

Abstract
PRMT5, which regulates gene expression by symmetric dimethylation of histones and non-histone target proteins, is overexpressed and plays a pathogenic role in many cancers. In diffuse large B cell lymphoma (DLBCL), the mechanisms of PRMT5 dysregulation and its role in lymphomagenesis remain largely unknown. Here we demonstrate that B cell receptor (BCR) signaling regulates PRMT5 expression in DLBCL cells. Immunohistochemical analysis reveals elevated levels of PRMT5 expression in DLBCL cases and in germinal center (GC) B cells when compared to naive B cells. PRMT5 can be induced in naive B cells by BCR stimulation. We discovered that BTK-NF-κB signaling induces PRMT5 transcription in activated B cell-like (ABC) DLBCL cells while BCR downstream PI3K-AKT-MYC signaling upregulates PRMT5 expression in both ABC and GCB DLBCL cells. PRMT5 inhibition inhibits the growth of DLBCL cells in vitro and patient derived xenografts. Genomic and biochemical analysis demonstrate that PRMT5 promotes cell cycle progression and activates PI3K-AKT signaling, suggesting a feedback regulatory mechanism to enhance cell survival and proliferation. Co-targeting PRMT5 and AKT by their specific inhibitors is lethal to DLBCL cell lines and primary cancer cells. Therefore, this study provides a mechanistic rationale for clinical trials to evaluate PRMT5 and AKT inhibitors for DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma arising from germinal center (GC) or post-GC center B cells1, 2. DLBCL includes two main molecular subtypes, termed activated B cell-like (ABC) and GC B cell-like (GCB), which demonstrate distinct biological and genetic characteristics and different clinical outcomes3–5. In more aggressive ABC DLBCL, NF-κB is constitutively activated by a variety of genetic alterations6–13, including somatic mutations targeting components of the B cell receptor (BCR) and Toll-like receptor (TLR) signaling pathways. For example, MYD88 mutations (mainly L265P) are present in ~40% of ABC DLBCL tumors, which promote cell survival by activating the NF-κB pathway and inducing production of IL-6 and/or IL-109. The NF-κB pathway can also be engaged by gain-of-function mutations of the BCR components CD79A and CD79B11 and the downstream signaling adaptor CARD1114. The active form of BCR signaling is required for the fitness of ABC DLBCL cells11, 15. BTK, a key component of the early BCR signaling pathway, is an effective drug target and its inhibitor ibrutinib has been used for the treatment of ABC DLBCL16, 17.
In GCB DLBCL, there are no highly recurrent mutations in the BCR signaling and NF-κB pathways. Rather, GCB DLBCL cells use antigen-independent tonic BCR signaling through the PI3K/AKT signaling pathway to promote their survival, similar to Burkitt lymphoma cells18, 19. PTEN, a negative regulator of PI3K, is lost in its expression in more than 50% of cases by a number of mechanisms including deletion, mutation, and amplification of the miR17–92 microRNA cluster20. One of the downstream targets of the PI3K pathway is MYC as re-expression of PTEN or inhibition of PI3K/AKT signaling in PTEN deficient cells reduces MYC expression20, 21. Targeting the PI3K signaling pathway has emerged as a therapeutic strategy in DLBCL22.
Arginine methylation is a common posttranslational modification that governs important cellular processes and impacts development, cell growth, proliferation, and differentiation23. Arginine methylation is catalyzed by protein arginine methyltransferases (PRMTs), which are classified as type I and type II enzymes responsible for the formation of asymmetric and symmetric dimethylarginine, respectively24. PRMT5 is the main type II enzyme that catalyzes symmetric dimethylarginine of histone proteins to induce gene silencing by generating repressive histone marks, such as H2AR3me2s, H3R8me2s, and H4R3me2s25–29. These histone modifications facilitate PRMT5 to form transcriptional repressive complexes, including those containing SIN3A/HDAC, MBD2/NURD, N-CoR/SMRT and DNMT3A29. PRMT5 can also methylate nonhistone proteins such as the transcription factors p53, E2F1 and p6530–32. PRMT5 deficiency leads to embryonic lethality due to the abrogation of pluripotent cells in mouse blastocysts33. PRMT5 expression is required for normal adult hematopoiesis in a PRMT5 conditional knockout mouse model34. A recent elegant biochemical and genetic study has demonstrated that PRMT5 methylates BCL6, regulates expression of BCL6 target genes, and therefore contributes to GC formation35.
A growing literature demonstrates a critical role of PRMT5 in tumorigenesis36–42. PRMT5 expression is upregulated in various cancers, including mantle cell lymphoma and DLBCL43–46. PRMT5 upregulation is associated with Epstein-Barr virus (EBV) infection41. Viral latent membrane protein 1 (LMP1) induces PRMT5 expression by driving the formation of an NF-κB suppressive complex, which inhibits transcription of the PRMT5 inhibitory microRNA9641. Given that less than 10% of DLBCL are EBV-positive47, the mechanisms underlying PRMT5 expression in DLBCL are still largely unknown.
Here, we investigated the role of BCR signaling in regulating PRMT5 expression in DLBCL. In both ABC and GCB DLBCL cells, the PI3K-AKT signaling pathway contributes to PRMT5 overexpression. Additionally, active BCR-BTK-NF-κB signaling in ABC DLBCL cells also upregulates PRMT5 expression. Using genetic and pharmacological approaches, we demonstrated that PRMT5 expression is required for the survival and proliferation of DLBCL cells in vitro and in vivo. We also revealed that co-targeting PRMT5 and AKT by their specific inhibitors synergistically inhibits the growth of DLBCL cell lines and primary cancer cells.
Methods
Cell lines and culture.
Doxycycline-inducible human DLBCL cell lines (HBL1, TMD8, U2932, SUDHL2, OCI-Ly10, Toledo, OCI-Ly7, SUDHL4, K422 and OCI-Ly7) that express the bacterial tetracycline repressor were engineered as described previously48. Doxycycline (20 ng/ml) was used for inducing the expression of genes of interest.
Gene knockout with an inducible 2-vector CRISPR-Cas9 knockout system.
pRSGT16-U6Tet-sg-HTS6C-CMV-TetRep-2A-TagRFP-2A-Puro (Cellecta) was used for generating sgRNA targeting specific gene and pR-CMV-Cas9–2A-Hygro (Cellecta) for generating Cas9-expressing cells.
Xenografts.
Male and female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) breeder pairs were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred under specific pathogen-free conditions in sterile ventilated racks in the animal care facility at the University of Wisconsin-Madison. All animal protocols were approved by the Animal Care and Use Committee. Tumor measurements were recorded 3 times a week, and volumes were calculated as previously described49. When mice became moribund or when tumors exceeded 20 mm in any direction, mice were euthanized as required by institutional protocols.
DLBCL patient derived xenografts (PDX).
DLBCL PDX model was established as described previously50. All experimental procedures and protocols (the animal IACUC protocol # 00001260 and the tissue collection protocol# Lab11–0342) were approved by the Institutional Animal Care and Use Committee and Institutional Review Boards of The University of Texas MD Anderson Cancer Center. Briefly, 10-week-old male NSG mice were housed in the animal research facility. 5 × 106 freshly isolated DLBCL cells were directly injected into the fetal bone chip of NSG-hu mice after the mice were anesthetized with 5% isoflurane vaporizer. Once tumor growth was detected in the first generation, tumor mass was monitored and then passaged. The passaged tumor equally grew and mice were assigned as 5 mice/group for in vivo treatment.
RNA-seq analysis.
Total RNA was extracted using RNeasy plus mini kit (Qiagen) according to the manufacturer’s protocol. RNA-seq libraries were prepared by using the Illumina TruSeq stranded mRNA LT sample preparation kit (Illumina). Sequencing was performed on Illumina Hiseq 2500 at 50-bp length. For the RNA analysis, raw reads were mapped to the human reference genome (UCSC hg19) by HISAT2 (v2.1), and differential expression analysis was done by StringTie (v1.3.4) and Ballgown51. Gene ontology analysis was performed by Panther Classification System (http://pantherdb.org/). Gene Set Enrichment Analysis (GSEA) was performed by GSEA software (V2.0) (http://software.broadinstitute.org/gsea/index.jsp). For the GSEA analysis, molecular signatures databases h.all.v5.2 symbols.gmt was used. RNAseq data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus and are accessible through GEO Series accession number: GSE115136.
Statistical analysis.
Two-tailed Student’s t-test was used to determine a significant difference. Results were presented as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, and ***P < 0.001 were used to show statistical significance.
Full details of the methods used and data analysis are presented in Supplementary Materials.
Results
PRMT5 expression is elevated in DLBCL and germinal center B cells
To determine the level of PRMT5 expression in DLBCL cells and compare it with normal B cells, we isolated naive B cells from human tonsils. Immunoblot analysis of naive B cells and several ABC and GCB DLBCL cell lines revealed that levels of PRMT5 expression were higher in DLBCL cell lines than that in naive B cells (Figure 1A). Consistent with these results, immunohistochemical (IHC) analysis of human tonsils and a tissue microarray of DLBCL patient samples demonstrated that PRMT5 expression in both Non-GCB and GCB DLBCL and germinal center B cells is elevated compared with non-tumor tissues and naive B cells (Figure 1B, 1C). However, there is no statistically significant difference in PRMT5 expression between the two subtypes of DLBCL. We also observed increased PRMT5 expression in mantle cell lymphoma patient samples when compared with normal lymph nodes (Figure S1A). The level of PRMT5 expression was relatively low in Burkitt lymphoma cell lines and Hodgkin lymphoma cell lines but similar in Jurkat T cells when compared with the GCB DLBCL cell line OCI-Ly1 (Figure S1B).
Figure 1. PRMT5 expression in DLBCL cell lines and tissues.
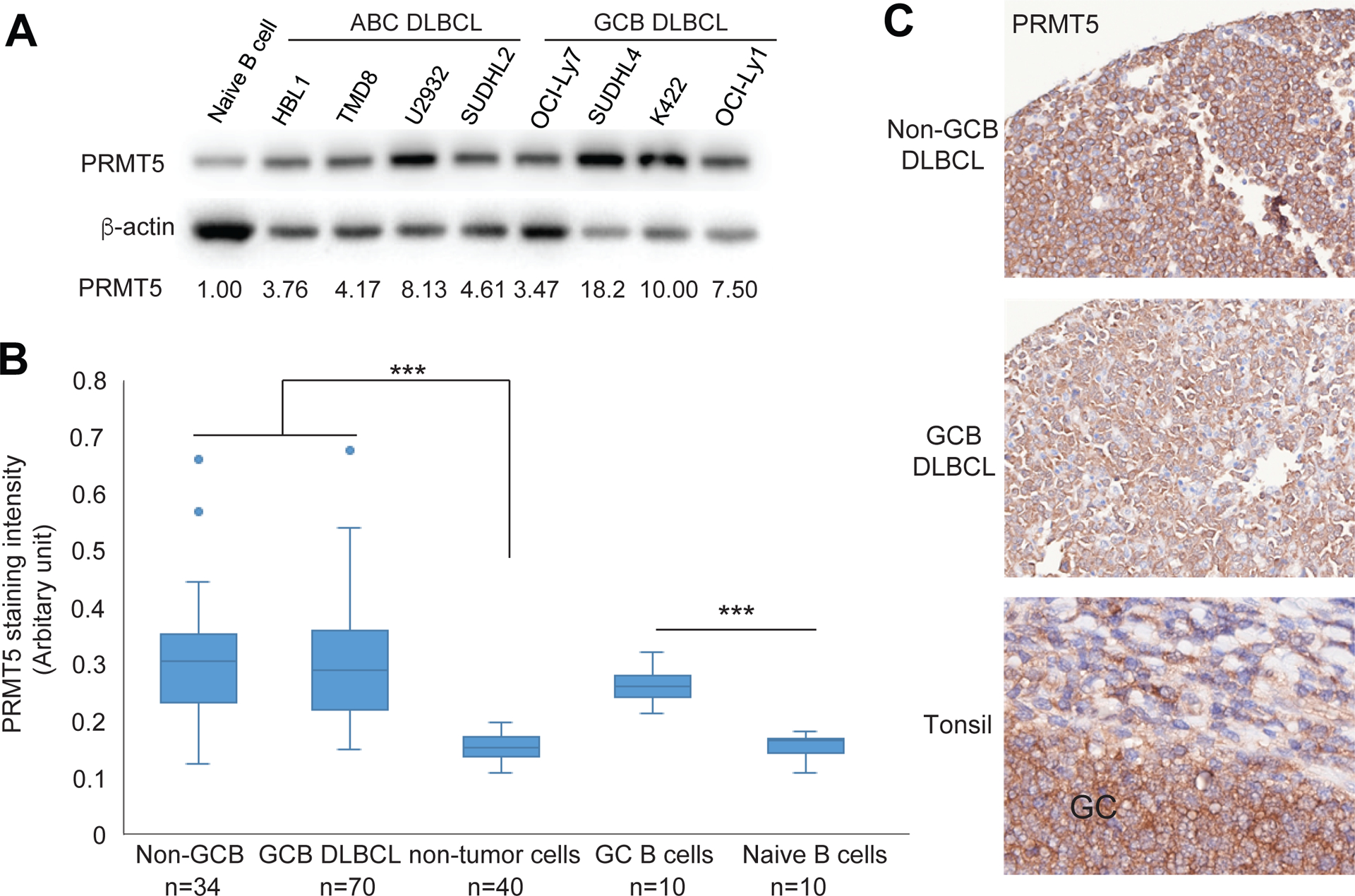
(A) Immunoblot analysis of PRMT5 expression in naive B cells and the indicated DLBCL cell lines. β-actin served as a loading control. (B) Immunohistochemical analysis of PRMT5 expression in DLBCL tumor tissues and human tonsils, analyzed with InformTM advanced image analysis software (student’s t-test, ***p<0.001). (C) Representative images of PRMT5 expression in a GCB DLBCL, a Non-GCB DLBCL, and a human tonsil (original magnification X400).
B cell receptor (BCR)-mediated canonical NF-κB pathway drives PRMT5 expression in ABC DLBCL cells
The above IHC analysis demonstrating increased PRMT5 expression in antigen experienced GC B cells prompted us to investigate the role of BCR signaling in regulating PRMT5 expression. Immunoblot analysis revealed higher levels of PRMT5 expression in tonsillar B cells than of naive B cells (Figure 2A). Notably, when BCR was engaged by IgM antibody in naive B cells, expression of PRMT5 along with IRF4, a known BCR downstream target, was significantly increased (Figure 2B). This result was further supported by IHC analysis, which showed a positive expression correlation of PRMT5 with BTK, a key component of the early BCR pathway (Figure 2C). To knock out BTK, we used an inducible CRISPR/Cas9 system that was created for our very recent study52. We induced BTK sgRNA expression with 20 ng/ml doxycycline in the ABC DLBCL cell line TMD8 and found a time-dependent reduction in BTK expression and PRMT5 expression (Figure S2A). After 6 days of BTK sgRNA induction, we observed a significant reduction of PRMT5 expression in another ABC DLBCL cell line HBL1 when compared with sgRNA uninduced control cells (Figure 2D), suggesting that PRMT5 is a downstream target of BTK. BCR-BTK mediated activation of the canonical NF-κB pathway is characteristic of the ABC subtype and an important pro-survival pathway in ABC DLBCL. Because this NF-κB pathway is not activated in GCB DLBCL, not surprisingly, BTK knockout did not change PRMT5 expression in two GCB DLBCL cell lines (Figure S2B).
Figure 2. PRMT5 expression driven by B cell antigen receptor signaling.
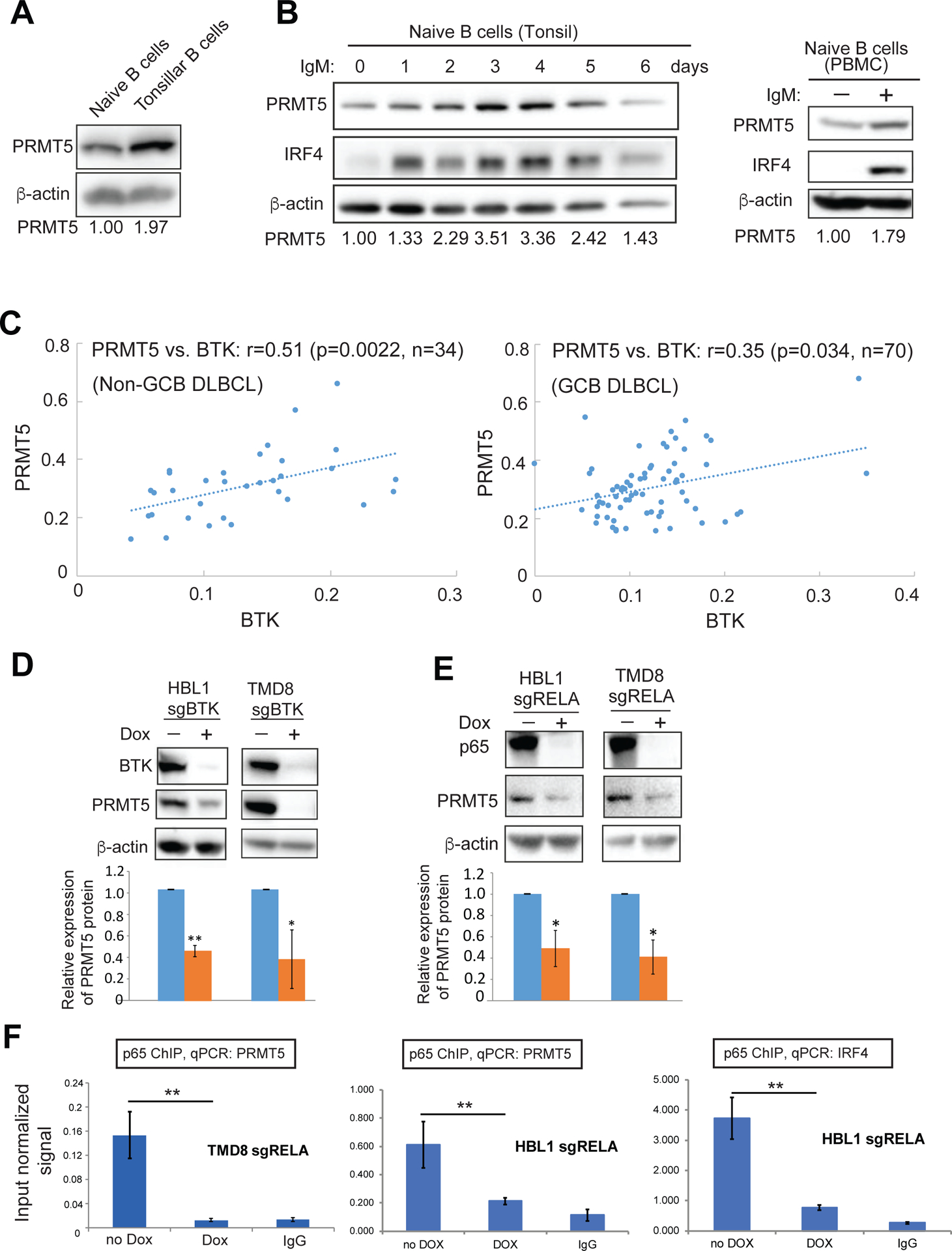
(A) Immunoblot analysis of PRMT5 expression in naive B cells and total tonsillar B cells isolated from the same human tonsil. (B) Immunoblot analysis of IgM antibody stimulated naive B cells from tonsils (left panel) and peripheral blood (right panel). (C) Correlation of protein expression by immunohistochemical analysis between BTK and PRMT5 in 104 DLBCL cases (34 Non-GCB DLBCL, 70 GCB DLBCL). (D) PRMT5 expression after knockout of BTK by sgRNA. BTK sgRNA was induced for expression with 20 ng/ml doxycycline for 6 days before immunoblot analysis. Error bars represent mean ± SD (*p<0.05, **p<0.01, n=3). (E) PRMT5 expression after knockout of p65 by sgRNA. p65 sgRNA was induced for expression with 20 ng/ml doxycycline for 7 days before immunoblot analysis. Error bars represent mean ± SD (*p<0.05, n=3). (F) p65 ChIP and qPCR for PRMT5 in DLBCL cells without or with 6 days of p65 sgRNA induction. IgG antibody served as a control. Error bars represent mean ± SD (**p<0.01, N =3).
Next, we used the above CRISPR-Cas9 system to knockout RelA/p65, which is an important component in the canonical NF-κB pathway and a critical transcription factor downstream of BTK6. Knockout of p65 led to a reduction in PRMT5 expression in TMD8 and HBL1 cells (Figure 2E), which was in a time-dependent manner (Figure S2C). Decreased PRMT5 expression by p65 sgRNA was not due to general cytotoxicity because the sgRNA expressing cells were viable after 6 days of induction (Figure S2D). The ENCODE data (GEO:GSM935478) showed that there are p65 binding sites around the transcription start site of PRMT5 in human lymphoblastoid cell line GM12878. For ChIP analysis, we designed primers around that region and indeed found p65 enrichment within the PRMT5 locus (−196, +3) in two ABC DLBCL cell lines (Figure 2G). PRMT5 was reduced in p65 knockout cells (Figure 2F). As expected, enrichment of the p65 target gene IRF4 was also reduced in p65 knockout cells (Figure 2F). Taken together, the results suggest that active BCR/BTK signaling contributes to upregulation of PRMT5 expression in ABC DLBCL cells.
Upregulation of PRMT5 expression by BCR-mediated PI3K/AKT signaling in both ABC and GCB DLBCL cells
In GCB DLBCL, NF-κB is not activated. Rather, GCB DLBCL cells use antigen-independent tonic BCR signaling through the PI3K/AKT signaling pathway to promote their survival53,18. The PI3K/AKT signaling pathway is also activated in ABC DLBCL cells due to the chronic antigen-dependent BCR signaling20, 21. MYC is a downstream target of this signaling pathway in DLBCL20, 21. AKT induces GSK3β phosphorylation, which leads to GSK3β autoinhibition54. Reduced GSK3β activity then stabilizes MYC protein since GSK3β phosphorylates MYC and facilitates MYC rapid proteolysis by the ubiquitin pathway55. A recent Eμ-myc mouse genetic study demonstrated that PRMT5 is a direct transcriptional target of MYC44.
Based on these findings, we hypothesized that the BCR-PI3K/AKT-MYC pathway upregulates the expression of PRMT5 in DLBCL. To test this hypothesis, we treated GCB and ABC DLBCL cell lines with the selective AKT inhibitor AZD536321. Indeed, AKT inhibition by AZD5363 diminished phosphorylation of GSK3β, a direct downstream target of AKT, and reduced expression of both MYC and PRMT5 in all six cell lines (Figure 3A, left panel). AZD5363 mediated reduction in expression of MYC and PRMT5 did not result from general cytotoxicity based on our cell apoptosis assay (Figure S3). Interestingly, a similar result was obtained in normal B cells when stimulated with an-IgM (Figure 3A, right panel), suggesting that PRMT5 regulation by the BCR-PI3K/AKT-MYC pathway is a general mechanism. MYC-mediated regulation of PRMT5 was confirmed by MYC knockout strategy in 2 ABC DLBCL cell lines (HBL1 and TMD8) and 2 GCB DLBCL cell lines (OCI-Ly1 and SUDHL4). Our time course experiments showed significant reduction in MYC expression after 5–6 days of MYC sgRNA induction (Figure S2E), which did not significantly reduce cell viability (Figure S2D). Indeed, PRMT5 expression significantly decreased in the cells expressing the MYC sgRNA (Figure 3B). We next performed MYC ChIP analysis using a pair of primers within the promoter region of PRMT5 (−321, −124), according to ENCODE data (GEO:GSM822290) in the human lymphoblastoid cell line GM12878. We found an enrichment of MYC on this promotor region of PRMT5 (Figure 3C). This is MYC specific since the enrichment signal was significantly diminished in MYC knockout cells (Figure 3C). Together, these data suggest PI3K/AKT signaling regulates PRMT5 expression through the AKT-GSK3β-MYC axis in both ABC and GCB DLBCL cells as well as in normal IgM activated B cells.
Figure 3. PI3K-AKT-MYC signaling regulates PRMT5 expression in DLBCL.
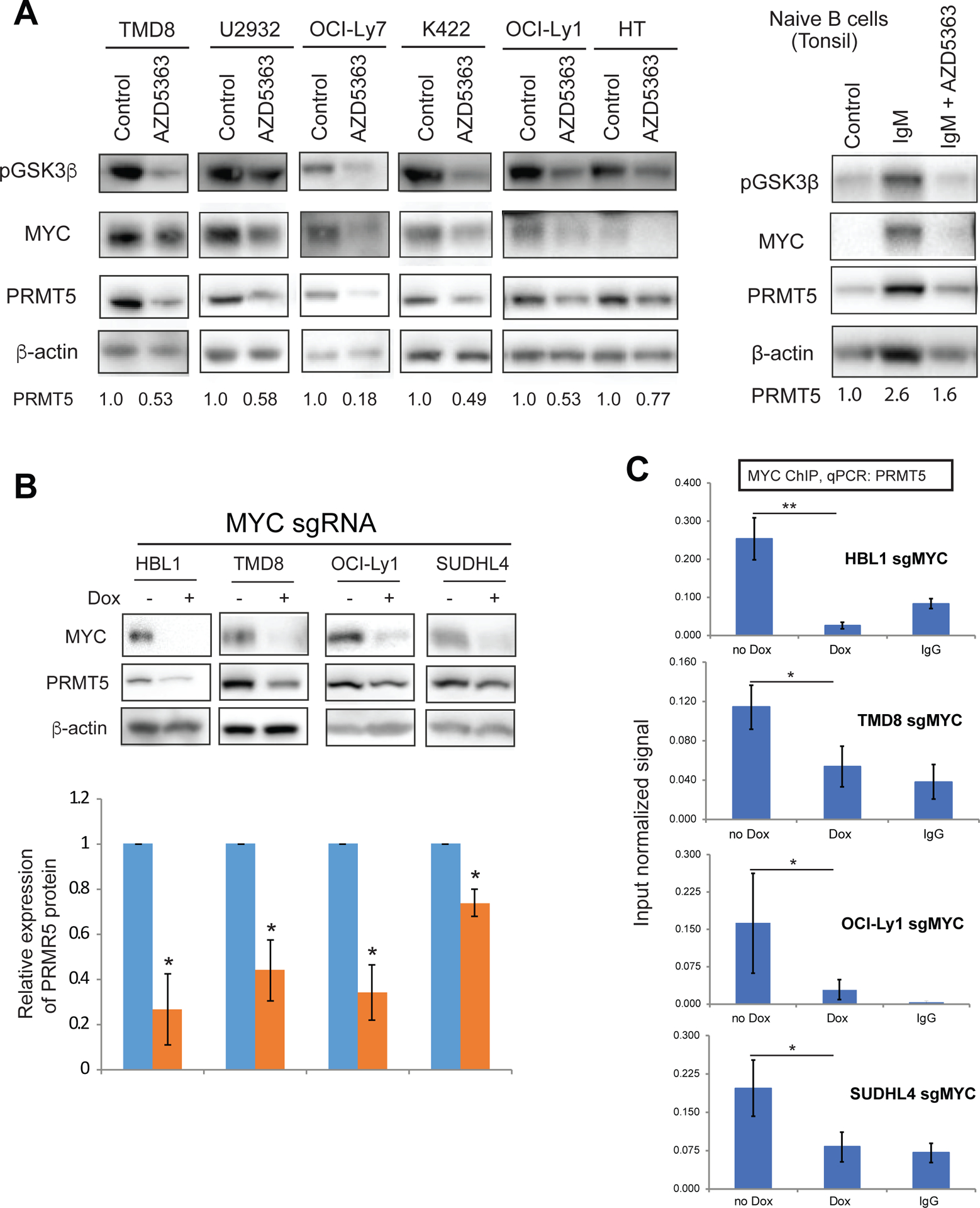
(A, left panel) Eexpression of phospho-GSK3β, MYC and PRMT5 by immunoblot analysis after 3-day treatment with 1 μM AKT inhibitor AZD5363 on 2 ABC DLBCL cell line (TMD8 and U2932) and 4 GCB DLBCL cell lines (OCI-Ly7, K422, OCI-Ly1 and HT). β-actin served as a loading control. Data are representative of 3 independent experiments. (A, right panel) Eexpression of phospho-GSK3β, MYC and PRMT5 by immunoblot analysis after 3-day treatment with 1μM AKT inhibitor AZD5363 on naive B cells when stimulated with 10 μg/ml anti-IgM. β-actin served as a loading control. Data are representative of 2 independent experiments. (B) PRMT5 expression after 6 days of MYC sgRNA induction with 20 ng/ml doxycycline. Error bars represent mean ± SD (*p<0.05, n=3). (C) MYC ChIP and qPCR for PRMT5 in DLBCL cells without or with 6 days of p65 sgRNA induction. IgG antibody served as a control. Error bars represent mean ± SD (N =3).
PRMT5 inhibition by its selective inhibitor EPZ015666 or its sgRNA inhibits the growth of DLBCL cells
To test whether highly expressed PRMT5 contributes to cancer cell survival, we used the selective PRMT5 inhibitor EPZ015666, which has previously showed significant efficacy in growth inhibition of mantle cell lymphoma cells both in vitro and in vivo43. We treated 4 ABC DLBCL and 4 GCB DLBCL cell lines with this inhibitor and observed functional impairment of PRMT5 because of diminished H4R3 symmetric dimethylation (Figure 4A), which subsequently led to a significant reduction in cell viability in all of the DLBCL cell lines tested (Figure 4B). For comparison, we also tested 4 mantle cell lymphoma cell lines in the same experimental setting and found two sensitive cell lines (Marver-1 and Z138) and two insensitive cell lines (Granta-519 and Jeko) (Figure 4B). We also confirmed insensitivity of Granta-519 and Jeko cell lines to PRMT5 inhibition with an extended observation time (Figure S4A). Given the important physiological role of PRMT5, there is the possibility of toxicity of EPZ015666 to normal B cells. To test that possibility, we isolated naive B cells from human tonsils and stimulated in vitro with IgM. The survival of EPZ015666 treated activated B cells was comparable to that of DMSO treated cells during 6 days of culture (Figure S4C), suggesting that PRMT5 inhibition is not toxic to normal B cells. Reduced number of viable DLBCL cells in the culture was found to be due to inhibition of cell cycle progression and cell proliferation (Figure 4C, Figure S4B) but unlikely through triggering of apoptosis (Figure S4D). These data demonstrate that PRMT5 function is required for the survival and proliferation of DLBCL cells. The insensitivity of normal B cells to the inhibition of PRMT5 suggests that PRMT5 could be a therapeutic target for DLBCL.
Figure 4. Inhibition of PRMT5 inhibits DLBCL cell proliferation.
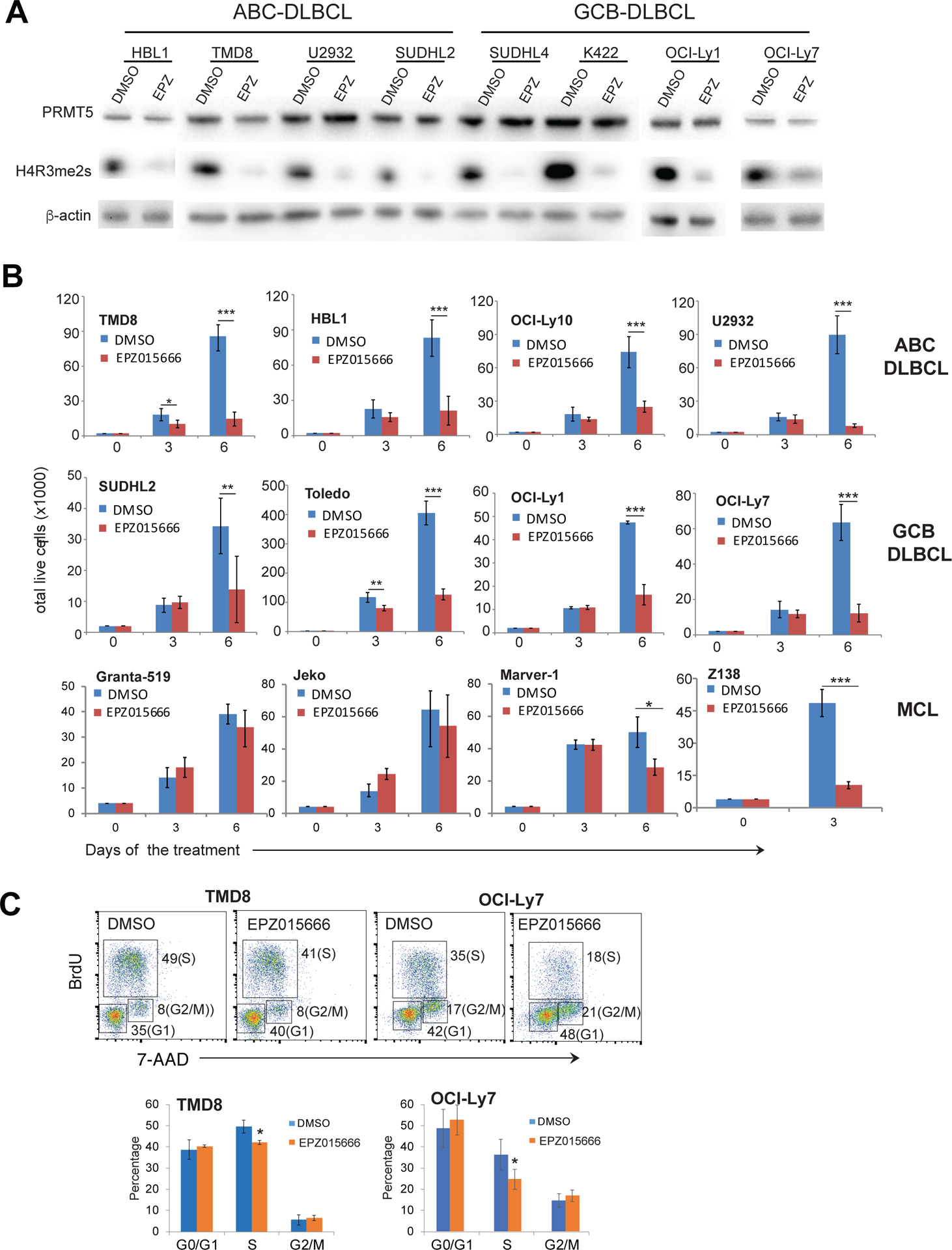
(A) Treatment of the indicated DLBCL cell lines for 5 days with 1 μM of the PRMT5 inhibitor EPZ015666 led to reduced symmetric dimethylation of histone H4R3. (B) Trypan blue dye exclusion viability assay of the indicated cell lines treated with 1 μM EPZ015666. Error bars represent mean ± SD (*p<0.05, ** p<0.01, *** p<0.001, N=3). (C) Cell cycle analysis of TMD8 and OCI-Ly7 cells after 6 days of treatment with 1 μM EPZ015666. Error bars represent mean ± SD (*p<0.05, N=3).
We then knocked out PRMT5 in ABC DLBCL (HBL1, TMD8) and GCB DLBCL (OCI-Ly7) cell lines by sgRNA. Immunoblot analysis indicated a near complete knockout of PRMT5 and a significant reduction of H4R3 dimethylation by sgPRMT5 (Figure 5A). The knockout was PRMT5 specific since the related members PRMT7 and PRMT9 were not affected (Figure 5A). Consistent with the above result, PRMT5 knockout by its sgRNA significantly reduced the number of viable cells when compared with control cells in which sgPRMT5 was not induced for expression (Figure 5B). We confirmed this as an on-target effect since overexpression of PRMT5 cDNA with mutations of the targeting sequence rescued the effect of sgPRMT5 on proliferation (Figure S5A). Consistent with the inhibitor result, sgPRMT5 expression inhibited cell cycle progression and cell proliferation (Figure S5B) but did not significantly induce apoptosis (Figure S4D). The antitumor effect of PRMT5 sgRNA was further revealed in xenograft mouse models of TMD8 and OCI-Ly7, where sgPRMT5 expression inhibited tumor growth and significantly reduced tumor volumes in 2 different DLBCL xenograft models (Figure 5C).
Figure 5. Knockout of PRMT5 inhibits DLBCL growth both in vitro and in vivo.
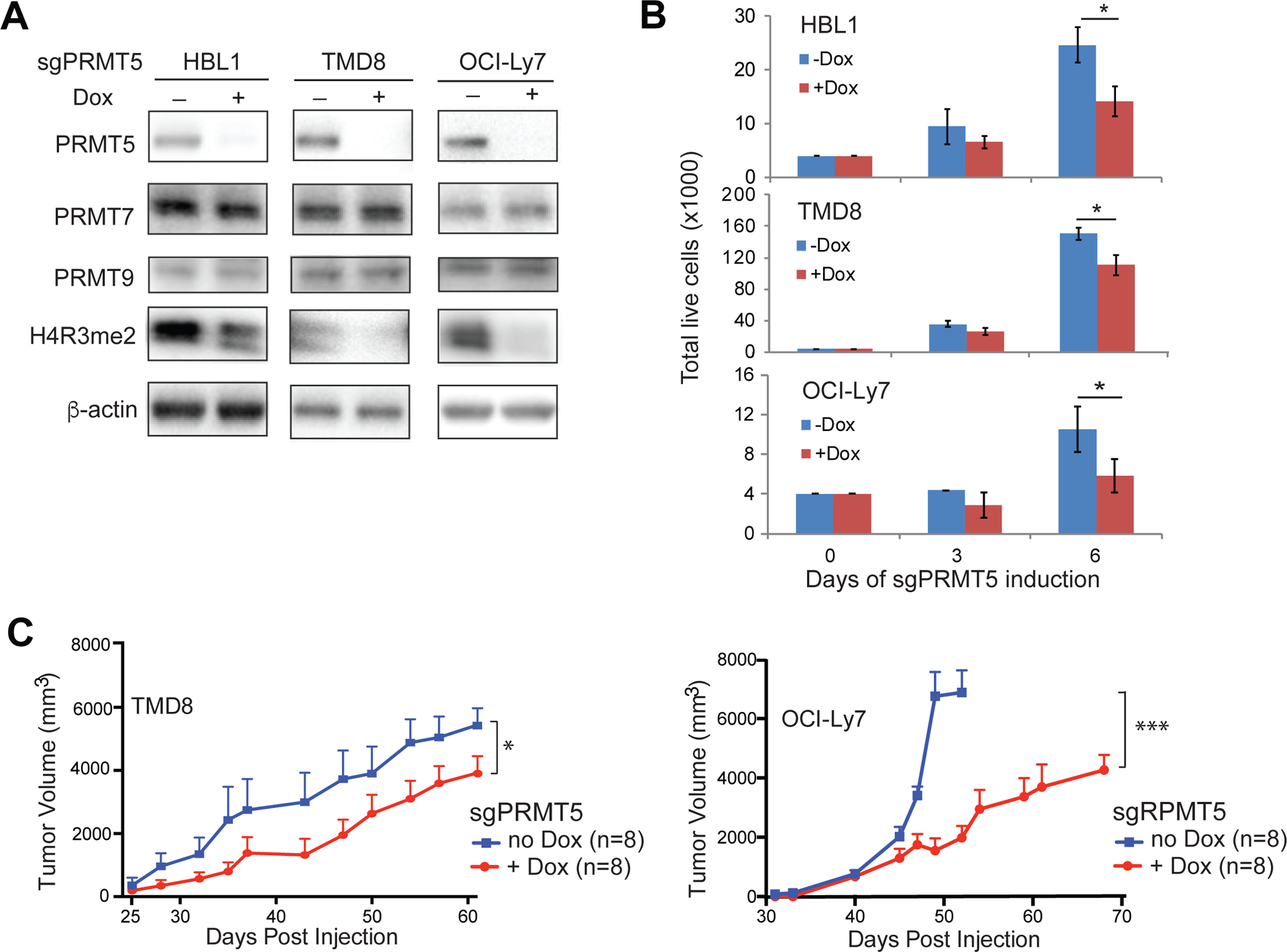
(A)Knockout of PRMT5 reduced symmetric dimethylation of histone H4R3. Immunoblot analysis of H4R3 dimethylation in the indicated cell lines after 6 days of PRMT5 sgRNA induction. β-actin served as a loading control. (B) Knockout of PRMT5 inhibited proliferation of DLBCL cells. Trypan blue dye exclusion viability assay of the indicated cell lines after 3 or 6 days of PRMT5 sgRNA induction. Error bars represent mean ± SD (*p<0.05, n=3). (C) Knockout of PRMT5 inhibited tumor growth in TMD8 and OCI-Ly7 xenograft mouse models. Error bars represent mean ± SD (*p<0.05, *** p<0.001, N=8).
To investigate the molecular mechanisms underlying tumor growth inhibition by PRMT5 sgRNA, we performed the whole genome transcriptome analysis by RNA-seq. PRMT5 sgRNA was induced for expression for either 3 days (partial knockout) or 5 days (complete knockout) (Figure S6A). Since TMD8 and OCI-Ly7 are two different DLBCL subtypes with their distinct gene expression profiles, it was not surprising that less than 10% of up- or down-regulated genes by PRMT5 sgRNA were overlapped (Figure S6B, Table S1). However, gene set enrichment analysis (GSEA) demonstrated common gene signatures enriched between the two cell lines upon PRMT5 knockout, including E2F targets, PI3K-AKT-mTOR signaling, glycolysis and cholesterol homeostasis (Figure 6A, Table S1, Figure S6C). Notably, among E2F1 target genes, CDK1, CDK4, PLK1, PLK4 and MYC were downregulated by PRMT5 sgRNA (Table S1). Immunoblot analysis verified cell cycle arrest by PRMT5 sgRNA, showing reduced expression of E2F1 while expression of the negative cell cycle regulators p53 and RBL2 was increased in the knockout cells (Figure 6B, top panel). PRMT5 sgRNA expression also reduced AKT phosphorylation (Figure 6B, bottom panel), suggesting a positive feedback loop between PRMT5 and PI3K-AKT signaling. These results suggest that PRMT5 regulates cell cycle progression and cellular metabolic pathways to promote DLBCL survival and proliferation.
Figure 6. Regulation of cell cycle progression and PI3K-AKT signaling by PRMT5.
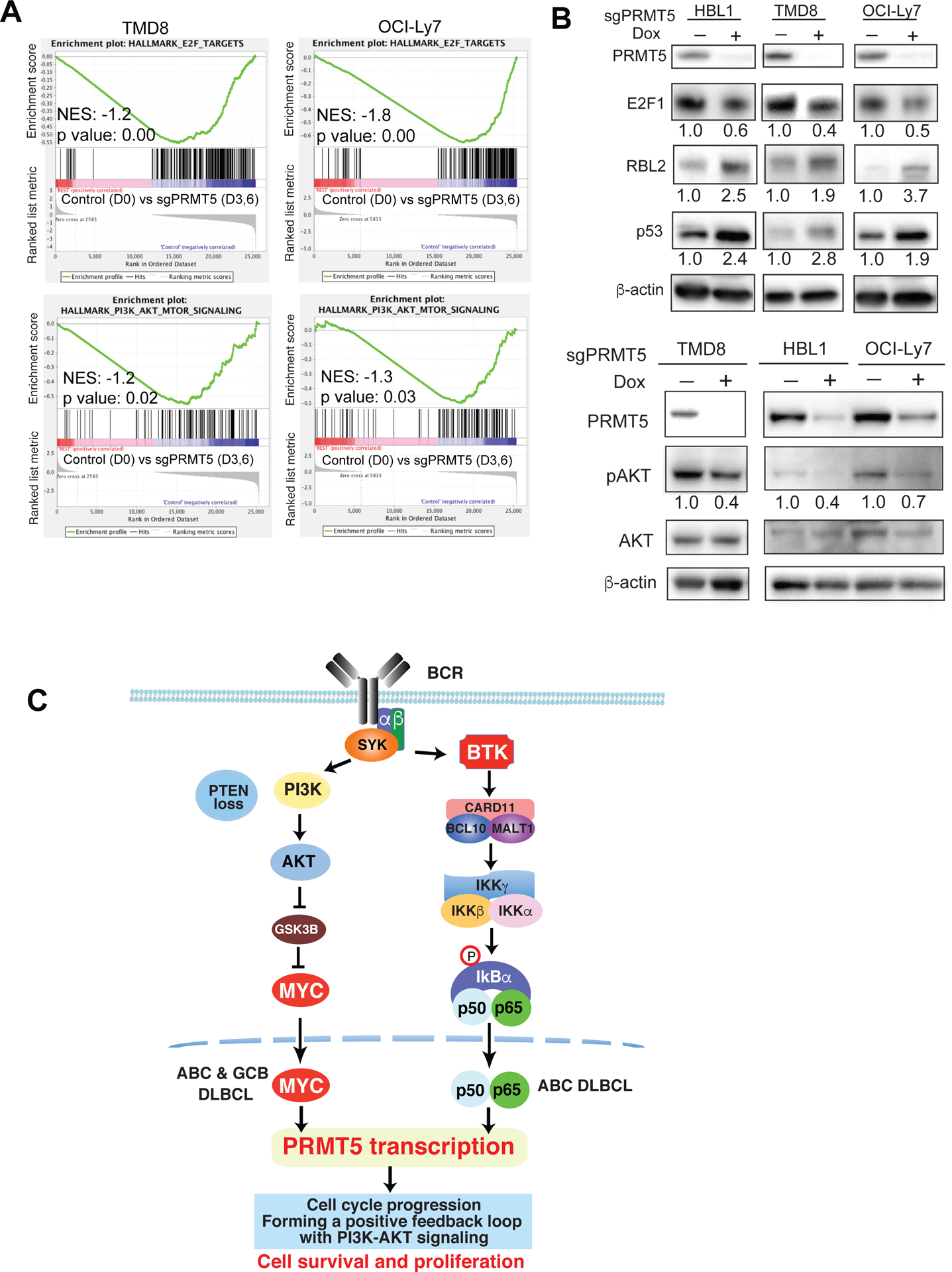
(A) Gene set enrichment analysis of E2F signature genes and PI3K-AKT-mTOR signaling signature genes in TMD8 and OCI-Ly7 cell line with PRMT5 knockout. (B, top panel) Expression level of the indicated cell cycle regulators by immunoblot analysis after PRMT5 knockout. β-actin served as a loading control. Data are representative of 3 independent experiments. (B, bottom panel) Eexpression of phospho-AKT and AKT by immunoblot analysis after PRMT5 knockout. β-actin served as a loading control. Data are representative of 3 independent experiments. (C) Model of PRMT5 regulation by BCR signaling in DLBCL. BCR downstream BTK-NF-κB signaling in ABC DLBCL and PI3K-AKT-GSK3β-MYC signaling in both ABC and GCB DLBCL lead to the upregulation of PRMT5. Overexpressed PRMT5 promotes cell cycle progression and forms a positive feedback loop with PI3K-AKT signaling to enhance cell survival and proliferation.
Co-targeting PRMT5 and AKT by their specific inhibitors in DLBCL
There has been a recent increase in the use of patient derived xenograft (PDX) engrafted into immune-compromised rodents such as NSG mice for preclinical modeling. For animal studies, we used a more potent PRMT5 inhibitor termed GSK3326595, which was recently used for mantle cell lymphoma xenografts56 and has led to a new clinical trial (NCT02783300). We first tested the drug in vitro and found a remarkable reduction in both AKT phosphorylation and symmetric dimethylation of histone H4R3 in all 4 cell lines after 3 days of treatment (Figure 7A, top panel). This result is consistent with the above PRMT5 sgRNA data. We then tested anti-tumor effects of GSK3326595 in a patient-derived xenograft model that we recently developed. Indeed, GSK3326595 treatment significantly inhibited the growth of DLBCL from a patient with ABC DLBCL and prolonged mouse survival (Figure 7B) while not affecting mouse body weight (Figure S8A) in a NSG xenograft model.
Figure 7. Co-targeting PRMT5 and AKT by GSK3326595 and AKT inhibitor V in DLBCL.
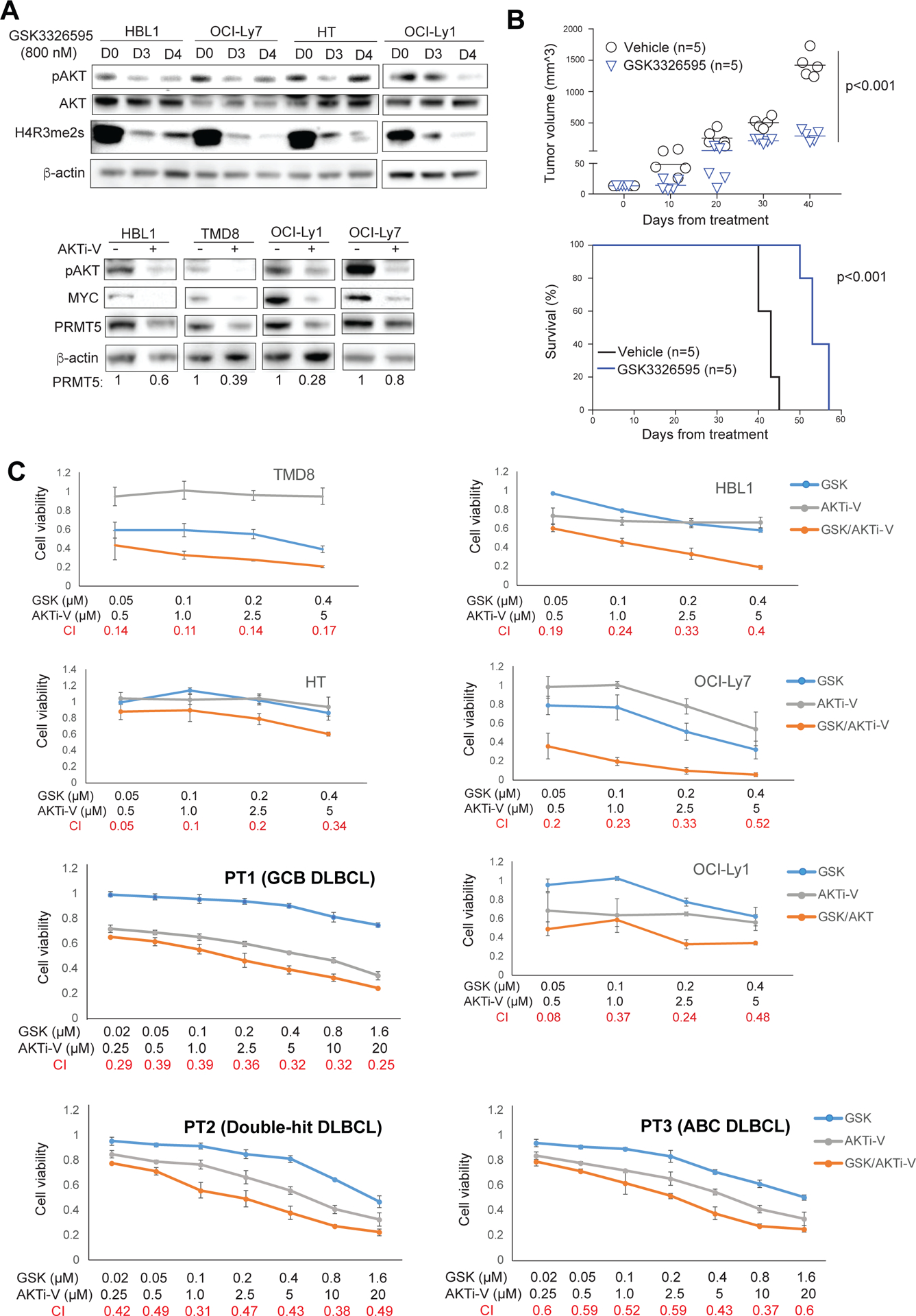
(A, top panel) Treatment of the indicated DLBCL cell lines for 3 or 4 days with 800 nM of the PRMT5 inhibitor GSK3326595 led to reduced Phospho-AKT and symmetric dimethylation of histone H4R3. β-actin served as a loading control. (A, bottom panel) Treatment of the indicated DLBCL cell lines for 2 days with 20 μM of AKT inhibitor V led to reduced phospho-AKT, MYC and PRMT5. β-actin served as a loading control. (B) DLBCL patient derived xenografts (PDX). 5 × 106 freshly isolated ABC DLBCL cells were directly injected into fetal bone chip of NSG-hu mice after the mice were anesthetized with 5% isoflurane vaporizer. Once tumor growth was detected in the first generation, tumor mass was monitored and then passaged. The passaged tumor equally grew and mice were assigned as 5 mice/group for in vivo treatment. Mice were administered vehicle control or GSK3226595 100mg/kg, oral gavage, twice per day for 35 consecutive days after 3 days of tumor engraftment. Tumor burden was calculated by measuring tumor volume (N=5; GSK3226595 vs vehicle, p=0.0000023). Survival curve was analyzed by the Kaplan-Meier method (N=5; GSK3226595 vs vehicle, p=0.00001). (C) CellTiter-Glo™ Luminescent Cell Viability Assay of the indicated DLBCL cell lines after 6-day treatment of the indicated concentrations of AKT inhibitor V or GSK3326595, or both, and CellTiter-Glo™ Luminescent Cell Viability Assay of primary cancer cells from 3 DLBCL patients after 3-day treatment of the indicated concentrations of AKT inhibitor V or GSK3326595, or both. Data indicate mean ± SD of triplicates. Combination index (CI) was calculated with CompuSyn software.
Given a positive regulatory loop between PRMT5 and AKT and the fact that PRMT5 maintains the fitness of DLBCL cells through regulating gene expression in multiple oncogenic and metabolic pathways, we hypothesized that PRMT5 inhibition by its specific inhibitor would enhance the antitumor effects of an AKT inhibitor in DLBCL. A recent preclinical study and our in vitro analysis demonstrated that AZD5363 is effective in killing those DLBCL cells that harbor PTEN mutations/deletions or lack PTEN expression21, which are largely restricted to GCB subtype20 (Figure S7). To test our hypothesis, we avoided using AZD5363 but used AKT inhibitor V (or triciribine) instead, an AKT inhibitor that is being used in clinical trials for solid cancers and other hematological malignancies57. Immunoblotting analysis showed that treatment with AKT inhibitor V dramatically reduced AKT phosphorylation, and MYC and PRMT5 expression in 2 ABC and 2 GCB DLBCL cell lines (Figure 7A, bottom panel). We then treated these cell lines and an additional GCB cell line (HT) with GSK3326595 or AKT inhibitor V alone or in combination and found synergistic cell killing of all 5 cell lines (Figure 7C). Synergism between the two drugs was also observed in primary cancer cells from 3 DLBCL patients, each with ABC, GCB and MYC/BCL-2 double-hit DLBCL (Figure 7C, Figure S8B). Therefore, our data suggest that co-targeting PRMT5 and AKT is a potential novel targeted combination therapeutic strategy with probable synergy in DLBCL.
Discussion
In this study, we have elucidated the molecular mechanisms of BCR signaling in regulating PRMT5 expression in DLBCL (Figure 6C). PRMT5 expression is elevated in DLBCL cells as well as in normal germinal center B cells. PRMT5 is induced upon BCR stimulation in naive B cells. BCR downstream PI3K signaling regulates PRMT5 expression in both ABC and GCB DLBCL cells through the AKT-GSK3β-MYC axis. In addition, active BCR-BTK-NF-κB signaling in ABC DLBCL cells also upregulates PRMT5 expression. Expression of PRMT5 is required for the fitness of DLBCL since genetic or pharmacological inhibition of PRMT5 inhibits the growth of DLBCL cells both in vitro and in vivo. PRMT5 promotes the survival and proliferation of these lymphoma cells, making it an attractive therapeutic target41, 43. In addition, disruption of a positive feedback regulatory loop between PRMT5 and AKT by their specific inhibitors has potential clinical implications.
Recent studies have demonstrated higher levels of PRMT5 expression in DLBCL and mantle cell lymphoma than in normal B cells41, 44–46. Our work extends these findings and describes an antigen-dependent PRMT5 expression mechanism. This mechanism applies to DLBCL cells as well as to normal B cells, including germinal center B cells and activated B cells. Notably, the level of PRMT5 expression is comparable between DLBCL and germinal center B cells. In addition, antigen stimulation dramatically increases PRMT5 expression in naive B cells either from tonsils or from peripheral blood. Despite elevated PRMT5 expression and its essential role in cell survival, our prognosis analysis revealed no relevance of PRMT5 expression to DLBCL patient outcomes (data not shown). This could be due to the fact that the level of PRMT5 expression is not distinguishable between GCB and Non-GCB DLBCL.
It is not well understood how PRMT5 maintains fitness of DLBCL cells. Our flow cytometric, biochemical and RNA-seq analyses have demonstrated that PRMT5 plays a positive role in cell cycle progression. Methylation of E2F1 by PRMT5 inhibits apoptosis and promotes cell cycle progression58. PRMT5 has also been shown to regulate the activity of p53 and its expression level30, 34. RBL2, which inhibits E2F function and cell cycle progression from G1 to S phase, is epigenetically silenced by PRMT5 in DLBCL46. Consistent with these findings, our immunoblot analysis revealed reduced expression of E2F1 in PRMT5 knockout cells while the level of RBL2 and p53 expression is increased. In support of these results, RNA-seq data showed that E2F1 target genes are downregulated in these knockout cells, including CDK1, CDK4, PLK1, PLK4 and MYC. PRMT5 can also promote DLBCL survival through interaction with, and methylation of, BCL635
GSEA also demonstrated that PRMT5 regulates the expression of genes that involve PI3K-AKT-mTOR signaling, glycolysis and cholesterol homeostasis. Interestingly, in addition to cell cycle signature enrichment, all these enriched metabolic signatures are also among the GSEA list of normal germinal center B cells (PRMT5 high) when compared with naive B cells (PRMT5 low) (Figure S6D), based on published RNA-seq data59. To date, our knowledge of PRMT5 in cellular metabolism is limited. A recent study demonstrated that PRMT5 is upregulated by high fat diet in the mouse liver and its enzymatic activity enhances hepatic mitochondrial biogenesis through activation of PI3K-AKT signaling60. Consistent with this finding, our data suggest that PRMT5 is required for AKT activation and forms a feedback regulatory loop with AKT to promote the survival and proliferation of DLBCL cells (Figure 6). Future work to gain more insights into the function of PRMT5 in cellular metabolism appears warranted.
Supplementary Material
Acknowledgement
We thank Dr. Scott McMurray for providing tonsils, Dr. Sameer Mathur for providing PBMCs, and Dr. Stephen Nimer for providing PRMT5 plasmid. This work was supported by the National Institutes of Health/National Cancer Institute (NIH/NCI) grant R01 CA187299 (L.R.), UW-Madison Forward Lymphoma Fund (L.L.), NIH/NCI grant K08 CA174750 and the MACC fund (C.M.C.). This work was also supported in part by NIH/NCI P30 CA014520 - UW Comprehensive Cancer Center Support.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008. January; 8(1): 22–33. [DOI] [PubMed] [Google Scholar]
- 2.Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol 2012; 30: 565–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–511. [DOI] [PubMed] [Google Scholar]
- 4.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med 2010. April 15; 362(15): 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008. November 27; 359(22): 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol 2010. June; 2(6): a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappa B activity is required for survival of activated B Cell-like diffuse large B cell lymphoma cells. J Exp Med 2001; 194(12): 1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008. April 1; 111(7): 3701–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011. February 3; 470(7332): 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Shaffer AL 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer cell 2012. June 12; 21(6): 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010. January 7; 463(7277): 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009. June 4; 459(7247): 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011. July 31; 43(9): 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008. March 21; 319(5870): 1676–1679. [DOI] [PubMed] [Google Scholar]
- 15.Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proceedings of the National Academy of Sciences of the United States of America 2015. November 03; 112(44): 13447–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015. August; 21(8): 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer cell 2017. June 12; 31(6): 833–843 e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 2017. August 24; 130(8): 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012. October 04; 490(7418): 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 2013. July 23; 110(30): 12420–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erdmann T, Klener P, Lynch JT, Grau M, Vockova P, Molinsky J, et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 2017. July 20; 130(3): 310–322. [DOI] [PubMed] [Google Scholar]
- 22.Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica 2017. November; 102(11): 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanc RS, Richard S. Arginine Methylation: The Coming of Age. Mol Cell 2017. January 05; 65(1): 8–24. [DOI] [PubMed] [Google Scholar]
- 24.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009. January 16; 33(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rho J, Choi S, Seong YR, Cho WK, Kim SH, Im DS. Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J Biol Chem 2001. April 06; 276(14): 11393–11401. [DOI] [PubMed] [Google Scholar]
- 26.Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004. November; 24(21): 9630–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 2009. March; 16(3): 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majumder S, Alinari L, Roy S, Miller T, Datta J, Sif S, et al. Methylation of histone H3 and H4 by PRMT5 regulates ribosomal RNA gene transcription. J Cell Biochem 2010. February 15; 109(3): 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci 2011. December; 36(12): 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008. December; 10(12): 1431–1439. [DOI] [PubMed] [Google Scholar]
- 31.Cho EC, Zheng S, Munro S, Liu G, Carr SM, Moehlenbrink J, et al. Arginine methylation controls growth regulation by E2F-1. EMBO J 2012. April 04; 31(7): 1785–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proceedings of the National Academy of Sciences of the United States of America 2013. August 13; 110(33): 13516–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tee WW, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P, et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev 2010. December 15; 24(24): 2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 2015. September; 125(9): 3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu X, Fernando TM, Lossos C, Yusufova N, Liu F, Fontan L, et al. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood 2018. November 8; 132(19): 2026–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013. January; 13(1): 37–50. [DOI] [PubMed] [Google Scholar]
- 37.Gu Z, Gao S, Zhang F, Wang Z, Ma W, Davis RE, et al. Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem J 2012. September 01; 446(2): 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao X, Zhao S, Liu T, Liu Y, Liu Y, Yang X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem 2013. March; 61(3): 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan F, Alinari L, Lustberg ME, Martin LK, Cordero-Nieves HM, Banasavadi-Siddegowda Y, et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res 2014. March 15; 74(6): 1752–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng X, Shao G, Zhang HT, Li C, Zhang D, Cheng L, et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017. March 02; 36(9): 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, Smith EM, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015. April 16; 125(16): 2530–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest 2016. October 03; 126(10): 3961–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015. June; 11(6): 432–437. [DOI] [PubMed] [Google Scholar]
- 44.Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015. July 02; 523(7558): 96–100. [DOI] [PubMed] [Google Scholar]
- 45.Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 2007. August 8; 26(15): 3558–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung J, Karkhanis V, Tae S, Yan F, Smith P, Ayers LW, et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) silencing. J Biol Chem 2013. December 6; 288(49): 35534–35547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci 2017. October 19; 372(1732). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006. May 4; 441(7089): 106–110. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Bouchlaka MN, Wolff J, Grindle KM, Lu L, Qian S, et al. FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene 2016. December 1; 35(48): 6223–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-Cell Lymphoma Patient-Derived Xenograft Models Enable Drug Discovery and Are a Platform for Personalized Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2017. August 1; 23(15): 4212–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 2016. September; 11(9): 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Wang F, Lu L, Zhu F, Huang S, Nomie K, et al. NR4A1 inhibition synergizes with ibrutinib in killing mantle cell lymphoma cells. Blood cancer journal 2017. November 23; 7(12): 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young RM, Shaffer AL 3rd, Phelan JD, Staudt LM. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol 2015. April; 52(2): 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul 2017. August; 65: 5–15. [DOI] [PubMed] [Google Scholar]
- 55.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 2003. December 19; 278(51): 51606–51612. [DOI] [PubMed] [Google Scholar]
- 56.Gerhart SV, Kellner WA, Thompson C, Pappalardi MB, Zhang XP, Montes de Oca R, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 2018. June 26; 8(1): 9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 2004. July 1; 64(13): 4394–4399. [DOI] [PubMed] [Google Scholar]
- 58.Zheng S, Moehlenbrink J, Lu YC, Zalmas LP, Sagum CA, Carr S, et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol Cell 2013. October 10; 52(1): 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer cell 2016. August 8; 30(2): 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang L, Liu J, Zhang XO, Sibley K, Najjar SM, Lee MM, et al. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J Biol Chem 2018. May 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.