

SUMMARY
Osteoprotegerin (OPG) inhibits the ability of receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) to stimulate the differentiation, activity, and survival of bone-resorbing osteoclasts. Genetic studies in mice show that osteocytes are an important source of RANKL, but the cellular sources of OPG are unclear. We use conditional deletion of Tnfrsf11b, which encodes OPG, from different cell populations to identify functionally relevant sources of OPG in mice. Deletion from B lymphocytes and osteocytes, two cell types commonly thought to supply OPG, has little or no impact on bone mass. By contrast, deletion of Tnfrsf11b from osteoblasts increases bone resorption and reduces bone mass to an extent similar to germline deletion, demonstrating that osteoblasts are an essential source of OPG. These results suggest that, in addition to producing new bone matrix, osteoblasts also play an active role in terminating the resorption phase of the bone remodeling cycle by suppressing RANKL activity.
Graphical Abstract
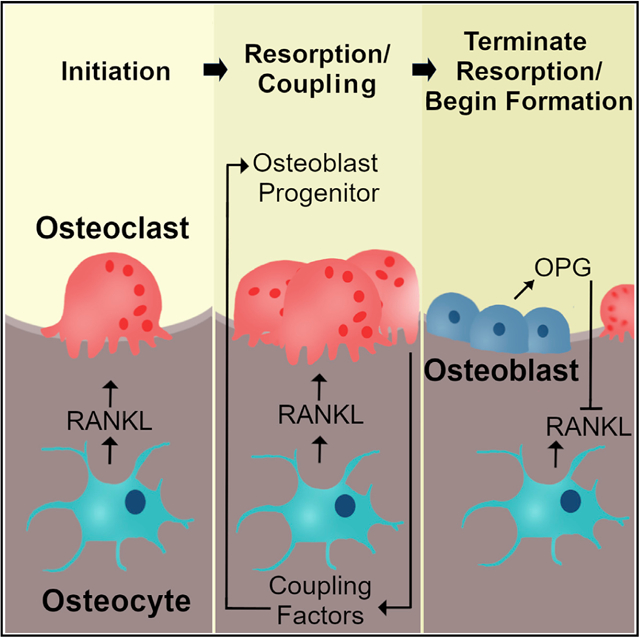
In Brief
Cawley et al. use conditional deletion of Tnfrsf11b, which encodes osteoprotegerin, in mice to demonstrate that osteoblasts actively suppress osteoclast formation by producing this RANKL antagonist. Circulating osteoprotegerin levels are unchanged in mice lacking Tnfrsf11b in osteoblasts, demonstrating that local production of osteoprotegerin is essential for control of bone resorption.
INTRODUCTION
Osteoclasts are bone-resorbing cells with key roles in several biological processes, including resorption of calcified cartilage during bone growth, tooth eruption, and remodeling of adult bone (Asagiri and Takayanagi, 2007; Teitelbaum, 2000). In each of these situations, osteoclasts must form at the correct place and time, and in appropriate numbers, to accomplish their physiological roles. The number of osteoclasts that form at a specific location depends on the availability of myeloid precursors and on differentiation and survival signals. Receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) is a member of the tumor necrosis factor (TNF) family and is essential for osteoclast differentiation, survival, and activity (Kong et al., 1999; Lacey et al., 1998). RANKL action is opposed by a soluble decoy receptor known as osteoprotegerin (OPG) (Simonet et al., 1997). Thus, the relative levels of RANKL and OPG are key factors that determine the number and activity of osteoclasts formed at a specific site.
Using conditional gene deletion in mice, we and others have shown that the majority of the RANKL required for osteoclast formation in remodeling cancellous bone is produced by osteocytes, which are former osteoblasts buried within bone matrix (Nakashima et al., 2011; Xiong et al., 2011, 2015). RANKL is produced initially as an integral membrane protein but can be cleaved by proteases to produce a functional soluble form (sRANKL) (Lacey et al., 1998). Recent studies demonstrate that the membrane-bound form of RANKL is responsible for the majority of RANKL functions, including osteoclast formation (Nagashima et al., 2017; Xiong et al., 2018). The finding that osteocytes support osteoclast formation via production of RANKL on their cell surface suggests that control of osteoclast formation is highly localized.
Mice and humans with a non-functional Tnfrsf11b gene, which encodes OPG, exhibit a profound increase in osteoclast number, low bone mass, and fractures (Bucay et al., 1998; Grasemann et al., 2017). Tnfrsf11b is expressed by cells of the osteoblast lineage, as well as by a wide variety of cells in other tissues such as lung, liver, blood vasculature, and lymphoid tissue (Simonet et al., 1997). Although it is often assumed that reciprocal regulation of RANKL and OPG in a specific cell type, such as osteocytes, initiates osteoclast formation, the cellular source of the OPG involved in suppressing osteoclast formation is unknown. Nonetheless, deletion of β-catenin from osteocytes is associated with reduced Tnfrsf11b expression in osteocytes and increased bone resorption, leading to the conclusion that this cell type is a major source of OPG (Kramer et al., 2010). By contrast, others have shown that B lymphocytes produce the majority of OPG mRNA in bone marrow cultures and that mice lacking B cells have elevated bone resorption, leading to the conclusion that B cells are an important source of OPG (Li et al., 2007).
Herein, we have used conditional deletion of Tnfrsf11b using a variety of different Cre-driver strains to identify important cellular sources of the OPG involved in suppressing osteoclast formation. Unexpectedly, we found that osteoblasts, but not osteocytes, are a major source of the OPG that suppresses osteoclast formation in cancellous bone. Our results also suggest that multiple cell types, including osteocytes, supply the OPG that protects cortical bone from resorption by osteoclasts.
RESULTS
OPG Produced by B Cells Does Not Suppress Bone Resorption
To perform cell-type-specific deletion of Tnfrsf11b, we first created a conditional allele for this gene. To accomplish this, we used gene editing to insert loxP sites upstream and downstream of exon 2 (Figure S1), since germline deletion of exon 2 results in complete loss of OPG function (Bucay et al., 1998; Mizuno et al., 1998). As expected from previous reports (Bucay et al., 1998; Mizuno et al., 1998), germline deletion of the Tnfrsf11bf/f allele using EIIa-Cre mice resulted in reduced cortical thickness and almost complete loss of cancellous bone in the femur at 5 weeks of age (Figure S1).
To determine the significance of OPG produced by B lymphocytes, we used CD19-Cre mice to delete the Tnfrsf11bf/f allele in the entire B cell lineage (Rickert et al., 1997). At 5 weeks of age, deletion of OPG from B cells did not alter cortical thickness or cancellous bone volume in the femur or spine, whereas Tnfrsf11b−/− mice exhibited low bone mass in both compartments (Figures 1A–1D). We observed similar results at 4 months of age (Figure S2). Circulating OPG and RANKL were unchanged by deletion of Tnfrsf11b from B cells at either age (Figure S2). Nonetheless, we confirmed that the CD19-Cre transgene led to effective and specific deletion of the Tnfrsf11bf/f allele from B cells (Figure S3). Based on these results, we conclude that OPG derived from B cells does not contribute to the suppression of bone resorption under physiological conditions.
Figure 1. Deletion of Tnfrsf11b from B Cells Does Not Alter Bone Mass.
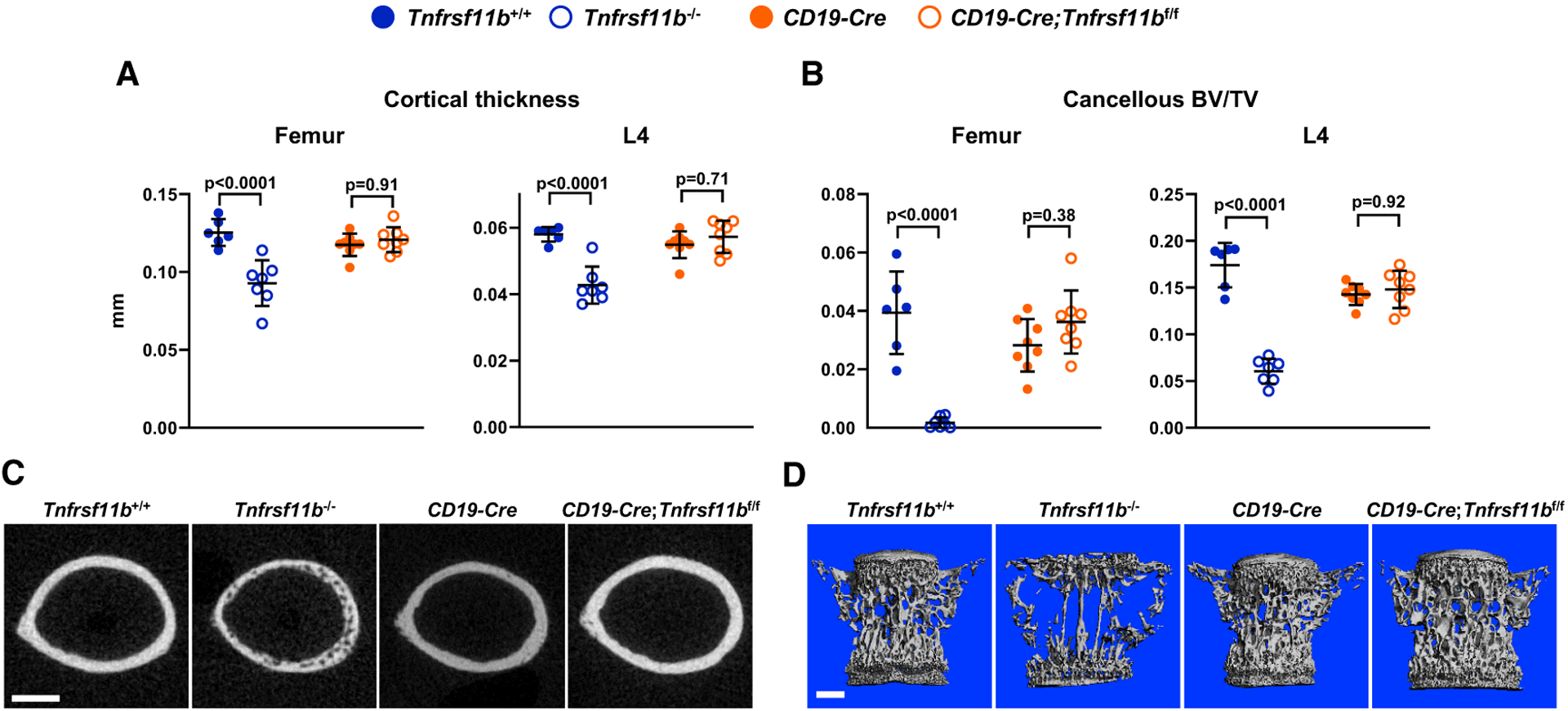
(A) Cortical thickness in the femur and L4 vertebra measured by micro-computed tomography (μCT) (n = 6–8).
(B) Cancellous bone volume over tissue volume (BV/TV) of femurs and L4 vertebra (n = 6–8).
(C) Cross sections of the femoral diaphysis viewed by μCT. Scale bar, 500 μm.
(D) μCT images of vertebral cancellous bone. Scale bar, 500 μm. All values are from 5-week-old female mice of the indicated genotypes, and bars are means ± SD. The indicated p values were determined by one-way ANOVA.
See also Figures S1–S3 and Data S1.
OPG Deletion from Osteoblasts and Osteocytes Reduces Bone Mass
We next deleted the Tnfrsf11bf/f allele using Dmp1-Cre transgenic mice, which delete target genes in osteoblasts and osteocytes (Xiong et al., 2015). Deletion of Tnfrsf11b exon 2 in osteocytes was confirmed using genomic DNA from osteocyte-enriched cortical bone (Figure S3). We also observed significant deletion in muscle tissue (Figure S3), as previously reported for this transgene (Piemontese et al., 2016).
At 5 weeks of age, cortical thickness in the femur and spine of female mice lacking Tnfrsf11b in Dmp1-Cre-targeted cells was similar to that of Tnfrsf11b−/− mice (Figures 2A and 2C). In this and subsequent analyses, we compared Dmp1-Cre;Tnfrsf11bf/f mice to their Tnfrsf11bf/f controls since there were no differences between the wild-type, Dmp1-Cre, and Tnfrsf11bf/f groups (Figure S4). Cancellous bone in the femur was also low, but not as low as in Tnfrsf11b−/− mice, whereas spinal cancellous bone volume was reduced by a similar magnitude in Dmp1-Cre;Tnfrsf11bf/f mice and Tnfrsf11b−/− mice (Figures 2B and 2D). Dmp1-Cre;Tnfrsf11bf/f mice displayed a slight increase in cortical porosity of the femur, but this was less extensive than in Tnfrsf11b−/− mice (Figures 2C and 2E). Similar changes in bone mass and structure were observed in male Dmp1-Cre;Tnfrsf11bf/f mice (Figure S4). Because the skeletal consequences of either germline or Dmp1-Cre-mediated deletion of Tnfrsf11b were similar between sexes, we focused our subsequent analyses on female mice.
Figure 2. Deletion of Tnfrsf11b from Mature Osteoblasts and Osteocytes Decreases Bone Mass.
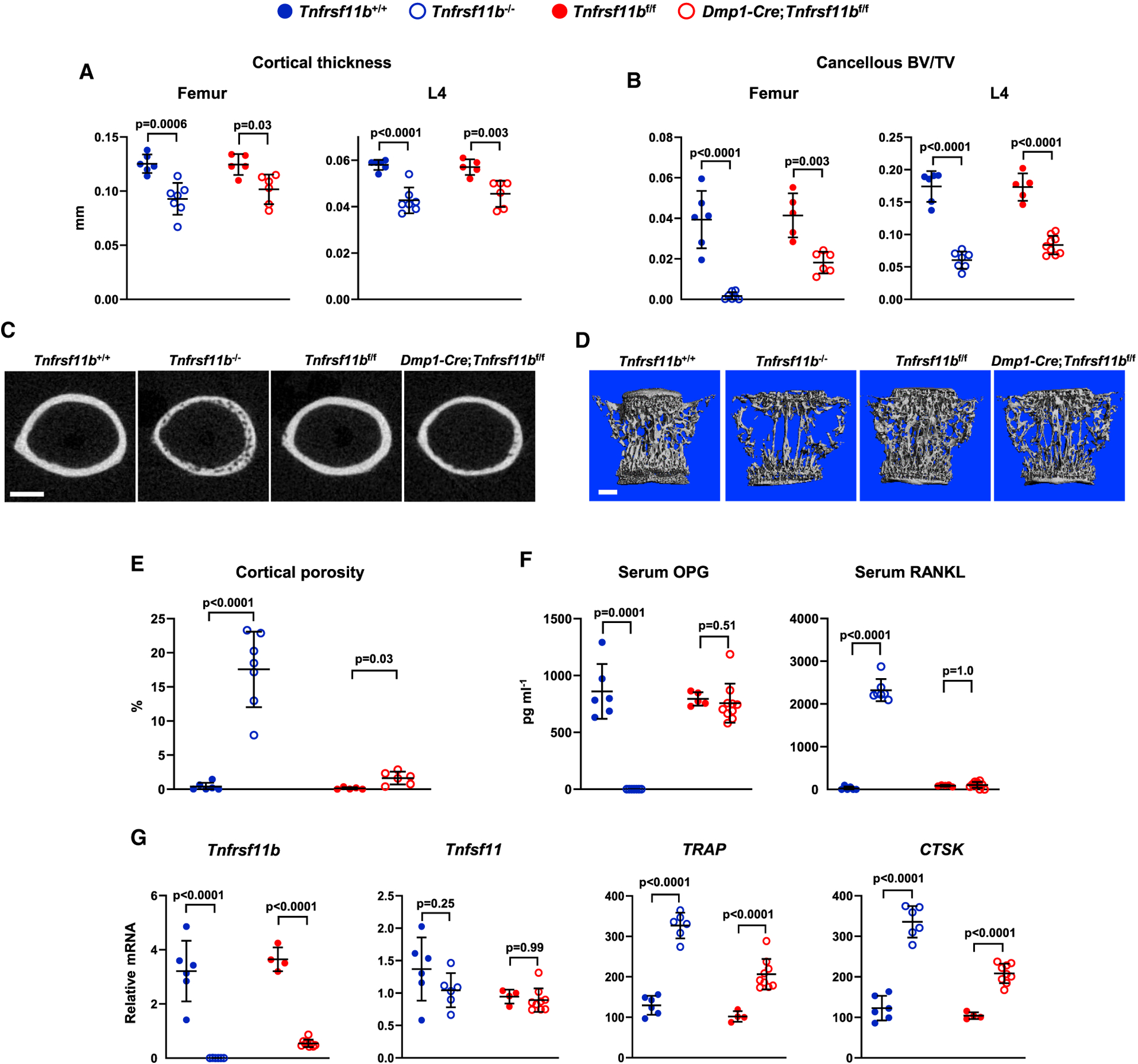
(A) Cortical thickness in the femur and L4 vertebra measured by μCT (n = 5–7).
(B) Cancellous BV/TV of femurs and L4 vertebra (n = 5–9).
(C) Cross sections of the femoral diaphysis viewed by μCT. Scale bar, 500 μm.
(D) μCT images of vertebral cancellous bone. Scale bar, 500 μm.
(E) Quantitative analysis of femoral cortical porosity measured by μCT (n = 5–7).
(F) Soluble OPG and RANKL in the serum measured by ELISA (n = 5–10).
(G) Tnfrsf11b, Tnfsf11, TRAP, and CTSK mRNA levels in tibial cortical bone (n = 4–9). All values are from 5-week-old female mice of the indicated genotypes, and bars are means ± SD. Cortical thickness and BV/TV values for Tnfrsf11b+/+ and Tnfrsf11b−/− mice are the same as in Figure 1. The indicated p values were determined by one-way ANOVA.
See also Figures S1, S3, and S4 and Data S1.
As expected, circulating OPG was undetectable in the serum of Tnfrsf11b−/− mice and, as previously reported (Nakamichi et al., 2007; Nakamura et al., 2003), OPG deletion resulted in a dramatic increase in the levels of detectable RANKL in the circulation (Figure 2F). By contrast, neither of these changes were observed in Dmp1-Cre;Tnfrsf11bf/f mice. Since the Dmp1-Cre;Tnfrsf11bf/f mice displayed levels of bone loss similar to Tnfrsf11b−/− mice, the normal levels of circulating OPG in these animals were unable to suppress bone resorption. In other words, under normal physiological conditions, circulating OPG does not appear to suppress bone resorption.
Analysis of gene expression in osteocyte-enriched cortical bone revealed the expected loss of Tnfrsf11b mRNA in Tnfrsf11b−/− mice as well as a notable loss in Dmp1-Cre;Tnfrsf11bf/f mice, indicating that Dmp1-Cre-targeted cells are the major source of OPG in this tissue (Figure 2G). We did not observe changes in expression of Tnfsf11, which encodes RANKL, in either Tnfrsf11b−/− or Dmp1-Cre;Tnfrsf11bf/f mice. Transcripts of genes highly expressed by osteoclasts were higher in both Tnfrsf11b−/− and Dmp1-Cre;Tnfrsf11bf/f mice, with greater increases in the Tnfrsf11b−/− mice, correlating with the greater degree of cortical porosity in the null mice (Figure 2G). Consistent with the gene expression results, histochemical staining for osteoclasts revealed increased abundance of osteoclasts in the cortical bone of both Tnfrsf11b−/− and Dmp1-Cre;Tnfrsf11bf/f mice (Figure S4).
Osteoblasts Are a Major Source of OPG Protecting Cancellous Bone
Results from the Dmp1-Cre-mediated deletion suggest that osteoblasts, osteocytes, muscle cells, or some combination of these are major sources of the OPG that suppress osteoclast formation. Skeletal muscle cell expression of Tnfrsf11b is ~800-fold lower than in osteoblast-lineage cells (Figure S4), arguing against a role for muscle cells in the suppression of bone resorption. Moreover, the inability of circulating OPG to suppress bone resorption strongly suggests that the cellular source of OPG must be located near the bone surface, also arguing against a role for myocytes. Therefore, we sought to distinguish between osteoblasts and osteocytes as sources of OPG.
To do this, we used Sost-Cre transgenic mice. The Sost-Cre transgene causes deletion in osteocytes, but not osteoblasts or bone lining cells (Xiong et al., 2015). In addition, this transgene causes recombination of conditional alleles in the majority of hematopoietic cells (Xiong et al., 2015). We first confirmed deletion of the Tnfrsf11bf/f allele in osteocyte-enriched cortical bone, which was as effective as it was in Dmp1-Cre;Tnfrsf11bf/f mice (Figure S3). We then examined bone mass at 5 weeks of age and found that Sost-Cre;Tnfrsf11bf/f mice displayed only a small reduction in cortical and cancellous bone compared with mice harboring only the Tnfrsf11bf/f allele (Figures 3A–3D). Cortical porosity was unchanged by deletion of Tnfrsf11bf/f using the Sost-Cre mice (Figures 3C and 3E). Consistent with the small effect on bone mass, deletion of Tnfrsf11bf/f in Sost-Cre mice slightly diminished expression of Tnfrsf11b and did not alter expression of osteoclast-specific genes (Figure 3F). These results, together with the results of the Dmp1-Cre-mediated deletion, support the conclusion that osteoblasts, but not osteocytes, are a major source of the OPG that suppresses bone resorption in growing mice.
Figure 3. Deletion of Tnfrsf11b from Osteocytes Results in a Mild Decrease in Bone Mass.
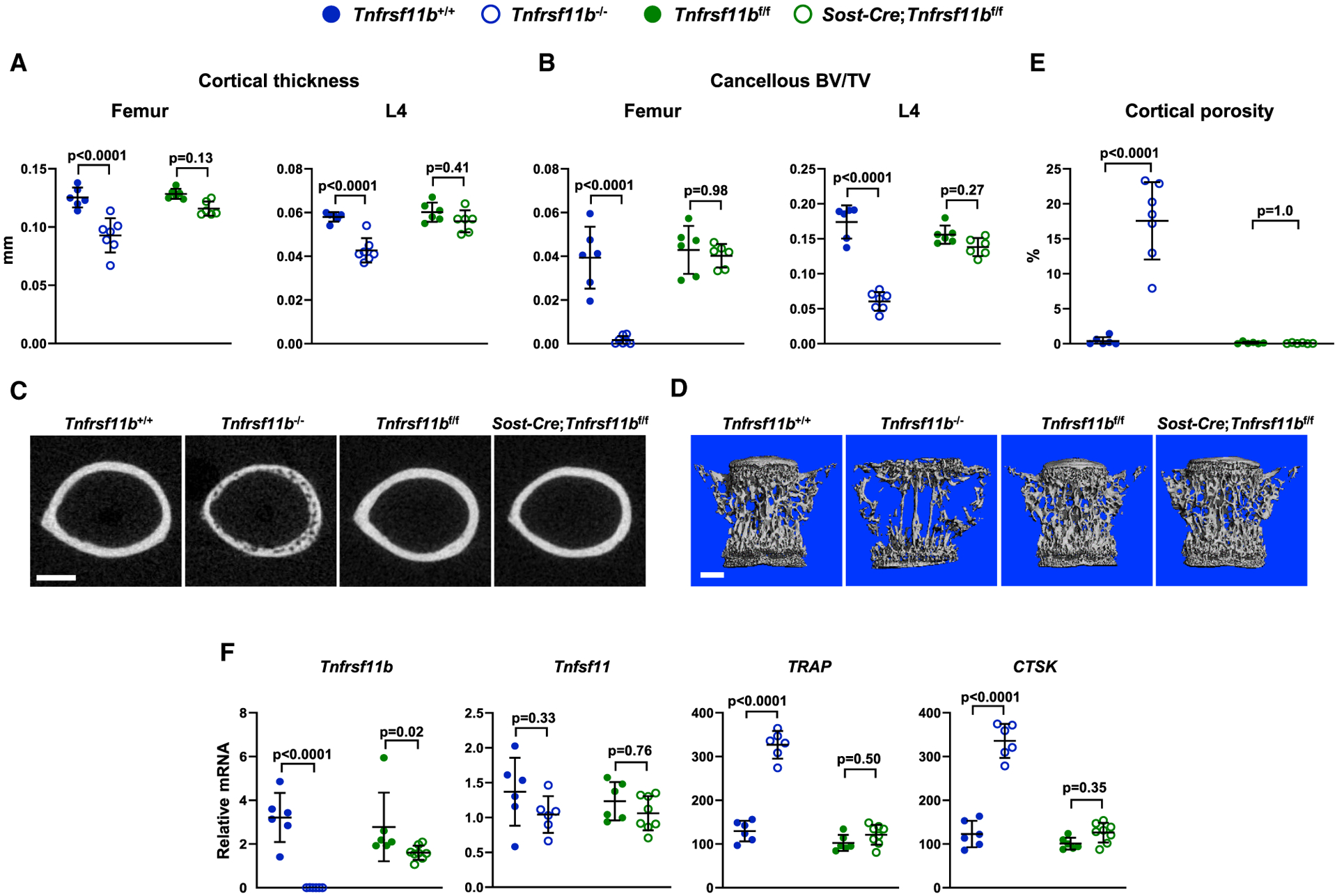
(A) Cortical thickness in the femur and L4 vertebra measured by μCT (n = 6–7).
(B) Cancellous BV/TV of femurs and L4 vertebra (n = 6–7).
(C) Cross sections of the femoral diaphysis viewed by μCT. Scale bar, 500 μm.
(D) μCT images of vertebral cancellous bone. Scale bar, 500 μm.
(E) Quantitative analysis of cortical porosity of femurs measured by μCT (n = 5–7).
(F) Tnfrsf11b, Tnfsf11, TRAP, and CTSK mRNA levels in tibial cortical bone (n = 6–8). All values are from 5-week-old female mice of the indicated genotypes, and bars are means ± SD. Cortical thickness, BV/TV, and cortical porosity values for Tnfrsf11b+/+ and Tnfrsf11b−/− mice are the same as in Figures 1 and 2. The indicated p values were determined by one-way ANOVA.
See also Figures S1 and S3 and Data S1.
To determine whether this is also the case in adult remodeling bone, we examined Tnfrsf11b−/−, Dmp1-Cre;Tnfrsf11bf/f, and Sost-Cre;Tnfrsf11bf/f mice, and their respective controls, at 4 months of age. At this age, Tnfrsf11b−/− mice exhibited reduced cortical thickness and an almost complete absence of cancellous bone in both the femur and spine (Figures 4A–4D). Similar results were seen with Dmp1-Cre-mediated deletion except that the reduction in femoral cortical bone was less severe than in Tnfrsf11b−/− mice. Sost-Cre-mediated deletion reduced femoral cortical thickness to an extent similar to Dmp1-Cre-mediated deletion, but it had a smaller effect on vertebral cortical thickness or cancellous bone volume at either site (Figures 4A–4D). Cortical porosity was evident in the femurs of Tnfrsf11b−/− mice, but to a lesser extent in Dmp1-Cre;Tnfrsf11bf/f mice, and an even lower extent in Sost-Cre;Tnfrsf11bf/f mice (Figures 4C and 4E). In contrast to the Tnfrsf11b−/− mice, porosity in the conditional knockout mice was not uniform but localized to the lateral, posterior, and medial regions of the cortex, which is evident upon visual examination of sections from all the mice in each group (Figure S5). Circulating OPG was slightly lower, and circulating RANKL slightly higher, in both Dmp1-Cre;Tnfrsf11bf/f and Sost-Cre;Tnfrsf11bf/f mice than in their respective controls, but these changes were much smaller than those in Tnfrsf11b−/− mice (Figure 4F).
Figure 4. Osteoblastic Tnfrsf11b Suppresses Resorption in Adult Mice.
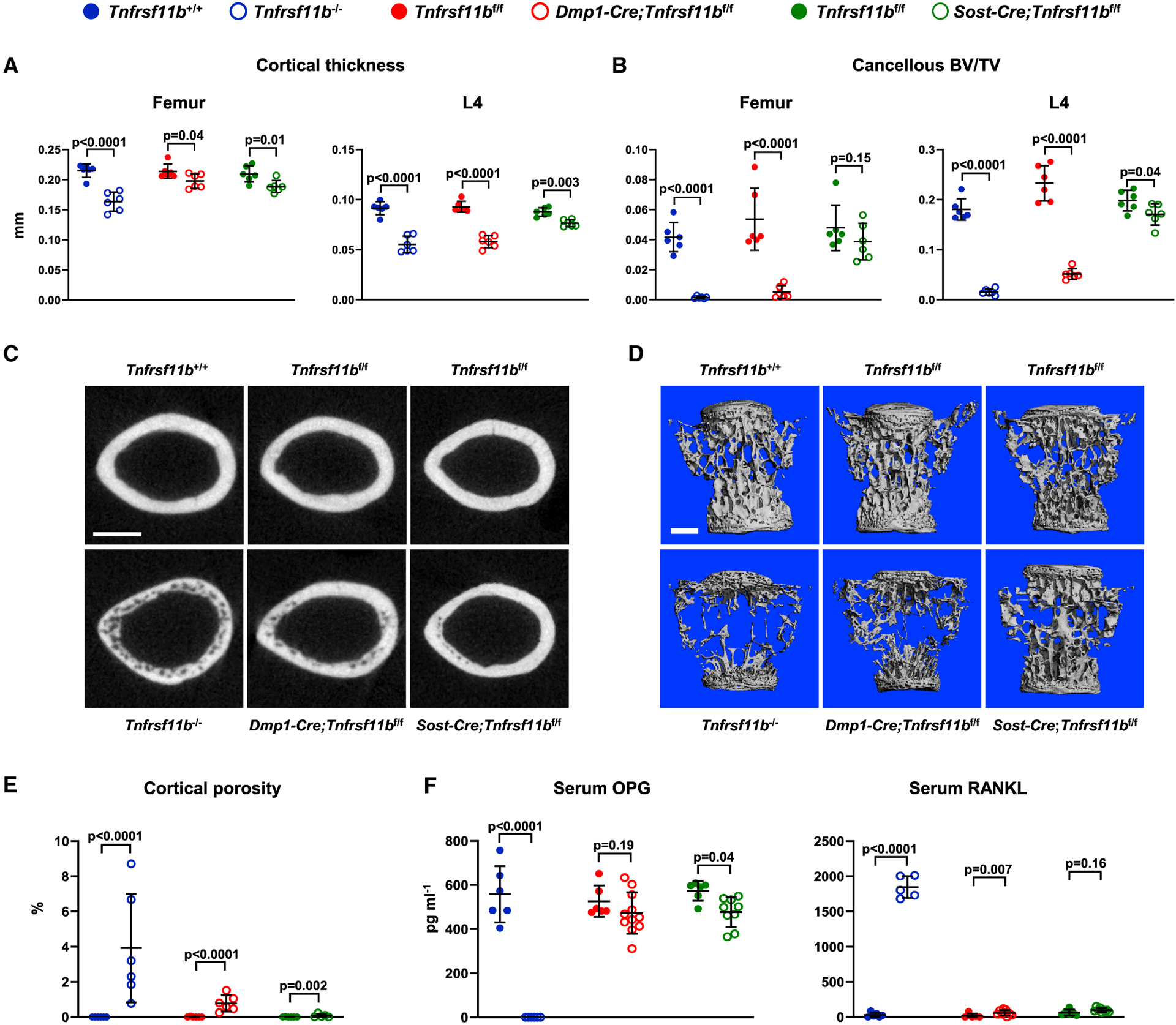
(A) Cortical thickness in the femur and L4 vertebra measured by μCT (n = 6).
(B) Cancellous BV/TV of femurs and L4 vertebra (n = 6).
(C) Cross sections of the femoral diaphysis viewed by μCT. Scale bar, 500 μm.
(D) μCT images of vertebral cancellous bone. Scale bar, 500 μm.
(E) Quantitative analysis of cortical porosity of femurs measured by μCT (n = 5–6).
(F) Soluble OPG and RANKL in the serum measured by ELISA (n = 6–11). All values are from 4-month-old female mice, and bars are means ± SD. The indicated p values were determined by one-way ANOVA.
See also Figures S1, S5, and S6 and Data S1.
Osteoblasts and Osteocytes Are Not the Source of OPG Protecting Auditory Ossicles
Tnfrsf11b−/− mice develop hearing loss due to osteoclastic resorption of auditory ossicles (Kanzaki et al., 2006; Zehnder et al., 2006). Gene expression analysis has suggested that the major source of OPG protecting these bones is the soft tissue surrounding the otic capsule rather than bone tissue (Zehnder et al., 2005). To determine whether osteoblasts or osteocytes contribute to the production of the OPG protecting auditory ossicles, we compared the structure of these bones in Tnfrsf11b−/− and Dmp1-Cre;Tnfrsf11bf/f, as well as their respective controls. While the ossicles were almost completely resorbed in 4-month-old Tnfrsf11b−/− mice, they appeared similar to controls in Dmp1-Cre;Tnfrsf11bf/f mice (Figure S6). Dmp1-Cre led to activation of a tdTomato Cre-reporter gene in osteocytes within the malleus, confirming activity of this transgene in osteoblastic cells in auditory ossicles (Figure S6). These results demonstrate that osteoblasts and osteocytes are not the source of OPG protecting auditory ossicles from resorption, consistent with earlier gene expression studies (Zehnder et al., 2005).
DISCUSSION
The main conclusion of our study is that mature osteoblasts are an essential source of the OPG that suppresses resorption of cancellous bone in both growing and adult mice. The cellular sources of the OPG that suppress resorption in cortical bone appear to be more complex, with contributions from osteoblasts, osteocytes, and other undetermined cell types, depending on the region of the cortical bone and the age of the mice. The latter finding is not unexpected since the needs for physiological bone resorption change with age and environmental conditions. For example, expansion of the medullary cavity via endocortical resorption is relatively slow in young adult mice but increases beginning at ~1 year of age (Ferguson et al., 2003).
If osteoblasts are a major source of OPG, it might be anticipated that low osteoblast number would be associated with reduced OPG levels in bone. We observed such a reduction in RANKL conditional knockout mice, which exhibit low numbers of both osteoclasts and osteoblasts (Fujiwara et al., 2016). One might also predict that changes in osteoblast number would be associated with subsequent changes in osteoclast number. While this clearly occurs in some situations, such as hyperparathyroidism or administration of anti-sclerostin antibody (O’Brien et al., 2005; Taylor et al., 2016), it does not occur in others, such as in mice with germline deletion of Sost or after conditional ablation of osteoblasts (Corral et al., 1998; Li et al., 2008). However, in each of these situations, the interactions between osteoblasts, osteocytes, and osteoclasts are complex, such that it is difficult to attribute changes in osteoclast abundance solely to changes in osteoblast abundance. Even in what might be considered the most straightforward situation, namely, conditional ablation of osteoblasts, loss of factors produced by osteoblast-lineage cells reduces the abundance of the hematopoietic progenitors of osteoclasts (Visnjic et al., 2001). Thus, whether or not changes in osteoblast number, and thus OPG levels, will lead to subsequent changes in osteoclast number likely depends on what factors or conditions are responsible for the change in osteoblast number.
We think that our findings are more relevant to understanding the sequence of events that occur locally within the basic multicellular unit (BMU) during the bone remodeling cycle. Specifically, production of OPG by mature osteoblasts may constitute a molecular signal that terminates the resorption phase of the remodeling cycle at a given site on the bone surface. In such a model, the arrival of osteoblasts at sites of bone resorption is driven by coupling factors produced by osteoclasts, such as leukemia inhibitory factor (LIF) (Koide et al., 2017). LIF production by osteoclasts may suppress Sost expression in nearby osteocytes, thereby allowing recruitment of osteoblast progenitors. Upon arrival and differentiation into mature osteoblasts, production of OPG by these cells may then inhibit further osteoclast production and activity at that site by blocking RANKL produced by osteocytes. In addition, continued production of OPG by osteoblasts would ensure that newly formed bone is protected from untimely resorption. Moreover, this model predicts that cancellous bone loss in the absence of OPG is due to prolongation of the resorption phase of the bone remodeling cycle rather than initiation of new BMUs. These findings suggest that a single cell type is not responsible for initiating and terminating bone resorption but that multiple cell types work together to perform a process that is self-limiting, at least under normal physiological conditions. Such a model is consistent with recent observations in human cortical bone that revealed the presence of osteoblasts and osteoclasts in close proximity within the reversal/resorption region of BMUs (Lassen et al., 2017).
Numerous in vitro and in vivo studies demonstrate that a given cell type can produce both RANKL and OPG (Kanzaki et al., 2001; O’Brien et al., 2002; Udagawa et al., 2000; Zhao et al., 2002). While reciprocal regulation of RANKL and OPG in a single cell type may be important for the control of resorption at certain sites, evidence from the current study, combined with RANKL conditional knockout studies, suggests that the cell type producing OPG in remodeling cancellous bone is distinct from the cell type that produces RANKL. This is highlighted by the finding that Dmp1-Cre-mediated deletion of Tnfsf11 did not result in skeletal changes at 5 weeks of age (Xiong et al., 2011), whereas Dmp1-Cre-mediated deletion of Tnfrsf11b clearly did (this study). The latter finding also suggests that in growing mice OPG produced by osteoblasts inhibits the action of RANKL produced by cells other than osteocytes, such as hypertrophic chondrocytes (Xiong et al., 2011).
Circulating OPG levels do not consistently correlate with bone mass or the rate of bone remodeling in human studies (Khosla et al., 2002; Rogers and Eastell, 2005; Rogers et al., 2002; Sarink et al., 2019). Similarly, we found that normal levels of circulating OPG could not suppress bone resorption in mice with deletion of OPG in osteoblasts and osteocytes. Consistent with these observations, two independent groups have demonstrated that OPG interaction with heparan sulfate proteoglycans on the cell surface is required for optimal inhibition of RANKL (Li et al., 2016; Nozawa et al., 2018). These studies support the idea that the local concentration of OPG near the cell surface is a key factor in its ability to block the activity of RANKL. Nonetheless, it is still possible that abnormally high levels of OPG in the circulation may lead to functional changes in bone resorption. This is evidenced by the suppression of bone resorption in transgenic mice that express high levels of OPG in the liver (Simonet et al., 1997). However, it is unclear whether any human conditions exist that result in circulating levels of OPG that are sufficient to suppress resorption.
Our results do not support the idea that OPG produced by B lymphocytes contributes to the suppression of bone resorption. Li et al. (2007) clearly demonstrated that B cells produce OPG and that μMT mice, which lack mature B cells, exhibit increased bone resorption, leading these authors to conclude that OPG produced by B cells suppresses bone resorption. One possible explanation for the different conclusions of the two studies is that cells earlier in the B cell lineage than those targeted by CD19-Cre produce OPG involved in suppressing osteoclastogenesis. However, evidence suggests that the CD19-Cre strain deletes genes at the pre-B cell stage, which is the stage at which B cell differentiation is halted in μMT mice (Kitamura et al., 1991; Rickert et al., 1997). Perhaps more importantly, the study by Li et al. (2007) demonstrated that OPG production by B cell precursors and immature B cells is minimal and that the majority of the OPG production within the B lymphocyte lineage is by mature B cells, which are efficiently targeted by CD19-Cre. Thus, targeting of different cell populations does not appear to explain the differences between the studies and the increase in bone resorption observed in μMT mice is likely due to factors other than loss of the OPG produced by B cells.
Femoral cortical thickness was considerably lower, and cortical porosity higher, in adult mice with germline deletion of Tnfrsf11b compared with mice with either Dmp1-Cre- or Sost-Cre-mediated deletion. This suggests that cells other than osteoblasts or osteocytes produce some of the OPG that suppresses resorption of femoral cortical bone. Multiple studies have documented expression of Tnfrsf11b by vascular endothelial cells (Collin-Osdoby et al., 2001; Malyankar et al., 2000). Moreover, a recent report has documented that murine femoral cortical bone contains numerous blood vessels that span the region between the endosteum and periosteum (Grüneboom et al., 2019). Future studies using a Cre-driver strain specific for vascular endothelial cells will be required to determine whether this cell type produces OPG involved in the protection of cortical bone.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles A. O’Brien (caobrien@uams.edu).
Materials Availability
The mouse line generated in this study (Tnfrsf11b-flox) is currently available from the Lead Contact. We have applied to deposit this line in the Mutant Mouse Resource and Research Center, MMRRC number 67367.
Data and Code Availability
This study did not generate or analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of Tnfrsf11b-flox mice
We generated a Tnfrsf11b conditional allele using gene-editing to insert loxP sites upstream and downstream of exon 2 (Figure S1). Plasmids encoding Cas9 and sgRNAs (pX330) and single-stranded DNA homology donors harboring the loxP sites were injected into the pronuclei of fertilized C57BL/6J mouse eggs at a final concentration of 5 ng/ml and 10 ng/ml, respectively. Microinjected eggs were then implanted into the oviducts of pseudopregnant ICR mice. F0 offspring were screened for the desired insertions using the following primers: OPG-L567-for 5′-TTGTTCACCTGTTGCCATCT-3′ and OPG-L567-rev 5′-CCATTCAATGATGTCCAGGAG-3′, product size 222 bp for the wild-type allele and 262 bp for the loxP allele; OPG-R2-geno-for2 5′-TCCGGAGTACTAACAATGAA GAGTT-3′ and OPG-R2-geno-rev2 5′-GTGGGGAATGTGAAAAGGAA-3′, product size 80 bp for the wild-type allele and 120 bp for the loxP allele. Offspring harboring the pX330 plasmid were identified by PCR and not used for further breeding. Breeding of the founder mouse and all subsequent breeding to maintain the line was performed with wild-type C57BL/6J mice.
Cell type-specific deletion of the Tnfrsf11b-flox allele
The Cre transgenic mice used in this study have been described previously: EIIa-Cre (Lakso et al., 1996), CD19-Cre (Rickert et al., 1997), Dmp1-Cre (Xiong et al., 2015), Sost-Cre (Xiong et al., 2015), and tdTomato Cre-reporter mice (Madisen et al., 2010). The experimental animals used in most of the studies were obtained by a two-step breeding strategy. Hemizygous Cre transgenic mice were crossed with Tnfrsf11bf/+ mice to generate Tnfrsf11bf/+ offspring with and without a Cre allele. These offspring were then inter-crossed to generate the following offspring: wild-type mice, mice hemizygous for a Cre allele, mice homozygous for the conditional allele, and mice homozygous for the conditional allele that were also hemizygous for a Cre allele. Offspring were genotyped for the Cre allele by PCR using the following primer sequences: Cre-for, 5′-GCGGTCTGGCAGTAAAAACTATC-3′, Cre-rev, 5′-GTGAAACAGCATTGCTGTCACTT-3′, product size 102 bp. The Dmp1-Cre and Sost-Cre driver strains exhibit partially penetrant germline deletion. Therefore, all offspring were screened for the presence of the deleted allele using the following primers: OPG-L567-for, 5′-TTGTTCACCTGTTGCCATCT-3′ and OPG-del-geno-rev, 5′-GGAATACGGGTTTCAGCTTTC-3′, product size 325 bp. All Cre-negative mice harboring a null Tnfrsf11b allele were excluded from the study as were any Cre-positive mice in which the null allele was detected in genomic DNA prepared from the liver, which is not targeted by either Dmp1-Cre or Sost-Cre. For experiments involving 5-week-old mice, both female and male mice were analyzed and the results from each sex were presented separately. For experiments involving 4-month-old mice, only female mice were analyzed and presented. A single experiment used 6-month-old male wild-type C57BL/6 mice (presented in Figure S4).
Animal housing and care
Mice were socially housed at 2–5 animals per cage using a blend of 1/4″ corncob bedding and white enrichment paper both produced by Andersons INC. Maumee, Ohio. The animal census was specific pathogen free based on the Division of Laboratory Animal Medicine’s exclusion list. Vendor health reports were monitored consistently to protect the resident animals. The mice were housed in an IVC housing system by Allentown called Nexgen Mouse 500. The mice cages were changed with the usage of an Allentown Phantom transfer station. The mice were provided ad libitum water and an irradiated Purina diet of either 5V5M (for breeders) or 5V5R (for maintenance). The temperature range in the room was 68–79 F with a set point of 71 ± 2, additionally the humidity range was 30%–70%. The room was on a 12:12 hour light cycle and the illumination was 364 lux measured 1 m from the floor. The UAMS Animal Care and Use Program has been consistently and fully accredited by AAALAC since 1973. Their OLAW Assurance is approved through 2025 and they are registered with USDA. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences.
METHOD DETAILS
Quantification of gene deletion
To quantify Tnfrsf11b gene deletion efficiency, the following custom TaqMan assay for exon 2 was used: forward, 5′-CCTTGCCCTGACCACTCTTAT-3′, reverse, 5′-GCTGCAATACACACACTCATCACT-3′, probe, 5′-FAM-ACGGACAGCTGGCACAC-NFQ-3′. The custom Tnfrsf11b assay was used in combination with a TaqMan copy number reference assay, Tfrc (catalog number 4458367). All TaqMan assays used in this study were obtained from Thermo-Fisher Scientific.
Flow cytometry
Bone marrow cells were collected by removing both ends of the femur and flushing out the cells with PBS containing 1% FBS and 1mM EDTA. Bone marrow cells were washed and stained with 2 μg/ml anti-CD19-APC (BD Biosciences) to identify B cells. After the cells were washed to remove unbound antibodies, the samples were sorted using a BD FACS Aria flow cytometer (BD Biosciences). The data were then analyzed using FlowJo Software (FlowJo, LLC, Ashland, OR). The guidance of Fluorescence Minus One (FMO) controls (BD Biosciences) was followed to draw appropriate gates for the cell populations.
RNA and genomic DNA isolation from osteocyte-enriched bone
Total RNA was prepared from osteocyte-enriched tibia shafts by homogenization in 1.5 mL of Trizol Reagent (Life Technologies), followed by centrifugation and isopropanol precipitation according to manufacturer’s instructions. Quantitation and determination of the 260/280 ratio of the extracted RNA were determined using a Nanodrop instrument (Thermo Scientific). 500ng of RNA was then used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies) according to manufacturer’s directions. For genomic DNA isolation, bone pieces were decalcified in 14% EDTA for 1 week. Decalcified osteocyte-enriched bone or soft tissues were digested with proteinase K (0.5 mg/ml in 10 mM Tris, pH 8.0, 100 mM NaCl, 20 mM EDTA, and 1% SDS) at 55 C overnight. Genomic DNA was then isolated by phenol/chloroform extraction and ethanol precipitation.
Quantification of sRANKL and OPG
Blood was collected by retro-orbital bleeding into microcentrifuge tubes, allowed to clot, and then centrifuged at 1500 × g for 10 minutes to separate serum from cells. Soluble RANKL and OPG in blood serum were measured using mouse Quantikine kits (R&D Systems) according to the manual provided by the manufacturer.
MicroCT
MicroCT scanning was used to measure cortical and trabecular architecture of the femur and fourth lumbar vertebra. Bones were dissected, cleaned of soft tissues, fixed in 10% Millonig’s formalin overnight, and transferred gradually from 70% to 100% ethanol. Dehydrated bones were then loaded into a 12.3-mm-diameter scanning tube and scanned by a μCT (model μCT40, Scanco Biomedical, Bruttisellen, Switzerland) to generate three-dimensional voxel images (1024 × 1024 pixels) of bone samples. A Gaussian filter (sigma = 0.8, support = 1) was used to reduce signal noise and a threshold of 200 was applied to all scans, at medium resolution (E = 55 kVp, I = 145 μA, integration time = 200 ms). For vertebra, the whole vertebral body was scanned and cortical bone and the primary spongiosa were excluded from the cancellous bone volume analyses manually. 100 slices above the anterior growth plate were analyzed for vertebral cortical thickness. For femurs, 151 slices above the growth plate of the distal femur were scanned for trabecular measurement and 18 slices centered on the mid-diaphysis were analyzed for cortical thickness. All trabecular measurements were made by drawing contours every 10–20 slices and using voxel counting for bone volume per tissue volume and sphere-filling distance transformation indices, without presumptions about the bone shape as a rod or plate for trabecular microarchitecture. Calibration and quality control were performed weekly using five density standards, and spatial resolution was verified monthly using a tungsten wire rod. Beam hardening correction was based on the calibration records.
Analysis of gene expression
cDNA was amplified by real-time PCR using TaqMan Universal PCR Master Mix (Life Technologies) as described previously (Xiong et al., 2011). The following TaqMan primer-probe sets were used: osteoprotegerin (Mm00435452_m1); RANKL (Mm00441908_m1); tartrate resistant acid phosphatase (Mm00475698_m1); Cathepsin K (m00484036_m1); and the house-keeping gene ribosomal protein S2 (forward, 5′- CCCAGGATGGCGACGAT-3′, reverse 5′- CCGAATGCTGTAATGGCGTAT-3′, probe 5′-FAM-TCCAGAGCAGGATCC-NFQ-3′ (O’Brien et al., 2004). Relative mRNA levels were calculated using the ΔCt method (Livak and Schmittgen, 2001).
Histology
Femurs were dissected, cleaned of soft tissues, fixed in 10% Millonig’s formalin overnight, and transferred gradually from 70% to 100% ethanol. Dehydrated bones were then plastic-embedded and 5-μm longitudinal sections were cut. After removal of plastic and rehydration, the sections were stained for tartrate-resistant acid phosphatase (TRAP) activity and counterstained with methyl green. Fluorescent imaging of tdTomato-positive osteocytes in the malleus was performed by fixing the malleus for 24 hr in 4% paraformaldehyde followed by whole mount and imaging using a Zeiss LSM 880 confocal microscope with Airyscan (Zeiss, Oberkochen, Germany).
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way ANOVA or Student’s t test were used to detect statistically significant treatment effects, after determining that the data were normally distributed and exhibited equivalent variances. In some cases, Wilcoxon rank-sum tests were used in place of Student’s t test and log, square root, or rank transformations were used to obtain normally distributed data in one-way ANOVA models. Pairwise comparisons and contrasts of genotypes were estimated in the ANOVA models. All t tests were two-sided. Tukey or Benjamini-Hochberg corrections were used for multiple comparison adjustments for each family of tests of pairwise comparisons and contrasts. Statistical analyses were performed using SAS version 9.4. Detailed results from all statistical analyses are contained in Data S1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat Anti-CD19 Monoclonal Anti-Mouse APC-Cy7 (Clone 1D3) | BD Biosciences | Cat#: 557655; RRID: AB_396770 |
| Critical Commercial Assays | ||
| Mouse TRANCE/RANK L/TNFSF11 Quantikine ELISA Kit | R&D Systems | Cat#: MTR00 |
| Mouse Osteoprotegerin/TNFRSF11B Quantikine ELISA Kit | R&D Systems | Cat#: MOP00 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6: Tnfrsf11bf/f | This paper | N/A |
| Mouse: C57BL/6: B6.FVB-Tg(EIIa-Cre)C5379Lmgd/J | The Jackson Laboratory | JAX: 003724 |
| Mouse: C57BL/6: Dmp1-Cre | Xiong et al., 2015 | N/A |
| Mouse: Mixed background: Sost-Cre | Xiong et al., 2015 | N/A |
| Mouse: C57BL/6: B6.129P2(C)-Cd19tm1(cre)cgn/J | The Jackson Laboratory | JAX: 006785 |
| Mouse: C57BL/6: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007909 |
| Oligonucleotides | ||
| See Table S1 for Primers | ||
| Recombinant DNA | ||
| Plasmid: pX330 | Addgene | Cat#: 42230 |
| Software and Algorithms | ||
| FlowJo | Becton, Dickinson and Company | https://www.flowjo.com/ |
Highlights.
Deletion of Tnfrsf11b from B cells or osteocytes has minimal effects on bone mass
Deletion of Tnfrsf11b from osteoblasts increases resorption and reduces bone mass
Circulating osteoprotegerin levels do not correlate with changes in bone resorption
Osteoblasts directly suppress osteoclast production by producing osteoprotegerin
ACKNOWLEDGMENTS
We thank P.E. Baltz and the staff of the UAMS Department of Laboratory Animal Medicine for technical support; M. Almeida, S.C. Manolagas, and R.L. Jilka for critique of the article; and M. Randolph for details regarding animal care. We also thank an anonymous reviewer for suggesting changes to the article’s title. This work was supported by the National Institutes of Health (R01AR049794 and P20GM125503), the Veterans Administration (I01 BX000294), and UAMS tobacco settlement funds.
Footnotes
DECLARATION OF INTERESTS
C.A.O. owns stock in Radius Health. The remaining authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108052.
REFERENCES
- Asagiri M, and Takayanagi H (2007). The molecular understanding of osteoclast differentiation. Bone 40, 251–264. [DOI] [PubMed] [Google Scholar]
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. (1998). osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 12, 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, and Osdoby P (2001). Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflamma-tory cytokines, and role in human osteoclastogenesis. J. Biol. Chem 276, 20659–20672. [DOI] [PubMed] [Google Scholar]
- Corral DA, Amling M, Priemel M, Loyer E, Fuchs S, Ducy P, Baron R, and Karsenty G (1998). Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc. Natl. Acad. Sci. USA 95, 13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson VL, Ayers RA, Bateman TA, and Simske SJ (2003). Bone development and age-related bone loss in male C57BL/6J mice. Bone 33, 387–398. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, and O’Brien CA (2016). RANKL (Receptor Activator of NFκB Ligand) Produced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J. Biol. Chem 291, 24838–24850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasemann C, Unger N, Hövel M, Arweiler-Harbeck D, Herrmann R, Schündeln MM, Müller O, Schweiger B, Lausch E, Meissner T, et al. (2017). Loss of Functional Osteoprotegerin: More Than a Skeletal Problem. J. Clin. Endocrinol. Metab 102, 210–219. [DOI] [PubMed] [Google Scholar]
- Grüneboom A, Hawwari I, Weidner D, Culemann S, Müller S, Henneberg S, Brenzel A, Merz S, Bornemann L, Zec K, et al. (2019). A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat. Metab 1, 236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki H, Chiba M, Shimizu Y, and Mitani H (2001). Dual regulation of osteoclast differentiation by periodontal ligament cells through RANKL stimulation and OPG inhibition. J. Dent. Res 80, 887–891. [DOI] [PubMed] [Google Scholar]
- Kanzaki S, Ito M, Takada Y, Ogawa K, and Matsuo K (2006). Resorption of auditory ossicles and hearing loss in mice lacking osteoprotegerin. Bone 39, 414–419. [DOI] [PubMed] [Google Scholar]
- Khosla S, Arrighi HM, Melton LJ 3rd, Atkinson EJ, O’Fallon WM, Dunstan C, and Riggs BL (2002). Correlates of osteoprotegerin levels in women and men. Osteoporos. Int 13, 394–399. [DOI] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kühn R, and Rajewsky K (1991). A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350, 423–426. [DOI] [PubMed] [Google Scholar]
- Koide M, Kobayashi Y, Yamashita T, Uehara S, Nakamura M, Hiraoka BY, Ozaki Y, Iimura T, Yasuda H, Takahashi N, and Udagawa N (2017). Bone Formation Is Coupled to Resorption Via Suppression of Sclerostin Expression by Osteoclasts. J. Bone Miner. Res 32, 2074–2086. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. (1999). OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323. [DOI] [PubMed] [Google Scholar]
- Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, and Kneissel M (2010). Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol 30, 3071–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, and Westphal H (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen NE, Andersen TL, Pløen GG, Søe K, Hauge EM, Harving S, Eschen GET, and Delaisse JM (2017). Coupling of Bone Resorption and Formation in Real Time: New Knowledge Gained From Human Haversian BMUs. J. Bone Miner. Res 32, 1395–1405. [DOI] [PubMed] [Google Scholar]
- Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, and Weitzmann MN (2007). B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109, 3839–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, et al. (2008). Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. [DOI] [PubMed]
- Bone Miner J. Res. 23, 860–869. [DOI] [PubMed] [Google Scholar]
- Li M, Yang S, and Xu D (2016). Heparan Sulfate Regulates the Structure and Function of Osteoprotegerin in Osteoclastogenesis. J. Biol. Chem 291, 24160–24171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyankar UM, Scatena M, Suchland KL, Yun TJ, Clark EA, and Giachelli CM (2000). Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem 275, 20959–20962. [DOI] [PubMed] [Google Scholar]
- Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, et al. (1998). Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem. Biophys. Res. Commun 247, 610–615. [DOI] [PubMed] [Google Scholar]
- Nagashima K, Sawa S, Nitta T, Tsutsumi M, Okamura T, Penninger JM, Nakashima T, and Takayanagi H (2017). Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat. Immunol 18, 675–682. [DOI] [PubMed] [Google Scholar]
- Nakamichi Y, Udagawa N, Kobayashi Y, Nakamura M, Yamamoto Y, Yamashita T, Mizoguchi T, Sato M, Mogi M, Penninger JM, and Takahashi N (2007). Osteoprotegerin reduces the serum level of receptor activator of NF-kappaB ligand derived from osteoblasts. J. Immunol 178, 192–200. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Udagawa N, Matsuura S, Mogi M, Nakamura H, Horiuchi H, Saito N, Hiraoka BY, Kobayashi Y, Takaoka K, et al. (2003). Osteoprotegerin regulates bone formation through a coupling mechanism with bone resorption. Endocrinology 144, 5441–5449. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, et al. (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med 17, 1231–1234. [DOI] [PubMed] [Google Scholar]
- Nozawa S, Inubushi T, Irie F, Takigami I, Matsumoto K, Shimizu K, Akiyama H, and Yamaguchi Y (2018). Osteoblastic heparan sulfate regulates osteoprotegerin function and bone mass. JCI Insight 3, e89624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Kern B, Gubrij I, Karsenty G, and Manolagas SC (2002). Cbfa1 does not regulate RANKL gene activity in stromal/osteoblastic cells. Bone 30, 453–462. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, and Weinstein RS (2004). Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145, 1835–1841. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Jilka RL, Fu Q, Stewart S, Weinstein RS, and Manolagas SC (2005). IL-6 is not required for parathyroid hormone stimulation of RANKL expression, osteoclast formation, and bone loss in mice. Am. J. Physiol. Endocrinol. Metab 289, E784–E793. [DOI] [PubMed] [Google Scholar]
- Piemontese M, Xiong J, Fujiwara Y, Thostenson JD, and O’Brien CA (2016). Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and is associated with reduced OPG expression in mice. Am. J. Physiol. Endocrinol. Metab 311, E587–E593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC, Roes J, and Rajewsky K (1997). B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25, 1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A, and Eastell R (2005). Circulating osteoprotegerin and receptor activator for nuclear factor kappaB ligand: clinical utility in metabolic bone disease assessment. J. Clin. Endocrinol. Metab 90, 6323–6331. [DOI] [PubMed] [Google Scholar]
- Rogers A, Saleh G, Hannon RA, Greenfield D, and Eastell R (2002). Circulating estradiol and osteoprotegerin as determinants of bone turnover and bone density in postmenopausal women. J. Clin. Endocrinol. Metab 87, 4470–4475. [DOI] [PubMed] [Google Scholar]
- Sarink D, Yang J, Johnson T, Chang-Claude J, Overvad K, Olsen A, Tjønneland A, Fournier A, Mancini FR, Kvaskoff M, et al. (2019). Reproductive and Lifestyle Factors and Circulating sRANKL and OPG Concentrations in Women: Results from the EPIC Cohort. Cancer Epidemiol. Biomarkers Prev 28, 1746–1754. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. (1997). Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319. [DOI] [PubMed] [Google Scholar]
- Taylor S, Ominsky MS, Hu R, Pacheco E, He YD, Brown DL, Aguirre JI, Wronski TJ, Buntich S, Afshari CA, et al. (2016). Time-dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone 84, 148–159. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL (2000). Bone resorption by osteoclasts. Science 289, 1504–1508. [DOI] [PubMed] [Google Scholar]
- Udagawa N, Takahashi N, Yasuda H, Mizuno A, Itoh K, Ueno Y, Shinki T, Gillespie MT, Martin TJ, Higashio K, and Suda T (2000). Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 141, 3478–3484. [DOI] [PubMed] [Google Scholar]
- Visnjic D, Kalajzic I, Gronowicz G, Aguila HL, Clark SH, Lichtler AC, and Rowe DW (2001). Conditional ablation of the osteoblast lineage in Col2.3deltatk transgenic mice. J. Bone Miner. Res 16, 2222–2231. [DOI] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, and O’Brien CA (2011). Matrix-embedded cells control osteoclast formation. Nat. Med 17, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, Bonewald L, Manolagas SC, and O’Brien CA (2015). Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 10, e0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Cawley K, Piemontese M, Fujiwara Y, Zhao H, Goellner JJ, and O’Brien CA (2018). Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun 9, 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehnder AF, Kristiansen AG, Adams JC, Merchant SN, and McKenna MJ (2005). Osteoprotegerin in the inner ear may inhibit bone remodeling in the otic capsule. Laryngoscope 115, 172–177. [DOI] [PubMed] [Google Scholar]
- Zehnder AF, Kristiansen AG, Adams JC, Kujawa SG, Merchant SN, and McKenna MJ (2006). Osteoprotegrin knockout mice demonstrate abnormal remodeling of the otic capsule and progressive hearing loss. Laryngoscope 116, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Zhang YK, Harris S, Ahuja SS, Bonewald LF, and Bonewald LF (2002). MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res 17, 2068–2079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.