

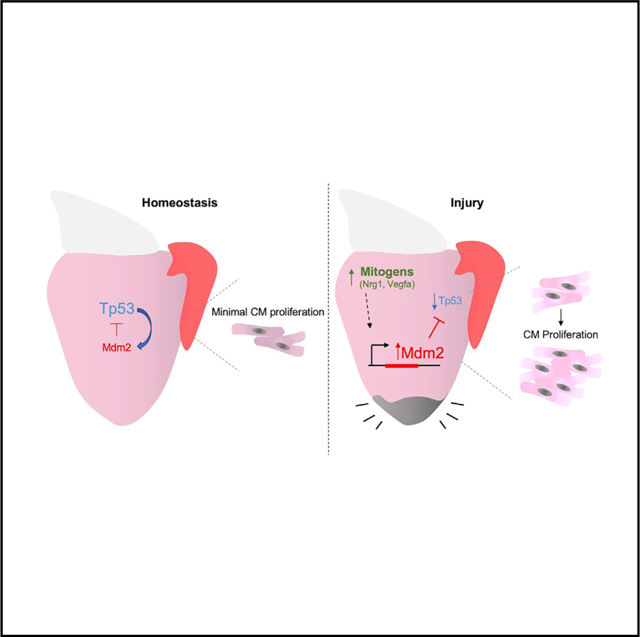
SUMMARY
Zebrafish regenerate heart muscle through division of pre-existing cardiomyocytes. To discover underlying regulation, we assess transcriptome datasets for dynamic gene networks during heart regeneration and identify suppression of genes associated with the transcription factor Tp53. Cardiac damage leads to fluctuation of Tp53 protein levels, concomitant with induced expression of its central negative regulator, mdm2, in regenerating cardiomyocytes. Zebrafish lacking functional Tp53 display increased indicators of cardiomyocyte proliferation during regeneration, whereas transgenic Mdm2 blockade inhibits injury-induced cardiomyocyte proliferation. Induced myocardial overexpression of the mitogenic factors Nrg1 or Vegfaa in the absence of injury also upregulates mdm2 and suppresses Tp53 levels, and tp53 mutations augment the mitogenic effects of Nrg1. mdm2 induction is spatiotemporally associated with markers of de-differentiation in injury and growth contexts, suggesting a broad role in cardiogenesis. Our findings reveal myocardial Tp53 suppression by mitogen-induced Mdm2 as a regulatory component of innate cardiac regeneration.
Graphical Abstract
In Brief
Zebrafish regenerate heart muscle by stimulating cardiomyocyte division. Shoffner et al. find that the Tp53 inhibitor Mdm2 is induced in cardiomyocytes by injury or experimental overexpression of cardiac mitogenic factors, suppressing the Tp53 network. tp53 mutations enhance cardiomyocyte proliferation during regeneration, whereas myocardial blockade of Mdm2 has opposite effects.
INTRODUCTION
Myocardial infarction (MI), a result of impaired blood flow to the heart and subsequent death of cardiomyocytes (CMs), continues to cause significant morbidity and mortality (Go et al., 2013). Enhancing the ability of MI victims to regenerate lost CMs and restore cardiac function is a major focus within the field of regenerative medicine (Sadek and Olson, 2020).
By contrast with mammals, adult zebrafish possess a high capacity to regenerate damaged or lost cardiac tissue following injury, through proliferation of spared, injury-escaping CMs (Kikuchi et al., 2010; Poss et al., 2002). Recently, a number of factors have been found to boost adult zebrafish CM proliferation if experimentally modulated during regeneration (Gemberling et al., 2015; Han et al., 2019; Karra et al., 2018; Missinato et al., 2018; Wu et al., 2016; Zhao et al., 2019). A subset of these factors, including Neuregulin1 (Nrg1), Vascular endothelial growth factor a (Vegfa), and vitamin D analogs (Gemberling et al., 2015; Han et al., 2019; Karra et al., 2018), have mitogenic effects even in the absence of injury. How these factors in particular exert their effects can enlighten the field, as they possess capacity as single entities to jumpstart a complex process of cardiogenesis, angiogenesis, and epicardial expansion, all seemingly without influences such as cell death, abrupt changes in tissue tension, and inflammation.
The tumor suppressor gene TP53, discovered 40 years ago, is the most widely mutated gene in human cancers (Lane and Crawford, 1979; Mantovani et al., 2019). Tp53 is a highly conserved transcription factor with critical roles in a variety of cell processes including cell cycle regulation, DNA repair, apoptosis, and senescence (Belyi et al., 2010; Berghmans et al., 2005; Fridman and Lowe, 2003; Hafner et al., 2019; Kruiswijk et al., 2015; Vousden and Lu, 2002). In CMs, Tp53 has been implicated as a key regulator of the cardiac transcriptome, and increased Tp53 levels have been shown to be associated with cardiac hypertrophy and remodeling (Mak et al., 2017; Nomura et al., 2018). Tp53 levels are typically kept low under homeostatic conditions, primarily through the activity of an E3 ubiquitin ligase, Mdm2. When Mdm2 is bound to Tp53 via its N-terminal Tp53 binding domain, its C-terminal ligase ubiquitinates Tp53 and targets it for proteosomal degradation (Chua et al., 2015; Michael and Oren, 2003; Nomura et al., 2017). These interactions between Tp53 and Mdm2 have been implicated in maintaining cardiac homeostasis in mice (Stanley-Hasnain et al., 2017). Mdm2-Tp53 binding has also been shown to physically inhibit the transactivational domain of Tp53 (Oliner et al., 1993). Following a cellular insult like ionizing radiation exposure, Mdm2 is prevented from binding to Tp53, allowing its accumulation and activation of target genes (Guo et al., 2013; Wade et al., 2013). As molecular damage resolves, Tp53 acts as its own negative regulator in a complex that directly increases mdm2 transcription (Berghmans et al., 2005; Chua et al., 2015; Pant et al., 2013). Additional, Tp53-independent roles for Mdm2 have been elucidated (Bohlman and Manfredi, 2014; Gu et al., 2009).
Here, a potential role of Tp53 during innate heart regeneration in zebrafish arose after bioinformatic assessment indicated that the network of Tp53-associated genes is suppressed during heart regeneration. We show evidence that this suppression is due to regulated induction of mdm2, which we observe in CMs during muscle regeneration as well as other cardiogenic events. Genetic reduction of Tp53 levels increased CM proliferation, either during heart regeneration or upon direct mitogen stimulation, whereas myocardial inhibition of Mdm2 decreased CM proliferation. Our experiments indicate that zebrafish heart regeneration is enabled by a mechanism in which injury-induced mitogens suppress Tp53.
RESULTS
Tp53 Protein Levels Transiently Decrease after Heart Injury
To uncover transcription factors that regulate gene expression during heart regeneration, we performed additional analyses of published RNA-sequencing (RNA-seq) datasets using Ingenuity Pathway Analysis (IPA) software, a curated database of literature-derived information on biological interactions. Comparisons were made between samples from hearts of uninjured zebrafish and those of zebrafish receiving a genetic ablation injury that destroys ~50% of all CMs (Kang et al., 2016; Krämer et al., 2014; Wang et al., 2011; Figures 1A, 1B, and S1A; Tables S1 and S2). In this injury model, CM death occurs uniformly throughout the heart over 5 to 7 days after incubation with tamoxifen (days post injury [dpi]; Wang et al., 2011). Thus, 7 dpi is considered an early post-injury stage and 14 dpi is more temporally removed from injury, with each time point displaying high indices of CM proliferation. The most significant activated upstream regulators included factors involved in inflammation, such as tumor necrosis factor (TNF) and interleukin-6 (IL6; Table S1). ErbB2 and Vegf signaling, known to impact CM proliferation (Bersell et al., 2009; D’Uva et al., 2015; Gemberling et al., 2015; Karra et al., 2018; Lai et al., 2017), were also found among the list of upstream regulators (Table S1). Several transcription factors were identified as possible suppressed upstream regulators (i.e., molecules whose suppression results in the observed gene expression changes), including KLF15 and RB1. Unexpectedly, among the most suppressed upstream regulators at 7 dpi was the downstream transcriptional network of Tp53 (Figures 1B and S1; Table S2). By 14 dpi, this was no longer the case (Table S2). To assess changes in Tp53 levels during heart regeneration, we performed western blotting on proteins isolated from whole hearts after CM ablation. Cardiac Tp53 levels trended as dynamic after injury, measuring on average at ~40% of uninjured levels at 7 dpi and recovering to levels greater than those of uninjured fish at 14 dpi (Figure 1C). Statistical tests reflected sample variability, but trends were consistent with the analyses of our transcriptome data and with a recent proteomics study assessing Tp53 levels by mass spectrometry after resection of the zebrafish ventricle (Ma et al., 2018).
Figure 1. Zebrafish Tp53 and mdm2 Expression Are Dynamic upon Heart Injury.
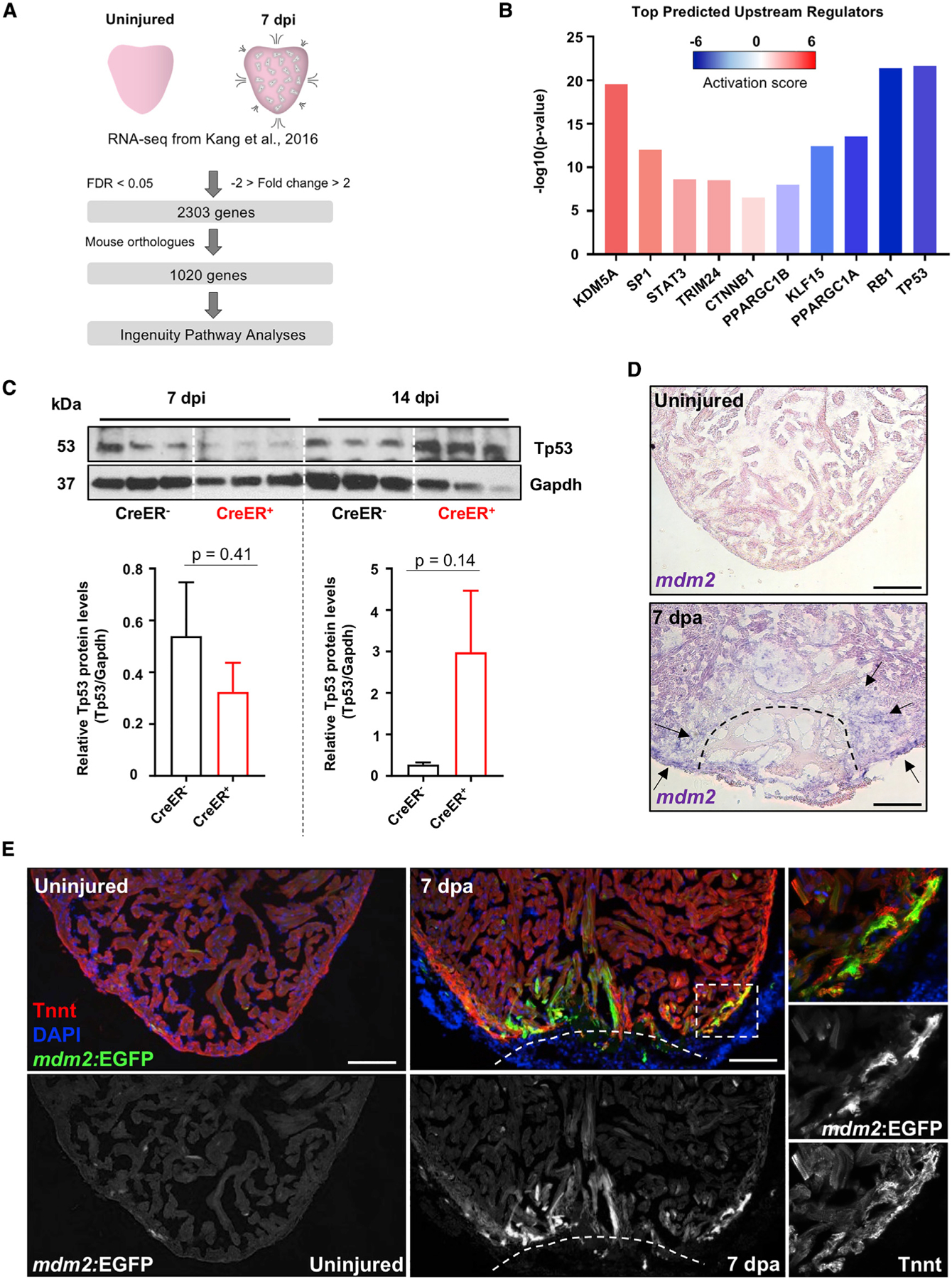
(A) Experimental design and bioinformatical data analysis.
(B) Selected top transcription factors acting as upstream regulators of differentially expressed genes in adult hearts 7 days after initiation (dpi) of CM ablation.
(C) Western blot on protein extract of control (β-actin2:loxp-mCherry-STOP-loxp-DTApd36), 7 dpi and 14 dpi (cmlc2:CreER; β-actin2:loxp-mCherry-STOP-loxp-DTApd36) ventricles, with quantification of Tp53 protein. n = 8–10 pooled hearts per sample. Data show mean ± SEM (unpaired t test).
(D) ISH for mdm2 expression (violet) in sections of uninjured and 7 days after resection (dpa) ventricles. Arrows identify sites of increased mdm2 expression following ventricle amputation.
(E) Section images of uninjured and 7 dpa mdm2:EGFP hearts. “Tnnt” marks CMs. Boxes correspond to the region magnified in the right panels. Scale bars: 100 μm.
Mdm2 is the primary regulator of Tp53. Based on our previously published transcriptome datasets, mdm2 transcripts are increased at 7 and 14 dpi at 1.7- and 2.6-fold those of uninjured levels, respectively (Figure S2B; Goldman et al., 2017; Kang et al., 2016). To visualize these changes, we performed in situ hybridization (ISH) using injured and regenerating hearts. mdm2 was rarely detectable in uninjured hearts. By contrast, increased mdm2 expression was detected in the cortical muscle layer near the injury site at 7 days following resection of the ventricular apex (days post amputation [dpa]), although variable and sometimes difficult to detect (Figure 1D). mdm2 expression remained detectable at 14 dpa but no longer by 30 dpa (Figure S2D). Additionally, induced genetic ablation of CMs led to mdm2 expression throughout the ventricular wall and trabecular compartment by 7 dpi (Figure S2C).
To more clearly define the spatiotemporal pattern of mdm2 expression during heart regeneration, we generated a BAC transgenic reporter line containing long sequence stretches upstream (80 kb) and downstream (122 kb) of the mdm2 start codon and flanking an enhanced green fluorescent protein (EGFP) cassette (TgBAC[mdm2:EGFPpd312], hereafter referred to as mdm2:EGFP; Figure S2E). As expected, based on the known requirement for Mdm2 during development (Chua et al., 2015), mdm2:EGFP fluorescence was evident throughout the entire bodies of developing larvae by 3 days post fertilization (dpf; Figure S2F). Adult mdm2:EGFP fish hearts displayed low or undetectable EGFP expression in the absence of injury, but EGFP fluorescence was sharply increased in both compact and trabecular muscle near the injury site upon ventricular resection at 7 and 14 dpa (Figures 1G and S2G). Thus, Tp53 levels, and ostensibly the expression of its direct and indirect target genes, are transiently suppressed during heart regeneration, concomitant with transcriptional induction of the Mdm2 negative regulator.
Genetic Modulation of Tp53 Controls CM Proliferation during Heart Regeneration
To investigate the function of Tp53 during regeneration, we resected the ventricular apices of zebrafish with null mutations in tp53 (tp53M214K) and quantified CM proliferation indices at 7 dpa (Berghmans et al., 2005). tp53M214K mutants showed a more than doubling (123%) increase in their average CM proliferation index (n = 8, 9; Figures 2A–2C). Transcriptome sequencing of uninjured and regenerating tp53M214K hearts revealed significant expression changes from wild types in a relatively small number of genes, 42 and 82, respectively, which represent a variety of cellular processes as characterized by gene ontology terms and IPA (Figure S2; Tables S3 and S4). These data suggest a function for Tp53 in restricting CM proliferation during regeneration.
Figure 2. Tp53 or Mdm2 Modulation Alters CM Proliferation during Heart Regeneration.
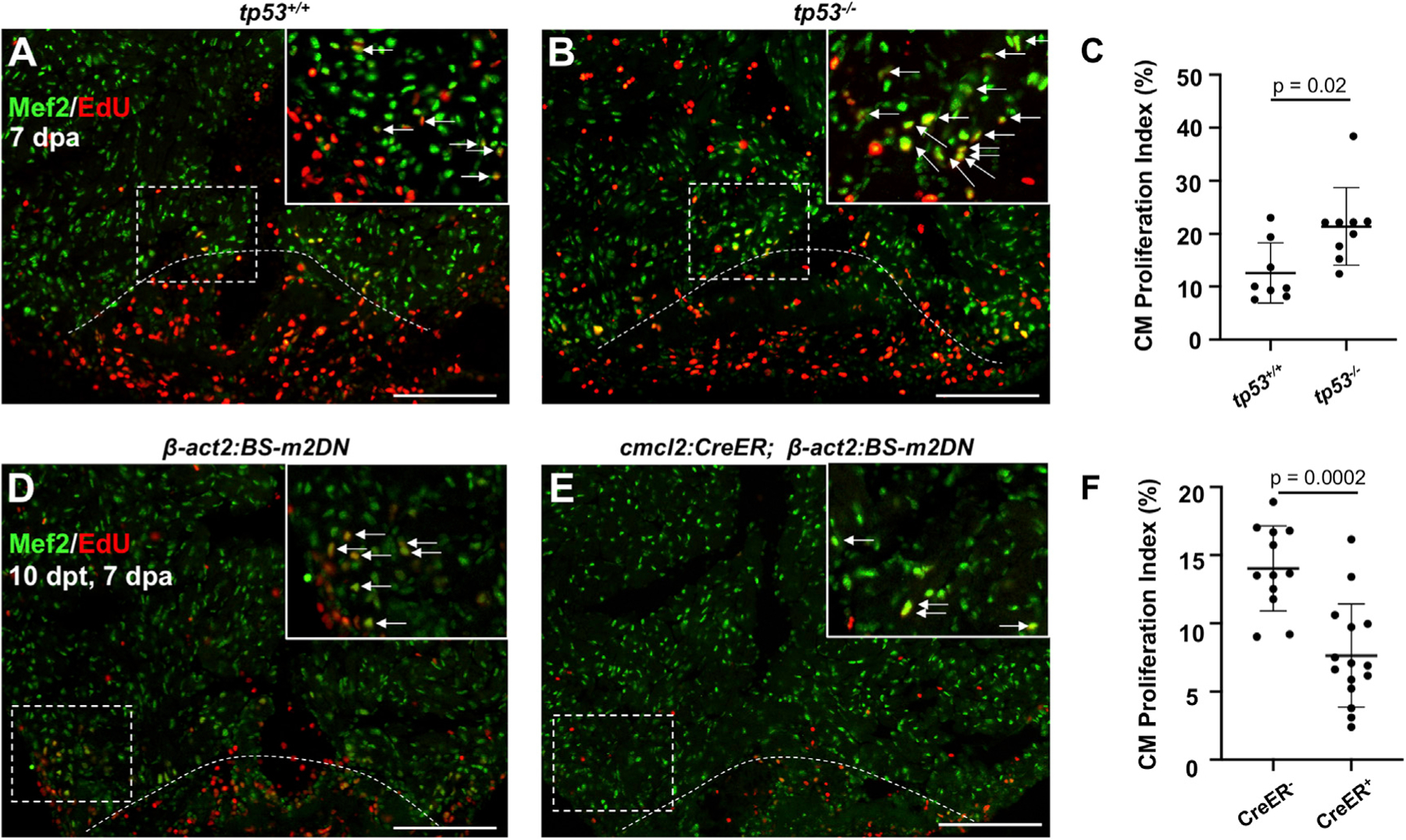
(A and B) Images of tp53+/+ and tp53−/− hearts at 7 days post ventricular apex (dpa) resection. Arrows indicate Mef2+/EdU+ nuclei.
(C) Quantification of CM proliferation at 7 dpa indicating an increased proliferation index in tp53−/− mutants. n = 8 and 9 for tp53+/+ and tp53−/−, respectively.
(D and E) Images of β-act2:BS-m2DN and cmlc2:CreER; β-act2:BS-m2DN hearts at 7 dpa and 10 days post tamoxifen (dpt) administration. Arrows indicate Mef2+/PCNA+ nuclei.
(F) Quantification of CM proliferation showing a reduction in the proliferation index animals expressing a dominant-negative Mdm2. n = 12 and n = 15 for CreER− and CreER+, respectively. Scale bars: 100 μm. Data show mean ± SEM (Mann-Whitney U test).
To increase Tp53 activity in heart muscle, we generated a transgenic line to express an Mdm2 isoform lacking its C-terminal ubiquitin ligase and shown to have dominant-negative activity in previous studies (Chua et al., 2015; Jones et al., 1995). We placed this m2DN cassette downstream of an actin promoter and a loxP-flanked fluorescent reporter gene and stop signal (Tg[β-actin2:loxP-BFP-STOP-loxP-dnMdm2–2A-mCherry]pd313, hereafter referred to as β-act2:BS-m2DN). Published homozygous mdm2 mutants are embryonic lethal (Chua et al., 2015), as were new mdm2pd314 mutants that we generated by CRISPR-based deletion, with no larvae surviving past 1 dpf (Figure S3). To evaluate the effects of inhibiting Mdm2 function on CM proliferation, we crossed β-act2:BS-m2DN with cmlc2:CreER fish, for 4-Hydroxytamoxifen (4-HT)-inducible, Cre-mediated release of m2DN expression in CMs (Figures 2D–2F and S3C). Adult cmlc2:CreER; β-act2:BS-m2DN and β-act2:BS-m2DN control clutchmates were treated with 4-HT 3 days before ventricular resection. cmlc2:CreER; β-act2:BS-m2DN animals demonstrated a ~50% reduction in CM proliferation at 7 dpa compared to controls (n = 12, 15; Figures 2D–2F). Together, these data indicate that suppression of Tp53-mediated gene regulation, at least in part by programmed induction of Mdm2, boosts CM proliferation upon cardiac injury in zebrafish.
CM Mitogens Trigger mdm2 Induction
Cardiac damage is a massive biologic insult, resulting in a constellation of cellular events including and not limited to inflammation, fibrinogenesis, angiogenesis, muscle de-differentiation, CM proliferation, and muscle patterning (González-Rosa et al., 2017; Sadek and Olson, 2020; Tzahor and Poss, 2017; Vujic et al., 2020). Previously, we reported that the extracellular factor Nrg1 causes overt CM proliferation when ectopically expressed in the adult heart (Gemberling et al., 2015). In this way, adult cardiogenesis is uncoupled from injury, enabling assessment of the impact of the mitogenic signal itself. To determine effects of Nrg1 overexpression on expression of mdm2, we first performed ISH on cmlc2:CreER; β-act2:BSNrg1 fish 14 days after induction of the nrg1 transgene. Whereas mdm2 was undetectable in control hearts as described above, nrg1 overexpression for 14 days led to strong and consistent mdm2 expression in CMs within the cortical wall by ISH (Figure S4A). Similarly, when the mdm2:EGFP transgene was crossed into the cmlc2:CreER; β-act2:BSNrg1 background, 14 days of nrg1 overexpression strongly induced EGFP throughout the cortical layer (Figure 3A).
Figure 3. Mitogens Activate mdm2 Regulatory Sequences and Have Increased Potency in tp53−/− Mutants.
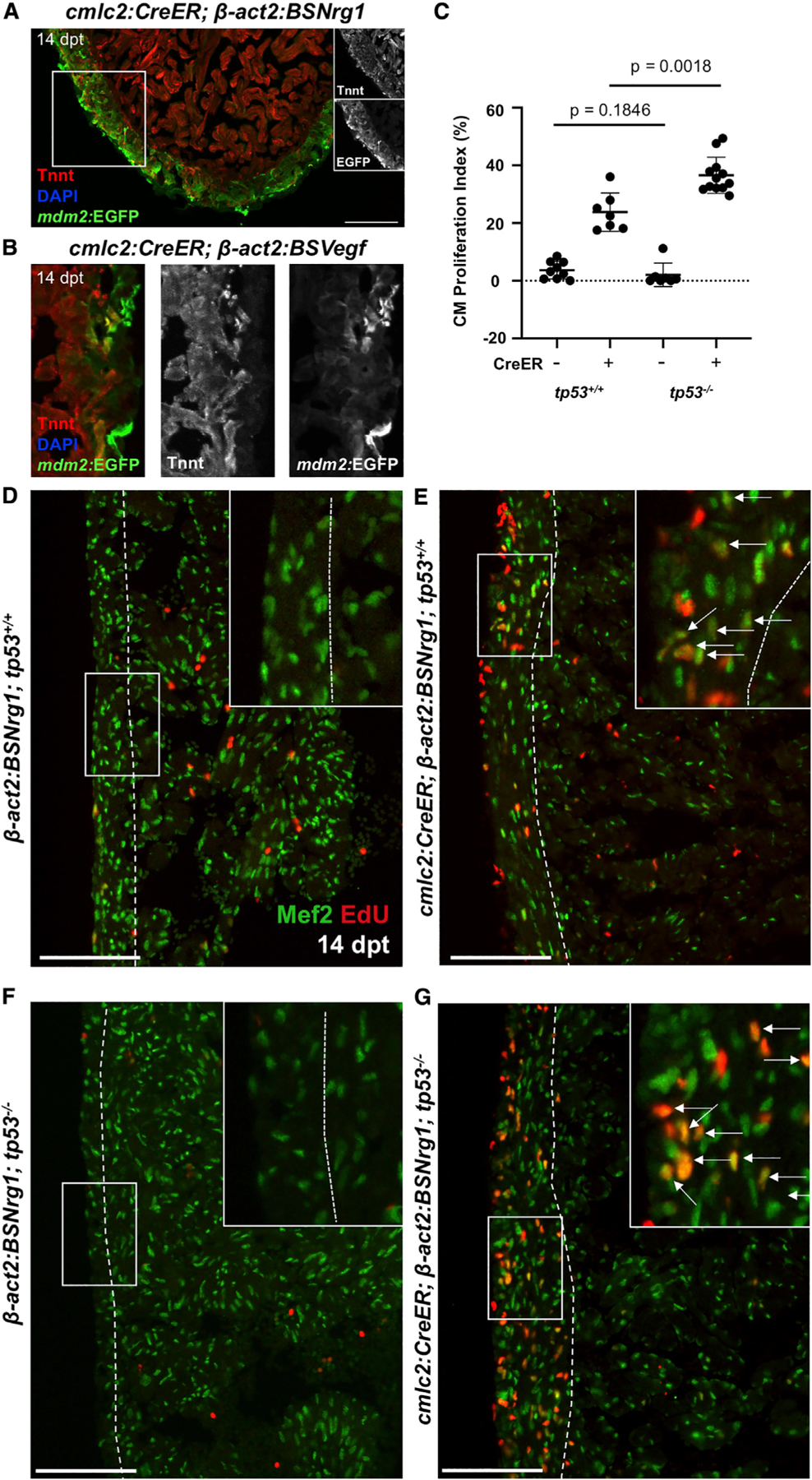
(A and B) mdm2:EGFP expression is activated in the ventricular wall after induced nrg1 (A) or vegfaa (B) overexpression (dpt, days post tamoxifen administration). High-magnification views only shown in (B).
(C) Quantification of CM EdU incorporation indices in ventricular walls of control (CreER−) or nrg1-overexpressing hearts at 14 dpt in tp53+/+ and tp53−/− backgrounds. n = 9 and n = 7 for tp53+/+ CreER− and CreER+, respectively. n = 7 and n = 12 for tp53−/− CreER− and CreER+, respectively.
(D–G) Section images of ventricular walls from groups in (C), indicating greater Nrg1-induced EdU incorporation in Mef2+ cells in tp53−/− ventricles. Boxes correspond to the region magnified in adjacent panels. Arrows indicate Mef2+EdU+ cells CMs. Scale bars: 100 μm. Data show mean ± SEM (Mann-Whitney U test).
Tp53 and Mdm2 are part of an autoregulatory feedback loop, conserved in zebrafish, with Tp53 binding to an upstream regulatory element of mdm2 to activate transcription (Berghmans et al., 2005; Pant et al., 2013). To test whether mdm2 induction with mitogenic stimulation reflects this mechanism of regulation, we first quantified proteins isolated from cmlc2:CreER; β-act2:BSNrg1 hearts and controls, finding that Tp53 protein levels were reduced upon 14 days of ectopic Nrg1 expression (Figures S4B and S4C). We then performed mdm2 ISH using cmlc2:CreER; βact2:BSNrg1; tp53M214K mutant zebrafish. As in wild-type zebrafish, ectopic Nrg1 expression increased mdm2 transcript levels throughout the outer cortical muscle layer in the presence or absence of functional Tp53 (Figure S4A; Berghmans et al., 2005). Taken together, these results suggest that mitogen stimulation induces mdm2 in a Tp53-independent fashion.
To examine effects of a second CM mitogen, we experimentally induced myocardial Vegfa overexpression, which hypervascularizes the adult zebrafish heart in the absence of injury but also has mitogenic effects on CMs (Karra et al., 2018). Visualization of EGFP in cmlc2:CreER, β-act2:BSVegfa, and mdm2:EGFP hearts revealed analogous findings as with nrg1 overexpression, with mdm2:EGFP fluorescence activated in areas of proliferating CMs (Figure 3B). Thus, two independent mitogens, having unique receptors and presumed mechanisms, lead cascades that trigger activation of myocardial transcription of mdm2 in the absence of injury.
To determine whether presence of Tp53 functionally impacts mitogen-stimulated CM proliferation, we crossed cmlc2:CreER; β-act2:BSNrg1 transgenes into the tp53M214K background. We found that, whereas 14 days of induced nrg1 overexpression sharply increased the cortical CM proliferation index in wild-type animals from baseline levels to ~24%, the index in tp53M214K animals was ~37%, representing a ~54% higher level (n = 7, 12; Figures 3C–3G). Together, these findings indicate that mitogen presence, either experimentally expressed or injury-induced, induces mdm2 and suppresses the Tp53 signaling network, and that this suppression of Tp53 has the effect of boosting CM proliferation.
mdm2 Regulation Occurs in Multiple Cardiogenic Contexts
Activation of regulatory sequences for the cardiogenic transcription factor gene gata4 is a visual marker for de-differentiated muscle in zebrafish (Kikuchi et al., 2010). gata4 activation also marks an emergent population of juvenile CMs from which the cortical muscle of the adult ventricular wall is derived (Gupta et al., 2013; Gupta and Poss, 2012), as well as CMs stimulated to proliferate upon transgenic Nrg1 stimulation (Gemberling et al., 2015). Sequence analysis revealed multiple predicted GATA4 binding sites in the mdm2 promoter region (Figure S4D). To determine if mdm2 induction is a general feature of highly cardiogenic tissue in zebrafish, we assessed co-localization between mdm2- and gata4-directed fluorescence in several contexts. We found that mdm2-directed EGFP fluorescence closely co-localized with gata4-directed dsRed fluorescence after ventricular resection injury (Figure 4A). mdm2:EGFP was also expressed in the same region as the form of myosin heavy chain that is prominent in embryonic heart muscle but diminished in adults upon resection (Sallin et al., 2015), but they were expressed in adjacent CMs and did not colocalize to the same expressing cells (Figure S4E). Moreover, mdm2-directed fluorescence was consistently present in a small domain at the base of the juvenile 4 weeks post fertilization (wpf) ventricle that partially overlaps with gata4-directed fluorescence. By 7 wpf, mdm2-directed EGFP fluorescence was evident in emerging and expanding cortical gata4+ CMs (Figures 4B and 4C). In addition, mdm2- and gata4-directed fluorescence were tightly co-localized in adult cmlc2:CreER; β-act2:BSNrg1; mdm2:EGFP; gata4:dsRed fish after 14 days of nrg1 overexpression (Figure 4D). These findings suggest a broad role for Tp53 suppression and mdm2 induction during particularly active cardiogenesis.
Figure 4. mdm2 Induction Is Associated with Highly Cardiogenic Areas during Heart Development and Regeneration.
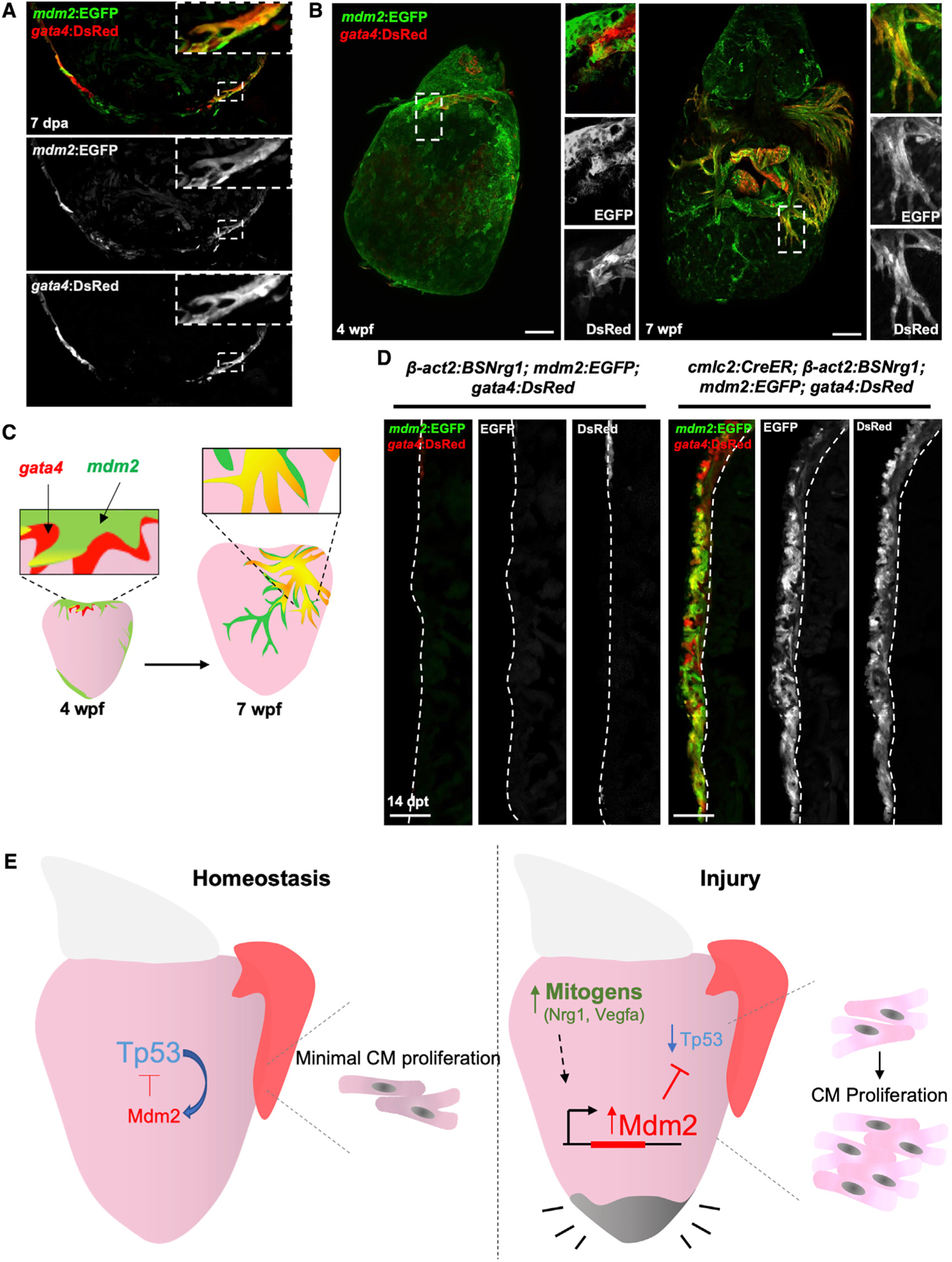
(A) Section confocal images of 7-dpa regenerating ventricles indicating co-localization of gata4- and mdm2-directed fluorescent reporter proteins. Boxes correspond to the region magnified in the panels.
(B) Whole mount images of juvenile hearts at 4 and 7 weeks post fertilization (wpf) showing gata4- and mdm2-directed fluorescence at the ventricular base at 4 wpf, and expanding on the surface muscle by 7 wpf.
(C) Cartoon showing co-localization of mdm2 and gata4 domains in emerging and proliferating CMs of the juvenile cortical wall.
(D) mdm2:EGFP and gata4:DsRed fluorescence are activated and co-localized in the ventricular walls of cmlc2:CreER; β-act2:BSNrg1 (right) ventricles by 14 days of tamoxifen-induced overexpression. Controls (without the cmlc2:CreER transgene) are shown at left. Dashed lines delineate cortical from trabecular muscle. Scale bars: 100 μm.
(E) Model of Mdm2-Tp53 regulation and its impact on CM proliferation after cardiac injury.
DISCUSSION
Here we report transient Tp53 suppression as a regulatory event that underlies and contributes to the strong cardiac regenerative response of adult zebrafish. Our findings indicate a mechanism in which mitogens present in injury sites activate myocardial expression of the negative Tp53 regulator Mdm2, which transiently reduces Tp53 levels and facilitates cell cycle entry (Figure 4E).
Tp53 has been implicated in other settings relevant to heart regeneration, most notably salamander limb regeneration (Yun et al., 2013). During limb regeneration, Tp53 protein levels are transiently suppressed early as the regeneration blastema forms, before recovering as new tissue is patterned. Moreover, modulation of Tp53 activity reduced the ability of limb cells to form a blastema and differentiate properly, with the authors proposing that Tp53 suppression helps post-mitotic limb cells re-enter the cell cycle (Yun et al., 2013). A direct relationship between Tp53 levels and cellular differentiation was also reported in the context of cellular reprogramming, when Tp53 mutant mouse fibroblasts showed increased capacity to undergo conversion to pluripotent cells (Hong et al., 2009; Kawamura et al., 2009; Li et al., 2009; Marión et al., 2009; Utikal et al., 2009). Furthermore, human somatic cells more readily undergo reprogramming upon acquisition of mutations in TP53 (Merkle et al., 2017). Our findings parallel these findings, as Tp53 suppression marked by mdm2 expression closely associates with markers of CM de-differentiation and division after injury. Thus, evidence is building toward a model in which Tp53 suppression broadly promotes de-differentiation across species and contexts.
Many potential regulators of Tp53 expression or activity exist in cardiac injury sites, including macromolecular damage, inflammation, and hypoxia. Each stimulus would be predicted to increase Tp53 levels and promote cell cycle arrest or apoptosis (Helton and Chen, 2007; Muñoz-Fontela et al., 2016; Williams and Schumacher, 2016). We speculate that an innate ability to counter this effect and reduce Tp53 impact, for instance by enhancing Mdm2 presence in response to injury, can facilitate division of source cells to an extent that it outcompetes scarring. Importantly, however, while injury itself might constitute a regulatory influence on Tp53 levels, we also show that mitogens can induce mdm2 expression and repress the Tp53 network. In fact, assessment of multiple cardiogenic contexts revealed mdm2 expression spatiotemporally localized with the areas of most active cardiogenesis. Mdm2 has pro-oncogenic effects that include Tp53 regulation but also other mechanisms (Marine and Lozano, 2010), and it is possible that induction of mdm2 expression and the suppression of its targets that facilitate de-differentiation during heart or limb regeneration are vestiges of the regulation that supports mammalian tumorigenic de-differentiation and cell division. Illuminating how mdm2 expression is regulated at the transcriptional level upon cardiac injury or mitogen stimulation can help resolve how key regenerative events like de-differentiation occur naturally without the threat of tumorigenesis.
Our findings along with those of others suggest that Tp53 occupies an important position in the transition of cell differentiation states, where it can play a crucial role during innate regeneration. As suppression of Tp53 can augment CM proliferation following injury or growth factor stimulation, we speculate that applications to safely and transiently reduce Tp53 network activity in CMs of MI patients might help provoke clinically significant regeneration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth Poss (kenneth.poss@duke.edu).
Materials Availability
Unique reagents generated in this study are available upon request.
Data and Code Availability
RNA-seq data is deposited in NCBI GEO database under accession number GSE146859.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Wild-type or transgenic zebrafish of the EK/AB strain were used for all experiments. β-actin2:loxp-mCherry-STOP-loxp-DTApd36 (Wang et al., 2011), β-actin2:loxp-mTagBFP-STOP-loxp-nrg1pd107 (Gemberling et al., 2015), β-actin2:loxp-mTagBFP-STOP-loxp-vegfaapd262 (Karra et al., 2018), cmlc2:CreERpd10 (Kikuchi et al., 2010), gata4:EGFP (Heicklen-Klein and Evans, 2004), gata4:ds-Red2pd28 (Karra et al., 2015), tp53M214K (Berghmans et al., 2005) transgenic mutant or fish have been previously described. Resection of ~20% of the cardiac ventricular apex was performed as described previously (Poss et al., 2002). To induce expression of nrg1 or vegfaa in CMs, adult cmlc2:CreER; β-act2:BS-nrg1 or β-actin2:loxp-mTagBFP-STOP-loxp-vegfaa zebrafish were treated for 24 hours with 5 μM tamoxifen. To induce recombination in adult cmlc2:CreER; βact2:BS-m2DN fish, animals were bathed 3 sequential days in 5 μM 4-HT (Sigma) for 10–12 hours. Heart injuries were performed 3 days after the final 4-HT treatment. To induce ablation of CMs, adult ZCAT animals were treated for 24 hours with 0.5 – 1.0 μM tamoxifen titrated for ablation of ~50% CMs (Wang et al., 2011). For EdU-incorporation experiments 10 mM EdU (Sigma) was injected intraperitoneally once daily for 3 days before collection. Procedures involving animals were approved by the Institutional Animal Care and Use Committee at Duke University.
METHOD DETAILS
Ingenuity pathway and gene ontology analyses
Pathway analysis of was performed using Ingenuity Pathway Analysis (IPA) software on the following published datasets: ventricle 7 dpi versus sham GSE75894 (Kang et al., 2016), ventricle 14 dpi versus sham GSE81865 (Goldman et al., 2017). Mouse orthologs of differentially expressed genes with p < 0.01 and −1 < log2FC < 1, retrieved via www.ensembl.org/biomart/martservice, were analyzed to find upstream regulators and pathways showing differential modulation upon injury. IPA analyses were performed with the following settings: Expression Value Type (Exp Log Ration), Reference set (Ingenuity Knowledge Base), Relationships to consider (Direct and Indirect Relationships), Interaction networks (70 molecules/network; 25 networks/analysis or 30 molecules/network; 25 or 10 network/analysis), Data Source (all), Confidence (Experimentally Observed), Species (Human, Mouse, Rat), Tissue & Cell Lines (all), Mutations (all) (Krämer et al., 2014). P values in Figure 1A measure the statistical significance of the overlap between differentially expressed genes and the genes under predicted control of a regulator, and activation scores infer the activation state of a regulator, either activating (positive) or inhibiting (negative). Gene Ontology analyses were performed with The PANTHER database (Protein Analysis Through Evolutionary Relationships, http://pantherdb.org).
RNA extraction and sequencing
Zebrafish ventricles were collected and placed in cold PBS while still pumping to help decrease intra-ventricular blood. Atrium and outflow tract were removed and ventricles (5 per sample) were homogenized in Trizol using a Tissue Lyser II (QIAGEN). RNA was extracted using the standard Trizol protocol, genomic DNA removed using RNA clean and Concentrator Kit (Zymo Research) and processed for preparation of libraries. Single-end 50-bp sequencing, with 30 million reads/sample was performed at BGI. RNA-Seq reads were trimmed by Trim Galore (0.6.4, with -q 15) and then mapped with TopHat (v 2.1.1, with parameters-b2-very-sensitive-no-coverage-search and supplying the UCSC danRer10 refSeq gene annotation) (Kim et al., 2013). Gene-level read counts were obtained using the htseq-count (v1.6.1) by the reads with MAPQ greater than 30. DESeq2 (v 1.26.0) was employed for differential expression analysis. Bioconductor package Biostrings software (v 2.56.0) was used to analyze predicted transcription factor binding motifs. The RNA-Seq data generated in this study have been deposited in the NCBI GEO database under accession number GSE146859.
Western blotting
Zebrafish ventricles were collected and placed in cold PBS while still pumping to help decrease intra-ventricular blood. Atrium and outflow tract were removed and ventricles (8–10 per sample) were homogenized in RIPA buffer containing Proteinase and Phosphatase inhibitor (Thermo Fisher Scientific #78442) and Phenylmethanesulfonyl fluoride solution (Sigma #93482). Samples were denatured at 95°C for 5 min, quantified and tissue lysates were analyzed on Mini-Protein tetra cell (Bio-Rad) using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in Tris/glycine/SDS buffer. After electrophoresis proteins were transferred to a PVDF membrane using the Mini-Protein tetra cell in Tris/glycine buffer (v/v). Membranes were blocked for 1 h at room temperature using 5% BSA in Tris-buffered saline and Tween-20 (TBST), then were incubated with primary anti-Tp53 antibody (GTX128135, 1:500) and anti-GAPDH (Proteintech 60004–1-IG, 1:500) overnight at 4°C. Membranes were incubated with appropriate HRP-conjugated secondary antibodies (Thermo Fisher Scientific), washed in TBST and developed with Pierce ECL western blotting substrate. Western blot signals were quantified as described in Davarinejad (2017).
Generation of transgenic zebrafish
A 2A-mCherry fragment was amplified from a g4DN template described previously (Gupta et al., 2013), generated using the primer sequences 2A-mCherry forward: CCGGCGCGCCT GCTACGAACTTCTCTCTGTTAAAGCAAGCAGGGGACGTGGAAGAAAACCCTGGTCCTATGGTGAGCAAGGGCGAGGAGGACAAC; 2A-mCherry reverse: GATATCGCGGCCGCTTACTTGTACAGCTCGTCCATGCC. The PCR product was ligated into the AscI/NotI site of the β-act2:loxP-TagBFP-STOP-lox-Pvector (Gupta et al., 2013). Mdm2 cDNA lacking the C-terminal RING finger domain was amplified using the primer sequences mdm2 forward: ATGGCAACAGAGAGTTGTTTAAGCAG; mdm2 dominant negative reverse: GCACGTAGCGGGAAGGC. The PCR product was ligated into the AgeI/AscI site of the β-act2:loxP-TagBFP-STOP-lox-P2A-mCherry. The full name of this transgenic line is Tg(β-actin2:loxP-mTagBFP-STOP- loxP-dnmdm2)pd313.
mdm2:EGFP. The translational start codon of mdm2 in the BAC clone CH211–209G11 (BACPAC Resources Center) was replaced with the EGFP sequence by Red/ET recombineering technology (GeneBridges) as described in Kikuchi et al. (2011). The entire construct was flanked with I-SceI sites allowing meganuclease mediated transgenesis as previously described (Thermes et al., 2002). After purifying the final BAC with nucleobond BAC 100 kit (Clontech) the BAC was co-injected along with I-SceI into one-cell embryos. A single founder was isolated and propogated. The full name of this transgenic line is TgBAC(mdm2:EGFP)pd312.
mdm2pd314 mutants were generated using injection of CRISPR/Cas9 and gRNAs at the one-cell stage as previously described (Tornini et al., 2017). Exon 3 was targeted using oligo 5′-GCGTAATACGACTCACTATAGGGCAATTGAAAAGCCTGTTAGAGGGTTTTAGAGCTAGAAATAGC-3′ (target sequence underlined). Intron 3 was targeted using oligo 5′-GCGTAATACGACTCACTATAGGGGGTGGGGTTTTACAACAACAGGGTTTTAGAGCTAGAAATAGC-3′ (target sequence underlined) resulting in a 1.7 kb deletion. Deletion lines were genotyped using primers: mdm2_KO_for 5′- GCAGTTCTCAGATCAGCAAGGTTG-3′; mdm2_prom_rev 5′- GCTGAAAAGTGACCTTCGCCATC-3′; and mdm2_geno_rev 5′-GGCAAATATCCAGATAGTGCCACC-3′. Wild-type embryos yield an expected amplicon of 455 bp, with homozygous mutants containing a single 180 bp amplicon. Heterozygotes produce 455 bp and 180 bp amplicons.
Histological analysis and imaging
Primary and secondary antibody staining for immunofluorescence was performed as described in Kikuchi et al. (2011). Antibodies used in this study were anti-troponin T (mouse; Lab Vision, 1:200), anti-Mef2 (rabbit; Abcam, 1:100), anti-EGFP, (rabbit; Thermo Fisher Scientific, 1:200), anti-EGFP (chicken; Aves Labs, 1:1000), anti-dsRed (rabbit; Clontech, 1:200), anti-myosin heavy chain N2.261 (mouse; Developmental Studies Hybridoma Bank, 1:50), Alexa Fluor 488 (chicken and rabbit; Life Technologies,1:200), Alexa Fluor 594 (mouse and rabbit; Life Technologies, 1:200). Confocal imaging was performed using a Zeiss LSM 700 or a Zeiss LSM 880 microscope. Tissue for CM proliferation was placed immediately into ice-cold 30% sucrose in PBS before being transferred to cold TFM (VWR). Hearts were flash frozen in dry ice ethanol bath. After sectioning hearts were fixed for 15 min with 3.7% formaldehyde (Sigma). Sections were stained for EdU, washed with PBS + 0.2% Triton (Sigma). Staining for Mef2, imaging, and counting were performed as previously described (Kikuchi et al., 2011). Proliferation indices were quantified using 3 sections per heart. Hearts for whole mount imaging were collected and fixed overnight in 4% paraformaldehyde at 4°C and imaged as previously described (Gupta et al., 2013).
mdm2 mRNA injections
Dominant negative mdm2 cDNA was transcribed and subcloned into the multicloning site of a pCS2+ vector containing an SP6 pro-motor and poly-A tail using ClaI and EcoRI restriction sites. mRNA was generated using the SP6 mMachine mMessage kit (Ambion) and 200 ng/uL was injected into single-cell embryos as described (Rosen et al., 2009).
QUANTIFICATION AND STATISTICAL ANALYSIS
Clutchmates were randomized into different treatment groups for each experiment. No animal or sample was excluded from the analysis. All experiments other than western blots were performed with at least two biological replicates. All statistical values are displayed as Mean ± Standard Deviation or Mean ± Standard Error of the Mean, as indicated in the figure legends. Statistical tests were calculated using two-tailed Student’s t tests or Mann-Whitney U tests.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Troponin T | Lab Vision | Cat# MS-295-PABX |
| Rabbit monoclonal anti-Mef2 | Abcam | Cat# ab197070 |
| Rabbit polyclonal anti-dsRed | Clontech | Cat# 632475 |
| Chicken polyclonal anti-GFP | Aves Labs | Cat# GFP-1020 |
| Rabbit polyclonal anti-GFP | Thermo Fisher Scientific | Cat # A-11122 |
| Mouse monoclonal anti-GAPDH | Proteintech | Cat # 60004–1-IG |
| Rabbit polyclonal anti-p53 | GeneTex | Cat# GTX128135 |
| Mouse monoclonal anti-Myosin heavy chain | Developmental Studies Hybridoma Bank | N2.261 |
| Alexa Fluor 594 Goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-11037 |
| Alexa Fluor 488 Goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-11034 |
| Alexa Fluor 594 Goat anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A-11032 |
| Alexa Fluor 488 Goat anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A-11001 |
| Alexa Fluor 488 Goat anti-Chicken IgG (H+L) | Thermo Fisher Scientific | Cat# A-11039 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# A-31430 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 65–6120 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Metronidazole | Sigma-Aldrich | Cat#M1547 |
| 5-ethynyl-2-deoxyuridine | Thermo Fisher Scientific | Cat#A10044 |
| DAPI | Thermo Fisher Scientific | Cat#D3571 |
| Alexa Fluor 488 Azide | Thermo Fisher Scientific | Cat#A10270 |
| Alexa Fluor 594 Azide | Thermo Fisher Scientific | Cat#C10617 |
| Deposited Data | ||
| RNA Sequencing | GEO database | GSE146859 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: Tg(β-actin2:loxP-mTagBFP-STOP- loxP-dnmdm2)pd313 | This paper | pd313 |
| Zebrafish: TgBAC(mdm2:EGFP)pd312 | This paper | pd312 |
| Zebrafish: Tg(tp53)M214K | Berghmans et al., 2005 | M214K |
| Zebrafish: Tg(β-actin2:loxp-mCherry-STOP-loxp-DTA)pd36 | Wang et al., 2011 | pd36 |
| Zebrafish: Tg(β-actin2:loxp-mTagBFP-STOP-loxp-nrg1)pd107 | Gemberling et al., 2015 | pd107 |
| Zebrafish: Tg(β-actin2:loxp-mTagBFP-STOP-loxp-vegfaa)pd262 | Karra et al., 2018 | pd262 |
| Zebrafish: Tg(cmlc2:CreER)pd10 | Kikuchi et al., 2010 | pd10 |
| Zebrafish: Tg(gata4:EGFP)ae1 | Heicklen-Klein and Evans, 2004 | ae1 |
| Zebrafish: Tg(gata4:dsRed2)pd28 | Karra et al., 2015 | pd28 |
| Software and Algorithms | ||
| Tophat2 | Kim et al., 2013 | Version 2.1.1 |
| Biostrings | Bioconductor | Version 2.56.0 |
| DESeq2 | Bioconductor | Version 1.26.0 |
| Ingenuity Pathway Analysis | QIAGEN, Krämer et al., 2014 | 830011 |
Highlights.
Tp53 network is transiently suppressed during zebrafish heart regeneration
The Tp53 inhibitor mdm2 is induced in regenerating cardiomyocytes
Tp53 or Mdm2 modulation can impact injury-induced cardiomyocyte proliferation
Mitogens can trigger mdm2 expression and suppress Tp53 in the absence of injury
ACKNOWLEDGMENTS
We thank Duke Zebrafish Shared Resource staff for zebrafish care. V.C. is supported by a SNSF Postdoctoral Fellowship. K.D.P. acknowledges research support from NHLBI (R35 HL150713), AHA, and Fondation Leducq.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108089.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, and Levine AJ (2010). The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol 2, a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CD, Morris JP, Liu TX, Schulte-Merker S, Kanki JP, et al. (2005). tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 102, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K, Arab S, Haring B, and Kühn B (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270. [DOI] [PubMed] [Google Scholar]
- Bohlman S, and Manfredi JJ (2014). p53-independent effects of Mdm2. Subcell. Biochem 85, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JS, Liew HP, Guo L, and Lane DP (2015). Tumor-specific signaling to p53 is mimicked by Mdm2 inactivation in zebrafish: insights from mdm2 and mdm4 mutant zebrafish. Oncogene 34, 5933–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, et al. (2015). ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol 17, 627–638. [DOI] [PubMed] [Google Scholar]
- Davarinejad H (2017). Quantifications of western blots with ImageJ. http://wwwyorkuca/yisheng/Internal/Protocols/ImageJpdf.
- Fridman JS, and Lowe SW (2003). Control of apoptosis by p53. Oncogene 22, 9030–9040. [DOI] [PubMed] [Google Scholar]
- Gemberling M, Karra R, Dickson AL, and Poss KD (2015). Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. eLife 4, e05871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. ; American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2013). Executive summary: heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 127, 143–152. [DOI] [PubMed] [Google Scholar]
- Goldman JA, Kuzu G, Lee N, Karasik J, Gemberling M, Foglia MJ, Karra R, Dickson AL, Sun F, Tolstorukov MY, and Poss KK (2017). Resolving heart regeneration by replacement histone profiling. Dev. Cell 40, 392–404 e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Burns CE, and Burns CG (2017). Zebrafish heart regeneration: 15 years of discoveries. Regeneration (Oxf.) 4, 105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Zhu N, Zhang H, Durden DL, Feng Y, and Zhou M (2009). Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 15, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Liew HP, Camus S, Goh AM, Chee LL, Lunny DP, Lane EB, and Lane DP (2013). Ionizing radiation induces a dramatic persistence of p53 protein accumulation and DNA damage signaling in mutant p53 zebrafish. Oncogene 32, 4009–4016. [DOI] [PubMed] [Google Scholar]
- Gupta V, and Poss KD (2012). Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 484, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Gemberling M, Karra R, Rosenfeld GE, Evans T, and Poss KD (2013). An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr. Biol 23, 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Bulyk ML, Jambhekar A, and Lahav G (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol 20, 199–210. [DOI] [PubMed] [Google Scholar]
- Han Y, Chen A, Umansky KB, Oonk KA, Choi WY, Dickson AL, Ou J, Cigliola V, Yifa O, Cao J, et al. (2019). Vitamin D stimulates cardiomyocyte proliferation and controls organ size and regeneration in zebrafish. Dev. Cell 48, 853–863 e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heicklen-Klein A, and Evans T (2004). T-box binding sites are required for activity of a cardiac GATA-4 enhancer. Dev. Biol 267, 490–504. [DOI] [PubMed] [Google Scholar]
- Helton ES, and Chen X (2007). p53 modulation of the DNA damage response. J. Cell. Biochem 100, 883–896. [DOI] [PubMed] [Google Scholar]
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, and Yamanaka S (2009). Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460, 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, and Bradley A (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208. [DOI] [PubMed] [Google Scholar]
- Kang J, Hu J, Karra R, Dickson AL, Tornini VA, Nachtrab G, Gemberling M, Goldman JA, Black BL, and Poss KD (2016). Modulation of tissue repair by regeneration enhancer elements. Nature 532, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karra R, Knecht AK, Kikuchi K, and Poss KD (2015). Myocardial NF-κB activation is essential for zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 112, 13255–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karra R, Foglia MJ, Choi WY, Belliveau C, DeBenedittis P, and Poss KD (2018). Vegfaa instructs cardiac muscle hyperplasia in adult zebrafish. Proc. Natl. Acad. Sci. USA 115, 8805–8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, and Izpisúa Belmonte JC (2009). Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, and Poss KD (2010). Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, and Poss KD (2011). Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 20, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, and Vousden KH (2015). p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol 16, 393–405. [DOI] [PubMed] [Google Scholar]
- Lai SL, Marín-Juez R, Moura PL, Kuenne C, Lai JKH, Tsedeke AT, Guenther S, Looso M, and Stainier DY (2017). Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. eLife 6, e25605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP, and Crawford LV (1979). T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, and Serrano M (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Tu C, Sheng Q, Yang Y, Kan Z, Guo Y, Shyr Y, Scott IC, and Lou X (2018). Dynamics of zebrafish heart regeneration using an HPLC-ESI-MS/MS approach. J. Proteome Res 17, 1300–1308. [DOI] [PubMed] [Google Scholar]
- Mak TW, Hauck L, Grothe D, and Billia F (2017). p53 regulates the cardiac transcriptome. Proc. Natl. Acad. Sci. USA 114, 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani F, Collavin L, and Del Sal G (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, and Lozano G (2010). Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 17, 93–102. [DOI] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, and Blasco MA (2009). A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460, 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, and Oren M (2003). The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol 13, 49–58. [DOI] [PubMed] [Google Scholar]
- Missinato MA, Saydmohammed M, Zuppo DA, Rao KS, Opie GW, Kühn B, and Tsang M (2018). Dusp6 attenuates Ras/MAPK signaling to limit zebrafish heart regeneration. Development 145, dev157206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Fontela C, Mandinova A, Aaronson SA, and Lee SW (2016). Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol 16, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura K, Klejnot M, Kowalczyk D, Hock AK, Sibbet GJ, Vousden KH, and Huang DT (2017). Structural analysis of MDM2 RING separates degradation from regulation of p53 transcription activity. Nat. Struct. Mol. Biol 24, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, Yamaguchi T, Tobita T, Naito AT, Ito M, et al. (2018). Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun 9, 4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, and Vogelstein B (1993). Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860. [DOI] [PubMed] [Google Scholar]
- Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintás-Cardama A, Hamir AN, and Lozano G (2013). The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 27, 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, and Keating MT (2002). Heart regeneration in zebrafish. Science 298, 2188–2190. [DOI] [PubMed] [Google Scholar]
- Rosen JN, Sweeney MF, and Mably JD (2009). Microinjection of zebrafish embryos to analyze gene function. J. Vis. Exp 25, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadek H, and Olson EN (2020). Toward the goal of human heart regeneration. Cell Stem Cell 26, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallin P, de Preux Charles AS, Duruz V, Pfefferli C, and Jaźwińska A (2015). A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Dev. Biol 399, 27–40. [DOI] [PubMed] [Google Scholar]
- Stanley-Hasnain S, Hauck L, Grothe D, Aschar-Sobbi R, Beca S, Butany J, Backx PH, Mak TW, and Billia F (2017). p53 and Mdm2 act synergistically to maintain cardiac homeostasis and mediate cardiomyocyte cell cycle arrest through a network of microRNAs. Cell Cycle 16, 1585–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J, and Joly J-S (2002). I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech. Dev 118, 91–98. [DOI] [PubMed] [Google Scholar]
- Tornini VA, Thompson JD, Allen RL, and Poss KD (2017). Live fate-mapping of joint-associated fibroblasts visualizes expansion of cell contributions during zebrafish fin regeneration. Development 144, 2889–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzahor E, and Poss KD (2017). Cardiac regeneration strategies: staying young at heart. Science 356, 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, and Hochedlinger K (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460, 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, and Lu X (2002). Live or let die: the cell’s response to p53. Nat. Rev. Cancer 2, 594–604. [DOI] [PubMed] [Google Scholar]
- Vujic A, Natarajan N, and Lee RT (2020). Molecular mechanisms of heart regeneration. Semin. Cell Dev. Biol 100, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Li YC, and Wahl GM (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Panáková D, Kikuchi K, Holdway JE, Gemberling M, Burris JS, Singh SP, Dickson AL, Lin YF, Sabeh MK, et al. (2011). The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 138, 3421–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AB, and Schumacher B (2016). p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med 6, a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Kruse F, Vasudevarao MD, Junker JP, Zebrowski DC, Fischer K, Noël ES, Grün D, Berezikov E, Engel FB, et al. (2016). Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev. Cell 36, 36–49. [DOI] [PubMed] [Google Scholar]
- Yun MH, Gates PB, and Brockes JP (2013). Regulation of p53 is critical for vertebrate limb regeneration. Proc. Natl. Acad. Sci. USA 110, 17392–17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ben-Yair R, Burns CE, and Burns CG (2019). Endocardial Notch signaling promotes cardiomyocyte proliferation in the regenerating zebrafish heart through Wnt pathway antagonism. Cell Rep. 26, 546–554 e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data is deposited in NCBI GEO database under accession number GSE146859.