kidney disease in patients with T2D, with or without heart failure [13,17–19].
Recently, we demonstrated that administration of the SGLT2 inhibitor EMPA, promoted glycosuria and inhibited vascular and renal impairments, without affecting increases in blood pressure in female db/db mice, a translationally relevant model of T2D [20]. In addition, EMPA inhibited enhanced renal expression of AGEs and periarterial and tubulointerstitial fibrosis (TF) in these diabetic mice. Interestingly, EMPA prevented the marked suppression of RECK (Reversion Inducing Cysteine Rich Protein with Kazal Motifs), a membrane-anchored MMP (matrix metalloproteinase) regulator that suppresses pro-fibrotic responses through inhibition of MMP activation [21,22]. Confirming those in vivo data, high glucose suppressed RECK expression in human PTEC and EMPA inhibited this deficiency suggesting that restoring RECK expression is renoprotective under chronic hyperglycemic conditions [20]. However, although the role of RECK deficiency in promoting angiogenesis [23] and cardiac fibrosis [21] has been reported previously, the mechanisms underlying RECK suppression and those targeted by EMPA in PTEC remain unknown.
Herein, we extend our previous work implicating RECK deficiency in the pathophysiology of DKD and its rescue by EMPA. Specifically, we show for the first time that the SGLT2 inhibitor, EMPA, attenuates HG-induced oxidative stress, RECK suppression and epithelial-to-mesenchymal transition (EMT) in cultured kidney proximal tubule cells (HK-2) and these salutary effects are associated with reductions in the expression of several proinflammatory and profibrotic mediators, including TRAF3IP2, NF-κB, p38MAPK and miR-21. Further, EMPA also prevented AGE/RAGE-induced TRAF3IP2 expression and RECK suppression.
2. Materials and methods
2.1. Reagents
EMPA was provided by Boehringer Ingelheim Pharma. d-glucose (#G8769), d-mannitol (M4125), polymyxin B sulfate (#P4932), tiron (4,5-Dihydroxy-1,3-benzenedisulfonic acid disodium salt, pyrocatechol-3,5-disulfonic acid disodium salt; D7389), TEMPOL (4-Hydroxy-TEMPO, 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, 4-hydroxy-TEMPO, 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl; #581599), DPI (diphenyleneiodonium chloride; D2926), hexadimethrine bromide (Polybrene; #H9268), protease inhibitor cocktail (#P8340), hydrogen peroxide solution (#16911), cOmplete™ Protease Inhibitor Cocktail (#11697498001), PhosSTOP™ (#4906845001), trypsin-EDTA solution (#T4049), trypan blue solution (#T8154), dimethyl sulfoxide (DMSO, #D8418),), and fatty acid free, globulin free albumin from human serum (#A3782) were purchased from Sigma-Aldrich (St. Louis, MO). SB239063 (trans-4-[4-(4-fluorophenyl)-5-(2-methoxy-4-pyrimidinyl)-1H-imidazol-1-yl] cyclohexanol; a potent and selective p38 MAPK inhibitor; #1962) was purchased from Tocris (Minneapolis, MN). The gp91 ds-tat (YGRKKRRQRRRCSTRIRRQL - NH2; #AS-63818) and its scrambled peptide control sgp91 ds-tat (YGRKKRRQRRRCLRITRQSR -NH2; #AS-63821) were purchased from AnaSpec (Fremont, CA, USA). One of the major sources of superoxide generation is NADPH oxidase (NOX). Binding of gp91phox to the p47-p67-p40phox complex is critical for NOX2 activation. A specific gp91 docking sequence (ds in gp91 ds, CSTRIRRQL) which is responsible for binding of gp91phox and gp47phox is linked to a specific 9-amino acid peptide of HIV (Human Immunodeficiency Virus) viral coat (HIV-tat, RKKRRQRRR), resulting in gp91 ds-tat that blocks the interaction of p47-p67-p40phox to disrupt Nox2 assembly and function. A scrambled sequence linked to HIV-tat served as a control (sgp91 ds-tat). The Nox1/Nox4 inhibitor GKT137831 (2–2(chlorophenyl)-4-[3-(dimethylamino) phenyl]-5-methyl-1H-pyrazolo[4,3-c] pyridine-3,6(2H,5H)-dione; #17164) was purchased from Cayman Chemical (Ann Arbor, MI). Restore™ Western blot Stripping Buffer (#21059), Vybrant® MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Cell Proliferation Assay Kit (#V13154), Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (#A22188), and Corning® BioCoat™ Matrigel® Migration Chambers with 8.0 μm polyethylene terephthalate (PET) membrane (#11563570) were purchased from Thermo Fisher Scientific. 96-well clear bottom, black-sided plates (#29444–008) were purchased from VWR Scientific (West Chester, PA). QCL-1000™Endpoint Chromogenic LAL (Limulus amoebocyte lysate) assay kit (#50–647 U) was purchased from Lonza. At the indicated concentrations and for the duration of treatment, the pharmacological inhibitors failed to modulate HK-2 morphology, viability or adherence to culture dishes (data not shown).
2.2. Culture of human proximal tubular epithelial cells
The human kidney-2 (HK-2) cells were purchased from ATCC (ATCC® CRL-2190™; Manassas, VA). HK-2 is an immortalized proximal tubule cell line derived from a single cell isolated from an adult male normal kidney. These cells were authenticated by ATCC®. The cells were cultured in DMEM/F-12 medium (Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12; Cat.# 11320033, Thermo Fisher Scientific) containing 5.6 mM glucose and supplemented with 10% heat-inactivated FBS, glutamine, HEPES buffer and penicillin-streptomycin in a humidified atmosphere containing 5% CO2 at 37 °C. At 80–90% confluency, cells were detached from the culture dishes by trypsin-EDTA, and plated for subculture. All experiments were performed between passages 3–12. At 70% confluency, the complete media was replaced with medium containing 1× ITS (Insulin-Transferrin-Selenite) liquid media supplement (Cat.# I3146, Sigma-Aldrich) for 16 h to arrest and synchronize cell growth. These quiescent (serum-starved) cells were then incubated with 25 mM d-glucose (high glucose). l-glucose (l-glucose 20 mM + d-glucose 5.6 mM) served as a control. Mannitol (mannitol, 20 mM + d-glucose, 5.6 mM) served as an osmolar control.
2.3. Preparation of advanced glycated human serum albumin (AGE-HAS)
AGE-HSA was prepared as previously described [24] by exposing fatty acid- and globulin-free human serum albumin (HSA) to 1 M d-glucose in 100 mM sodium phosphate buffer (pH 7.4) containing 200 U/ml of penicillin, 200 μg/mL streptomycin, 80 μg/ml of gentamycin, and 1.5 mM of PMSF at 37 °C in the dark for 60 days, and dialyzed for 16 h against PBS. As a control, HSA was subjected to the same procedure, but without exposure to d-glucose. Fluorescence was measured at excitation/emission wavelengths of 370/440 nm in a spectrofluorophotometer, and the concentrations of AGE-HSA and HSA were determined by the method of Bradford. HAS and AGE-HSA were each used at 100 μg/ml. The preparations contained < 0.02 EU/ml of endotoxin as determined by the QCL-1000™Endpoint Chromogenic LAL (Limulus Amoebocyte Lysate) assay.
2.4. Quantitation of superoxide by chemiluminescence
Superoxide (O2−) generation was quantified using the lucigenin-enhanced chemiluminescence assay as previously described [25]. Briefly, HK-2 cells (1 × 104) were rinsed twice in PBS and suspended in Krebs/HEPES buffer (in mM: 115 NaCl, 20 HEPES, 1.17 K2HPO4, 1.17 MgSO4, 4.3 KCl, 1.3 CaCl2, 25 NaHCO3, and 11.7 glucose, pH 7.4) containing NADPH (10−4 M), and incubated in a 37 °C water bath for 20 min. Lucigenin (5 × 10−6 M) and high glucose (HG; 25 mM for 15 min) were added. After equilibrating for 30 min at 37 °C, the tubes were placed in a Sirius luminometer (Berthold Detection Systems, Pforzheim, Germany). Luminescence was measured for 10 s with a delay of 5 s. After subtracting background luminescence, results were expressed as relative light units (RLU)/s/104 cells. In a subset of experiments, cells were treated with Tiron, a vitamin E derivative and a superoxide scavenger at 100 μM, Tempol, a cell permeable free radical scavenger and a superoxide dismutase mimetic at 1 μM in DMSO, gp91 ds-tat, a peptide composed of gp91phox of Nox2 sequence linked to HIV-tat peptide to facilitate cellular uptake at 1 μM for 1 h, DPI, an inhibitor of NOXes and DUOXes at 10 μM in DMSO for 30 min or EMPA at 0.5 μM in DMSO for 15 min prior to HG addition (25 mM for 15 min). sgp91 ds-tat containing the scrambled gp91phox sequence tagged to HIV-tat peptide and DMSO (vehicle) served as controls.
2.5. Detection of hydrogen peroxide by Amplex Red® assay
Hydrogen peroxide (H2O2) levels were quantified using the Amplex Red assay [26]. HK-2 cells were treated with HG for 30 min. H2O2 production was measured in the presence of horseradish peroxidase (0.1 unit/ml) and Amplex Red (50 μM). Fluorescence was recorded at 530 nm excitation and 590 nm emission wavelengths (CytoFluor II; Applied Biosystems, Foster City, CA). Standard curves were generated using known concentrations of H2O2. The results are expressed as μM H2O2 produced/104 cells. Studies were also performed with GKT137831, a Nox1/4 dual inhibitor, at 5 μM in DMSO for 15 min, DPI, and EMPA prior to HG addition.
2.6. Adeno and lentiviral transduction
Adenovirus containing the full-length mouse RECK ORF (GenBank accession # NM_016678.2) under control of the CMV promoter (Ad.RECK) was previously described. Ad.GFP served as a control. HK-2 cells were transduced with Ad.RECK at the indicated multiplicity of infection (moi) for 24 h. Lentiviral shRNA targeting TRAF3IP2 and p65 have also been previously described. RAGE shRNA lentiviral particles (sc-36,374-V) were purchased from Santa Cruz Biotechnology, Inc. For lentiviral infection, HK-2 cells at 50–60% confluency were infected with the indicated shRNA at a moi of 0.5 for 48 h in complete media. Lentiviral shRNA against eGFP (SHC005V; Sigma-Aldrich) served as a control. The viral transduction had no off-target effects, and at the indicated moi and duration of treatment, failed to modulate HK-2 adherence, shape, and viability (trypan blue dye exclusion; data not shown).
2.7. miRNA expression, mimics, inhibitors, controls, and transfections
For miRNA expression, small RNA-enriched total RNA was isolated using the mirVana™ miRNA Isolation Kit (Ambion®, Austin, TX). Expression levels of miR-21 were analyzed by RTqPCR using miRNA TaqMan® probes (hss-miR-21, Assay ID: 000397; Thermo Fisher Scientific). Human miR21 inhibitor (Assay ID: MH10206), inhibitor negative control (Invitrogen™ mirVana™ miRNA Inhibitor, Negative Control #1; #4464076), human miR-21 mimic (Assay ID: MC10206) and mimic negative control (Invitrogen™ mirVana™ miRNA Mimic, Negative Control #1, #4464058) were all purchased from Thermo Fisher Scientific. HK-2 cells were transfected with the indicated mimic, mimic negative control, inhibitor or inhibitor negative control (80 nM) using the Neon® transfection system (MPK-5000; Invitrogen, Waltham, MA). HK-2 cells were microporated (pulse voltage: 1300 V; pulse width: 20 ms; pulse number: 2; the tip type: 10 μl) and then cultured for 24 h. SMC showed transfection efficiency of 46% with only 3% cell death as determined using the pEGFP-N1 vector. Transfections at the indicated concentration and for the duration of treatment failed to significantly modulate HK-2 adherence, shape, and viability (trypan blue dye exclusion; data not shown). Induction or inhibition of miR-21 was confirmed by Northern blotting as previously described using radiolabeled StarFire oligonucleotide probes [27]. Radiolabeling of miRNA antisense oligonucleotides was performed with a StarFire labeling kit (IDT, Coralville, IA), according to the manufacturer’s instructions. Small RNA-enriched total RNA (20 mg) was resolved through a 12.5% urea-polyacrylamide gel under denaturing conditions, transferred onto GeneScreen Plus membranes (PerkinElmer, Waltham, MA), UV-cross-linked, pre-hybridized, hybridized, and washed as described. U6 served as a loading control.
2.8. NF-κB and p38 MAPK activation
Activation of NF-κB was analyzed as previously described [25] by immunoblotting using cleared whole cell homogenates and activation-specific antibodies against p65 (Ser536). Total p65 served as a loading control. Similarly, p38 MAPK activation was analyzed by immunoblotting using cleared whole cell homogenates and antibodies against phospho-p38 MAPK (Thr180/Tyr182). Total p38 MAPK served as a loading control.
2.9. mRNA expression
Total RNA was isolated using the TRIzol method, treated with DNase, and 1 μg of DNA-free total RNA reverse transcribed using the Quantitect cDNA Synthesis Kit (Qiagen). Gene expression was analyzed by RT-qPCR using a TaqMan® probes (thermo Fisher Scientific-Applied Biosystems) for MMP2 (Hs01548727-m1), TIMP3 (Hs00165949-m1), and 18S (Hs03003631-g1) using Eppendorf Realplex4 system. All data were normalized to corresponding 18S rRNA, and expressed as fold change relative to untreated controls. Data are shown as fold change (2-ΔΔCt).
2.10. Immunoblotting and ELISA
Preparation of whole cell homogenates, immunoblotting, detection of the immunoreactive bands by enhanced chemiluminescence, and quantification by densitometry were performed as described previously [21]. Briefly, protein extracts of HK-2 cultures were separated on a 4–15% precast gel, and electrophoresed at 100 V. Separated proteins were transferred onto PVDF membranes. After blocking, membranes were incubated with TRAF3IP2 (1:400; NB 100–56,740, Novus Biologicals), RECK (1:1000; catalog# 3433, Cell Signaling Technology, Inc./CST), cleaved caspase-3 (1 μg/ml; #9664, CST), caspase-3 (1:5000; ab32499, abcam), p65 (1:1000; #3033; CST), p-p65 (1:1000; #3031; CST), ASK1 (1:1000; #3762, CST), p-p38 MAPK (1:1000; #4631, CST), p38 MAPK (1:1000; #8690, CST), RAGE (1:1000; #6996; CST), MMP2 (1:500; ab97779, abcam), TIMP3 (1:1000; ab39184, abcam), E-cadherin (1:500; ab15148, abcam), fibronectin (1 μg/ml; catalog# ab23750, abcam), α-SMA (0.25 mg/ml; MAB1420, R&D Systems), vimentin (1:1000; ab92547, abcam) for 1 h at room temperature followed overnight at 4 °C. α tubulin (1:1000; #2144, CST) and GAPDH (1:1000, catalog# sc-25,779, Santa Cruz Biotechnology, Inc) served as invariant controls. After rinsing in TTBS (Tris-buffered saline containing 0.05% Tween 20) for 30 min and incubated in horseradish peroxidase-conjugated secondary antibodies in 5% milk in TTBS for 1 h. Following a 30-min rinse in TTBS, blots were incubated in chemiluminescent substrate (SuperSignal Pico West; Pierce) supplemented with 5% Super-Signal Femto (Pierce) and exposed in a gel doc imager, the intensities of immunoreactive bands were quantified by NIH Image J software. The results were normalized to corresponding tubulin or GAPDH, and presented as a fold change.
Secreted cytokine levels were quantified by commercially available ELISA kits. Briefly, 24 h after treatment, equal amounts of culture supernatants were used to quantify IL-1β (IL-1 beta Human ELISA kit, High Sensitivity, #BMS224HS, Thermo Fisher Scientific, Analytical Sensitivity: 0.05 pg/ml; Assay range: 0.16–10 pg/ml), IL-6 (Human IL-6 Quantikine ELISA Kit, #D6050, R&D Systems; Sensitivity: 0.7 pg/ml; Assay Range: 3.1–300 pg/ml), TNFα (Human TNF-alpha Quantokine HS ELISA, #HSTA00E, R & Systems; Sensitivity: 0.049 pg/ml, Assay range: 0.2–10 pg/ml) and MCP-1 (Human MCP-1 ELISA Kit, #ab179886, abcam; Sensitivity: 1.26 pg/ml; Assay range: 4.7–300 pg/ml) levels.
2.11. MMP2 activity assay
At 70% confluency, the complete media was replaced with medium containing 1× ITS media supplement and incubated overnight. The quiescent HK-2 cells were then incubated with HG (25 mM) for 12 h. MMP2 activity in equal amounts of culture supernatants was analyzed according to manufacturer’s instructions using a SensoLyte Plus 520 MMP2 Assay Kit *Fluorometric and Enhanced Sensitivity*(#AS-72224; AnaSpec, Fremont, CA). Following immunocapture of MMP2 by immobilized anti-MMP-2 antibody, its proteolytic activity was measured by 5-FAM/QXL® 520 FRET peptide substrate by monitoring the fluorescent signal at Ex/Em = 490/520 nm.
2.12. Cell migration assay
HK-2 cell migration was quantified as described previously using BioCoat™ Matrigel™ migration chambers and 8.0-μm pore polyethylene terephthalate membranes with a thin layer of Matrigel™ basement membrane matrix [1–3]. Cultured HK-2 cells were trypsinized and suspended in RPMI + ITS (1×), and 1 ml containing 2.0 × 105 cells/ml was layered on the coated insert filters. Cells were stimulated with HG (25 mM). The lower chamber contained medium with 10% serum. Plates were incubated at 37 °C for 12 h. The membranes were washed with PBS, and non-invading cells on the upper surface were removed using cotton swabs. HK-2 cells invading into and through the Matrigel™ matrix were quantified by MTT assay as previously described [28]. Numbers of HK-2 cells migrating in response to HG were normalized to those of untreated cells and expressed as fold change from untreated. Migration assays were carried out 6 times.
2.13. Analysis of epithelial mesenchymal transition (EMT) biomarkers
To analyze HG-induced EMT, quiescent HK-2 cells were incubated HG (25 mM) for 48 h, and the expression levels of biomarkers such as the epithelial marker E-cadherin, and the mesenchymal markers fibronectin, α-SMA and vimentin by immunoblotting.
2.14. Cell death detection
To determine whether transduction of viral vectors, pharmacological inhibitors or overexpression of mutant proteins affected cell viability, cell death was analyzed in cleared whole cell homogenates by immunoblotting using antibodies that specifically detect cleaved caspase-3 levels. Total caspase-3 served as a loading control.
2.15. Statistical analysis
Results are reported as the mean ± SE. Differences in outcomes were determined using one-way ANOVA and Dunnett’s post hoc t-tests for paired comparisons and were considered significant when p < .05. All assays were performed at least three times and error bars in figures indicate the S.E. Further, although representative immunoblots are shown in select figures, the intensity of immunoreactive bands from three independent experiments were semi quantified by densitometry and summarized in accompanying bar graphs and displayed as ratios or fold changes from untreated or respective controls. The numbers at the bottom of each panel in figures denote lane numbers.
3. Results
3.1. EMPA inhibits high glucose (HG)-induced superoxide and hydrogen peroxide generation without altering cell viability
We first investigated whether high glucose (HG) induces oxidative stress in renal proximal tubular cells, and whether EMPA inhibits oxidative stress. We used HK-2 cells, a human proximal tubular epithelial cell line, as an in vitro model. Quiescent HK-2 cells were incubated with 25 mM glucose, and superoxide generation was quantified after 15 min by Cytochrome C assay. Mannitol served as an osmotic control. The results in Fig. 1 show that treatment with 25 mM glucose (HG), but not mannitol, significantly enhanced superoxide generation (Fig. 1A), and this effect was markedly inhibited by the superoxide scavenger Tiron and the membrane permeable superoxide dismutase inhibitor Tempol (Fig. 1B). Moreover, gp91 ds-tat, which is known to block the NOX2 subunit assembly and DP1, the NOX and flavoprotein inhibitor, similarly reduced HG-induced superoxide generation. Importantly, EMPA pretreatment markedly inhibited HG-induced superoxide generation, indicating that HG is a potent inducer of superoxide, and its prooxidant effects are blunted by EMPA. Similar to its effects on superoxide generation, HG, but not its osmotic control mannitol, induced hydrogen peroxide generation (Fig. 1C), and this effect was inhibited by the Nox1/4 inhibitor GKT137831, the NOX and flavoprotein inhibitor DPI (Fig. 1D). Importantly, EMPA pretreatment blunted HG-induced hydrogen peroxide generation (Fig. 1D). Although Tiron, Tempol, gp91 ds-tat, GKT and EMPA all inhibited superoxide and hydrogen peroxide generation, they reduced oxidative stress without modulating cell viability, as evidenced by no changes in the cleaved caspase-3 levels (Fig. 1E). However, exogenous addition of supraphysiological concentrations of hydrogen peroxide induced cell death (Fig. 1E, last lane), and served as a positive control. Together, these results indicate that EMPA inhibits HG-induced oxidative stress in HK-2 cells (Fig. 1).
Fig. 1.
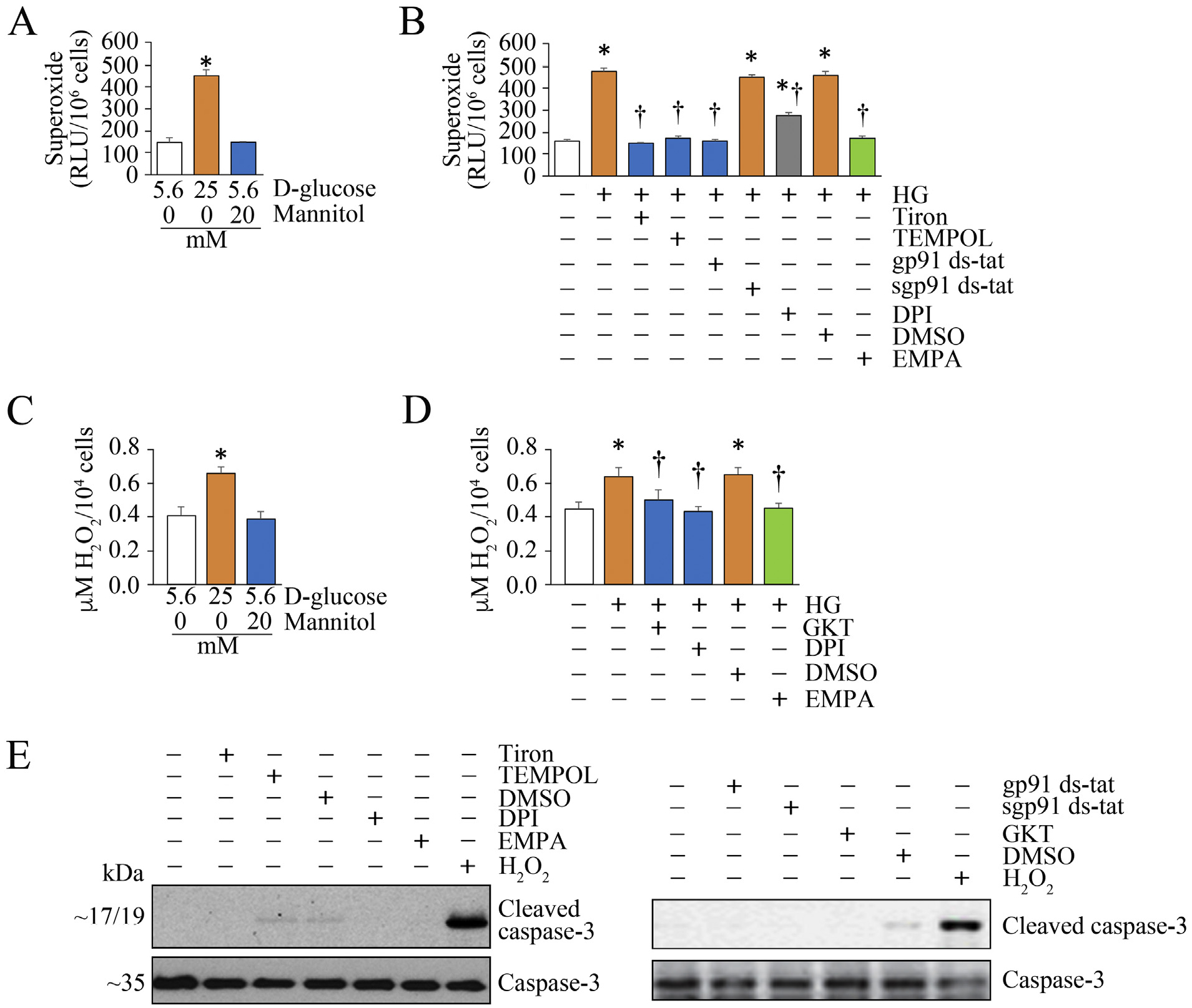
Empagliflozin (EMPA) suppresses high glucose (HG)-induced oxidative stress in HK-2 cells. A, B, HG (25 mM), but not its osmotic control mannitol, induces superoxide generation in HK-2 cells, and is inhibited by EMPA and other free radical inhibitors. At 70% confluency, the complete media was replaced with medium containing 0.5% BSA, and cultured for 24 h. The quiescent cells were then treated high d-glucose (25 mM) for 15 min. Superoxide anion (O2-•) generation was quantified by cytochrome c assay. Mannitol served as a control (A). In a subset of experiments (B), the quiescent HK-2 cells were incubated with Tiron (100 μM), Tempol (1 μM in DMSO), gp91 ds-tat (1 μM for 1 h), DPI (10 μM in DMSO for 30 min) or EMPA (0.5 μM in DMSO for 15 min; green) prior to HG addition (25 mM for 15 min). sgp91 ds-tat (scrambled) and DMSO (vehicle) served as controls. Superoxide generation was quantified as in A. C, D, HG, but not its osmotic control, mannitol (C), induces H2O2 generation (D), and is inhibited by EMPA and other free radical inhibitors. Quiescent HK-2 cells, as in panel A, were treated with HG (25 mM) or its osmotic control mannitol for 30 min (A). H2O2 generation was quantified by Amplex Red assay. In a subset of experiments, quiescent HK-2 cells were treated with EMPA, GKT (5 μM in DMSO for 15 min) or DPI prior to HG (25 mM for 30 min) addition. H2O2 generation was quantified by Amplex Red assay. E, Inhibition of oxidative stress fails to affect cell viability. Quiescent HK-2 cells treated with EMPA, Tiron, Tempol, gp91 ds-tat, DPI, EMPA, sgp91 ds-tat or DMSO alone for 8 h were analyzed for cell viability by analyzing cleaved caspase-3 levels by immunoblotting. Total caspase-3 served as a loading control (n = 3). H2O2 (100 mM for 8 h) served as a positive control. Bars represent means ± SEM. A-D, n = 6. E, n = 3. * P < at least 0.05 versus mannitol or 5.6 mM d-glucose, † P < at least 0.05 versus HG.
3.2. HG induces TRAF3IP2 expression in HK-2 cells via oxidative stress
TRAF3IP2 is a proinflammatory adapter molecule and an upstream regulator of multiple transcription factors known to positively regulate various inflammatory mediators. We have previously demonstrated that oxidative stress induces TRAF3IP2 expression in cardiac fibroblasts and endothelial cells [29,30]. Therefore, we investigated whether HG induces TRAF3IP2 expression in HK-2 cells, and whether the induction is dependent on oxidative stress. Indeed, HG upregulated TRAF3IP2 expression in as little as 30 min followed by a return to baseline at the 6 h time point with a subsequent return to high levels at the 48 h time point. Further, pretreatment with Tiron, Tempol, DPI, gp91 ds-tat, and GKT all reduced TRAF3IP2 expression, as did EMPA pretreatment (Fig. 2B and C). Taken together, these results indicate that HG induces TRAF3IP2 expression in HK-2 cells in a biphasic manner and, in part, via oxidative stress (Fig. 2).
Fig. 2.
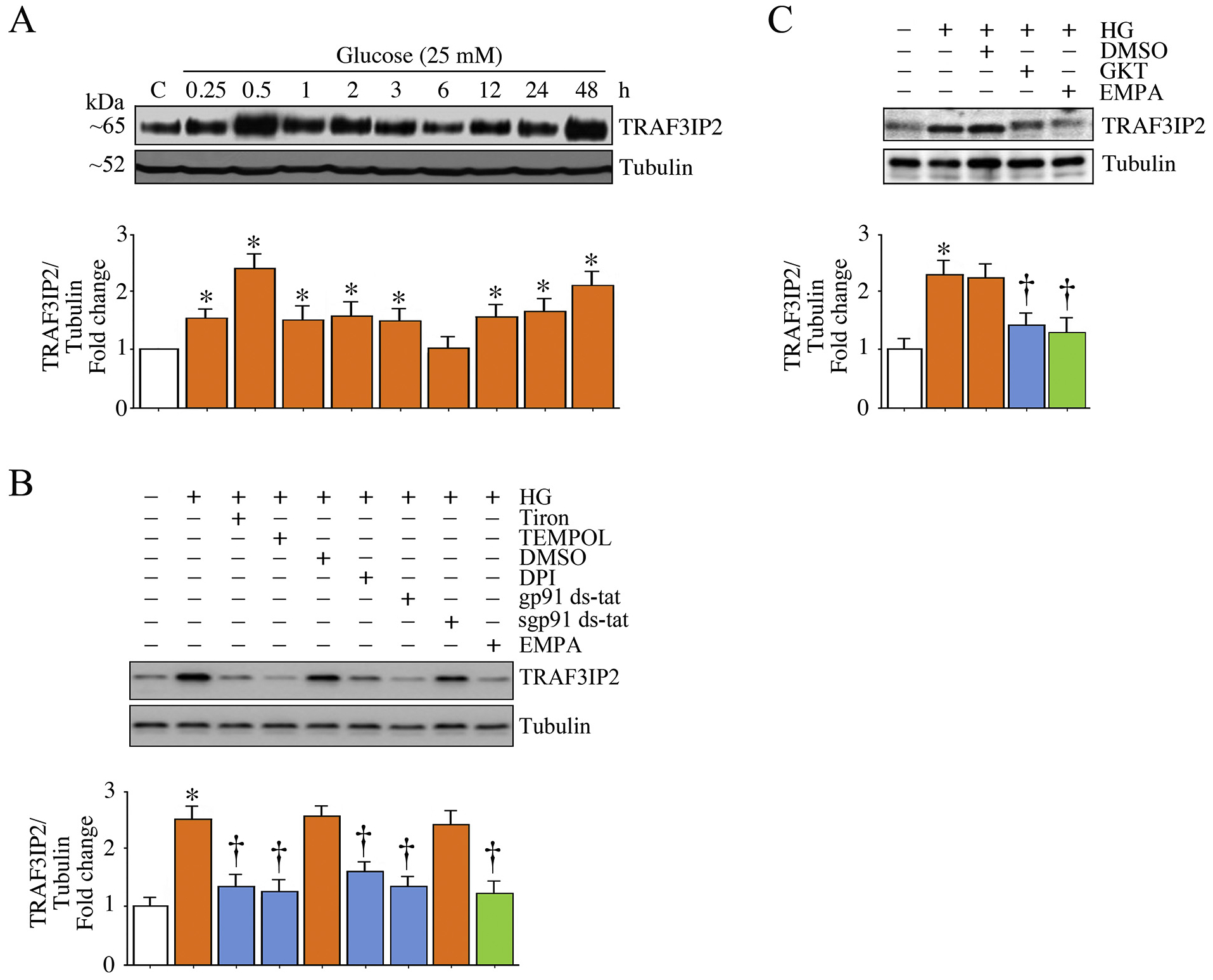
EMPA inhibits high glucose (HG)-induced oxidative stress-dependent TRAF3IP2 expression in HK-2 cells. A, HG induces TRAF3IP2 expression in a biphasic manner. Quiescent HK-2 cells incubated with high glucose (25 mM) for the indicated time periods were analyzed for TRAF3IP2 expression by immunoblotting. Tubulin served as a loading control. B, C, HG induces TRAF3IP2 expression via oxidative stress. Quiescent HK-2 cells were incubated with Tiron, Tempol, DPI, gp91 ds-tat, EMPA (B) or GKT 137831 (5 mM in DMSO for 15 min; C) or prior to HG addition for 30 min. TRAF3IP2 expression was analyzed as in panel A. A-C, The intensity of immunoreactive bands from three independent experiments was semi quantified by densitometry and summarized in the accompanying bar graphs. * P < .05 versus Con; † P < at least 0.05 versus HG.
3.3. EMPA inhibits HG-induced NF-κB activation in HK-2 cells
NF-κB is an oxidative stress-responsive nuclear transcription factor that transcriptionally regulates various inflammatory mediators and matrix-degrading metalloproteinases. Since TRAF3IP2 is an upstream regulator of NF-κB, we next investigated whether targeting oxidative stress and TRAF3IP2 inhibits HG-induced NF-κB activation. More importantly, we determined whether EMPA inhibits HG-induced NF-κB activation. Since site-specific phosphorylation plays a critical role in NF-κB transactivation, we analyzed phosphorylation of p65, a main subunit of the NF-κB dimer, by immunoblotting using activation-specific antibodies. Indeed, the data in Fig. 3A show that HG induces NF-κB activation in HK-2 cells in a time-dependent manner, as evidenced by a rapid increase in phosphorylated levels of p65 at the Ser536 residue, with peak level detected at 1 h that persisted through the 6 h time point. Further, shRNA-mediated TRAF3IP2 knockdown attenuated HG-induced NF-κB activation (Fig. 3B), as did EMPA pretreatment (Fig. 3B). Confirming the role of oxidative stress in NF-κB activation, treatment with Tiron and Tempol inhibited HG-induced NF-κB activation, as did pretreatment with the Nox2 inhibitor gp91 ds-tat and the flavoprotein inhibitor DPI (Fig. 3D). Importantly, the reduction in NF-κB activation following TRAF3IP2 knockdown was not due to reduced cell viability as indicated by no change in active/cleaved caspase-3 levels (Fig. 3E). Collectively, these results indicate that HG rapidly induces NF-κB activation in HK-2 cells, an effect that persists for at least 6 h. Moreover, HG-induced NF-κB activation is oxidative stress and TRAF3IP2 dependent. More importantly, EMPA effectively inhibited HG-induced NF-κB activation (Fig. 3B–D). Together, these results indicate that HG is a potent inducer of oxidative stress/TRAF3IP2-dependent NF-kB activation in HK-2 cells, an effect inhibited by EMPA (Fig. 3).
Fig. 3.
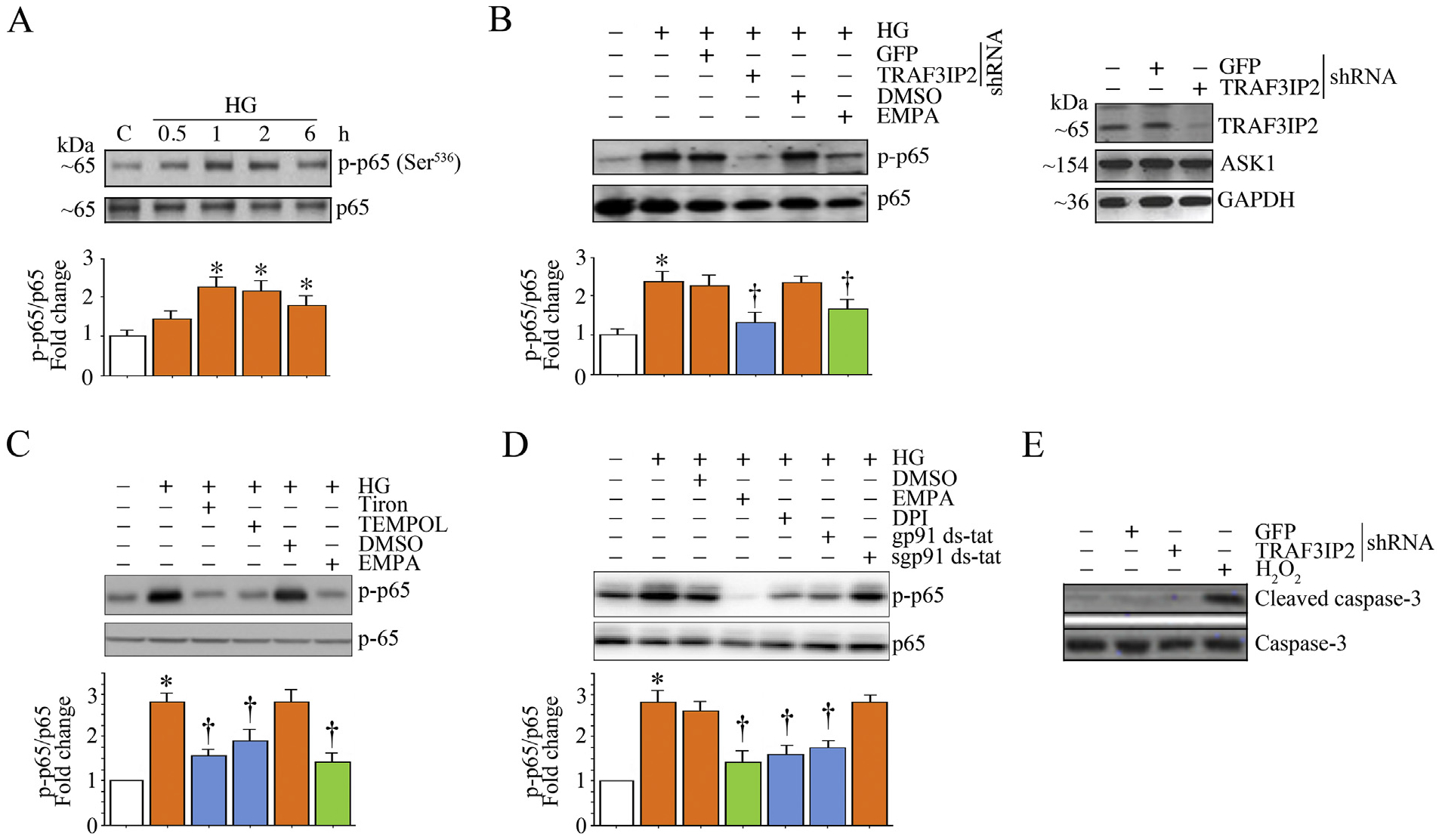
EMPA inhibits high glucose (HG)-induced oxidative stress and TRAF3IP2-dependent NF-κB activation in HK-2 cells. A, HG induces time-dependent NF-κB activation. Quiescent HK-2 cells were incubated with HG (25 mM) for the indicated time periods. NF-κB activation was analyzed by phospho-p65 levels in cleared whole cell lysates. Total p65 served as a loading control. B, HG-induced NF-κB activation is blunted by TRAF3IP2 silencing or EMPA. HK-2 cells were transduced with lentiviral TRAF3IP2 shRNA (moi 0.5 for 48 h) or incubated with EMPA prior to HG addition (25 μM for 1 h). Lentiviral shRNA against GFP or DMSO served as respective controls. Total and phospho-p65 levels were analyzed as in panel A. Knockdown of TRAF3IP2 was confirmed by immunoblotting and is shown on the right. C, D, HG-induced NF-κB activation is oxidative stress dependent. Quiescent HK-2 cells were incubated with Tiron, Tempol or EMPA (C), DPI or gp91 ds-tat (D) prior to HG addition (25 mM for 1 h). Total and phospho-p65 levels were analyzed as in panel A. E, Silencing TRAF3IP2 does affect viability of HK-2 cells. HK-2 cells transduced with lentiviral TRAF3IP2 shRNA (moi 0.5 for 48 h) were analyzed for cell death by immunoblotting using antibodies that detect active (cleaved) and total caspase-3 levels (n = 3). H2O2 (100 μM for 8 h) served as a positive control. A-D, The intensity of immunoreactive bands from three independent experiments was semi quantified by densitometry and summarized in the accompanying bar graphs. * P < .05 versus Con; † P < at least 0.05 versus HG.
3.4. EMPA suppresses HG-induced p38 MAPK activation and inflammatory cytokine expression
Among the three main stress-activated kinases, activation of p38 MAPK has been shown to regulate the expression of various inflammatory mediators and MMPs [31,32]. Moreover, p38 MAPK has been shown to regulate transcriptional activity of NF-κB, suggesting a crosstalk between NF-κB and p38 MAPK [33]. In fact, combined activation of NF-κB and p38 MAPK has been shown to amplify inflammatory signaling. Therefore, we analyzed p38 MAPK activation by quantifying activation (phospho)-specific p38 MAPK levels. We further investigated whether HG induces inflammatory cytokine expression. Indeed, our data show that HG rapidly and significantly increased phospho-p38 MAPK levels that gradually decreased over the 6 h-study period (Fig. 4A). Further, TRAF3IP2 knockdown reduced activation of p38 MAPK, as did pretreatment with EMPA (Fig. 4B). Moreover, inhibitors of oxidative stress, such as Tiron, Tempol, and gp91 ds-tat, all inhibited HG-induced p38 MAPK activation in HK-2 cells (Fig. 4C). Similarly, pretreatment with SB239063, a potent and selective p38 MAPK inhibitor, reduced HG-induced p38 MAPK activation, without affecting cell viability (Fig. D-E). Importantly, EMPA suppressed HG-induced oxidative stress-dependent p38 MAPK activation (Fig. 4B–C). Further, while HG significantly increased IL-1β, IL-6, TNFα and MCP-1 secretion by HK-2 cells, TRAF3IP2 knockdown and EMPA each markedly suppressed their secretion (Fig. 4F). Taken together, these results indicate that HG rapidly induces p38 MAPK activation and inflammatory cytokine expression in HK-2 cells, in part, via TRAF3IP2. More importantly, EMPA effectively inhibits HG-induced p38 MAPK activation and inflammatory cytokine expression (Fig. 4).
Fig. 4.
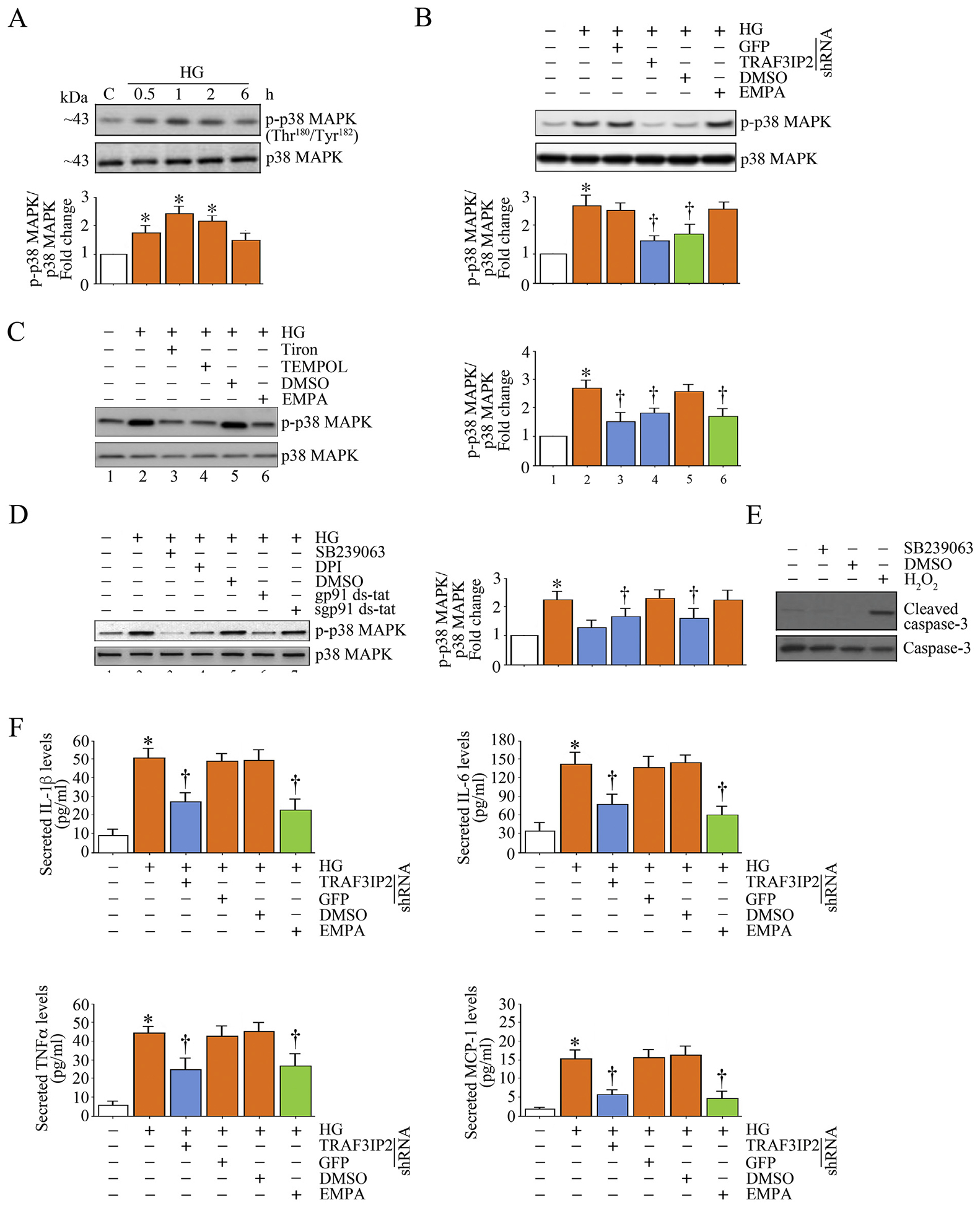
EMPA suppresses high glucose (HG)-induced p38 MAPK activation and inflammatory cytokine expression in HK-2 cells. A, HG induces time-dependent p38 MAPK activation. Quiescent HK-2 cells were incubated with HG (25 mM) for the indicated time periods. Activation of p38 MAPK was analyzed by immunoblotting using cleared whole cell lysates and activation-specific antibodies. Total p38 MAPK served as a loading control. B, HG induces p38 MAPK activation via TRAF3IP2, and is inhibited EMPA. HK-2 cells were transduced with lentiviral TRAF3IP2 shRNA (moi 0.5 for 48 h) or incubated with EMPA (0.5 μM in DMSO for 15 min) prior to HG addition (25 μM for 1 h). Lentiviral shRNA against GFP or DMSO served as respective controls. Total and phospho-p38 MAPK levels were analyzed as in panel A. C, D, HG-induced p38 MAPK activation is oxidative stress dependent. Quiescent HK-2 cells were incubated with Tiron, Tempol or EMPA (C), SB239063 (10 μM in DMSO for 1 h), DPI or gp91 ds-tat (D) prior to HG addition (25 mM for 1 h). sgp91 ds-tat and DMSO served as controls. Total and phospho-p38 MAPK levels were analyzed as in panel A. E, Pharmacological inhibition of p38 MAPK fails to affect HK-2 cell viability. HK-2 cells treated with SB239063 (10 μM in DMSO for 1 h) were analyzed for cell death by quantifying cleaved caspase-3 levels by immunoblotting (n = 3). H2O2 served as a positive control. F, Silencing TRAF3IP2 and EMPA each inhibit HG-induced proinflammatory cytokine secretion. HK-2 cells were transduced with lentiviral TRAF3IP2 shRNA (moi 0.5 for 48 h) or incubated with EMPA prior to HG addition (25 μM for 24 h). Secreted cytokine levels in equal amounts of culture supernatants were quantified by ELISA. The intensity of immunoreactive bands from three independent experiments is semi quantified by densitometry and summarized in the accompanying bar graphs. * P < .05 versus Con, † P < at least 0.05 versus HG.
3.5. EMPA prevents HG-induced miR-21-dependent RECK suppression in HK-2 cells
Both NF-κB and p38 MAPK, either alone or in combination, have been shown to positively regulate the induction of inflammatory mediators and MMPs [31–33]. RECK is a MMP inhibitor, and we have previously reported that IL-17-mediated human aortic smooth muscle cell migration is characterized by increased TRAF3IP2-dependent miR-21 induction and miR-21-dependent RECK suppression [34]. Since p38 MAPK and NF-κB regulate miR-21 induction [35] and as miR-21 plays a role in kidney fibrosis [36], we next investigated whether HG induces miR-21 expression and whether its induction is NF-κB and p38 MAPK dependent in HK-2 cells. The data in Fig. 5A show that HG did indeed induce miR-21 expression in HK-2 cells, an effect markedly inhibited by TRAF3IP2 or p65 knockdown or pretreatment with the p38 MAPK-selective inhibitor SB239063. Importantly, EMPA inhibited HG-induced miR-21 expression in HK-2 cells. Since RECK is one of the targets of miR-21 and RECK inhibits the activation MMPs that play a role in cell migration, we next investigated whether HG regulates RECK via miR-21. Therefore, we incubated quiescent HK-2 cells with HG for various periods of time. RECK expression was analyzed by immunoblotting. The results show that HG suppresses RECK expression in a time-dependent manner, with a significant reduction seen as early as 1 h (Fig. 5B). RECK levels were further reduced at 6, 12, and 24 h, and increased after 48 h. Further, knockdown of TRAF3IP2 or p65 partially prevented HG-induced RECK suppression (Fig. 5C). Similarly, the p38 MAPK-selective inhibitor, SB23906, restored RECK expression (Fig. 5D). Importantly, pretreatment with EMPA prevented HG-induced RECK suppression (Fig. 5E). Since miR-21 is known to negatively regulate RECK expression, we next determined whether miR-21 inhibition restores RECK expression. Indeed, data in Fig. 5F show that treatment with the miR-21 inhibitor (miR-21-I; inhibition of miR-21 expression is shown in panel G, left hand panel), but not its negative control, restored HG-induced RECK suppression. As a proof-of-concept that miR-21 does inhibit RECK expression, we next investigated whether a miR-21 mimic by itself inhibits RECK basal expression. As seen in Fig. 5F (last two lanes), transfection with miR-21 mimic suppressed RECK expression. Notably, although both the miR-21 inhibitor and the miR-21 mimic suppressed RECK expression they did so without affecting cell viability, as seen by no increase in cleaved caspase-3 levels (Fig. 5H). Together, these results indicate that HG suppresses RECK expression in part via TRAF3IP2, p38 MAPK and NF-κB -dependent miR-21 induction, and these effects were prevented by EMPA (Fig. 5).
Fig. 5.
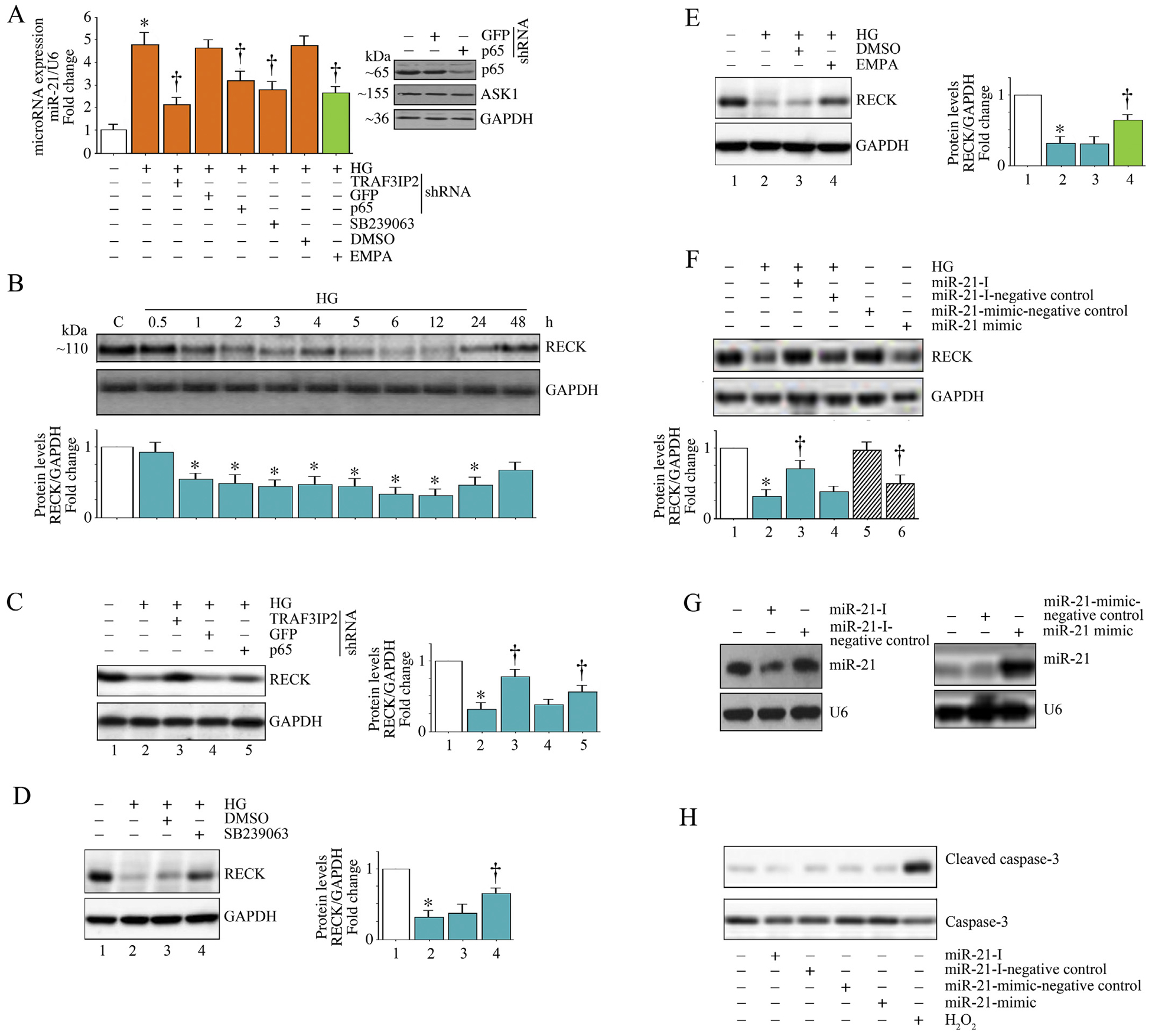
EMPA prevents high glucose (HG)-induced miR-21 induction and RECK suppression in HK-2 cells. A, HG induces miR-21 expression via TRAF3IP2, NF-κB and p38 MAPK, and is inhibited by EMPA. HK-2 cells were transduced with lentiviral TRAF3IP2 or NF-κBp65 shRNA, or pretreatment SB239063 or EMPA, prior to HG addition (25 mM for 2 h), miR-21 expression was analyzed by advanced miRNA assay. Knockdown of p65 was confirmed by immunoblotting, and is shown on the right. B, HG suppresses RECK expression in HK-2 cells. Quiescent HK-2 cells were treated with HG (25 mM) for the indicated time periods. RECK protein levels were analyzed by immunoblotting. GAPDH served as an invariant control. C, Silencing TRAF3IP2 or NF-κBp65 prevents HG-induced RECK suppression. HK-2 cells transduced with lentiviral TRAF3IP2 or NF-κBp65 shRNA prior to HG addition were analyzed for miR-21 expression after 6 h as in panel A. D, Inhibition of p38 MAPK prevents HG-induced RECK suppression. Quiescent HK-2 cells were treated with SB239063 prior to HG addition (25 mM for 6 h) were analyzed for RECK expression by immunoblotting. E, EMPA prevents HG-induced RECK suppression. Quiescent HK-2 cells were treated with EMPA prior to HG addition (25 mM for 6 h) were analyzed for RECK expression by immunoblotting. F, G, While forced expression of miR-21 inhibitor prevents HG-induced RECK suppression, ectopic expression of miR-21 mimic by itself suppresses basal RECK expression. HK-2 cells transduced with the miR-21 inhibitor for 48 h were treated with HG (25 mM for 6 h) and then analyzed for RECK expression by immunoblotting. miR-21-I-Scrambled served as a control. In a separate set of experiments, HK-2 cells transduced with miR-21 mimic for 48 h were analyzed RECK expression by immunoblotting. miR-21-I-Scrambled served as a control. Inhibition of miR-21 following miR-21-I treatment and induction of miR-21 following transduction of miR-21-mimic were confirmed by Northern blotting (G). H, Transduction of miR-21-Inhibitor or miR-21-mimic or their Scrambled controls do not affect HK-2 cell viability. HK-2 cells transduced with miR-21-Inhibitor or miR-21-mimic or their Scrambled controls for 48 h were analyzed for cell death by quantifying cleaved caspase-3 levels by immunoblotting. Total caspase-3 levels served as a loading control. B–F, The intensity of immunoreactive bands from three independent experiments is semi quantified by densitometry and summarized in the accompanying bar graphs. * P < .05 versus Con, † P < at least 0.05 versus HG.
3.6. EMPA prevents HG-induced MMP2 expression and activation in HK-2 cells
Induction and/or activation of MMPs play a crucial role in renal interstitial fibrosis [37,38]. Under pathological conditions, including diabetic nephropathy, MMPs breakdown extracellular matrix and promote migration of interstitial fibroblasts. Therefore, we investigated whether HG induces MMP2 expression, and whether HG-induced MMP2 expression is inhibited by EMPA. Results in Fig. 6A show that HG indeed induced MMP2 mRNA expression in a time dependent manner, with induction seen as early as 1 h that peaked at 2 h. Further, HG-induced MMP2 mRNA expression was inhibited by TRAF3IP2 and p65 knockdown, and pre-treatment with the p38 MAPK-specific inhibitor SB239063 (Fig. 6B). Additionally, targeting TRAF3IP2, p65 and p38 MAPK each reduced HG-induced MMP2 activity (Fig. 6C). Importantly, EMPA inhibited HG-induced MMP2 mRNA expression (Fig. 6B) and activity (Fig. 6C). Together, these data indicate that EMPA inhibits TRAF3IP2, p65, and p38 MAPK-mediated MMP2 expression and activation in HK-2 cells (Fig. 6).
Fig. 6.
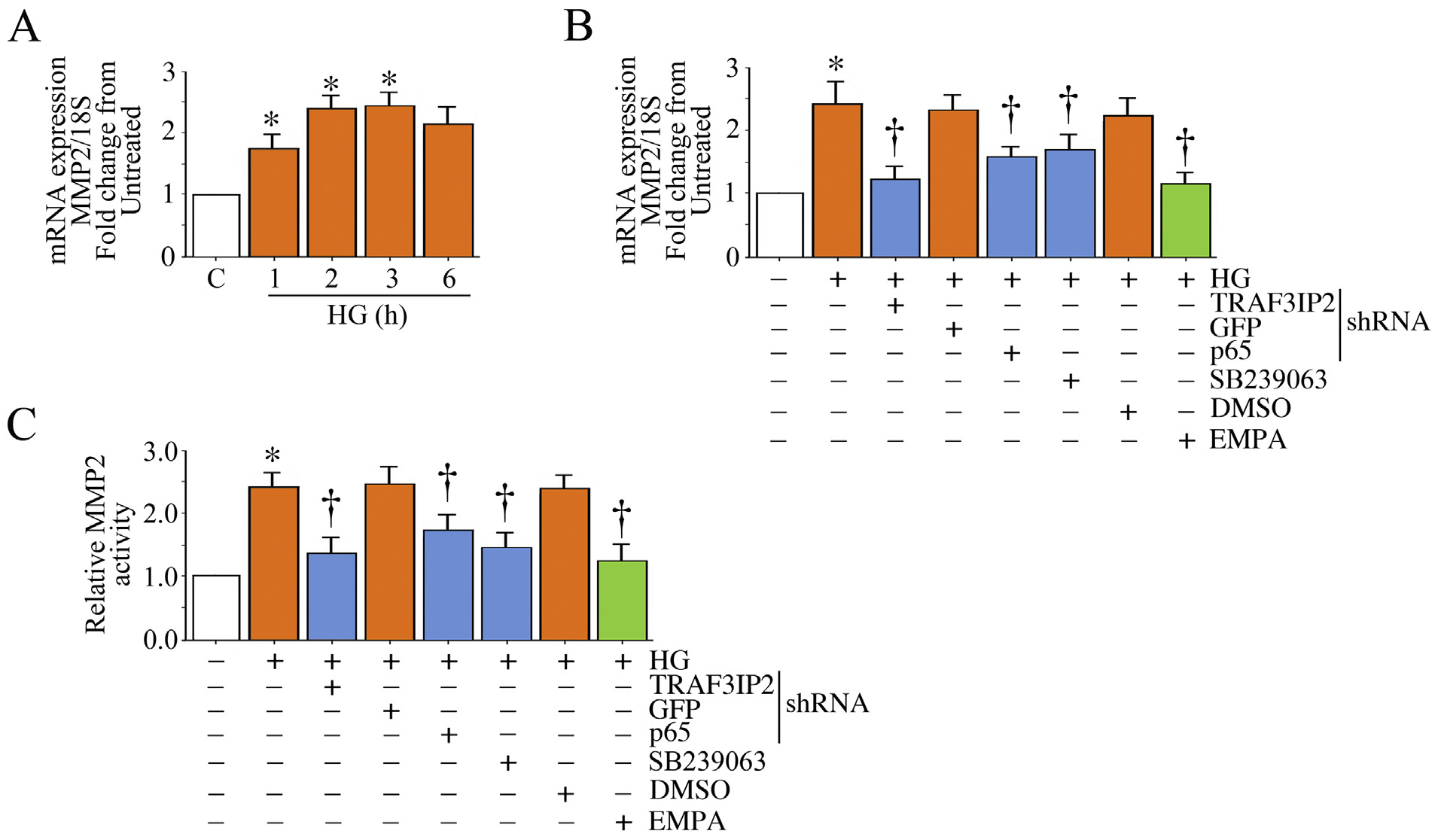
EMPA inhibits High glucose (HG)-induced MMP2 activation in HK-2 cells. A, HG induces MMP2 mRNA expression. Quiescent HK-2 cells were treated with HG (25 mM) for the indicated time periods. MMP2 mRNA expression was analyzed by RT-qPCR using a validated TaqMan® probe. 18S served as an invariant control. B, C, HG induces MMP-2 mRNA expression (B) and activation (C) in part via TRAF3IP2, NF-κB and p38 MAPK, and is inhibited by EMPA. HK-2 cells transduced with lentiviral TRAF3IP2 or p65 shRNA or incubated with SB2239063 or EMPA prior to HG addition (25 mM for 2 h) were analyzed MMP2 mRNA expression by RT-qPCR. MMP-2 activity was analyzed in equal amounts of culture supernatants by Sensolyte® Plus 520 MMP-2 Activity Assay Kit according to the manufacturer’s instructions. * P < at least 0.05 versus Con, † P < at least 0.05 versus HG.
3.7. EMPA and ectopic expression of RECK each inhibit HG-induced MMP2 activation
We and others have previously demonstrated that RECK is an inhibitor of multiple MMPs, including MMP2, in various cell types [21]. Since HG suppressed RECK (Fig. 5B) and induced MMP2 activation (Fig. 6C) in HK-2 cells, we next investigated whether ectopic expression of RECK or pretreatment with EMPA inhibit HG-induced MMP2 activation. Data in Fig. 7A show that transduction of an adenoviral vector expressing full length RECK (Ad.RECK, Fig. 7, left hand panel), but not control GFP (Fig. 7A, right hand panel), increased RECK protein levels in a dose-dependent manner, with peak levels detected at an moi of 10. Therefore, in all subsequent experiments, Ad.RECK was used at moi10. Interestingly, ectopic expression of RECK did not inhibit HG-induced MMP2 mRNA expression or total protein levels (Fig. 7B). On the other hand, Ad.RECK markedly suppressed MMP2 activity (Fig. 7C). Moreover, ectopic expression of RECK and a pharmacological inhibitor of MMP2 each inhibited HG-induced HK-2 cell migration (Fig. 7D). The MMP2 inhibitor reduced HG-induced HK-2 migration without affecting cell viability (Fig. 7E). Importantly, EMPA inhibited HG-induced HK-2 cell migration (Fig. 7D). Since TIMP3 has functions somewhat similar to RECK with respect to MMP inhibition, and is also inhibited by miR-21 induction, we also determined whether HG suppresses TIMP3 and whether ectopic expression of RECK prevents its suppression. Results in Fig. 7B show that similar to its effects on RECK (Fig xx), HG suppressed TIMP3 mRNA expression and protein levels. However, ectopic expression of RECK had no effect in HG-induced TIMP3 suppression (Fig. 7B). Together these results indicate that EMPA inhibits HG-induced MMP2 activation and HK-2 cell migration (Fig. 7).
Fig. 7.
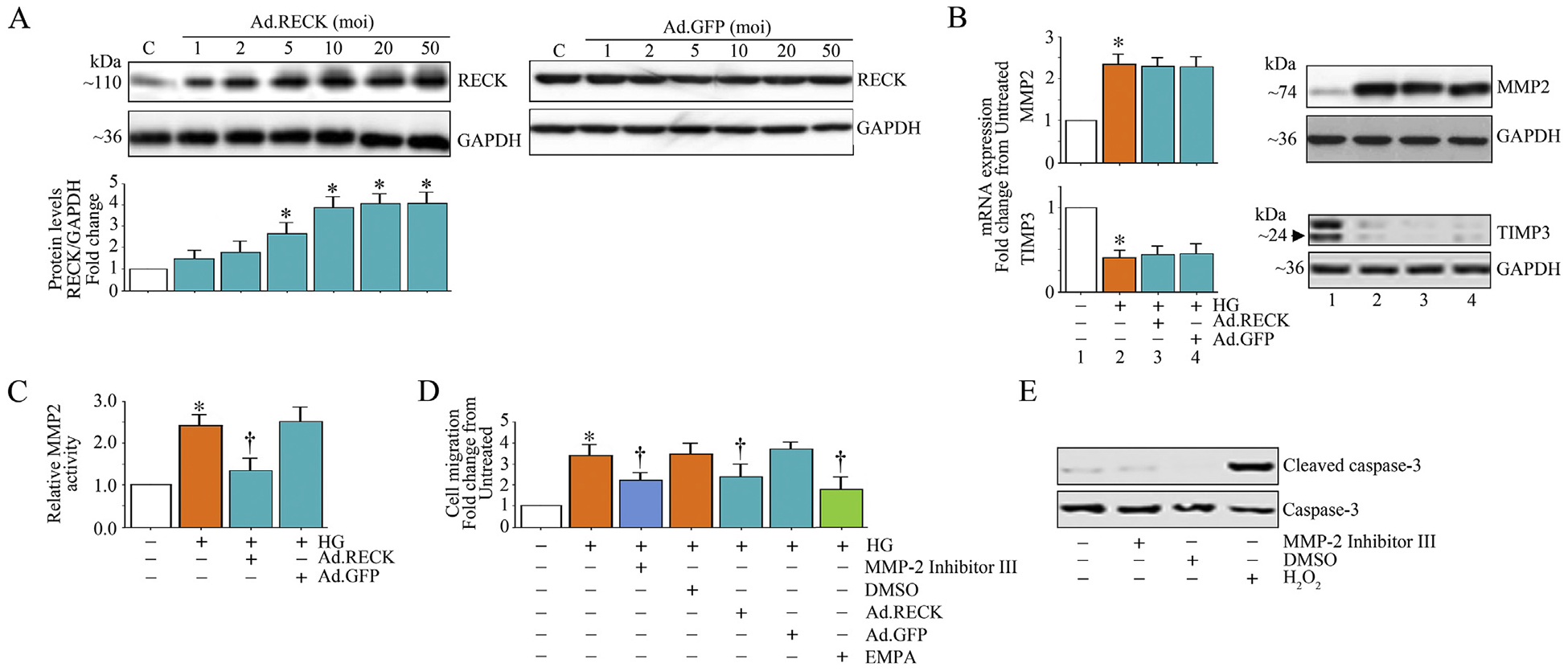
EMPA or ectopic expression of RECK suppresses high glucose (HG)-induced MMP2 activity and HK-2 cell migration. A, Adenoviral transduction of RECK (Ad.RECK; left), but not control GFP (Ad.GFP; right), increases RECK expression in a dose-dependent manner. HK-2 cells transduced with Ad.RECK or Ad.GFP at the indicated moi for 24 h were analyzed for RECK expression by immunoblotting. B, Ectopic expression of RECK does not affect HG-induced differential regulation of MMP-2 and TIMP-3 mRNA and protein expression. HK-2 cells transduced with Ad.RECK (moi 10 for 24 h) were incubated with HG (25 mM) for 2 h, and then analyzed for mRNA (left; RT-qPCR) and protein (right; immunoblotting) expression of MMP-2 and TIMP-3. C, Ectopic expression of RECK suppresses HG-induced MMP-2 activity. HK-2 cells transduced with adenoviral RECK (moi10 for 24 h) were incubated with HG (25 mM for 12 h). MMP-2 activity was analyzed by Sensolyte® Plus 520 MMP-2 Activity Assay Kit according to the manufacturer’s instructions using equal amounts of culture supernatants. D, Ectopic expression of RECK or pretreatment with the MMP-2 inhibitor or EMPA inhibits HG-induced HK-2 cell migration. HK-2 cells transduced with Ad.RECK (moi10 for 24 h) or treated with the MMP-2 Inhibitor II () or EMPA () prior to HG addition (25 mM for 12 h) were analyzed for cell death by quantifying cleaved and total caspse-3 levels by immunoblotting. * P < at least 0.05 versus Con, † P < at least 0.05 versus HG.
3.8. EMPA inhibits HG-induced epithelial-to-mesenchymal transition
Renal TF often leads to end-stage renal disease (ESRD). Kidney fibrosis is characterized by the activation of interstitial fibroblasts, potentially derived from tubular epithelial cells that have undergone epithelial-to-mesenchymal transition (EMT). During EMT, kidney tubular cells lose their epithelial phenotype and acquire features of mesenchymal cells such as downregulation of the epithelial marker E-cadherin, and induction of mesenchymal markers such as fibronectin, α-SMA and vimentin. Therefore, we investigated whether HG promotes EMT of HK-2 cells, and whether EMPA inhibits this phenomenon. Indeed, HG suppressed E-cadherin expression (Fig. 8A), but induced the expression of fibronectin (Fig. 8B), α-SMA (Fig. 8C) and vimentin (Fig. 8D). Importantly, EMPA restored E-cadherin expression, and suppressed HG-induced fibronectin, α-SMA and vimentin. Together, these results indicate that EMPA inhibits HG-induced EMT of HK-2 cells (Fig. 8).
Fig. 8.
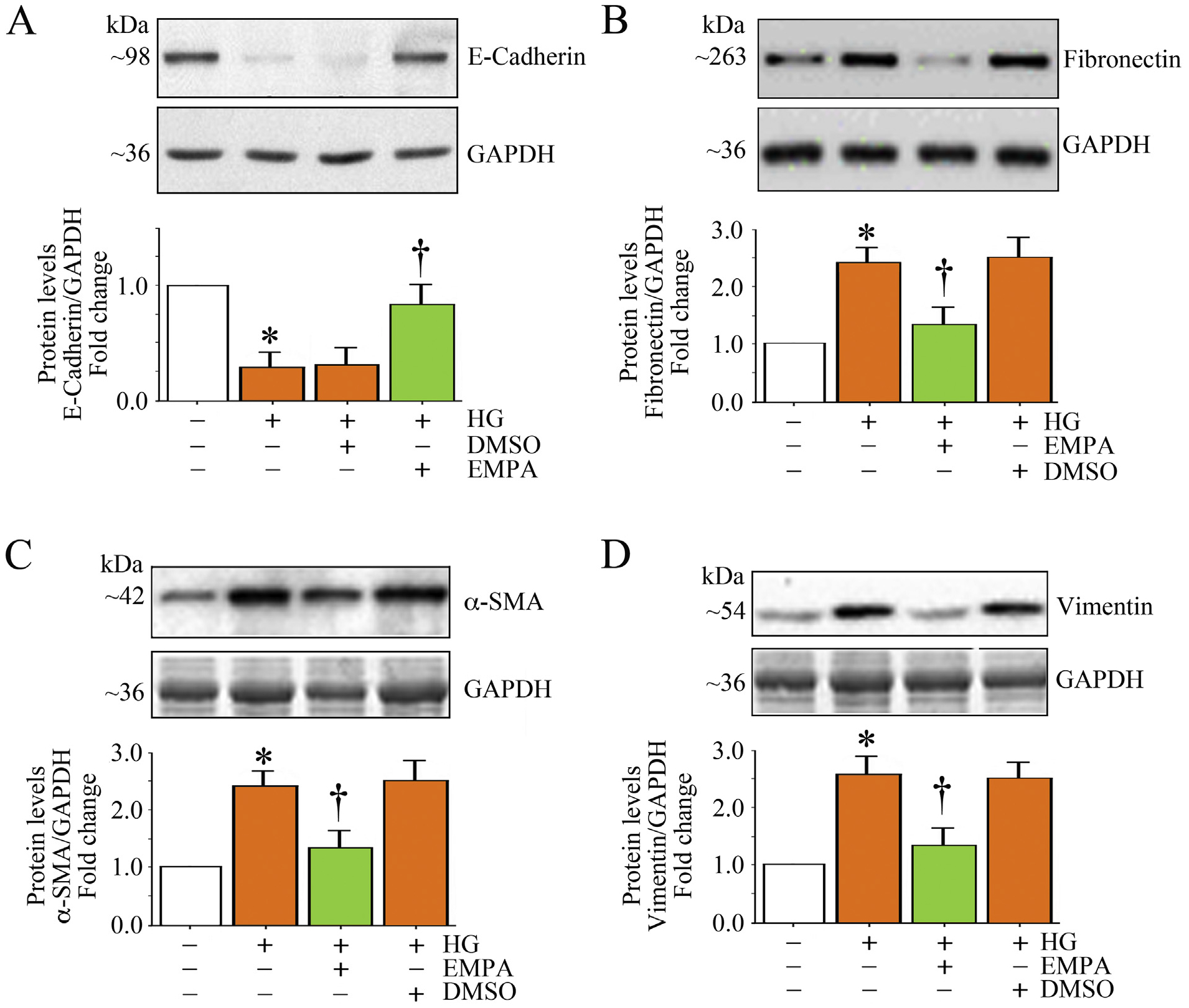
EMPA blunts high glucose (HG)-induced differential regulation of EMT markers in HK-2 cells. A-D, Quiescent HK-2 cells treated with EMPA prior to HG addition (25 mM for 3 h) were analyzed for E-cadherin (A), fibronectin (B), α-SMA (C) and vimentin (D) protein expression by immunoblotting. A-D, The intensity of immunoreactive bands from three independent experiments was semi quantified by densitometry and summarized in the respective lower panels. * P < .05 versus Con, † P < at least 0.05 versus HG.
3.9. EMPA prevents AGE-HSA/RAGE-induced TRAF3IP2 expression and RECK suppression in HK-2 cells
During persistent hyperglycemia, glucose forms adducts with plasma proteins by glycation, a non-enzymatic process. For example, glycated albumin contributes to various diabetes-associated complications such as neuropathy, retinopathy and nephropathy [39]. Similar to hyperglycemia, glycated albumin via RAGE (Receptor for Advanced Glycation End Products) also induces oxidative stress and inflammation in various cell types [40]. Therefore, we investigated whether glycated albumin regulates TRAF3IP2 and RECK expression via RAGE in HK-2 cells, and whether regulation is prevented by EMPA. Indeed, incubation with advanced glycated human serum albumin (AGE-HSA) induced TRAF3IP2 expression in a time-dependent manner, with significant induction seen as early as 30 min. Its expression further increased at 1 h, remained at these high levels until 6 h, and declined thereafter (Fig. 9A). In contrast, and similar to HG, treatment with AGE-HSA suppressed RECK expression in HK-2 cells (Fig. 9A) in a time-dependent manner, with significant suppression seen at 1 h. Its expression was further decreased at 2 h, and remained at these low levels until 48 h (Fig. 9A). Moreover, lentiviral-mediated RAGE knockdown prevented AGE-HSA-induced TRAF3IP2 expression. Importantly, pretreatment with EMPA prevented these changes; suppressed TRAF3IP2, but restored RECK expression (Fig. 9B). Together, these results indicate that, similar to HG, its metabolite AGE-HSA/RAGE induces TRAF3IP2 expression, but suppresses RECK in HK-2 cells, and EMPA prevents these effects (Fig. 9).
Fig. 9.
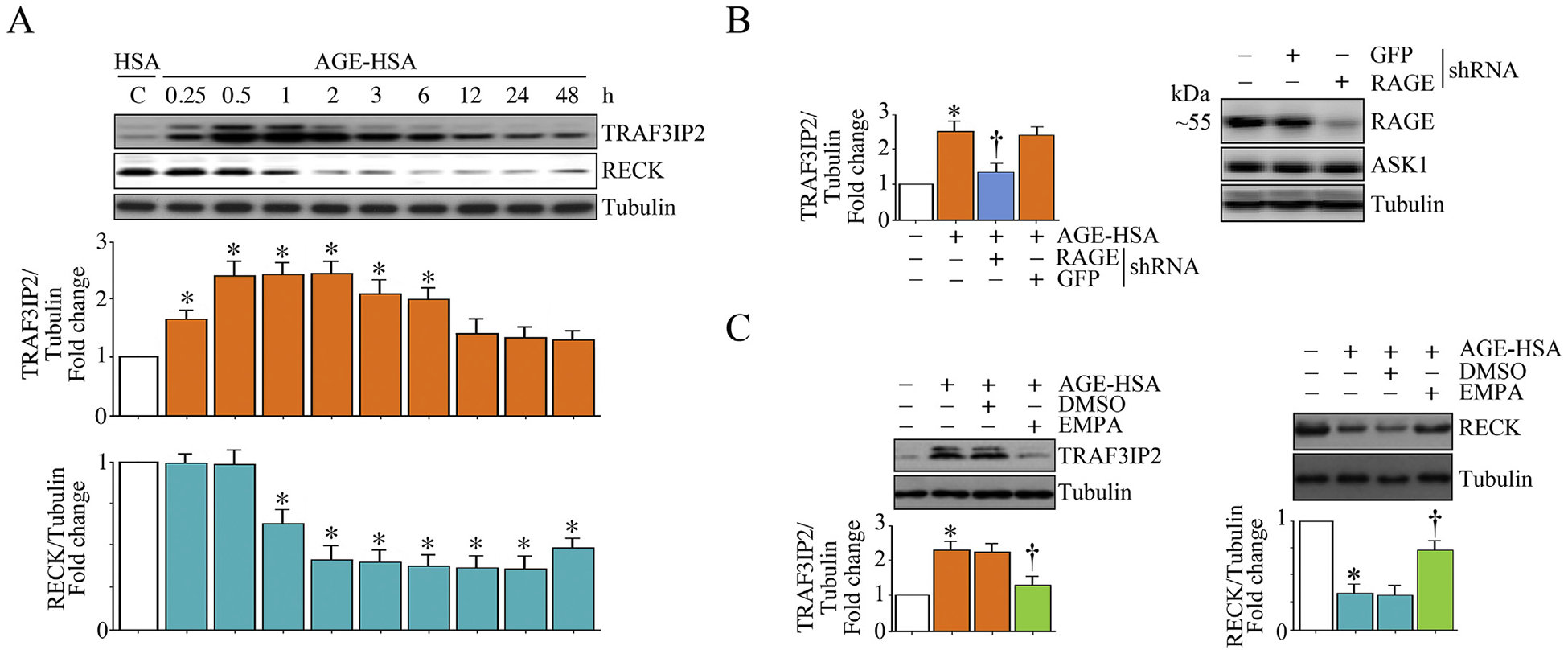
EMPA prevents AGE/RAGE-induced TRAF3IP2 expression and RECK suppression in HK-2 cells. A, AGE-HSA induces TRAF3IP2, but suppresses RECK expression in a time-dependent manner. Quiescent HK-2 cells incubated with AGE-HSA (50 μg/ml) for the indicated time periods were analyzed for TRAF3IP2 and RECK expression by immunoblotting. Unmodified normal HSA at 50 μg/ml served as a control. B, AGE-HSA induces TRAF3IP2 expression via RAGE. HK-2 cells transduced with lentiviral particles expressing RAGE shRNA were incubated with AGE-HSA (50 mg/ml) for 1 h. TRAF3IP2 expression was analyzed by immunoblotting. Lentiviral particles expressing GFP shRNA served as a control. Knockdown of RAGE was confirmed by immunoblotting and is shown on the right. ASK1 served as an off-target control. C, EMPA prevents AGE-HAS-induced TRAF3IP2 expression and RECK suppression. Quiescent HK-2 cells were incubated with EMPA () prior to AGE-HAS () addition for 1 h (TRAF3IP2, left hand panel) or 6 h (RECK, right hand panel). TRAF3IP2 and RECK expression were analyzed by immunoblotting. A, C, The intensity of immunoreactive bands from three independent experiments was semi quantified by densitometry and summarized in the respective lower panels. * P < at least 0.05 versus HSA, †P < at least 0.05 versus AGE-HSA ± DMSO.
4. Discussion
According to the National Institute of Diabetes and Digestive and Kidney Diseases, over thirty million people in the US suffer from diabetes, and approximately 661,000 Americans suffer from kidney failure. Recent cardiovascular safety trials of antihyperglycemic agents that inhibit the sodium-glucose cotransport-2 (SGLT2) transporter have demonstrated reductions in major adverse cardiovascular events and reductions in hospitalization for heart failure, as well as progression of DKD [14,18,41]. We recently reported that db/db mice, a clinically relevant model of type II diabetes, develop TF that is ameliorated by the SGLT2 inhibitor, empagliflozin (EMPA) [20]. In that study we reported renal suppression of RECK, a membrane-anchored endogenous MMP inhibitor and anti-fibrotic mediator, as well as in human kidney proximal tubule epithelial cells (HK-2) exposed to high glucose (HG), effects that were prevented by the SGLT2 inhibitor, empagliflozin (EMPA) both in vivo and in vitro. The purpose of this study was to further examine the molecular mechanisms underlying hyperglycemia-induced RECK suppression, its potential contribution to kidney fibrosis, and activation, migration and proliferation of proximal tubular cells, as well as epithelial mesenchymal transition to determine whether these pathological mechanisms occur in a clinically relevant model of human PTEC. Importantly, we determined whether EMPA could prevent activation and migration of proximal tubule cells.
Here we show for the first time that SGLT2 inhibitor, EMPA, attenuates HG-induced oxidative stress, RECK suppression, EMT and migration of cultured kidney proximal tubule epithelial cells (HK-2). We further show that these salutary effects of EMPA are associated with reductions in expression of several proinflammatory and profibrotic mediators, including TRAF3IP2, NF-κB, p38MAPK, miR-21, and IL-1β, IL-6, TNF-α and MCP-1. Moreover, similar to HG, its metabolite AGE, via RAGE, induced TRAF3IP2 expression, but suppressed RECK, effects that were inhibited by EMPA. These results suggest the therapeutic potential of EMPA in kidney fibrosis and nephropathy under chronic hyperglycemic conditions, such as DKD (Fig. 10).
Fig. 10.
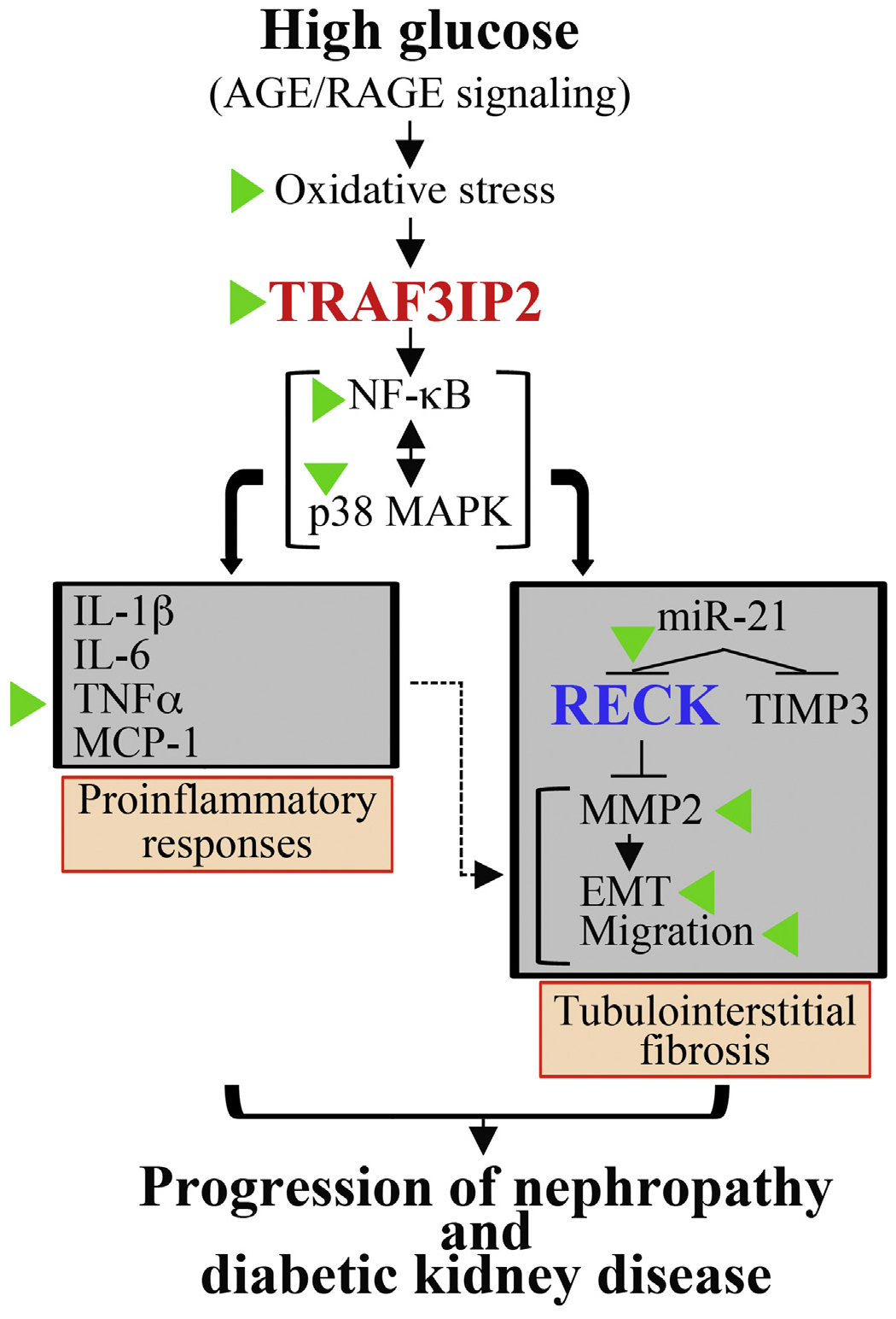
Schema showing the signal transduction pathway involved in high glucose (HG)-induced HK-2 cell proinflammatory response, epithelial-to-mesenchymal transition (EMT) and migration, and those molecules or events targeted by EMPA (indicated by green arrowheads). HG and AGE/RAGE induce oxidative stress as evidenced by increased generation of superoxide and hydrogen peroxide, oxidative stress-dependent TRAF3IP2 upregulation, and TRAF3IP2-dependent NF-κB and p38 MAPK activation, proinflammatory cytokine and miR-21 induction. Further, miR-21 suppresses RECK (and TIMP3) leading to activation of MMP2 that promotes EMT and cell migration that is further exacerbated by the proinflammatory response (indicated by a dashed line). We hypothesize that the illustrated signal transduction pathway leading to high glucose-induced oxidative stress, TRAF3IP2 induction, inflammation, MMP activation and RECK suppression contribute, in part, to progression of diabetic kidney disease. Similar to HG, its metabolite AGE also induces TRAF3IP2, but suppresses RECK. Importantly, EMPA has therapeutic potential in diabetic kidney disease by preventing HG and AGE-induced TRAF3IP2 expression and RECK suppression.
We and others have previously demonstrated that TRAF3IP2 plays a critical role in HG-induced endothelial dysfunction [24,29]. Although this cytoplasmic adapter molecule is essential in the signal transduction of IL-17, its induction/activation has been reported in the absence of IL-17 [26,42]. In fact, TRAF3IP2 expression is regulated by various cis-regulatory elements, including IRF-1, AP-1 and c/EBP in its proximal promoter region [24], all of which are positively regulated by oxidative stress [26], suggesting that oxidative stress is both upstream and downstream of TRAF3IP2 signaling. Our current data support this premise, and show that HG induced TRAF3IP2 expression in HK-2 cells via oxidative stress, as evidenced by reduced expression of TRAF3IP2 in cultures pretreated with various free radical scavengers and anti-oxidants, including NOX2 and NOX4 inhibitors.
Our data show that targeting TRAF3IP2 inhibits activation of NF-κB and the stress-activated kinase p38 MAPK, both of which are essential in the induction of various immune and inflammatory mediators, including cytokines, chemokines, adhesion molecules and MMPs. NF-κB is a nuclear transcription factor and has been shown to play a pathological role in DKD. The NF-κB family includes p50, p52, p65, c-Rel and RelB transcription factors which form either homo or heterodimers, with p65/p65 homodimers and p50/p65 heterodimers being the most predominant. Importantly, site-specific phosphorylation of p65 plays a role in its transactivation. Our data show that HG induced phosphorylation of p65 at Ser536 and in a time-dependent manner, with increased levels detected as early as 30 min after exposure to HG. Of note, genetic deletion of NF-κB in PTEC protected mice from acute kidney injury by suppressing inflammatory cell infiltration and tubular apoptosis [43]. Similarly, siRNA-mediated targeting of IKKβ, an upstream regulator of NF-κB, has shown protection in ischemia-reperfusion-induced acute kidney injury [44]. Since TRAF3IP2/IKKβ physical association leads to NF-κB activation, we hypothesize that TRAF3IP2 may be a better therapeutic target in DKD compared to targeting its downstream signaling intermediates IKK and NF-κB.
Our data also show that targeting TRAF3IP2 inhibits HG-induced p38 MAPK activation in HK-2 cells. Among the stress-activated kinases, ERK, JNK and p38 MAPK, persistent activation of p38 MAPK has consistently been shown to play a pathological role in kidney inflammation and injury. Like NF-κB, activation of p38 MAPK induces the expression of various inflammatory mediators, including cytokines, chemokines and MMPs in kidney. Importantly, pharmacological inhibition of p38 MAPK exerts renoprotective effects, including ischemia-reperfusion-induced kidney injury by targeting IL-1β, TNF-α, and MCP-1 expression and inflammatory cell infiltration [43]. A crosstalk has also been described between NF-κB and p38 MAPK in further amplifying inflammatory signaling. Moreover, activation of NF-κB and p38 MAPK is also implicated in migration and proliferation. Since the adapter molecule TRAF6 plays a role in p38 MAPK activation, and as TRAF3IP2 possesses two TRAF binding sites, we hypothesize that targeting TRAF3IP2 is renoprotective by suppressing activation of NF-κB and p38 MAPK. Importantly, EMPA treatment effectively attenuated HG-induced NF-κB and p38 MAPK activation, suggesting the potential of EMPA in inhibiting DKD.
Both NF-κB and p38 MAPK regulate the expression of various MMPs that contribute to renal inflammation and fibrosis. MMPs degrade extracellular matrix and promote migration of differentiated PTEC under pathological conditions, contributing to adverse matrix remodeling and renal inflammation. Among various MMPs, the gelatinases MMP2 and MMP9 are implicated in PTEC migration. Interestingly, both of these MMPs are NF-κB-responsive genes, and are also regulated by p38 MAPK. In fact, our data show that HG induces MMP2 mRNA expression in HK-2 cells, in part, via TRAF3IP2, NF-κB and p38 MAPK. HG also induced MMP2 protein levels and enhanced its activity. Moreover, a MMP2-specific inhibitor attenuated HG-induced MMP2 activity and HK-2 cell migration, without affecting cell viability. More importantly, EMPA pretreatment inhibited HG-induced MMP2 mRNA expression and activity, further supporting the therapeutic potential of EMPA in DKD.
Interestingly, while inducing MMP2 expression, HG markedly suppressed RECK expression in PTEC. HG suppressed RECK expression in a time-dependent manner, with significant reduction seen as early as 1 h post-treatment. RECK levels remained low until 24 h, and slightly increased after 48 h, indicating that RECK expression is chronically suppressed during hyperglycemic conditions, possibly contributing to increased MMP2 activity, as RECK is known to physically associate and block MMP2 maturation and activity [23]. Since an inverse correlation was observed between MMP2 and RECK expression in HK-2 cells under hyperglycemic conditions, we next investigated whether RECK gain-of-function would limit MMP2 activity. Indeed, RECK gain-of-function inhibited HG-induced MMP2 activity, without affecting its mRNA expression or total protein levels and inhibited HK-2 cell migration. We previously reported that specificity protein 1 (SP1), which is induced by hyperglycemia, suppresses RECK and induces MMP2 activation in response to Ang II in adult mouse cardiac fibroblasts [21,22]. Our future studies will explore whether the SP1 contributes to progression of renal fibrosis and whether EMPA inhibits its activation. Importantly, silencing TRAF3IP2 and EMPA pretreatment each prevented HG-induced RECK suppression and HK-2 cell migration. Since differentiation and migration of PTEC contribute to renal fibrosis, our results again suggest the therapeutic potential of EMPA in DKD, a condition characterized by excessive interstitial fibrosis.
Since HG suppressed RECK expression, we next investigated potential mechanisms underlying its down regulation under hyperglycemic conditions. RECK expression is regulated at both the transcriptional and post-transcriptional level and includes acetylation, methylation and modulation by microRNAs. In fact, multiple micro-RNAs, including miR-21, target RECK 3’UTR and repress its expression. We have previously reported that Angiotensin-II, a RAAS member, induces AP-1-dependent miR-21 expression and RECK suppression in cardiac fibroblasts [27]. In those studies, we demonstrated that miR-21 inhibition and RECK overexpression each inhibited Angiotensin-II-induced cardiac fibroblast migration. Since HG suppressed RECK in HK-2 cells, we next investigated whether HG-induced RECK suppression is mediated by miR-21. Indeed, while a miR-21 inhibitor prevented HG-induced RECK suppression, transfection with a miR-21 mimic, by itself, suppressed RECK expression, indicating that one of the mechanisms by which HG suppresses RECK expression in HK-2 cells is via miR-21 induction. Though we have not investigated the time course of miR-21 induction, we hypothesize that the chronic suppression in RECK expression following HG addition, is due to persistent induction of miRs that negatively regulate RECK expression. Our future studies will investigate this possibility, as well as the potential involvement of other HG-associated RECK targeting miRNAs. Of particular interest in this regard is miR-221, which has been identified as a regulator of fibrosis in renal tubular compartments [45]. Interestingly, TIMP3, which shares similar functions as RECK, is also a target of miR-21 and miR-221 [46], and its expression is also suppressed by HG at both mRNA and protein levels. However, RECK overexpression failed to restore the expression of TIMP3.
Epithelial-to-mesenchymal transition (EMT) precedes PTEC migration. Though the role of EMT in extracellular matrix deposition and inflammatory cytokine expression is well characterized in vitro, its role in vivo in TF is controversial and dependent on the model of renal injury and inflammation. Several markers have been analyzed to characterize EMT in PTEC, including the expression of E-cadherin, N-cadherin, α-SMA, fibronectin, vimentin and collagen I. Importantly, during EMT, PTEC lose the expression of E-cadherin, an epithelial marker, and gain the expression of several mesenchymal markers, including N-cadherin, fibronectin, α-SMA and vimentin. Our data show that HG, while suppressing E-cadherin expression, upregulated the expression of fibronectin, α-SMA and vimentin in PTEC. More importantly, EMPA reversed these effects, indicating that EMPA may exert its therapeutic potential in DKD by suppressing EMT, PTEC migration and the excess deposition of extracellular matrix. In fact, we recently demonstrated that EMPA treatment suppresses kidney fibrosis in T2D female db/db mice [20]. In that study, while the diabetic mice expressed increased levels of collagen Iα1, collagen IIIα1 and fibronectin in kidneys, EMPA treatment repressed their expression, indicating that SGLT2 inhibition by EMPA exerts anti-fibrotic effects in DKD. Supporting our results, a recent report demonstrated that dapagliflozin, another SGLT2 inhibitor, suppressed HG-induced EMT in HK-2 cells [47].
In our previous published reports we demonstrated induction of TRAF3IP2 by the proinflammatory cytokine IL-18, advanced oxidation protein products (AOPP), Angiotensin-II and aldosterone, all of which increase its expression, in part, via ROS generation [24,26,48,49]. Interestingly, in those studies the increase in TRAF3IP2 expression was monophasic, whereas HG-induced TRAF3IP2 expression observed in the present study is biphasic in HK-2 cells. A limitation of this study is that we have not yet investigated the mechanisms responsible for this biphasic expression pattern. Nonetheless, we hypothesize that the initial upregulation in TRAF3IP2 expression is due to rapid generation of superoxide and hydrogen peroxide in response to HG, and the later increase or upregulation may be due to inflammatory cytokines and RAAS components induced in response to HG.
Abundant evidence supports a critical role for TGFβ in TF by promoting EMT of PTEC and this has been reviewed in detail [50]. TGFβ enhances the expression of various mesenchymal markers, including α-SMA, vimentin and collagen I in these cells. Interestingly, TGFβ also signals via NF-κB and mediates miR-21-dependent profibrotic effects in the kidney following ureteral obstruction [51]. Since TRAF3IP2 is an upstream regulator of NF-κB and miR-21, we hypothesize that targeting TRAF3IP2 blunts TGFβ-induced NF-κB/miR-21 induction and the excess deposition of extracellular matrix. However, in the current study we did not investigate the effects of the interactions between TGFβ and TRAF3IP2 in the regulation of HG-induced proinflammatory and fibrotic responses, and consider it as a limitation.
A third limitation relates to gender-dependent effects of hyperglycemia. In the present study, we utilized HK-2 cells, derived from a male individual, and observed HG-induced regulation of TRAF3IP2 and RECK. In contrast, we previously demonstrated upregulation of TRAF3IP2 in cardiac tissue from western diet fed female mice associated with hyperglycemia [52]. Additionally, we demonstrated regulation of RECK by EMPA in hyperglycemic female db/db mice [20]. Thus, the mechanisms explored in the current study and our previous work raise the possibility that the salutary effects of EMPA may be relevant to both males and females. We will further explore these limitations in future studies.
Recent studies suggest that other targets of SGLT2 inhibitors may contribute to the changes in parameters investigated in this study in response to HG [53]. This is an important and ongoing area of interest and hypotheses and data are emerging to suggest that the cardioprotective effects of SGLT2 inhibitors may be due, in part, to their inhibitory effects on NHE1 in the heart [53–57]. This is highly relevant given that enhanced activation of NHEs are associated with glucose intolerance, DKD and heart failure, conditions that often coexist in diabetic individuals [57]. Indeed, recent studies have shown that the major SGLT2 inhibitors, EMPA, dapagliflozin and canaglifozin, inhibit NHE1 in cardiomyocytes and have additional SGLT2-independent salutary effects in myocardial constituent cells, including endothelial cells, vascular smooth muscle cells and fibroblasts [54]. In kidneys, NHEs have been shown to be abundantly expressed, specifically in PTEC [58]. HK-2 cells, the cell line used in the present study, are also shown to express high levels of NHEs [59]. Thus, it is reasonable to hypothesize that, in addition to their glucose lowering effects, SGLT2 inhibitors exert renoprotective effects via suppression of NHEs [55,56]. Notably, HG is shown to induce NHE1 expression and subsequent apoptosis of HK-2 cells [59]. Moreover, inhibition of NHE3 has been shown to attenuate AGE-induced renal dysfunction [60]. Collectively, these reports suggest that EMPA exerts renoprotective effects in PTEC (HK-2) through inhibition of NHE1 and NHE3, which will be investigated in our future studies.
In summary, our data show for the first time that the SGLT2 inhibitor, EMPA, attenuates HG-induced oxidative stress, MMP2 activation, RECK suppression in cultured kidney proximal tubule cells (HK-2), resulting in suppressed EMT and migration. Importantly, these changes are associated with reductions in the expression of several proinflammatory and profibrotic mediators, including TRAF3IP2, NF-κB, p38MAPK, miR-21, IL-1β, IL-6, TNFα and MCP1. Furthermore, EMPA prevented AGE/RAGE-induced TRAF3IP2 expression and RECK suppression. Together, these data further support the therapeutic potential of EMPA in DKD via novel mechanisms involving TRAF3IP2 inhibition and RECK induction.
Acknowledgements
This work was supported with resources and facilities at the Harry S. Truman Memorial Veterans Hospital in Columbia, MO. VGD is the guarantor of this study.
Funding
This work was supported by an unrestricted research grant from Boehringer Ingelheim Pharma to VGD, the U.S. Department of Veterans Affairs, Office of Research and Development-Biomedical Laboratory Research and Development (ORD-BLRD) Service Award I01-BX004220 and Research Career Scientist Award (IK6BX004016) to BC, and by the Intramural Research Program of the NIH to US.
Abbreviations:
- AGE
advanced glycation end products
- AGSI
average grey scale intensities
- AOPP
advanced oxidation protein products
- α-SMA
alpha smooth muscle actin
- ColIα1
collagen Iα1
- ColIIIα1
collagen IIIα1
- CVD
cardiovascular disease
- DKD
diabetic kidney disease
- DMSO
dimethylsulfoxide
- DPI
diphenyleneiodonium
- EMPA
empagliflozin
- EMPA-REG OUTCOME TRIAL
Empagliflozin, Cardiovascular Outcomes and Mortality in Type 2 Diabetes trial
- eNOS
endothelial nitric oxide synthase
- EMT
epithelial-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GKT137831
NOX1/4 inhibitor
- HG
high glucose
- HK-2
human kidney proximal tubular epithelial cells
- IKKβ
inhibitor of nuclear factor kappa-B kinase subunit beta
- p-p38MAPK
phospho-p38 mitogen-activated protein kinase
- miR-21
microRNA-21
- MMP
matrix metalloproteinase
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
- 3-NT
3-nitrotyrosine
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PSS
physiological salt solution
- PSR
picrosirius red
- PTEC
proximal tubular epithelial cells
- RECK
reversion inducing cysteine rich protein with Kazal motifs
- RAAS
renin-angiotensin-aldosterone system
- RAGE
receptor for AGE
- REL
REL protooncogene
- RelB
RELB protooncogene
- SGLT2
sodium-glucose co-transporter 2
- SP1
specificity protein 1
- Tempol
4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
- TF
tubulointerstitial fibrosis
- TIMP
tissue inhibitor of MMP
- TIRON
disodium 4,5-dihydroxy-1,3-benzenedisulfo-nate
- TRAF3IP2
TRAF3-interacting protein 2
- TGF-β
transforming growth factor-β
Footnotes
Declarations/disclosure statement
VGD has received unrestricted research support from Boehringer Ingelheim. No other potential conflicts of interest relevant to this article were reported.
Ethics approval and consent to participate
This study was approved by the Subcommittee for Animal Safety of the Harry S Truman Veterans Administration and the Institutional Animal Care and Use Committee of the University of Missouri.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Competing Interest
Funding was provided, in part, by Boehringer Ingelheim Pharma to VGD.
References
- [1].Alicic RZ, Rooney MT, Tuttle KR, Diabetic kidney disease: challenges, progress, and possibilities, Clin. J. Am. Soc. Nephrol 12 (12) (2017) 2032–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hashimoto Y, et al. , Polyol pathway and diabetic nephropathy revisited: early tubular cell changes and glomerulopathy in diabetic mice overexpressing human aldose reductase, J. Diabetes Investig 2 (2) (2011) 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yan LJ, Redox imbalance stress in diabetes mellitus: role of the polyol pathway, Anim. Model Exp. Med 1 (1) (2018) 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nistala R, Whaley-Connell A, Resistance to insulin and kidney disease in the cardiorenal metabolic syndrome; role for angiotensin II, Mol. Cell. Endocrinol 378 (1–2) (2013) 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bettiga A, et al. , The modern Western diet rich in advanced glycation end-products (AGEs): an overview of its impact on obesity and early progression of renal pathology, Nutrients 11 (8) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zeni L, et al. , A more tubulocentric view of diabetic kidney disease, J. Nephrol 30 (6) (2017) 701–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vallon V, The proximal tubule in the pathophysiology of the diabetic kidney, Am. J. Physiol. Regul. Integr. Comp. Physiol 300 (5) (2011) (p. R1009–22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vallon V, Thomson SC, Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition, Diabetologia 60 (2) (2017) 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vallon V, et al. , SGLT2 mediates glucose reabsorption in the early proximal tubule, J. Am. Soc. Nephrol 22 (1) (2011) 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vallon V, The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus, Annu. Rev. Med 66 (2015) 255–270. [DOI] [PubMed] [Google Scholar]
- [11].Hatanaka T, et al. , Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice, Pharmacol. Res. Perspect 4 (4) (2016) e00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee KA, et al. , Effect of Empagliflozin, a selective sodium-glucose Cotransporter 2 inhibitor, on kidney and peripheral nerves in Streptozotocin-induced diabetic rats, Diabetes Metab. J 42 (4) (2018) 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kawanami D, et al. , SGLT2 inhibitors as a therapeutic option for diabetic nephropathy, Int. J. Mol. Sci 18 (5) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Neal B, et al. , Canagliflozin and cardiovascular and renal events in type 2 diabetes, N. Engl. J. Med 377 (7) (2017) 644–657. [DOI] [PubMed] [Google Scholar]
- [15].Heerspink HJ, et al. , Canagliflozin slows progression of renal function decline independently of glycemic effects, J. Am. Soc. Nephrol 28 (1) (2017) 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McMurray JJV, et al. , Dapagliflozin in patients with heart failure and reduced ejection fraction, N. Engl. J. Med 381 (2019) 1995–2008. [DOI] [PubMed] [Google Scholar]
- [17].Butler J, et al. , Empagliflozin improves kidney outcomes in patients with or without heart failure, Circ. Heart Fail 12 (6) (2019) e005875. [DOI] [PubMed] [Google Scholar]
- [18].Wanner C, et al. , Empagliflozin and progression of kidney disease in type 2 diabetes, N. Engl. J. Med 375 (2016) 323–334. [DOI] [PubMed] [Google Scholar]
- [19].Mayer GJ, et al. , Analysis from the EMPA-REG OUTCOME((R)) trial indicates empagliflozin may assist in preventing the progression of chronic kidney disease in patients with type 2 diabetes irrespective of medications that alter intrarenal hemodynamics, Kidney Int. 96 (2) (2019) 489–504. [DOI] [PubMed] [Google Scholar]
- [20].Aroor AR, et al. , Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury, Cardiovasc. Diabetol 17 (1) (2018) 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Siddesha JM, et al. , Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK, J. Mol. Cell. Cardiol 65 (2013) 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Somanna NK, et al. , Histone deacetyltransferase inhibitors Trichostatin A and Mocetinostat differentially regulate MMP9, IL-18 and RECK expression, and attenuate angiotensin II-induced cardiac fibroblast migration and proliferation, Hypertens. Res 39 (10) (2016) 709–716. [DOI] [PubMed] [Google Scholar]
- [23].Oh J, et al. , The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis, Cell 107 (6) (2001) 789–800. [DOI] [PubMed] [Google Scholar]
- [24].Venkatesan B, et al. , CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction, Cell. Signal 25 (1) (2013) 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Valente AJ, et al. , OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators, Free Radic. Biol. Med 70 (2014) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Valente AJ, et al. , TRAF3IP2 mediates interleukin-18-induced cardiac fibroblast migration and differentiation, Cell. Signal 25 (11) (2013) 2176–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Siddesha JM, et al. , Docosahexaenoic acid reverses angiotensin II-induced RECK suppression and cardiac fibroblast migration, Cell. Signal 26 (5) (2014) 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mummidi S, et al. , Metformin inhibits aldosterone-induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt-induced cardiac fibrosis in vivo, J. Mol. Cell. Cardiol 98 (2016) 95–102. [DOI] [PubMed] [Google Scholar]
- [29].Padilla J, et al. , TRAF3IP2 mediates high glucose-induced endothelin-1 production as well as endothelin-1-induced inflammation in endothelial cells, Am. J. Physiol. Heart Circ. Physiol 314 (2017) H52–H64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Erickson JM, et al. , Targeting TRAF3IP2 by genetic and interventional approaches inhibits ischemia/reperfusion-induced myocardial injury, J. Biol. Chem 292 (2017) 2345–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma FY, Liu J, Nikolic-Paterson DJ, The role of stress-activated protein kinase signaling in renal pathophysiology, Braz. J. Med. Biol. Res 42 (1) (2009) 29–37. [DOI] [PubMed] [Google Scholar]
- [32].Adhikary L, et al. , Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy, Diabetologia 47 (7) (2004) 1210–1222. [DOI] [PubMed] [Google Scholar]
- [33].Saha RN, Jana M, Pahan K, MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65, J. Immunol 179 (10) (2007) 7101–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mummidi S, et al. , RECK suppresses interleukin-17/TRAF3IP2-mediated MMP-13 activation and human aortic smooth muscle cell migration and proliferation, J. Cell. Physiol 234 (12) (2019) 22242–22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zaman MS, et al. , Up-regulation of microRNA-21 correlates with lower kidney cancer survival, PLoS One 7 (2) (2012) e31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zarjou A, et al. , Identification of a microRNA signature in renal fibrosis: role of miR-21, Am. J. Physiol. Renal. Physiol 301 (4) (2011) F793–F801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zakiyanov O, et al. , Matrix Metalloproteinases in renal diseases: a critical appraisal, Kidney Blood Press Res. 44 (3) (2019) 298–330. [DOI] [PubMed] [Google Scholar]
- [38].Zhao H, et al. , Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases, World J. Nephrol 2 (3) (2013) 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Huh JH, et al. , Glycated albumin is a more useful glycation index than HbA1c for reflecting renal tubulopathy in subjects with early diabetic kidney disease, Diabetes Metab. J 42 (2018) 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Martinez Fernandez A, et al. , Pro-oxidant and pro-inflammatory effects of glycated albumin on cardiomyocytes, Free Radic. Biol. Med 144 (2019) 245–255. [DOI] [PubMed] [Google Scholar]
- [41].Zinman B, Lachin JM, Inzucchi SE, Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes, N. Engl. J. Med 374 (11) (2016) 1094. [DOI] [PubMed] [Google Scholar]
- [42].Leonardi A, et al. , CIKS, a connection to Ikappa B kinase and stress-activated protein kinase, Proc. Natl. Acad. Sci. U. S. A 97 (19) (2000) 10494–10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Marko L, et al. , Tubular epithelial NF-kappaB activity regulates ischemic AKI, J. Am. Soc. Nephrol 27 (9) (2016) 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wan X, et al. , Small interfering RNA targeting IKKbeta prevents renal ischemiareperfusion injury in rats, Am. J. Physiol. Renal Physiol 300 (4) (2011) F857–F863. [DOI] [PubMed] [Google Scholar]
- [45].Nishi H, Angiopoietin-like protein 2 and kidney fibrosis: lessons from knockout mice, Kidney Int. 89 (2) (2016) 272–274. [DOI] [PubMed] [Google Scholar]
- [46].Fiorentino L, et al. , Regulation of TIMP3 in diabetic nephropathy: a role for microRNAs, Acta Diabetol. 50 (6) (2013) 965–969. [DOI] [PubMed] [Google Scholar]
- [47].Huang F, et al. , Dapagliflozin attenuates renal tubulointerstitial fibrosis associated with type 1 diabetes by regulating STAT1/TGFbeta1 signaling, Front. Endocrinol. (Lausanne) 10 (2019) 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Valente AJ, et al. , CIKS (Act1 or TRAF3IP2) mediates angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy, J. Mol. Cell. Cardiol 53 (1) (2012) 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Somanna NK, et al. , Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2, Cell. Signal 27 (10) (2015) 1928–1938. [DOI] [PubMed] [Google Scholar]
- [50].Hills CE, Squires PE, The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy, Cytokine Growth Factor Rev. 22 (3) (2011) 131–139. [DOI] [PubMed] [Google Scholar]
- [51].Zhong X, et al. , Smad3-mediated upregulation of miR-21 promotes renal fibrosis, J. Am. Soc. Nephrol 22 (9) (2011) 1668–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Aroor AR, et al. , Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice, Cardiovasc. Diabetol 16 (1) (2017) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Uthman L, et al. , Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation, Diabetologia 61 (3) (2018) 722–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Uthman L, et al. , Direct cardiac actions of sodium glucose Cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients, Front. Physiol 9 (2018) 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Arjun S, Bell RM, SGLT2 inhibitors: reviving the sodium-hydrogen exchanger cardioprotection hypothesis? Cardiovasc. Res 115 (10) (2019) 1454–1456. [DOI] [PubMed] [Google Scholar]
- [56].Packer M, et al. , Effects of sodium-glucose Cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action, JAMA Cardiol. 2 (9) (2017) 1025–1029. [DOI] [PubMed] [Google Scholar]
- [57].Packer M, Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure, Circulation 136 (16) (2017) 1548–1559. [DOI] [PubMed] [Google Scholar]
- [58].Parker MD, Myers EJ, Schelling JR, Na+-H+ exchanger-1 (NHE1) regulation in kidney proximal tubule, Cell. Mol. Life Sci 72 (11) (2015) 2061–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wu Y, et al. , Oxidative stress-activated NHE1 is involved in high glucose-induced apoptosis in renal tubular epithelial cells, Yonsei Med. J 57 (5) (2016) 1252–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li P, et al. , Inhibition of NA(+)/H(+) Exchanger 1 attenuates renal dysfunction induced by advanced glycation end products in rats, J. Diabetes Res 2016 (2016) 1802036. [DOI] [PMC free article] [PubMed] [Google Scholar]