

Abstract
The coenzyme NAD+ is critical in cellular bioenergetics and adaptive stress responses. Its depletion has emerged as a fundamental feature of aging that may predispose to a wide range of chronic diseases. Maintenance of NAD+ levels is important for cells with high energy demands and for proficient neuronal function. NAD+ depletion is detected in major neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases, cardiovascular disease and muscle atrophy. Emerging evidence suggests that NAD+ decrements occur in various tissues during aging, and that physiological and pharmacological interventions bolstering cellular NAD+ levels might retard aspects of aging and forestall some age-related diseases. Here, we discuss aspects of NAD+ biosynthesis, together with putative mechanisms of NAD+ action against aging, including recent preclinical and clinical trials.
Keywords: NAD+, metabolism, autophagy, neurodegenerative disorder, mitophagy, aging, stem cell, DNA repair, clinical application
NAD+ Plays a Key Role in Human Health
Nicotinamide adenine dinucleotide (NAD+) is a cofactor for numerous enzymes involved in cellular energy metabolism, and for adaptive responses of cells to bioenergetic and oxidative stress. Over 110 years ago, NAD+ was discovered as a “cozymase” necessary for fermentation (Supplementary Figure S1) [1–5]. In his 1930 Nobel lecture, Dr. Hans von Euler-Chelpin stated that “cozymase (NAD+) is one of the most widespread and biologically important activators within the plant and animal world” [6]. NAD+ is a necessary cofactor for many metabolic pathways such as glycolysis, fatty acid β-oxidation and the tricarboxylic acid cycle, while the reduced form of NAD+ (NADH) is a primary hydride donor in the production of ATP via anaerobic glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) [7]. Recently, the importance of NAD+ has expanded from a key element in intermediate metabolism to a critical regulator of multiple cell signaling pathways, and is now a major player in aging and age-related diseases [8–11].
Mounting evidence links compromised NAD+ status to the hallmarks of aging. Among the age-related cellular processes that may predispose to disease, impaired autophagy has emerged as an important component [12, 13]. Indeed, recent studies in human cells and animal models unveil a novel role for NAD+ in autophagy and mitophagy, in which impairments in lysosome-targeting and -recycling mechanisms result in the accumulation of damaged molecules and mitochondria, leading to cell dysfunction and/or death [14]. In the present article, we discuss the interconnected roles of autophagy (especially mitochondrial autophagy or ‘mitophagy’), mitochondrial maintenance, DNA repair, and cell death relative to NAD+ metabolism. We summarize new insight on pathways of NAD+ biosynthesis and consumption, and highlight how NAD+ depletion might contribute to mammalian aging. We also examine how supplementation of NAD+ precursors might constitute a promising therapeutic strategy to counter aging-associated pathologies and/or accelerated aging.
NAD+ Biosynthesis and Consumption
Cellular NAD+ Levels
NAD+ is produced in all eukaryotic cells. The basal intracellular NAD+ concentration can be up to 800 μM in yeast [15], 100–400 μM in human HEK293 cells [16, 17], and approximately 0.2 mmol/kg in mouse tibialis anterior muscle [18]. New methods have been developed to enable the detection of subcellular NAD+ levels (Box 1). NAD+ is consumed in many catabolic pathways: in the cytosol, NAD+ is reduced to NADH by lactate dehydrogenase (LDH) during anaerobic glycolysis [7]. In mitochondria, the three tricarboxylic acid cycle enzymes, isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (α-KGDH), and malate dehydrogenase (MDH), reduce NAD+ to NADH. NADH serves as the primary source of reducing equivalents for complex I (NADH dehydrogenase) of the electron transport chain (ETC) to fuel OXPHOS, generating NAD+, ultimately reducing oxygen to H2O and producing ATP [15]. Exercise and diet can affect NAD+ concentration in various tissues. For example, 6 weeks of exercise was shown to improve glucose tolerance and increase muscle NAD+ in a mouse model of high-fat diet (HFD)-induced obesity[19, 20]. Studies in both mice and humans have shown that exercise can change circulating NAD+ in a biphasic manner with moderate intensity exercise increasing, and strenuous exercise reducing, NAD+ [21]. In addition, while a high-fat diet decreases muscle NAD+ in HFD-induced obese mice, exercise and caloric restriction can increase NAD+ in the muscle and liver of obese or aged mice, respectively [19, 20]. Thus, intracellular NAD+ is regulated by many cellular activities including oxidative phosphorylation, mitochondrial metabolism, transcription and signaling, but can also be significantly influenced by diet, exercise, and other health conditions.
Box 1. NAD+ Measurement, Subcellular Concentrations, and Molecular Precursors.
Different assays are used to detect NAD+, including high-performance liquid chromatography (HPLC)-based methods (e.g., HPLC/MALDI/MS) [16] and NAD+/NADH enzymatic cycling assays [28] (including commercial kits). The recent development of genetically encoded fluorescent biosensors, such as SoNar and a biosensor with a bipartite NAD+-binding domain, allows the imaging of relative levels of free NAD+ in subcellular compartments [17, 120]. For example, in HEK293T cells, the concentration of NAD+ is similar in the cytoplasm and nucleus (approximately 100 μM), and higher in the mitochondria (230 μM) [17]. These levels are consistent with other reports demonstrating that in highly metabolically active, post-mitotic cells such as neurons, mitochondria have higher NAD+ levels than other subcellular compartments [9, 121]. Moreover, the nuclear and cytoplasmic NAD+ pools are interchangeable, while the mitochondrial pool is relatively isolated, although a substantial decrease in the cytosolic pool may influence the mitochondrial pool [17].
NAD+ precursors include nicotinamide (NAM), nicotinic acid (NA), tryptophan (Trp), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN). Niacin (vitamin B3; NA and NAM) is abundant in eggs, fish, meat, diary, some vegetables, and whole grains. Milk is a source of NR [122], and NMN is present in various types of food including broccoli (0.25–1.12 mg/100 g), avocado (0.36–1.60 mg/100g), and beef (0.06–0.42 mg/100g) [44]. It should also be noted that NAD+, NADH, NADPH and NADP are present in many foods, and that ingested NAD+ can be metabolized to precursors, including NR (mild digestion), NMN (by intestinal brush border cells [22]), NAM, NA (deaminated from NAM by gut bacterial nicotinamidase), followed by absorption in the intestinal epithelial cells and transfer to the blood [123] (Figure 1). The gut can also directly absorb these NAD+ precursors from foods and dietary supplements.
NAD+ Biosynthesis
In mammals, cellular NAD+ is synthesized from a broad variety of dietary sources including NAD+ itself (it is metabolized in the gut, then synthesized again in cells), and from one or more of its major precursors: tryptophan (Trp), nicotinic acid (NA), nicotinamide riboside (NR), nicotinamide mononucleotide (NMN) and nicotinamide (NAM) [22]. Food sources of NAD+ and its precursors are summarized in Box 1. Depending on the bioavailability of the precursors, there are three pathways for the synthesis of NAD+ in cells: i) from Trp by the de novo biosynthesis pathway or kynurenine pathway; ii) from NA in the Preiss-Handler pathway; and iii) from NAM, NR and NMN in the salvage pathway (Figure 1).
Figure 1. NAD+ Biosynthetic Pathways.
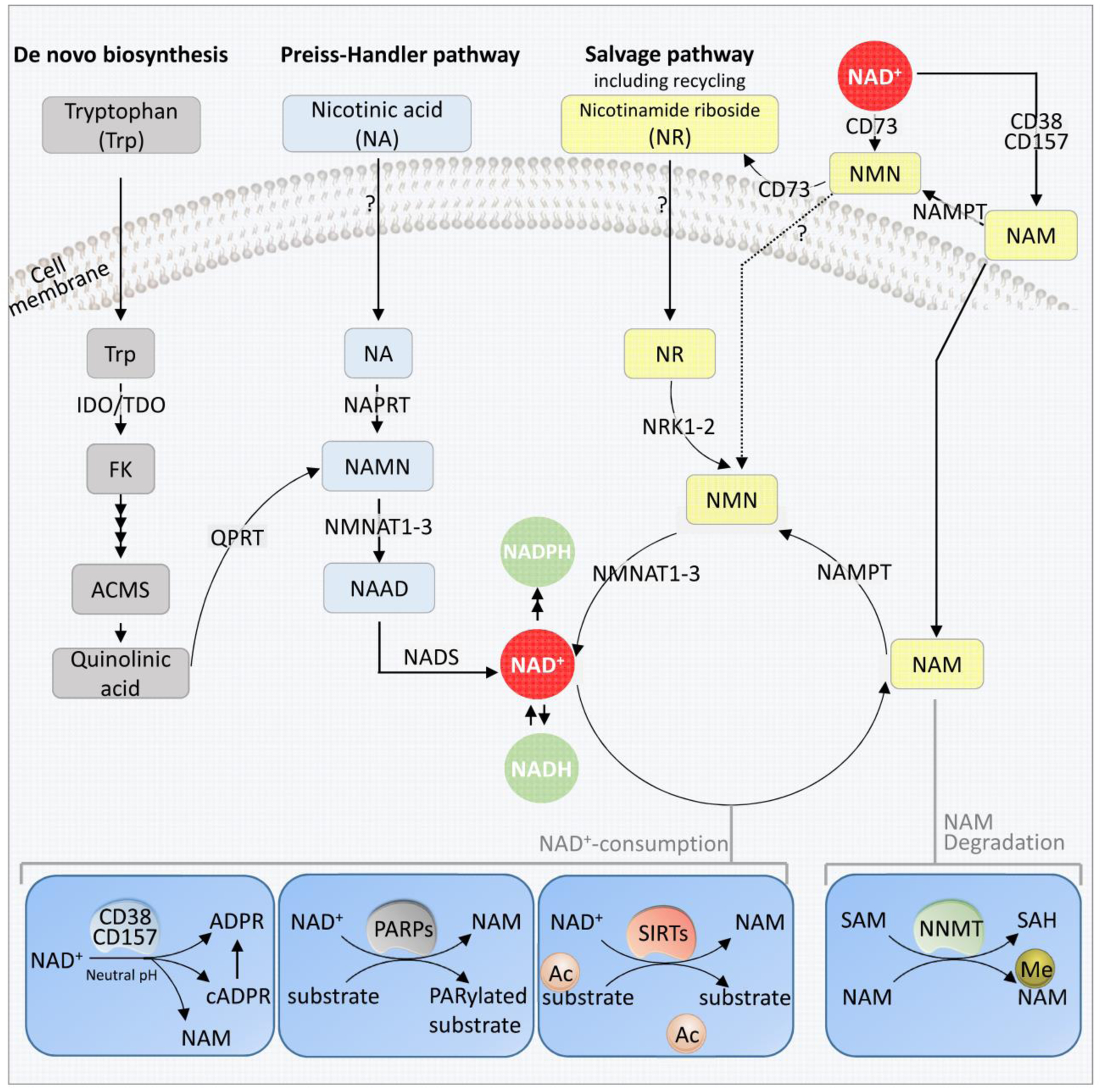
NAD+ is synthesized via three major pathways in mammals; the de novo biosynthesis from Tryptophan (Trp) (to the left) in a total of 8 steps. Four steps are shown in this figure, including the conversion of tryptophan to formylkinurenine (FK), and a spontaneous reaction conversion of ACMS to quinolinic acid (Qa). Qa is then converted to NAMN by QPRT. The second pathway is the Preiss-Handler pathway initiated by the conversion of nicotinic acid (NA) to NAMN by NAPRT. NANM, an intermediate in both the de novo biosynthesis and the Preiss-Handler pathway is then converted to form NAAD by NMNATs. The link between Trp and NA shown here is the human pathway. The last step of these pathways is the conversion of NAAD to NAD+ by NADS. The third pathway is the Salvage pathway, generating NAD+ from nicotinamide riboside (NR), which also includes the recycling of NAM back to NAD+ via NMN. Extracellularly, NAD+ or NAM can be converted to NMN, which is in turn dephosphorylated to NR, possibly by CD73. NR is transported into the cell via unknown mechanism (possibly nucleoside transporters), where it is phosphorylated by the NRK1 or NRK2 forming NMN. NMN is then converted to NAD+ by NMNATs. The dashed lines indicate that the mechanism and involved proteins are not known. The lower part of the figure shows the major NAD+ consuming enzymes. From left: The cyclic ADP-ribose synthases (cADPRSs) CD38 and CD157 hydrolyze NAD+ to NAM; in addition, CD38 can degrade NMN to NAM, removing NMN from the NAD+ synthesis. PARPs, especially PARP1 and PARP2 use NAD+ as a co-substrate to PARylate target proteins, generating NAM as a by-product. The deacetylation activity of SIRTs SIRT1, SIRT3 and SIRT6 depend on NAD+, generating NAM as a byproduct, which can inhibit the activity of SIRTs. The enzyme NNMT methylates NAM, using SAM as a methyl-donor. This removes NAM from recycling, and indirectly affects NAD+ levels.
Abbreviations: ACMS: 2-amino-3-carboxymuconate semialdehyde; cADPRSs: cyclic ADP-ribose synthases; FK: formylkinurenine; IDO: indoleamine 2,3-dioxygenase; Me-NAM: Methylated nicotinic acid mononucleotide; NA: nicotinic acid; NAAD: nicotinic acid adenine dinucleotide; NAD+: nicotinamide adenine dinucleotide; NADS: NAD+ synthase; NAM: nicotinamide; NAMN: nicotinic acid mononucleotide; NAMPT: nicotinamide phosphoribosyl transferase (including extracellular and intracellular ones); NAPRT: nicotinic acid phosphoribosyl-transferase; NMN: nicotinamide mononucleotide; NMNATs: nicotinic acid mononucleotide transferases; NR: nicotinamide riboside; NRKs: nicotinamide riboside kinases; PARPs: Poly(ADP-ribose) polymerases; QPRT: quinolinate phosphoribosyltransferases; SIRTs: sirtuins; SAM: S-adenosyl methionine; TDO: tryptophan 2,3-dioxygenase; Trp: tryptophan.
The de novo biosynthesis pathway and the Preiss-Handler pathway are well characterized (recently reviewed elsewhere [8, 9]). Here, we focus on the salvage pathway which is important from a translational research perspective because it produces NAD+ from the precursors NAM, NMN and NR; these have shown potential as dietary supplements to increase intracellular NAD+ levels (see below). The major steps and key enzymes in this pathway are mentioned in Box 2.
Box 2. The NAD+ Salvage Pathway and its Major Enzymes.
The salvage pathway describes the formation of NAD+ from precursors including NR, NMN and NAM. NR can be transported into cells via nucleoside transporters though a NR-specific transporter has not yet been identified in mammals. In the cytoplasm, NR is phosphorylated by nicotinamide riboside kinases (NRK1–2) to form NMN [122]. NRKs are not expressed in mitochondria suggesting that the phosphorylation step may likely take place in the cytosolic compartment [124]. In addition, extracellular interconversion of NAD+ and its precursors may occur (Figure 1). Extracellular NMN can be converted from NAD+ and NAM, with the latter in a Nicotinamide phosphoribosyl-transferase (NAMPT) (extracellular)-dependent manner. NMN can either enter the cells directly, though a mechanism that is still unclear, or be converted to NR before transport into cells [124].
NAMPT (intracellular) first converts NAM to NMN, which is then adenylated to NAD+ by NMNATs. NMNATs are the final step in NAD+ synthesis and are also involved in the Preiss-Handler pathway. In humans, there are three NMNAT isoforms with different sub-cellular localizations. NMNAT1 is localized in the nucleus, and shows the highest expression in skeletal muscle, heart, kidney, liver and pancreas [125]. NMNAT2 is primarily found in the cytosol, tethered to the outer membrane of the Golgi apparatus by palmitoylation. NMNAT3 is localized to mitochondria, and is expressed at high levels in skeletal muscle and heart tissue [125]. Thus, intracellular NMN can be converted to NAD+ in the nucleus, cytoplasm, or the mitochondria by one of the three NMNATs. It is still unclear how cytoplasmic NMN is imported into mitochondria.
NAM is not only an ingested NAD+ precursor, but is also a byproduct of the degradation of cellular NAD+ by NAD+-consuming enzymes (detailed below). In mammalian cells, nicotinamide N-methyltransferase (NNMT) removes NAM from the NAD+ biosynthesis pathway through methylation of NAM, which is further degraded into N-methyl-2-pyridone-5-carboxamide (Me2PY) and N-methyl-4-pyridone-5-carboxamide (Me4PY) [77].
While there is inherent redundancy among the three NAD+ synthetic pathways, distinct functions can be ascribed to the importance of specific metabolites and/or the tissue-specific expression of some of the enzymes in each pathway. The nicotinic acid mononucleotide transferases (NMNATs) -- necessary for both the Preiss-Handler pathway and the salvage pathway (Figure 1)-- play pivotal roles in embryonic development in mice [23] and in neuroprotection across species [23–25]. Nmnat1−/− mice exhibit embryonic lethality [23] and studies in Drosophila suggest a general neuroprotective function for NMNAT. Specifically, Overexpression of NMNAT in a Drosophila model of spinocerebellar ataxia 1 (SCA1)-induced neurodegeneration indicated that NMNAT could inhibit activity-induced neurodegeneration, injury-induced axonal degeneration, as well as SCA1-induced neurodegeneration [24]. In addition, mice lacking nicotinamide phosphoribosyltransferase (Nampt), a key enzyme in the salvage pathway, also exhibit embryonic lethality [26]. Furthermore, murine neurons and pancreatic β cells express low levels of Nampt protein compared to other cells and pancreatic β cells have been reported to be particularly vulnerable to NAMPT inhibition [26, 27]. The importance of NAMPT has also been noted in mice with muscle-specific Nampt-depletion; these mice experience fiber degeneration and loss of strength and endurance, whereas lifelong Nampt overexpression increases NAD+ and improves the physical function of aged mice [28]. Of note, the three major NAD+-synthesizing pathways use different substrates for the generation of NAD+; thus, the production of NAD+ may be dependent on both substrate availability as well as the local levels of synthesizing proteins (e.g. NAMPT, NMNATs, etc.).
NAD+-consuming Enzymes
NAD+ is a co-enzyme for three groups of enzymes: i) deacetylases in the sirtuin family (SIRTs); ii) ADP-ribosyltransferases, including poly(ADP-ribose) polymerases (PARPs); and iii) cyclic ADP-ribose synthases (cADPRSs) (Figure 1). SIRTs are a group of NAD+-dependent deacetylases and ADP-ribosyltransferases, which promote mitochondrial homeostasis, neuronal survival, stem cell rejuvenation, and prevent certain aspects of the aging process, such as neurodegeneration, loss of stem cells and mitochondrial dysfunction [29–31]. ADP-ribosylation is an important protein posttranslational modification, which affects DNA repair, epigenetic regulation, neurodegeneration, and aging [14]. ADP-ribosylation of proteins is executed by ADP-ribosyltransferases through transfer of the ADP-ribose moiety from NAD+ to target substrates. PARPs are prominent members of the ADP-ribosyltransferase family, consisting of 17 different enzymes in mammals [32]. PARPs transfer the first ADP-ribose unit from NAD+ to target proteins, followed by sequential addition of ADP-ribose units to the preceding ones to form poly(ADP-ribose) polymers (PARs) [32]. The cADPRSs include CD38 and its homolog CD157, in mammals and birds: CD38 and CD157 are transmembrane proteins, localized to the plasma membrane and to membranes of intracellular organelles including the mitochondria, nucleus and endoplasmic reticulum [33]. CD38 is expressed in immune cells, liver, testis, kidney and brain [33]. It plays important roles in several physiological processes such as nuclear Ca2+ homeostasis, immunity, inflammation, as well as glucose and lipid homeostasis [34–36]. cADPRSs can function as glycohydrolases, or NADases, hydrolyzing NAD+ to NAM and ADP-ribose [9]. These NAD+-consuming enzymes regulate a spectrum of cellular activities including mitochondrial maintenance, DNA repair and stem cell rejuvenation, processes which are critical for cellular health. (see also recent reviews [9, 11, 35]).
Cross-talk among NAD+-consuming Enzymes: Sirtuins, PARPs and cADPRs
The different classes of NAD+ consuming enzymes compete for bioavailable NAD+, which affects their cellular functions in human health. Thus, hyper-activity of one protein might limit the activities of the others, and conversely, inhibition of one protein may increase the NAD+ pool for the others [35]. For example, SIRT1 activity can be substantially decreased when there is excessive PARP1 activation, and PARP1 or PARP2 deletion in human kidney cells and in in vivo models (Parp2−/− mice) has been reported to increase SIRT1 activity [37, 38]. Persistent activation of PARP1, caused by DNA damage, has resulted in a greater than 50% decrease in cellular NAD+ in DNA repair-deficient primary rat neurons and human neuroblastoma cells[14, 39]. Moreover, in DNA repair-deficient human neuroblastoma cells, as well as in mouse and C. elegans models, cellular NAD+ and Sirt1 activity have been reported to increase after treatment with Parp1 inhibitors (3-aminobenzamide, 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone) or after supplementation with NAD+ precursors (NR, NMN) [14, 39]. These studies suggest that PARP1 inhibition may constitute a potential therapeutic target to sustain cellular NAD+, and to maintain sirtuin activity.
CD38 is another major NADase in tissues. In APP/PS1 mice, CD38-depletion and increased NAD+ led to neuroprotection, as evidenced from a reduction of Aβ aggregates in the brain, which was associated with improved learning [40]. In addition, CD38-deficient mice showed protection against HFD-induced obesity and metabolic syndrome, exhibiting higher metabolic rates compared to wild-type mice [41]. This resistance to diet-induced obesity has been attributed, at least in part, to NAD+-dependent activation of Sirt1 and the mitochondrial regulator peroxisome proliferator-activated receptor γ coactivator (PGC-1α) [41]. Of note, CD38-deficient mice show significant neuroprotection in the brain despite high levels of PARylation [42], indicating that the available NAD+ appears to be sufficient in enabling the activity of Parp1, Sirt1 and other NAD+-dependent enzymes. A recent study showed an age-dependent increase of CD38 in murine tissues, and documented (using CD38 knockout mice) that CD38 was required for the observed age-related NAD+ decline [43]. This is interesting as CD38 not only consumes NAD+, but can also degrade the NAD+ precursor NMN [43]. Thus, there appears to be a delicate balance between NAD+ consumption and bioavailability, which is important for cellular function and survival. However, further studies on the functional significance of the interconnected networks between NAD+ consuming enzymes is warranted.
NAD+ in Aging and Age-related Diseases
Age-dependent Decrease of NAD+ in Animals and Humans
Mice display an age-dependent decrease of NAD+ in multiple organs including brain, liver, muscle, pancreas, adipose tissue and skin [14, 44–46]. In C. elegans, an age-dependent reduction of NAD+ has also been reported [14, 39]. There is also evidence of decreased NAD+ in aged human tissues. Specifically, in vivo NAD+ assays have been used to demonstrate that intracellular NAD+ declines with age in the human brain [47]. Also, NAD+ in post-pubescent males and females negatively correlates with age [48]. Together, these data suggest that there may be a universal age-dependent decrease of cellular NAD+ across species. However, it is not clear whether this is due to increased NAD+ consumption and/or decreased production.
NAD+ Replenishment can Improve Lifespan and Healthspan
The effects of different NAD+ precursors including NR and NMN on the lifespan and healthspan of yeast, C. elegans, Drosophila and mice have been investigated. One study showed that 10 μM NR could extend the replicative lifespan of wild-type yeast by more than 10 generations [15]. In this yeast model, two NAD+ synthetic pathways appeared to be necessary for the NR-induced lifespan extension, the tNrk1 and the Urh1/Pnp1/Meu1 pathways [15]. In C. elegans, 500 μM NR extended the average lifespan of wild-type worms (N2) via the SIR-2.1 (ortholog to mammalian SIRT1) pathway [39]. For Drosophila, no information is available regarding a direct effect of NAD+ precursors on lifespan, but genetic overexpression of an NAD+ synthetic enzyme nicotinamidase (D-NAAM) has been reported to extend lifespan [49]. D-NAAM, an ortholog of yeast PNC1, functions in the NAD+ salvage pathway and converts NAM to NA [49]. In Drosophila, overexpression of D-NAAM can increase the NAD+/NADH ratio, as well as the mean and maximal lifespan of up to 30% in a Sir2-dependent manner [49]. Notably, NR has been shown to improve mouse lifespan; even when administered late in life. C57BL/6J mice, at ~2 years of age, were given NR, resulting in a significant increase in lifespan (5%) [46]. Supplementation with NAD+ precursors not only extends lifespan but also improves healthspan in yeast, flies, worms and mice, as shown from various features, including improved mitochondrial health, muscle strength and motor function. [15, 39, 46, 49–51]. In summary, these data suggest that NAD+ replenishment can delay normal aging in laboratory animal models.
NAD+ may also delay the onset of aging in some premature aging diseases. These are a group of rare diseases in which patients exhibit aging features at a younger age. DNA repair impairment is a cause for many of these diseases, and some patients exhibit severe neurodegeneration, as in the case of Xeroderma pigmentosum group A (XPA), Cockayne syndrome (CS), and Ataxia-telangiectasia (A-T) [14]. XPA is caused by mutations in the XPA gene, which participates in nucleotide excision DNA repair (NER) [52, 53]. The etiology of CS has been related to mutations in two proteins, CS group A (CSA) and CS group B (CSB) [54, 55]. A-T is a multifaceted disease caused by mutations in A-T mutated protein (ATM), a master regulator of the DNA damage response. ATM plays a key role in DNA double strand break repair (DSBR). Interestingly, all three premature aging diseases show mitochondrial dysfunction and NAD+ depletion (demonstrated in C. elegans, mice and human cells) [14]. Furthermore, NAD+ replenishment, using NR and/or NMN, improves lifespan and healthspan of C. elegans worms in relevant models of XPA, A-T and CS, relative to controls [14, 39, 56]. Remarkably, NR extended the lifespan in an Atm−/− mice (B6;129S4-Atmtm1Bal/J), which typically perish at 3~5 months of age. Specifically, NR supplementation was given at 12 mM in drinking water after weaning, resulting in 80% survival at 10 months of age [39]. In C. elegans, NAD+ supplementation has been shown to improve neuronal DNA repair through deacetylation of the DNA repair protein Ku70, and to restore mitochondrial homeostasis via the mitophagy regulator NIX (DCT-1 in C. elegans) [39]. The DCT-1 mechanism of mitochondrial homeostasis was implicated based on evidence that DCT-1 co-localized with mitochondria in Atm-1 worms [39]. Of note, NR and NMN exhibited similar beneficial effects in A-T models of C. elegans and mice [39]. In another example, mice hypomorphic for BubR1(a mitotic checkpoint kinase), also presented signs of premature aging, and mice overexpressing BubR1 exhibit an extended lifespan [57]. This is pertinent because loss of BubR1 during aging has can be caused with NAD+ depletion and decreased Sirt2 activity, unable to deacetylate BubR1 (normally Sirt2 deacetylates BubR1 targeting it for ubiquitination and degradation) [57]. Both overexpression of Sirt2 or NMN treatment increased the lifespan of the BubR1 mice, suggesting that BubR1 stabilization may be important to achieve increased lifespan [57]. In summary, restoration of intracellular NAD+ has been shown to improve lifespan and healthspan in normal and prematurely aged laboratory organisms, but whether these findings can be translated to humans remains unproven.
NAD+ and Neurodegeneration
Age is the greatest risk factor for neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and hearing loss [58]. Recent studies in AD animal models suggest that Aβ plaques, tau tangles, and mitochondrial dysfunction (due to compromised mitophagy) are among the key features in AD [59]. Strategies to increase intracellular NAD+ are considered as novel potential therapeutic interventions in AD. For example, three months of NR treatment has been documented to attenuate cognitive deterioration through Aβ reduction in the cortex and hippocampus in a mouse model (AD mouse crossed with a PCG-1α KO mouse) [60]. The study suggests that NR treatment can promote PGC-1α activity and induce the ubiquitin proteasome system, leading to degradation of Aβ aggregates [60]. Similarly to NR, NMN has also been found to ameliorate mitochondrial dysfunction, and neuronal death in APP/PS1 mice, as evidenced from restored oxygen consumption rates, increased levels of Sirt1 and PGC-1α, and normalization of morphology of brain mitochondria from NMN-treated APP/PS1 mice [61]. NMN has also been found to protect against Aβ oligomer-induced cognitive impairment, neuronal death and cognitive dysfunction in a rat model of AD [62]. In another study, treatment with a different NAD+ precursor, NAM, delayed pathology and cognitive decline in 3xTg AD mice, through upregulation of neuronal bioenergetics, including neuroplasticity-involved kinases and transcription factors, as well as by improving autophagy processing (reduced autophagosome accumulation mediated by enhanced lysosome/autolysosome acidification) [63]. At the molecular level, NAD+-dependent reduction of AD phenotypes may be attributed to upregulation of autophagy/mitophagy because NAD+/SIRT1 are able to upregulate autophagy through deacetylation of major autophagic proteins Atg5, Atg7, and Atg8 in human cells, murine neurons and C. elegans [39]. Furthermore, NAD+/SIRT1 has been shown to upregulate mitophagy through the forkhead box-O3 (FOXO3)-NIX (BNIP3L/DCT-1) axis in C. elegans and in calorically-restricted mice, or indirectly through an interaction between PGC-1α and Parkin, as evidenced from co-expression of PGC-1α and Parkin in murine cortical neurons that led to improved mitochondrial biogenesis and mitophagic activity [39, 64–66]. Accordingly, upregulation of autophagy/mitophagy has been reported to clear Aβ plaques, tau tangles, and damaged mitochondria in mice, leading to improved mitochondrial function and neuronal survival [59, 63, 67].
PD appears to involve impairment of mitochondrial complex I, as well as compromised mitophagy in vulnerable neurons of the brain, including midbrain dopaminergic neurons [68]. There are several promising studies that suggest that NAD+ precursors may have therapeutic potential in PD. Early clinical studies showed that supplementation (intravenous infusion or oral capsules) with the reduced form of NAD+ (NADH) improved motor disability in PD patients [69]. Recently, in models of PD in vitro (rotenone treated PC12 cells) and in vivo, in Drosophila bearing pink1 mutations, studies have shown that NR and NAM administration could ameliorate PD phenotypes [70, 71]. For example, dietary NAM (5 mM) could rescue thoracic defects and inhibit the loss of dopaminergic neurons in pink1 mutant Drosophila, possibly through maintenance of a healthy mitochondrial pool, as evidenced from improved mitochondrial morphology and respiration [71]. However, further studies of NAD+ precursor treatments in PD mice are necessary to elucidate the underlying molecular mechanisms and functional roles of NAD+ in PD pathology.
Hearing loss is a common feature associated with advanced age, and noise exposure is a major cause of hearing loss [72]. Intense noise exposure can result in direct mechanical damage (acoustic trauma) to cochlear hair cells, and can trigger delayed death of these auditory sensory neurons. NR administration (intraperitoneal injection) has been found to protect mice from transient and permanent noise-induced hearing loss and spiral ganglia neurite degeneration, as shown by retraction of neurite ends from inner hair cells in the cochlea [73]. These effects appear to be mediated by NAD+-dependent mitochondrial Sirt3, since the effect of NR was reduced in Sirt3-deficient mice [73]. This role of Sirt3 is supported by studies showing that caloric restriction prevents hearing loss in wild-type mice, but not in Sirt3-deficient mice [74]. Moreover, ototoxicity, caused by chemical damage to the inner ear, has been reported to be prevalent in cancer patients receiving cisplatin chemotherapy [75, 76]. Cisplatin causes DNA damage by crosslinking the two DNA strands. Increased NAD+ has been reported to prevent cisplatin-induced cochlear damage through suppression of oxidative stress, DNA damage and inflammatory responses in mice, as shown by a decrease in γ-H2AX signals, increased Sirt1 activity, and activation of p53 and NF-κB, proteins involved in inflammation in cochlear tissue [75, 76]. These studies support the hypothesis that NAD+ supplementation might be able to provide a putative treatment option to preserve hearing during normal aging, or in individuals undergoing chemotherapeutic treatment. However, rigorous testing will be required to determine if NAD+ supplementation might modify the effectiveness of the chemotherapeutic treatment.
NAD+ can Mitigate Age-associated Muscle Atrophy and Metabolic Disorders in Laboratory Animal Models
Studies in C. elegans and mice suggest that NAD+ supplementation can delay the onset of muscle atrophy, vision loss, as well as certain age-related diseases that might include metabolic disease, heart dysfunction, and cancer [39, 44, 50, 73, 77–82]. In the Key Table (Table 1 A and B), we summarize the known organismal benefits of NAD+ supplementation for specific animal models, NAD+ precursor doses, and putative molecular mechanisms underlying these improvements.
Table 1. Known Benefits And Mechanisms of Action of NAD+ Precursors in Animal Models.
(A). Human/Worms. (B) Mice
| Tissue | Supplement dose | Benefits | Pathways affected | Ref | |
|---|---|---|---|---|---|
| Human | Neurons | NR 500 μM | Decreased AT pathology & restored mitochondrial function in ATM KD cells. | ↑ NAD+, SIRT1 activation, BDNF levels & CREB activation, ↓ DNA damage. | [38] |
| Blood | Acipimox 250 mg/3x/d NR 1000 mg/d/7d | ↓ triglycerides & glucose in plasma of type 2 diabetes patients. Restored levels of NEFA. | 2-fold ↑ NAD+, 45-fold ↑ NAAD. | [11] [7] | |
| Muscle | Acipimox 250 mg/3x/d | Improved mt function in type 2 diabetes patients. ↑ Lipid content in patient skeletal muscle, due to ↑ NEFA. | Gene sets affected were similar to those affected by NR & NMN in animal models. | [11] [6] | |
| Worm | Neurons | NR 500 μM | Improved long- & short-term memory in Atm worms. | ↑ NAD+, activation of sir-2.1, ↑ CREB, HSP-6. | [38] |
| Muscle | NR 500 μM | Improved mt network in Atm worms. | ↑ NAD+, sir-2.1 activation, CREB, HSP-6. | [38] [45] [76] | |
| Lifespan | NR 500 μM NAM 200 μM NR 500 μM | ↑ lifespan & improved fitness with age in wild-type N2 worms. ↑ lifespan of Atm-worms. |
↑ NAD+, sir-2.1, CREB. HSP-6, mt content & ATP, improved metabolism. | [38] [44] | |
| Species | Tissue | Supplement dose | Benefits | Pathways affected | Refs |
| Human | Neurons | NR 500 μM | Decreased AT pathology & restored mitochondrial function in ATM KD cells. | Increased NAD+ level, SIRT1 activation, BDNF level & CREB activation, decreased DNA damage. | [38] |
| Blood | Acipimox 250 mg/3x/d NR 1000 mg/d/7d | Decreased triglycerides & glucose in plasma of type 2 diabetes patients. Restored levels of NEFA. | Two fold increased NAD+, 45-fold increased NAAD. | [117] | |
| Muscle | Acipimox 250 mg/3x/d | Improved mitochondrial (mt) function in type 2 diabetes patients. Lipid content in the skeletal muscle of the patients was increased, due to increased NEFA. | Gene sets affected were similar to those affected by NR & NMN in animal models. | [116] | |
| Mice | Brain | NR 400 mg/kg/d NR 250 mg/kg/d NR or NMN 12 mM | Increased neurogenesis. Attenuated cognitive deterioration, increased synaptic plasticity, & reduced Aβ production. NR abolished metabolic profile of cerebellum from ATM KO mice. NR & NMN improved mt morphology & health, leading to neuron protection. |
Increased NAD+, activation of SIRT1 & PGC-1α & degradation of BACE1. Increased LC3-II & altered fission/fusion balance. | [45] [59] |
| Ear/cochlear | NR 1000 mg/kg/2x/d | Prevented transient & permanent hearing loss (noise-induced) by preventing degeneration of spiral ganglia neurites. | Increased NAD+ levels & SIRT3 activation. | [38] [72] | |
| Eye | NMN, 100 or 300 mg/kg/d | Prevented age-associated decline of rod & cone cell function. Increased tear production. | Likely due to increased NAD+ level & sirtuin activity. | [43] | |
| Muscle | NR 400 mg/kg/d NR 750 mg/kg/d NMN 500 mg/kg/d NMN 300 mg/kg/d | Increased NAD+ level & mt content, prevented mt myopathy. Increased lifespan of Trf1hrt /hrt mutants & improved cardiac function. Reversed age-associated muscle atrophy & inflammation, impaired insulin signaling, & insulin-stimulated glucose uptake. Improved mitonuclear signaling & mt function. |
SIRT1 & SIRT3 activation, & their targets FOXO1, SOD2, PGC1a, UPRmt, FGF21. Improved mt function & mitophagy mediated via SIRT1 & EglN1. Increased mtDNA encoded proteins/nuclear encoded proteins. | [43] [76] [45] [84] [80] [49] | |
| Stem cells | NR 400 mg/kg/d | Rejuvenated muscle stem cells. Attenuated senescence in neuronal & melanocyte stem cells. | Activation of UPRmt, prohibitin pathways | [45] [76] | |
| Fat | BAT: NR 400 mg/kg/d WAT: IP NMN 500 mg/kg/d | Increased mt content & mt respiratory capacity, reduced fat mass. Restored NAD+ levels in diabetic mice & normalized glucose tolerance. |
Increased NAD+ levels, SIRT1 & SIRT3 activation. | [36] [37] [84] [50] | |
| Liver | NR 400 mg/kg/d, 500 mg/kg/d NMN 500 mg/kg/d, 300 mg/kg/d | Increased oxidative profile, biogenesis, content & activity of mt, & decreased tumorigenesis & DNA damage. Prevented fatty liver & inflammation induced by high-fat high-sucrose diet. Improved hepatic insulin sensitivity, decreased oxidative stress, & improved inflammatory response, immune response, & lipid metabolism. |
SIRT1 & SIRT3 activation, & SIRT1 target genes including AKT, prevention of NAFLD, decreased DNA damage. | [43] [50] [84] [85] | |
| Pancreas | NMN 500 mg/kg/d | Improved glucose-stimulated insulin secretion. | Increased NAD+ level & SIRT1 activation. | [50] | |
| Lifespan | NR 400 mg/kg/d 12 mM NR throughout life | Increased lifespan of mice. Increased lifespan of ATM KO mice. |
Increased NAD+ level, SIRT1 & prohibition activity, improved mt function & mitophagy, DNA repair, & anti-cancer potential. | [45] [38] | |
| Worms | Neurons | NR 500 μM | Improved long- & short-term memory in ATM worms. | Increased NAD+, activation of sir-2.1, increased CREB, HSP-6. | [38] |
| Muscle | NR 500 μM | Improved mt network in ATM worms. | Increased NAD+ level, sir-2.1 activation, CREB, HSP-6. | [38] [45] [76] | |
| Lifespan | NR 500 μM NAM 200 μM NR 500 μM | Increased lifespan & improved fitness with age in wild-type N2 worms. Increased lifespan of ATM-worms. |
Increased NAD+ level, sir-2.1, CREB. HSP-6, mt content & ATP, improved metabolism. | [38] [44] | |
| Tissue | Supplement dose | Benefits | Pathways affected | Ref | |
| Mice | Brain | NR 400 mg/kg/d NR 250 mg/kg/d NR or NMN 12 mM | ↑ neurogenesis ↓ cognitive deterioration & Aβ production. ↑ synaptic plasticity NR abolished metabolic profile of cerebellum from Atm KO mice. NR & NMN improved mt morphology & health, leading to neuron protection. |
↑ NAD+, activation of Sirt1 & PGC-1α & degradation of Bace1. ↑ LC3-II & altered fission/fusion balance. |
[45] [59] |
| Ear/cochlear | NR 1000 mg/kg/2x/d | Prevented transient & permanent hearing loss (noise-induced) by preventing degeneration of spiral ganglia neurites. | ↑ NAD+ & Sirt3 activation. | [38] [72] | |
| Eye | NMN, 100 or 300 mg/kg/d | Prevented age-associated decline of rod & cone cell function. Increased tear production. | Likely due to ↑ NAD+ level & sirtuin activity. | [43] | |
| Muscle | NR 400 mg/kg/d NR 750 mg/kg/d NMN 500 mg/kg/d NMN 300 mg/kg/d | ↑ NAD+ & mt content, prevented mt myopathy. ↑ lifespan of Trf1hrt /hrt mutants & improved cardiac function. Reversed age-associated muscle atrophy & inflammation, impaired insulin signaling, & insulin-stimulated glucose uptake. Improved mitonuclear signaling & mt function. |
Sirt1 & Sirt3 activation, & their targets Foxo1, Sod2, PGC1α, UPRmt, Fgf21. Improved mt function & mitophagy mediated via Sirt1 & EglN1. ↑ mtDNA encoded proteins/nuclear encoded proteins. |
[43] [76] [45] [84] [80] [49] | |
| Stem cells | NR 400 mg/kg/d | Rejuvenated muscle stem cells. Attenuated senescence in neuronal & melanocyte stem cells. | Activation of UPRmt, prohibitin pathways | [45] [76] | |
| Fat | BAT: NR 400 mg/kg/d WAT: IP NMN 500 mg/kg/d | ↑ mt content & mt respiratory capacity, reduced fat mass. Restored NAD+ levels in diabetic mice & normalized glucose tolerance. | ↑ NAD+ levels, Sirt1 & Sirt3 activation. | [36] [37] [84] [50] | |
| Liver | NR 400 mg/kg/d, 500 mg/kg/d NMN 500 mg/kg/d, 300 mg/kg/d | ↑ oxidative profile, biogenesis, content & activity of mt, & decreased tumorigenesis & DNA damage. Prevented fatty liver & inflammation induced by high-fat high-sucrose diet. Improved hepatic insulin sensitivity, decreased oxidative stress, & improved inflammatory response, immune response, & lipid metabolism. |
Sirt1 & Sirt3 activation, & Sirt1 target genes including Akt, prevention of NAFLD, decreased DNA damage. | [43] [50] [84] [85] | |
| Pancreas | NMN 500 mg/kg/d | Improved glucose-stimulated insulin secretion. | ↑ NAD+ & Sirt1 activation. | [50] | |
| Lifespan | NR 400 mg/kg/d 12 mM NR throughout life | ↑ lifespan of mice. ↑ lifespan of Atm KO mice. |
↑ NAD+, Sirt1 & prohibition activity, improved mt function & mitophagy, DNA repair, & anti-cancer potential. | [45] [38] |
Abbreviations: mt: mitochondrial; ↑ increase; ↓ decrease
Skeletal muscle mass and strength is reduced with aging as a result of muscle atrophy, leading to significant susceptibility to injury and reduced quality of life [83]. In the mdx mouse model of Duchenne’s muscular dystrophy that exhibits low muscle NAD+, NR protects against disease progression through maintenance of muscle stem cell function and regeneration, where Sirt1-dependent enhancement of mitochondrial function and energetics as well as a reduction of PARylation have been implicated [46, 77]. In another study, muscle-specific depletion of Nampt−/− in mice (mNampt−/− mice) caused myocyte necrosis and progressive loss of muscle function, as well as inducing pro-inflammatory and regenerative transcriptional programs coincident with alterations in glucose metabolism [28]. In this model, oral NR supplementation reversed deficits in muscle mass, strength, and exercise capacity of mNampt−/− mice to wild type levels [28]. In combination, these studies suggest that NAD+ supplementation can inhibit muscular dystrophy in mice, which may have interesting implications for aged individuals.
NAD+ replenishment has also been shown to have beneficial affects against obesity and/or other metabolic diseases. NR can enhance oxidative metabolism and protect mice against HFD-induced obesity and non-alcoholic fatty liver disease by activating Sirt1 and Sirt3, and enhancing the energy expenditure and oxygen consumption rate of mitochondria [84][85]. NR induced a Sirt1 and Sirt3-dependent mitochondrial unfolded protein response, thereby improving mitochondrial metabolism (increasing mitochondrial complex formation and activity) [85]. NR administration (via an osmotic pump) lowered serum insulin levels and serum cholesterol concentrations in the KK/H1J mouse model of type 2 diabetes [86]. Other NAD+ precursors, such as NMN (delivered intraperitoneally), have been reported to improve diet- and age-induced diabetes in 2-year-old mice by enhancing hepatic insulin sensitivity and antioxidative responses, including the production of glutathione S-transferase, among others [51]. NAD+ precursors can also act partly through Sirt1 activation, as demonstrated from an abrogated effect of NMN via Sirt1 inhibition with EX527 (a Sirt1-specific inhibitor) [51]. NMN administration has also ameliorated the impairment in glucose tolerance and glucose-stimulated insulin secretion in Nampt+/− mice [26].
The aforementioned data strongly suggest that NAD+ replenishment may have systemic benefits in aged laboratory animals and disease models in mice, although further testing is required. Importantly, translational human intervention studies with NAD+ supplementation are already in progress (https://clinicaltrials.gov/ct2/results?term=nad%2B&Search=Search).
Potential NAD+-mediated Mechanisms to Counter Aging
While the phenotypic studies of NAD+ replenishment on aging have been extensively explored in laboratory animals, the exact molecular mechanisms responsible for their beneficial effects are not yet understood. To dissect the interconnected mechanisms of the multi-faceted functions of NAD+ to aging, we can associate NAD+ to most of the nine ‘hallmarks of aging’ [58] (Table 2). We also consider autophagy impairment as a NAD+-related hallmark of aging.
Table 2.
NAD+ and the Hallmarks Of Aging.
| Hallmarks of aging | Pathways & conditions affected by changes in NAD+ levels | Ref |
|---|---|---|
|
[11] [38] [14] [55] | |
|
||
|
[9] [8] [44] [59] [49] | |
|
[45] [76] | |
|
[26] [38] [44] [39] [59] | |
|
[14] [84] [40] [50] [49] | |
|
[9] [40] [49] [50] [84] | |
|
[72] [25] [40] [50] | |
|
[8] [45] [47] | |
|
[105] |
Abbreviations: mt: mitochondrial; ↑ increase; ↓ decrease
Effects of NAD+ Supplementation on DNA Repair
Cumulative evidence indicates that impaired genomic maintenance may causally contribute to aging. An age-dependent accumulation of DNA damage occurs in humans, possibly due to impaired DNA repair [87]. This suggests that maintenance of efficient DNA repair may delay the onset of aging and age-related diseases [88, 89]. NAD+ replenishment can improve DNA repair in cells, C. elegans, and mice. An early study reported that suitable doses of NAM (~3 mM) significantly enhanced DNA repair in gamma-irradiated XP cells in vitro [90]. In line with this finding, studies in human aortas suggest that the NAM-consuming enzyme NAMPT plays a significant role in DNA repair to maintain genome integrity, as demonstrated from an increase in DNA oxidative DNA lesions and DNA double strand breaks in murine Nampt-deficient murine smooth muscle cells [91]. In addition to NAM, NA and NR also improve DNA repair in vitro. Indeed, studies revealed increased DNA repair in vitro in peripheral blood mononuclear cells, where NA treatment increased DNA repair efficiency and decreased micronuclei numbers following X-ray irradiation, and in murine Atm-deficient neurons, following NR administration[39, 92]. NAD+ supplementation can also increase DNA repair in vivo as wild-type mice treated with NR showed decreased DNA damage, as evidenced from lower global PARylation and γ-H2AX foci relative to controls [46, 77]. As previously mentioned, NAD+ replenishment can also improve aging features of some DNA repair-deficient premature aging disorders, XPA, CS, and A-T, such as neurodegeneration, possibly through a mechanism of upregulation/activation of DNA repair and mitophagy [14, 39, 93]. For example, due to the dysfunction of the DSBR protein ATM, accumulation of DNA damage occurs in both nuclear, as well as mitochondrial genomes, as revealed from human patient fibroblasts and brain-tissue from Atm-deficient mice [94, 95]. NR supplementation improved genomic stability in murine Atm-deficient neurons and C. elegans models of A-T, at least partially through Sirt1/Sirt6-dependent DSBR, upon protection against ionizing irradiation [39]. In neurons, there is no homologous recombination (HR)-based DSBR, only non-homologous end joining (NHEJ)-based DSBR [96]. In murine Atm-deficient neurons, NR increases DNA-PKC-associated NHEJ through deacetylation of Ku70 [39]. These findings suggest that enhancing NAD+ bioavailability might also be able to target and increase DNA repair.
NAD+ Maintains Mitochondrial Health
Mitochondrial abundance and quality are pivotal for health, and mitochondrial dysfunction is a hallmark of aging, detected in a broad spectrum of age-associated diseases [97]. NAD+ replenishment can inhibit age-dependent mitochondrial decline or production in both C. elegans and mice [45, 46]. For example, dietary NR treatment can compensate for a respiratory chain defect and reverse exercise intolerance in a mitochondrial disease mouse model Sco2KOKI [98]. NAD+ can also bolster mitochondrial function by enabling mitochondrial biogenesis and mitophagy; mitochondrial biogenesis and respiration are induced by PGC-1α through transcriptional upregulation of Nrf1 and Nrf2 in mouse myoblasts [99]. Thus, NAD+ replenishment has been documented to induce mitochondrial biogenesis through the NAD+/Sirt1-PGC1α pathway in aged mice, murine muscle stem cells and C. elegans [45, 46]. Indeed, NAD+ can promote the removal of damaged/dysfunctional mitochondria via mitophagy, as detailed below.
NAD+ in Autophagy Induction
Autophagy plays multi-faceted roles in health and aging, including maintenance of cellular homeostasis, cellular energy (especially during nutrient starvation), neuroprotection, and anti-inflammation [13, 67, 100]. Compromised autophagy is a common signature of aging and compromised autophagy contributes to age-related diseases, such as AD and PD [13, 35, 67]. Upregulation of autophagy can inhibit disease progression in animal models of AD and PD (3xTgAD model, Tg2675 model) [59, 60, 63, 67]. Moreover, mutations of proteins in the autophagy pathway, including Atg1 (unc-1 in C. elegans) and other proteins encoded by the Atg genes, can lead to premature aging in yeast, nematodes and Drosophila, and treatment with autophagy inhibitors can reduce their lifespan [101–103].
Autophagy induction may delay the onset of aging and potentially slow down the initiation or progression of age-related diseases [13]. For example, upregulation of autophagy inhibits major markers of aging, including inflammation and senescence. Indeed, autophagy can inhibit inflammation through the regulation of immune mediators and their interaction with innate immune signaling pathways by removing endogenous inflammasome agonists [104]. In senescent cells, adenosine monophosphate-activated protein kinase (AMPK) can be inactivated; however, pharmacological activation of AMPK has been found to inhibits cellular senescence through NAD+/SIRT1 induction and autophagy upregulation [105]. At the organismal level, pharmacological or genetic upregulation of autophagy has been reported to extend healthspan and lifespan in laboratory animal models [13]. Impaired autophagy might occur both upstream and downstream of other aging hallmarks including loss of proteostasis, and stem cell exhaustion. Moreover, mitophagy, which plays a major role in mitochondrial maintenance increases in an age-dependent manner [66].
NAD+ plays a fundamental role in the initiation of autophagy and is likely a key regulator in the molecular mechanism of autophagy induction. For instance, the NAD+/SIRT1 signaling pathway stimulates autophagy, and several findings support a crucial role for SIRT1 in autophagy: i) SIRT1 is required for autophagy induction and SIRT1 overexpression increases autophagic flux; ii) the SIRT1 activator resveratrol induces autophagy; and iii) autophagy is required for the lifespan prolonging effect of SIRT1 [13, 106, 107]. Several mechanisms have been implicated in NAD+/SIRT1-mediated autophagy. For instance, the NAD+/SIRT1 pathway may upregulate macro-autophagy through the deacetylation of autophagy proteins (Atgs) including Atg5, Atg7, and Atg8/LC3 [108]. NAD+/SIRT1 may also stimulate autophagy/mitophagy through activation of the AMPK pathway: activated AMPK can upregulate autophagy/mitophagy through the phosphorylation of ULK1, the human autophagy protein 1 (hATG1) [109]. Because of the strong association between NAD+ and AMPK [18, 105, 110], it is likely that NAD+ may also induce autophagy/mitophagy through activation of AMPK. In agreement, the upregulation of the rate-limiting enzyme in NAD+ synthesis, Nampt, induces autophagy through the NAD+/Sirt1 pathway in primary rat cortical neurons [111]. Specifically, NAD+/Sirt1 can activate AMPK in primary rat cortical neurons, which then phosphorylates Tuberous Sclerosis Complex 2 (Tsc2) at Ser1387, leading to inhibition of the autophagy inhibitor mTOR [111]. In addition, Sirt1 can deacetylate Foxo1, leading to upregulation of the autophagic protein Rab7, which mediates late autophagosome-lysosome fusion in murine cardiomyocytes [112]. In addition to NR, another NAD+ precursor, NAM, can also induce mitophagy/autophagy in human fibroblasts, murine cortical neurons and 3xTg AD mice [63, 113].
Recent studies suggest that NR can induce mitophagy in both wild-type mice and C. elegans, and in some DNA repair-deficient premature aging diseases, including XPA, A-T, and CS. Specifically, nuclear DNA damage can induce mitochondrial dysfunction, which could be a common cause of the neurodegenerative phenotypes seen in XPA, A-T, and CS. [14, 39, 93]. We recently performed cross-species studies using C. elegans, mice, and human patient cells and observed increased mitochondrial ROS and accumulation of damaged mitochondria in models of XPA, A-T, and CS [14, 39, 93]. More specifically, in atm-1 C. elegans, NR treatment was shown to ameliorate mitochondrial dysfunction through mitophagy in a SIR2.1-DAF16-DCT-1-dependent manner [39]. The mammalian orthologues of DAF16 and DCT-1 are FOXO3 and NIX (BNIP3L), and this pathway is conserved in mammalian cells as SIRT1-FOXO3-BNIP3L-dependent mitophagy is necessary to protect against hypoxia-induced mitochondrial damage, (revealed in mice) [64]. As mentioned above, NAD+ replenishment can also improve DNA repair, and thus, we suggest that NAD+ likely serves to link nuclear DNA repair and mitochondrial maintenance [35, 108]. Also, NAD+ can stimulate autophagy/mitophagy, which in certain models and across species, can help delay aging and extend longevity.
Clinical Translation
NAD+ precursors can delay aging and counteract a broad spectrum of age-related disease; however, the most important unanswered question is whether their beneficial effects will translate to humans. Preclinical and clinical safety assessments of some NAD+ precursors in mice, rats, and humans have or are being conducted. In mice, there is no detectable toxicity of short-term (500 mg /kg body weight/day for 14 days) [14] or long-term NR treatment (~400 mg/kg/day NR treatment in drinking water for 6 weeks, or 570–590 mg/kg/day for over 10 months) [28, 39]. The dose of 570–590 mg/kg/day in mice is equivalent to 3.19–3.30 g in humans based on weight [114]. Studies in rats show that there are no observed adverse effects of 300 mg/kg/day, and the lowest dose of NR which can induce observed adverse effects was 1000 mg/kg/day with target organs for toxicity assessment being the liver, kidney, ovaries, and testes [115]. Similarly, there is no detectable toxicity of short-term (500 mg/kg/day, i.p. injection for 7 consecutive days) [50] or 12-month long NMN treatment in wild-type C57BL/6N mice (100 mg/kg/day or 300 mg/kg/day) [44]. Consequently, the low toxicity of NAD+ precursors in mammals, may render these good candidates for clinical intervention.
Acipimox is a nicotinic acid analog, and a 2015 report showed that it improved skeletal muscle mitochondrial function in patients with type 2 diabetes [116]. In the first controlled clinical trial of NR, researchers demonstrated that the compound is safe for humans and increased blood levels of NAD+ were detected relative to control subjects [117]. This trial involved six healthy men and six healthy women. Each participant received a single oral dose of 100 mg, 300 mg, or 1,000 mg of NR with a seven-day gap between doses. The data indicate that NR administration increased NAD+ in a dose-dependent manner with no serious side effects at any tested dose [117, 118]. 1000 mg/day of NR taken orally resulted in a 2.7-fold increase of NAD+, and 45-fold increase of the NAD+ intermediate, NAAD [118]. Studies in mice also suggest that NR is more effective at increasing intracellular NAD+ than other NAD+ precursors, such as NAM and NA [118]. In 2016, a follow-up clinical trial was initiated with 140 healthy adults (40–60 year of age), examining the benefits of 8-weeks NR treatment [118]. However, the results are not yet available. Some recent clinical trials are designed to evaluate the efficacy of NAD+ supplementation for the treatment of metabolic and age-related diseases. Over 10 clinical studies have assessed or are currently assessing the safety and efficacy of NR (Box 3). It will be exciting to see whether NAD+ supplementation has any effects on some of these human conditions going forward.
Box 3. Clinician’s Corner.
NAD+ depletion may contribute to a wide spectrum of age-predisposed diseases, including neurodegenerative diseases, muscle atrophy, and progeria. Recent progress in animal studies support the hypothesis that NAD+ replenishment may inhibit metabolic diseases, Alzheimer’s disease, hearing loss, and muscle atrophy, among others. However, further research elucidating the molecular mechanisms of the functions of NAD+ precursor in delaying aging is warranted.
Over 10 clinical studies have or are currently assessing the safety and efficacy of NR in humans. In addition, to a focus on safety (ClinicalTrials.gov identifier number: NCT02678611), pharmacokinetics (NCT02300740, NCT02191462), bioavailability (NCT02712593), there are trials focusing on metabolic disturbance (NCT02689882), aging (NCT02921659, NCT02950441), obesity, diabetes, or coronary artery disease (NCT02835664, NCT02812238, NCT02303483), concussion (NCT02721537) and mild cognitive impairment (NCT02942888). Some of these clinical trials have been completed but have not yet been published. The first NMN clinical trial was launched in Japan in 2016, with a focus on safely and bioavailability of NMN in 10 healthy humans [126]. In addition to NR and NMN, there are some clinical studies on NAM. Based on the benefits of NAM on AD mice [63], a safety study of NAM to treat human AD, 50 participants aged from 50–95, has just ended (NCT00580931). Even though some NAD+ precursors (NR, NMN, niacin/vitamin B3) are available in the market as dietary supplements, results from these clinical studies will determine the broad applications of NAD+ in the aging field.
However, many questions remain. What are the therapeutic doses of NR/NMN needed for different diseases in clinical trials? Does long-term supplementation with NR/NMN have any side effects in humans? If NR/NMN shows clinical benefit, what other clinical studies or combinational drugs should be pursued.
Concluding Remarks
The increasing population of older individuals presents a serious socioeconomic burden for families, societies, and the health care system. Lifestyle interventions, such as a healthy diet, fasting, and exercise are ways to improve health and quality of life [35, 59, 119]. However, not every individual at risk to develop an age-associated ailment may be willing or able to follow these lifestyle interventions. Therefore, the beneficial effects of NAD+ precursors discussed in this review may potentially promote healthy aging and delay various age-related diseases. Especially encouraging is the evidence demonstrating that NAD+ replenishment is beneficial in multiple organs with varying disease conditions. The observations that NAD+ replenishment delays or prevents muscle atrophy, hearing loss and cognitive decline are remarkable. Moreover, preclinical data suggest that NAD+ precursor treatment could be a promising therapeutic strategy to improve clinical characteristics of the AD phenotype. Moreover, NAD+ precursors such as NR and NMN are relatively safe and orally bioavailable. Thus, NAD+ precursors may serve as promising candidates to combat normal aging and age-related disease. However, extensive research is warranted to validate their potential, particularly in humans.
While many studies on NAD+ precursors are ongoing, major questions remain to be addressed (see Outstanding Questions and Box 3). What predictive value does the alteration in NAD+ levels have in normal aging and age-related disorders? Further studies evaluating alterations in cellular NAD+ as a hallmark of aging are necessary. This task requires the development of new technologies to simultaneously detect NAD+ and its metabolites in humans. Moreover, the pleotropic role of NAD+ in human physiology is complex and requires further mechanistic insight. For example, the tissue and sub-cellular specificity of NAD+ precursors need to be carefully evaluated. It is also possible that some unforeseen side-effects may present in certain human populations. Thus, highly stringent and carefully designed clinical trials are necessary to ensure safety. Indeed, a decline in NAD+ concentrations may have numerous roles in human physiology, some of which we have only begun to understand. Some of these roles appear to be important in the aging process and are likely to be important drivers of aging and age-related dysfunction. With careful scientific evaluation, NAD+ replenishment strategies might serve as a promising multi-functional approach to improve the quality of life for an increasingly aged population.
Outstanding questions.
What are the precise molecular mechanisms by which NAD+ acts on mitochondrial homeostasis?
What molecular mechanisms underlie the autophagy/mitophagy-inducing activity of NAD+?
In addition to deacetylation of major autophagic proteins (such as Atg-5, Atg-7, and Atg-8) and upregulation of certain autophagic proteins (e.g., DCT-1), are there other molecular mechanisms whereby NAD+ modulates autophagy/mitophagy?
How are NA and NR transported into the cytosol from the extracellular milieu?
What cell membrane transport does NR use to enter the cells?
Which are the intra-cellular transporters for different NAD+ precursors, especially for NR and NMN?
Supplementary Material
Trends.
Recent discoveries have demonstrated an age-dependent decrease of cellular/tissue NAD+ levels in laboratory animal models. Moreover NAD+ depletion has been linked to multiple hallmarks of aging.
In premature aging animal models, NAD+ levels are decreased, while NAD+ replenishment can improve lifespan and healthspan through DNA repair and mitochondrial maintenance.
Mitochondrial autophagy (mitophagy) plays a major role in clearance of damaged/dysfunctional mitochondria, and compromised mitophagy has been linked to metabolic disorders, neurodegeneration (including Alzheimer’s disease (AD) and Parkinson’s disease (PD)) in addition to aging, and other age-related diseases.
New evidence suggests that NAD+ precursors, such as nicotinamide and nicotinamide riboside, may forestall pathology and cognitive decline in mouse models of AD.
NAD+ supplementation can inhibit multiple aging features in animal models. This highlights essential roles for NAD+ in maintaining healthy aging, and suggest that NAD+ repletion may have broad benefits in humans.
Acknowledgements
We acknowledge the valuable work of the many investigators whose published articles we were unable to cite owing to space limitations. We appreciate personal communications of NAD+ consumption with Dr. Charles M. Brenner. We thank Drs. Rachel Abbotts and Beverly Baptiste for critical reading of the manuscript, and Marc Raley for generation of the figures. This research was supported by the Intramural Research Program of the National Institute on Aging, including a 2015-2016, 2016-2017 NIA intra-laboratory grant (EFF/VAB). EFF was supported by HELSE SOR-OST RHF (Project No: 2017056) and the Research Council of Norway (Project No: 262175). SL was sponsored by The Oticon Foundation, Ib Henriksen’s Foundation, Her Majesty Queen Margrethe II’s Travel Grant and The Velux Foundation.
Glossary
- Nicotinamide adenine dinucleotide
(NAD), major coenzyme/compound in all human cells, exists in oxidized (NAD+) and reduced (NADH) forms. NAD+ plays major roles in cellular energy metabolism, adaptive responses of cells to bioenergetic and oxidative stress, and aging.
- Autophagy
evolutionarily conserved process where cytoplasmic substrates are engulfed in an autophagic vesicle, fused to lysosomes, followed by degradation and recycling. Autophagy is necessary for cellular homeostasis through a balance with apoptosis and inflammation. Compromised autophagy occurs in many age-related diseases.
- Premature aging syndromes (accelerated aging/progeria)
are rare diseases in which patients show aspects of aging at a very early age.
- Cockayne syndrome
rare premature aging disease with progressive neurodegeneration caused predominantly by mutations in genes encoding two DNA repair proteins, Cockayne syndrome group A (CSA) or Cockayne syndrome group B (CSB).
- Xeroderma pigmentosum (XP)
rare autosomal-recessive disorder characterized by severe sun sensitivity and skin cancer. The etiology of XP is caused by mutation of genes encoding a group of DNA repair proteins, XP genes.
- Ataxia telangiectasia
(A-T) rare, untreatable, recessive inherited human disease characterized by severe neuromotor dysfunction, telangiectasia, sterility, cancer, and hypersensitivity to ionizing radiation. It is caused by mutations in the ATM gene, encoding ATM kinase, a regulator of the DNA damage response that is critical for genomic stability, telomere maintenance, and DNA double strand break repair
- Hallmarks of aging
concepts or cellular/organismal processes all influencing and contributing to the process of aging.
- Mitophagy
specialized form of autophagy which regulates the turnover of damaged and dysfunctional mitochondria.
- Caloric restriction
diet where the organism (e.g. mice) are not fed ad libitum. This restriction of food intake has been shown to increase lifespan.
- De novo biosynthesis pathway/kynurenine pathway
pathway from where NAD+ is produced from Tryptophan.
- Preiss-Handler pathway
pathway from where NAD+ is produced from nicotinic acid.
- Salvage pathway
primary pathway from where NAD+ is produced from NR, NMN or NAM.
- APP/PS1 mice
AD model; mice express human Aayloid precursor protein (APP) and human Presilin 1 (PS1). The mice show severe Aβ plaque formation and neurodengeration/cognitive impairments.
- Metabolic syndrome
a group of risk factors and pathologies, including heart disease Diabetes, obesity, stroke, and other.
- PARylation
posttranslational modification performed by PARPs, mainly PARP1 in mammalian cells, via use of NAD+. PARylation is also named Poly(ADP-ribosylation)
- Nucleotide Excision Repair
(NER) DNA repair pathway repairing bulky DNA adducts introduced in DNA by UV irradiation, environmental toxins and certain antitumor agents.
- DNA damage response
cell’s response to DNA damage. DNA damage activates a wide range of cellular processes enabling the cell to survive and maintain genome integrity.
- Aβ plaques
are formed from cleaved APP, called Amyloid beta (Aβ), aggregating to large rafts of proteins, and being toxic to the neurons.
- Tau tangles
are rafts or aggregates of Tau proteins, often hyperphosphorylated, causing dysfunction and cell death of affected neurons.
- 3xTg AD mice
AD model. The mice express the human genes APP and PS1 as the APP/PS1 mouse, and also human Tau. The mice show AD-like phenotypes, similar to APP/PS1 mice, including tau tangles and fibrils in the brain.
- Parkin
E3 ubiquitin ligase; mutations in PARKIN, together with PINK1, (a serine/threonine kinase), are a leading cause of PD. PARKIN and PINK1 are also involved in the process of mitophagy.
- Duchenne’s muscular dystrophy
genetic disorder caused by the lack of dystrophin, which results in muscle weakness and degeneration.
- Nampt−/− mice
do not express NAMPT, a key protein in the NAD+ salvage pathway. They show embryonic lethality.
- γ-H2AX foci
occur when the histone H2A variant H2AX is being phosphorylated rapidly after a double strand break induction in DNA. Because the phosphorylation of H2AX occurs rapidly after induction of DSBs and correlates well with DSBs, it is often used as a DSB marker.
- Homologous recombination (HR)-based DSBR
repair process of double strand breaks by the repair pathway HR. HR is dependent on the cell cycle.
- Non-homologous end joining (NHEJ)-based DSBR
is the only DSBR pathway in post replicative cells, including neurons.
- Mdx mouse model
model of Duchenne’s muscle dystrophy with a phenotype resembling that of human patients, including muscle weakness and degeneration.
- KK/H1J mouse model
often used to study metabolic syndromes because it presents inherited glucose intolerance and insulin resistance, which result in hyperglycemia. KK/HlJ mice have a strong tendency to develop type 2 diabetes (T2D) in response to certain dietary regimens (eg, high-fat diet) and aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer
The Bohr laboratory has CRADA arrangements with ChromaDex and GlaxoSmithKline.
REFERENCES
- 1.Nobel Lectures C−. (1966. (https://www.nobelprize.org/nobel_prizes/chemistry/laureates/1907/buchner-facts.html)) Eduard Bunchner-Facts, Elsevier Publishing Company. [Google Scholar]
- 2.Harden A and Young WJ (1906) The alcoholic ferment of yeast-juice. Part II.-The coferment of yeast-juice. Proc. R. Soc. Lond. B (78), 369–375. [Google Scholar]
- 3.James LK (1993) Nobel Laureates in Chemistry 1901–1992, Merck& Co., Inc.. [Google Scholar]
- 4.P.D. B(1970) The enzymes (Third edition), Academic Press. [Google Scholar]
- 5.Kornberg A and Pricer WE Jr. (1950) On the structure of triphosphopyridine nucleotide. J Biol Chem 186 (2), 557–67. [PubMed] [Google Scholar]
- 6.Euler-Chelpin HV (1930) Fermentation of sugars and fermentative enzymes Nobel Lecture. [Google Scholar]
- 7.Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12 (10), 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verdin E (2015) NAD+ in aging, metabolism, and neurodegeneration. Science In press. [DOI] [PubMed] [Google Scholar]
- 9.Canto C et al. (2015) NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 22 (1), 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalkiadaki A and Guarente L (2015) The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. [DOI] [PubMed] [Google Scholar]
- 11.Bonkowski MS and Sinclair DA (2016) Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17 (11), 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerr JS et al. (2017) Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci 40 (3), 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubinsztein DC et al. (2011) Autophagy and aging. Cell 146 (5), 682–95. [DOI] [PubMed] [Google Scholar]
- 14.Fang EF et al. (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157 (4), 882–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belenky P et al. (2007) Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129 (3), 473–84. [DOI] [PubMed] [Google Scholar]
- 16.Yang H et al. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130 (6), 1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cambronne XA et al. (2016) Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352 (6292), 1474–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canto C et al. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458 (7241), 1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uddin GM et al. (2016) Head to Head Comparison of Short-Term Treatment with the NAD(+) Precursor Nicotinamide Mononucleotide (NMN) and 6 Weeks of Exercise in Obese Female Mice. Front Pharmacol 7, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell SJ et al. (2016) Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab 23 (6), 1093–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukuwatari T et al. (2001) Elevation of blood NAD level after moderate exercise in young women and mice. J Nutr Sci Vitaminol (Tokyo) 47 (2), 177–9. [DOI] [PubMed] [Google Scholar]
- 22.Verdin E (2015) NAD(+) in aging, metabolism, and neurodegeneration. Science 350 (6265), 1208–13. [DOI] [PubMed] [Google Scholar]
- 23.Conforti L et al. (2011) Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. Febs j 278 (15), 2666–79. [DOI] [PubMed] [Google Scholar]
- 24.Zhai RG et al. (2008) NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452 (7189), 887–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilley J and Coleman MP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8 (1), e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Revollo JR et al. (2007) Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 6 (5), 363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai S and Yoshino J (2013) The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab 15 Suppl 3, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frederick DW et al. (2016) Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab 24 (2), 269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa T and Guarente L (2011) Sirtuins at a glance. J Cell Sci 124 (Pt 6), 833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramsey KM et al. (2008) Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7 (1), 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai S and Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24 (8), 464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouleau M et al. (2010) PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10 (4), 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aksoy P et al. (2006) Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345 (4), 1386–92. [DOI] [PubMed] [Google Scholar]
- 34.Adebanjo OA et al. (1999) A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol 1 (7), 409–14. [DOI] [PubMed] [Google Scholar]
- 35.Fang EF et al. (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17 (5), 308–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escande C et al. (2013) Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62 (4), 1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai P et al. (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13 (4), 461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai P et al. (2011) PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab 13 (4), 450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang EF et al. (2016) NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab 24 (4), 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blacher E et al. (2015) Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann Neurol 78 (1), 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbosa MT et al. (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J 21 (13), 3629–39. [DOI] [PubMed] [Google Scholar]
- 42.Long A et al. (2017) CD38 Knockout Mice Show Significant Protection Against Ischemic Brain Damage Despite High Level Poly-ADP-Ribosylation. Neurochem Res 42 (1), 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camacho-Pereira J et al. (2016) CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab 23 (6), 1127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mills KF et al. (2017) Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mouchiroud L et al. (2013) The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154 (2), 430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. [DOI] [PubMed] [Google Scholar]
- 47.Zhu XH et al. (2015) In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 112 (9), 2876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massudi H et al. (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7 (7), e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balan V et al. (2008) Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem 283 (41), 27810–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes AP et al. (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155 (7), 1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshino J et al. (2011) Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 14 (4), 528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugitani N et al. (2016) XPA: A key scaffold for human nucleotide excision repair. DNA Repair (Amst) 44, 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.States JC et al. (1998) Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat 12 (2), 103–13. [DOI] [PubMed] [Google Scholar]
- 54.Karikkineth AC et al. (2017) Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev 33, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehmann AR (1982) Three complementation groups in Cockayne syndrome. Mutat Res 106 (2), 347–56. [DOI] [PubMed] [Google Scholar]
- 56.Scheibye-Knudsen M et al. (2014) A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20 (5), 840–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.North BJ et al. (2014) SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. Embo j 33 (13), 1438–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Otin C et al. (2013) The hallmarks of aging. Cell 153 (6), 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kerr JS et al. (2017. In press) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong B et al. (2013) Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol Aging 34 (6), 1581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long AN et al. (2015) Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol 15, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X et al. (2016) Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res 1643, 1–9. [DOI] [PubMed] [Google Scholar]
- 63.Liu D et al. (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34 (6), 1564–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kume S et al. (2010) Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120 (4), 1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng L et al. (2017) Parkin functionally interacts with PGC-1alpha to preserve mitochondria and protect dopaminergic neurons. Hum Mol Genet. [DOI] [PubMed] [Google Scholar]
- 66.Palikaras K et al. (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521 (7553), 525–8. [DOI] [PubMed] [Google Scholar]
- 67.Menzies FM et al. (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16 (6), 345–57. [DOI] [PubMed] [Google Scholar]
- 68.Scarffe LA et al. (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37 (6), 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Birkmayer JG et al. (1993) Nicotinamide adenine dinucleotide (NADH)--a new therapeutic approach to Parkinson’s disease. Comparison of oral and parenteral application. Acta Neurol Scand Suppl 146, 32–5. [PubMed] [Google Scholar]
- 70.Lu L et al. (2014) Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp Ther Med 8 (3), 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lehmann S et al. (2016) Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol Open. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hammer MS et al. (2014) Environmental noise pollution in the United States: developing an effective public health response. Environ Health Perspect 122 (2), 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown KD et al. (2014) Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab 20 (6), 1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Someya S et al. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143 (5), 802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim HJ et al. (2015) Nicotinamide adenine dinucleotide: An essential factor in preserving hearing in cisplatin-induced ototoxicity. Hear Res 326, 30–9. [DOI] [PubMed] [Google Scholar]
- 76.Kim HJ et al. (2014) Augmentation of NAD(+) by NQO1 attenuates cisplatin-mediated hearing impairment. Cell Death Dis 5, e1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryu D et al. (2016) NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med 8 (361), 361ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noseworthy JH et al. (2000) Multiple sclerosis. N Engl J Med 343 (13), 938–52. [DOI] [PubMed] [Google Scholar]
- 79.Shindler KS et al. (2007) SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci 48 (8), 3602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gujar AD et al. (2016) An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci U S A 113 (51), E8247–e8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu W et al. (2015) Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep 13 (3), 533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Picciotto NE et al. (2016) Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15 (3), 522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goodpaster BH et al. (2006) The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61 (10), 1059–64. [DOI] [PubMed] [Google Scholar]
- 84.Canto C et al. (2012) The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15 (6), 838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gariani K et al. (2016) Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63 (4), 1190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee HJ et al. (2015) Nicotinamide Riboside Ameliorates Hepatic Metaflammation by Modulating NLRP3 Inflammasome in a Rodent Model of Type 2 Diabetes. J Med Food 18 (11), 1207–13. [DOI] [PubMed] [Google Scholar]
- 87.Weissman L et al. (2007) Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 35 (16), 5545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maynard S et al. (2015) DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb Perspect Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.White RR and Vijg J (2016) Do DNA Double-Strand Breaks Drive Aging? Mol Cell 63 (5), 729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riklis E et al. (1988) Increased radioprotection attained by DNA repair enhancement. Pharmacol Ther 39 (1–3), 311–22. [DOI] [PubMed] [Google Scholar]
- 91.Watson A et al. (2017) Nicotinamide Phosphoribosyltransferase in Smooth Muscle Cells Maintains Genome Integrity, Resists Aortic Medial Degeneration and Is Suppressed in Human Thoracic Aortic Aneurysm Disease. Circ Res. [DOI] [PubMed] [Google Scholar]
- 92.Weidele K et al. (2017) The NAD+ precursor nicotinic acid improves genomic integrity in human peripheral blood mononuclear cells after X-irradiation. DNA Repair (Amst) 52, 12–23. [DOI] [PubMed] [Google Scholar]
- 93.Scheibye-Knudsen M et al. (2014) A High Fat Diet and NAD+ Rescue Premature Aging in Cockayne Syndrome. Cell Metab 20 (5), 840–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shiloh Y and Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14 (4), 197–210. [PubMed] [Google Scholar]
- 95.Sharma NK et al. (2014) Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia. DNA Repair (Amst) 13, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iyama T and Wilson DM 3rd (2013) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12 (8), 620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fang EF et al. (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cerutti R et al. (2014) NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 19 (6), 1042–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu Z et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98 (1), 115–24. [DOI] [PubMed] [Google Scholar]
- 100.Marino G et al. (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15 (2), 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matecic M et al. (2010) A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet 6 (4), e1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toth ML et al. (2008) Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4 (3), 330–8. [DOI] [PubMed] [Google Scholar]
- 103.Lee JH et al. (2010) Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327 (5970), 1223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deretic V et al. (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13 (10), 722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Han X et al. (2016) AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 15 (3), 416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee IH et al. (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105 (9), 3374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morselli E et al. (2011) Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 192 (4), 615–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fang EF and Bohr VA (2016) NAD+: The convergence of DNA repair and mitophagy. Autophagy, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Egan DF et al. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331 (6016), 456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lettieri Barbato D et al. (2013) FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis 4, e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang P et al. (2012) Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8 (1), 77–87. [DOI] [PubMed] [Google Scholar]
- 112.Hariharan N et al. (2010) Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 107 (12), 1470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jang SY et al. (2012) Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem 287 (23), 19304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fang EF et al. (2016) NAD+ replenishment improves lifespan and healthspan in Ataxia telangiectasia models via mitophagy and DNA repair. Cell Metabolism 24 (4), 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Conze DB et al. (2016) Safety assessment of nicotinamide riboside, a form of vitamin B3. Hum Exp Toxicol. [DOI] [PubMed] [Google Scholar]
- 116.van de Weijer T et al. (2015) Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 64 (4), 1193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Trammell SA et al. (2016) Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7, 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.ChromaDex, ChromaDex Initiates Second Human Clinical Study on NIAGEN® -The World’s First and Only Commercially Available Form of Nicotinamide Riboside (NR), in: ChromaDex (Ed.) ChromaDex, 2016. [Google Scholar]
- 119.Longo VD and Mattson MP (2014) Fasting: molecular mechanisms and clinical applications. Cell Metab 19 (2), 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao Y et al. (2015) SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab 21 (5), 777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ying W et al. (2005) NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res 79 (1–2), 216–23. [DOI] [PubMed] [Google Scholar]
- 122.Bieganowski P and Brenner C (2004) Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117 (4), 495–502. [DOI] [PubMed] [Google Scholar]
- 123.Bogan KL and Brenner C (2008) Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 28, 115–30. [DOI] [PubMed] [Google Scholar]
- 124.Nikiforov A et al. (2011) Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 286 (24), 21767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Berger F et al. (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280 (43), 36334–41. [DOI] [PubMed] [Google Scholar]
- 126.Tsubota K (2016) The first human clinical study for NMN has started in Japan. Aging and Mechanisms of Disease 2 (16021), 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.