

Abstract
Nitrogen fixation, the six-electron/six-proton reduction of N2, to give NH3, is one of the most challenging and important chemical transformations. Notwithstanding the barriers associated with this reaction, significant progress has been made in developing molecular complexes that reduce N2 into its bioavailable form, NH3. This progress is driven by the dual aims of better understanding biological nitrogenases and improving upon industrial nitrogen fixation. In this review, we highlight both mechanistic understanding of nitrogen fixation that has been developed, as well as advances in yields, efficiencies, and rates that make molecular alternatives to nitrogen fixation increasingly appealing. We begin with a historical discussion of N2 functionalization chemistry that traverses a timeline of events leading up to the discovery of the first bona fide molecular catalyst system and follow with a comprehensive overview of d-block compounds that have been targeted as catalysts up to and including 2019. We end with a summary of lessons learned from this significant research effort and last offer a discussion of key remaining challenges in the field.
Graphical Abstract
1. INTRODUCTION TO THE FIELD OF N2 REDUCTION CATALYSIS
Catalytic nitrogen fixation is an essential chemical transformation in both biology and industry as it represents the primary means by which nitrogen (N2) from the air becomes bioavailable. This review focuses on the development and study of synthetic molecular catalysts that mediate the catalytic conversion of nitrogen to ammonia (N2-to-NH3, often abbreviated as the nitrogen reduction reaction or N2RR) in the presence of acid and reductant under moderate temperatures and pressures.
1.1. Motivation for New Ammonia Synthesis Catalyst Technologies
Conventional ammonia synthesis (i.e., Haber-Bosch) is among the most significant technological advances of the 20th century and has been critical to sustained global population growth.1 However, with operating pressures of 150–250 bar and temperatures of 400–500 °C, it has high cost demands for infrastructure leading to centralization of the manufacturing process and thus requires a global distribution system.2 This large-scale distribution and the necessary temperature and pressure to form NH3 from N2 and H2 over a solid-state Fe catalyst necessitates significant fossil fuel input with related high carbon dioxide (CO2) emissions. While estimates vary, approximately 1–2% of annual global energy consumption is accounted for by conventional ammonia synthesis, with some 4% of global methane (CH4) and 60% of global hydrogen going into its production. The generation of the needed hydrogen via steam-reforming (providing for ~72% global ammonia production) accounts for nearly 0.5 gigatons of CO2 released annually.3 In addition, other environmental consequences from fertilizer use are severe, including surface and groundwater pollution from runoff, eutrophication of freshwater systems, and massive killing of aquatic organisms in coastal regions that comprise so-called dead zones due to depleted oxygen.4,5 These consequences could be mitigated if ammonia synthesis were electrified,6 and hence produced on scale, and on demand, in a distributed fashion. On demand distributed production of fertilizer could offset use (and hence production) of fertilizer that is sourced conventionally and could also be generated locally and at a rate that increases net absorption by crops (versus runoff), offering a possible environmental benefit to conventional practices in fertilizer acquisition and use.7
Although ammonia is a commodity chemical produced primarily for fertilizer (on a massive scale, ~150 million metric tons annually), it has also been identified as a promising alternative fuel. It is highly storable, easily liquefied, and has an energy density approaching half that of gasoline far exceeding that of compressed hydrogen. It can also be used as a fuel within an internal combustion engine (ICE) or via a solid-oxide fuel cell (SOFC). Moreover, globally substantial ammonia transport infrastructure and related safe-handling protocols already exist.8 For these reasons, ammonia synthesis is an extremely attractive target for electrification, especially via renewable energy technologies, requiring major advances in catalyst development. N2RR electrification would enable surplus energy in the grid, at times of excess supply, to be converted to fertilizer and/or to a storable and transportable fuel, particularly desirable in areas where wind and solar resources are vast. The eventual realization of an “ammonia fuel economy” that can contribute to diverse future energy strategies, along with technologies for on-site and on-demand ammonia fertilizer generation, will require breakthrough research discoveries in catalysis.9
1.2. Inspiration for Organometallic and Inorganic Chemistry
In contrast to the forcing conditions required in the Haber–Bosch process, certain microorganisms can fix N2 under ambient conditions, using extensive hydrolysis of adenosine triphosphate (ATP) to power the delivery of H+ and e− equivalents to N2. These enzymes may hold important clues as to how H+/e− currency, potentially derived from photosynthetic water splitting, could be efficiently delivered to N2 via an appropriate catalyst. Housed within any given nitrogen-fixing organism are conserved sets of proteins—the nitrogenase enzymes—that bind and convert N2-to-NH3. Nitrogenases appear to require iron as an essential transition metal and typically contain molybdenum (FeMo-nitrogenase, most common form), with either vanadium (VFe-nitrogenase) or Fe (FeFe-nitrogenase) being assembled (and functionally active) in the absence of Mo.10,11 FeMo-nitrogenase was the first to be discovered and has been by far the most widely studied.12,13 In addition to various exogenous cofactors required for its function, this enzyme consists of an Fe-protein that delivers reducing equivalents and a MoFe-protein.10 The latter contains two structurally unique clusters. The first is the P-cluster (Fe8S7), which serves as an electron relay to the Fe-protein. The second is the M-cluster (MoFe7S9C-homocitrate), an inorganic FeMo cofactor (FeMoco, Figure 1) that mediates the catalytic bond-breaking and making steps.14
Figure 1.

X-ray structure of the FeMo-cofactor active site in the MoFe protein (PDB: 1M1N); blue = N, red = O, yellow = S, brown = Fe, gray = C, purple = Mo.13
Inorganic chemists have long puzzled (and engaged in spirited debate!) over how this cofactor operates15–19 and, in collaboration with biochemists, microbiologists, structural biologists, and spectroscopists, have pursued molecular model systems as a means of constraining hypotheses regarding viable inorganic mechanisms for catalytic N2RR. This has proven to be a remarkably rich research area. The list of talented chemists who have significantly contributed to what is now an enormous body of knowledge reads like a “Who’s Who of Inorganic Chemistry.” Fortunately, much of this work has been reviewed previously.15,20–29
The goal of this review is to be comprehensive only with respect to the comparatively recent body of literature pertaining to catalytic N2RR that is mediated by nominally well-defined synthetic complexes, in the presence of H+/e− sources. We acknowledge at the outset that much of the fascinating literature preceding such catalyst discoveries will not be detailed, except in cases where introducing background is needed to set the stage for the catalyst discoveries that will be covered. One such case is the early and pioneering work of Joseph Chatt and his co-workers.15,30 Given how central the research and ideas he and his team espoused were to the development of the broader field of N2RR catalysis, we felt it appropriate to briefly summarize some of this historical context here. The interested reader should consult a number of excellent reviews for a deeper dive into this and other early literature.15,31–34
2. DISCUSSION OF THE CHATT CYCLE
Coordination chemists began to think seriously about alternative catalyst technologies to the Haber–Bosch ammonia synthesis in the early 1960s (Figure 2). Several factors had set the stage; key among these was that, in 1963, the British Agricultural Council, led by Secretary Sir Gordon Cox, himself a coordination chemist by training, appointed the British inorganic chemist Joseph Chatt to oversee a multidisciplinary research unit dedicated toward understanding the mechanism of biological nitrogen fixation.35 It was by this time presumed that Fe and Mo were present within the active site of the single nitrogenase that was then known.36 Refreshingly, the Secretary of Agriculture must have intuited that, at its heart, the mechanism of nitrogen fixation was an inorganic chemistry problem. This took imagination and foresight, as it was not until two years later (1965) that Allen and Senoff reported their landmark (and fortuitous) discovery that N2 could coordinate as a ligand to a transition metal, via the isolation and characterization of [(NH3)5Ru(N2)]2+ (Figure 2).37,38 Hence, from the outset, the systematic study of inorganic and organometallic complexes for N2RR was established as an area of bioinorganic model chemistry.
Figure 2.

Timeline (1930 to present) of selected advances in nitrogen fixation catalysis by synthetically well-defined complexes.18,37,48–56
Fundamental studies toward understanding the binding, activation, and conversion of N2 to protonated intermediates and/or products at well-defined transition metal centers were deemed essential to helping formulate and constrain hypotheses concerning the biological process. The Unit of Nitrogen Fixation, first located at the University of London but soon thereafter relocated to Sussex, was highly innovative in its approach and comprised not just chemists but also microbiologists, biochemists, and geneticists, working collectively.39
Prior to his appointment leading the Unit of Nitrogen Fixation, Chatt (with Duncanson), contemporaneously with Dewar,40 played a key role in defining and generalizing the bonding of olefins to transition metals, describing the interaction by both sigma donation (from the olefin π-electrons to the metal) and π-bonding (from the metal to the olefin π-antibonding orbital).41 This type of bonding is today often referred to as the Dewar–Chatt–Duncanson model.42 This proposal paralleled what had already been developed for metal carbonyls43 and anticipated the type of bonding interactions in metal dinitrogen complexes.
Following Allen and Senoff’s isolation of a terminally bound Ru(N2) complex,38 it was not much of a stretch to postulate that synthetic metal complexes might be able to bind and catalyze N2RR under suitable conditions. A surge of relevant research activity thus followed, not just in the UK but around the world, and within just a decade profound progress was made.15,33,34 For instance, Shilov and his collaborators in the former Soviet Union reported the exciting discovery that certain transition metal mixtures containing, for example, molybdenum precursors mixed with Mg(OH)2, could in fact catalyze N2 reduction to N2H4 and NH3 in alcohol/water mixtures in the presence of reducing agents such as sodium amalgam.20,44–47 But it was the work of Chatt and his team at Sussex, via their careful, rigorous preparation and study of M(N2) and M(NxHy) complexes, that most directly laid the groundwork for the well-defined synthetic N2RR catalysts that have emerged thus far.15,32
2.1. The Chatt Cycle As It Is Commonly Known
While M(N2) complexes for a range of TMs (e.g., Ru, Re, Os, Co) were known by the end of the 1960s, much of the early biomimetic work focused on Mo(N2) (and related W(N2)) complexes.32–34,57 Since the work of Bortels in 1930, it was long-held dogma that molybdenum was essential to nitrogen fixation48 and this fact, combined with the practical reality that so many M(NxHy) complexes featuring Mo (and W) proved accessible, helped focus model chemistry research in this area. Relatedly, biochemical experiments had suggested that nitrogenase activity was highly sensitive to alteration at or near the molybdenum site, such as at the homocitrate ligand.10 Chatt’s team established N2 binding and activation at Mo (and W), and showed that the coordinated N2 ligand could be protonated to release NH3 (and hydrazine) in variable yield (as high as 90% for W(PMe2Ph)4(N2)2 with H2SO4, assuming one N2 equiv is released as gas)58 depending on a range of factors (Figure 3). What is more, the group was able to identify a number of M(NxHy) complexes, including M(N),59 M(NNH2),60–64 M(NHNH2),65 M(NNH),60,66,67 M(NNH3)68,69 species (M = Mo or W), that likewise underwent protonation to release NH3 in similar yields.
Figure 3.

Protonation of M(cis-N2)2(PMe2Ph)4 and M(NxHy); M = Mo or W.15
These findings led Chatt to propose a simple scenario in which triple functionalization at Nβ leads to the release of NH3 and a nitride intermediate. This scenario is now known as the Chatt cycle, and a simplified version is depicted below (Figure 4).30 Worth noting is that while examples of the types of M(NxHy) species invoked in the Chatt cycle could be generated, most typically these species were not characterized in the same formal state of oxidation as invoked in the catalytic scheme.26 It would not be until Schrock’s discoveries some 30 years later50,70 that well-characterized examples of the proposed catalytic Mo(NxHy) intermediates, in an oxidation/protonation state matching the envisaged catalytic cycle, were discovered (Figure 11). Critically, this triamidoamine-ligated Mo system would also prove to be the first catalyst for nitrogen fixation, thereby cementing the importance of the Chatt cycle, sometimes formulated as the Schrock or “distal” cycle in the contemporary literature, in the field. Equally important, Chatt and his team underscored via their work the value of preparing and carefully studying well-defined M(NxHy) model complexes to test the validity of possible pathways for nitrogen fixation.
Figure 4.

Scheme demonstrating the distal (or Chatt) cycle for nitrogen fixation. In the modern literature, this cycle is also sometimes referred to as the Schrock cycle. Ligands on molybdenum are omitted for clarity.30
Figure 11.

(Top) Protonation reactions of (HIPTN3N)Mo(L) (L = CO or N2). For the CO analogue, pulse EPR (ENDOR) data indicated protonation at an amido arm; for N2, the site of protonation remains unknown;102 (middle) protonation/reduction reactions of the L = N2 and N analogues gave rise to [Mo]-NNH and [Mo]-NH3+; (bottom) reduction of [[Mo]-NH3]+ under N2 gives [Mo]-N2 in ~10% yield; [Mo] = (HIPTN3N)Mo.99
2.2. Alternative Mechanistic Scenarios for NH3 Formation
While it is the aforementioned cycle for which Chatt is most often credited, he and his co-workers also envisaged alternative scenarios that could account for NH3 (and N2H4) production via intermediates not included on the so-called Chatt pathway. For example, protonation of an M(NHNH2) or M(NH2NH2) intermediate can release hydrazine or ammonia (Figure 5).65 It is perhaps ironic that, although a M(NHNH2) species is not commonly referred to as a “Chatt-type” intermediate, it was his group that first prepared such a species (for W) and considered its direct relevance to NH3 release.61
Figure 5.

An alternative to the Chatt pathway that can account for productive NH3 formation.
Although Schrock’s best known contribution to N2-fixation chemistry, the (HIPTN3N)Mo system (HIPTN3N = 3,5-(2,4,6-i-Pr3C6H2)2C6H3NCH2CH2]3N3−), appears to proceed via a distal reaction pathway, during the mid-1980s, Schrock and co-workers also reported important results regarding alternative mechanisms for NH3 formation in Cp*-Group VI (Cp* = C5Me5−) compounds. During their study of the general reactivity of (Cp*)M (M = Mo, W) complexes with NxHy ligands, they found that protonation reactions of the N2 complexes gave (primarily) NH3, but studies suggested that the pathway for NH3 release was unlikely to involve the terminal nitride, M(N), intermediate.71 Instead, protonation of the hydrazido(2−) complex, (Cp*)W(Me)3(NNH2), occurred at Nα to give [(Cp*)W(Me)3(η2-NHNH2)]OTf —a reversible process in the presence of base. This is an interesting transformation, as one can ask whether the kinetic site of protonation of (Cp*)W(Me)3(NNH2) is at Nα or Nβ, and whether protonation occurs prior to, or following, isomerization of the nitrogenous ligand from an η1- to η2-form (Figure 6). A subsequent study by Norton provided compelling evidence for initial protonation at Nβ to generate [(Cp*)W(Me)3(NNH3)]+, followed by isomerization to give [(Cp*)W(Me)3(η2-NHNH2)]+.72 While the W(NNH3)+ species in this system was not derived from N2, the aforementioned transformations underscore that a so-called Chatt (or distal) intermediate can isomerize to an alternating intermediate W(η2-NHNH2).72
Figure 6.

Protonation of the hydrazido(2−) complex, (Cp*)W(Me)3(NNH2), can initially occur at Nα or Nβ, but ultimately gives [(Cp*)W(Me)3(η2-NHNH2)]+—a reversible process in the presence of NEt3. It remains unclear whether protonation occurs at the η1- or η2-form of the hydrazido(2−) species.72
With the (Cp*)M platform, the Schrock group could also synthesize hydrazine intermediates [(Cp*)M(Me)3(NH2NH2)]+. Reduction of these intermediates with sodium amalgam led to the formation of NH3 and (Cp*)M(NH). Indeed, reacting this platform with excess acid and reductant in the presence of hydrazine led to the catalytic formation of NH3 in yields up to 95% (note that the maximum yield for hydrazine disproportionation is 67%).73,74 This work, thus, conclusively demonstrated that NH3 formation does not necessarily proceed via the Chatt cycle, but rather that formally “alternating” intermediates M(NHNH2) and M(NH2NH2) can also lead to NH3 formation. The potential of such intermediates to contribute to hydrazine formation will be discussed later in the context of catalysis with Fe complexes.
Much of the work from Chatt was pursued in the absence of exogenous reductant and thus necessarily focused on zerovalent Mo and W species that could themselves provide the electrons required for nitrogen fixation. Nonetheless, Chatt, as early as 1969, noted that “the reduction of complexed (di)nitrogen requires specific reagents or conditions, and these may be just as elusive as the first nitrogen complexes.”75
Schrock’s early work is not only important for its initial elucidation of hybrid mechanisms for NH3 formation, but also for its preliminary efforts to perform protonolysis experiments in the presence of external reductants. Perhaps inspired by the success of Shilov and his collaborators in achieving catalytic N2 reduction with transition metal mixtures and amalgam reductants,44,76,77 Schrock and co-workers performed protonolysis experiments using [LutH]Cl (LutH = lutidinium) of μ-bridged Group VI dinitrogen complexes, such as [(Cp*)Mo(Me)3]2(μ-N2), in the presence of ZnHg amalgam.78 Although these experiments only gave 0.62–0.72 equiv of NH3, this work provided a viable approach for future studies that would yield catalysis.
In fact, the study of nitrogen fixation has revealed that the choice of acid and reductant is critical to catalyst performance. In many ways, the reagents are as much a part of the nitrogen fixation story as the catalysts themselves, with certain systems demonstrating dramatically different results and, by implication, different mechanisms when distinct reagent cocktails are supplied.18,51,79–81 Combinations of compatible acid and reductant have moved the field forward and allowed for the discovery of new catalysts. Indeed, in recent years attention has focused how the choice of reagents also controls the overall thermochemistry of the N2RR catalysis, providing a framework toward improvement of the thermal efficiency of the catalysis. In the next section, we will detail the two successive breakthroughs made by Schrock and co-workers. The first was the application of the (HIPTN3N)Mo ligand to support a series of Chatt-cycle intermediates in the relevant oxidation states70 and the second, the discovery that these intermediates could be turned over by application of an appropriate reductant and acid,50 a combination that went on to find broad application in the field.51,82–84
Another pathway for dinitrogen fixation concerns the formal scission of dinitrogen to two metal-bound nitrides followed by the transfer of proton and electron equivalents (via PCET or otherwise) to release ammonia (vide infra in Section 4.3 for an example from Nishibayashi and co-workers). This transformation has been shown feasible for a number of transition metal complexes, typically following formation of a μ2-bimetallic N2 adduct that undergoes N–N bond cleavage upon addition of a reducing agent.85–90 A classic example is shown in Figure 7 and concerns a Mo tris(amido) complex, (N(R)Ar)3Mo.91,92 This species binds N2 at room temperature reversibly to form (N(R)Ar)3Mo(N2), with the μ2-N2 bridged derivative, [(N-(R)Ar)3Mo]2(μ-N2), being formed at −35 °C over a period of days. Warming this complex results in formation of two terminally bonded nitride complexes, (N(R)Ar)3Mo(N).92,93
Figure 7.

N2 splitting by a Mo tris(anilide) complex; an alternative pathway to the formation of ammonia from μ-N2 complexes.
3. SCHROCK’S TRIS(AMIDO)AMINE Mo: THE FIRST WELL-DEFINED CATALYST SYSTEM
3.1. Azatrane Mo Complexes Relevant to N2 Fixation
With an eye toward nitrogen fixation, in 1994, Kol et al. showed that triamidoamine ligands [(RNCH2CH2)3N]3− where R = C6F5 could be used to gain access to molybdenum complexes of potential relevance to N2RR (Figure 8).94 A number of relevant molybdenum complexes were accessed, including a terminal nitride (((C6F5NCH2CH2)3N)Mo(N)), methylimido cation ([((C6F5NCH2CH2)3N)Mo(NCH3)]+), as well as both bridged (((C6F5NCH2CH2)3N)Mo]2(μ-N2) and terminal ([((C6F5NCH2CH2)3N)Mo(N2)]− dinitrogen complexes. The latter of these was shown to be nucleophilic at Nβ, undergoing facile silylation by iPr3SiCl to give [(C6F5NCH2CH2)3N]Mo-N=N−Si(iPr)3. Silyl-substituted-triamidoamine ligands have also been studied (vide infra for-an example using Ti - Figure 69)—with reaction of MoCl3(THF)3 and [(Me3SiNCH2CH2)3N]Li3 giving the paramagnetic μ-N2 complex, [(Me3SiNCH2CH2)3N]Mo]2(μ-N2) in 10% yield. In 1997, O’Donoghue et al. showed that a related TMS anion, {[(TMSNCH2CH2)3N]Mo-N=N}2(Mg(THF)2) reacted with FeCl2 to give {[(TMSNCH2CH2)3N]Mo-N=N}3(μ3-Fe).95,96 These contributions laid the groundwork for further exploring azatrane-Mo complexes as competent for the binding and activation of dinitrogen, as well as the stabilization of potential N2-fixation intermediates.
Figure 8.

(Top) Reactions of a Mo triamidoamine complex having C6F5 anilido groups provided access to complexes of potential relevance to N2RR;94 (bottom) reaction of an analogous Mo complex with TMS anilido groups with FeCl2 afforded a μ3-Fe bridging via N2 to three triamidoamine Mo centers.95
Figure 69.

Synthesis and characterization data of triamidoamine-Ti complexes relevant to catalytic nitrogen fixation.55
Recognizing that TMS- and C6F5- substituted tren [(RNCH2CH2)3N]3− ligands underwent deleterious side-reactivity (due to Si−N bond cleavage and the sensitivity of C6F5 groups to nucleophilic reagents), in 2001 Greco and Schrock designed a new route to access aryl-substituted triamidoamine ligands by Buchwald—Hartwig amination.97 A small library of ligand candidates was constructed including monoaryl (C6H5, 4–F-C6H4, 2,4,6-Me3C6H3) and triaryl (3,5-Ph2C6H3, 3,5-(4-tBuC6H3)2C6H3) derivatives that could be readily metalated with Mo (Figure 9). In a subsequent contribution, Greco and Schrock illustrated that a variety of different MoIIN2 complexes could be synthesized in a reductant-dependent fashion. These aryl-substituted triamidoamine Mo complexes (RPhN3N, R = H; 4-F; 2,4,6-Me; 3,5-Ph; 3,5-(4-tBu)Ph) all feature fairly activated N2 complexes (1740 cm–1 < νNN < 1815 cm−1). Accordingly, these species could be silylated or alkylated to provide (RPhN3N)Mo(NNR) (R = SiMe3 or Me) species. The alkyl diazenidos, (RPhN3N)Mo(NNCH3), can be reacted further with alkylating agents (e.g., CH3OTf, CH3OTs) to form cationic hydrazidos [(RPhN3N)Mo(NN(CH3)2] + (Figure 9). Similarly, reaction of (RPhN3N)Mo(NNTMS) with alkylating reagents provides [(RPhN3N)Mo(NN(CH3)(TMS)]+. Efforts to access neutral molybdenum hydrazido complexes were not successful, but reduction of [(RPhN3N)Mo(NN(CH3)2]+ with NaHg amalgam, provided the Mo(Vl) nitride complex, (RPhN3N)Mo(N) (and presumably a fixed nitrogen product).98
Figure 9.

Synthesis of alkylated Mo diazenido and hydrazido complexes en route to a Mo nitride compound in a triamidoamine Mo complex with Ar = 3,5-Ph2C6H3 or 3,5-(4-tBuC6H4)2C6H3 ligands.98
To prevent undesired bimolecular pathways problematic to an N2 reduction cycle, the highly encumbering 3,5-(2,4,6-iPr3C6H2)2C6H3 (HIPT = HexaIsoPropylTerphenyl) derivative was prepared (Figure 10).70 On this ligand, an impressive suite of N2-derived monometallic molybdenum complexes were prepared (vide infra), including (HIPTN3N)Mo(N2), [(HIPTN3N)Mo(N2)] −, (HIPTN3N)Mo(NNH), and [(HIPTN3N)Mo(NNH2)]+. Additional, species potentially relevant to an N2-fixation cycle could be accessed with azide as the N atom source, (HIPTN3N)Mo(N) and (HIPTN3NMo(NH). Lastly, [(HIPTN3N)Mo(NH3)]+ was accessed by treatment of (HIPTN3N)Mo(Cl) with NH3 in the presence of NaBArF4.
Figure 10.

Synthesis of a series of monometallic molybdenum NxHy complexes on 3,5-(2,4,6-iPr3C6H2)2C6H3 (HIPT) ligated Mo; [Mo] = (HIPTN3N)Mo.70
With these species in hand, Schrock’s team explored the possibility of catalysis with soluble acid and reducing equivalents.99 Treatment of (HIPTN3N)Mo(N2) with 1.0 equiv of [LutH]BArF4 and 2.0 equiv of Cp2Co (E1/2 = −1.3 V vs Fc/Fc+) provided (HIPTN3N)Mo(NNH) nearly quantitatively (Figure 10).100 Intriguingly, (HIPTN3N)Mo(N2) reacted directly with neither [LutH]BArF4or Cp2Co, but rapidly reacted with the combination of these reagents. The Schrock group suggested that N-amido protonation could proceed reduction followed by proton transfer. This hypothesis has found indirect support from theoretical work101 and ENDOR studies. ENDOR revealed that treatment of (HIPTN3N)Mo(CO) with [LutH]+ results in protonation of an N-amido arm, but similar behavior was not confirmed for (HIPTN3N)Mo(N2) (Figure 11).102 It has been hypothesized that this reactivity profile suggests a degree of concertedness in the transfer of the proton and electron to form the new N–H bond. In this review, we will refer to such reactions as proton-coupled electron transfer (PCET) reactions.103,104 Two specific suggestions have been made. Chirik and co-workers suggested that reduction of pyridinium acids by the metallocenes results in pyridinyl radicals with weak N–H bonds that could therefore mediate the conversion of (HIPTN3N)Mo(N2) to (HIPTN3N)Mo(NNH) (and potentially other N–H bond forming steps).105,106 Although such a mechanism could be operative under catalytic conditions, it is known that triethylammonium is also efficacious for this reaction in a stoichiometric fashion. The Peters group has suggested with the support of computation that instead these acids protonate the metallocene species on the Cp(*) ring resulting in species with weak C–H bonds that can then mediate conversion of the N2 species to the NNH species via a PCET reaction (vide infra).80
If a larger quantity of [LutH]BArF4 (7.0 equiv) and Cp2Co (8.2 equiv) was reacted with (HIPTN3N)Mo(N2), then [(HIPTN3N)Mo(NH3)]+ was formed in 60% yield. Furthermore, the reaction of (HIPTN3N)Mo(N) with [LutH]BArF4 (3.5 equiv) and Cp2Co (4.2 equiv) gave [(HIPTN3N)Mo(NH3)]+ in 80% yield, which could be liberated with Bu4NCl and NEt3 to afford 0.88(2) equiv of NH3. Closing the catalytic cycle involved the reduction of [(HIPTN3N)Mo- NH3]+ to (HIPTN3N)Mo(NH3), the release of NH3, and the binding of N2. However, [(HIPTN3N)Mo(NH3)]+ is quite reducing, so the reaction with Cp2Co only converted 10% of the starting material to the desired (HIPTN3N)Mo(N2) complex (Figure 11).99
3.2. Description of the System and Key Findings
Building on this work, Yandulov and Schrock published the first example of N2 reduction catalysis mediated by a well-defined, synthetic catalyst in 2003 (Figure 12).50 In particular, they observed that using the more reducing Cp*2Cr (E1/2 = −1.47 V vs Fc+/0) allowed for complete reduction of [(hIPTN3N)Mo(NH3)]+. Likewise, slow addition of reductant (6 h, syringe pump) and solvent selection (heptane) were needed to attenuate background H2 formation. With these conditions, it was found that (HIPTN3N)Mo(N2), (HIPTN3N)Mo(NNH), (HIPTN3N)Mo(N), or [(HIPTN3N)Mo(NH3)]+, when combined with Cp*2Cr (36 equiv) and [LutH]BArF4 (48 equiv), were all competent (pre)catalysts for N2 reduction, generating between 7.56 and 8.06 equiv of NH3 (Figure 12). At the time, this selectivity (~66%) was second only to nitrogenase (~75%).50
Figure 12.

Catalytic N2RR cycle depicting (HIPTN3N)Mo intermediates and their efficacy as (pre)catalysts for N2RR using [LutH]BArF4 (48 equiv) and Cp*2Cr (36 equiv); [Mo] = (HIPTN3N)Mo.50,107 Characterized compounds are shown in purple and unobserved complexes in gray.
A series of experiments were performed in order to interrogate the catalytic reaction. (HIPTN3N)Mo(NNH) was reacted with 1 equiv of [LutH]BArF4 (Figure 13).100 This was found to give a 44:56 equilibrium mixture of (HIPTN3N)Mo(NNH) and [(HIPTN3N)Mo(NNH2)]+ (Keq = 1.6). Similarly, protonation of (HIPTN3N)Mo(N) using [LutH]BArF4 gave a 3:1 mixture of (HIPTN3N)Mo(N) and [(HIPTN3N)Mo(NH)]+.100 These results suggest that weaker acids would not be suitable for the catalysis.
Figure 13.

(Top) [Mo]-NNH and [LutH]+ are in equilibrium with [Mo]-NNH2+ and Lut (Keq = 1.6); (bottom) protonation of [Mo]N gives a mixture of the starting material and [Mo]NH+, which upon reduction, decayed to give [Mo]N and [Mo]-NH2; [Mo] = (HIPTN3N)Mo.99
Consistent with these weak predicted bond strengths, reduction of [(HIPTN3N)Mo(NNH2)]+ by Cp2Co did not yield the expected (HIPTN3N)Mo(NNH2), but rather the formation of NH3, (HIPTN3N)Mo(NNH), and (HIPTN3N)Mo(N). Similarly, reduction of the cationic imide with Cp*2Cr initially gave a species tentatively assigned as the neutral imide, (HIPTN3N)Mo(NH), which then decayed to (HIPTN3N)Mo(N) and (HIPTN3N)Mo(NH2) in solution. These results are consistent with calculations by Chirik and co-workers that suggest that these species (the neutral hydrazido and neutral imide) have the weakest N–H bonds on this platform.106 In contrast, the alkylated congeners, (HIPTN3N)Mo(NNEt2) and (HIPTN3N)Mo(NEt), are stable, but efforts to achieve the catalytic reduction of N2 to NEt3 with [Et3O]BArF4 and Cp*2Cr were unsuccessful (Figure 14).108 While many factors may of course be at play that impede catalysis, one possible conclusion is that the net exchange of H atoms between nitrogen fixation intermediates may be relevant to achieving overall catalysis in N2RR. This is clearly speculative. Observations regarding the productive net transfer of H atoms between nitrogen fixation intermediate and the enhanced stability of alkylated congeners have been made on the silyl-anchored triphosphine system from Peters and co-workers (vide infra).109
Figure 14.
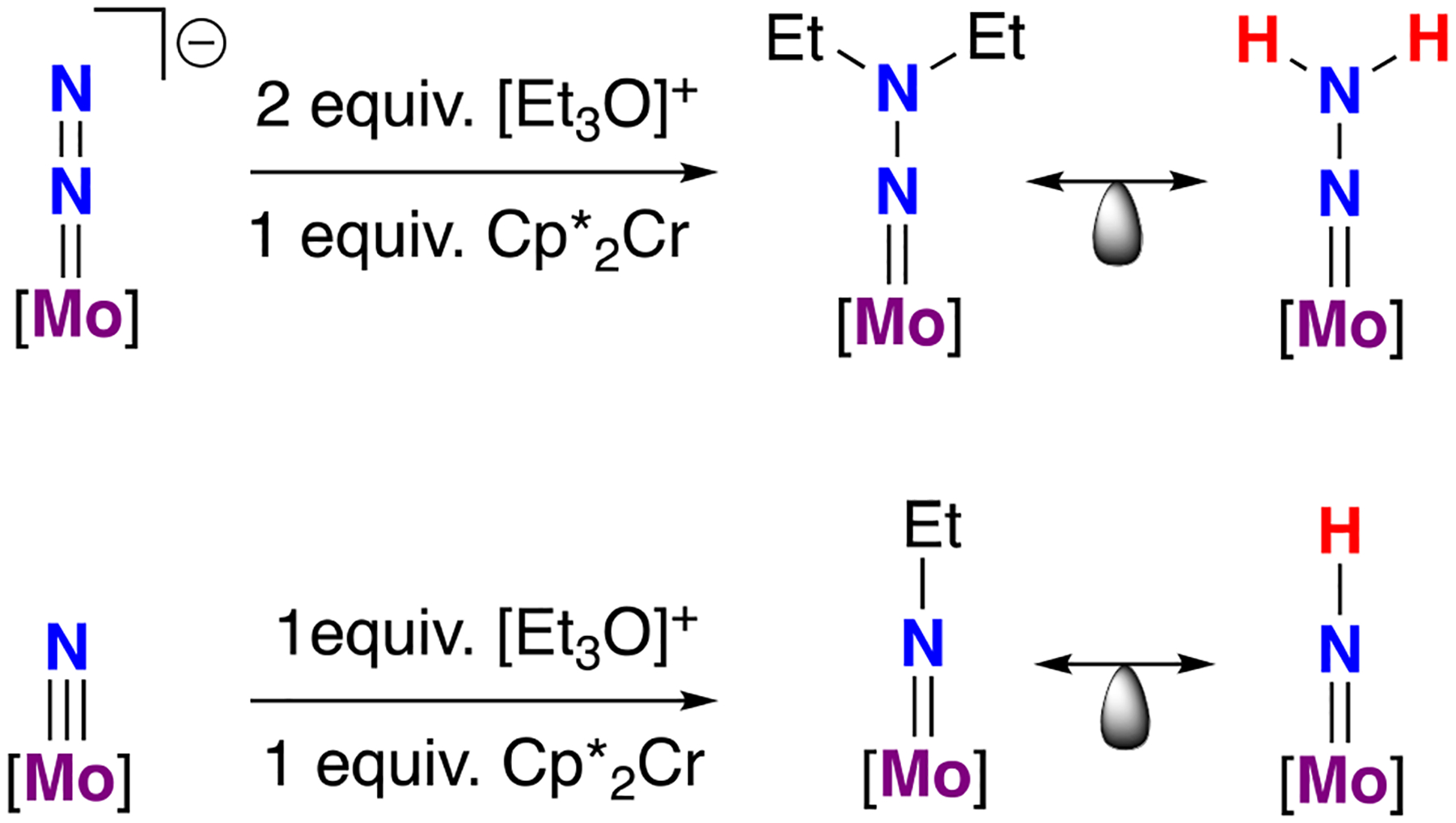
Use of an alkylating agent provided access to neutral [Mo]-NNEt2 and [Mo]-NEt; these species serve as structural analogues of the highly reactive R = H variants; [Mo] = (HIPTN3N)Mo.99,108
At the end of catalysis, [(HIPTN3N)Mo(NH3)]+ was the only Mo-containing species present, suggesting that the slow steps of the catalysis involve the reduction of this species and then exchange with N2. Consistent with this observation, the reduction potential of this species (−1.51 V vs Fc+/0 in THF) is slightly lower than that of the employed reductant, Cp*2Cr (–1.47 V vs Fc+/0 in THF).110 Exchange of N2 for NH3 in (HIPTN3N)Mo(NH3) was determined to undergo an associative mechanism on the basis of variable pressure N2 experiments (kobs = 2.4 fold faster at a 15 psi N2 overpressure). Indeed, performing N2RR at a pressure of 30 psi (instead of 15 psi) provides a small, but measurable, increase in ammonia yield (71% vs 63%) over a 6 h addition and 55% versus 45% over a 3 h addition time.110 Under catalytic conditions, dissociated NH3 is anticipated to be trapped via protonation, which should drive the reaction toward the N2 complex. Consistent with this, the rate of the substitution reaction was significantly enhanced by inclusion of triphenylborane as an NH3 trap.110 Computational studies by Reiher and co-workers have found support for the involvement of a six-coordinate (HIPTN3N)Mo(NH3)(N2) intermediate (as opposed to a five-coordinate species with a dissociated ligand arm) in the exchange process.101,111 Accessing states where there is rapid exchange of NH3 for N2 remains a general challenge for N2RR catalysts and accordingly is an ideal area for future catalyst design.
Schrock and co-workers performed X-ray diffraction analysis on (HIPTN3N)Mo(NNH), [(HIPTN3N)Mo(NNH2)]+, [(HIPTN3N)Mo(NH)]+, and [(HIPTN3N)Mo(NH3).100 The accumulated structural data show that the Mo–Namine distance varies as a function of the Mo axial ligand; as the N–N bond is weakened and cleaved on going from [HIPTN3N]Mo(N2) to [HIPTN3N]Mo(N), a lengthening in the Mo–Namine by ~0.19 Å is observed. An even more dramatic lengthening of the metal-axial ligand distance has been observed in the borane-anchored trisphosphine Fe system from Peters and co-workers.112–114 This suggests that flexibility of this interaction is perhaps critical for allowing catalysts to stabilize both π-accepting intermediates and π-donating intermediates and/or low-valent and high-valent states.
Given the formation of H2, presumably from Mo(H) species, during (HIPTN3N)Mo catalyzed nitrogen fixation, the reactivity of Mo(H) and Mo(H2) complexes was also of interest.99,107 Intriguingly, (HIPTN3N)Mo(H) could be prepared from the diazenido derivative, (HIPTN3N) Mo(NNH), via an effective “β-H elimination” (k1 = 2.2 × 10−6 s−1) (Figure 15). This is a very unusual transformation that potentially warrants further study. The use of the hydride species as a (pre)catalyst produced 7.65(3) equiv of NH3, suggesting that it could be readily returned to a catalytically on-path intermediate. Schrock and co-workers demonstrated that reaction of (HIPTN3N)Mo(H) with [LutH]BArF4 led to the formation of [(HIPTN3N)Mo(Lut)]+, which can presumably be reduced and bind N2 to rejoin the catalytic cycle. The performance of the (HIPTN3N)Mo(H) species as a precatalyst contrasts with that of other M(H) (M = Fe, Co, Os) species. In those cases, the M(H) species have uniformly performed more poorly as precatalysts than the related N2 complex (vide infra).53,54,115,116
Figure 15.

β-H elimination from the diazenido derivative, [Mo]–NNH, generated [Mo]–H; [Mo] = (HIPTN3N)Mo.99
The H2 complex, (HIPTN3N)Mo(H2), could be formed via the reaction of (HIPTN3N)Mo(N2) with H2 over 2–3 days (Figure 16).107 Exchange of N2 for H2 occurred at a rate of k = 3.4 × 10−6 s−1 and was independent of the H2 pressure, indicating a rate-limiting dissociation of N2 prior to H2 coordination, consistent with the proposed dissociative exchange of N2 and 15N2.99 Experiments for the reverse reaction (i.e., formation of (HIPTN3N)Mo(N2) from (HIPTN3N)Mo(H2) found the rate to be 1.0 × 10−6 s−1 (t1/2 = 8 days)). Use of (HIPTN3N)Mo(H2) as a (pre)catalyst provided ammonia in 52% yield relative to 60–65% (from (HIPTN3N)Mo(N2)). Experiments carried out with dihydrogen injected (~32 equiv and 64 equiv of H2 with respect to Mo) using (HIPTN3N)Mo(N) as the catalyst, showed that only 1 equiv of NH3 was formed, presumably from the nitride ligand.107 Cumulatively, these data suggest that H2 poisons the N2 fixation catalysis. This presents a significant challenge to achieving higher turnover numbers with this catalyst. Indeed, the ability to manage H2 formation is ultimately going to be an issue of concern in any catalytic nitrogen fixation system that is not perfectly selective for NH3.
Figure 16.

Generation of [Mo]–H2 and a plausible decay pathway involving intramolecular deprotonation to a terminal (HIPTN3NH)Mo(H) species; [Mo] = (HIPTN3N)Mo.107
3.3. Use of EPR for the Study of (HIPTN3N)Mo Complexes
Electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) have enabled the characterization of a number of (HIPTN3N)Mo complexes. These have included (HIPTN3N)Mo(NH3),117 (HIPTN3N)Mo(N2),102,117–119 (hIPTN3N)Mo(CO),102,117 the amido-protonated analogue, [(HIPTN3N(H))Mo(CO)]+102 (vide supra) as well as the product of H2 with (HIPTN3N)Mo(N2), the Mo(III) anion, [(HIPTN3N)Mo(H)]−.119 More recently, Schrock, Hoffman, and Neese studied the electronic and geometric structures of [(HIPTN3N)Mo(N)]–-and (HIPTN3N)Mo(NH) by irradiation of their oxidized congeners with γ-rays at 77 K.120 Collectively, these studies serve to provide useful spectroscopic models to which more complicated [Mo]-containing clusters can be compared; several of these open-shell compounds are not characterizable by other means.
3.4. Evidence in Support of the Chatt/Schrock Cycle from Theory
Several studies that bear on the theoretical mechanism of N2 fixation by the (HIPTN3N)Mo system have been published (Figure 17),101,121–127 and in 2008, theoretical and experimental data for this system were compared by Schrock.128 A brief summary is provided here. Early theoretical studies tended to simplify the catalyst structure to reduce computational expense. For example, Morokuma129 and Cao126 performed studies on truncated models that were not referenced to the catalytically relevant acid and reductant sources. In 2005, Tuczek and co-workers reinvestigated the complete cycle, modeling the HIPT portion of the ligand as hydrogen but using the relevant acid, [LutH]+ and reductant, Cp*2Cr sources as energetic references.121 Independently, Reiher101,122 and Neese & Tuczek124 later performed a series of DFT calculations taking the entire HIPT ligand moiety into account, requiring models of ~280 atoms. In broad terms, these data corroborated the cycle reported above (Figure 12). The following key points are recapitulated with numerical values taken from Nees and Tuczek (B3LYP/def2-TZVP)124—this study also summarized and compared its findings with those from other studies.
Figure 17.
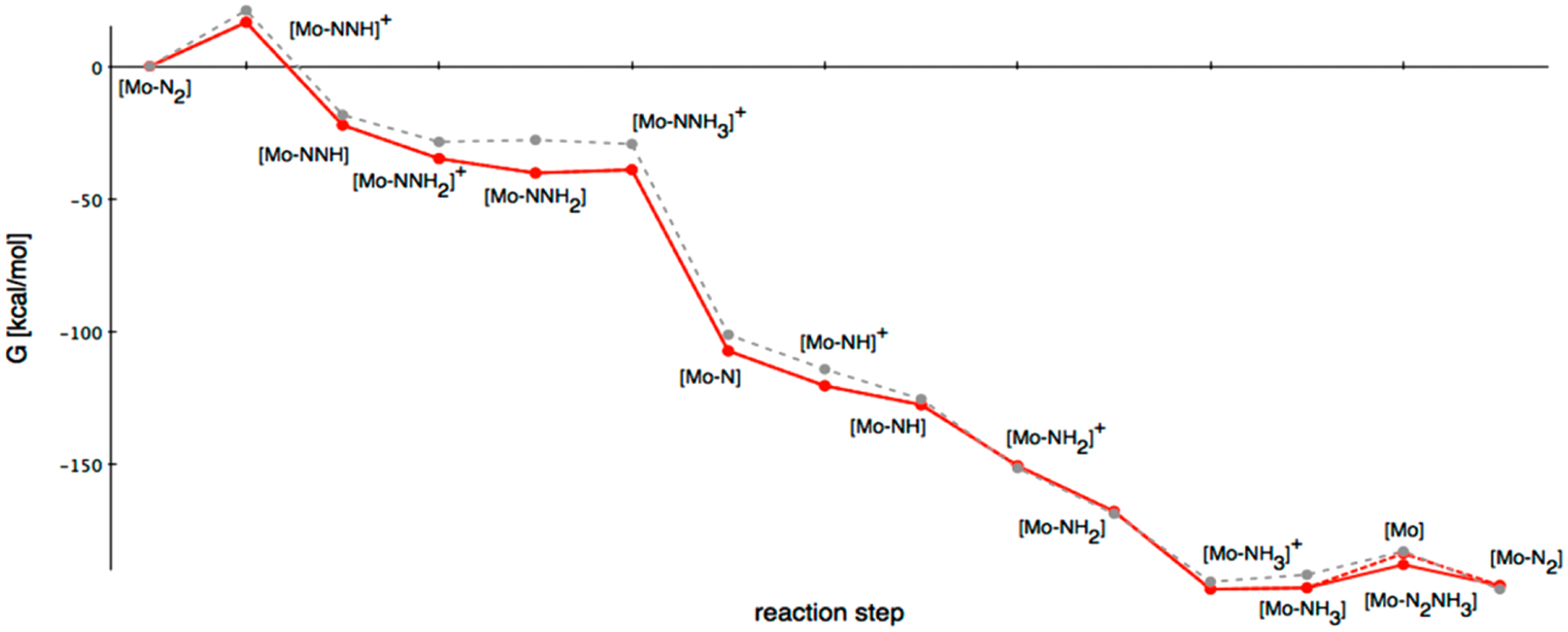
Reproduced with permission from ref 124. Copyright 2015 American Chemical Society. Red: Gibbs free enthalpy ΔG° scheme of the Schrock cycle calculated with the B3LYP functional and the def2-TZVP basis set including solvent correction. Gray: calculations by Studt et al.121
Importantly, all on path intermediates were predicted to be neutral or positively charged; the formation of negatively charged intermediates are predicted to be highly unlikely.
Protonation of (HIPTN3N)Mo(N2) was calculated to be the most endergonic reaction step of the reaction (ΔG = +16.6 kcal mol−1) with protonation of an N-amido ligand arm being more favorable; this species has been characterized spectroscopically for (HIPTN3N)Mo(CO).102 Subsequent reduction, followed by protonation, was proposed to yield [(HIPTN3NH)Mo(NNH)]+. Later computational work from the Chirik group and Peters group have proposed PCET pathways to form the diazenido complex directly from (HIPTN3NH)Mo(N2) as a plausible first step in the catalysis.80,105,106
The most exergonic step of the reaction was N–N bond cleavage upon reduction of [(HIPTN3N)Mo(NNH3)]+ (ΔG = −68.4 kcal·mol−1).124
Consistent with experiment, reduction of the ammine cation, [(HIPTN3N)Mo(NH3)]+ to the neutral species by Cp*2Cr was essentially thermoneutral (ΔGcalc = +0.5 kcal·mol−1, ΔGexp = +0.9 kcal·mol−1).124
The dissociation of NH3 from (HIPTN3N)Mo(NH3) is endergonic (ΔG = +13.0 kcal·mol−1). Subsequent N2 coordination was exergonic (ΔG = −12.1 kcal·mol−1), rendering the net exchange slightly uphill (ΔG = +0.9 kcal·mol−1).124 This calculation agrees with the observed Keq value of 0.1 for this reaction, which corresponds to ΔG = +1.1 kcal·mol−1.124 An associative mechanism was also considered for this process, with calculation of the putative, six-coordinate (HIPTN3N)Mo(NH3)(N2) species being +8.8 kcal·mol−1 higher in energy than (HIPTN3N)Mo(NH3) and free N2.124
A necessary shortcoming of these (and most) DFT studies is that they cannot consider all of the species/interactions that might be relevant in the catalytic mixture. Hence, their primary value lies in their ability to shed light on specific questions given certain assumptions. So as to explore secondary interactions that might be relevant to the Schrock system, Reiher et al. studied “one-pot” models (where acid (acid = [LutH]+) and base (base = a Mo compound) were initially placed approximately 8 Å from one another). This study predicted hydrogen bonded complexes between (HIPTN3N)Mo(N) or (HIPTN3N)Mo(N2) and [LutH]+, indicating that the acid source is sufficiently small to approach the basic part of the molecule through voids made by the three HIPT ligands.122 A related hydrogen-bonded complex between [LutH]+ and a Mo(N) species in a different N2-fixation relevant complex (vide infra) was crystallographically characterized by Schrock and co-workers in 2019 (Figure 18).130 That complex underwent net PCET on treatment with reductant. These collective data support the possibility that a multisite PCET reaction could be relevant to diazenido formation in the (HIPTN3N)Mo system. In this proposal, a hydrogen bonded [(HIPTN3N)Mo(N2)—LutH]+ complex would transfer the proton only upon electron transfer from Cp*2Cr (Figure 17).
Figure 18.

(Left) A depiction of a hydrogen bonded complex between (HIPTN3N)Mo(N2) and [LutH]+ identified in a “one-pot” calculation;122 (right) a depiction of a crystallographically characterized hydrogen bonded complex between (Ar2N3)Mo(N) (Ar = 2,6-diisopropylphenyl) and [LutH]+.130
3.5. Second Generation Schrock Systems
Despite extensive efforts invested in improving the TON of the original (HIPTN3N)Mo system, reactivity (e.g., protonolysis) of the Mo-amido linkages and ligand exchange appears to plague it and related systems. These efforts are summarized here. By 2004, three new ligand variants appeared: two of which gauged sterics (tBu and CH3 in place of iPr) and one that gauged electronics (Br in place of H at the para position of the middle terphenyl ring) (Figure 19). For each ligand, the Mo(N) complex was prepared and its efficacy for catalytic N2RR was tested. The t-butyl analogue produced only 1.06 equiv of NH3 using Cp*2Cr (36 equiv) and [LutH]BArF4 (48 equiv), while the methyl analogue gave only 1.49 equiv of NH3. The p-Br analogue provided 6.4–7.0 equiv of NH3, slightly less than the standard HIPT-based Mo catalyst system.131
Figure 19.

A survey of second generation complexes tested for N2RR by Schrock and co-workers. Reported yields were found using the optimized conditions for the original catalytic result: [LutH]+ (48 equiv), Cp*2Cr (36 equiv), heptane, room temperature.131–134
In another iteration of ligand design, asymmetrical tren ligands were prepared in which one of the three HIPT groups was replaced by (1) a 3,5-disubstitutedbenzene-3,5-Me2C6H3, 3,5-(CF3)2C6H3, or 3,5-(MeO)2C6H3; (2) a 3,5-disubstituted-pyridine-3,5-Me2NC5H3 or −3,5-Ph2NC5H3 or (3) a 2,4,6-trisubstitutedbenzene-2,4,6-Me3C6H2 or −2,4,6-iPr3C6H2 (Figure 19).132 For each ligand, the Mo(N) complex was prepared and probed for competency in catalyzing N2RR. For these candidates, 0.2–2.0 equiv of NH3 was found.
In 2010, Chin and Schrock outlined the synthesis of a system comprising pyrrolide rather than anilide donor groups, which were pursued in hopes of attenuating undesired aminolysis in the presence of acid (a limitation of the HIPT system) (Figure 19).133 Diamido(2-mesityl)pyrrolyl complexes of the type ((ArN)2Pyr)Mo(X) (Ar = C6F5, 3,5-Me2C6H3, or 3,5-tBu2C6H3, X = Cl or NMe2), having two amide and one pyrrole group were prepared. The degree of N2 activation was attenuated in [((ArtBuN2)Pyr)]Mo(N2)]− (νNN = 2012 cm−1 vs 1990 cm−1 [(HIPTN3n)Mo(N2)]−). Perhaps relatedly, ((ArtBuN2)Pyr)Mo(N) proved to be a much poorer catalyst for N2RR (1.02 ± 0.12 equiv of NH3) with no evidence for N2 uptake during catalysis.
In a subsequent study, Reithofer and Schrock developed a HIPT derivative incorporating pendant nitrogens (2,5-diiso-propylpyrrolyl, DPPN3N) in the secondary coordination sphere (Figure 19).134 The hypothesis was that if (HIPTN3N)Mo undergoes decomposition by competitive protonation at the Mo–Namide bond, then providing Brønsted basic groups in the secondary coordination would prevent that and allow a greater population of catalyst to remain on-cycle. In this vein, (DPPN3N)Mo(Cl), (DPPN3N)Mo(N2), and [(DPPN3N)Mo(N2)]− precursors were prepared similarly to those for the HIPT system (vide supra). For (DPPN3N)Mo(N2), υnn = 1993 cm−1, indicating a similar degree ofactivation when compared to (HIPTN3N)Mo(N2). Of the Schrock Mo(N2) compounds discussed in this review, this is the only one that undergoes associative N2 substitution. In a standard experiment, treatment of (DPPN3N)Mo(14N2) with 15N2 showed a first order dependence on [15N2]. This conclusion is rationalized by virtue of having a smaller five-membered pyrrole-based ligand platform that allows for associative substitution, cf. a bulkier six-membered 3,5-substituted terphenyl-group. Other species, including (DPPN3N)Mo(NNH), [(DPPN3N)Mo(NNH2)]+, (DPPN3N)Mo(N), [(DPPN3N)Mo(NH)]+, and [(DPPN3N)Mo(NH3)]+, were also prepared. Efforts toward N2 reduction using (DPPN3N)Mo(N) as the (pre)catalyst produced 2.53 ± 0.35 equiv of NH3 making it formally catalytic.
In 2018, Wickramasinghe and Schrock again endeavored to maintain catalyst efficiency by limiting Mo–Namide protonation or dissociation (Figure 20). In this effort, they synthesized a calix[6]azacryptand ligand (CAC(OMe)3) that tethered the three HIPT triamidoamine ligand arms together.135 The X-ray structure of the nitride complex, (CAC(OMe)3)Mo(N), confirmed that the three-amido arms were locked in the desired configuration. Unfortunately, this system was not active for catalytic N2 fixation, producing only 1.28 equiv of NH3 using [Ph2NH2]OTf (96 equiv) and KC8 (84 equiv). The yield under the standard conditions used for (HIPTN3N)Mo were not reported.
Figure 20.

A calix[6]azacryptand ligand designed by the Schrock group aimed at protecting the reactive Mo active site for use in N2RR.135
3.6. Schrock Work on W, Cr, and V
The Schrock group explored the (HIPTN3N)M platforms (M = W, Cr, and V) for catalytic N2RR, but without success.136–138 In the case of tungsten, the preparative chemistry was found to be very similar to that for Mo (vide supra), with many candidate intermediates of N2RR being readily synthesized (Figure 21). However, attempts to demonstrate catalytic nitrogen fixation resulted in only stoichiometric yields of NH3 (1.3–1.5 equiv). The authors attributed the inability of the (HIPTN3N)W system to serve as an N2RR catalyst to a problematic redox couple for the ammonia adduct: [(HIPTN3N)W(NH3)]+/0.136 It is much harder to reduce this species than its Mo analogue (−2.06 V vs Fc+/0 compared to −1.63 V vs Fc+/0).100 Furthermore, chemical reduction does not lead to the neutral ammonia or N2 adduct hinting at undesired side reactivity.84
Figure 21.

Synthesis of candidate NxHy intermediates on (HIPTN3N) W relevant to N2 fixation. Note: this system produces only stoichiometric yields of NH3 (1.3−1.5 equiv).136
In the case of Cr, (HIPTN3N) complexes of both CrIII and CrII proved unable to bind N2. This contrasts directly with the behavior of the Mo and W complexes, which readily form dinitrogen complexes at these redox states, but is consistent with the lack of known CrII or CrIII dinitrogen complexes. The authors speculate that the relatively slow binding of CO to the HIPTN3N-complexes of CrII and CrIII compared to that observed for Mo and W could be due to the high-spin nature of the Cr centers. The nitride complex, (HIPTN3N)Cr(N), was prepared via thermolysis of the corresponding azide complex (Figure 22). Reductive protonation of this species under their standard catalytic conditions led to the formation of 0.8 equiv of NH3. This result suggests that downstream reduction steps are viable for (HIPTN3N)Cr, but presumably the lack of N2 uptake short-circuits viable catalysis.137
Figure 22.

Synthesis of (HIPTN3N)Cr(N) and (HIPTN3N)V(NH) and their reductive protonation to give 0.8 and 0.76 equiv of NH3, respectively. These results demonstrate that catalytic N2RR is not accessible with these species, but downstream functionalization reactions are productive.137,138
To compare V, the complex (HIPTN3N)V(THF) was prepared. Dinitrogen binding was only observed upon reduction to the anionic state, [(HIPTN3N)V(N2)]−, which required the use of KC8 (as opposed to Cp*2Cr). Attempts to use the latter as a precatalyst for nitrogen fixation under the standard Schrock-type conditions were unsuccessful (0.2 equiv of NH3). The (HIPTN3N)V(NH) complex could be prepared either via reaction of the V(THF) species with methylaziridine (Figure 22) or via reaction of the V(NH3) with base and oxidant. Its use as a precatalyst led to the formation of 0.78 equiv of NH3 akin to results obtained with (HIPTN3N)Cr(N) discussed above. This again points to the challenge of N2-uptake, at least using the original catalytic conditions with Cp*2Cr as the reductant.138 A computational study of this system posited several reasons to explain its inefficiency. In particular, the study pointed to the lower basicity of the various N2-fixation intermediates which might lead to increased preference for ligand protonation, the lesser thermodynamic driving force for N–N bond cleavage, and the persistent challenge of exchanging NH3 for N2.139
3.7. Third Generation Schrock Systems (Tridentate Platforms)
Recently, Schrock and co-workers have exploited a geometrically distinct, diamido(pyridine) pincer framework for Mo catalyzed N2 fixation (Figure 22).140 In 2017, they reported that the diamido(pyridine) ligand, [2,6-(ArNCH2)2NC5H3]2− (Ar2N3, Ar = 2,6-diisopropylphenyl), can be used to prepare (Ar2N3)Mo(N)(OtBu). Reaction of this nitride with 108 equiv of [Ph2NH2]OTf and 54 equiv of Cp*2Co (conditions discovered in the context of Fe-catalyzed N2RR, (vide infra) gave 7.9 equiv of NH3. By contrast, [Ar2N3]Mo(N)(OC6F5) was not competent for catalysis (1.3 equiv of NH3 under same conditions). The authors suggested that the tBuO fragment in the more active species might be more readily converted to a Mo–OH or Mo(O) unit, which is the catalytically active species.
A series of stoichiometric experiments were performed to shed light on the mechanism by which (Ar2N3)Mo(N)(OtBu) facilitates N2RR (Figure 24). Reaction of this species with the relevant acid, [H2NPh2]OTf, was found to protonate a ligand Mo-N amido linkage. By contrast, use of the weaker/bulkier (and catalytically ineffective) acid, [LutH]OTf, was found to provide an adduct species via a hydrogen-bond to the nitride (vide supra).130 Treatment of either of these species with Cp2Co resulted in the reformation of (Ar2N3)Mo(N)(OtBu), presumably via the loss of a half-equivalent of H2, though this was not confirmed. The authors speculated that this reaction pointed to the possibility of PCET reactivity during N2RR, but this has not been confirmed.
Figure 24.

Reaction of (Ar2N3)Mo(N)(OtBu) with [H2NPh2]+ results in protonation of a Mo-Nanilido arm whose product is shown to the right and use of [LutH]+ results in hydrogen-bond formation with the nitride shown on the left. Following reduction, each of these complexes releases H2 to provide the starting material shown in the center.130
4. NISHIBAYASHI’S LOW-VALENT Mo-PHOSPHINE SYSTEMS
4.1. Description of the Original System and Key Findings
In 2010, Nishibayashi and co-workers introduced a dinuclear molybdenum system as an N2 fixation (pre)catalyst (Figure 25).51 Specifically, a PNP-based (PNP = 2,6-bis(di-tert-butyl-phosphinomethyl)pyridine) complex (PNP)MoCl3 was prepared and reduced to the corresponding dinitrogen Mo(0) complex, [(PNP)Mo(N2)2]2(μ-N2), through reaction with NaHg amalgam (terminal υNN= 1936 cm−1). Use of adamantly-or isopropyl-substitutents in lieu of the tert-butyl groups did not provide the analogous Mo(0) complexes, highlighting the critical role of P-substituent. In a trial experiment, N2 reduction was assayed using [LutH]OTf (96 equiv) and Cp2Co (72 equiv) as proton and electron sources, in toluene at room temperature. Notably, a solution of the reductant was added slowly over the course of the reaction by syringe pump to prevent rapid recombination of electron and proton sources. Altogether, this mixture produced 11.8 equiv of NH3 (6.9 equiv of per Mo); using larger quantities of acid and reductant (266 equiv and 288 equiv, respectively) produced 23.2 equiv of NH3 (11.6 equiv per Mo center). Similar activity was found using Cp*2Cr, though use of less reducing Cp2Cr produced no NH3. The optimal acid was found to be [LutH]OTf, with Cl− and BArF4− counteranions providing significantly less NH3 (0.7 and 2.7 equiv, respectively). Other Mo complexes, such as cis-Mo(Me2Ph)4(N2) and trans-Mo(N2)(dppe)2 (dppe = 1,2-bis(diphenylphosphino)-ethane), give only stoichiometric quantities of NH3. To probe catalytic intermediates, [(PNP)Mo(N2)2]2(μ-N2) was treated with [HOEt2]BF4 in pyridine, providing the cationic hydrazido adduct, [(PNP)Mo(N-NH2)(F)(pyr)]BF4 (pyr = pyridine, Figure 25). Early on, the authors posited that this reaction proceeds through a mononuclear Mo complex, invoking a series of six stepwise proton and electron transfers, though this hypothesis was later subjected to refinement (vide infra). Notwithstanding, in 2011, this represented the most active catalytic method of accessing NH3 directly from N2 using soluble acid and reducing equivalents with a molecular species.
Figure 25.

Nishibayashi’s first generation, [(PNP)Mo(N2)2]2}(μ-N2) N2RR catalyst. In these reactions, Cp2Co was added via syringe pump and the acid counterion had a marked effect on the performance. Protonation of [(PNP)Mo(N2)2]2}(μ-N2) using [HOEt2]BF4 provided a hydrazido(2−) Mo complex.51
Further studies were performed using phosphine adducts of the aforementioned PNP-pincer complexes.141 In a follow-up contribution, Arashiba et al. showed that reduction of the corresponding (PNP)MoCl3 precursors with NaHg amalgam in the presence of PMe2R (R = Me or Ph) provided (PNP)Mo(N2)2(PMe2R) (Figure 26). Initial protonation studies using excess sulfuric acid (H2SO4) revealed that these derivatives produced 1.38 and 0.85 equiv of NH3, respectively. Under catalytic conditions, the PMe2Ph derivative produced only a small amount of NH3 (0.2 equiv).
Figure 26.

Protonation studies of six-coordinate (PNP)Mo(N2)2(PMe2R) complexes gave 1.38 (R = Me) and 0.85 (R = Ph) equiv of NH3.141
Examination of phosphine R group also provided information into the nature of PNP coordination mode (Figure 27).141 In this way, a variety of unsymmetrical PNP-based ligands were prepared, featuring R = tBu groups positioned on one phosphine arm and one of R′ = Ad, tBu, iPr, or Cy on the other. In all cases, the corresponding (PNP)MoCl3 precursors were accessed and reduced under standard NaHg amalgam conditions, giving binuclear [Mo] complexes having one of two geometries. In the first, the two terminal N2 units remain characteristically trans-disposed (R = tBu, R′ = Ad or Ph) and in the second, the two terminal N2 groups are mixed; both -trans and -cis geometries were found; both geometries feature a bridging N2 unit. Subjecting the four of these catalysts to a solution of [LutH]OTf (96 equiv) and addition of Cp2Co (72 equiv) via syringe pump over 1 h produces between 1 and 14 equiv of NH3/Mo center—the greatest yields were observed for the catalyst containing R′ = tBu or Ad groups. Naturally, the phosphine R groups have a profound influence on downstream Mo(NxHy) adducts, causing either enhanced or decreased activity. This work highlights the subtle nuances of ligand design and shows that larger R groups are more adept at protecting the reactive Mo core.
Figure 27.

Modification of ligand phosphine substituent gave different Mo2(μ-N2) isomers. For R = tBu, R′ = Ad or Ph, trans-/trans- is observed, while for R = tBu, R′ = iPr or Cy, trans-/cis-is observed. The R = tBu, R′ = Ad substituted phosphine provides the highest yield of NH3 (14 equiv) during N2RR.141
In an effort to develop catalytic N2RR using N2 and H2 under mild reaction conditions, the Nishibayashi group studied the reactivity of high-valent molybdenum complexes with hydride reagents to generate on-cycle catalytic dinitrogen complexes (Figure 28).142 For this reason, Arashiba et al. showed that reduction of [PNP]MoCl3 (R = tBu) with KC8 produces a μ-N2 bridged dimolybdenum precursor, {[PNP]Mo(Cl)2]2}(μ-N2) (Figure 27). Treatment of this precursor with KHBEt3 results in formation of the dinitrogen complex, {[PNP]Mo(N2)2]2}(μ-N2) as well as 2 equiv of H2. Reaction of {[PNP]Mo(Cl)2]2}(μ-N2) with excess H2SO4 provides no reduced nitrogen-based products (cf. 0.6 equiv of NH3, 0.06 equiv of N2H4 for {[PNP]Mo(N2)2]2}(μ-N2)). This reaction demonstrates that high-valent [Mo]-halide bonds can be cleaved by way of hydride reagents, allowing for access to on-path [Mo]-N2 complexes by way of H2 elimination.
Figure 28.

On-cycle (PNP)Mo(N2) complexes can be generated through H2 evolution from their respective halide precursors.142
In 2013, a modified ligand was reported where the ligand pincer phosphine donors were changed to arsine, a donor featuring a larger element of poorer σ-donating/π-accepting ability (Figure 29).143 The ANA pincer ligand was prepared and coordinated to molybdenum; reduction of this precursor provided a cis-/trans-N2 dimolybdenum complex having υNN = 1904 cm−1 for the μ-N2 ligand (cf. υNN = 1890 cm−1 for the PNP analogue, {[PNP]Mo(N2)2]2}(μ-N2) R = tBu). In the presence of PMe3, a phosphine complex, [ANA]Mo(N2)2PMe3 was isolated. Despite producing similar amounts of NH3 (cf. the PNP system) when exposed to excess H2SO4 (ca. 0.6 equiv of NH3), catalysis was nonproductive using {[ANA]Mo(N2)2]2}-(μ-N2) R = tBu) and varying equivalents of Cp2Co and [LutH]OTf, producing only 2 equiv of NH3. This data point contrasts with the PNP-based system where 12 equiv of NH3 was observed under similar conditions. The previously formed unsymmetrical phosphine complexes that contained trans-/cis-N2 units were also ineffective for catalysis (R = tBu, R′ = iPr (3 equiv of NH3), Cy (1 equiv of NH3)).
Figure 29.

Arsine analogues of the PNP class of ligands have been prepared, though Mo complexes of these ligands are not active for catalytic N2RR under the conditions shown.143
4.2. Mechanistic Conclusions That Can Be Drawn and Uncertainties
Next, the effect of substitution at the 4-position of such PNP ligands having PtBu2 groups was explored (Figure 30).82 The installation of both electron-donating and -withdrawing groups was carried out, with the hypothesis that electron-donating groups would increase N2 activation and thus accelerate Nβ protonation. From this design perspective, five candidate [(4-XPNP)Mo(N2)2]2(μ-N2) complexes were prepared having X = Ph, Me3Si, tBu, Me, and OMe groups at the 4-position. Structurally, no distinct differences were observed from the parent (X = H) system. As judged by IR spectroscopy, substitution of more electron-donating groups provides a more activated N2 entity (υNN = 1932 cm−1 for X = OMe vs 1944 cm−1 for X = H). All of these species are catalytically active for NH3 formation, resulting in 28 and 31 equiv of NH3 for the tBu- and Me-substituted PNP ligands, respectively, using 216 equiv of Cp2Co and 288 equiv of [LutH]OTf. The greatest amount of NH3 is formed when R = OMe (34 equiv) under the same conditions.
Figure 30.

Ligand modifications have been targeted by changing the ligand 4-X group. The reaction scheme depicts the first three steps relevant to catalytic N2RR as calculated by DFT (1) Nβ protonation, (2) N2 dissociation, and (3) triflate coordination.82
The study of N2 reduction catalysis using density functional theory provided insight into the high barrier by which initial Nβ protonation occurs (Figure 30). This process involves three steps, (1) protonation to give a Mo diazenido, Mo(NNH), followed by (2) displacement of the trans-N2 ligand, and (3) attack of a triflate (OTF−) unit—the final two steps offsetting the high endothermicity of the first. In terms of phosphine R group, installation of tBu best stabilizes the protonated diazenido intermediate and was found to have the lowest energy pathway. Substitution of an electron-donating group at the PNP 4-position is also thought to accelerate Nβ protonation due to N2 activation.
To test the importance of reductant concentration on the catalytic behavior of the 4-OMe catalyst, Cp2Co was added over a longer period of time (5 h vs 1 h) and gave a greater amount of NH3 (36 equiv), suggesting a longer catalyst lifetime. Increasing the amount of acid ([LutH]OTf: 480 equiv) and reductant (Cp2Co: 360 equiv) provides 52 equiv of NH3. In terms of reductant and acid optimization, it was found that Cp*2Cr has a negligible effect on reaction outcome, while use of [4-ClLutH]OTf (pKa = 5.46 in H2O) provides only a stoichiometric amount of NH3. Use of a weaker acid, [4-CH3LutH]OTf (pKa = 7.55 in H2O) gives only 10 equiv of NH3. Given the highly active nature of the 4-OMe catalyst, mechanistic studies comparing the 4-OMePNP catalyst and the parent PNP catalyst were carried out. From these, it was determined that when X = H the reaction is first order in Mo, while when X = OMe the reaction is zeroth order in Mo, suggesting different rate-determining steps (the reaction order with respect to reductant was found to be first order in both cases). At present, it is unclear how such a mechanism can be zeroth order in Mo. Dihydrogen yields were also measured, showing that X = OMe not only produces the highest quantity of NH3, but also the lowest quantity of H2. Performing the reaction under a partial H2 atmosphere also results in lower activity for NH3 formation for the 4-OMe catalyst (96:4 N2/H2 — 23 equiv of NH3), indicating that H2 inhibits NH3 formation—a conclusion that has been drawn for other synthetic nitrogenases (N2ases).107 Overall, substitution at the 4-position of the PNP ligand was found to have a marked effect on N2 reduction selectivity, allowing for rate enhancement of the first protonation event and suppression of molecular hydrogen formation.
In 2014, Tanaka et al. explored the use of mononuclear molybdenum nitrides for catalytic N2RR (Figure 31).83 Treatment of Mo(Cl)3(THF)3 with Me3SiN3 at 50 °C for 1 h, followed by addition of the PNP ligand, gave (PNP)Mo(N)-(Cl)2, which served as a synthetic precursor to related species including (PNP)Mo(N)(Cl), [(PNP)Mo(N)(Cl)]OTf, and [(PNP)Mo(NH)(pyr)(Cl)]OTf (pyr = pyridine). Of these compounds, only (PNP)Mo(N)(Cl) and the oxidized product, [(PNP)Mo(N)(Cl)]OTf, exhibited catalytic efficacy (6.6 and 7.1 equiv of NH3).
Figure 31.

Mononuclear PNP-ligated molybdenum nitrides are active for catalytic N2 fixation using [LutH]+ (48 equiv) and Cp2Co (36 equiv).83
In view of this result, DFT calculations were performed to assess the relevancy of [(PNP)Mo(N)]OTf or related complexes to the observed catalysis83 (Figure 32). The initial pathway was calculated to be similar to that outlined above (Figure 30), with a dinuclear molybdenum complex being protonated initially, and then breaking up upon a third (reductive) protonation step to liberate NH3 and a terminal nitride product, [(PNP)Mo(N)]OTf. The latter is a competent precatalyst, and, following a series of reductive protonation steps, (PNP)Mo(NH3) is generated and combines with (PNP)Mo(N2)3 to regenerate a μ-bridged N2 complex; NH3 can be replaced by N2 in this species to restart the catalytic cycle. (PNP)Mo(N2)3 itself is not thought to be catalytically competent. Instead, the authors suggest that the terminal N2 ligand that undergoes monoprotonation (the first step of the cycle) must be that of a dimolybdenum complex, with electron transfer from an adjacent Mo center preactivating the Mo(N2) fragment. This is an interesting hypothesis that provides an opportunity for future experimental studies.
Figure 32.

Complete calculated N2RR cycle for Nishibayashi’s dinuclear Mo2 complexes. [Mo] = (PNP)Mo(N2)3.83
Redox-active subunits have also been incorporated within the PNP ligand framework by the Nishibayashi group to canvass possible advantages of placing a redox relay close to the catalytic active site (Figure 33).144 The direct incorporation of metallocene subunits such as ferrocene and ruthenocene, in the trans-/trans-N2 (PNP)dimolybdenum framework, was found to have a negligible effect on the degree of N2 activation as judged by IR spectroscopy; derivatives featuring ethylene or phenylene linkers to the metallocene subunits also showed similar activation. Cyclic voltammetry revealed marked shifts in the E1/2 associated with the Fe(II/III) couple compared to free ferrocene, consistent with electronic communication between Fe and Mo; spectroelectrochemistry demonstrated low energy transitions in the near-IR at around 800–1800 nm, consistent with Fe-to-Mo charge transfer (MMCT). The degree of electronic communication was weaker when a linking (ethylene or phenylene) group was present.
Figure 33.

A survey of Nishibayashi’s Mo complexes for catalytic N2RR Fc = ferrocene, EtFc = ethylferrocene.83,84,144–146
In terms of catalysis, it was found that the ferrocene-appended catalyst gave 37 equiv of NH3, compared with 23 equiv of NH3 under the same conditions where X = H (Figure 33). Control experiments employing the X = H parent catalyst in the presence of exogenously added ferrocene gave only 21 equiv of NH3, suggesting that inclusion of the redox-active subunit may have some benefit. From a DFT study, one step that was suggested to be dramatically affected by inclusion of a metallocene within the catalyst framework concerns reduction of the hydrazidium, [Mo(NNH3)]+, complex, enabling release NH3 and Mo(N). Azaferrocene-based PNP Mo complexes have also been employed to survey their efficacy for catalytic N2 fixation, but they were not successful under the conditions explored.145
PNN-based Mo complexes have also been studied by Nishibayashi and co-workers, especially for derivatives featuring the PNN ligand 2-(di-ferf-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine (Figure 32).146 (PNN)Mo(N2)2(PMe2R) (R = Ph or Me) derivatives were accessed by reduction of the (PNN)MoCl3 precursor and treatment with PMe2R. The PNN ligand was observed to be a stronger donor ligand than the corresponding PNP ligand, as judged by N2 stretching frequencies (1877 cm−1 vs 1915 cm−1 for the PNP ligand). However, treatment of these complexes, or the corresponding nitride derivatives, with excesses of reductants and acids (e.g., 36 equiv of Cp2Co, 48 equiv of [LutH]OTf failed to reveal catalytic activity (Figure 33)). Other PNP ligands, such as N,N′-bis(di-ferf-butyl-phosphino)-2,6-diaminopyridine and 2,6-bis(diadamantyl-phosphinomethyl)pyridine, have also been used to access terminal molybdenum nitrides of the type [(PNP)Mo(N)(Cl)]BArF4; however, these adducts produced little NH3 (1.3 and 1.8 equiv., respectively) when exposed to 36 equiv of Cp2Co and 48 equiv of [LutH]OTf.147
PPP-pincer ligands were next targeted as promising candidates for catalysis (Figure 33).84 Despite difficulties in synthesizing the N2-bridged dimolybdenum PPP-pincer complex, the terminal nitrides, (PPP)Mo(N)(Cl) and [(PPP)Mo(N)(Cl)]BArF4—akin to those prepared in the PNP systems83—were accessible. Exposure of the cationic Mo nitride to [LutH]OTf (48 equiv) and 36 equiv of Cp*2Co produced 6.1 equiv of NH3, whereas Cp*2Cr and Cp2Co were far less effective reductants. However, the use of [ColH]OTf (ColH = 2,4,6-trimethylpyridinium) as the acid produced 9.6 equiv of NH3, and the addition of much higher amounts of reductant and acid (540 equiv of Cp*2Co and 720 equiv of [ColH]OTf), produced as many as 63 equiv of NH3.
4.3. Catalyst Redesign and Keys to Achieving Higher Turnover Numbers
In 2017, Nishibayashi and co-workers proposed that reactive terminal (PNP)Mo(N) could be formed via cleavage of a bridging dinitrogen unit, thus circumventing the formation of early protonation intermediates such diazenido, hydrazido, and hydrazidium (Figure 34).81 Use of (PNP)Mo(I)3 as a precursor afforded access to (PNP)Mo(N)(I) following reduction by Cp*2Co. By contrast, reduction of the chloride-containing species, (PNP)MoCl3, with NaHg amalgam produced [(PNP)Mo(N2)2]2(μ-N2), thus highlighting the importance of the Mo-halide starting material and reductant. Using (PNP)Mo(I)3 as a (pre)catalyst, treatment with standard loadings of 36 equiv of Cp*2Co and 48 equiv of [ColH]OTf gave 10.9 equiv of NH3, while (PNP)Mo(N)(I) gave 12.2 equiv of NH3. Under similar conditions, [(PNP)Mo(N2)2]2(μ-N2) gave 7.1 equiv of NH3. [LutH]OTf was not applicable as an acid source when using (PNP)Mo(I)3 as the (pre)catalyst.
Figure 34.

Proposed pathway for reductive cleavage of N2 to form a terminal nitride, (PNP)Mo(N)(I), of relevance to catalytic N2RR. The observed reactivity profile is I (50.7 equiv of NH3) > Br (40.5 equiv of NH3) > Cl (24.4 equiv of NH3).81
Much higher loadings established the viability of much higher, and truly impressive turnover numbers. For example, the use of 2160 equiv of Cp*2Co and 2880 equiv of [ColH]OTf gave the highest amount of NH3 (415 equiv), more than 35 times greater yield than achieved by [(PNP)Mo(N2)2]2(μ-N2) under similar acid and reductant loadings. A lower amount of ammonia was produced using (PNP)Mo(Br)3 (40.5 equiv of NH3) or (PNP)Mo(Cl)3 (24.4 equiv of NH3) when compared with (PNP)Mo(I)3 (50.7 equiv of NH3; [catalyst] = 0.002 mmol). In all cases, the catalysis showed little competing dihydrogen formation.
The proposed catalytic cycle, which warrants additional experimental support, invokes reduction of (PNP)Mo(X)3 to give [(PNP)]Mo(X)]2(μ-N2), from which (PNP)Mo(N)(X) is then formed (Figure 34). Following a series of stepwise reductive protonation steps (PNP)Mo(NH3)(X) is generated, which then loses NH3 and (re)binds N2 to restart the catalytic cycle. This cycle is interesting to consider by comparison to a distal (or alternating) pathway for N2 reduction via discrete one-electron/one-proton steps, as it features fewer protonated intermediates. Moreover, this work points to the viability of halide precatalysts for generating on path intermediates.
The Nishibayashi group has more recently reported catalytic studies with a variety of molybdenum triiodide complexes containing nearly all of the PNP permutations discussed above, (RPNP)Mo(I)3, including different phosphine substituents (R = H, R′ = tBu, Ad, iPr, or Ph) or different groups at the 4-position of the central ring of the ligand (X = Ph, Me, OMe, Fc, or Rc, R′ = tBu).148 Interestingly, and in contrast to the Mo2(μ-N2) complexes discussed previously, the highest quantity of NH3 was obtained when X = Ph. To rationalize this observation, it was proposed that (PNP)Mo(I)3 and [(PNP)Mo(N2)2]2(μ-N2) operate via different rate-determining steps (RDS). In the molybdenum iodide system, electron-withdrawing groups are thought to promote an RDS involving reduction of a cationic “Mo(NHy)” fragment, while in the Mo2(μ-N2) system, electron-donating groups are thought to promote an RDS involving Nβ protonation.
PCP-type pincer ligands, comprised of both N-heterocyclic carbene and diphosphine donors, have also been explored by the Nishibayashi group in an effort to evaluate N2RR catalyst performance (Figure 35).149 Two such ligands: 1,3-bis(2-(di-tert-butylphosphino)methyl)benzimidazol-2-ylidene (BImPCP) and 1,3-bis(2-(di-tert-butylphosphino)ethyl)imidazol-2-ylidene (ImPCP) were prepared, from which (rPCp)moCl3 (R = Im or BIm) precursors were accessed. Reduction using NaHg amalgam provided the μ-N2 molybdenum complexes [(RPCP)Mo(N2)2]2(μ-N2), for which the IR spectrum provides signals at 1978 cm−1 and 1911 cm−1. The catalytic utility of these compounds was next interrogated using a mixture of [LutH]OTf (96 equiv) and metallocene (72 equiv); the metallocene was added in toluene via syringe pump. After optimization, it was found that pairing larger amounts of reductant (Cp*2Cr: 1440 equiv) and acid ([LutH]OTf: 1920 equiv) with the 5,6-dimethylbenzimidazol-2-ylidene catalyst afforded the maximum (up to 115 equiv) amount of ammonia.
Figure 35.

PCP-type pincer ligands featuring N-heterocyclic carbene donors are useful for N2RR in the presence of [ColH]OTf (96 equiv) and reductant (72 equiv); Cp*2Cr was found to be most effective for both classes of catalyst.149
In an effort to use H2O as a proton source, Nishibayashi and co-workers have explored the combination of coupling wazter oxidation with nitrogen fixation (Figure 36).150 In a model study, water oxidation was performed using ceric ammonium nitrate ([Ce(NO3)6](NH4)2) and a Ru complex, Ru(bda)-(isoq)2 (bda = 2,2′-bipyridine-6,6′-dicarboxylate, isoq = isoquinoline) to generate O2 and acid. Addition of lutidine formed [LutH]+, which upon combination with Cp2Co in the presence of [(PNP)Mo(N2)2]2(μ-N2), generated 8.5 equiv of NH3; this compares well with 9.7 equiv of NH3 that was observed when purified [LutH]OTf was used. Using larger amounts of reagent resulted in up to 17.1 equiv of NH3. Visible light-driven water oxidation was also studied using Na2S2O8 as a sacrificial oxidizing reagent and [Ru(bpy)3]OTf (bpy = 2,2′-bipyridine) as the photosensitizer. Using the same catalyst, 6.0 equiv of NH3 was observed following sequestration of the solvated HOTf with lutidine; it was proposed that sulfate inhibits the formation of NH3 from N2.
Figure 36.

Efforts toward using H2O as a proton source in N2RR. Water oxidation was performed using [Ru(bpy)3]OTf2 as a photooxidant, peroxydisulfate as a sacrificial reductant, and a Ru complex, [Ru(bda)-(isoq)2] (bda = 2,2′-bipyridine-6,6′-dicarboxylate, isoq = isoquinoline), as the water oxidation catalyst. The generated acid was trapped using lutidine to give [LutH]+, which enabled N2RR catalysis by (PNP)Mo.150
4.4. New Conditions Lead to Remarkable Rate and Efficiency
Very recently, Nishibayashi and co-workers have exploited SmI2 as a reductant compatible with polar, protic H+ donors (e.g., alcohols or water) (Figure 37).79 Related approaches have been gaining in popularity for the reduction of organic molecule substrates owing to the dramatic O–H bond weakening that occurs upon coordination of the alcohol or water to the Sm(II) center.151–154 This is an exciting result that opens up a new approach for the study of N2RR catalysts that shows great promise. Indeed, the selectivity for NH3 and total turnover number demonstrated in this recent report are remarkable. Using the aforementioned (BMimPCP)Mo(Cl)3 precatalyst, reaction of N2 with SmI2 and ethylene glycol generates NH3 at a turnover frequency (TOF) of 117 min−1. Water can also be used in this reaction; treatment of N2 with a [Mo]-precatalyst, SmI2 (14 400 equiv), and H2O (14 400 equiv) in THF at room temperature for 4 h gives 4350 equiv of NH3 (91%).
Figure 37.

Remarkable efficiencies were achieved using a Sm/alcohol mixture for N2RR. This reaction is proposed to occur via PCET. Use of H2O (14400 equiv) and SmI2 (14 400 equiv). gave ca. 4350 equiv of NH3.79
5. ACHIEVING NITROGEN FIXATION CATALYSIS AT Fe
For decades, Mo had played center stage in the modeling of biological nitrogenases, owing in part to its early discovery within the MoFe variant.48 The initially unexpected presence of Mo, the sensitivity of nitrogenase performance to alterations near Mo, and the early success in developing model systems, led to a strong emphasis on this metal as being the site of N2RR. Then, in 1986, it was discovered that there was a vanadium-dependent nitrogenase, VFe,155 and subsequently in 1988, the existence of an all Fe biological nitrogenase was disclosed.156 Subsequent study of the biological nitrogenases has led to the conclusion that they are likely structurally (and potentially mechanistically) similar.11,36,157–159 In particular, recent spectroscopic and crystallographic evidence has begun to emerge in both the MoFe and VFe nitrogenases that the site of N2-binding and functionalization is likely to be at an Fe site. In particular, both the Rees and Einsle groups have obtained structures that demonstrate sulfide-loss and ligand binding between Fe2 and Fe6 (Figure 38).160,161 Meanwhile, Hoffman, Seefeldt, and co-workers have established that reductive elimination of bridging iron hydrides may be essential to N2 binding in all three biological nitrogenases.158,159,162
Figure 38.

Structure of a recently reported, proposed intermediate of VFe-nitrogenase, featuring removal of a bridging sulfide as SH− and the identification of a μ2-bridging light atom in its place. The light atom (X) has been hypothesized to be the N atom of an imido ligand.161 Further studies are needed to validate this assignment.
The discovery of new nitrogenases expanded the focus not only from Mo to other metals, but also from NH3 to another N2 fixation product, N2H4. In 1991, Dilworth and co-workers discovered that the VFe-nitrogenase, unlike MoFe-nitrogenase, released N2H4 as a product of N2 reduction, particularly at higher temperature.163 In this initial publication, they proposed that VFe-nitrogenase can access two routes to fixed nitrogen products from a common hydrazido(2−) ligand. The first is a further Nβ-functionalization (forming M(NNH3)) and subsequent N-N bond cleavage to release NH3, in a Chatt-like mechanism, while the other pathway would involve double Nα-functionalization leading to M(NHNH2) then M(NH2NH2) and ultimately N2H4 release (Figure 39). This proposal disappeared relatively quickly from the literature (though it has been recently revisited by Peters and co-workers, and termed a “hybrid” mechanism, vide infra) and was replaced by a mechanistic scenario typically referred to as the alternating mechanism. In this latter mechanism, the second functionalization occurs at Nα rather than Nβ, leading to a characteristic diazene (HNNH) intermediate. Indeed, this mechanism was popularized by the Hoffman group after the observation that diazene (and related derivatives) were substrates of FeMo-nitrogenase and led to the formation of a cofactor-bound intermediate that could be detected by ENDOR spectroscopy.164,165 They hypothesized that further reduction of diazene to hydrazine would occur via alternating Nβ- and Nα-functionalization. In a general sense, such a hydrazine-bound intermediate could then be released, protonated to release N2H5+, or undergo protolytic cleavage to form NH3 and M(NH2). Still, to date, no fixed NxHy intermediate has been definitively assigned in a biological nitrogenase and mechanistic uncertainty remains.
Figure 39.

(Left) Outer cycle depicts the distal or Chatt-cycle for N2RR.30 Central intermediate (highlighted in blue) represents a “hybrid” mechanism for N2RR, which may be relevant in both biological163 and synthetic109,114 nitrogenases; (right) the alternating mechanism for the reduction of N2 to NH3 or N2H4.164,165
In response to the growing evidence for the biological importance of Fe and mechanistic diversity in nitrogen fixation, inorganic chemists have articulated both multi-Fe166,167 and single-Fe168 hypotheses for nitrogen fixation, as well as approaches to interrogate mechanisms for nitrogen fixation that account for both NH3 and N2H4 formation.25,109 To support these hypotheses, researchers have developed a number of systems to achieve N2-binding and functionalization, as well as the synthesis and interconversion of potential late-stage intermediates.26,29,169–171 However, the lack of early progress on both protonation reactions of Fe(N2) complexes that led to identifiable intermediates or the development of catalytic systems for N2-to-NH3 conversion that featured Fe, left a notable gap for the field until relatively recently.17
Since 2013, there has been a surge in the discovery of catalytically active systems (Figure 40)18,80,115,116,172–175 and the identification of protonated intermediates from Fe(N2) complexes.109,113,176,177 Indeed, many of the types of species and reactivity patterns of synthetic, homogeneous Mo model chemistry in the context of nitrogen fixation have now been demonstrated for Fe as well. This body of work adds chemical evidence to the growing weight of biological evidence that Fe is capable of N2-to-NH3 conversion when supplied with the appropriate H+/e− source and unravels the historical bias that Mo is somehow unique in its ability to mediate N2RR via cycles such as those shown in Figure 39.16,17
Figure 40.

Homogeneous Fe complexes reported to mediate catalytic N2-to-NH3 conversion; Dipp = 2,6-diisopropylphenyl.18,115,116,172,173,175,178–180 **(depe)2Fe(N2) (depe = diethylphosphinoethane) is instead selective for N2H4.174
5.1. Catalysis with a Trisphosphineborane Fe Species
In 2013, the Peters group reported that a tris-(isopropylphosphino(o-phenylene)borane iron complex, (P3B)Fe, was capable of catalytic nitrogen fixation.18 This atrane system was initially targeted for catalytic nitrogen fixation due to its ability to stabilize a variety of nitrogen fixation intermediates. The P3B ligand reacts with FeBr2 in the presence of Fe powder to form (P3B)Fe(Br), a valuable synthon.112 This complex could then be reduced by stoichiometric sodium naphthalenide to give the Fe(N2) complex (νNN = 2011 cm−1, Figure 41). Reaction with excess sodium naphthalenide led to formation of the anionic Fe(N2) complex (νNN = 1877 cm−1), and encapsulation of the sodium with 12-crown-4 afforded a separated ion pair (νNN = 1918 cm−1). In addition to activating π-accepting ligands, (P3B)Fe was capable of stabilizing π-donating imido ligands (i.e., (P3B)Fe(NR)) formed via reaction of (P3B)Fe(N2) with organoazides.112,113
Figure 41.

Interconversion of different nitrogen fixation intermediates on the (P3B)Fe platform.112,113,176,182
The functionalization of [(P3B)Fe(N2)]− with electrophiles also led to the formation of Fe–N multiply bonded species.181 In particular, reaction with TMSCl led to formation of the silyldiazenido, the solid-state structure of which reveals significant lengthening of the Fe–B interaction and the N–N bond and a contracted Fe–N bond. This flexibility of the Fe–B interaction in response to the apical ligand and redox state of the Fe has emerged as a key factor in stabilizing intermediates throughout the N2 fixation process (vide infra). The anionic silyldiazenido could be readily synthesized via reduction of this complex with Na/Hg. Likewise, difunctionalization of the N2 ligand could be achieved by reaction of (P3B)Fe(Br) with excess Na/Hg and the disilylating reagent, 1,2-bis(chlorodimethyl)-silylethane. DFT calculations performed on this disilylhydrazido(2−) complex suggested that it has structural features similar to (P3B)Fe(NR) species. As expected, calculations of the bond orders indicated that across the series: (P3B)Fe(N2) → [(P3B)Fe(N2)]− → (P3B)Fe(NNTMS) → [(P3B)Fe(NNTMS)]− → (P3B)Fe(NN(SiR3)2) the Fe–N bond order increased from ~0.5 to 2, with the N–N bond order concomitantly decreasing from ~2.6 to 1.2. Considering the silyl groups as proton surrogates, these results suggested that (P3B)Fe might support early nitrogen fixation intermediates.
Next, late nitrogen fixation intermediates were targeted for synthesis using the (P3B)Fe platform.182 Synthesis of the low-coordinate, cationic complex [(P3B)Fe]+ provided access to a number of species that would be candidate downstream intermediates of catalytic nitrogen fixation, including [(P3B)Fe(N2H4)]+, [(P3B)Fe(NH3)]+, and (P3B)Fe(NH2). Low temperature protonation of (P3B)Fe(NH2) with HBArF4 led to the quantitative formation of [(P3B)Fe(NH3)]+. Reductive N2-for-NH3 substitution was then established by addition of KC8 to [(P3B)Fe(NH3)]+, which provided (P3B)Fe(N2) quantitatively. These reactions demonstrated the final key steps of a hypothetical nitrogen-fixing cycle and suggested the possibility that HBArF4 and KC8 might be competent proton and electron sources to drive such a process, at least insofar as the productive cycle could compete with background hydrogen evolution.
In 2013, it was discovered that the reaction of [(P3B)Fe(N2)]− with 6 equiv of HBArF4 at low temperature, followed by warming and then adding 6 equiv of 1,8-bis(dimethylamino)naphthalene (Proton Sponge) resulted in the generation of ~35% [(P3B)Fe(NH3)]+, ~40% [(P3B)Fe]+, as well as several unidentified Fe-containing products.18 Low temperature reaction with acid followed by KC8 led to the regeneration of [(P3B)Fe(N2)]−, suggesting that the system might indeed be amenable to catalysis (Figure 41). After screening a variety of solvents, temperatures, reductants, and acids, it was found that catalytic yields of NH3 (7.0 ± 1 equiv of NH3 per Fe, 44 ± 6% based on H+ used) could be formed in diethyl ether at −78 °C using HBArF4 and KC8 as the proton and electron sources, respectively. This provided the first demonstration that Fe could catalyze N2-to-NH3 conversion. Worth underscoring is that it was surprising that HBArF4 and KC8 could provide an effective acid and reductant cocktail. It would have been reasonable to presume that essentially all of the acid would be reduced by KC8 to produce H2. While such a reaction does occur, at −78 °C in Et2O it is comparatively slow and the [(P3B)Fe(N2)]− catalyst is sufficiently competent making N2RR kinetically competitive.173
Additional mechanistic studies of this catalytic system were undertaken using kinetics and freeze-quench Müssbauer spectroscopy.173 The catalytic reaction was found to be first order in Fe but zeroth order in acid, suggesting that the rate-limiting step likely involved either a reduction step or a ligand exchange process. The initial rate of NH3 formation was found to be 1.2 ± 0.1 mol of NH3 mol of Fe1− min−1, which was among the fastest rates for a synthetic system, despite the fact that the reaction was carried out at −78 °C.79 Catalytic reactions with preadded NH3 demonstrated that the reaction was product-inhibited. This is perhaps not surprising given the slow kinetics of NH3 and N2 exchange observed in (HIPTN3N)Mo systems110 and points to a challenge for developing systems that can achieve substantially higher turnover numbers. Interestingly, (P3B)Fe was shown to catalyze both NH3 and H2 formation. This suggested that to achieve improved catalysis it would be necessary not only by limiting background reactivity between the acid and the reductant, but also to develop systems with improved intrinsic selectivity for NH3.
Addition of further substrate (HBArF4 and KC8) following a standard catalytic run with (P3B)Fe furnished additional NH3, suggesting that catalytically active “(P3B)Fe” material was still present. To explore this further, freeze-quench Müssbauer spectra of the catalytic reaction at various time points were used to evaluate the evolution of the catalyst speciation. Spectra at early times (~5 min) revealed the formation of a previously characterized hydride/borohydride complex, (P3μ-B-H)Fe(H)(L) (L = N2 or H2). These derivatives were originally accessed via the addition of H2 to (P3B)Fe(N2) (Figure 41).183 At later reaction times (~25 min), the original [(P3B)Fe(N2)− (pre)catalyst had reformed, consistent with the use of a modest excess of reductant. This result was in apparent contrast with initial results that had indicated (P3μ-B-H)Fe(H)(N2) was inactive for catalysis; indeed, it was presumed it might be a catalyst poison. Its in situ generation and subsequent consumption, regenerating a substantial quantity of the [(P3B)Fe(N2)]− (pre)catalyst, suggested it to instead be an off-path intermediate that ties-up the catalyst. Consistent with this idea, it was shown that the addition of a toluene cosolvent to help solubilize (P3μ-B-H)Fe(H)(N2) in Et2O at −78 °C significantly improved its catalytic activity (up to 5.6 ± 0.9 equiv of NH3 per Fe). This result supported that (P3μ-B-H)Fe(H)(N2) can be converted back into an on-cycle intermediate and is therefore an off-path resting state of the system (Figure 42).
Figure 42.

Reaction scheme showing synthetic conversions of the catalytic resting state, (P3μ-B-H)Fe(H)(N2), including an α-hydride elimination step that leads to release of a hydrazine surrogate following acidic workup.173,175,184
Understanding how the catalytic resting state, (P3μ-B-H)Fe(H)(N2) returns to an on-cycle intermediate is of interest (Figure 42). One demonstrated pathway is that the sequential reaction of (P3μ-B-H)Fe(H)(N2) with HBArF4 and KC8 in 3:1 Et2O/toluene at low temperature led to the reformation of [(P3B)Fe(N2)]− (presumably accompanied by H2 loss). It was also found that the reaction of (P3μ-B-H)Fe(H)(N2) with KC8 led to the formation of a dianionic species, [(P3μ-B-H)Fe(H)(N2)]2−, in which one phosphine donor is dissociated from the iron center. The reaction of this dianionic species with silyl electrophiles at low temperature led to the formation of a disilylhydrazido(2−) species, (P3μ-B-H)Fe(H)(NNSi2). Upon warming, the Fe–H group was observed to migrate to Nα giving a hydrazido(1−) ligand. The resulting hydrazido(1−) ligand could then be protolytically released as a hydrazine derivative by reaction with HBArF4 in the presence of tert-butylisocyanide. This result,184 along with those from other groups,185 suggests that hydrides may be productively incorporated into fixed-N products, in addition to being lost as H2.
It has recently been discovered that irradiation of (P3μ-B-H)Fe(H)(N2) with a Hg lamp for 10 min at −78 °C results in formation of (P3B)Fe(N2) along with (P3μ-B-H)Fe(H2)(H). Such a process can improve overall catalysis by photo inducing an off-path species, (P3μ-B-H)Fe(H)(N2) to return to an on-path species, (P3B)Fe(N2). Irradiation of catalytic runs led to an increased total turnover number (~+150%) for a given loading of acid and reductant using [P3BFe(N2)]− as the (pre)catalyst.175
In the original catalytic report,18 Peters and co-workers had noted that EPR spectra of the reaction of [(P3B)Fe(N2)]− with excess acid, in the absence of added reductant or base, led to a new and intense S = 1/2 EPR signature (~85% of the Fe present via spin-quantification). This new species that was tentatively ascribed to [(P3B)Fe(NNH2)]+, or possibly an isomer such as [(P3B)Fe(NHNH)]+. To elucidate the chemical structure of this EPR-active intermediate, pulse EPR and XAS spectroscopic studies were undertaken.113 Consistent with retention of the (P3B)Fe-core, pulse EPR techniques revealed couplings to three phosphorus nuclei and a single boron nucleus. Additionally, protons (one more strongly and one more weakly coupled) and two coupled nitrogen nuclei were also identified. These data were consistent with formulation as either a hydrazido(2−) or an asymmetrically coordinated diazene (HNNH) ligand. X-ray absorption spectroscopy (XAS) revealed a short Fe–N bond (1.64 Å), highly consistent with bond lengths previously observed for disilylhydrazido(2−) and related imido species in the P3BFe-system.112,181 Thus, this species was assigned as [P3BFe(NNH2)]+.113 It was the first iron species to be characterized in which an N2 ligand had been protonated to an Fe(NxHy) product, and the product is clearly an intermediate of a distal pathway.
The distinction between limiting distal and alternating pathways has already been discussed and is an issue that has been much discussed with respect to Fe- and Mo-mediated nitrogen fixation, both with respect to the enzymatic process and in synthetic catalysts. While interconversion between these limiting pathways may also be operative (vide infra),22 probing whether N–N bond cleavage occurred early (forming a nitride and ammonia) or late (forming an amide and ammonia) in this catalyst system was of interest. The ability to generate [(P3B)Fe(NNH2)]+ led to studies to probe the viability of an early N–N cleavage step to generate [(P3B)Fe(N)]+, consistent with a distal pathway. The reaction of [(P3B)Fe(NNH2)]+ with excess acid did not lead to N–N bond cleavage, likely because [(P3B)Fe(NNH2)]+ is a very poor base. Thus, its one-electron reduced congener was targeted. To achieve this, [(P3B)Fe(N2)]− was further reduced by KC8 to its dianionic derivative, [(P3B)Fe(N2)]2− (νNN = 1836 cm−1). Reaction of this species at −135 °C in 2-MeTHF with excess HBArF4 or HOTf for 15 min led to the clean formation of (P3B)Fe(NNH2), characterized in situ via XAS (Fe–N bond length of 1.65(2) Å) and Mössbauer spectroscopy.176 Its methylated analogue, (P3B)Fe(NNMe2), was similarly characterized and proved sufficiently stable to also obtain its solid-state crystal structure.114 Protonation of [(P3B)Fe(N2)]2− with larger excesses of acid, and for longer reaction times, led to formation of a new species in the Mössbauer spectrum with ≥50% spectroscopic yield. The low isomer shift (−0.15 mms−1) and the very large quadrupole splitting (6.2 mm s−1) have previously been demonstrated to be diagnostic for tetrahedral, iron(IV) nitrides (Figure 43).186–188 XAS characterization of [(P3B)Fe(N)]+, and comparison with its [(P3B)Fe(N2)]2−, (P3B)Fe(NNH2), and (P3B)Fe(NNMe2) relatives, confirmed its expectedly short Fe–N bond distance (1.54(2) Å) owing to its Fe-to-N triple bond. It was additionally established that formation of [(P3B)Fe(N)]+ occurred concomitant with NH3 formation (36.0 ± 0.5% isolated yield). The yield of NH3 could be increased by sequential reaction of [(P3B)Fe(N2)]− with HOTf and Cp*2Co (73 ± 7%). These results demonstrated, for the first time, the viability of an Fe-mediated distal pathway for N2-to-NH3 conversion, more than four decades after such a pathway was first by Chatt.15
Figure 43.

Representative tetrahedral Fe(IV) nitride complexes.176,187–189
Regardless of which pathway(s) are viable for overall N2-to-NH3 conversion, a linchpin intermediate is the initial product of pronation, M(NNH). Parent diazenidos (sometimes referred to as imides) of this type are very rare and prior to recent work with iron had only been reliably characterized on select Mo and W systems.99,136,190,191 Germane to the iron systems being discussed here, efforts to isolate and crystallize a parent Fe(NNH) species have been unsuccessful owing to the presumed high reactivity of such species; the N–H bonds are predicted to be very weak for systems such as (P3B)Fe (vide infra)80,192 and accordingly the addition of stoichiometric acid to [(P3B)Fe(N2)]−, even at very low temperature, led to net oxidation of the iron complex to (P3B)Fe(N2) with concomitant release of 0.5 equiv of H2. In the presence of excess acid, the presumed (P3B)Fe(NNH) intermediate is trapped by H+ to produce ([(P3B)Fe(NNH2)]+ faster than it can bimolecularly expel H2.113 Silyl diazenidos have proven more stable and have been prepared and crystallographically characterized on (P3B)Fe181 (and other ligated Fe species, vide infra).172,193–196
In an effort to slow the bimolecular decay of the (P3B)Fe(NNH), Peters and co-workers prepared a (P3B)Fe analogue with increased steric shielding by replacing the isopropyl substituents on the phosphines with bulky, electron-rich aryl substituents (3,5-diisopropyl,4-methoxyphenyl) (Figure 44). An anionic N2 complex of this new ligand (ArP3BFe) was prepared by analogy to the parent system. Although the N2 ligand in [(ArP3B)Fe(N2)]− is slightly less activated than the parent system (νNN = 1937 cm−1 vs 1905 cm−1), protonation with excess acid and warming led to the observation of NH3 (~0.25 equiv of NH3 per Fe), confirming the viability of N2-to-NH3 conversion with this platform. Reaction of [(ArP3B)Fe(N2)]− with 1 equiv of HBArF4, via mechanical mixing of a 2-MeTHF glass, afforded a new S = 1/2 species that was characterized via CW and pulse EPR spectroscopy. Using a range of isotopically labeled derivatives, and also via comparison with a more stable isoelectronic and isostructural silyldiazenido analogue, (ArP3B)Fe(NNSiMe3), a definitive assignment of the species as (ArP3B)Fe(NNH) could be made. The distal proton showed strong coupling to the unpaired spin (aiso(1H) = 16.5 MHz). Consistent with prior results,113 annealing these samples to −78 °C led to decay of the signature for (ArP3B)Fe(NNH) and (primarily) the formation of (ArP3B)Fe(N2), presumably via loss of H2. However, annealing such a sample in the presence of excess acid showed its gradual evolution to [(ArP3B)Fe(NNH2)]+; the latter features similar hyperfine parameters to the previously characterized species [(P3B)Fe(NNH2)]+.113
Figure 44.

Comparison of reactivity of (A) [(P3B)Fe(N2)]− and (B) [(ArP3B)Fe(N2)]− with acid. The bottom figure was taken from ref 177 and shows the reaction progress of [(ArP3B)Fe(N2)]− with excess HBArF4 in 2-Me-THF, as monitored by CW X-band EPR. Simulations for discrete species are shown in red. Spectra show the progression from (A) pure [(ArP3B)Fe(N2)]−. (B) Mixture of[(ArP3B)Fe(N2)]− and (ArP3B)Fe(NNH), on addition of 1 equiv of HBArF4 for 15 min at 138 K. (C) Same as in spectrum B, but instead with 2.3 equiv of HBArF4 present, showing full conversion to (ArP3B)Fe(NNH). An identical spectrum was obtained on mixing for 30 min in the presence of 1 equiv of HBArF4. (D) Reaction mixture from trace C, after warming to 195 K for 30 s and then rapidly freeze quenching in liquid N2. Trace shows a mixture of (ArP3B)Fe(NNH) and [(ArP3B)Fe(NNH2)]+. (E) Reaction mixture from trace D after warming to 195 K for 90 s, showing complete conversion to [(ArP3B)Fe(NNH2)]+. Reactions with up to 5 equiv of HBArF4 provided identical spectra. Acquisition parameters for all spectra: temperature = 77 K; MW frequency = 9.44 GHz; MW power = 6.44 mW; modulation amplitude = 0.1 mT; conversion time = 5.12 ms·E.113,177 EPR spectrum reproduced with permission from ref 176. Copyright 2017 American Chemical Society.
This study completed the characterization of early (and highly reactive) nitrogen fixation intermediates on tris(phosphine)borane Fe systems (Figure 45). Combined with the earlier studies on late-stage nitrogen fixation intermediates,182 many (P3B)Fe(NxHy) intermediates of a plausible N2 fixation cycle are known (Figure 41). Work remains to generate and characterize a few intermediates in the middle of the cycle (shown in light gray; e.g., (P3B)Fe(NH)) and to study their respective interconversion to later stage species. Of special interest is to further explore the possibility of hybrid mechanisms wherein distal intermediates convert to alternating intermediates that can then undergo a later-stage N–N cleavage event, indicated by dashed line and discussed further below.
Figure 45.

A proposed N2RR cycle for Peters’ (P3B)Fe platform. Characterized compounds are shown in green and unobserved complexes in gray. Solid arrows indicate a distal mechanism of nitrogen fixation, while dashed arrows indicate hybrid mechanisms of nitrogen fixation.109,112–114,176,182 Note that steps are shown as discrete e− and H+ transfer steps, though concerted PCET steps may also be operative.
5.2. (P3B)Fe: More Efficient Catalysis at Lower Overpotential
In 2017, Peters and co-workers found that the reaction of [(P3B)Fe]+ at −78 °C in Et2O under an N2 atmosphere with excess Cp*2Co and [Ph2NH2]OTf led to significantly enhanced efficiency for NH3 production (up to 72 ± 3%, along with trace amounts of N2H4) (Figure 46).80 This result was fascinating as it demonstrated the overall efficiency could be improved with reagents that provide significantly less net driving force for the overall transformation (ΔΔHf= 77 kcal·mol−1 NH3 vs ≥156 kcal·mol−1 NH3 with KC8 and HBArF4). Also noteworthy, freeze-quench Müssbauer spectroscopy of the catalytic reaction under these new conditions failed to show any of the borohydride/hydride species, (P3μ-B-H)Fe(H)(L) (L = H2 or N2), which, as discussed above, dominated the speciation of the catalyst at early times when using KC8 and HBArF4. However, high-spin FeII species (presumably decomposition products) are formed, which were posited to be a consequence of the presence of a more coordinating triflate counteranion. Furthermore, the observation of [(P3B)Fe(N2)]− at early reaction times by freeze-quench Müssbauer and EPR spectroscopy suggested that protonation under these new conditions is slow, consistent with the highly insolubility of the acid under the catalytic conditions. However, employing a less coordinating and more soluble acid, [Ph2NH2]BArF4, led to poorer catalytic performance (42 ± 6% efficiency). These data suggest that acid strength, concentration, and counteranion all play roles in determining the selectivity for NH3 versus H2.
Figure 46.

Comparison of performance for N2RR by [(P3B)Fe]+ depending on the acid/reductant.18,80,173 For discussion of overpotential see Section 8.3.
In a follow-up study, the effect of the pKa on the selectivity for NH3 versus H2 was studied by using variably substituted anilinium triflate acids.197 Notably, despite the known importance of pH on the selectivity of the MoFe protein,198 the effect of pKa on selectivity had not been previously addressed with a model system. The ease of pKa tunability with anilinium acids, and their compatibility with N2RR catalysis with [(P3B)Fe]+, made such a study possible. NH3 selectivities greater than 70% were observed with acids that have pKa’s in THF between 2 and 4. Moving to stronger acids decreased catalytic performance, while weaker acids showed a steady decline in performance, with only trace NH3 being observed with [4-MeOPhNH3]OTf (pKa = 8.8 in THF). This behavior is in fact quite similar to that observed for the MoFe protein itself.198
Efforts to protonate [(P3B)Fe(N2)]− at low temperature with [2,6-ClPhNH3]OTf, which is very effective in catalysis, did not lead to productive N–H bond formation at −78 °C in Et2O, contrasting with the aforementioned results with HBArF4, where [(P3B)Fe(NNH2)]+ is generated. Using the more soluble acid [2,6-ClPhNH3]BArF4 enabled low temperature protonation to be observed (i.e., formation of [(P3B)Fe(NNH2)]+) and the observation of NH3 upon warming.113 These data suggest that the mechanism when soluble (BArF4−) acids are employed (maximum efficiency of ~45%) is likely different than when insoluble (OTf−) acids (maximum efficiency of ~75%) are employed.
To explain the lack of productive N–H bond formation in the absence of reductant and insoluble acids, it was suggested that under these conditions, PCET mechanisms become relevant, whereas with HBArF4 and KC8 proton transfer-electron transfer (PT-ET) mechanisms likely dominate. Discussion of PCET mechanisms for N–H bond formation in the context of homogeneous nitrogen fixation catalysis had previously been limited to reactions mediated by pyridinyl radicals;103,105,106 as such mechanisms were not relevant to this work, the authors explored the possibility of metallocene protonation to generate PCET reagents. Using DFT calculations, the authors demonstrated that even with pyridinium acids, reduction of the pyridinium rings is less favorable than proton transfer from the pyridinium to the metallocene reductant.80 The relevant metallocenes (Cp2Co, Cp*2Cr, and Cp*2Co) are all predicted by DFT to undergo ring-protonation to generate species that have homolytically very weak C–H bonds (BDEC–H ≈ 30 kcal·mol−1, Figure 47), suggesting they should behave as competent PCET donors.80,197
Figure 47.

Bond dissociation enthalpies of the thermodynamically preferred protonation isomer for three commonly used reductants in N2RR (Cp2Co, Cp*2Cr, and Cp*2Co). The site of protonation and the C–H bond dissociation enthalpy of the resulting product has been confirmed experimentally for Cp*2Co.80,199
If protonation of the metallocene is critical for N–H bond formation under these types of conditions, then one would expect it to have a marked effect on NH3 selectivity. As such, DFT was used to interrogate the ability of three different acids to protonate Cp*2Co ([2,6-ClPhNH3]OTf, high experimental efficiency for N2RR; [2,6-MePhNH3]OTf, moderate efficiency; [4-OMePHNH3]OTf, low efficiency). Although all of these reactions were predicted to occur with a low barrier, the reaction was found to be uphill with the least efficient acid, downhill with the most efficient acid, and thermoneutral for the moderately efficient acid. Thus, the experimental catalytic efficiency data correlate with the predicted availability of the protonated metallocene.197
Pulse EPR spectroscopy was recently used to experimentally verify that Cp*2Co undergoes both endo- and exoring protonation to form [(Cp*)(η4-C5Me5H)Co]+. Furthermore, thermochemical measurements confirmed that the exoisomer possessed a homolytically very weak C–H bond (BDFEC–H < 29 kcal·mol−1), consistent with earlier computational predictions (Figure 48).80 This experimental work also unveiled that the protonated metallocene, [(Cp*)(η4-C5Me5H)Co]+, can be reduced by one electron at a very mild potential (−0.62 V vs Fc+/0) to generate (Cp*)(η4-C5Me5H)Co; the latter features a very hydridic C–H bond (ΔG(H−) < 41 kcal·mol−1). This result hints at the possibility that hydride (H−) transfer might also play a role in catalysis, an idea that warrants further study.
Figure 48.

Thermochemistry of protonated Cp*2Co species that have been hypothesized to be relevant to PCET and hydride transfer in N2RR.199
Given the ubiquity of metallocenes in high efficiency N2-fixation catalysis, such as that of the Mo systems discussed above,50,51,84,149 it seems plausible that metallocene-mediated N–H bond formation, via PCET and/or hydride transfer processes, could be an important and previously overlooked pathway in N2RR. The very recently reported catalytic results from the Nishibayashi group utilizing the known PCET donor mixture SmI2 and H2O/ethylene glycol offers added support to this suggestion.79
5.3. A Catalyst with an Fe–C Bond
In 2014, the Peters group reported a related Fe catalyst for nitrogen fixation featuring a carbon-anchored tris(phosphine) atrane ligand, (P3C)Fe.115 This complex was of particular interest to be able to explore a model catalyst in which the N2 substrate and a C atom are trans-disposed. Such an arrangement has been proposed upon initial binding of N2 to either Fe2 or Fe6 of the FeMo-cofactor in MoFe-nitrogenase, which features an interstitial carbide.13,200 The possibility that hemilability of the interstitial light atom could play a key role in N2-binding at Fe in the cofactor was suggested by the Peters group as early as 2005168 and was the subject of an in depth iron model study with a tris(phosphino)alkyl ligand, CSiP3Ph, in 2013 (Figure 49).194 The 2013 study demonstrated that the Fe–Capical in the model complex is highly sensitive to the overall charge (see the [(CSiP3Ph)Fe(CO)]n+ complexes (Fe–C = 2.303 Å in the anion, 2.236 Å in the neutral, and 2.138 Å in the cation). Indeed, the anionic charge state of both the CO and N2 complexes feature unusually long Fe–Capical bonds. Although the anionic N2 complex is silylated to form (CSiP3Ph)Fe(NNSiiPr3), the system was not catalytically active with the reagents tested at the time (KC8 and HBArF4).
Figure 49.

Reductive formation of [(CSiP3Ph)Fe(N2)]− and subsequent silylation chemistry.194
In contrast, the (P3C)Fe system showed the ability to bind N2 across three oxidation states (Figure 50), akin to its isostructural analogue, (P3Si)Fe, featuring Si in place of C as the anchoring atom.193 As with(CSiP3Ph)Fe(CO),194 the Fe–C bond distance contracted upon oxidation, opposite to what is observed with (P3B)Fe complexes.114,201 Nonetheless, (P3C)Fe also showed catalytic N2RR when exposed to excess HBArF4 and KC8.115
Figure 50.

Trends in Fe–C bond distance for a redox series of (P3C)Fe(N2) complexes.115 Related datafor [(P3B)Fe(N2)]− Figure 64.
Exposing [(P3C)Fe(N2)]− at −78 °C in Et2O to excess HBArF4 and KC8 led to the formation of 4.6 ± 0.8 equiv of NH3 per Fe. Under these conditions, a significant amount of the iron(II) hydride complex, (P3C)Fe(H)(N2), was observed to form, which itself proved inactive as a precatalyst.173 Thus, it was suggested that hydride formation limits the overall turnover. This contrasts with the aforementioned hydride-resting state, (P3μ-B-H)Fe(H)(L) (L = H2 or N2), of the (P3B)Fe-system, which can re-enter the catalytic cycle. These results (and those of other groups)50,53,107,116,173,178 highlight the importance of reversibly formed hydride intermediates in the context of the synthetic N2-fixation catalyst design. Indeed, it may be that the fact that nitrogenase enzymes also display hydrogenase activity essential to their biological function; if hydride intermediates cannot liberate H2, the active site can get stuck in an inactive hydride state and becomes effectively poisoned.173
5.4. Catalysis and Mechanism of a Tris(phosphino)silyl Fe Species
The Peters group has extensively explored the chemistry of a silyl-anchored tris(phosphine)iron system, (P3Si)Fe (Figure 51). Indeed, the study of such species predated the discovery of Fe-mediated N2RR catalysis in 2013.18,193,202 In many regards, the N2 activation and functionalization chemistry of the (P3Si)Fe-system are very similar to that of (P3B)Fe. This includes its demonstrated ability to support N2 complexes in multiple oxidation states,193,202 to form complexes with late-stage nitrogen fixation intermediates,193 to undergo redox-induced expulsion of NH3 with concomitant binding of N2,193 and also N2 silylation193 and protonation109 at the distal N atom of [(P3Si)Fe(N2)]−. However, in the presence of excess HBArF4 and KC8, the (P3Si)Fe-system is far more selective for the hydrogen evolution reaction (HER) than for N2RR (max efficiency for NH3 5 ± 3% with HBArF4/KC8). Nevertheless, under a sufficiently high loading of acid (1500 equiv of HBArF4) and reductant (1800 equiv of KC8), [(P3Si)Fe(N2)]− can be confirmed to be a catalyst for N2RR (3.8 ± 0.8 equiv NH3 per Fe).173 A similar lack of selectivity for N2RR is also observed with [Ph2NH2]OTf/Cp*2Co (1.2 ± 0.1 equiv of NH3 per Fe, 6 ± 1% NH3).80
Figure 51.

Reactivity of (P3Si)Fe hydrazido(2−) complexes.109
The difference in reactivity between [(P3Si)Fe(N2)]− and its [(P3B)Fe(N2)]− and [(P3C)Fe(N2)]− analogues poses interesting questions regarding the underlying cause of their divergent selectivities. [(P3Si)Fe(N2)]− and [(P3B)Fe(N2)]− have been studied in the most detail and allow for some insightful comparisons. Two key differences between these platforms emerge when interrogating their ability to stabilize Fe–N multiply bonded species. Whereas imido species of the type [(P3B)Fe(NR)]0/+ are highly stable,112–114,181 related imides for (P3Si)Fe are highly reactive, have only been characterized at very low temperature, and display hydrogen atom abstraction reactivity and imide/nitrene coupling reactivity to generate azoarenes (ArN = NAr).203 This difference in stability of imido-like species might be pertinent to nitrogen fixation by considering the respective stabilities and reactivity profiles of imide, Fe(NH) and “imido-like” hydrazido(2−) intermediates, Fe(NNH2). Surprisingly, the hydrazido(2−) complex, [(P3Si)Fe(NNH2)]+, was accessible via double protonation of [(P3Si)Fe(N2)]− at low temperature and could be isolated and crystallographically characterized.109 [(P3B)Fe(NNH2)]+ has not proven stable enough to isolate.
One-electron reduction of [(P3Si)Fe(NNH2)]+ by Cp*2Co generated neutral, S = 1/2 (P3Si)Fe(NNH2) as a highly reactive intermediate (Figure 51). Its characterization was aided by the generation of its far more stable (and crystallographically characterized) alkylated analogue, (P3Si)Fe(NNMe2). Addition of 0.5 equiv of Cp*2Co to [(P3Si)Fe(NNH2)]+ led to the generation of 0.5 equiv of [(P3Si)Fe(N2H4)]+ and 0.5 equiv of (P3Si)Fe(N2). These data are consistent with a scenario in which (P3Si)Fe(NNH2) is generated in situ and reacts, via a net transfer of two H atoms, with remaining [(P3Si)Fe(NNH2)]+. These data are moreover consistent with the observation that reaction of (P3Si)Fe(N2) with excess Cp*2Cr and [HNiPr2Et]BF4 led to the formation of N2H4 in 13% yield.204 One possibility for the reduced selectivity of (P3Si)Fe for N2-to-NH3 conversion is that the enhanced reactivity at Nα in (P3Si)Fe complexes, for example, via hydrogen atom transfer (HAT), can prevent the system from efficiently traversing a distal pathway that appears to be accessible for (P3B)Fe.176
The question of selectivity for NH3 versus N2H4 is of general interest with Fe catalysts. Every known Fe-catalyst (vide infra) has demonstrated an ability to form both NH3 and N2H4, with the primary product varying based on catalyst and condition. However, prior to this report, the formation of an alternating pathway intermediate directly from N2 was unprecedented for Fe. This led the authors to propose an alternative N2-fixation mechanism, dubbed the hybrid mechanism because it begins in a distal fashion and then on the third functionalization switches to an alternating pattern, similar to the pathway proposed by Dilworth and co-workers in the context of N2H4 formation on VFe nitrogenase.163 A recent study of methylhydrazido (2−) intermediates on (P3B)Fe may offer some insight into the question of NH3 versus N2H4 selectivity. The enhanced stability of these methylated (vs protonated) species allowed for a redox series (P3BFe(NNMe2)+/0/−) to be studied with a number of spectroscopic methods (i.e., Müssbauer, XAS, nuclear resonance vibrational spectroscopy, UV–vis spectroscopy), which were complemented by theoretical methods (DFT and complete active calculations). These methods revealed that for [(P3B)Fe(NNMe2)]+, the NNMe2 unit is best described in one of its canonical forms, namely, isodiazene. However, in the neutral and anionic complexes, the ligand is best described as a hydrazyl radical(1−) antiferromagnetically coupled to the Fe center. A hydrazyl radical should be reactive at both Nβ and Nα, potentially explaining the source of mixed selectivity (NH3 and N2H4) observed upon (P3B)Fe mediated N2RR in the presence of Cp*2Co and [Ph2NH2]OTf. Spectroscopic measurements suggest that the isoelectronic (P3Si)Fe(NNH2) has a similar electronic structure, consistent with the observed net transfer of protons and electrons reactivity to yield hydrazine.114 This relative distribution of electron density between Nα and Nβ may be a significant and general factor controlling NH3 versus N2H4 selectivity and warrants further study.
5.5. Theoretical Work on Atrane-Fe Systems
Phukan and co-workers performed a computational study comparing the potential for (P3B)Fe-and (P3Si)Fe for N2RR.205 They correctly predicted a greater preference of the silyl system to undergo protonation at Fe, which would lead to unproductive H2 formation. They also predicted smaller barriers for productive N2 functionalization steps on the boratrane platform. However, they suggested that functionalization would occur in an alternating rather than distal pathway, which is inconsistent with current experimental observations.109,113 Because it is a truncated ligand platform in which the phenylene linkers were replaced with ethylene linkers and the isopropyl substituents with methyl substituents, it is hard to draw reliable connections between this study and experimental observations. In addition to large conformational differences between the theoretically predicted structures and experimental observations due potentially to steric and electronic differences in the ligands, spin states are also systematically overestimated in the study.
In 2017, Peters and co-workers published a computational study evaluating the propensity of (P3E)Fe (E = B, C, Si) to release H2 during catalysis (Figure 52).192 By contrast to most studies that posit metal hydrides as the source of catalyzed H2 formation during nitrogen fixation, this study suggested that the source of H2 might be early nitrogen fixation intermediates, owing to their weak (<50 kcal·mol−1) N–H bonds. In support of this hypothesis, this study found that the N–H bond strengths in (P3E)Fe(NNH2) are correlated with the overall selectivity for NH3 versus H2 formation, with complexes that build up and feature weaker N–H bonds giving greater yields of H2. These bond strengths were correlated with the cumulative bond indices of the E–Fe–N–N unit. The more flexible the Fe–E interaction, the more Fe–N and N–N bonding is preserved as the N2 is functionalized, and the stronger the resulting N–H bonds. Species with stronger N–H bonds are thus predicted to be less prone to bimolecular H2 release.192 Important to note is that this study was almost exclusively based on thermodynamic arguments. Kinetic barriers for specific reaction steps, which can be challenging to reliably predict, especially in the absence of experimental data for calibration, were not computed. There is a need to directly measure some of the kinetic barriers for reaction steps in this Fe-mediated catalysis to better guide theoretical work. Such measurements are likely to be technically very challenging due to the highly reactive nature of the early stage (P3B)Fe(NxHy) intermediates predicted to influence overall selectivity in this study.
Figure 52.

Overview of predicted bimolecular HER and N2RR pathways for (P3E)Fe(NNHy) species and estimated BDFEN–H values. Reproduced with permission from ref 192. Copyright 2018 American Chemical Society.
Li and co-workers have published two recent computational studies on N2RR at (P3E)Fe-complexes (E = B, Si, N). In the first study, they used complete active space (CAS) calculations to interrogate the electronic structures of (P3B)Fe(NxHy) complexes in detail.206 They found that protonation from the [(P3B)Fe(N2)]− to the Fe(NNH) and then to the Fe(NNH2)+ leads to successive lengthening and weakening of the Fe–B bond. This suggestion had been previously advanced by the Peters group to explain the ability of this platform to support both low-valent (e.g., [Fe(N2)]2−) and high-valent (e.g., [Fe(N)]+) intermediates, and corroborated by a wealth of crystallographically characterized Fe–B distances.112–114,176,181 Li and co-workers, akin to a suggestion by Peters and co-workers,176 suggested that this kind of buffering effect could also be relevant to the FeMo-cofactor in nitrogenase, or in heterogeneous catalysts that feature single-metal atoms on supports.
More recently, the Li group has evaluated the relative influence of having apical B, C, or N atoms on the catalytic system by interrogating how the Fe–E bond indices evolve over the catalytic cycle.207 In the case of both B and C, they found significant changes in the bond indices throughout the catalytic cycle. For the anionic C-based ligand, an increased bond order was observed upon protonation, and a decreased bond order was observed following reduction, consistent with experimentally observed bond lengthening in the [(P3C)Fe(N2)]+/0/− series. By contrast, the neutral borane-based ligand was predicted to show the opposite behavior, consistent with experimentally observed bone lengths.114,181,201 Furthermore, this study predicted that the interaction between a Lewis basic N atom anchor and Fe would minimal throughout the catalytic cycle and is thereby unable to buffer the oxidation state changes experienced at Fe. However, the “peripheral” ligand, namely, the phosphine donors and the phenylene linkers, play a significant role in buffering the redox changes at Fe. From their studies, the authors suggested that catalysis with an N-anchored ligand might be achieved by installing electron-withdrawing −CF3 groups at the para-position (relative to the apical N atom linchpin of the ligand), so as to enhance the electronic buffering capacity of the tris(phosphine)amine ligand.207
Visser and co-workers have investigated the thermodynamics and kinetics of [(P3B)Fe(N2)]− protonation by [(Et2O)2H]+.208 Consistent with spectroscopic results, double protonation at the distal nitrogen was found to be kinetically and thermodynamically more favorable than formation of a diimide-type intermediate (e.g., (P3B)Fe(η2-HNNH)). They then determined that protolytic N–N bond cleavage should be possible following either 0, 1, or 2 electron transfers, suggesting the thermodynamic plausibility of multiple mechanisms for NH3 formation. Unfortunately, this report systematically overestimates the stability of high spin states (e.g., they predict that the S = 3/2 state of [(P3B)Fe(NNH2)]+ to be more stable than the experimentally observed S =1/2 ground state by 20 kcal·mol−1). This makes it difficult to draw firm conclusions from the work.
Oláh, Szilvási, and co-workers have interrogated the Gibbs free energy profile of nitrogen fixation catalysis on (P3E)Fe (E = B, Si).209 They identified that these systems could access both a distal and a hybrid distal-to-alternating mechanism, with the key intermediate being (P3E)Fe(NNH2). Consistent with experimental results,109,114,176,204 the Si system was predicted to have a greater preference for the hybrid distal-to-alternating pathway. Furthermore, this theoretical study identified [(P3Si)Fe(N2H4)]+ as a thermodynamic sink that is difficult to escape once formed (i.e., unfavorable electron transfer, proton transfer, and Fe–N bond cleavage); however, they suggested that PCET to [(P3Si)Fe(N2H4)]+ to form NH3 and [(P3Si)Fe(NH2)]+ could rapidly consume such an intermediate. An identified challenge for these iron catalyst systems is that all of the free energy provided by the reagents is consumed as early as the formation of [(P3E)Fe(NH3)]+, which precedes catalyst turnover. A similar problem has been identified computationally and experimentally for Schrock’s (HIPTN3N)Mo system,128 suggesting this maybe a general challenge in homogeneous N2 fixation catalysts, offering a potential area for improvement via rational catalyst design.
5.6. Nishibayashi’s Fe Systems
An Fe catalyst was developed by the Nishibayashi group featuring a pyrrole-based PNP ligand.116 Although no fixed-N products were observed when they used conditions similar to those developed for their Mo catalysts (i.e., Cp2Co, [LutH]OTf, room temperature),51 when they explored the original conditions reported for (P3B)Fe (i.e., KC8, HBArF4, −78 °C in Et2O)18 they detected catalytic yields of fixed-N products. In contrast to the (P3B)Fe system, Nishibayashi and co-workers observed catalytic yields of not only NH3 but also some N2H4. Furthermore, the selectivity for NH3 versus N2H4 was affected by the solvent, with THF enhancing the yield of N2H4 relative to Et2O. The electronic structure of the most efficient precatalyst, (PNP)Fe(N2), was studied by Walter and co-workers via SQUID magnetometry, Mössbauer, and EPR spectroscopy and found to feature a well-isolated S = 1/2 ground state.210
Notably, the Nishibayashi group found that two potential products of (PNP)Fe(N2) protonation, (PNP)Fe(H) and a species resulting from protonation at the pyrrole-backbone (which DFT predicted to be the thermodynamic product of protonation), were also precatalysts for N2RR, albeit with diminished efficiency (Figure 54). Again, this observation underscores the importance of forming hydridic species, in this case either Fe(H) or C–H of the dearomatized pyrrole, reversibly if N2RR catalysis is to be productive, thereby avoiding an off-path sink.
Figure 54.

Catalytic N2RR efficiencies for all of Nishibayashi’s (PNP)Fe systems. aThese complexes were only tested under higher loading conditions with KC8 (200 equiv) and HBArF4 (184 equiv).29,116,178
Given the diminished efficiency of the pyrrole-protonated precatalyst, the Nishibayashi group synthesized two new catalysts that feature methyl and phenyl substitution at the 3- and 4-positions of pyrrole (Figure 54).178 While phenyl substitution did not lead to improved catalysis, methyl substitution improved the catalyst lifetime (max fixed-N product: 26.1 ± 2 vs 17.9 ± 2; max efficiency: 42 ± 8% vs 37 ± 4%).116,178 Markedly, reaction of these (3,4-MePNP)Fe(N2) complexes with HBArF4 resulted in protonation at the 2-position of the pyrrole backbone rather than the 3-position. Thus, methyl substitution prevented protonation at the 3-and 4-position, but the electron-rich pyrrole ring remained the thermodynamically favored site of protonation.178
Efforts to completely prevent pyrrole ligand protonation by replacing the pyrrole with a carbazole were unsuccessful (Figure 54).179 Reduction of the carbazole-containing (CarPNP)Fe(Cl) species led to bridged, rather than terminal dinitrogen complexes, which may retard protonation steps at N2.211 Possibly as a consequence of slower kinetics, these N2-bridged species were not catalysts for N2RR.
5.7. A Non-Phosphine Fe System for Catalysis
The low coordination numbers of the iron centers in the FeMo-cofactor, especially prior to the reassignment of its structure with an interstitial light atom,13 when the iron centers were presumed to be three-coordinate has inspired inorganic chemists.212,213 Holland and co-workers, in particular, have advanced the hypothesis that low-coordinate Fe centers would be particularly competent for activating N2. Although Holland and co-workers have not yet demonstrated catalytic N2RR with their β-diketiminate system, they have demonstrated important results regarding N–N bond cleavage and N–H bond formation on this platform.90,211,214–217
However, to date, the only iron system featuring coordination numbers less than four reported to facilitate catalytic N2-to-NH3 conversion is the 2-coordinate complex (CAAC)2Fe (CAAC = cyclic alkyl amino carbene, Figure 55), which goes three-coordinate upon N2 binding.172 Interestingly, (CAAC)2Fe is stable to reduction at room temperature, but entropy-controlled equilibrium N2 binding (Keq = 0.2 at 298 K) to (CAAC)2Fe facilitated reduction at low temperature to [(CAAC)2Fe(N2)]− (νNN = 1,850 cm−1). The low N2 stretching frequency correlates with strong N2 activation. Accordingly, [(CAAC)2Fe(N2)]− reacted productively with silyl electrophiles to yield the silyldiazenido complex (CAAC)2Fe(NNSiMe3). Consistent with temperature-controlled binding of N2 (and hence reduction), the reaction temperature had a significant impact on catalytic efficiency when (CAAC)2Fe was exposed to excess HBArF4 and KC8 (0.4 equiv of NH3 at RT, 0.3 equiv of NH3 at −50 °C, 0.9 equiv of NH3 at −78 °C, 3.3 equiv of NH3 at −95 °C; average values given).
Figure 55.

Catalytic conditions for (CAAC)2Fe. Equilibrium binding of N2 at −80 °C and access to a (CAAC)2Fe(NNTMS) via reductive silylation; Dipp = 2,6-diisopropylpheyl.172
5.8. Historic Work on Bis-Diphosphine Fe Complexes in Nitrogen Fixation
In 2016, the Ashley group demonstrated the catalytic formation of hydrazine (N2H4) using (depe)2Fe(N2) as the (pre)catalyst (depe = 1,2-bis(diethylphosphino)ethane).174 The stage for this result was set by the rich history of bis-diphosphine–Fe-N2 systems. We first describe early and mechanistically relevant details prior returning to the fascinating catalysis reported by Ashley and co-workers.
Leigh and co-workers first suggested that a dmpe-supported (dmpe = 1,2-bis(dimethylphosphino)ethane) Fe complex, (dmpe)2Fe(N2) (νNN = 1975 cm−1), liberated fixed-N produts (up to 20%) upon treatment with H2SO4 (Figure 56). However, efforts to protonate the N2/hydride species, [(dmpe)2Fe(N2)-(H)]+ (νNN = 2,074 cm−1) at N2, with H2SO4 were largely unsuccessful (<4% NH3).218 Employing HCl as the acid source instead allowed for recovery of the starting material trans-(dmpe)2Fe(Cl)2 in about 80% yield, hinting that such systems might ultimately be amenable to catalysis.219 Nevertheless, these results were later called into question due to the lack of fixed-N products observed from protonolysis reactions with the structurally related complex (depe)2Fe(N2).220,221 Later work definitively established that protonation of (dmpe)2Fe(N2) by HCl does not result in the formation NH4+.222 However, these results remain important as they provided motivation for the study of nitrogen fixation using these types of complexes. Furthermore, Leigh’s suggestion that complexes of the type: (diphosphine)2Fe(N2) could yield fixed-N products was ultimately proven correct by Ashley and co-workers, the key being replacing HCl with HOTf.223
Figure 56.

Reactivity of (dmpe)2Fe (dmpe = 1,2-bis(dimethylphosphino)ethane) complexes.218,223–229
Much of the early mechanistic work on (diphosphine)2Fe-species relevant to N2 fixation was performed with the ligands dmpe and DMeOPrPE (1,2-bis[di(methoxypropyl)-phosphino]ethane). Although much of this work was performed contemporaneously, the dmpe system will be discussed first, with distinct features of the DMeOPrPE system then being introduced.
The η2-N2H4 hydrazine adduct complex [(dmpe)2Fe(N2H4)]2+ was synthesized by reaction of trans-(dmpe)2Fe(Cl)2 with excess N2H4 in THF (Figure 56).229 Reaction of trans-(dmpe)2Fe(Cl)2 with excess hydrazine in the presence of KC8 led to the formation of an η2-bound diazene complex, (dmpe)2Fe(η2-HNNH), in 17% yield. The latter complex is structurally (and electronically) unusual and was proposed to form via reduction of the aforementioned [(dmpe)2Fe(N2H4)]2+ complex to an (dmpe)2Fe(N2H4) product that then loses H2 to form the observed Fe-diazene. The observation of cis-(dmpe)2Fe(H)2 as a minor byproduct is at least consistent with the in situ formation of H2. Although the diazene species can be isolated by crystallization, it ultimately decomposed in solution to afford [(dmpe)2Fe]2(μ-dmpe) with the loss of the diazene fragment. Consistent with the known decomposition pathways of free diazene, monitoring the decay of the 15N-labeled diazene complex by NMR spectroscopy confirmed the formation of 15NH3 and 15N2.
Additional work by Field and co-workers revealed that the diazene complex could be synthesized in improved yield (55%) via deprotonation of the hydrazine complex by KOtBu (Figure 57).224 This reaction was reversible with [LutH]OTf able to protonate the diazene complex to the hydrazine complex. Reaction of the 15N-hydrazine complex with Schlosser’s base (KOtBu and tBuLi) under a 14N2 atmosphere led to the formation of (dmpe)2Fe(15N2), as confirmed by NMR and IR spectroscopy, suggesting that under certain conditions, the bound diazene could effectively lose H2 without dissociating from the metal. DFT calculations performed on the diazene complex led the authors to propose an Fe0 oxidation state with backbonding to a neutral diazene fragment, as opposed to charge-transfer to form a hydrazido(2−) ligand bound to an FeII center; however, this assignment has been questioned.230
Figure 57.

(Top) Acid/base chemistry that interconverts hydrazine, hydrazido(1−), and diazene ligands on (DMeOPrPE)2Fe;232,233 (bottom) potential N2RR intermediates of the form (dxpe)2Fe(H)(L) species (x = m or e, L = H2, N2, NH3, and N2H4) can be synthesized under aqueous conditions.224,231,234,235
The potential relevance of Fe(H) intermediates to nitrogen fixation, both within this system and in biological nitrogen fixation, led Field and co-workers to also consider the coordination of nitrogen fixation intermediates (NxHy) to Fe(H) species. The reaction of trans-(dmpe)2Fe(Cl)(H) with excess N2H4 in the presence of NaBPh4 led to the isolation of trans-[(dmpe)2Fe(N2H4)(H)]BPh4 in moderate yield (36%). Reaction of trans-[(dmpe)2Fe(15N2H4)(H)]BPh4 with KOtBu formed (dmpe)2Fe(η2-H15N15NH) and trace amounts of (dmpe)2Fe(15N2) and (dmpe)2Fe(H)2, consistent with previous results.224 The hydrazine complex was unstable in solution, decomposing to trans-[(dmpe)2Fe(NH3)H]BPh4, trans-[(dmpe)2Fe(N2)(H)]BPh4, and free N2. The amide complex, trans-(dmpe)2Fe(NH2)(H) was also accessible by reaction of trans-(dmpe)2Fe(Cl)H with NaNH2 (63% yield). It could be converted into the ammonia complex, trans-[(dmpe)2Fe(NH3)H]+, via protonation with H2O or fluorene,231 thus demonstrating that most of the potential intermediates of a N2RR cycle can be synthesized in the presence of iron hydrides.
The reactivity of (dmpe)2Fe(N2) with electrophiles has been recently revisited (Figure 58). Field and co-workers found that reaction with methyl electrophiles (i.e., MeOTf, MeOTs) methylated the Fe center trans to the N2 ligand. In contrast, silylation followed by quenching with HOTf led to the formation of NH3 (17 ± 3%), suggesting N2-based functionalization. Yields of NH3 could be increased by the addition of KOtBu and Cp*2Co (51 ± 2% NH3).222 Ashley and co-workers also explored the reactivity of (dmpe)2Fe(N2) and found that under a vacuum this species dimerizes to form [(dmpe)2Fe]2(μ-N2) (νNN = 1933 cm−1). While acidification of (dmpe)2Fe(N2) with HCl formed only trace NH3, fixed-N yields could be increased by using HOTf (up to 9.1% N2H4).223 The Ashley group also noted that by cyclic voltammetry (CV), (dmpe)2Fe(N2) decomposes upon oxidation to the putative [(dmpe)2Fe(N2)]+, but for (depe)2Fe(N2) this oxidation occurred reversibly at −2.0 V vs Fc+/0.
Figure 58.

Treatment of (dmpe)2Fe(N2) with electrophiles.222
The mechanism of nitrogen fixation using (dmpe)2Fe(N2) has been studied computationally by the Tyler group.230 In these calculations, HOTf was the reference acid, and the electrons for N2RR were provided by oxidation of (dmpe)2Fe(N2) to [(dmpe)2Fe]2+. Protonation of (dmpe)2Fe(N2) was found to be most favorable at Fe (ΔE = −58 kcal·mol−1), but it is known that the reaction of trans-(dmpe)2Fe(N2)(H) with protons did not lead to the formation of fixed-N products, so they considered only the further reactivity of the kinetic protonation product [(dmpe)2Fe(NNH)]+ (ΔE = −18 kcal·mol−1). Their key observation was that although protonation of [(dmpe)2Fe(NNH)]+ to give the dicationic trans-diazene (HNNH) and isodiazene (NNH2) species were similar in energy, ΔE = −2 and −6 kcal·mol−1, respectively, the two-electron reduction of the isodiazene was predicted to be much more unfavorable than that of the trans-diazene (ΔE = 18 kcal·mol−1 vs −7 kcal·mol−1). Thus, a Chatt-type mechanism was considered unlikely; this is consistent with the ultimate discovery that (depe)2Fe selectively catalyzes the formation of N2H4.174
Although the core bis-phosphinoethane architecture is maintained between dmpe and DMeOPrPE, certain features are distinct to the alkyl-ether containing phosphine substituents that warrant comment. With the DMeOPrPE ligand, one can envision the possibility of secondary-sphere hydrogen bonding interactions between NxHy intermediates and the ethereal O-substituents of the ligands—[(DMeOPrPE)2Fe(N2H4)]2+ demonstrates one such interaction (Figure 57). It is likely that similar interactions exist in other nitrogen fixation intermediates on this platform, but crystallographic evidence is limited. Reaction of this hydrazine complex with excess HOTf leads to N2H5+ (64%), NH4+ (21%), and N2. The authors attributed the formation of NH4+ to hydrazine disproportionation.232 It is possible that hydrogen bonding plays a key role in stabilizing intermediates such as the hydrazido(1−) on this platform, allowing for the reversible, stepwise interconversion of the hydrazine and diazene complexes.233
Another interesting feature of the DMeOPrPE system is that the alkyl ether substituents render the complexes water-soluble.236–238 Unfortunately, although trans-[(DMeOPrPE)2Fe(N2)(H)]+ could be formed, the hydride was too basic to be deprotonated in aqueous solvent and thus access to the key nitrogen-fixing intermediate, (DMeOPrPE)2Fe(N2), was not available.239 These results are nonetheless of interest as being able to perform catalytic nitrogen fixation under aqueous conditions remains an important goal for the field; recent results from the Nishibayashi group have begun to address this issue.79,150
Tyler and co-workers have also shown that with (DMeOPrPE)2Fe, H2 can serve as a source of electrons for nitrogen fixation. This was achieved by synthesizing trans-[(DMeOPrPE)2Fe(H)(H2)]+ from reaction of trans-(DMeOPrPE)2Fe(Cl)2 with H2 in the presence of base. Substitution of the H2 ligand for N2 occurred upon exposure to an atmosphere of N2, and deprotonation of the resulting trans-[(DMeOPrPE)2Fe(H)(N2)]+ by KOtBu generated (DMeOPrPE)2Fe(N2) (Figure 57). Reaction of the latter species with acid provided fixed-N products, for which the reducing equivalents were derived from H2.240 Despite exciting advances by a variety of groups aimed at the goal of utilizing H2 as the source of both protons and electrons in catalytic nitrogen fixation (akin to Haber–Bosch),88,185,241,242 this overall target remains unrealized.
5.9. Achieving Catalysis with (depe)2Fe(N2)
Although much of the mechanistic work concerning bis-diphosphine systems has been performed with dmpe and DMeOPrPE, depe can also be used to form related N2 complexes.220,243 Given its enhanced selectivity for the formation of fixed-N products on protonation (26% yield vs 9% yield for dmpe) and the reversibility of its 0/1+ redox couple, (depe)2Fe(N2) was pursued as a (pre)catalyst for nitrogen fixation. The use of Cp*2Co and [Ph2NH2]OTf in diethyl ether at −78 °C was found to be particularly effective for the catalytic fixation of N2 (up to 24.5 ± 0.5 equiv of N2H4, Figure 59). The use of THF as the reaction solvent, [Ph2NH2]BArF4 as the acid, or KC8 as the reductant was all found to be deleterious.
Figure 59.

Catalytic N2 fixation by (depe)2Fe(N2) using Cp*2Co (270 equiv) and [Ph2NH2]OTf (360 equiv).174
Ashley and co-workers made several observations during this work that led them to hypothesize that NH3 was forming via a catalyst decomposition pathway. First, although some NH3 forms irrespective of acid loading, it never exceeded 1 equiv (relative to catalyst). Second, the catalyst was not active for further nitrogen fixation upon reloading of substrate, but most of the quantified NH3 (~ 80%) originated from an unidentified Fe-species that was in solution at the end of catalysis. Lastly, under the reaction conditions, N2H4 was rapidly protonated to form insoluble [N2H5]OTf, which control reactions demonstrated was stable under the relevant conditions. These data suggested that NH3 cannot originate from the same pathway as N2H4. The question of whether there are different operative pathways to fixed-N products and what the consequence of that is for selectivity/turnover number is worthy of further interrogation.
Ashley and co-workers also suggested that [(depe)2Fe(N2H4)](OTf)2 cannot be an on-path intermediate of N2RR due to its poor catalytic performance. However, (pre)catalyst insolubility173 and the deleterious reactivity of certain intermediates at high concentration54 have been previously postulated as reasons why even on-path intermediates can lead to anomalously low yields when employed as (pre)catalysts. Hence, caution in interpreting this observation is warranted.
Subsequently, the reactivity of (depe)2Fe(N2) with electrophiles has been studied (Figure 60). In 2017, Szymczak and co-workers interrogated a “push-pull” hypothesis for N2 activation with this system. They found that (depe)2Fe(N2) reacted with a variety of Lewis acids (e.g., boranes and alkali metals) to form persistent adducts that significantly activated the N2 ligand (Δνnn ≈ − 50 to −170 cm−1). Solid-state characterization of the tris-(pentafluorophenyl)borane adduct revealed an elongated N–N distance, a shorter Fe–N distance, and an N–N–B angle of 137.0(3)°, all of which suggested a significant interaction between the boron p-orbital and the N–N π* orbital (Figure 60). The 11B NMR and 15N NMR spectroscopic data confirmed the interaction to be persistent in solution, with the latter also suggesting that there was increased electron density on Nβ. The degree of activation of the N2 ligand was found to correlate with the acceptor number244,245 of the Lewis acid employed. Interestingly, the B(C6F5)3-adduct was found to react selectively (~95%) with protons at Nβ to produce [(depe)2Fe(NNHBAr3)]+ (BAr3 = tris(pentafluorophenyl)borane), in direct contrast to the reaction of (depe)2Fe(N2) with protons which primarily led to the formation of Fe-hydrides. [(depe)2Fe(NNHBAr3)]+ has an N–N bond length of 1.252(8) Å and a νNN of 1519 cm−1, suggesting it is best formulated as a hydrazido(2−) ligand (Figure 60).246
Figure 60.

Observed reactivity of (depe)2Fe(N2) with electrophiles occurred at Nβ.195,246,247 The relationship between this reactivity and the catalytic reactionz remains unclear.
It was also found that (depe)2Fe(N2) reacted with TMSCl in the presence of KBArF4 to form a cationic silyldiazenido complex, [(depe)2Fe(NNSiMe3)]+ (Figure 60). DFT calculations suggested that this species is a good model of the first intermediate of catalytic nitrogen fixation, [(depe)2Fe(NNH)]+.195 It was subsequently found that one-electron reduction of [(depe)2Fe(NNTMS)]+ caused a disproportionation reaction that resulted in formation of (depe)2Fe(N2) and (depe)2Fe(NNTMS2).247 These data are consistent with the previous suggestion by the Peters group that the N–E (E = H, SiR3) bonds in early N2 fixation intermediates are weak.80,192 As with the report from the Szymczak group, difunctionalization of the coordinated N2 ligand was observed at Nβ rather than in an alternating fashion, which diverges from the observed formation of an η2-diazene ligand formed via deprotonation of the N2H4 complex and calculations.224,230 Thus, whether protonation reactions uniquely favor an alternating pathway, if the hydrazido ligand (NNH2) can rearrange to the diazene ligand (HNNH) or if hydrazine is formed via a hybrid mechanism, a possible scenario that has been considered by several groups,109,163 remains an open question.
5.10. Triphos-Fe Systems
Peters and co-workers recently reported two triphosphine-Fe hydride species, (PP2)Fe(H)2 and [(PP2)Fe(H)](μ-N2) (PP2 = bis(o-diisopropylphosphino-phenyl)phenylphosphine) that were (pre)catalysts for N2-to-NH3 conversion (Figure 61).175 Despite only moderate efficiency for ammonia (up to 7.5 ± 0.8% versus HER), these examples highlighted that irradiation of the catalytic reaction mixture can significantly enhance performance (18.1 ± 0.8% efficiency). The enhancement was even more significant at higher loadings of substrate (3000 equiv of HBArF4 and 3600 equiv of KC8), with photolysis increasing the yields from 24.5 ± 1.2 equiv of per Fe to 66.7 ± 4.4 equiv of per Fe (272%). These results are of particular interest not only to improving catalyst importance, but also to understanding the dihydride/N2 exchange process suggested to occur at the FeMo cofactor,19 which can also be triggered by irradiation.162
Figure 61.

Pathways for HER, N2RR, and hypothesized origins of the light enhancement for (PP2)Fe.175
Mechanistic experiments were employed to interrogate the origin of light enhancement. One possibility considered was that a mononuclear iron species is on-path for catalysis and that the dinuclear species undergoes photodissociation. However, freeze-quench EPR spectroscopy did not support this hypothesis. Interrogation of the catalytic reaction mixture after warming revealed that the main product (~90%) was (PP2)Fe(N2)(H)2 (Figure 61). Experiments using this dihydride as the (pre)catalyst led to slightly poorer efficiency for NH3 (5.2 ± 0.2% efficiency), whereas photolysis of such reactions recovered the same catalytic activity (8.9 ± 0.9 equiv per Fe, 17.8 ± 1.8% efficiency). This suggested that perhaps these species (i.e., dinuclear [(PP2)Fe(H)](μ-N2) and (PP2)Fe(N2)(H)2 proceed via a unified mechanism. It had been previously demonstrated that photolysis of (dmpe)2Fe(H)2 leads to H2 elimination and N2 binding.225–228 Similarly, low-temperature irradiation of (PP2)Fe(N2)(H)2 also released H2, leading to N2 binding.196
Efforts to detect protonated N2 species using these Fe(H) species proved unsuccessful. Instead, it seemed that such species might be involved in a parallel, unproductive hydrogen evolution cycle. Furthermore, (PP2)Fe(N2)2 was prepared and underwent protonation at Fe (and not N2) to product iron-bound hydride species. However, (PP2)Fe(N2)2 could be reduced by KC8 to structurally unusual monoanionic ([(PP2)Fe(N2)]−) (Figure 62) and dianionic ([(PP2)Fe(N2)]2−)complexes. [(PP2)Fe(N2)]2− underwent facile silylation to give the stable anionic diazenido complex, [(PP2)Fe(NNSiMe3)]−. However, oxidation of this latter species, or reaction of [(PP2)Fe(N2)]− with trimethylsilyl chloride, formed an unstable, neutral diazenido complex (presumed to be (PP2)Fe(NNSiMe3)) that was characterized at low temperature by EPR spectroscopy. Over time, this complex decayed to form P3Fe(N2)(SiMe3), via net silyl migration. This result highlights that migration steps that shift the electrophile to the metal can occur even when N2 is the kinetic site of functionalization. A similar issue has been discussed for (HIPTN3N)Mo.99
Figure 62.

N2 silylation reactions have been demonstrated with (PP2)Fe. The migration of SiMe3 from Nβ to Fe is particularly noteworthy.196
The nitrogen fixation chemistry of a related triphos-Fe system (cyclohexyl rather than isopropyl substituents on phosphorus) has been reported by Mézailles and co-workers (Figure 63). Notably, they found that an FeI hydride species was not stable (with isopropyl an N2-bridged dinuclear FeI hydride species forms), but rather that it disproportionated to form an FeII dihydride and Fe0 decomposition products. This highlights the effect that subtle changes in the ligand can have on species of potential relevance to N2RR. Nonetheless, they found similar efficiencies for nitrogen fixation as did the Peters group under analogous conditions (200 equiv of KC8 and 200 equiv of HBArF4 - up to 3.6 ± 0.3 equiv of NH3 per Fe, 5.4 ± 0.4% efficiency) and also note that the dihydride complex showed slightly poorer activity (2.7 ± 0.3 equiv of NH3 per Fe, 4.1 ± 0.4% efficiency).180
Figure 63.

Nitrogen fixation using a cyclohexyl-substituted tris(phosphino) (PP2)Fe complex.180
6. BEYOND Mo-AND Fe-PROGRESS TOWARD N2-to-NH3 CATALYSIS USING OTHER METALS
The catalytic systems described thus far have featured either Mo or Fe, and it is these two metals, and the extensive studies that have been undertaken to explore the mechanisms by which they fix N2, that have largely driven this field of catalysis forward. While Mo and Fe are known to be the biologically relevant metals in MoFe-nitrogenase, there is no reason a priori to anticipate that these should be the only metals able to mediate catalytic N2-to-NH3 conversion. Instead, catalytic studies focusing first on Mo and then Fe have played a dominant role in the advance of the field due to many inorganic chemists primarily rooting their explorations in the context of modeling biological nitrogenases.26,57,171,248,249 With the growing availability of different acid/reducing equivalent cocktails, conditions, and methods for analysis, it has become possible to systematically screen the efficacy of other transition metal systems. As a result, in the last five years exciting examples of N2-to-NH3 conversion catalysis by a number of metals, most of which are not biologically relevant (the exception being V), have been reported. As this occurs, design principles essential to this type of N2-to-NH3 conversion catalysis are beginning to emerge.
6.1. Evidence for Catalysis with a Tris(phosphino)borane Co System
As a complement to the Fe model work done with the P3B, P3C, and P3Si ligands, the N2RR capacity of the corresponding Co complexes has been explored (Figure 64). In all three cases, Co(N2) complexes could be prepared. An anionic [Co(N2)]− complex was only accessible for the (P3B)Co-system, which correlates with the extra valence electron provided by Co compared to Fe (i.e., [(P3B)Co(N2)]− is an 18-electron complex, whereas [(P3Si)Co(N2)]− and [(P3C)Co(N2)]− would be 19-electron complexes).52,202,250 Despite the extra valence electron in [(P3B)Co(N2)]−, the [(P3B)Co(N2)]0/− reduction potential was found to be anodically shifted by ~200 mV compared to [(P3B)Fe(N2)]0/− (Figure 64), suggesting the reduction potential is largely influenced by overall charge. Compared to the isoelectronic, [(P3C)Fe(N2)]− and [(P3Si)Fe(N2)]− (νNN for E = C, 1905 cm−1; E = Si, 1920 cm−1), the N2 ligand in [(P3B)Co(N2)]− (νNN = 1978 cm−1) is significantly less activated, consistent with poorer backbonding correlated with the greater electronegativity and less diffuse dorbitals for Co compared to Fe. When [(P3B)Co(N2)]− was subjected to excess HBArF4 and KC8 in Et2O at −78 °C, greater than stoichiometric amounts of NH3 (2.4 ± 0.3 equiv of NH3 per Co) were produced. While inefficient, any NH3 yield reliably greater than 2.0 per Co confirms that catalysis is operative; the NH3 yields were carefully reproduced to ensure this was the case. Hence, this study established the first example of a synthetic catalytic N2RR system featuring a nonbiologically relevant metal. The NH3 yields using [(P3B)Co(N2)]− under these conditions fall between those observed when using [(P3C)Fe(N2)]− or [(P3Si)Fe(N2)]− (4.6 ± 0.8 equiv of NH3per (p3c)Fe and 0.8 ± 0.5 equiv of NH3 per (P3Si)Fe) under the same conditions. The neutral derivatives (P3C)Co(N2) and (P3Si)Co(N2) gave ≤0.1 equiv of NH3 per Co under the same conditions, consistent with the general trend that access to an anionic [(P3e)M(N2)]− for these atrane scaffolds correlates with N2RR catalysis. The catalytic efficiency of these same Co complexes has also been surveyed using excess Cp*2Co and [Ph2NH2]OTf (Figure 64). With these reagents, [(P3b)Co(N2)]− yielded essentially the same amount of NH3 as [(P3Si)Fe(N2)]− (1.1 ± 0.4 equiv of NH3 per Co vs 1.2 ± 0.1 equiv of NH3 per Fe). Once again, neutral (P3Si)Co(N2) proved ineffective for mediating catalytic nitrogen fixation (0 equiv of NH3) under these conditions.80
Figure 64.

Electrochemical and spectroscopic comparison of [(P3B)Co(N2)]− and [(P3B)Fe(N2)]−. Maximum N2RR efficiencies are compared using KC8/HBArF4 and [Ph2NH2]OTf/Cp*2Co.52,80,173
These results, when considered collectively, suggest that the total electron count in the E–M linkage plays a key role in determining catalytic efficiency ({(P3B)Fe}8 ≫ {(P3C)Fe}9 ≈ {(P3B)Co}9 ≈ {(P3Si)Fe}9 ≫ {(P3Si)Co}10 = {(P3C)Co}10). This observation suggests the possibility that the more electron-deficient systems, such as {(P3Si)Mn}8 or {(P3C)Mn}8, could be promising targets for catalytic nitrogen fixation. Such systems have yet to be explored. Also, in accord with the observation that access to an overall anionic N2 complex is critical for catalysis, DFT-calculated charge maps show that in [(P3B)Co(N2)]− significantly more negative partial charge (δ) was localized on Nβ (δ = −0.51) than in its formally isoelectronic but neutral counterpart (P3C)Co(N2) (δ = −0.39).52
6.2. Improved Catalysis with (PNP)Co
Nishibayashi and co-workers have also studied Co-based N2RR catalysis (Figure 65)53 using the same bis-(phosphino)pyrrole platform they exploited previously for Fe (Figure 54).116 They found that stirring (PNP)Co with excess HBArF4 and KC8 at −78 °C in Et2O led to N2 fixation efficiencies similar to the Fe congener, both at lower (33 ± 1% NH3 per Co vs 37 ± 4% NH3 per Fe) and higher (max fixed-N Yield = 17.9 ± 1.0 for Co and 17.9 ± 0.8 for Fe) loadings of acid and reductant. The use of other solvents (e.g., methyl tert-butyl ether and THF) or reductants (e.g., K, Cp*2Co) led to suboptimal results. The authors argued that the significant improvement in N2RR by this (PNP)Co catalyst system as compared to [(P3B)Co(N2)]− points to an advantage of moving to a lower valence electron count system. (PNP)Co(N2) is a 16-electron complex (excluding π-donation from the pyrrole ring), and hence access to its anion [(PNP)Co(N2)]−, and possibly reduced downstream intermediates, may occur more readily than for the [(P3B)Co(N2)]− system. Clearly, other factors may also be at play.
Figure 65.

Transformation of (PNP)Co(H) to (PNP)Co(N2) via protonation, and their respective competency as precatalysts for N2 fixation using KC8 (40 equiv) and HBArF4 (38 equiv) in Et2O at −78 °C.53
As with (PNP)Fe(H),116 employing (PNP)Co(H) as the (pre)catalyst led to slightly decreased NH3 yields, though it was established that, at least at room temperature, the reaction of (PNP)Co(H) with HBArF4, and then KC8, led to the release of H2 and reformation of (PNP)Co(N2). Efforts to identify Co speciation under catalytic conditions led only to the identification of(PNP)Co(N2), suggesting the platform is fairly robust. Because of the observation that N2H4 forms in some reactions, (PNP)Co(N2) was reacted with 4 equiv of N2H4 in the presence of excess HBArF4 and KC8 under an Ar atmosphere. This resulted in the generation of 0.8 equiv of NH3 and recovery of 2.4 equiv of N2H4, with no observation of N2 formation. These data suggest that the Co complex was able to mediate the reduction of N2H4 to NH3, suggesting that N2H4 is a plausible intermediate and may be the source of some, or all, of the observed NH3.
6.3. Os and Ru Catalysts with a Tris(phosphino)silyl Ligand
As discussed above for (P3E)M(N2) complexes, accessing an anionic charge state appears to be important to N2RR activity, at least under catalytic conditions surveyed thus far. Thus, for N2RR to be achieved with the P3Si complexes of the heavier Group 8 metals, Ru and Os, electronically (and structurally) unusual zerovalent examples need to be accessible (Figure 66).251 Such a study is attractive as it affords an isoelectronic and isostructural set of [(P3Si)M(N2)]− group 8 complexes to help delineate factors most important to catalysis when proceeding down a group. The anionic, zerovalent complexes [(P3Si)Ru(N2)]− and [(P3Si)Os(N2)]− were thus prepared and tested for their catalytic performance in N2-to-NH3 conversion using KC8/HBArF4 and Cp*2Co/[Ph2NH2]OTf (Figure 66).54 With KC8 and HBArF4, it was found that [(P3Si)Fe(N2)]−, [(P3Si)Ru(N2)]−, and [(P3Si)Os(N2)]− gave 0.8 ± 0.5, 4.3 ± 0.3, and 1.6 ± 0.3 equiv of NH3 per M, respectively. By contrast, with Cp*2Co and [Ph2NH2]OTf, it was found that [(P3Si)Fe(N2)]−, [(P3Si)Ru(N2)]−, and [(P3Si)Os(N2)]− gave 1.4 ± 0.3, 0.8 ± 0.5, and 7.1 ± 0.6 equiv of NH3 per M, respectively. These results demonstrated the first examples of N2RR catalysis by Ru and Os.
Figure 66.

Comparison of physical properties and catalytic N2RR performance for known [(P3Si)M(N2)]n complexes (M = Co, Fe, Ru, Os; n = 0 or −1).
Particular attention was devoted to the system that was most efficient using the milder [Ph2NH2]OTf and Cp*2Co conditions, [(P3Si)Os(N2)]−. Compared to the (P3B)Fe system, there was a lesser decrease in overall efficiency (46% to 24%) as the loading of the acid and reductant was increased (36-fold in this case), resulting in the demonstration of a higher maximum turnover number (120 ± 11 equiv of NH3 per Os). This observation suggests potentially enhanced stability for the heavier Os analogue. Also, and in direct contrast to results described previously with (P3B)Fe,197 the yield of NH3 was found to be relatively invariant to the pKa of the acid employed, suggesting that the overall mechanism, or some limiting step, might be distinct between the two systems.197 Compared to the (P3B)Fe system, more soluble acid sources, such as HOTf, [Ph2NH2]BArF4, and [H3NPh]BArF4, were more detrimental to the overall N2RR performance of [(P3Si)Os(N2)]−, in this case leading to substoichiometric yields of NH3.
The poorer performance of the Os catalyst when using ether soluble acids may be accounted for by the rapid generation of hydrides. [(P3Si)Os(N2)]− reacted with HBArF4 at −78 °C to form (P3Si)Os(N2)(H) and (P3Si)Os(H)3; these two hydride complexes were demonstrated to be inactive as (pre)catalysts for N2RR. Potentially, hydride formation is diminished with the less soluble triflate-based acids, facilitating N2RR by comparison to HER.
Performing protonation reactions of [(P3Si)Os(N2)]− at even lower temperature (−135 °C, Figure 67) formed the cationic hydrazido(2−) complex [(P3Si)Os(NNH2)]+. This reactivity was similar to that observed for [(P3Si)Fe(N2)]− and [(P3B)Fe(N2)]−.109,113 Isolated [(P3Si)Os(NNH2)]+ was reacted with 46 equiv of [Ph2NH2]OTf and 50 equiv of Cp*2Co at −78 °C in Et2O (i.e., the catalytic conditions), but only 2.6 equiv of NH3 was generated. Under these conditions, [(P3Si)Os(N2)]− produced 7.1 ± 0.6 equiv of NH3. The decreased catalytic performance by what is presumed to be an on-path intermediate could be explained by the high reactivity of [(P3Si)Os(NNH2)]+, and also and its one electron reduced congener (P3Si)Os(NNH2). In this scenario, [(P3Si)Os(NNH2)]+ is present at much higher concentrations at early stages when used as the precatalyst, whereas it likely never builds up during catalysis when [(P3Si)Os(N2)]− is employed as the (pre)catalyst. The ability of [(P3Si)Os(NNH2)]+ (or (P3Si)Os(NNH2)) to undergo undesired reaction steps, for example, bimolecular H2 release or disproportionation to hydride species is thus higher than under the standard catalytic conditions. Alternatively, the kinetic insolubility of [(P3Si)Os(NNH2)]+ may hinder its performance; a similar explanation has been posited as an explanation for the poor catalytic performance of (P3B)Fe(N2) and (P3B–H)Fe(H)(N2).18,173
Figure 67.

Comparison of the reactivity of [(P3Si)M(N2)]− (M = Fe or Os) complexes with excess acid at different temperatures.54,109
Given the aforementioned observation that anionic [(P3E)M(N2)]− complexes are most effective for facilitating the first protonation step, the reduction of (P3Si)Os(N2) was attempted with excess Cp*2Co. Curiously, it was found that this reduction occurred reversibly, but only upon cooling; no reaction between (P3Si)Os(N2) and Cp*2Co occurred at room temperature. This fact suggests that the redox reaction is entropy-controlled. For Fe and Ru, the [(P3Si)M(N2)]0/− couple is more negative, suggesting that the key, anionic complexes, [(P3Si)M(N2)]− may be inaccessible with Cp*2Co, potentially accounting for their poor performance under these conditions. This observation is also important to keep in mind with respect to the factors that lead to better N2RR performance at low temperature by [(P3E)M(N2)]− and a variety of other systems.
6.4. Establishing Catalysis with V
Until recently, vanadium was the only metal ion known to be present in a functionally active nitrogenase enzyme (VFe-N2ase),155 which had not been successfully incorporated into a synthetic catalyst for N2RR.252 With the discovery of more successful conditions for N2RR, it was possible to establish the efficacy of early metals such as V (and also Ti; vide infra). Specifically, Nishibayashi and co-workers reported in 2018 that using the same PNP ligand (2,5-bis(dialkylphosphinomethyl)-pyrrolide)) that they had previously exploited for N2RR mediated by Fe (Figure 54)116 and Co (Figure 65)53 could be exploited to achieve N2RR by V in the presence of excess KC8 and HBArF4 at low temperature (Figure 68).56 Inclusion of an aryloxide ligand, an approach also recently adopted by Schrock and co-workers in an diamidopyridine-pincer-ligated Mo system,140 proved necessary for N2RR.
Figure 68.

Synthesis of pyrrole-based (PNP)V precatalysts for catalytic N2RR.56
The maximum catalytic efficiency achieved by this V system (56 ± 3%) at low loadings was slightly superior to those reported for other catalysts using KC8 and HBArF4 at low temperature.18,53,116,173 However, increased turnover numbers (up to fixed-N yield of 16.0 ± 1.4) were only accessible with a significant attenuation in efficiency (23 ± 1.5%). The parent amide complex, (PNP)V(NH2)(OAr), also proved an effective (pre)catalyst, consistent with (but not necessitating) its intermediacy along the catalytic cycle.
6.5. Nishibayashi’s Work on PNP with Other Metals
Related to the above study, Nishibayashi and co-workers recently reported the formation of PNP-ligated Ti and Zr dinitrogen complexes.253 It was found that the use of Cp as an additional ligand facilitated N2 binding upon reduction of halide precursors. Formation of PNP- and Cp-ligated Ti and Zr–N2 complexes has been reported previously.254 These bridging N2 complexes were then tested for catalytic activity, both with KC8/HBArF4 and Cp*2Co/[Ph2NH2]OTf; in all cases, less than 1.4 fixed-N atom equivalents were observed.
6.6. Triamidoamine Ligands on Ti
Triamidoamine-ligated Ti complexes have recently been reported to be competent for catalytic N2-to-NH3 conversion (Figure 69).55 Like Schrock and co-workers previously,255 Liddle and co-workers found that the trimethylsilyl substituted triamidoamine ligand (TMSN3N) gave rise to a dinuclear μ-N2 complex [(TMSN3N)Ti]2(μ-N2) (Figure 69). However, with Ti upon reduction the bridging motif was retained and a dianionic complex [[(Me3SiN3N)Ti]2(μ-N2)}2− was formed, in which the N–N bond lengthened (1.121(6)−1.315(3) Å) and weakened dramatically (ΔνNN = −500 cm−1). Even after encapsulation of the K+ ions with crown ether, the N2 ligand was still highly activated. Indeed, DFT calculations suggested that the N2 ligand is reduced to a hydrazido(2−) state in {[[Me3SiN3N]Ti]2(N2)]2−. Reaction of [[(Me3SiN3N)Ti]2(μ-N2)]2− in pentane with ethereal HCl led to substantial fixed-N yields (0.88 equiv of N2H4 and 0.13 equiv of NH3). In contrast, reaction of the neutral complex, [(Me3SiN3N)Ti]2(μ-N2), with ethereal HCl produced only 0.03 equiv of N2H4 and 0.01 equiv of NH3.55
It was found that by pairing KC8 with phosphonium acids [R3PH]+ (R = Cy, nBu, tBu; pKa = 9.7, 8.4, and 8.0 in CH3NO2, respectively)256 catalytic NH3 formation could be observed.55 The best efficiencies reported (17%) were achieved with I− as the counteranion and Et2O as the solvent. Of mechanistic interest was the suggestion by the authors that the end-on bridging N2 mode was critical for functionalization. This mode of binding often leads to significant activation of the N2 unit but is typically presumed to kinetically retard electrophilic functionalization.211,257 The authors suggested that such a bridged state can be protonated in the present system to form a bridging hydrazine complex that is then released, with the final reduction to NH3 mediated by the acid and reductant. The authors did suggest caution with respect to this hypothesis, noting it warrants further investigation to rule out the transient formation of a Ti(N) species. The recent suggestion by the Nishibayashi group that the inclusion of I− in their catalytic reactions biases the mechanism toward cleavage of a bridging N2 ligand to form two Mo(N) species81 underscores that such caution is warranted until/if more data become available.
If it ultimately proves true that μ-N2 complexes in this Ti-system are active for nitrogen functionalization, then known (RN3N)M–N2–M(NN3R) complexes (M = V, W, Mo)98,255,258,259 may warrant further study for applications in catalytic N2RR. Furthermore, the pairing of KC8 with the weak, insoluble phosphonium iodide acids suggests that known systems that rely on KC8 should be reinvestigated for their activity with this acid source. Previously, all systems that used KC8 relied on the use of the highly soluble and very strong acid HBArF4,18,52–54,56,116 which can lead to rapid and undesired metal protonations, or background H2 evolution.
7. ELECTROCATALYTIC NITROGEN FIXATION
The vast majority of studies of homogeneous molecular catalysts that mediate N2RRhave utilized chemical reductants. However, replacement of chemical reductants with an electrode is an attractive route for developing new N2RR technologies. Such an approach could enable distributed fertilizer production strategies as a complement to Haber–Bosch. Also, if one considers that ammonia is a high energy density liquid in its easily condensed form, it becomes a very attractive solar fuel target if its synthesis can be efficiently driven by renewable energy resources.260–264 While there is a great deal of work ahead to move this realm of N2RR catalysis forward, recent results have demonstrated that electrocatalytic nitrogen fixation with well-defined molecular catalysts is indeed possible. These studies will hopefully be complementary to the burgeoning field of heterogeneous electrocatalytic N2RR6,262,264 and the more recently demonstrated bioelectrocatalytic N2RR.265,266
7.1. Early Results on Mo and W
Early work on the electrosynthesis of ammonia from nitrogen using coordination complexes was motivated by the early work of Chatt and co-workers.15 In particular, Chatt and co-workers had demonstrated that the protonolysis of (dmpe)2W(N2)2 (dmpe = dimethylphosphinoethane) in alcoholic solvents led to the formation of nearly 1 equiv of NH3.58 Thereafter, Pickett and co-workers demonstrated the first electrosynthesis of ammonia with a molecular complex using this same system.267 Protonation of (dmpe)2W(N2)2 with p-toluenesulfonic acid (TsOH) led to quantitative formation of the hydrazido(2−) complex, [(dmpe)2W(NNH2)(OTs)]+ (Figure 70). Cyclic voltammograms of the cationic hydrazido complex featured an irreversible reduction at −2.37 V vs Fc+/0.268 Exhaustive electrolysis of this species at −2.6 V vs Fc+/0 at a mercury-pool electrode consumed 2 faradays. Product analysis showed the formation of 0.22−0.24 mol of NH3 and 0.01−0.02 mol of N2H4 per mol of [(dmpe)2W(NNH2)(OTs)]+ (Figure 70). These results were confirmed both by 15N isotopic labeling and a variety of control reactions.
Figure 70.

Electrosynthetic cycle for the formation of NH3 from N2 by a (dppe)2W complex.267
Post electrolysis, 31P NMR and IR spectroscopy and cyclic voltammetry measurements confirmed that the starting (dmpe)2W(N2)2 complex was regenerated in 85−95% yield.268 The use of a platinum electrode led to similar fixed-N yields, but a lower recovery of the W(N2)2 complex. As no excess acid was added to these experiments and an aprotic solvent (THF) was used, the additional protons necessary to form NH3 were likely derived from the starting complex. Consistent with this hypothesis, the cationic diazenido complex [(dmpe)2W(NNH)]+ was observed by multinuclear NMR spectroscopy and cyclic voltammetry during the electrolysis. Thus, the maximum amount of NH3 that could have been formed is 0.66 mol, so the overall electrolysis process was ~36% efficient. These data are consistent with a pattern, in which net H atom transfer between NxHy intermediates is critical to engendering productive N2 fixation (see previous discussion of Schrock’s (HIPTN3N)Mo(NNH2)/(NH)100 and Peters’ [(P3Si)Fe(NNH2)]0/+).109 Cycling the process three times by first electrolyzing and then adding acid to the solution led to the formation of 0.73 mol of NH3 in total per mole of starting [(dmpe)2W(NNH2)(OTs)]+.267
These systems were further investigated by Becker and co-workers.269 They studied related Mo and W phosphine complexes via cyclic voltammetry with the aim of evaluating their applicability to electrocatalytic N2RR. Cyclic voltammograms of the dinitrogen complexes, (dppe)2M(N2)2 (dppe = diphenylphosphinoethane, M = Mo, W), did not reveal any redox processes. However, as observed by Pickett and co-workers, the cationic hydrazido species, [(dppe)2Mo(X)-(NNH2)]+ (X = Br, F, HSO4) and [(dppe)2W(Br)(NNH2)]+, featured irreversible reduction events. Cyclic voltammograms of the hydrazido species in the presence of an inorganic acid (3–5 equiv) led to an observable increase in current (10–20%), suggesting the possibility of an electrocatalytic process (i.e., HER or N2RR). Therefore, controlled potential electrolysis (CPE) was performed in the presence of inorganic acids (3–5 equiv). However, yields of NH3 were lower than those observed by Pickett and co-workers, with the observed NH3 yields for OTs− > HSO4− > Br−. While yields of NH3 from CPE were never high enough to establish bona fide electrocatalysis, they were somewhat enhanced by running the experiment under N2, but also under CO. These observations led the authors to suggest that while atmospheric N2 was not being incorporated into NH3, the electroreduction of one of the intermediates was enhanced by loss of the X-type ligand and binding of a π-acid.
Relatedly, Shilov and co-workers reported a number of studies looking at the electrocatalytic reduction of N2 by Mo salts in methanolic solution at a Hg drop electrode.20 In these studies, they primarily interrogated the activity of ill-defined metal salt mixtures that are not further described here. However, after discovering that phosphatidyl choline (a phosphine-capped fatty acid) enhanced the reactivity of their Mo salts,76 they tested the effect of adding (PPh2Me)4Mo(N2)2 and (dppe)2Mo(N2)2 to their CPE experiments.77 The inclusion of these complexes did not lead to any enhancement in NH3 formation.
The cumulative work by these groups suggests that while these bis-diphosphine Mo and W complexes are suitable for the stepwise electrosynthesis of NH3, they are not capable of electrocatalytic NH3 formation, at least under the conditions thus far studied.77
7.2. Achieving True Electrocatalysis with (P3B)Fe
In 2016, Peters and co-workers studied the effect of acid addition, HBArF4, to cyclic voltammograms of [(P3B)Fe]+ (conditions: −45 °C, 0.1 M [Na]BArF4 in Et2O).Upon addition of 5 equiv of HBArF4, only minimal current enhancement was observed at the [(P3B)Fe]+/(P3B)Fe(N2) couple (−1.5 V vs Fc+/0), but a more substantial enhancement was observed at the [(P3B)Fe(N2)]0/−couple (−2.2 V vs Fc+/0). Analysis of this current enhancement is likely complicated by competing catalytic hydrogen evolution and N2RR. A CPE experiment with a vitreous carbon-working electrode at −2.6 V vs Fc+/0 with 10 equiv of HBArF4 led to the formation of NH3 (0.5 equiv, FE = 18%) and H2 (FE = 58%). A further CPE experiment at −2.3 V vs Fc+/0 in the presence of 50 equiv of HBArF4 formed 2.2 equiv of NH3 per Fe (FE = 25%) and 6.6 equiv of H2 per Fe (FE = 48%). This result placed this system on the precipice of electrocatalytic formation of NH3 as any reliable NH3 yield above 2.0 equiv per Fe requires a catalytic process.173 Significant effort was made to improve the yield of NH3, including changes in the acid (i.e., more acid, batchwise acid addition, constant acid addition) and the ratio of the electrode surface area to the working compartment solution volume (i.e., smaller cell geometries, different carbon morphologies).197 These efforts did not result in substantial improvement to the overall turnover number (TON ≤ 2.6 equiv of NH3 per Fe).
Following the discovery that chemical catalysis could be mediated at significantly higher efficiencies by Cp*2Co and [Ph2NH2]OTf80 than by KC8 and HBArF418,173 efforts to adapt these new conditions to electrocatalysis were undertaken. Cyclic voltammograms of [(P3B)Fe]BArF4 at −35 °C in 0.1 M [Na]BArF4 solution changed significantly upon addition of 10 equiv of [Ph2NH2]OTf. Notably, the [(P3B)Fe]+/(P3B)Fe(N2) couple observed previously disappeared and was replaced by an irreversible feature at ~ −1.9 V vs Fc+/0. Cyclic voltammograms of [(P3B)Fe]BArF4 with added [TBA]OTf suggest this feature arose from the reduction of (P3B)Fe(OTf). Scanning the cyclic voltammogram further in the cathodic direction in the presence of [Ph2NH2]OTf revealed a substantial catalytic enhancement at the [(P3B)Fe(N2)]0/−couple. CPE experiments under these conditions (−35 °C, Et2O, 0.1 M [Na]BArF4, 50 equiv of [Ph2NH2]OTf) led to a similar yield of NH3 (2.6 ± 0.3 equiv per Fe, 24 ± 5% FE) as observed with HBArF4.
Given the evidence for the role of a protonated metallocene, [Cp*(exo/endo-η4-C5Me5H)Co]+, in N–H bond formation during the chemical catalysis,80,197,199 it was explored whether the addition of cocatalytic [Cp*2Co]BArF4 additive might enhance the yield of NH3. As expected, cyclic voltammograms of [Cp*2Co]BArF4 at −35 °C in 0.1 M [Na]BArF4 solution revealed the reversible [Cp*2Co]+/0 couple at −2.0 V versus Fc+/0. Addition of 10 equiv of [Ph2NH2]OTf led to the observation of a catalytic enhancement at this reduction event. CPE experiments with [Cp*2Co][BArF4] in the absence of [(P3B)Fe]BArF4 resulted in catalytic H2 formation (FE = 75%) but no NH3 formation. Cyclic voltammograms with [(P3B)Fe]BArF4, [Cp*2Co]BArF4, and [Ph2NH2]OTf also showed the onset of a catalytic wave at ~ −2.0 V versus Fc+/0. CPE experiments (−2.1 V vs Fc+/0) in the presence of [(P3b)Fe]BArF4 and 1 equiv of [Cp*2Co][BArF4] provided unequivocal evidence for electrocatalytic nitrogen fixation (4.0 ± 0.6 equiv of NH3 per Fe/Co, 28 ± 5% FE, Figure 71). The use of higher amounts of [Cp*2Co]BArF4 did not enhance the yields or improve the FE. However, it is important to note that in CPE experiments with 5 equiv of [Cp*2Co]BArF4, addition of further substrate after completion of the initial CPE experiment enhanced the yield of NH3 from 4.0 ± 0.6 equiv of NH3 per Fe to 5.5 ± 0.9 equiv of NH3 per Fe. XPS experiments on the electrode at the end of the CPE indicated a modest degree of Fe decomposition but provided no evidence for Co decomposition. Furthermore, CPE experiments performed after rinsing the electrode did not lead to the formation of NH3. In sum, this system represents the first example of electrocatalytic N2RR by a well-defined coordination complex.
Figure 71.

First demonstration of molecular, electrocatalytic N2RR enabled by the use of cocatalytic [Cp*2Co]+. Conditions: glassy carbon, Et2O, −35 °C, −2.1 V versus Fc+/0, 0.1 M [Na]BArF4, X equiv of [Cp*2Co]BArF4.197
To compare the effect of using an electrode as compared to a chemical reductant, N2 fixation experiments were performed with [(P3B)Fe]BArF4 in the presence of 108 equiv of [Ph2NH2]OTf and 54 equiv of Cp*2Co at −35 °C in 0.1 M [Na]BArF4 Et2O. Only 1.8 ± 0.7 equiv of NH3 per Fe was formed (Figure 72). This result is much lower than the 12.8 ± 0.5 equiv of NH3 per Fe observed under the standard chemical catalytic conditions,80 potentially due to the higher temperature (−35 °C vs −78 °C) and/or higher polarity solvent (0.1 M [Na]BArF4 Et2O versus Et2O). More significantly, the performance with the chemical reductant is poorer than that with the electrochemical reductant under analogous conditions (4.0 ± 0.6 equiv of NH3 per Fe).197 This observation suggests that under certain conditions electrocatalysis can be superior to chemical catalysis. However, challenges in developing better electrocatalytic systems remain, as the conditions that have thus far worked best (insoluble acid, low dielectric media, and low temperature) are not conducive to standard electrochemical techniques. Nevertheless, these results provide a springboard for future work in electrocatalytic N2RR.
Figure 72.

Reproduced with permission from ref 197. Copyright 2018 American Chemical Society. (A) Cyclic voltammograms of 10 equiv of [Ph2NH2]OTf (gray trace), 1 equiv of [Cp*2Co]BArF4 (Cp*2Co+) (yellow trace), 1 equiv of Cp*2Co+ with 10 equiv of [Ph2NH2]OTf (green trace), and [(P3B)Fe]+ with 1 equiv of Cp*2Co+ and 10 equiv of [Ph2NH2]OTf (red trace); (B) Cyclic voltammograms of 10 equiv of [Ph2NH2]OTf (gray trace), 1 equiv of [(P3B)Fe]+ (dark blue trace), 1 equiv of [(P3B)Fe]+ with 10 equiv of [Ph2NH2]OTf (light blue trace), and 1 equiv of [(P3B)Fe]+ with 1 equiv of [Cp*2Co]+ and 10 equiv of [Ph2NH2]OTf (red trace). All voltammograms are collected in 0.1 M [Na]BArF4 solution in Et2O at −35 °C using a glassy carbon working electrode and externally referenced to the Fc+/0 couple. Scan rate is 100 mV/s.
7.3. Work on (Cp)xTi-Species
In one of the first reports regarding the fixation of N2-to-NH3 under ambient conditions by metal complexes, Shur and Vol’pin found that the use of Cp as a ligand dramatically enhanced the performance of TiIV complexes for N2 fixation by comparing Ti(Cl)4 with (Cp)2Ti(Cl)2.270 This report spawned substantial interest in (Cp)2Ti(Cl)2 for nitrogen fixation; however, the mechanism of N2 activation and fixation remains disputed.271–275 Nonetheless, (Cp)Ti-complexes are among the most highly investigated systems for electrosynthetic nitrogen fixation (Figure 73).
Figure 73.

Comparison of conditions and results for the electrosynthesis of NH3 from N2 by (Cp)2Ti(Cl)2.276–278
The first report of (Cp)2Ti(Cl)2 in the context of electrocatalytic nitrogen fixation was by Becker and co-workers, who studied its ability to reduce N2 in MeOH and THF at platinum and glassy carbon electrodes with catechol as the proton source. This system was found to be more efficient than with other titanium salts, but yields were nonetheless very poor, with the best results achieved in the presence of a Mg2+ additive (0.0145 equiv of NH3 per Ti and 0.28% Faradaic efficiency (FE), Figure 73).276
A more recent report from Yoon and co-workers examined the electroreduction of (Cp)2Ti(Cl)2 in MeOH, H2O, and wet THF with LiCl electrolyte (Figure 73). They found that the maximal rate of NH3 formation (9.5 × 10−10 mol·cm−2·s−1·M (Cp)2Ti(Cl)2−1) occurred in water at −1 V vs RHE, and that the maximal FE for NH3 formation (0.95%) occurred at −2 V vs RHE in THF. Yields with respect to Ti were not reported.277
Masuda and co-workers have looked at the effect of using ionic liquids as the solvent and electrolyte on the electrosynthesis of NH3 by (Cp)2Ti(Cl)2. When (Cp)2Ti(Cl)2 was immobilized in a solid polymer electrolyte cell with an ionic liquid (Pyr4FAP) as the supporting material and water as the proton source, they found that the introduction of N2 led to significant current enhancement in the cyclic voltammogram at the reductive feature corresponding to (Cp)2Ti(Cl)2 reduction to (Cp)2Ti(Cl) (Figure 73). However, they also noted that most of the current enhancement stems from hydrogen evolution mediated by a Ti(N2) complex rather than nitrogen fixation. Nonetheless, CPE experiments at −1.5 V vs Fc+/0 revealed a significant improvement in the yield of NH3 with little change in the FE (0.27 equiv of NH3 per Ti, 0.2% FE). This represented a notable improvement over the previous conditions, given the lower applied bias used along with a weaker acid source (H2O vs catechol). The authors attributed the formation of NH3 at lower applied bias to enhanced N2 binding afforded by the ionic liquid environment. In a subsequent report, they demonstrated that in this ionic liquid, TiIII can bind N2, which has not been observed in traditional solvents (i.e., MeOH, THF, H2O).279 These observations suggest that the use of ionic liquids may provide a promising strategy for electrocatalytic nitrogen fixation by helping to both polarize the N2 molecule and to activate it for binding/reduction, and in limiting proton reduction. More recent work from Masuda and co-workers has demonstrated that the related dimeric species, {(Cp)2Ti(μ-Cl)}2, can provide higher yields and FEs for nitrogen fixation (up to 0.34 equiv of NH3 per Ti and up to 1.44% FE).278
7.4. (Electro)synthesis of NH3 with p-block Metals
In a series of papers, Furuya and co-workers examined the potential of different phtahlocyanines loaded on to gas-diffusion electrodes for nitrogen fixation. CPE experiments with Fe-substituted phthalocyanine at −0.6 V vs RHE in 1 M Na2SO4 led to an initial FE of 1.6%, but a sharp reduction to 0.1% was observed after 10 min.280 Interrogation of the effect of different electrolytes on the efficiency of the electrocatalysis led to the observation that potassium electrolytes were uniformly superior to sodium electrolytes, with 1 M KOH giving the best performance with retention of >0.1% efficiency for at least 30 min.281 However, when a variety of phthalocyanines were tested (M = H, Ti, Fe, Co, Ni, Pd, Pt, Cu, Zn, Al, Ga, In, Sb, Sn), the best and most stable FE was observed with Sn (max 1.8%, 1.2% at 25 min).282 However, more recent work on Sn-phthalocyanine has demonstrated that it is not an electrocatalyst for N2RR, but rather that the detected NH3 comes from the decomposition of contaminants in the starting materials.283 This and other recent work exploring the confounding sources of NH3 in electrocatalytic N2RR by materials284 illustrate the importance of careful control reactions for establishing the source of NH3 in studies toward electrocatalytic N2RR.
Recently, Berben and co-workers reported the electrosynthesis of NH3 with an aluminum complex, notable as the first example of the formation of fixed-N products by a main group metal.285 Cyclic voltammograms of the pyridine-diimine (PDI) ligated Al complex (Figure 74) in the presence of 4-dimethylaminopyridinium and N2 led to the observation of enhanced current. CPE at −1.16 V versus SCE yielded 21% NH3 and 16% H2 (the poor overall FE was not unexplained). The source of the NH3 was confirmed to be N2 via isotopic labeling. The lack of turnover was explained by product inhibition; the starting complex was demonstrated chemically to be unstable to NH3. Interestingly, no N2 complex is known for this platform, so more work is needed to begin to elucidate an overall mechanism (Figure 74) for this process. The authors asserted that the overall reduction reaction is occurring at a significantly lower overpotential than other known, catalytic systems. However, their analysis used pKip values as substitutes for pKa values in nonpolar solvents. The arbitrary zero point of the pKip scale in these solvents creates a significant bias against those systems operating in nonpolar solvents (i.e., Schrock and Nishibayashi’s original Mo work).50,51 Hence, caution in this interpretation is warranted. Regardless, this report, along with recent work from Braunschweig and co-workers describing reductive capture of N2 by two borylene units and subsequent protonation to a hydrazine derivative, provides inspiration for future work on nitrogen fixation by p-block elements (Figure 74).286
Figure 74.

(Top) Preliminary, posited mechanism for the electrosynthesis of NH3 by a (PDl)Al complex;285 (bottom) stoichiometric N2 binding and protonation at a (CAAC)B species; Dipp = 2,6-diisopropylphenyl, Dur = 2,3,5,6-tetramethylphenyl.286
8. MECHANISTIC INSIGHTS FOR FUTURE CATALYST DESIGN
Having discussed aspects of synthetic nitrogenases that have been developed to date, we finish this review by summarizing some lessons we think will be useful to consider in designing future catalysts.
-
1
Balancing sterics: Key to the design of efficient N2 fixation catalysts is the preparation of molecular species with the appropriate sterics. Sterics can control the rate or nature of dimer formation. For example, see work from the Nishibayashi group on the effect of phosphine substituents on cis- vs trans-dimer formation (Figure 27),141 the control that wingtip bulk has over the formation of terminal vs bridged N2 species in triamidoamine Mo chemistry from Schrock and co-workers.98,255 The mechanism (and presumably rate) of reactant/product exchange is also dependent on sterics. For example, we have noted that among the Schrock systems that (DPPN3N)Mo(N2) undergoes associative rather than dissociative N2 exchange.134 Similarly the site (and presumably rate) of ligand protonation is controlled by sterics. One clear example comes from Nishibayashi and co-workers, in which they prevent protonation in the 3,4-positions of the pyrrole ligand in their (PNP)Fe ligand by introducing methyl substitution (Figure 54).178 Lastly, sterics can be important for controlling the rate of bimolecular decomposition. This includes the rate of formation and degradation of intermediates. This idea is highlighted in ability to observe a diazenido intermediate, Fe(NNH), only when using a highly sterically encumbering ligand (ArP3B) (Figure 44). Notably, while this system is better at stabilizing this highly reactive intermediate, it is a poorer catalyst than the parent system, (P3B)Fe.177 Thus, a Goldilocks approach to sterics is necessary for achieving optimal functionality, as is the case for a variety of catalytic reactions.
-
2
The design of innocent yet activating ligands: One factor that dictates the efficacy of N2-to-NH3 reduction catalysis is linked to the ease by which protonation occurs at Nβ. Activation of N2 proceeds by cooperative donation from a transition metal or main group compound and the given ligand set through inductive effects. The importance of N2 activation is reflected in (i) the need to access [(HIPTN3N)Mo(N2)]− for protonation at Nβ to occur - (HIPTN3N)Mo(N2) is not protonated at Nβ by catalytically relevant acids (Figure 10),102 (ii) the catalytic enhancement that is observed for the (PNP)Mo catalyst upon 4-OMe substitution (Figure 30),82 and (iii) in the necessity of accessing an anionic [(P3E)M(N2)]− state (M = Fe E = B, C, Si; M = Ru, Os E = Si) to observe N2RR catalysis with (P3E)M species (Figure 66).54,80 Again, protonation of Nβ of neutral (P3E)M(N2) species has not been observed. On the other hand, highly donating ligands that enhance the electron richness of the metal center also render it harder to reduce, thereby necessitating stronger reductants to achieve the low-valent states necessary to initiate catalysis. Furthermore, more donating ligands are often more Bronsted basic and are thus prone to protonolysis, thereby limiting turnover due to degradation of the catalyst. In this respect, neutral phosphine ligands appear to be far superior to amide ligands for enhancing overall stability and thus turnover.50,79,116,173 It is also worth highlighting the utility of adaptable ligand frameworks that help to stabilize a variety of formal metal oxidation states and [M]-NxHy bond orders—this concept is highlighted by Peters’ use of boratrane and silatrane-containing ligands, for example109,113,114,181,193
-
3
Tuning N2 Reduction Overpotentials with different reagent combinations: Central to the reduction of N2 are the reagents that are employed for catalysis. From this perspective, it is noteworthy that no single set of catalytic conditions can be applied to all of the catalytic systems that have been reported to date. Indeed, a number of electron sources (metallocene,50 KC8,18 electrode,197 Sm(II)79) and proton sources (pyridiniums,50 protonated ether,18 aniliniums,174 phosphoniums,55 water/alcohols79) have all been employed. The particular combination of acid and reductant, along with other relevant reaction conditions, defines the net overpotential for the catalytic reaction. It has long been known in heterogeneous catalysis that overpotential is correlated with reaction rate;287 more recently, it has been increasingly recognized in related fields (ORR, CO2RR, HER, etc.) that similar constraints exist,288–292 and some theoretical approaches, and physical intuition, suggest that a similar relationship exists in catalysts for homogeneous N2RR.293
Notable in N2RR is also that the acids and reductants typically only work in particular pairs. For example, metallocenes and pyridinium acids effectively promote N2RR catalysis with Nishibayashi’s Mo systems (Figure 33),51 as do Sm(II) and water/alcohols (Figure 37), but combining Sm(II) and pyridinium acids has been shown to be ineffective.79 Similarly, both KC8/HBArF4 and Cp*2Co/[Ph2NH2]OTf are efficient N2 fixation cocktails with the (P3B)Fe-system,18,80 but the combination of Cp*2Co and HBArF4 proves ineffective. Indeed, changing these reagent pairs can have a larger impact on efficiency, TON, and TOF than does changing the catalyst itself (Figure 66). This is an important point. Whereas it may seem less exciting to a budding coordination chemist to survey reagents than to design an elegant new catalyst system supported by a new ligand, the development of reagent combinations for canvassing nitrogen fixation will drive the field forward at an accelerated pace.
In the past few years, defining the overpotential supplied by different reagent cocktails in catalytic N2RR has become increasingly a point of focus. Two means of measuring the overpotential have been discussed in the literature: the first, more traditional approach, is to determine the thermodynamic potential of nitrogen fixation for a given solvent at a given pKa (or pH).285,294 The overpotential, or η, is then the difference between this value and the applied potential. This approach is useful in facilitating comparisons with heterogeneous N2RR electrocatalysts and for defining the potential of the most reducing intermediate in the N2RR cycle. However, developing accurate overpotential values in the apolar solvents that to date have been commonly used for chemical nitrogen fixation (i.e., heptane, toluene, Et2O) is not feasible. These values are, however, well-defined for H2O and acetonitrile,295 with extension to THF being seemingly possible.296 A second approach that has been developed as a measure of “overpotential,” typically noted as ΔΔGf/ΔΔHf, in the literature takes the approach of comparing the energetic input to the energy of formation for NH3, in which N2 is fixed using H2 as the source of protons and electrons (Figure 75).79,80,105 Thus, this metric provides for a ready comparison to the Haber–Bosch process, in which H2 is employed to achieve N2-to-NH3 conversion. To define ΔΔGf, one first needs to know the effective bond strength of the reagent combination used (i.e., Cp*2Co and [Ph2NH2]OTf) as defined by eq 1 (where CG is a solvent-dependent thermodynamic constant accounting for the energy of formation for H• from H+ and e− in solution).295 Comparing this effective BDFE with the energy of H• formation derived from H2, found by dividing the BDFE(H2) by two, defines the extra energy being used in the system to generate each N–H bond. To get the overpotential (ΔΔGf) per mol of NH3, one then multiplies by three to account for the three N–H bonds in NH3 (eq 2).
| (1) |
| (2) |
Figure 75.

N2 overpotentials calculated as a function of catalyst reagent cocktail using eqs 1 and 2. All of the pKa values and reduction potentials are literature values for these reagents in acetonitrile.297–299 Consequently, the Cg and BDFE(H2) in acetonitrile were also used.295 The exception is for Sm/H2O for which aqueous values are used, and the values are approximated due to the inner-sphere nature of the Sm/H2O interaction. The bottom portion of the figure provides a sample overpotential calculation.154
Again, the necessity of a well-defined pKa scale and a CG constant also limits the accuracy of this approach to particular solvents (e.g., H2O, MeCN, THF, etc.). This approach also does not account for the excess energy being supplied by the conversion of NH3 to NH4+, which readily occurs under most of the catalytic conditions discussed in this review. However, the appeal of this approach lies in the potential insight it provides into the N–H bond strengths of key N2RR intermediates (i.e., [M]-NNH or [M]-NH).
-
4
Reaction conditions: Nitrogen fixation catalysis is sensitive to solvent, temperature, and reagent concentration. In many cases, these factors control the interplay between catalyst-mediated nitrogen fixation, catalyst-mediated hydrogen evolution, and background hydrogen evolution. Different strategies have been employed in an effort to control these factors, including performing reactions at lower temperature,18,116,174 the use of nonpolar solvents,50,51 and the slow addition of reagents.50,51 Further, in most cases discussed herein, at least one catalyst component (acid or reductant) has been less soluble than the other in an effort to attenuate the rate of reactions between them (e.g., HER). The importance of these factors can be highlighted by dramatic differences in efficiencies. For example, this is demonstrated by some of Nishibayash’s systems, for variable rates of reductant addition have led to improved catalysis (Figure 25),51 and also in the strong correlation of performance of the (CAAC)2Fe-catalyst system with temperature (Figure 55).172 Nonetheless, many of these factors, and other potentially important factors (e.g., N2 pressure, NH3 concentration) remain unexplored for most of these platforms. Furthermore, continue to develop conditions that would be more highly compatible with electrocatalysis, namely, higher solvent polarity, higher reaction temperatures (especially for non-Mo catalysts), and conditions that do not require slow addition of the acid (especially for Mo catalysts).
-
5
A role for PCET: Although the ability of PCET reactions to mediate stoichiometric N–H bond forming reactions in noncatalytic platforms has been extensively demonstrated,105,217,300–302 discussions on the mechanism for nitrogen fixation by homogeneous catalysts had traditionally focused on stepwise ET-PT or PT-ET pathways.80,106,128 In the past few years, there has been increased discussion of PCET steps as a possible mechanism for the N–H bond forming steps in catalytic systems with a particular emphasis on its critical role in the formation of intermediates with weak N–H bonds (BDFEN–H < 50 kcal·mo−1). Proposed species of relevance to PCET in nitrogen fixation catalysis have included pyridinyl radicals,103,105,106 protonated metallocenes (Figure 47);80,197,199 encounter complexes of a reductant with an acid and a nitrogenous ligand that are hydrogen-bonded (MS-PCET) (Figure 18),103,140 and alcoholic or aquo complexes of SmI2 (Figure 37).79 This recent work suggests that a fresh look at earlier mechanistic assumptions may be warranted, but the more important conclusion to draw is that PCET reactions may offer a path forward for developing catalyst systems that operate at lower net overpotentials and improved rates.
9. FUTURE OUTLOOK: REMAINING CHALLENGES
We next provide an overview of some of the important research opportunities that remain for N2RR catalysis mediated by synthetic complexes.
-
6
N2 fixation with improved selectivity: Nishibayashi and co-workers have recently demonstrated two different cases, (PPP)Mo with Cp*2Co/[ColH]OTf81 and (PCP)Mo with SmI2/HOCH2CH2OH (Figure 37),79 the ability to achieve >90% selectivity for NH3. These results represent significant improvements on the maximum efficiency of these catalysts in the presence of the original Schrock conditions (Cp*2Cr/[ColH]OTf) with which (PNP)Mo achieved ~50% selectivity for NH351 and (PCP)Mo achieved ~66% selectivity.149 The improved selectivity of the new conditions comes at the cost of greater energetic input. Thus, the search for high selectivity systems under mild conditions continues.
-
7
N2 fixation conditions: Reaction temperature must also be considered. Although all Mo catalyst systems currently operate at room temperature under an atmosphere of N2, all of the other N2RR catalyst systems studied are far more efficient at low temperature (≤−78 °C) than at room temperature.172,173 This represents an opportunity for improvement and will be particularly important with respect to electrocatalytic N2RR, where low reaction temperatures can significantly attenuate accessible current densities. On the other hand, the potential benefits of elevated N2 pressures in N2RR catalysis have not been explored, though hints from the N2-silylation literature suggest it could be a powerful tool for improving catalysis.303 Computational studies suggest that the low driving force for the exchange of NH3 for N2 is an issue in both (HIPTN3N)Mo124 and (P3E)Fe (E = Si, B).209 Additional studies exploring the possibility of using solvent media, including ionic liquids, to increase the availability or reactivity of N2 is also of interest.279
-
8
N2 fixation with more biologically relevant platforms: Despite steady advances in nitrogenase model chemistry, current synthetic nitrogenases appear to involve key bond-making and breaking events at a single metal center, with the presumed exception of Nishibayashi’s pincer-ligated catalysts under specific conditions.304 None of these synthetic catalysts yet feature inorganic sulfide or carbide ligands as are present in the cofactors of nitrogenases, although progress toward this aim has been made.249,305 As efforts to understand the nature of biological nitrogen fixation continue, the development of catalysts featuring more biologically faithful coordination environments will be of significant interest to further constrain and test the feasibility of mechanistic hypotheses. Such studies should also ultimately consider secondary sphere influences.132,306
-
9
N2 fixation product selectivity: One of the fascinating areas of mechanistic diversity in nitrogen fixation catalysis is the differing selectivity for NH3 and N2H4. Thus far, all Mo catalysts have been proposed to operate through a distal mechanism (Figures 12 and 32),110,304 or an initial N≡N bond cleavage in a specific instance (Figure 34).79,81 All Fe catalysts appear to form at least some amount of NH3 and N2H4.80,116,174 Despite this, mechanistic studies that demonstrate a strictly alternating pathway for an active catalyst are lacking. Mechanistic work has, by contrast, pointed to the viability of a hybrid distal-to-alternating pathway.109 Therefore, increased attention toward identifying a system that traverses an alternating pathway, and/or further elucidating hybrid pathways that lead to N2H4 formation, are warranted. Given the utility of N2H4-based derivatives as fuels, catalysts that are selective for the direct reduction of N2-to-N2H4 may find niche applications. Furthermore, biological nitrogenases display mechanistic diversity with respect to their selectivity for NH3 and N2H4.163 Thus, understanding which mechanisms are compatible from a synthetic catalyst perspective will help in considering hypotheses within a biological framework.
-
10
N2 fixation using light: Recent research from Peters and co-workers has demonstrated that N2RR turnover can be enhanced via irradiation of the system (Figure 61).175,196 This observation serves as a potentially important pathway for the removal of catalytically inactive hydride species, via H2 loss, which in turn increases the availability of catalytically active species. Such studies may also have implications with respect to understanding the release of H2 and binding of N2 at biological nitrogenases, a process that can also be light-triggered.162 Furthermore, recent work from Nishibayashi and co-workers has begun to consider how known nitrogen fixation platforms can be integrated with known water photooxidation processes (i.e., H2O → O2 + 4 H+ + 4e−) (Figure 36).150 However, there remains significant space for development in this area, which is critical to the future integration of N2 fixation with renewable energy conversion technologies.
-
11
N2 fixation using electrocatalysis: The development of electrocatalytic nitrogen fixation systems is critical to the field’s future. In this arena, N2 fixation lags significantly behind other multielectron reduction processes such as hydrogen evolution,307,308 O2 reduction,309 or carbon dioxide fixation.310 Progress in the realm of N2RR catalysis has been hindered by the significant kinetic requirement of biasing a system that is sufficiently reducing to catalyze N2RR and does so in preference to HER. This challenge also confronts progress in heterogeneous N2RR electrocatalyst studies.6,262 Furthermore, N2RR catalysis has typically been studied under conditions that are poorly suited to electrocatalysis (i.e., low-temperature, low dielectric solvent, heterogeneous acids). A recent study by Peters and co-workers shows that these challenges can be overcome to demonstrate electrocatalysis and comparatively high Faradaic efficiencies (Figure 72).197 Indeed, this study underscores that electrode-mediated N2RR catalysis can be superior to chemical reductant-mediated catalysis, when compared under identical conditions. Developing new electrocatalytic systems for N2RR catalysis and comparing and contrasting their mechanisms with chemical catalysis systems presents an important area for exploration.
-
12
N2 fixation in water: A significant goal for the field is to drive N2RR with the electrons and protons of water. Progress here is needed to advance the use of ammonia as a no-carbon solar fuel. Progress in this context has been limited for synthetic N2RR catalysts. There have been some studies that offer a path for further development. For example, DMeOPrPE-ligated Fe complexes bind N2 in water (Figure 57),236 and protons ultimately derived from water oxidation have been used for N2-fixation catalysis (Figure 36).150 Nevertheless, the remaining challenges are significant. Approaches that may lead to progress in this area will include the development of more water-soluble (and stable) catalysts, or the use of micelles that stabilize hydrophobic catalysts in water; the latter has been demonstrated by Shilov and co-workers.76 The protic nature of H2O cautions that its competitive reduction to H2 is a serious consideration for aqueous N2-fixation catalysis. In this regard, the development of catalysts that operate at lower net overpotentials will be essential.
-
13
An assessment of best N2RR practices: The accurate detection of NH3 (or N2H4) is not without complication. In the context of electrocatalytic N2RR, several papers have appeared recently addressing potential sources of NH3 contamination.284,311 Many of these factors, such as trace contamination of the N2 gas and impure catalysts/reagents, are heightened in the set-ups used for heterogeneous electrocatalytic N2RR, or cases where very small amounts of NH3 are produced. Nonetheless, caution is warranted when analyzing homogeneous N2RR catalysts as well. Of particular importance are both control experiments both without catalyst and without N2 to detect potential sources of contamination, as well as isotopic labeling experiments to verify the source of the NH3/N2H4. As always, it is essential that analytical rigor be employed when quantifying these products.
10. SUMMARY AND CONCLUSIONS
In this review, we have offered a lens into the field N2-to-NH3 catalysis (N2RR) mediated by synthetic complexes. While we have focused almost exclusively on systems for which bona fide catalysis has been demonstrated, we began by discussing the pioneering studies of Chatt and co-workers as well as others who laid the early groundwork for this field (Figure 3). The groundbreaking work of Schrock and co-workers, who in 2003 demonstrated the first example of N2RR catalysis via a well-defined tri(amido)amine Mo system (Figure 12), has been discussed in detail.50 This system still represents one of the most important for the field because of the thorough characterization of many catalytic intermediates that was undertaken. The discussion of Schrock’s system was followed by a description of Nishibayashi’s low-valent Mo-phosphine systems. These systems are related to the original Chatt-type Mo systems in that they feature low valent molybdenum complexes with phosphine donors, and they have proven highly amenable to tuning via manipulations of the ligand (Figure 33). Mononuclear Mo nitrides in particular have proven to be effective (pre)catalysts for N2 fixation in this context. Most recently, catalysis with unprecedented efficiency, turnover number, and turnover frequency has been realized via SmI2 as a reductant in combination with protic solvents.79 Many of the mechanistic details of the latter system remain to be explored.
We next focused on Fe-mediated N2RR catalysis, which was first reported in 2013 by Peters and co-workers via a tris(phosphine)borane iron system (Figure 45). This represented a significant advance; prior to this report, the field had focused heavily on Mo-systems, which biased mechanistic proposals relevant to biological nitrogen fixation. Significant experimental and theoretical work for this system has been undertaken to place the mechanism/s by which it operates on firm footing. As for the Schrock system, many candidate intermediates and interconversions have been studied, with many of the early intermediates being highly unstable and requiring low temperatures for characterization. Of particular interest are comparative studies with related (P3C)Fe- and (P3Si)Fe-systems, which have helped to identify important principles that determine catalyst selectivity. With the optimal combination of reductant and acid at low temperature the (P3B)Fe system can catalyze N2RR with selectivity for NH3 approaching 80%. Also, studies of these iron systems have underscored a role for PCET in N2RR catalysis (Figure 47); such processes may be operative in other systems, including Mo. A number of other Mo and Fe catalysts were also discussed, including a structurally unusual bis(carbene)-iron system, (CAAC)2Fe (Figure 55), whose efficacy for N2RR catalysis becomes apparent only at −95 °C, and also a bis-diphosphine-iron that is selective for N2H4 instead of NH3 (Figure 59).
Recent studies have been undertaken with a host of other metals, most of which (except V) are not found in nitrogenases. These have included Co, Ru, Os, V, and Ti. Particular emphasis has been placed here on the mechanistic understanding that can be obtained via comparisons of these systems to the Mo or Fe catalysts discussed in the earlier sections. A discussion of historic and more recent efforts toward electrocatalytic nitrogen fixation with transition metal complexes has also been presented, including a discussion of the first (and still only) molecular system, (P3B)Fe+, that mediates bona fide N2RR electrocatalysis (Figure 72). Other systems that show progress here, and especially those featuring main group (e.g., Al/B) rather than transition metal active sites, were also highlighted (Figure 74).
Lastly, in an effort to highlight some of the lessons drawn from this review, a bulleted discussion of specific insights important to future catalyst designs and, also, remaining challenges is presented. We hope these ideas help researchers devise new catalysts, and reagent/condition combinations, to continue the rapid progress now being made in this important realm of catalysis.
Figure 23.

A diamido(pyridine) pincer ligand used by the Schrock group for Mo-catalyzed N2RR, Ar = 2,6-diisopropylphenyl.140
Figure 53.

Reproduced with permission from ref 209. Copyright 2018 American Chemical Society. Gibbs free enthalpy, ΔG°, scheme of the (P3Si)Fe cycle calculated with the B3LYP functional and the def2-TZVP basis set.
ACKNOWLEDGMENTS
The authors are grateful to NIH (GM-070757) for ongoing support of their work exploring (Ln)Fe(NxHy) model systems of biological nitrogenases and the Department of Energy (DOE-0235032) for supporting their research towards electrocatalytic N2RR systems. M.W.D. acknowledges NSERC (Banting PDF award to MWD), and M.W.D./M.J.C. thank the Resnick Sustainability Institute at Caltech for fellowships. Dr. Cooper Citek, Dr. Pablo G. Barros, Javier Fajardo Jr., Nina X. Gu, Dr. Heejun Lee, Dr. Alonso Rosas, Dirk J. Schild are thanked for providing helpful discussion.
Biographies
Dr. Matthew J. Chalkley received his B.S. from Yale University with exceptional distinction in Chemistry in 2013. There he undertook research with Prof. Nilay Hazari on PdI dimers as a Barry Goldwater Scholar. He then received a Fulbright Fellowship to perform research in the group of Prof. Karsten Meyer at the University of Erlangen-Nuremberg for one year. His Ph.D. was supervised by Prof. Jonas Peters at Caltech and supported by an NSF Graduate fellowship, a Caltech Environmental Microbial Interactions fellowship, and a Resnick Sustainability Institute fellowship. He is currently a postdoctoral scholar with Prof. William DeGrado at the University of California, San Francisco. His interests include catalysis and bioinorganic and organometallic chemistry.
Prof. Marcus W. Drover received his B.Sc. (Hons.) degree in 2012 from Memorial University of Newfoundland. In 2016, he earned his doctorate in chemistry from the University of British Columbia (UBC) under the cosupervision of Profs. Laurel L. Schafer and Jennifer A. Love. At UBC, Marcus was an NSERC Vanier scholar (2013–2016) and in 2016 was awarded the Chemical Institute of Canada’s Award for Graduate Research in Inorganic Chemistry. In 2014, Marcus was also awarded a Michael-Smith visiting fellowship for research abroad at the University of Oxford under the supervision of Prof. Andrew S. Weller. In January 2017, Marcus began work as a joint Resnick Prize Postdoctoral Fellow in Sustainability Science and NSERC Banting Fellow at Caltech under the guidance of Prof. Jonas C. Peters. Currently, Marcus is an assistant professor at the University of Windsor in Ontario, Canada. His interests include inorganic, organometallic, and main group chemistry.
Prof. Jonas C. Peters was born in 1971 in Chicago, Illinois. In 1993, he received his Bachelor of Science degree in chemistry at the University of Chicago, where doing undergraduate research with Prof. Gregory Hillhouse first piqued his interest in the transition metal chemistry of nitrogenous ligands such as HN=NH and HNO. Jonas then spent a year as a Marshall Scholar at the University of Nottingham, UK, working with Prof. James J. Turner, FRS, detecting short-lived transients by rapid time-resolved methods. In the fall of 1994, Jonas began his doctoral studies under the direction of Prof. Christopher C. Cummins at the Massachusetts Institute of Technology. Jonas’ research focused on the activation and functionalization of small molecules using low coordinate tris-amido molybdenum and titanium complexes. After receiving his Ph.D. in inorganic chemistry in 1998, Jonas was a Miller Fellow at the University of California, Berkeley under the guidance of Prof. T. Don Tilley, where he began to design and synthesize new phosphine ligands for metal-mediated bond activation reactions. Jonas began as Assistant Professor in the Division of Chemistry and Chemical Engineering at Caltech in August of 1999, was promoted to Associate Professor in 2004, and to Professor of Chemistry in 2006. In July of 2007, he relocated to the MIT Department of Chemistry as the W. M. Keck Professor of Energy, and then returned to Caltech in January 2010 as Bren Professor of Chemistry. He is also the Director of the Resnick Sustainability Institute at Caltech. His research focuses on the structure, bonding, and catalytic activity of inorganic compounds, particularly ones of relevance to global C, N, and O cycles and renewable energy applications.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Matthew J. Chalkley, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States.
Marcus W. Drover, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States.
Jonas C. Peters, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States.
REFERENCES
- (1).Smil V Global Population and the Nitrogen Cycle. Sci. Am 1997, 277, 76–81. [Google Scholar]
- (2).Schlögl R Catalytic Synthesis of Ammonia—A “Never-Ending Story”? Angew. Chem., Int. Ed 2003, 42, 2004–2008. [DOI] [PubMed] [Google Scholar]
- (3).Appl M Ammonia http://www.iipinetwork.org/wp-content/Ietd/content/ammonia.html#technology-resources (accessed Jun 22, 2019).
- (4).Mosier AR Environmental Challenges Associated with Needed Increases in Global Nitrogen Fixation. Nutr. Cycling Agroecosyst 2002, 63, 101–116. [Google Scholar]
- (5).Canfield DE; Glazer AN; Falkowski PG The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. [DOI] [PubMed] [Google Scholar]
- (6).Kyriakou V; Garagounis I; Vasileiou E; Vourros A; Stoukides M Progress in the Electrochemical Synthesis of Ammonia. Catal Today 2017, 286, 2–13. [Google Scholar]
- (7).Comer BM; Fuentes P; Dimkpa CO; Liu Y-H; Fernandez CA; Arora P; Realff M; Singh U; Hatzell MC; Medford AJ Prospects and Challenges for Solar Fertilizers. Joule 2019, 3, 1578–1605. [Google Scholar]
- (8).Potential Roles of Ammonia in a Hydrogen Economy https://www.energy.gov/eere/fuelcells/downloads/potential-roles-ammonia-hydrogen-economy (accessed Jun 22, 2019).
- (9).Valera-Medina A; Xiao H; Owen-Jones M; David WIF; Bowen PJ Ammonia for Power. Prog. Energy Combust. Sci 2018, 69, 63–102. [Google Scholar]
- (10).Burgess BK; Lowe DJ Mechanism of Molybdenum Nitrogenase. Chem. Rev 1996, 96, 2983–3011. [DOI] [PubMed] [Google Scholar]
- (11).Eady RR Structure–Function Relationships of Alternative Nitrogenases. Chem. Rev 1996, 96, 3013–3030. [DOI] [PubMed] [Google Scholar]
- (12).Spatzal T; Aksoyoglu M; Zhang LM; Andrade SLA; Schleicher E; Weber S; Rees DC; Einsle O Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Einsle O; Tezcan FA; Andrade SLA; Schmid B; Yoshida M; Howard JB; Rees DC Nitrogenase MoFe-Protein at 1.16 Angstrom Resolution: A Central Ligand in the FeMo-Cofactor. Science 2002, 297, 1696–1700. [DOI] [PubMed] [Google Scholar]
- (14).Howard JB; Rees DC Structural Basis of Biological Nitrogen Fixation. Chem. Rev 1996, 96, 2965–2982. [DOI] [PubMed] [Google Scholar]
- (15).Chatt J; Dilworth JR; Richards RL Recent Advances in Chemistry of Nitrogen-Fixation. Chem. Rev 1978, 78, 589–625. [Google Scholar]
- (16).Leigh GJ So That’s HowIt’s Done - Maybe. Science 2003, 301, 55–56. [DOI] [PubMed] [Google Scholar]
- (17).Schrock RR Nitrogen Reduction: Molybdenum Does It Again. Nat. Chem 2011, 3, 95–96. [DOI] [PubMed] [Google Scholar]
- (18).Anderson JS; Rittle J; Peters JC Catalytic Conversion of Nitrogen to Ammonia by an Iron Model Complex. Nature 2013, 501, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hoffman BM; Lukoyanov D; Yang ZY; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bazhenova TA; Shilov AE Nitrogen-Fixation in Solution. Coord. Chem. Rev 1995, 144, 69–145. [Google Scholar]
- (21).Hidai M; Mizobe Y Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev 1995, 95, 1115–1133. [Google Scholar]
- (22).MacKay BA; Fryzuk MD Dinitrogen Coordination Chemistry: On the Biomimetic Borderlands. Chem. Rev 2004, 104, 385–401. [DOI] [PubMed] [Google Scholar]
- (23).Hazari N Homogeneous Iron Complexes for the Conversion of Dinitrogen into Ammonia and Hydrazine. Chem. Soc. Rev 2010, 39, 4044–4056. [DOI] [PubMed] [Google Scholar]
- (24).MacLeod KC; Holland PL Recent Developments in the Homogeneous Reduction of Dinitrogen by Molybdenum and Iron. Nat. Chem 2013, 5, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tanabe Y; Nishibayashi Y Developing More Sustainable Processes for Ammonia Synthesis. Coord. Chem. Rev 2013, 257, 2551–2564. [Google Scholar]
- (26).Peters JC; Mehn MP Bio-Organometallic Approaches to Nitrogen Fixation Chemistry In Activation of Small Molecules; Tolman WB, Ed.; Wiley-VCH, 2006; pp 81–119. [Google Scholar]
- (27).Roux Y; Duboc C; Gennari M Molecular Catalysts for N2 Reduction: State of the Art, Mechanism, and Challenges. ChemPhysChem 2017, 18, 2606–2617. [DOI] [PubMed] [Google Scholar]
- (28).Stucke N; Flöser BM; Weyrich T; Tuczek F Nitrogen Fixation Catalyzed by Transition Metal Complexes: Recent Developments. Eur. J. Inorg. Chem 2018, 2018, 1337–1355. [Google Scholar]
- (29).Nishibayashi Y Development of Catalytic Nitrogen Fixation Using Transition Metal–Dinitrogen Complexes under Mild Reaction Conditions. Dalton Trans 2018, 47, 11290–11297. [DOI] [PubMed] [Google Scholar]
- (30).Pickett CJ The Chatt Cycle and the Mechanism of Enzymic Reduction of Molecular Nitrogen. JBIC, J. Biol. Inorg. Chem 1996, 1, 601–606. [Google Scholar]
- (31).Postgate J The Origins of the Unit of Nitrogen Fixation at the University of Sussex. Notes Rec. R. Soc. London 1998, 52, 355–362. [Google Scholar]
- (32).Chatt J; Leigh GJ Nitrogen Fixation. Chem. Soc. Rev 1972, 1, 121–144. [Google Scholar]
- (33).Allen AD; Harris RO; Loescher BR; Stevens JR; Whiteley RN Dinitrogen Complexes of the Transition Metals. Chem. Rev 1973, 73, 11–20. [Google Scholar]
- (34).Sellmann D Dinitrogen-Transition Metal Complexes: Synthesis, Properties, and Significance. Angew. Chem. Int. Ed. Engl 1974, 13, 639–649. [Google Scholar]
- (35).Leigh GJA Celebration of Inorganic Lives: Interview of Joseph Chatt (University of Sussex). Coord. Chem. Rev 1991, 108, 1–25. [Google Scholar]
- (36).Eady RR Current Status of Structure Function Relationships of Vanadium Nitrogenase. Coord. Chem. Rev 2003, 237, 23–30. [Google Scholar]
- (37).Allen AD; Senoff CV Nitrogenopentammineruthenium(II) Complexes. Chem. Commun 1965, 24, 621–622. [Google Scholar]
- (38).Allen AD; Bottomley F; Harris RO; Reinsalu VP; Senoff CV Ruthenium Complexes Containing Molecular Nitrogen. J. Am. Chem. Soc 1967, 89, 5595–5599. [Google Scholar]
- (39).Eaborn C; Leigh GJ Joseph Chatt C. B. E. Biographical Mem. Fellows R. Soc 1996, 42, 96–110. [Google Scholar]
- (40).Dewar JSA Review of the n-Complex Theory. Bull. Soc. Chim. Fr 1951, 18, C71–C79. [Google Scholar]
- (41).Chatt J; Duncanson LA Olefin Co-Ordination Compounds. Part III. Infra-Red Spectra and Structure: Attempted Preparation of Acetylene Complexes. J. Chem. Soc 1953, 0, 2939–2947. [Google Scholar]
- (42).Crabtree RH The Organometallic Chemistry of the Transition Elements; John Wiley & Sons: New York, 2001. [Google Scholar]
- (43).Cable JW; Sheline RK Bond Hybridization and Structure in the Metal Carbonyls. Chem. Rev 1956, 56, 1–26. [Google Scholar]
- (44).Shilov A; Denisov N; Efimov O; Shuvalov N; Shuvalova N; Shilova A New Nitrogenase Model for Reduction of Molecular Nitrogen in Protonic Media. Nature 1971, 231, 460–461. [DOI] [PubMed] [Google Scholar]
- (45).Denisov NT; Shilov AE; Shuvalova NI; Panova TP Mechanism of Electron Transfer in Dinitrogen Fixation in the System Ti(III)–Mo(III). React. Kinet Catal. Lett 1975, 2, 237–241. [Google Scholar]
- (46).Shilov AE; Shilova AK; Vorontsova TA Molybdenum Complexes as Catalysts for the Reduction of Molecular Nitrogen in Protic Media. React. Kinet. Catal. Lett 1975, 3, 143–148. [Google Scholar]
- (47).Pospíšil L; Didenko LP; Shilov AE The Electrochemical Properties of Nitrogen-Fixating Mo(III) and the Reduction of Mo(V) in Alkaline Methanolic Media Influenced by Mg2+ Ions. J. Electroanal. Chem. Interfacial Electrochem 1986, 197, 305–316. [Google Scholar]
- (48).Bortels H Molybdän als Katalysator bei der biologischen Stickstoffbindung. Arch. Microbiol 1930, 1, 333–342. [Google Scholar]
- (49).Chatt J; Heath GA; Richards RL The Reduction of Ligating Dinitrogen to Yield a Ligating N2H2 Moiety. J. Chem. Soc., Chem. Commun 1972, 18, 1010–1011. [Google Scholar]
- (50).Yandulov DV; Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- (51).Arashiba K; Miyake Y; Nishibayashi Y A Molybdenum Complex Bearing PNP-Type Pincer Ligands Leads to the Catalytic Reduction of Dinitrogen into Ammonia. Nat. Chem 2011, 3, 120–125. [DOI] [PubMed] [Google Scholar]
- (52).Del Castillo TJ; Thompson NB; Suess DLM; Ung G; Peters JC Evaluating Molecular Cobalt Complexes for the Conversion of N to NH3. Inorg. Chem 2015, 54, 9256–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kuriyama S; Arashiba K; Tanaka H; Matsuo Y; Nakajima K; Yoshizawa K; Nishibayashi Y Direct Transformation of Molecular Dinitrogen into Ammonia Catalyzed by Cobalt Dinitrogen Complexes Bearing Anionic PNP Pincer Ligands. Angew. Chem. Int. Ed 2016, 55, 14291–14295. [DOI] [PubMed] [Google Scholar]
- (54).Fajardo J Jr.; Peters JC Catalytic Nitrogen-to-Ammonia Conversion by Osmium and Ruthenium Complexes. J. Am. Chem. Soc 2017, 139, 16105–16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Doyle LR; Wooles AJ; Jenkins LC; Tuna F; McInnes EJL; Liddle ST Catalytic Dinitrogen Reduction to Ammonia at a Triamidoamine-Titanium Complex. Angew. Chem. Int. Ed 2018, 57, 6314–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sekiguchi Y; Arashiba K; Tanaka H; Eizawa A; Nakajima K; Yoshizawa K; Nishibayashi Y Catalytic Reduction of Molecular Dinitrogen to Ammonia and Hydrazine Using Vanadium Complexes. Angew. Chem., Int. Ed 2018, 57, 9064–9068. [DOI] [PubMed] [Google Scholar]
- (57).Chatt J Molybdenum in Nitrogen-Fixation. J. Less-Common Met 1974, 36, 429–435. [Google Scholar]
- (58).Chatt J; Pearman AJ; Richards RL Reduction of Mono-Coordinated Molecular Nitrogen to Ammonia in a Protic Environment. Nature 1975, 253, 39–40. [Google Scholar]
- (59).Chatt J; Dilworth JR Molybdenum(VI) and Molybdenum(V) Nitrido-Complexes and a μ-Nitrido-Molybdenumrhenium Complex. J. Chem. Soc., Chem. Commun 1974, 13, 517–518. [Google Scholar]
- (60).Chatt J; Heath GA; Richards RL Diazene-N-(Di-Imide) And Hydrazido-(2−)N-(Aminoimido) (Aminoimido) Complexes - Addition of Acids to Dinitrogen Complexes. J. Chem. Soc., Dalton Trans 1974, 19, 2074–2082. [Google Scholar]
- (61).Chatt J; Pearman AJ; Richards RL Conversion of Dinitrogen in Its Molybdenum and Tungsten Complexes into Ammonia and Possible Relevance to Nitrogenase Reaction. J. Chem. Soc., Dalton Trans 1977, 19, 1852–1860. [Google Scholar]
- (62).Chatt J; Pearman AJ; Richards RL Hydrazido(2−)-Complexes of Molybdenum and Tungsten Formed from Dinitrogen Complexes by Protonation and Ligand Exchange. J. Chem. Soc., Dalton Trans 1978, 12, 1766–1776. [Google Scholar]
- (63).Chatt J; Hussain W; Leigh GJ; Neukomm H; Pickett CJ; Rankin DA The Mechanism of the Secondary Alkylation of Dinitrogen and the Relationship Between Structure and Reactivity in the Alkylation of Coordinated Dinitrogen. J. Chem. Soc., Chem. Commun 1980, 21, 1024–1025. [Google Scholar]
- (64).Anderson SN; Fakley ME; Richards RL; Chatt J Hydrazido(2−)-Complexes as Intermediates in the Conversion of Ligating Dinitrogen into Ammonia and Hydrazine. J. Chem. Soc., Dalton Trans 1981, 9, 1973–1980. [Google Scholar]
- (65).Chatt J; Pearman AJ; Richards RL Preparation and Oxidation, Substitution, and Protonation Reactions of Trans-Bis-(Dinitrogen)Tetrakis(Methyldiphenylphosphine)Tungsten. J. Chem. Soc., Dalton Trans 1977, 21, 2139–2142. [Google Scholar]
- (66).Chatt J; Pearman AJ; Richards RL Diazenido-Complexes of Molybdenum and Tungsten. J. Chem. Soc., Dalton Trans 1976, 15, 1520–1524. [Google Scholar]
- (67).Chatt J; Pearman AJ; Richards RL Diazenido (Iminonitrosyl) (N2H), Hydrazido(2−) (N2H2), and Hydrazido(1−) (N2H3) Ligands as Intermediates in the Reduction of Ligating Dinitrogen to Ammonia. J. Organomet. Chem 1975, 101, C45–C47. [Google Scholar]
- (68).Galindo A; Hills A; Hughes DL; Richards RL A Complex of the Hydrazidium (N–NH3+) Ligand: X-Ray Structure of [WCl(NNH3)(PMe3)4]Cl2. J. Chem. Soc., Chem. Commun 1987, 24, 1815–1816. [Google Scholar]
- (69).Galindo A; Hills A; Hughes DL; Richards RL; Hughes M; Mason J Protonation Reactions of Dinitrogen Complexes of Molybdenum and Tungsten with PMe3 as Co-Ligand -X-Ray Structure of the Hydrazidium Complex [WCl(NNH3)(PMe3)4]Cl2. J. Chem. Soc., Dalton Trans 1990, 1, 283–288. [Google Scholar]
- (70).Yandulov DV; Schrock RR Reduction of Dinitrogen to Ammonia at a Well-Protected Reaction Site in a Molybdenum Triamidoamine Complex. J. Am. Chem. Soc 2002, 124, 6252–6253. [DOI] [PubMed] [Google Scholar]
- (71).Murray RC; Schrock RR Preparation of an Unsubstituted Hydrazido(1−) Complex and an Authentic High Oxidation State Ditungsten Dinitrogen Complex. J. Am. Chem. Soc 1985, 107, 4557–4558. [Google Scholar]
- (72).Wagenknecht PS; Norton JR Mechanism of Transition-Metal-Mediated Nitrogen-Fixation-Where Does the 3rd Proton Go. J. Am. Chem. Soc 1995, 117, 1841–1842. [Google Scholar]
- (73).Vale MG; Schrock RR Synthesis and Reactions of Monomeric Hydrazine and Hydrazido Complexes That Contain the Cp*MoMe3 Core. Inorg. Chem 1993, 32, 2767–2772. [Google Scholar]
- (74).Schrock RR; Glassman TE; Vale MG Cleavage of the Nitrogen-Nitrogen Bond in a High Oxidation State Tungsten or Molybdenum Hydrazine Complex and the Catalytic Reduction of Hydrazine. J. Am. Chem. Soc 1991, 113, 725–726. [Google Scholar]
- (75).Chatt J; Dilworth JR; Richards RL; Sanders JR Chemical Evidence Concerning the Function of Molybdenum in Nitrogenase. Nature 1969, 224, 1201–1202. [DOI] [PubMed] [Google Scholar]
- (76).Didenko LP; Gavrilina OK; Yablonskaya EE; Shilova AK; Shilov AE Phospholipid-Dependent Catalytic Dinitrogen Reduction in the Presence of Molybdenum Complexes. Nouv. J. Chim 1983, 7,605–611. [Google Scholar]
- (77).Didenko LP; Gavrilov AB; Shilova AK; Strelets VV; Shilov AE Dinitrogen Fixation. Fast Amalgam and Electrochemical Catalytic Dinitrogen Reduction at Ambient Temperature and Pressure. Nouv. J. Chim 1986, 10, 583–588. [Google Scholar]
- (78).Schrock RR; Kolodziej RM; Liu AH; Davis WM; Vale MG Preparation And Characterization Of Two High Oxidation-State Molybdenum Dinitrogen Complexes - [MoCp*Me3]2(μ-N2) And [MoCp*Me3](μ-N2)[WCp’Me3]. J. Am. Chem. Soc 1990, 112, 4338–4345. [Google Scholar]
- (79).Ashida Y; Arashiba K; Nakajima K; Nishibayashi Y Molybdenum-Catalysed Ammonia Production with Samarium Diiodide and Alcohols or Water. Nature 2019, 568, 536–540. [DOI] [PubMed] [Google Scholar]
- (80).Chalkley MJ; Del Castillo TJ; Matson BD; Roddy JP; Peters JC Catalytic N2-to-NH3 Conversion by Fe at Lower Driving Force: A Proposed Role for Metallocene-Mediated PCET. ACS Cent. Sci 2017, 3, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Arashiba K; Eizawa A; Tanaka H; Nakajima K; Yoshizawa K; Nishibayashi Y Catalytic Nitrogen Fixation via Direct Cleavage of Nitrogen-Nitrogen Triple Bond of Molecular Dinitrogen under Ambient Reaction Conditions. Bull. Chem. Soc. Jpn 2017, 90, 1111–1118. [Google Scholar]
- (82).Kuriyama S; Arashiba K; Nakajima K; Tanaka H; Kamaru N; Yoshizawa K; Nishibayashi Y Catalytic Formation of Ammonia from Molecular Dinitrogen by Use of Dinitrogen-Bridged Dimolybdenum–Dinitrogen Complexes Bearing PNP-Pincer Ligands: Remarkable Effect of Substituent at PNP-Pincer Ligand. J. Am. Chem. Soc 2014, 136,9719–9731. [DOI] [PubMed] [Google Scholar]
- (83).Tanaka H; Arashiba K; Kuriyama S; Sasada A; Nakajima K; Yoshizawa K; Nishibayashi Y Unique Behaviour of Dinitrogen-Bridged Dimolybdenum Complexes Bearing Pincer Ligand towards Catalytic Formation of Ammonia. Nat. Commun 2014, 5, 3737–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Arashiba K; Kinoshita E; Kuriyama S; Eizawa A; Nakajima K; Tanaka H; Yoshizawa K; Nishibayashi Y Catalytic Reduction of Dinitrogen to Ammonia by Use of Molybdenum-Nitride Complexes Bearing a Tridentate Triphosphine as Catalysts. J. Am. Chem. Soc 2015, 137, 5666–5669. [DOI] [PubMed] [Google Scholar]
- (85).Clentsmith GKB; Bates VME; Hitchcock PB; Cloke FGN Reductive Cleavage of Dinitrogen by a Vanadium Diamidoamine Complex: The Molecular Structures of [V(Me3SiN{CH2CH2NSiMe3}2(μ-N)]2 and K[V(Me3SiN{CH2CH2NSiMe3}2(μ-N)]2. J. Am. Chem. Soc 1999, 121, 10444–10445. [Google Scholar]
- (86).Kawaguchi H; Matsuo T Dinitrogen-Bond Cleavage in a Niobium Complex Supported by a Tridentate Aryloxide Ligand. Angew. Chem., Int. Ed 2002, 41, 2792–2794. [DOI] [PubMed] [Google Scholar]
- (87).Fryzuk MD; MacKay BA; Patrick BO Hydrosilylation of a Dinuclear Tantalum Dinitrogen Complex: Cleavage of N2 and Functionalization of Both Nitrogen Atoms. J. Am. Chem. Soc 2003, 125, 3234–3235. [DOI] [PubMed] [Google Scholar]
- (88).Pool JA; Lobkovsky E; Chirik PJ Hydrogenation and Cleavage of Dinitrogen to Ammonia with a Zirconium Complex. Nature 2004, 427, 527–530. [DOI] [PubMed] [Google Scholar]
- (89).MacKay BA; Patrick BO; Fryzuk MD Hydroalumination of a Dinuclear Tantalum Dinitrogen Complex: N-NBond Cleavage and Ancillary Ligand Rearrangement. Organometallics 2005, 24, 3836–3841. [Google Scholar]
- (90).Rodriguez MM; Bill E; Brennessel WW; Holland PL N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Laplaza CE; Cummins CC Dinitrogen Cleavage by a 3-Coordinate Molybdenum(III) Complex. Science 1995, 268, 861–863. [DOI] [PubMed] [Google Scholar]
- (92).Laplaza CE; Johnson MJA; Peters JC; Odom AL; Kim E; Cummins CC; George GN; Pickering IJ Dinitrogen Cleavage by Three-Coordinate Molybdenum(III) Complexes: Mechanistic and Structural Data. J. Am. Chem. Soc 1996, 118, 8623–8638. [Google Scholar]
- (93).Cui Q; Musaev DG; Svensson M; Sieber S; Morokuma K N2 Cleavage By 3-Coordinate Group-6 Complexes - W(III) Complexes Would-Be Better Than Mo(III) Complexes. J. Am. Chem. Soc 1995, 117, 12366–12367. [Google Scholar]
- (94).Kol M; Schrock RR; Kempe R; Davis WM Synthesis of Molybdenum and Tungsten Complexes That Contain Triamidoamine Ligands of the Type (C6F5NCH2CH2)3N and Activation of Dinitrogen by Molybdenum. J. Am. Chem. Soc 1994, 116, 4382–4390. [Google Scholar]
- (95).O’Donoghue MB; Zanetti NC; Davis WM; Schrock RR Fixation” of Dinitrogen by Molybdenum and the Formation of a Trigonal Planar Iron–Tris[Molybdenum(Dinitrogen)] Complex. J. Am. Chem. Soc 1997, 119, 2753–2754. [Google Scholar]
- (96).O’Donoghue MB; Davis WM; Schrock RR Derivatization of Dinitrogen by Molybdenum in Triamidoamine Complexes. Inorg. Chem 1998, 37, 5149–5158. [Google Scholar]
- (97).Greco GE; Schrock RR Synthesis of Triamidoamine Ligands of the Type (ArylNHCH2CH2)3N and Molybdenum and Tungsten Complexes That Contain an [(ArylNCH2CH2)3N]3− Ligand. Inorg. Chem 2001, 40, 3850–3860. [DOI] [PubMed] [Google Scholar]
- (98).Greco GE; Schrock RR Synthesis, Structure, and Electrochemical Studies of Molybdenum and Tungsten Dinitrogen, Diazenido, and Hydrazido Complexes That Contain Aryl-Substituted Triamidoamine Ligands. Inorg. Chem 2001, 40, 3861–3878. [DOI] [PubMed] [Google Scholar]
- (99).Yandulov DV; Schrock RR; Rheingold AL; Ceccarelli C; Davis WM Synthesis and Reactions of Molybdenum Triamidoamine Complexes Containing Hexaisopropylterphenyl Substituents. Inorg. Chem 2003, 42, 796–813. [DOI] [PubMed] [Google Scholar]
- (100).Yandulov DV; Schrock RR Studies Relevant to Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum Triamidoamine Complexes. Inorg. Chem 2005, 44, 1103–1117. [DOI] [PubMed] [Google Scholar]
- (101).Schenk S; Le Guennic B; Kirchner B; Reiher M First-Principles Investigation of the Schrock Mechanism of Dinitrogen Reduction Employing the Full HIPTN3N Ligand. Inorg. Chem 2008, 47, 3634–3650. [DOI] [PubMed] [Google Scholar]
- (102).Kinney RA; McNaughton RL; Chin JM; Schrock RR; Hoffman BM Protonation of the Dinitrogen-Reduction Catalyst [HIPTN3N]MoIII Investigated by ENDOR Spectroscopy. Inorg. Chem 2011, 50,418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Munisamy T; Schrock RR An Electrochemical Investigation of 1ntermediates and Processes 1nvolved in the Catalytic Reduction of Dinitrogen by [HIPTN3N]Mo (HIPTN3N = (3,5-(2,4,6-i-Pr3C6H2)2C6H3NCH2CH2)3N). Dalton Trans 2012, 41, 130–137. [DOI] [PubMed] [Google Scholar]
- (104).van der Ham CJM; Koper MTM; Hetterscheid DGH Challenges in Reduction of Dinitrogen by Proton and Electron Transfer. Chem. Soc. Rev 2014, 43, 5183–5191. [DOI] [PubMed] [Google Scholar]
- (105).Pappas I; Chirik PJ Catalytic Proton Coupled Electron Transfer from Metal Hydrides to Titanocene Amides, Hydrazides and 1mides: Determination of Thermodynamic Parameters Relevant to Nitrogen Fixation. J. Am. Chem. Soc 2016, 138, 13379–13389. [DOI] [PubMed] [Google Scholar]
- (106).Bezdek MJ; Pappas I; Chirik PJ Determining and Understanding N-H Bond Strengths in Synthetic Nitrogen Fixation Cycles In Nitrogen Fixation; Nishibayashi Y, Ed.; Topics in Organometallic Chemistry; Springer International Publishing: Cham, 2017; pp 1–21. [Google Scholar]
- (107).Hetterscheid DGH; Hanna BS; Schrock RR Molybdenum Triamidoamine Systems. Reactions 1nvolving Dihydrogen Relevant to Catalytic Reduction of Dinitrogen. Inorg. Chem 2009, 48, 8569–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Kupfer T; Schrock RR Alkylation of Dinitrogen in [(HIPTNCH2CH2)3N]Mo Complexes (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3). J. Am. Chem. Soc 2009, 131, 12829–12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Rittle J; Peters JC An Fe-N2 Complex That Generates Hydrazine and Ammonia via Fe = NNH2: Demonstrating a Hybrid Distal-to-Alternating Pathway for N2 Reduction. J. Am. Chem. Soc 2016, 138, 4243–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Weare WW; Dai X; Byrnes MJ; Chin JM; Schrock RR; Müller P Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Proc. Natl. Acad. Sci U. S. A 2006, 103, 17099–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Schenk S; Kirchner B; Reiher M A Stable Six-Coordinate Intermediate in Ammonia–Dinitrogen Exchange at Schrock’s Molybdenum Catalyst. Chem. - Eur. J 2009, 15, 5073–5082. [DOI] [PubMed] [Google Scholar]
- (112).Moret M-E; Peters JC Terminal 1ron Dinitrogen and 1ron 1mide Complexes Supported by a Tris(Phosphino)Borane Ligand. Angew. Chem., Int. Ed 2011, 50, 2063–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Anderson JS; Cutsail GE; Rittle J; Connor BA; Gunderson WA; Zhang L; Hoffman BM; Peters JC Characterization of an Fe≡N–NH2 Intermediate Relevant to Catalytic N2 Reduction to NH3. J. Am. Chem. Soc 2015, 137, 7803–7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Thompson NB; Oyala PH; Dong HT; Chalkley MJ; Zhao J; Alp EE; Hu M; Lehnert N; Peters JC Electronic Structures of an [Fe(NNR2)]+/0/−Redox Series: Ligand Noninnocence and Implications for Catalytic Nitrogen Fixation. Inorg. Chem 2019,58, 3535–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Creutz SE; Peters JC Catalytic Reduction of N2 to NH3 by an Fe-N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc 2014, 136, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Kuriyama S; Arashiba K; Nakajima K; Matsuo Y; Tanaka H; Ishii K; Yoshizawa K; Nishibayashi Y Catalytic Transformation of Dinitrogen into Ammonia and Hydrazine by Iron-Dinitrogen Complexes Bearing Pincer Ligand. Nat. Commun 2016, 7, 12181–12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).McNaughton RL; Roemelt M; Chin JM; Schrock RR; Neese F; Hoffman BM Experimental and Theoretical EPR Study of Jahn–Teller-Active [HIPTN3N]MoL Complexes (L = N2, CO, NH3). J. Am. Chem. Soc 2010, 132, 8645–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).McNaughton RL; Chin JM; Weare WW; Schrock RR; Hoffman BM EPR Study of the Low-Spin [d3; S = ½],Jahn–Teller-Active, Dinitrogen Complex of a Molybdenum Trisamidoamine. J. Am. Chem. Soc 2007, 129, 3480–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Kinney RA; Hetterscheid DGH; Hanna BS; Schrock RR; Hoffman BM Formation of {[HIPTN3N]Mo(III)H}− by Heterolytic Cleavage of H2 as Established by EPR and ENDOR Spectroscopy. Inorg. Chem 2010, 49, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Sharma A; Roemelt M; Reithofer M; Schrock RR; Hoffman BM; Neese F EPR/ENDOR and Theoretical Study of the Jahn–Teller-Active [HIPTN3N]MoVL Complexes (L = N−, NH). Inorg. Chem 2017, 56, 6906–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Studt F; Tuczek F Energetics and Mechanism of a Room-Temperature Catalytic Process for Ammonia Synthesis (Schrock Cycle): Comparison with Biological Nitrogen Fixation. Angew. Chem. Int. Ed 2005, 44, 5639–5642. [DOI] [PubMed] [Google Scholar]
- (122).Reiher M; Le Guennic B; Kirchner B Theoretical Study of Catalytic Dinitrogen Reduction under Mild Conditions. Inorg. Chem 2005, 44, 9640–9642. [DOI] [PubMed] [Google Scholar]
- (123).Schenk S; Reiher M Ligands for Dinitrogen Fixation at Schrock-Type Catalysts. Inorg. Chem 2009, 48, 1638–1648. [DOI] [PubMed] [Google Scholar]
- (124).Thimm W; Gradert C; Broda H; Wennmohs F; Neese F; Tuczek F Free Reaction Enthalpy Profile of the Schrock Cycle Derived from Density Functional Theory Calculations on the Full [MoHIPTN3N] Catalyst. Inorg. Chem 2015, 54, 9248–9255. [DOI] [PubMed] [Google Scholar]
- (125).Le Guennic B; Kirchner B; Reiher M Nitrogen Fixation under Mild Ambient Conditions: Part I—The Initial Dissociation/Association Step at Molybdenum Triamidoamine Complexes. Chem. - Eur. J 2005, 11, 7448–7460. [DOI] [PubMed] [Google Scholar]
- (126).Cao Z; Zhou Z; Wan H; Zhang Q Enzymatic and Catalytic Reduction of Dinitrogen to Ammonia: Density Functional Theory Characterization of Alternative Molybdenum Active Sites. Int. J. Quantum Chem 2005, 103, 344–353. [Google Scholar]
- (127).Hölscher M; Leitner W DFT Investigation of the Potential of [H-M{(NHCH2CH2)3X}] Catalysts (M = Mo, Ru, Os; X = N, P) for the Reduction of N2 to NH3 by H2. Eur. J. Inorg. Chem 2006, 2006, 4407–4417. [Google Scholar]
- (128).Schrock RR Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum: Theory versus Experiment. Angew. Chem. Int. Ed 2008, 47, 5512–5522. [DOI] [PubMed] [Google Scholar]
- (129).Khoroshun DV; Musaev DG; Morokuma K Sigma Trans Promotion Effect in Transition Metal Complexes: A Manifestation of the Composite Nature of Binding Energy. Mol. Phys 2002, 100, 523–532. [Google Scholar]
- (130).Hickey AK; Wickramasinghe LA; Schrock RR; Tsay C; Müller P Protonation Studies of Molybdenum(VI) Nitride Complexes That Contain the [2,6-(ArNCH2)2NC5H3]2− Ligand (Ar = 2,6-Diisopropylphenyl). Inorg. Chem 2019, 58, 3724–3731. [DOI] [PubMed] [Google Scholar]
- (131).Ritleng V; Yandulov DV; Weare WW; Schrock RR; Hock AS; Davis WM Molybdenum Triamidoamine Complexes That Contain Hexa- Tert -Butylterphenyl, Hexamethylterphenyl, or p-Bromohexaisopropylterphenyl Substituents. An Examination of Some Catalyst Variations for the Catalytic Reduction of Dinitrogen. J. Am. Chem. Soc 2004, 126, 6150–6163. [DOI] [PubMed] [Google Scholar]
- (132).Weare WW; Schrock RR; Hock AS; Müller P Synthesis of Molybdenum Complexes That Contain “Hybrid” Triamidoamine Ligands, [(Hexaisopropylterphenyl-NCH2CH2)2NCH2CH2N-Aryl]3−, and Studies Relevant to Catalytic Reduction of Dinitrogen. Inorg. Chem 2006, 45, 9185–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Chin JM; Schrock RR; Müller P Synthesis of DiamidoPyrrolyl Molybdenum Complexes Relevant to Reduction of Dinitrogen to Ammonia. Inorg. Chem 2010, 49, 7904–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Reithofer MR; Schrock RR; Müller P Synthesis of (DPPNCH2CH2)3N3− Molybdenum Complexes (DPP = 3,5-(2,5-Diisopropylpyrrolyl)2C6H3) and Studies Relevant to Catalytic Reduction of Dinitrogen. J. Am. Chem. Soc 2010, 132, 8349–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Wickramasinghe LA; Schrock RR; Tsay C; Müller P Molybdenum Complexes That Contain a Calix[6]Azacryptand Ligand as Catalysts for Reduction of N2 to Ammonia. Inorg. Chem 2018, 57, 15566–15574. [DOI] [PubMed] [Google Scholar]
- (136).Yandulov DV; Schrock RR Synthesis of Tungsten Complexes That Contain Hexaisopropylterphenyl-Substituted Triamidoamine Ligands, and Reactions Relevant to the Reduction of Dinitrogen to Ammonia. Can. J. Chem 2005, 83, 341–357. [Google Scholar]
- (137).Smythe NC; Schrock RR; Müller P; Weare WW Synthesis of [(HIPTNCH2CH2)3N]Cr Compounds (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3) and an Evaluation of Chromium for the Reduction of Dinitrogen to Ammonia. Inorg. Chem 2006, 45, 7111–7118. [DOI] [PubMed] [Google Scholar]
- (138).Smythe NC; Schrock RR; Müller P; Weare WW Synthesis of [(HIPTNCH2CH2)3N]V Compounds (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3) and an Evaluation of Vanadium for the Reduction of Dinitrogen to Ammonia. Inorg. Chem 2006, 45, 9197–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Guha AK; Phukan AK Why Vanadium Complexes Perform Poorly in Comparison to Related Molybdenum Complexes in the Catalytic Reduction of Dinitrogen to Ammonia (Schrock Cycle): A Theoretical Study. Inorg. Chem 2011, 50, 8826–8833. [DOI] [PubMed] [Google Scholar]
- (140).Wickramasinghe LA; Ogawa T; Schrock RR; Müller P Reduction of Dinitrogen to Ammonia Catalyzed by Molybdenum Diamido Complexes. J. Am. Chem. Soc 2017, 139, 9132–9135. [DOI] [PubMed] [Google Scholar]
- (141).Arashiba K; Sasaki K; Kuriyama S; Miyake Y; Nakanishi H; Nishibayashi Y Synthesis and Protonation of Molybdenum– and Tungsten–Dinitrogen Complexes Bearing PNP-Type Pincer Ligands. Organometadics 2012, 31, 2035–2041. [Google Scholar]
- (142).Arashiba K; Kuriyama S; Nakajima K; Nishibayashi Y Preparation and Reactivity of a Dinitrogen-Bridged Dimolybdenum-Tetrachloride Complex. Chem. Commun 2013, 49, 11215–11217. [DOI] [PubMed] [Google Scholar]
- (143).Tanabe Y; Kuriyama S; Arashiba K; Miyake Y; Nakajima K; Nishibayashi Y Preparation and Reactivity of Molybdenum-Dinitrogen Complexes Bearing an Arsenic-Containing ANA-Type Pincer Ligand. Chem. Commun 2013, 49, 9290–9292. [DOI] [PubMed] [Google Scholar]
- (144).Kuriyama S; Arashiba K; Nakajima K; Tanaka H; Yoshizawa K; Nishibayashi Y Nitrogen Fixation Catalyzed by Ferrocene-Substituted Dinitrogen-Bridged Dimolybdenum-Dinitrogen Complexes: Unique Behavior of Ferrocene Moiety as Redox Active Site. Chem. Sci 2015, 6, 3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Kuriyama S; Arashiba K; Nakajima K; Tanaka H; Yoshizawa K; Nishibayashi Y Azaferrocene-Based PNP-Type Pincer Ligand: Synthesis of Molybdenum, Chromium, and 1ron Complexes and Reactivity toward Nitrogen Fixation. Eur. J. Inorg. Chem 2016, 2016,4856–4861. [Google Scholar]
- (146).Arashiba K; Nakajima K; Nishibayashi Y Synthesis and Reactivity of Molybdenum-Dinitrogen Complexes Bearing PNN-Type Pincer Ligand: Molybdenum-Dinitrogen Complexes with PNN-Type Pincer Ligand. Z. Anorg. Attg. Chem 2015, 641, 100–104. [Google Scholar]
- (147).Kinoshita E; Arashiba K; Kuriyama S; Eizawa A; Nakajima K; Nishibayashi Y Synthesis and Catalytic Activity of Molybdenum-Nitride Complexes Bearing Pincer Ligands: Molybdenum-Nitride Complexes Bearing Pincer Ligands. Eur. J. Inorg. Chem 2015, 2015, 1789–1794. [Google Scholar]
- (148).Itabashi T; Mori I; Arashiba K; Eizawa A; Nakajima K; Nishibayashi Y Effect of Substituents on Molybdenum Triiodide Complexes Bearing PNP-Type Pincer Ligands toward Catalytic Nitrogen Fixation. Dalton Trans 2019, 48, 3182–3186. [DOI] [PubMed] [Google Scholar]
- (149).Eizawa A; Arashiba K; Tanaka H; Kuriyama S; Matsuo Y; Nakajima K; Yoshizawa K; Nishibayashi Y Remarkable Catalytic Activity of Dinitrogen-Bridged Dimolybdenum Complexes Bearing NHC-Based PCP-Pincer Ligands toward Nitrogen Fixation. Nat. Commun 2017, 8, 14874–14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Tanabe Y; Arashiba K; Nakajima K; Nishibayashi Y Catalytic Conversion of Dinitrogen into Ammonia under Ambient Reaction Conditions by Using Proton Source from Water. Chem. - Asian J 2017, 12, 2544–2548. [DOI] [PubMed] [Google Scholar]
- (151).Chciuk TV; Flowers RA Proton-Coupled Electron Transfer in the Reduction of Arenes by SmI2–Water Complexes. J. Am. Chem. Soc 2015, 137, 11526–11531. [DOI] [PubMed] [Google Scholar]
- (152).Chciuk TV; Anderson WR; Flowers RA Proton-Coupled Electron Transfer in the Reduction of Carbonyls by Samarium Diiodide-Water Complexes. J. Am. Chem. Soc 2016, 138, 8738–8741. [DOI] [PubMed] [Google Scholar]
- (153).Chciuk TV; Anderson WR; Flowers RA 1nterplay between Substrate and Proton Donor Coordination in Reductions of Carbonyls by SmI2–Water Through Proton-Coupled Electron-Transfer. J. Am. Chem. Soc 2018, 140, 15342–15352. [DOI] [PubMed] [Google Scholar]
- (154).Kolmar SS; Mayer JM SmI2(H2O)n Reduction of Electron Rich Enamines by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2017, 139, 10687–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Robson RL; Eady RR; Richardson TH; Miller RW; Hawkins M; Postgate JR The Alternative Nitrogenase of Azotobacter Chroococcum Is a Vanadium Enzyme. Nature 1986, 322, 388–390. [Google Scholar]
- (156).Chisnell JR; Premakumar R; Bishop PE Purification of a Second Alternative Nitrogenase from a NifHDK Deletion Strain of Azotobacter Vinelandii. J. Bacteriol 1988, 170, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Krahn E; Weiss B; Kröckel M; Groppe J; Henkel G; Cramer S; Trautwein A; Schneider K; Müller A The Fe-Only Nitrogenase from Rhodobactercapsulatus: Identification of the Cofactor, an Unusual, High-Nuclearity Iron-Sulfur Cluster, by Fe K-Edge EXAFS and 57Fe Mössbauer Spectroscopy. JBIC, J. Biol. Inorg. Chem 2002, 7, 37–45. [DOI] [PubMed] [Google Scholar]
- (158).Harris DF; Lukoyanov DA; Shaw S; Compton P; Tokmina-Lukaszewska M; Bothner B; Kelleher N; Dean DR; Hoffman BM; Seefeldt LC Mechanism of N Reduction Catalyzed by Fe-Nitrogenase Involves Reductive Elimination of H2. Biochemistry 2018, 57,701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Harris DF; Lukoyanov DA; Kallas H; Trncik C; Yang Z-Y; Compton P; Kelleher N; Einsle O; Dean DR; Hoffman BM; et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism To Achieve N2 Reduction. Biochemistry 2019, 58, 3293–3301. [DOI] [PubMed] [Google Scholar]
- (160).Spatzal T; Perez KA; Einsle O; Howard JB; Rees DC Ligand Binding to the FeMo-Cofactor: Structures of CO-Bound and Reactivated Nitrogenase. Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A Bound Reaction Intermediate Sheds Light on the Mechanism of Nitrogenase. Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
- (162).Lukoyanov D; Khadka N; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Reductive Elimination of H2 Activates Nitrogenase to Reduce the N≡N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc 2016, 138, 10674–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Dilworth MJ; Eady RR Hydrazine Is a Product of Dinitrogen Reduction by the Vanadium-Nitrogenase from Azotobacter Chroococcum. Biochem. J 1991, 277, 465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Barney BM; Lukoyanov D; Yang TC; Dean DR; Hoffman BM; Seefeldt LC A Methyldiazene (HN = N-CH3)-Derived Species Bound to the Nitrogenase Active-Site FeMo Cofactor: Implications for Mechanism. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17113–17118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Barney BM; McClead J; Lukoyanov D; Laryukhin M; Yang TC; Dean DR; Hoffman BM; Seefeldt LC Diazene (HN = NH) Is a Substrate for Nitrogenase: Insights into the Pathway of N2 Reduction. Biochemistry 2007, 46, 6784–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Coucouvanis D; Han J; Moon N Synthesis and Characterization of Sulfur-Voided Cubanes. Structural Analogues for the MoFe3S3 Subunit in the Nitrogenase Cofactor. J. Am. Chem. Soc 2002, 124, 216–224. [DOI] [PubMed] [Google Scholar]
- (167).Vela J; Stoian S; Flaschenriem CJ; Münck E; Holland PL A Sulfido-Bridged Diiron(II) Compound and Its Reactions with Nitrogenase-Relevant Substrates. J. Am. Chem. Soc 2004, 126, 4522–4523. [DOI] [PubMed] [Google Scholar]
- (168).MacBeth CE; Harkins SB; Peters JC Synthesis and Characterization of Cationic Iron Complexes Supported by the Neutral Ligands NPi-Pr3, NArPi-Pr3, and NSt-Bu3 Can. J. Chem 2005, 83, 332–340. [Google Scholar]
- (169).Crossland JL; Tyler DR Iron-Dinitrogen Coordination Chemistry: Dinitrogen Activation and Reactivity. Coord. Chem. Rev 2010, 254, 1883–1894. [Google Scholar]
- (170).McWilliams SF; Holland PL Dinitrogen Binding and Cleavage by Multinuclear Iron Complexes. Acc. Chem. Res 2015, 48, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Leigh GJ Update: Chemistry - Biological Nitrogen Fixation and Model Chemistry. Science 1997, 275, 1442–1442. [Google Scholar]
- (172).Ung G; Peters JC Low-Temperature N2 Binding to Two-Coordinate L2Fe0 Enables Reductive Trapping of L2FeN2− and NH3 Generation. Angew. Chem., Int. Ed 2015, 54, 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Del Castillo TJ; Thompson NB; Peters JC A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mossbauer Data, and a Hydride Resting State. J. Am. Chem. Soc 2016, 138, 5341–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Hill PJ; Doyle LR; Crawford AD; Myers WK; Ashley AE Selective Catalytic Reduction of N2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc 2016, 138, 13521–13524. [DOI] [PubMed] [Google Scholar]
- (175).Buscagan TM; Oyala PH; Peters JC N2-to-NH3 Conversion by a Triphos-Iron Catalyst and Enhanced Turnover under Photolysis. Angew. Chem., Int. Ed 2017, 56, 6921–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Thompson NB; Green MT; Peters JC Nitrogen Fixation via a Terminal Fe(IV) Nitride. J. Am. Chem. Soc 2017, 139, 15312–15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Nesbit MA; Oyala PH; Peters JC Characterization of the Earliest Intermediate of Fe-N2 Protonation: CW and Pulse EPR Detection of an Fe-NNH Species and Its Evolution to Fe-NNH2+. J. Am. Chem. Soc 2019, 141, 8116–8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Sekiguchi Y; Kuriyama S; Eizawa A; Arashiba K; Nakajima K; Nishibayashi Y Synthesis and Reactivity of Iron–Dinitrogen Complexes Bearing Anionic Methyl- and Phenyl-Substituted Pyrrole-Based PNP-Type Pincer Ligands toward Catalytic Nitrogen Fixation. Chem. Commun 2017, 53, 12040–12043. [DOI] [PubMed] [Google Scholar]
- (179).Higuchi J; Kuriyama S; Eizawa A; Arashiba K; Nakajima K; Nishibayashi Y Preparation and Reactivity of Iron Complexes Bearing Anionic Carbazole-Based PNP-Type Pincer Ligands toward Catalytic Nitrogen Fixation. Dalton Trans 2018, 47, 1117–1121. [DOI] [PubMed] [Google Scholar]
- (180).Cavaillé A; Joyeux B; Saffon-Merceron N; Nebra N; Fustier-Boutignon M; Mézailles N Triphos–Fe Dinitrogen and Dinitrogen–Hydride Complexes: Relevance to Catalytic N2 Reductions. Chem. Commun 2018, 54, 11953–11956. [DOI] [PubMed] [Google Scholar]
- (181).Moret ME; Peters JC N2 Functionalization at Iron Metallaboratranes. J. Am. Chem. Soc 2011, 133, 18118–18121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Anderson JS; Moret ME; Peters JC Conversion of Fe–NH2 to Fe–N2 with Release of NH3. J. Am. Chem. Soc 2013,135, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Fong H; Moret M-E; Lee Y; Peters JC Heterolytic H2 Cleavage and Catalytic Hydrogenation by an Iron Metallaboratrane. Organometallics 2013, 32, 3053–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Deegan MM; Peters JC Electrophile-Promoted Fe-to-N2 Hydride Migration in Highly Reduced Fe(N2)(H) Complexes. Chem. Sci 2018, 9, 6264–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Shima T; Hu S; Luo G; Kang X; Luo Y; Hou Z Dinitrogen Cleavage and Hydrogenation by a Trinuclear Titanium Polyhydride Complex. Science 2013, 340, 1549–1552. [DOI] [PubMed] [Google Scholar]
- (186).Hendrich MP; Gunderson W; Behan RK; Green MT; Mehn MP; Betley TA; Lu CC; Peters JC On the Feasibility of N2 Fixation via a Single-Site FeI/FeIV Cycle: Spectroscopic Studies of FeI(N2)FeI, FeIV≡N, and Related Species. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17107–17112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Vogel C; Heinemann FW; Sutter J; Anthon C; Meyer K An Iron Nitride Complex. Angew. Chem., Int. Ed 2008, 47, 2681–2684. [DOI] [PubMed] [Google Scholar]
- (188).Scepaniak JJ; Vogel CA; Khusniyarov MM; Heinemann FW; Meyer K; Smith JM Synthesis, Structure, and Reactivity of an Iron(V) Nitride. Science 2011, 331, 1049–1052. [DOI] [PubMed] [Google Scholar]
- (189).Betley TA; Peters JC A Tetrahedrally Coordinated L3Fe–Nx Platform That Accommodates Terminal Nitride (FeIV≡N) and Dinitrogen (FeI–N2–FeI) Ligands. J. Am. Chem. Soc 2004,126, 6252–6254. [DOI] [PubMed] [Google Scholar]
- (190).Lehnert N; Tuczek F The Reduction Pathway of End-on Coordinated Dinitrogen. I. Vibrational Spectra of Mo/W–N2, – NNH, and – NNH2 Complexes and Quantum Chemistry Assisted Normal Coordinate Analysis. Inorg. Chem 1999, 38, 1659–1670. [DOI] [PubMed] [Google Scholar]
- (191).Lehnert N; Tuczek F The Reduction Pathway of End-On Coordinated Dinitrogen. II. Electronic Structure and Reactivity of Mo/W–N2, – NNH, and – NNH2 Complexes. Inorg. Chem 1999, 38, 1671–1682. [DOI] [PubMed] [Google Scholar]
- (192).Matson BD; Peters JC Fe-Mediated HER vs N2RR: Exploring Factors That Contribute to Selectivity in P3EFe(N2) (E = B, Si, C) Catalyst Model Systems. ACS Catal 2018, 8, 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Lee Y; Mankad NP; Peters JC Triggering N2 Uptake via Redox-Induced Expulsion of Coordinated NH3 and N2 Silylation at Trigonal Bipyramidal Iron. Nat. Chem 2010, 2, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Rittle J; Peters JC Fe–N2/CO Complexes That Model a Possible Role for the Interstitial C Atom of FeMo-Cofactor (FeMoco). Proc. Natl. Acad. Sci. U. S. A 2013, 110, 15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Piascik AD; Hill PJ; Crawford AD; Doyle LR; Green JC; Ashley AE Cationic Silyldiazenido Complexes of the Fe(Diphosphine)2(N2) Platform: Structural and Electronic Models for an Elusive First Intermediate in N2 Fixation. Chem. Commun 2017, 53, 7657–7660. [DOI] [PubMed] [Google Scholar]
- (196).Schild DJ; Peters JC Light Enhanced Fe-Mediated Nitrogen Fixation: Mechanistic Insights Regarding H2 Elimination, HER, and NH3 Generation. ACS Catal 2019, 9, 4286–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Chalkley MJ; Del Castillo TJ; Matson BD; Peters JC Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring pKa Effects and Demonstrating Electrocatalysis. J. Am. Chem. Soc 2018, 140, 6122–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Pham DN; Burgess BK Nitrogenase Reactivity: Effects of pH on Substrate Reduction and Carbon Monoxide Inhibition. Biochemistry 1993, 32, 13725–13731. [DOI] [PubMed] [Google Scholar]
- (199).Chalkley MJ; Oyala PH; Peters JC Cp* Noninnocence Leads to a Remarkably Weak C–H Bond via Metallocene Protonation. J. Am. Chem. Soc 2019, 141, 4721–4729. [DOI] [PubMed] [Google Scholar]
- (200).Lancaster KM; Roemelt M; Ettenhuber P; Hu Y; Ribbe MW; Neese F; Bergmann U; DeBeer S X-Ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Chalkley MJ; Peters JC A Triad of Highly Reduced, Linear Iron Nitrosyl Complexes: {FeNO}8–10. Angew. Chem 2016, 128, 12174–12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Whited MT; Mankad NP; Lee YH; Oblad PF; Peters JC Dinitrogen Complexes Supported by Tris(Phosphino)Silyl Ligands. Inorg. Chem 2009, 48, 2507–2517. [DOI] [PubMed] [Google Scholar]
- (203).Mankad NP; Müller P; Peters JC Catalytic N-N Coupling of Aryl Azides to Yield Azoarenes via Trigonal Bipyramid Iron-Nitrene Intermediates. J. Am. Chem. Soc 2010, 132, 4083–4084. [DOI] [PubMed] [Google Scholar]
- (204).Mankad NP; Whited MT; Peters JC Terminal FeI–N2 and FeII⋯H–C Interactions Supported by Tris(Phosphino)Silyl Ligands. Angew. Chem. Int. Ed 2007, 46, 5768–5771. [DOI] [PubMed] [Google Scholar]
- (205).Gogoi U; Guha AK; Phukan AK Tracing the Route to Ammonia: A Theoretical Study on the Possible Pathways for Dinitrogen Reduction with Tripodal Iron Complexes. Chem. - Eur. J 2013, 19, 11077–11089. [DOI] [PubMed] [Google Scholar]
- (206).Lu J-B; Ma X-L; Wang J-Q; Liu J-C; Xiao H; Li J Efficient Nitrogen Fixation via a Redox-Flexible Single-Iron Site with Reverse-Dative Iron → Boron σ Bonding. J. Phys. Chem. A 2018, 122, 4530–4537. [DOI] [PubMed] [Google Scholar]
- (207).Jiang Y-F; Ma X-L; Lu J-B; Wang J-Q; Xiao H; Li J N2 Reduction on Fe-Based Complexes with Different Supporting Main-Group Elements: Critical Roles of Anchor and Peripheral Ligands. Small Methods 2019, 3, 1800340–1800346. [Google Scholar]
- (208).Kaczmarek MA; Malhotra A; Balan GA; Timmins A; de Visser SP Nitrogen Reduction to Ammonia on a Biomimetic Mononuclear Iron Centre: Insights into the Nitrogenase Enzyme. Chem. - Eur. J 2018, 24, 5293–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Benedek Z; Papp M; Oláh J; Szilvási T Identifying the Rate-Limiting Elementary Steps of Nitrogen Fixation with Single-Site Fe Model Complexes. Inorg. Chem 2018, 57, 8499–8508. [DOI] [PubMed] [Google Scholar]
- (210).Ehrlich N; Kreye M; Baabe D; Schweyen P; Freytag M; Jones PG; Walter MD Synthesis and Electronic Ground-State Properties of Pyrrolyl-Based Iron Pincer Complexes: Revisited. Inorg. Chem 2017, 56, 8415–8422. [DOI] [PubMed] [Google Scholar]
- (211).McWilliams SF; Bill E; Lukat-Rodgers G; Rodgers KR; Mercado BQ; Holland PL Effects of N2 Binding Mode on Iron-Based Functionalization of Dinitrogen to Form an Iron(III) Hydrazido Complex. J. Am. Chem. Soc 2018, 140, 8586–8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Holland PL Low-Coordinate Iron Complexes as Synthetic Models of Nitrogenase. Can. J. Chem 2005, 83, 296–301. [Google Scholar]
- (213).Betley TA; Peters JC Dinitrogen Chemistry from Trigonally Coordinated Iron and Cobalt Platforms. J. Am. Chem. Soc 2003, 125, 10782–10783. [DOI] [PubMed] [Google Scholar]
- (214).Smith JM; Sadique AR; Cundari TR; Rodgers KR; Lukat-Rodgers G; Lachicotte RJ; Flaschenriem CJ; Vela J; Holland PL Studies of Low-Coordinate Iron Dinitrogen Complexes. J. Am. Chem. Soc 2006, 128, 756–769. [DOI] [PubMed] [Google Scholar]
- (215).Chiang KP; Bellows SM; Brennessel WW; Holland PL Multimetallic Cooperativity in Activation of Dinitrogen at Iron-Potassium Sites. Chem. Sci 2014, 5, 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).MacLeod KC; Vinyard DJ; Holland PL A Multi-Iron System Capable of Rapid N2 Formation and N2 Cleavage. J. Am. Chem. Soc 2014, 136, 10226–10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).MacLeod KC; McWilliams SF; Mercado BQ; Holland PL Stepwise N–H Bond Formation from N2-Derived Iron Nitride, Imide and Amide Intermediates to Ammonia. Chem. Sci 2016, 7, 5736–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Hall DA; Leigh GJ Reduction of Dinitrogen Bound at an Iron(0) Centre. J. Chem. Soc. Dalton Trans 1996, 17, 3539–3541. [Google Scholar]
- (219).Hills A; Hughes DL; Jimenez-Tenorio M; Leigh GJ; Rowley AT Bis[1,2-Bis(Dimethylphosphino)Ethane]-Dihydrogenhydrido-Iron(II) Tetraphenylborate as a Model for the Function of Nitrogenases. J. Chem. Soc., Dalton Trans 1993, 20, 3041–3049. [Google Scholar]
- (220).Komiya S; Akita M; Yoza A; Kasuga N; Fukuoka A; Kai Y Isolation of a Zerovalent Iron Dinitrogen Complex With 1,2-Bis(Diethylphosphino)Ethane Ligands. J. Chem. Soc., Chem. Commun 1993, 787–788. [Google Scholar]
- (221).Hirano M; Akita M; Morikita T; Kubo H; Fukuoka A; Komiya S Synthesis, Structure and Reactions of a Dinitrogen Complex of Iron(0), [Fe(N2)(Depe)2] (Depe = Et2PCH2CH2PEt2). J. Chem. Soc. Dalton Trans 1997, 19, 3453–3458. [Google Scholar]
- (222).Field LD; Hazari N; Li HL Nitrogen Fixation Revisited on Iron(0) Dinitrogen Phosphine Complexes. Inorg. Chem 2015, 54, 4768–4776. [DOI] [PubMed] [Google Scholar]
- (223).Doyle LR; Hill PJ; Wildgoose GG; Ashley AE Teaching Old Compounds New Tricks: Efficient N2 Fixation by Simple Fe(N2)(Diphosphine)2 Complexes. Dalton Trans 2016, 45, 7550–7554. [DOI] [PubMed] [Google Scholar]
- (224).Field LD; Li HL; Magill AM Base-Mediated Conversion of Hydrazine to Diazene and Dinitrogen at an Iron Center. Inorg. Chem 2009, 48, 5–7. [DOI] [PubMed] [Google Scholar]
- (225).Sacco A; Aresta M Nitrogen Fixation: Hydrido- and Hydrido-Nitrogen-Complexes of Iron(II). Chem. Commun 1968, 20, 1223–1224. [Google Scholar]
- (226).Aresta M; Giannoccaro P; Rossi M; Sacco A Nitrogen Fixation. II. Dinitrogen-Complexes of Iron. Inorg. Chim. Acta 1971, 5, 203–206. [Google Scholar]
- (227).Whittlesey MK; Mawby RJ; Osman R; Perutz RN; Field LD; Wilkinson MP; George MW Transient and Matrix Photochemistry of Fe(Dmpe)2H2 (Dmpe = Me2PCH2CH2PMe2): Dynamics of C-H and H-H Activation. J. Am. Chem. Soc 1993, 115, 8627–8637. [Google Scholar]
- (228).Van der Sluys LS; Eckert J; Eisenstein O; Hall JH; Huffman JC; Jackson SA; Koetzle TF; Kubas GJ; Vergamini PJ; Caulton KG An Attractive Cis-Effect of Hydride on Neighbor Ligands: Experimental and Theoretical Studies on the Structure and Intramolecular Rearrangements of Fe(H)2(η2-H2)(PEtPh2)3. J. Am. Chem. Soc 1990, 112, 4831–4841. [Google Scholar]
- (229).Field LD; Li HL; Dalgarno SJ; Turner P The First Side-on Bound Metal Complex of Diazene, HN = NH. Chem. Commun 2008, 14, 1680–1682. [DOI] [PubMed] [Google Scholar]
- (230).Yelle RB; Crossland JL; Szymczak NK; Tyler DR Theoretical Studies of N2 Reduction to Ammonia in Fe(Dmpe)2N2. Inorg. Chem 2009, 48, 861–871. [DOI] [PubMed] [Google Scholar]
- (231).Fox DJ; Bergman RG Synthesis of an Iron Parent Amido Complex and a Comparison of Its Reactivity with the Ruthenium Analog. Organometallics 2004, 23, 1656–1670. [Google Scholar]
- (232).Crossland JL; Zakharov LN; Tyler DR Synthesis and Characterization of an Iron(II) η2-Hydrazine Complex. Inorg. Chem 2007, 46, 10476–10478. [DOI] [PubMed] [Google Scholar]
- (233).Crossland JL; Balesdent CG; Tyler DR Intermediates in the Reduction of N2 to NH3: Synthesis of Iron η2 Hydrazido(1−) and Diazene Complexes. Dalton Trans 2009, 23, 4420–4422. [DOI] [PubMed] [Google Scholar]
- (234).Field LD; Li HL; Dalgarno SJ; Jensen P; McIntosh RD Synthesis and Characterization of Iron(II) and Ruthenium(II) Hydrido Hydrazine Complexes. Inorg. Chem 2011, 50, 5468–5476. [DOI] [PubMed] [Google Scholar]
- (235).Wiesler B; Tuczek F; Nather C; Bensch W [FeHCl-(C10H24P2)2]. Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1998,54, 44–46. [Google Scholar]
- (236).Miller WK; Gilbertson JD; Leiva-Paredes C; Bernatis PR; Weakley TJR; Lyon DK; Tyler DR Precursors to Water-Soluble Dinitrogen Carriers. Synthesis of Water-Soluble Complexes of Iron(II) Containing Water-Soluble Chelating Phosphine Ligands of the Type 1,2-Bis(Bis(Hydroxyalkyl)Phosphino)Ethane. Inorg. Chem 2002, 41, 5453–5465. [DOI] [PubMed] [Google Scholar]
- (237).Gilbertson JD; Szymczak NK; Tyler DR H2 Activation in Aqueous Solution: Formation of Trans-[Fe(DMeOPrPE)2H(H2)]+ via the Heterolysis of H2 in Water. Inorg. Chem 2004, 43, 3341–3343. [DOI] [PubMed] [Google Scholar]
- (238).Gilbertson JD; Szymczak NK; Crossland JL; Miller WK; Lyon DK; Foxman BM; Davis J; Tyler DR Coordination Chemistry of H2 and N2 in Aqueous Solution. Reactivity and Mechanistic Studies Using Trans-FeII(P2)2X2-Type Complexes (P2 = a Chelating, Water-Solubilizing Phosphine). Inorg. Chem 2007, 46, 1205–1214. [DOI] [PubMed] [Google Scholar]
- (239).Tyler DR Mechanisms for the Formation of NH3, N2H4, and N2H2 in the Protonation Reaction of Fe(DMeOPrPE)2N2 DMeOPrPE = 1,2-Bis[Bis(Methoxypropyl)Phosphino]Ethane. Z. Anorg. Allg. Chem 2015, 641, 31–39. [Google Scholar]
- (240).Gilbertson JD; Szymczak NK; Tyler DR Reduction of N2 to Ammonia and Hydrazine Utilizing H2 as the Reductant. J. Am. Chem. Soc 2005, 127, 10184–10185. [DOI] [PubMed] [Google Scholar]
- (241).Nishibayashi Y; Iwai S; Hidai M Bimetallic System for Nitrogen Fixation: Ruthenium-Assisted Protonation of Coordinated N2 on Tungsten with H2. Science 1998, 279, 540–542. [DOI] [PubMed] [Google Scholar]
- (242).Fryzuk MD; Love JB; Rettig SJ; Young VG Transformation of Coordinated Dinitrogen by Reaction with Dihydrogen and Primary Silanes. Science 1997, 275, 1445–1447. [Google Scholar]
- (243).Bancroft GM; Mays MJ; Prater BE A New Route to Molecular Nitrogen Complexes. J. Chem. Soc. D 1969, 11, 585–585. [Google Scholar]
- (244).Mayer U; Gutmann V; Gerger W The Acceptor Number — A Quantitative Empirical Parameter for the Electrophilic Properties of Solvents. Monatsh. Chem 1975, 106, 1235–1257. [Google Scholar]
- (245).Beckett MA; Strickland GC; Holland JR; Sukumar Varma K A convenient NMR Method for the Measurement of Lewis Acidity at Boron Centres: Correlation of Reaction Rates of Lewis Acid Initiated Epoxide Polymerizations with Lewis Acidity. Polymer 1996, 37, 4629–4631. [Google Scholar]
- (246).Geri JB; Shanahan JP; Szymczak NK Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc 2017, 139, 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Piascik AD; Li R; Wilkinson HJ; Green JC; Ashley AE Fe-Catalyzed Conversion of N2 to N(SiMe3)3 via an Fe-Hydrazido Resting State. J. Am. Chem. Soc 2018, 140, 10691–10694. [DOI] [PubMed] [Google Scholar]
- (248).Schrock RR Reduction of Dinitrogen. Proc. Natl.Acad. Sci. U. S. A 2006, 103, 17087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Čorić I; Holland PL Insight into the Iron-Molybdenum Cofactor of Nitrogenase from Synthetic Iron Complexes with Sulfur, Carbon, and Hydride Ligands. J. Am. Chem. Soc 2016, 138, 7200–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Suess DLM; Tsay C; Peters JC Dihydrogen Binding to Isostructural S = 1/2 and S = 0 Cobalt Complexes. J. Am. Chem. Soc 2012, 134, 14158–14164. [DOI] [PubMed] [Google Scholar]
- (251).Takaoka A; Gerber LCH; Peters JC Access to Well-Defined Ruthenium(I) and Osmium(I) Metalloradicals. Angew. Chem. Int Ed 2010, 49, 4088–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Tanabe Y; Nishibayashi Y Recent Advances in Nitrogen Fixation upon Vanadium Complexes. Coord. Chem. Rev 2019, 381, 135–150. [Google Scholar]
- (253).Sekiguchi Y; Meng F; Tanaka H; Eizawa A; Arashiba K; Nakajima K; Yoshizawa K; Nishibayashi Y Synthesis and Reactivity of Titanium- and Zirconium-Dinitrogen Complexes Bearing Anionic Pyrrole-Based PNP-Type Pincer Ligands. Dalton Trans 2018, 47, 11322–11326. [DOI] [PubMed] [Google Scholar]
- (254).Fryzuk MD; Haddad TS; Mylvaganam M; McConville DH; Rettig SJ End-on versus Side-on Bonding of Dinitrogen to Dinuclear Early Transition-Metal Complexes. J. Am. Chem. Soc 1993, 115, 2782–2792. [Google Scholar]
- (255).Shih K-Y; Schrock RR; Kempe R Synthesis of Molybdenum Complexes That Contain Silylated Triamidoamine Ligands. A μ-Dinitrogen Complex, Methyl and Acetylide Complexes, and Coupling of Acetylides. J. Am. Chem. Soc 1994, 116, 8804–8805. [Google Scholar]
- (256).Henderson Wm. A.; Streuli CA The Basicity of Phosphines. J. Am. Chem. Soc 1960, 82, 5791–5794. [Google Scholar]
- (257).Manriquez JM; Sanner RD; Marsh RE; Bercaw JE Reduction of Molecular Nitrogen to Hydrazine - Structure of a Dinitrogen Complex of Bis(Pentamethylcyclopentadienyl)Zirconium(II) and an N-15 Labeling Study of Its Reaction with Hydrogen-Chloride. J. Am. Chem. Soc 1976, 98, 3042–3044. [Google Scholar]
- (258).Kokubo Y; Yamamoto C; Tsuzuki K; Nagai T; Katayama A; Ohta T; Ogura T; Wasada-Tsutsui Y; Kajita Y; Kugimiya S; et al. Dinitrogen Fixation by Vanadium Complexes with a Triamidoamine Ligand. Inorg. Chem 2018, 57, 11884–11894. [DOI] [PubMed] [Google Scholar]
- (259).Scheer M; Müller J; Schiffer M; Baum G; Winter R Pnictides as Symmetrically Bridging Ligands in Novel Neutral Complexes. Chem. - Eur. J 2000, 6, 1252–1257. [DOI] [PubMed] [Google Scholar]
- (260).Jewess M; Crabtree RH Electrocatalytic Nitrogen Fixation for Distributed Fertilizer Production? ACS Sustainable Chem. Eng 2016, 4, 5855–5858. [Google Scholar]
- (261).Chen JG; Crooks RM; Seefeldt LC; Bren KL; Bullock RM; Darensbourg MY; Holland PL; Hoffman B; Janik MJ; Jones AK; et al. Beyond Fossil Fuel–Driven Nitrogen Transformations. Science 2018, 360, 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Shipman MA; Symes MD Recent Progress towards the Electrosynthesis of Ammonia from Sustainable Resources. Catal Today 2017, 286, 57–68. [Google Scholar]
- (263).Ostermann N; Siewert I Electrochemical N2 Splitting at Well-Defined Metal Complexes. Curr. Opin. Electrochem 2019, 15, 97–101. [Google Scholar]
- (264).Martín AJ; Shinagawa T; Pérez-Ramírez J Electrocatalytic Reduction of Nitrogen: From Haber-Bosch to Ammonia Artificial Leaf. Chem 2019, 5, 263–283. [Google Scholar]
- (265).Milton RD; Cai R; Abdellaoui S; Leech D; De Lacey AL; Pita M; Minteer SD Bioelectrochemical Haber–Bosch Process: An Ammonia-Producing H2/N2 Fuel Cell. Angew. Chem. Int. Ed 2017, 56, 2680–2683. [DOI] [PubMed] [Google Scholar]
- (266).Chen H; Cai R; Patel J; Dong F; Chen H; Minteer SD Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates. J. Am. Chem. Soc 2019, 141, 4963–4971. [DOI] [PubMed] [Google Scholar]
- (267).Pickett CJ; Talarmin J Electrosynthesis of Ammonia. Nature 1985, 317, 652–653. [Google Scholar]
- (268).Pickett CJ; Ryder KS; Talarmin J Electron-Transfer Reactions in Nitrogen Fixation. Part 2. The Electrosynthesis of Ammonia: Identification and Estimation of Products. J. Chem. Soc. Dalton Trans 1986, 1453–1457. [Google Scholar]
- (269).Becker JY; Avraham S Nitrogen Fixation: Part III. Electrochemical Reduction of Hydrazido (−NNH2) Mo and W Complexes. Selective Formation of NH3 under Mild Conditions. J. Electroanal. Chem. Interfacial Electrochem 1990, 280, 119–127. [Google Scholar]
- (270).Vol’pin ME; Shur VB Nitrogen Fixation by Transition Metal Complexes. Nature 1966, 209, 1236–1236. [Google Scholar]
- (271).Brintzinger H Formation of Ammonia by Insertion of Molecular Nitrogen into Metal-Hydride Bonds. II. Di-μ-Imino-Bis(Dicyclopentadienyltitanium(III)) as a Product of the Reaction between Di-μ-Hydrido-Bis(Dicyclopentadienyltitanium(III)) and Molecular Nitrogen. J. Am. Chem. Soc 1966, 88, 4307–4308. [Google Scholar]
- (272).Brintzinger H Formation of Ammonia by Insertion of Molecular Nitrogen into Metal-Hydride Bonds. III. Considerations on the Properties of Enzymatic Nitrogen-Fixing Systems and Proposal of a General Mechanism. Biochemistry 1966, 5, 3947–3950. [Google Scholar]
- (273).Brintzinger H Formation of Ammonia by Insertion of Molecular Nitrogen into Metal-Hydride Bonds. I. the Formation of Dimeric Dicyclopentadienyltitanium(III) Hydride as an Intermediate in the Vol’pin-Shur Nitrogen-Fixing System. J. Am. Chem. Soc 1966, 88, 4305–4307. [Google Scholar]
- (274).Maskill R; Pratt JM Kinetics of the Reaction of N2 with Titanium Cyclopentadienyl Complexes. Chem. Commun. (London) 1967, 0,950–951. [Google Scholar]
- (275).Bayer E; Schurig V Stickstoff-Fixierung und Reduktion zu Ammoniak mit metallorganischen Katalysatoren. Chem. Ber 1969,102, 3378–3390. [Google Scholar]
- (276).Becker JY; Avraham Tsarfaty S; Posin B Nitrogen Fixation: Part I. Electrochemical Reduction of Titanium Compounds in the Presence of Catechol and N2 in MeOH or THF. J. Electroanal. Chem. Interfacial Electrochem 1987, 230, 143–153. [Google Scholar]
- (277).Jeong E-Y; Yoo C-Y; Jung CH; Park JH; Park YC; Kim J-N; Oh S-G; Woo Y; Yoon HC Electrochemical Ammonia Synthesis Mediated by Titanocene Dichloride in Aqueous Electrolytes under Ambient Conditions. ACS Sustainable Chem. Eng 2017, 5, 9662–9666. [Google Scholar]
- (278).Katayama A; Inomata T; Ozawa T; Masuda H Electrochemical Evaluation of Titanocenes in Ionic Liquids with Non-coordinating and Coordinating Anions and Application for NH3 Synthesis. ChemElectroChem 2017, 4, 3053–3060. [Google Scholar]
- (279).Katayama A; Inomata T; Ozawa T; Masuda H Ionic Liquid Promotes N2 Coordination to Titanocene(III) Monochloride. Dalton Trans 2017, 46, 7668–7671. [DOI] [PubMed] [Google Scholar]
- (280).Furuya N; Yoshiba H Nitrogen Fixation Using a Gas-Diffusion Electrode Loaded with Iron Phthalocyanine. Denki Kagaku Oyobi Kogyo Butsuri Kagaku 1989, 57, 261–262. [Google Scholar]
- (281).Furuya N; Yoshiba H Electroreduction of Nitrogen to Ammonia on Gas-Diffusion Electrodes Modified by Fe-Phthalocyanine. J. Electroanal. Chem. Interfacial Electrochem 1989, 263, 171–174. [Google Scholar]
- (282).Furuya N; Yoshiba H Electroreduction of Nitrogen to Ammonia on Gas-Diffusion Electrodes Loaded with Inorganic Catalyst. J. Electroanal. Chem. Interfacial Electrochem 1990, 291, 269–272. [Google Scholar]
- (283).Shipman MA; Symes MD A Re-Evaluation of Sn(II) Phthalocyanine as a Catalyst for the Electrosynthesis of Ammonia. Electrochim. Acta 2017, 258, 618–622. [Google Scholar]
- (284).Andersen SZ; Čolić V; Yang S; Schwalbe JA; Nielander AC; McEnaney JM; Enemark-Rasmussen K; Baker JG; Singh AR; Rohr BA; et al. A Rigorous Electrochemical Ammonia Synthesis Protocol with Quantitative Isotope Measurements. Nature 2019, 570, 504–508. [DOI] [PubMed] [Google Scholar]
- (285).Sherbow TJ; Thompson EJ; Arnold A; Sayler RI; Britt RD; Berben LA Electrochemical Reduction of N2 to NH3 at Low Potential by a Molecular Aluminum Complex. Chem. - Eur. J 2019, 25, 454–458. [DOI] [PubMed] [Google Scholar]
- (286).Légaré M-A; Bélanger-Chabot G; Dewhurst RD; Welz E; Krummenacher I; Engels B; Braunschweig H Nitrogen Fixation and Reduction at Boron. Science 2018, 359, 896–900. [DOI] [PubMed] [Google Scholar]
- (287).Tafel J Über Die Polarisation bei Kathodischer Wasser-stoffentwicklung. Z. Phys. Chem 1905, 50U, 641–712. [Google Scholar]
- (288).Pegis ML; McKeown BA; Kumar N; Lang K; Wasylenko DJ; Zhang XP; Raugei S; Mayer JM Homogenous Electrocatalytic Oxygen Reduction Rates Correlate with Reaction Overpotential in Acidic Organic Solutions. ACS Cent. Sci 2016, 2, 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Pegis ML; Wise CF; Koronkiewicz B; Mayer JM Identifying and Breaking Scaling Relations in Molecular Catalysis of Electrochemical Reactions. J. Am. Chem. Soc 2017,139, 11000–11003. [DOI] [PubMed] [Google Scholar]
- (290).Costentin C; Savéant J-M Towards an Intelligent Design of Molecular Electrocatalysts. Nat. Rev. Chem 2017, 1, 1–8. [Google Scholar]
- (291).Klug CM; Cardenas AJP; Bullock RM; O’Hagan M; Wiedner ES Reversing the Tradeoff between Rate and Overpotential in Molecular Electrocatalysts for H2 Production. ACS Catal 2018, 8, 3286–3296. [Google Scholar]
- (292).Huo PF; Uyeda C; Goodpaster JD; Peters JC; Miller TF Breaking the Correlation between Energy Costs and Kinetic Barriers in Hydrogen Evolution via a Cobalt Pyridine-Diimine-Dioxime Catalyst. ACS Catal 2016, 6, 6114–6123. [Google Scholar]
- (293).Wang Y; Montoya JH; Tsai C; Ahlquist MSG; Nørskov JK; Studt F Scaling Relationships for Binding Energies of Transition Metal Complexes. Catal. Lett 2016, 146, 304–308. [Google Scholar]
- (294).Lindley BM; Appel AM; Krogh-Jespersen K; Mayer JM; Miller AJM Evaluating the Thermodynamics of Electrocatalytic N2 Reduction in Acetonitrile. ACS Energy Lett 2016, 1, 698–704. [Google Scholar]
- (295).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Cappellani EP; Drouin SD; Jia G; Maltby PA; Morris RH; Schweitzer CT Effect of the Ligand and Metal on the pKa Values of the Dihydrogen Ligand in the Series of Complexes [M(H2)H(L)2]+, M = Fe, Ru, Os, Containing Isosteric Ditertiaryphosphine Ligands, L. J. Am. Chem. Soc 1994, 116, 3375–3388. [Google Scholar]
- (297).Kaljurand I; Kütt A; Sooväli L; Rodima T; Mäemets V; Leito I; Koppel IA Extension of the Self-Consistent Spectrophoto-metric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem 2005, 70, 1019–1028. [DOI] [PubMed] [Google Scholar]
- (298).Robbins JL; Edelstein N; Spencer B; Smart JC Syntheses and Electronic Structures of Decamethylmetallocenes. J. Am. Chem. Soc 1982, 104, 1882–1893. [Google Scholar]
- (299).Abdur-Rashid K; Fong TP; Greaves B; Gusev DG; Hinman JG; Landau SE; Lough AJ; Morris RH An Acidity Scale for Phosphorus-Containing Compounds Including Metal Hydrides and Dihydrogen Complexes in THF: Toward the Unification of Acidity Scales. J. Am. Chem. Soc 2000, 122, 9155–9171. [Google Scholar]
- (300).Scepaniak JJ; Young JA; Bontchev RP; Smith JM Formation of Ammonia from an Iron Nitrido Complex. Angew. Chem., Int. Ed 2009, 48, 3158–3160. [DOI] [PubMed] [Google Scholar]
- (301).Pappas I; Chirik PJ Ammonia Synthesis by Hydrogenolysis of Titanium-Nitrogen Bonds Using Proton Coupled Electron Transfer. J. Am. Chem. Soc 2015, 137, 3498–3501. [DOI] [PubMed] [Google Scholar]
- (302).Scheibel MG; Abbenseth J; Kinauer M; Heinemann FW; Würtele C; de Bruin B; Schneider S Homolytic N–H Activation of Ammonia: Hydrogen Transfer of Parent Iridium Ammine, Amide, Imide, and Nitride Species. Inorg. Chem 2015, 54, 9290–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Kendall AJ; Johnson SI; Bullock RM; Mock MT Catalytic Silylation of N2 and Synthesis of NH3 and N2H4 by Net Hydrogen Atom Transfer Reactions Using a Chromium P4 Macrocycle. J. Am. Chem. Soc 2018, 140, 2528–2536. [DOI] [PubMed] [Google Scholar]
- (304).Tanaka H; Nishibayashi Y; Yoshizawa K Interplay between Theory and Experiment for Ammonia Synthesis Catalyzed by Transition Metal Complexes. Acc. Chem. Res 2016, 49, 987–995. [DOI] [PubMed] [Google Scholar]
- (305).Ohki Y; Uchida K; Tada M; Cramer RE; Ogura T; Ohta T N2 Activation on a Molybdenum–Titanium–Sulfur Cluster. Nat. Commun 2018, 9, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Creutz SE; Peters JC Exploring Secondary-Sphere Interactions in Fe–NxHy Complexes Relevant to N2 Fixation. Chem. Sci 2017, 8, 2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).McKone JR; Marinescu SC; Brunschwig BS; Winkler JR; Gray HB Earth-Abundant Hydrogen Evolution Electrocatalysts. Chem. Sci 2014, 5, 865–878. [Google Scholar]
- (308).Bullock RM; Appel AM; Helm ML Production of Hydrogen by Electrocatalysis: Making the H–H Bond by Combining Protons and Hydrides. Chem. Commun 2014, 50, 3125–3143. [DOI] [PubMed] [Google Scholar]
- (309).Zhang W; Lai W; Cao R Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin- and Corrole-Based Systems. Chem. Rev 2017, 117, 3717–3797. [DOI] [PubMed] [Google Scholar]
- (310).Francke R; Schille B; Roemelt M Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev 2018, 118, 4631–4701. [DOI] [PubMed] [Google Scholar]
- (311).Tang C; Qiao S-Z How to Explore Ambient Electrocatalytic Nitrogen Reduction Reliablyand Insightfully. Chem. Soc. Rev 2019, 48, 3166–3180. [DOI] [PubMed] [Google Scholar]