

The mitophagy receptor FUNDC1 interacts with FBXL2 to preserve mitochondrial Ca2+ homeostasis and cardiac function in obesity.
Abstract
Defective mitophagy is causally linked to obesity complications. Here, we identified an interaction between mitophagy protein FUNDC1 (FUN14 domain containing 1) and receptor subunit of human SCF (SKP1/cullin/F-box protein) ubiquitin ligase complex FBXL2 as a gatekeeper for mitochondrial Ca2+ homeostasis through degradation of IP3R3 (inositol 1,4,5-trisphosphate receptor type 3). Loss of FUNDC1 in FUNDC1−/− mice accentuated high-fat diet–induced cardiac remodeling, functional and mitochondrial anomalies, cell death, rise in IP3R3, and Ca2+ overload. Mass spectrometry and co-immunoprecipitation analyses revealed an interaction between FUNDC1 and FBXL2. Truncated mutants of Fbox (Delta-F-box) disengaged FBXL2 interaction with FUNDC1. Activation or transfection of FBXL2, inhibition of IP3R3 alleviated, whereas disruption of FBXL2 localization sensitized lipotoxicity-induced cardiac damage. FUNDC1 deficiency accelerated and decelerated palmitic acid–induced degradation of FBXL2 and IP3R3, respectively. Our data suggest an essential role for interaction between FUNDC1 and FBXL2 in preserving mitochondrial Ca2+ homeostasis and cardiac function in obese hearts.
INTRODUCTION
Sustained obesity prompts ample chronic noncommunicable diseases such as insulin resistance, type 2 diabetes mellitus, coronary heart disease, hypertension, atherosclerosis, and stroke, resulting in high cardiovascular disease morbidity and mortality (1). Obesity is associated with unfavorable myocardial structural and functional changes accompanied by mitochondrial injury, a pathological condition commonly known as obesity cardiomyopathy (2–5). Several hemodynamic, neurohormonal, and metabolic factors are shown to participate in the etiology of obesity cardiomyopathy through oxidative stress, lipotoxicity, inflammation, autophagy failure, cell death, and activation of adrenergic and renin-angiotensin aldosterone systems (2–9). Nonetheless, precise cues for obesity cardiomyopathy remain elusive, making it somewhat challenging to engage effective therapeutic intervention for cardiac anomalies in obesity.
A number of clinical and experimental studies have unveiled a key role of deranged autophagy, in particular mitophagy, in cardiovascular abnormalities in metabolic disorders including obesity, insulin resistance, and type 2 diabetes mellitus (2, 8, 10, 11). Mitophagy, the selective encapsulation and clearance of long-lived or damaged mitochondria, serves as a primary mitochondrial quality control machinery for mitochondrial quantity, metabolic reprogramming, energy metabolism, and cell differentiation (12). Genetic or acquired defects in mitophagy are linked with neurodegenerative gastrointestinal and cardiovascular diseases (12). Both receptor-mediated and Parkin-dependent mitophagy have been identified with B-cell lymphoma 2 (BCL2)/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) and FUN14 domain containing 1 (FUNDC1) comprising receptor-mediated mitophagy (12, 13). BNIP3 is a mitochondrial outer membrane protein with the property of facilitating mitophagy. FUNDC1-mediated mitophagy is turned on in response to hypoxia to remove long-lived or damaged mitochondria, favoring cell survival (12, 14). Evidence from our laboratory and others has revealed a vital role of FUNDC1 in the maintenance of cardiovascular homeostasis in pathological conditions including heart failure, cardiac aging, myocardial infarction, and ischemia-reperfusion injury (14–19). However, controversial findings were noted for the role of FUNDC1 receptor-mediated mitophagy under metabolic stress such as diabetes and obesity (16, 20). To this end, the present study evaluated the effect of FUNDC1 ablation on cardiac remodeling and contractile defect in high-fat (HF) diet–induced obesity, hoping to better understand the precise role of receptor-mediated mitophagy in obesity heart disease. Given that metabolic diseases including obesity is often tied with derangement of mitochondrial Ca2+ and mitochondrial integrity (21, 22), mitochondrial Ca2+ homeostasis was monitored with a focus on inositol 1,4,5-trisphosphate [IP3 receptors (IP3Rs)] located on the endoplasmic reticulum (ER) to feed Ca2+ to mitochondria to promote oxidative phosphorylation (23, 24). Result from our study revealed that FUNDC1 interacts with FBXL2 [receptor subunit of human SCF (SKP1/cullin/F-box protein) ubiquitin ligase complex] to govern IP3R3 levels, mitochondrial Ca2+ load and integrity, and, ultimately, cardiac homeostasis in the face of obesity. FUNDC1 deficiency accentuated cardiac remodeling, contractile dysfunction, apoptosis, autophagy defect, and mitochondrial injury due to loss of FBXL2-mediated IP3R3 degradation and thus mitochondrial Ca2+ overload in obesity.
RESULTS
Effects of diet-induced and genetic obesity on mitophagy pathways and genes in the heart
To discern altered biological pathways in hearts following HF diet intake, adult Sprague-Dawley rats were fed 60% HF diet for 20 weeks. Heart tissues were collected and subjected to RNA sequencing (RNA-seq) analysis. Differentially expressed genes (DEGs) between low-fat (LF) and HF fed rats were analyzed by Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. A total of 569 DEGs were identified, and KEGG pathway analysis showed that many of these DEGs are from Ca2+ signaling, autophagy, and mitophagy pathways (Fig. 1, A and B), suggesting a key role of these biological pathways in the onset of obesity-related cardiac complications. Ca2+ signaling, autophagy, and mitophagy pathways were further analyzed to reveal individual gene components governing these biological processes (Fig. 1C). To elucidate the participation of mitophagy in obesity heart diseases, heart samples from ob/ob and db/db genetically predisposed obese models, HF diet (60% fat diet intake for 20 weeks)–induced obese rats and obese [body mass index (BMI) > 28] humans were scrutinized. Results presented in Fig. 1 (D to G) show overtly down-regulated mitophagy protein markers including FUNDC1, Parkin, and BNIP3, to various degrees, in heart tissues from genetically predisposed or diet-induced rodents as well as obese human subjects (with the exception of unchanged BNIP3 in ob/ob mouse hearts).
Fig. 1. HF intake–affected pathways and genes in hearts using RNA-seq and effect of obesity on mitophagy markers in the heart.
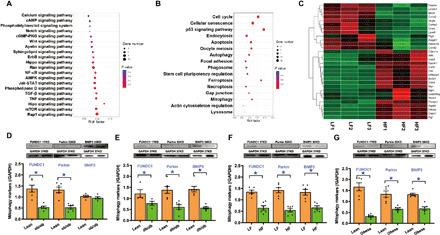
(A to C) Rats were fed HF (60% fat) diet for 20 weeks, and cardiac tissues were collected and subjected to RNA-seq. (A) Signal transduction pathway of KEGG. (B) Cellular processes of KEGG. (C) Heatmap displaying relative expression of autophagy and Ca2+ signaling pathway–related genes in the heart (n = 3). (D) Mitophagy protein profiles (FUNDC1, Parkin, and BNIP3) in adult ob/ob mouse hearts. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) Mitophagy protein profiles in adult db/db mouse hearts. (F) Mitophagy protein profiles in adult rat hearts following 20 weeks of HF intake. (G) Mitophagy protein profiles in lean and obese human heart samples. Mean ± SEM; n = 4 to 6 group, *P < 0.05 between indicated groups.
General features and echocardiographic properties of wild-type and FUNDC1−/− mice fed LF or HF diet
Next, we evaluated the effect of 20-week HF diet (60% fat) intake on biometric, morphological, and echocardiographic properties in wild-type (WT) and FUNDC1−/− mice. As shown in Fig. 2A, consumption of an HF diet intake caused an overt separation in body weight starting from week 7 in a comparable manner in WT and FUNDC1−/− mice. Although food intake was similar in all mouse groups (by calorie or weight determination), HF diet intake resulted in glucose intolerance, elevated plasma insulin, increased homeostatic model assessment for insulin resistance (HOMA-IR) index, and serum triglycerides without affecting fasting blood glucose levels in WT and FUNDC1−/− mice, in a comparable manner (Fig. 2, B to I). HF diet intake overtly promoted cardiac hypertrophy (gross heart weight and cardiomyocyte area) along with elevated mRNA levels of the prohypertrophic genes atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and GATA binding protein 4 (GATA4), as well as interstitial fibrosis, with a more pronounced effect in FUNDC1−/− mice (Fig. 2, J to O). FUNDC1 ablation itself did not exert any effect on body and heart weight or size, nor did it affect global metabolic profiles [intraperitoneal glucose tolerance test (IPGTT), serum triglycerides, plasma insulin, or HOMA-IR] and cardiac geometry (cardiomyocyte cross-sectional area, prohypertrophic gene expression, and fibrosis).
Fig. 2. HF diet intake–induced body weight gain, glucose intolerance, plasma profile, and cardiac remodeling in WT and FUNDC1−/− mice.
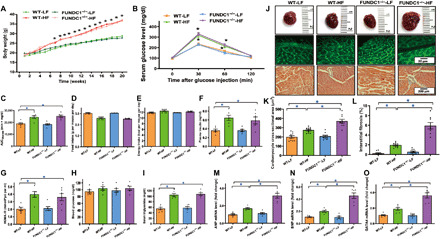
(A) Body weight gain over 20-week feeding period. (B) Intraperitoneal glucose tolerance test (IPGTT). (C) Area underneath IPGTT curve (AUC). (D) Food intake (by weight). (E) Food intake (by calorie). (F) Plasma insulin. (G) HOMA-IR index. (H) Fasting blood glucose. (I) Serum triglycerides. (J) Gross images, lectin, or Masson’s trichrome staining in hearts from LF or HF diet–fed mice. (K) Pool data of cardiomyocyte cross-sectional area. (L) Quantitative analysis of interstitial fibrotic area (as a percentage of entire cardiac region). (M) ANP mRNA levels. (N) BNP mRNA levels. (O) GATA4 levels. Mean ± SEM; n = 7 to 10 mice per group, *P < 0.05 between indicated groups. Photo credits: Taken by Jun Ren, Zhongshan Hospital, Fudan University.
Echocardiographic assessment revealed that HF diet intake caused an enlarged left ventricular (LV) end-systolic diameter (LVESD), LV mass and heart weights, decreased fractional shortening, and ejection fraction in the absence of notable changes in LV wall thickness, septal thickness, LV end-diastolic diameter (LVEDD), and heart rate. Although FUNDC1 ablation itself failed to alter cardiac geometry or function under LF diet intake, it exacerbated HF intake–induced changes in echocardiographic indices while unmasking enlarged LVEDD (Fig. 3).
Fig. 3. HF diet intake–induced changes in echocardiographic indices in WT and FUNDC1−/− mice.
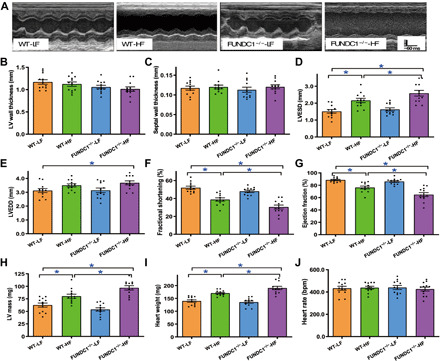
(A) Representative echocardiographic images from all four experimental groups. (B) LV wall thickness. (C) Septal thickness. (D) LV ESD. (E) LV EDD. (F) Fractional shortening. (G) Ejection fraction. (H) LV mass. (I) Heart weight. (J) Heart rate. bpm, beats per minute. Mean ± SEM; n = 7 to 9 mice per group, *P < 0.05 between indicated groups.
Effect of FUNDC1 deletion on HF diet–induced cardiomyocyte contractile, intracellular Ca2+, and mitochondrial Ca2+ responses
Neither chronic HF diet intake nor FUNDC1 ablation exhibited any effect on cardiomyocyte cell length. HF diet intake decreased peak shortening (PS), maximal velocity of shortening/relengthening (±dL/dt), and prolonged relengthening duration [time-to-90% relengthening (TR90)] without affecting shortening duration [time to PS (TPS)]. FUNDC1 ablation significantly accentuated HF diet–induced cardiomyocyte contractile anomalies without any notable effect itself (fig. S1, A to F). To explore potential mechanisms of action involved in FUNDC1 ablation-elicited beneficial effect against HF diet–induced cardiomyocyte dysfunction, intracellular and mitochondrial Ca2+ handling was evaluated using the fluorescence dye fura-2 and Rhod-2, respectively. Our data suggested that HF diet intake provoked an overt rise in basal mitochondrial (but not intracellular) Ca2+, decreased electrically stimulated rise in intracellular Ca2+ (ΔFFI), and prolonged intracellular Ca2+ clearance, with a more pronounced effect in cardiomyocytes from FUNDC1−/− mice. FUNDC1 ablation elicited little effect on intracellular or mitochondrial Ca2+ handling (fig. S1, G to J).
Effect of FUNDC1 ablation on HF diet–induced mitochondrial injury and cell death
Transmission electron microscopy was used to evaluate ultrastructure of sarcomere and mitochondria. Data in Fig. 4A show pronounced cytoarchitectural aberrations such as mitochondrial swelling, fragmentation of cristae, distortion of sarcomeres, and myocardial filaments in HF diet–fed mouse myocardium and that the response was accentuated by FUNDC1 ablation without notable ultrastructural change from FUNDC1 ablation itself. HF intake increased mitochondrial number and area (% area or per mitochondrion) (Fig. 4, B to D), down-regulated levels of Uncoupling protein 2 (UCP2) (Fig. 4E), and mitochondrial respiration complexes (Fig. 4, F to H) while promoting mitochondrial O2− production (Fig. 4, I and J), the effects of which were augmented by FUNDC1 deletion with little effect from FUNDC1 ablation alone. Our assessment further revealed that HF intake promoted myocardial cell death including apoptosis, necroptosis, and necrosis, as well as mitochondrial injury (shown as decreased aconitase activity) (Fig. 4, K to O). HF diet intake fostered myocardial apoptosis, in particular mitochondrial apoptosis, as evidenced by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay, inflammation [shown as interleukin-1β (IL-1β)], mitochondrial translocation of Bax into cytosolic fractions, and Bcl2 levels, with a more pronounced effect in FUNDC1−/− mice. FUNDC1 ablation itself did not affect levels of apoptotic or proinflammatory markers (Fig. 5, A to F). Evaluation of necroptosis and necrosis using receptor-interacting protein (RIP)–mediated cell death, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and lactate dehydrogenase (LDH) revealed that HF diet intake promoted RIPK1 (RIP kinase 1)/RIPK3–mediated necroptotic and necrotic cell death, the effect of which was overtly accentuated by FUNDC1 ablation with little effect of ablation in the absence of HF intake (Fig. 4, K to N).
Fig. 4. Effect of HF intake on myocardial ultrastructure, mitochondrial integrity, mitochondrial respiration, cell death, and mitochondrial O2− production in WT and FUNDC1−/− mice.
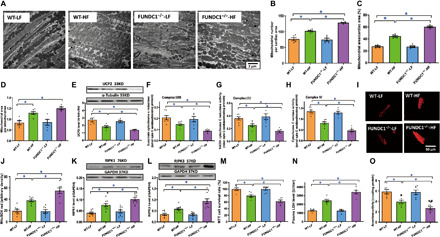
(A) Mitochondria and sarcomere ultrastructure using transmission electron microscopy. (B) Mitochondrial number by image area. (C) Mitochondrial area (% total area). (D) Mitochondrial area (per mitochondrion). (E) UCP2 levels. (F to H) Mitochondrial respiration (complex II/III, complex I/III, and complex IV). (I) Representative MitoSOX Red fluorescence images. (J) Pooled data of mitochondrial O2− levels using MitoSOX Red. (K) Receptor-interacting protein (RIP)–associated cell death marker RIPK1. (L) RIP cell death marker RIPK3. (M) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay in cardiomyocytes. (N) Plasma lactate dehydrogenase (LDH) levels. (O) Mitochondrial aconitase activity. Insets: Representative immunoblots depicting UCP2, RIPK1, and RIPK3 using respective antibodies. GAPDH was used as the loading control. Mean ± SEM, n = 5 to 6 images or visual fields per group, *P < 0.05 between indicated groups.
Fig. 5. Effect of HF intake on apoptosis, inflammation, mitochondrial apoptosis, and ER proteins including IP3Rs, SERCA2a, and RyR2 levels in hearts from WT and FUNDC1−/− mice.
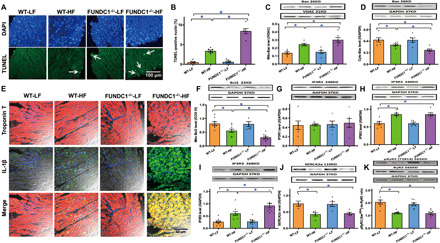
(A) Representative 4′,6-diamidino-2-phenylindole (DAPI) (nucleus) and TUNEL (apoptosis) staining in four experimental groups (arrowheads denote apoptosis). (B) Pooled data of TUNEL apoptosis. (C) Mitochondrial fraction of Bax. (D) Cytosolic fraction of Bax. (E) Immunofluorescence detection of IL-1β. (F) Mitochondrial Bcl2. (G) IP3R1. (H) IP3R2. (I) IP3R3. (J) SERCA2a [sarco(endo)plasmic reticulum Ca2+-ATPase 2a]. (K) Pan and phosphorylated RyR2. Insets: Representative immunoblots depicting levels of Bax, Bcl2, IP3Rs, SERCA2a, and pan and phosphorylated RyR2 using respective antibodies. GAPDH was used as the loading control. Mean ± SEM, n = 8 to 9 mice per group, *P < 0.05 between indicated groups.
Effect of FUNDC1 ablation on HF diet–induced changes in autophagy and mitophagy
Obesity is often associated with dampened autophagy and mitophagy (2, 8, 25–27). Our data also revealed suppressed autophagy levels in hearts from HF diet–fed mice, as manifested by decreased microtubule-associated protein 1 light chain 3 beta II (LC3BII)-to-LC3BI ratio, Beclin1, autophagy-related gene (Atg) 7, Atg5, and increased SQSTM1/p62 (p62). FUNDC1 ablation augmented HF-induced autophagy loss with little effect by itself (Fig. 6, A to E). Mitophagy levels were also suppressed in HF diet–fed murine hearts (decreased mitophagy markers Parkin, Pink1, FUNDC1, and BNIP3), the effect of which was unaffected by FUNDC1 ablation (with exception of FUNDC1) (Fig. 6, F to I). These findings support a unique role of FUNDC1-mediated mitophagy in HF diet–induced myocardial geometric and functional changes.
Fig. 6. Effect of HF intake on autophagy and mitophagy protein markers in WT and FUNDC1−/− mice and evidence of FUNDC1-FBXL2 interaction.
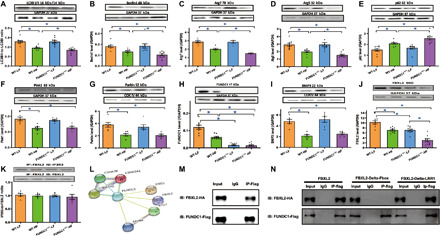
(A) LC3BI/II, (B) Beclin1, (C) Atg7, (D) Atg5, (E) p62, (F) Pink1, (G) Parkin, (H) FUNDC1, and (I) BNIP3. Insets: Representative immunoblots (IB) depicting levels of LC3BI/II, Beclin1, Atg7, Atg5, p62, Pink1, Parkin, FUNDC1, and BNIP3 using respective antibodies. GAPDH or COXIV (mitochondria) was used as the loading control. (J) Levels of FBXL2. (K) IP analysis for FBXL2-IP3R3 interaction. Bar graph depicts ratio of immunoprecipitated IP3R3 and FBXL2. (L) Co-IP mass spectrometry identified FBXL2 as an interacting protein of FUNDC1. (M) IP analysis of FBXL2 tagged with hemagglutinin (HA) (FBXL2-HA) and FUNDC1 tagged with Flag (FUNDC1-Flag) in H9c2 cells. IgG, immunoglobulin G. (N) IP analysis of FBXL2 domain mutations (Delta-F-box or Delta LRR1, HA-tagged) using FUNDC1-Flag in H9c2 cells. Mean ± SEM, n = 9 mice per group, *P < 0.05 between indicated groups.
Effect of FUNDC1 ablation on HF diet–induced changes in ER proteins and FBXL2
HF diet intake overtly up-regulated levels of the ER Ca2+ regulatory proteins IP3R2 and IP3R3 while down-regulating levels of sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) and impaired ryanodine receptor (RyR2) activity (T2814 phosphorylation) without affecting IP3R1 and pan RyR2. Although FUNDC1 ablation did not alter levels of IP3Rs, SERCA2a, and pan or phosphorylated RyR2, it overtly accentuated HF diet–induced up-regulation of IP3R3 without affecting IP3R2, IP3R1, SERCA2a, and RyR2 (either pan or phosphorylated levels) (Fig. 5, G to K). Given the reported role of F-box protein FBXL2, the receptor subunit of one the 69 human SCF ubiquitin ligase complex, in binding IP3R3 to target it for degradation (22), levels of FBXL2 were evaluated. The results show overtly down-regulated FBXL2 levels with sustained HF diet intake, with a more pronounced drop in FUNDC1−/− mice. FUNDC1 deletion itself also down-regulated FBXL2 (Fig. 6J). Next, co-immunoprecipitation (co-IP) examination revealed unaltered interaction between FBXL2 and IP3R3 with HF diet intake or FUNDC1 ablation or both (Fig. 6K). We examined possible interacting partners of FUNDC1 by coupling IP with mass spectrometry. Flag-tagged FUNDC1 (FUNDC1-Flag) was overexpressed in H9c2 cells, which were then lysed and immunoprecipitated with an anti-flag antibody. Liquid chromatography–mass spectrometry (LC-MS) analysis was used to identify proteins recovered in the IP. As a result, FBXL2 as well as a few autophagy related proteins were recovered to specifically co-immunoprecipitate with FUNDC1-Flag (Fig. 6L). Interaction between FUNDC1 and FBXL2 was further consolidated using co-IP in H9c2 cells (Fig. 6M).
FBXL2 binding domain for FUNDC1-FBXL2 binding modality
To discern the potential interaction interface between FUNDC1 and FBXL2, PRISM tool (http://cosbi.ku.edu.tr/prism) was applied to predict their potential interaction interfaces. According to the prediction results from PRISM, FUNDC1 and FBXL2 may form protein interaction interface in F-box (with CYS-33, GLN-37, LYS-40, ASN-43, and LEU-47 as the interaction hotspots) and LRR1 (with ARG-75 as the interaction hotspot) domains of FBXL2 (Fig. 7A). To explore such interaction, truncated mutants of FBXL2 were constructed according to the predicted interaction interfaces with the deletion of either F-box (Delta-F-box) or LRR1(Delta-LRR1) domain (Fig. 7B). Truncated variants of hemagglutinin (HA)–tagged FBXL2 plasmids were transfected with FUNDC1-Flag plasmid in H9c2 cells. Co-IP assays showed that mutation of F-box domain disengaged the interaction of FBXL2 with FUNDC1. However, deletion of LRR1 failed to diminish the interaction between FUNDC1 and FBXL1, suggesting a vital role for the F-box domain of FBXL2 in the direct interaction with FUNDC1 (Fig. 6N). To further evaluate whether HF diet intake alters FUNDC1-FBXL2 interaction, co-IP was executed using rat heart samples following 20-week HF intake (60% calorie from fat, same samples as described in Fig. 1, A to C). These results failed to identify any difference in the FUNDC1-FBXL2 interaction between the two fat diet feeding conditions (Fig. 7C).
Fig. 7. FBXL2 binding domain and bioinformatics prediction for FUNDC1-FBXL2 binding modality.
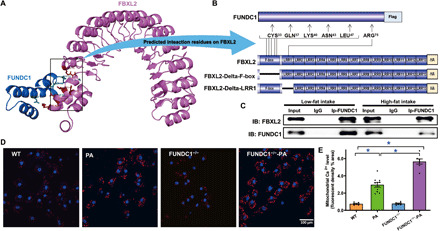
(A) Structure-based protein interaction interface analysis between FUNDC1 and FBXL2. Cartoon represents the predicted FBXL2-FUNDC1 complex structure, where the interaction hotspot residues are labeled. (B) Schematic diagram of domain deletion of FBXL2-HA for co-IP experiments, and the predicted interaction sites on FBXL2 with FUNDC1. One interaction hotspot (ARG-75) is settled on the LRR1 domain of FBXL2, while the others (CYS-33, GLN-37, LYS-40, ASN-43, and LEU-47) are all from the F-box domain. Boxes indicate exons; lines indicate deleted domains. (C) IP analysis of FBXL2 and FUNDC1 in heart tissues from LF- and HF-fed WT mouse groups. (D) Representative images displaying mitochondrial Ca2+ fluorescence in neonatal cardiomyocytes from WT and FUNDC1−/− mice challenged with palmitic acid (PA, 0.5 mM for 8 hours). (E) Pooled data of mitochondrial Ca2+ levels. Mean ± SEM, n = 7 to 10 cell batches per group, *P < 0.05 versus between indicated groups.
Effect of FUNDC1 loss on palmitic acid–induced mitochondrial Ca2+ overload, mitochondrial damage, and cardiomyocyte dysfunction through a FBXL2-IP3R3–mediated mechanism
Considering the essential role of mitochondrial Ca2+ and mitochondrial integrity in cardiomyocyte function (24, 28, 29), mitochondrial Ca2+, mitochondrial membrane potential (ΔΨm), and cardiomyocyte function were assessed in palmitic acid–challenged cardiomyocytes isolated from WT and FUNDC1−/− mice. Data in Fig. 7 (D and E) show levels of mitochondrial Ca2+ evaluated using a fluorescence probe in neonatal mouse cardiomyocytes challenged with palmitic acid (0.5 mM) for 8 hours. Palmitic acid significantly elevated mitochondrial Ca2+ levels with a more pronounced action in FUNDC1−/− cardiomyocytes. Furthermore, the cationic lipophilic probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi- dazolylcarbocyanine iodide (JC-1) was used to monitor ΔΨm in cardiomyocytes from WT and FUNDC1−/− mice challenged with palmitic acid (0.5 mM for 8 hours). ΔΨm was assessed using the ratio between red (aggregated JC-1) and green (monomeric JC-1) fluorescence. Quantitative analysis showed that palmitic acid significantly lowered the JC-1 ratio with a more pronounced drop in FUNDC1−/− group, indicating a fall in ΔΨm and mitochondrial injury. The FBXL2 agonist BC-1258 and the IP3R3 inhibitor 2-Aminoethoxydiphenyl borate (2-APB) obliterated palmitic acid–induced collapse in ΔΨm in WT and FUNDC1−/− groups. Moreover, disruption of FBXL2 localization with GGTi-2418, a geranylgeranyl transferase inhibitor, sensitized palmitic acid–induced loss of ΔΨm without eliciting notable effect itself (fig. S2, A to D). Assessment of cardiomyocyte function revealed that palmitic acid compromised cardiomyocyte function (decreased PS, ±dL/dt, prolonged TPS, and TR90), with a more pronounced effect in FUNDC1−/− group. Reminiscent of the ΔΨm response, the FBXL2 agonist BC-1258 and the IP3R3 inhibitor 2-APB negated palmitic acid–induced cardiomyocyte dysfunction in WT and FUNDC1−/− groups. Likewise, disruption of FBXL2 localization with GGTi-2418 accentuated palmitic acid–induced cardiomyocyte dysfunction (fig. S2, E to I).
Effect of FUNDC1 on palmitic acid–induced cytochrome C release through a FBXL2-IP3R3–mediated mechanism
To evaluate the role of palmitic acid– and FUNDC1-elicted responses in mitochondrial integrity and contractile function, cytochrome C levels were monitored in neonatal mouse cardiomyocytes challenged with palmitic acid (0.5 mM for 8 hours) (8). A cohort of cardiomyocytes was transfected with FUNDC1 overnight before challenge with palmitic acid in the presence or absence of pharmacological inhibitors. Compared to the control group, palmitic acid promoted cytochrome C release from mitochondria to cytosol along with nuclear cytochrome C buildup. These adverse effects were effectively reversed by FUNDC1 transfection, FBXL2 activator BC-1258, or the IP3R3 inhibitor 2-APB, with little effect from FUNDC1 vector or inhibitors themselves. Disruption of FBXL2 localization with GGTi-2418 negated FUNDC1-mediated benefits against palmitic acid effects on cytochrome C release. These effects were supported by assessment of nuclear and cytosolic cytochrome C levels using Western blot analysis (fig. S3, A to D).
Effect of FUNDC1 on palmitic acid–induced change in mitochondrial structure
In light of the cardinal role of mitochondrial abnormalities in obesity- or lipotoxicity-induced cardiac injury, we evaluated the role of FUNDC1 in the regulation of mitochondrial homeostasis in the setting of palmitic acid–induced lipotoxicity. Mitochondrial morphology was assessed using immunofluorescence. Palmitic acid exposure (0.5 mM, 8 hours) caused mitochondria from neonatal mouse cardiomyocytes to divide into fragments with reduced mitochondrial length. FUNDC1 transfection, the FBXL2 activator BC-1258, and the IP3R3 inhibitor 2-APB preserved mitochondrial network structure. However, disruption of FBXL2 localization with GGTi-2418 nullified FUNDC1-induced protective actions against palmitic acid–elicited mitochondrial fragmentation (low mitochondrial length). None of the pharmacological inhibitors used here nor FUNDC1 viral vector itself affected mitochondrial structure in the absence of palmitic acid exposure (Fig. 8, A and B).
Fig. 8. Effect of FUNDC1 transfection and/or FBXL2 stimulation on palmitic acid–induced mitochondrial structure in neonatal mouse cardiomyocytes in the presence or absence of FBXL2 localization inhibitor GGTi-2418.
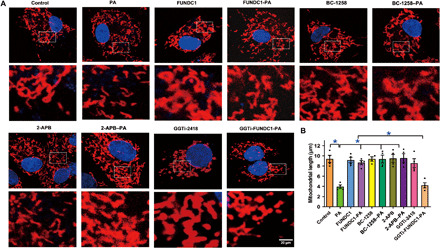
Neonatal cardiomyocytes were transfected with FUNDC1 overnight before incubation with palmitic acid (0.5 mM) for another 8 hours in the presence or absence of FBXL2 activator BC-1258 (10 μg/ml), the IP3R3 inhibitor 2-APB (30 μM), or the FBXL2 colocalization inhibitor GGTi-2418 (15 μM) before assessment of mitochondrial structure using the confocal microscopy. (A) Representative mitochondrial immunofluorescence. (B) Quantification of mitochondrial length. Mean ± SEM, n = 8 cells per group. *P < 0.05 between indicated groups.
Effect of FUNDC1 on palmitic acid–induced change in mitochondrial apoptosis and IP3R levels
To discern the effect of FUNDC1 in lipotoxicity-induced mitochondrial injury, we evaluated the role of FUNDC1 in lipotoxicity-induced mitochondrial apoptosis, disturbed ΔΨm, and IP3R levels. As shown in Fig. 9 (A to D), palmitic acid (0.5 mM, 8 hours) promoted loss in ΔΨm and mitochondrial apoptosis (increased mitochondrial Bax levels and decreased levels of Bcl2 and cytosolic Bax), the effects of which were accentuated by FUNDC1 ablation and reversed by FUNDC1 or FBXL2 transfection. Neither FUNDC1 viral vector nor ablation of FUNDC1 affected levels of Bax or Bcl2 in the absence of palmitic acid challenge. Likewise, palmitic acid up-regulated levels of IP3R2 and IP3R3 without affecting that of IP3R1. FUNDC1 ablation accentuated palmitic acid–induced rise in IP3R3 (but not IP3R2), and the effects were restored to control levels by FUNDC1 or FBXL2 viral vector. Neither FUNDC1 or FBXL2 viral vector nor ablation of FUNDC1 itself affected levels of IP3Rs in the absence of palmitic acid challenge (Fig. 9, E to G). These data support a role for the FUNDC1-FBXL2-IP3R3 axis in lipotoxicity-induced mitochondrial injury.
Fig. 9. Effect of FUNDC1 or FBXL2 transfection on palmitic acid–induced changes in mitochondrial apoptosis, ΔΨm, and IP3Rs in neonatal cardiomyocytes.
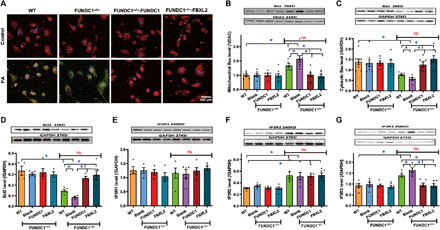
Neonatal cardiomyocytes from WT or FUNDC1−/− mice were transfected with FUNDC1 or FBXL2 overnight before the incubation with palmitic acid (0.5 mM) for an additional 8 hours. (A) Representative fluorescence probing of ΔΨm. (B) Mitochondrial Bax levels. (C) Cytosolic Bax levels. (D) Bcl2 levels. (E) IP3R1 levels. (F) IP3R2 levels. (G) IP3R3 levels. Insets: Representative immunoblots depicting levels of Bax, Bcl2, IP3R1, IP3R2, and IP3R3 using specific antibodies. GAPDH or COXIV (for mitochondria) was used as the loading control. Mean ± SEM, n = 8 cells per group, *P < 0.05 between indicated groups.
Stabilization of FBXL2 and IP3R3 in the face of lipotoxicity challenge
IP3R3 is a key ER protein governing Ca2+ delivery from ER into mitochondria (24). Therefore, levels of IP3R3 and its degrading protein FBXL2 might explain the regulatory role of FUNDC1 against HF intake or lipotoxicity-induced mitochondrial Ca2+ overload and mitochondrial injury. Pulse-chase analysis showed an overt time-dependent drop in FBXL2 levels upon palmitic acid challenge, the effects of which were accentuated by FUNDC1 ablation and reversed by FUNDC1 viral transfection. Moreover, palmitic acid exposure also imposed a time-dependent loss in IP3R3 protein level, the effect of which was absent with FUNDC1 ablation whereas unaffected by viral transfection of FUNDC1 or FBXL2. Application of the protease inhibitor MG132 effectively nullified palmitic acid–induced degradation in FBXL2 and IP3R3 in various experimental settings (Fig. 10, A to D). These findings indicate that FUNDC1 positively and negatively preserves the stabilization of FBXL2 and IP3R3, respectively, through proteasomal degradation.
Fig. 10. Determination of stabilization of FBXL2 and IP3R3 using pulse-chase assay in neonatal cardiomyocytes from WT and FUNDC1−/− mice transfected with FUNDC1 or FBXL2 viral vector.
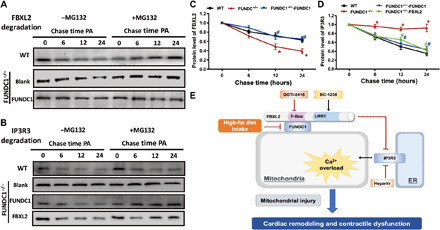
(A) Western blots analysis of palmitic acid (0.5 mM)–induced time-dependent FBXL2 degradation (0 to 24 hours) in neonatal cardiomyocytes from WT or FUNDC1−/− mice with or without FUNDC1 transfection. A cohort of cardiomyocytes was incubated with the proteasomal inhibitor MG132 throughout the course of study. (B) Western blots analysis of palmitic acid (0.5 mM)–induced time-dependent IP3R3 degradation (0 to 24 hours) in neonatal cardiomyocytes from WT or FUNDC1−/− mice with or without FUNDC1 or FBXL2 transfection. A cohort of cardiomyocytes was incubated with MG132 throughout the study. (C) Pooled data of FBXL2 degradation in the absence of MG132. (D) Pooled data of IP3R3 degradation in the absence of MG132. (E) Diagram depicting proposed mechanism for the role of FUNDC1 in HF diet–induced changes in cardiac remodeling and contractile defects. HF intake suppressed FUNDC1–mediated mitophagy, leading to disturbed interaction between FUNDC1 and FBXL2, thus prompting destabilization of FBXL2. FBXL2 loss facilitates IP3R3-mediated Ca2+ mobilization into mitochondria (mitochondrial Ca2+ overload) and RIP-mediated cell death, resulting in mitochondrial injury and cardiac geometric and functional anomalies.
DISCUSSION
The salient findings from our present investigation revealed that deficiency in the mitophagy membrane receptor FUNDC1 overtly accentuated HF diet intake–induced myocardial remodeling and cardiac contractile dysfunction without affecting HF diet–induced obesity, as well as insulin, lipid, and glucose metabolism. FUNDC1-mediated maintenance of myocardial structural and functional responses appears to be dependent on its interaction with F-box protein FBXL2, which stabilizes FBXL2 to promote degradation of IP3R3 (but not other ER proteins such as IP3R1/2, SERCA2a, and RyR2) and mitochondrial integrity (Ca2+ overload, mitochondrial membrane depolarization, mitochondrial apoptosis, and mitochondrial division). Deficiency in FUNDC1, such as occurs in obesity, accentuates HF diet–induced myocardial structural and functional anomalies through disengaging the F-box protein FBXL2, resulting in diminished degradation of ER Ca2+ regulatory protein IP3R3, mitochondrial Ca2+ overload, mitochondrial membrane depolarization, mitochondrial apoptosis, necroptosis, and necrosis, as well as loss of mitochondrial integrity and function. These findings collectively support a rather unique role for FUNDC1 as a therapeutic target in obesity-induced cardiac anomalies.
Diet-induced obesity promotes unfavorable myocardial structural and functional changes including cardiac hypertrophy, interstitial fibrosis, compromised cardiac contractility, and prolonged diastole in concert with intracellular Ca2+ derangement (2, 8, 30). This is in line with our observations of cardiac hypertrophy, prohypertrophic gene expression (ANP, BNP, and GATA4), interstitial fibrosis, enlarged LVESD, reduced fractional shortening, ejection fraction, PS, ±dL/dt, and prolonged TR90 following chronic HF diet intake. A number of biological pathways were identified as enriched in HF diet–fed rat hearts. Of particular relevance, enriched KEGG pathways were closely associated with regulation of apoptosis, mitophagy, and Ca2+ signaling, in line with our recent report in diet-induced obese hearts (31). A heatmap of DEGs displayed several down-regulated and up-regulated autophagy and Ca2+ signaling regulatory components in obese hearts such as UCP2, Bcl2l1, and Atg4d. Our experimental results further show interrupted intracellular Ca2+ homeostasis, apoptosis, necroptosis, necrosis, inflammation, mitochondrial O2− production, membrane depolarization, and mitochondrial injury (mitochondrial ultrastructure, mitochondrial apoptosis, compromised respiration, aconitase, and UCP2) following diet-induced obesity, denoting a vital role for intracellular Ca2+ dysregulation, in particular mitochondrial Ca2+ overload and injury in the onset and progression of obesity cardiomyopathy. Increased mitochondrial apoptosis along with elevated necroptosis (RIPK1/3) and necrosis (MTT and LDH) in conjunction with collapsed ΔΨm may be due to mitochondrial permeation transition pore opening in the face of HF diet challenge. More intriguingly, deficiency in the mitophagy receptor FUNDC1 markedly accentuated HF diet–induced myocardial remodeling (along with prohypertrophic genes), contractile anomalies, apoptosis, and other forms of cell death, inflammation, and mitochondrial injury. These results convincingly support an obligatory role for FUNDC1 in the preservation of cardiac homeostasis in diet-induced obesity. In concert with cardiac contractile defects, intracellular Ca2+ mishandling (mitochondrial Ca2+ overload) and ultrastructural changes, decreased UCP2, mitochondrial respiration, and aconitase activity along with elevated mitochondrial size or number and O2− production were noted following fat diet intake, the effect of which was exacerbated by FUNDC1 deficiency. HF intake promoted down-regulation of F-box protein FBXL2 (possibly due to loss of FUNDC1 in obesity), with a more pronounced drop in FUNDC1−/− mice. With respect to ER Ca2+ regulatory machineries, cardiac up-regulation of IP3R3 and IP3R2 receptors, down-regulation of SERCA2a, and decreased RyR2 activity (T2814 phosphorylation) caused by HF feeding were noted in HF diet–fed mouse hearts and are responsible for feeding Ca2+ from the ER to mitochondria, resequestration of Ca2+ into ER, and releasing Ca2+ from ER/sarcoplasmic reticulum stores, respectively (32). Consistent with our present findings, earlier work demonstrated decreased cardiac SERCA levels and RyR2 Ca2+ release as well as elevated hepatic IP3R1 and IP3R2 in metabolic diseases including diet-induced obesity (33, 34). The disparity in IP3Rs subtypes may be related to tissue specificity. In our hands, FUNDC1 ablation accentuated HF diet–induced changes in IP3R3 but not IR3R2, SERCA2a, or RyR2 activity, suggesting a rather unique role for IP3R3 in FUNDC1-mediated regulation of feeding of Ca2+ from ER into mitochondria, mitochondrial Ca2+ load, and mitochondrial homeostasis. This is supported by our observation of mitochondrial Ca2+ overload in response to HF diet intake or palmitic acid exposure, with a much more pronounced rise with FUNDC1 deficiency. Recent research showed that IP3R3 functions as a binding substrate for FBXL2 (the only F-box protein co-immunoprecipitated with IP3R3) (22). Our results show that HF intake did not overtly affect co-IP binding between IP3R3 and FBXL2. However, FBXL2 was down-regulated with HF intake or FUNDC1 ablation, with an additive effect between the two. In vitro pulse-chase analysis showed that palmitic acid provoked a time-dependent proteasomal degradation of FBXL2, with a more pronounced effect in FUNDC1−/− cardiomyocytes. Moreover, FUNDC1 transfection nullified palmitic acid– or FUNDC1 ablation–induced FBXL2 destabilization. These data likely support the notion that FUNDC1 may stabilize FBXL2. Likewise, palmitic acid–induced degradation of IP3R3 was nearly absent with ablation of FUNDC1, an effect reversed by FBXL2 transfection. These findings indicated that lipotoxicity may effectively promote MG132-dependent proteosomal degradation of FBXL2, resulting in the preservation of IP3R3, in a FUNDC1-dependent manner. This notion is also supported by our in vitro observations that the FBXL2 activator BC-1258 or the IP3R inhibitor 2-APB–mitigated palmitic acid–induced mechanical and mitochondrial injuries, whereas incubation with the FBXL2 colocalization inhibitor nullified FUNDC1-induced benefit against cardiac lipotoxicity.
Derangements of mitophagy, a conservative machinery for mitochondrial quality control through disposal and recycling of injured mitochondria, is evident in obesity complications including cardiomyopathy (2). FUNDC1 preserves mitochondrial homeostasis via mitophagy-mediated ridding of toxic proteins, lipids, and organelles. FUNDC1 deficiency accentuated HF diet intake–induced cardiac remodeling and contractile dysfunction without affecting Pink1-Parkin or BNIP3-mediated mitophagy components, suggesting a unique role of FUNDC1-mediated mitophagy in HF diet–induced cardiac abnormalities. Using MS, PRISM tool, co-IP, and truncated mutation techniques, we observed that FUNDC1 interacts with FBXL2 on Delta-Fbox but not LRR1 (Delta-LRR1), to stabilize FBXL2. Moreover, this interaction is unaffected by chronic HF intake. Given the reported role of IP3R3 as a binding substrate for FBXL2 (35), levels of FBXL2 appear to be essential for degradation of the ER-mitochondrial Ca2+ feeding protein IP3R3 and, therefore, mitochondrial Ca2+ overload, apoptosis, and injury, as observed in this study (Fig. 10E). This notion also received convincing support from our in vitro evidence using gain and loss of function as well as pharmacological regulators targeting on FUNDC1, FBXL2, and IP3R3. It is noteworthy that FUNDC1 protein was also reported to reside on mitochondria-associated ER membranes (MAMs) to regulate Ca2+ release from ER to mitochondria (16). These investigators noted co-IP between FUNDC1 and IP3R2, suggesting the possible role of FUNDC1 as a springboard between IP3R2 and mitochondria. Data from our current study did not favor a major role of IP3R2 (along with SERCA2a, RyR2, and IP3R1) in FUNDC1 deficiency–accentuated cardiac dysfunction in fat diet–induced obesity. The apparent discrepancy between the earlier report (16) and our current study (which denotes an IP3R3-mediated mechanism) suggests that FUNDC1 possibly regulates mitochondrial Ca2+ homeostasis through direct (e.g., IP3R2) or indirect (e.g., FBXL2-mediated IP3R3) interactions with specific ER proteins at different sites (e.g., MAMs). Moreover, involvement of MAMs (where FUNDC1 may reside) adds the complexity of FUNDC1 biological function in different disease settings. MAMs display distinct roles in various pathological settings, with enrichment of MAMs being detrimental in metabolic diseases (20, 34), whereas MAMs exert cardioprotection in heart failure (16).
In summary, findings from this investigation provide evidence that FUNDC1 interacts with FBXL2 to preserve cardiac homeostasis in the face of HF diet–induced obesity. Through FBXL2-IP3R3–dependent regulation of mitochondrial Ca2+ and mitochondrial integrity (depicted in Fig. 10E), FUNDC1 deficiency accentuated HF diet–induced cardiac anomalies including cardiac remodeling, mechanical defect, and intracellular (in particular mitochondrial) Ca2+ mishandling. These findings support the concept that FUNDC1-mmediated mitophagy may serve as a possible target for drug therapy for heart dysfunction in patients with obesity. Further investigation is warranted to unveil the precise mechanism behind FUNDC1-mediated regulation of F-box protein on FBXL2 and mitochondrial Ca2+ mobilization in a clinically relevant setting of obesity heart abnormalities.
MATERIALS AND METHODS
Experimental animals, fat diet feeding, and IPGTT
The experimental procedures were approved by Zhongshan Hospital, Fudan University (Shanghai, China) Institutional Animal Use and Care Committee and were in compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. In brief, 4-month-old male WT and FUNDC1 knockout (FUNDC1−/−) mice were randomly assigned to an LF (10% of calorie from fat, Research Diets Inc., New Brunswick, NJ) or an HF (60% of total calorie from fat, product D12492) diet for 20 weeks (8). Blood glucose, plasma insulin, and serum triglyceride levels were measured using a commercial glucose meter and enzyme-linked immunosorbent assay commercial kits. Following the 20-week feeding regimen, mice were fasted for 12 hours before glucose challenge [2 g/kg body weight, intraperitoneally (i.p.)]. Blood glucose levels were determined using an Accu-Chek glucose analyzer immediately before or 30, 60, and 120 min thereafter. Area under the curve was generated using trapezoidal analysis (8, 36).
Human samples
Cardiac samples were obtained from unsuccessful cardiac transplants from lean (BMI = 24.3 ± 0.9, n = 4) or obese (BMI = 30.3 ± 0.8, n = 4) donors. The protocol was approved by the Sun Yat-Sen Memorial Hospital Ethics Committee.
mRNA sequencing
mRNA sequencing was performed by Novogene (Beijing, China). Standard Illumina protocols were used for sequencing. The FastQC software was used to exam the raw data. The reads (quality score > 28) were mapped to rat genome, according to the genome annotation, and uniquely localized reads were used for the DEG analysis. Gene Ontology and KEGG pathway enrichment analyses were performed using the OmicShare tools, a free online platform for data analysis (www.omicshare.com/tools).
Echocardiographic assessment
Cardiac structure and function were evaluated in anesthetized [ketamine (80 mg/kg) and xylazine (12 mg/kg), i.p.] mice using a two-dimensional guided M-mode echocardiography (Vevo 2100, FUJIFILM VisualSonics, Toronto, ON, Canada) equipped with a 22 - to 55-MHz linear transducer (MS550D, FUJIFILM VisualSonics). LV dimensions were recorded from three consecutive cycles. Fractional shortening was calculated from LVEDD and LVESD using the equation of (LVEDD − LVESD)/LVEDD. Ejection fraction and heart rate were also calculated by Vevo 2100 echocardiography (8, 36).
Histological examination
After anesthesia, hearts were removed and placed in 10% neutral-buffered formalin for 24 hours before fixation in paraffin. Myocardial sections (5 μm) were stained with fluorescein isothiocyanate–conjugated wheat germ agglutinin. Cardiomyocyte cross-sectional areas were calculated on a digital microscope (×400) using the ImageJ (version 1.34S) software. Masson’s trichrome staining was used to estimate interstitial fibrosis. Percentage of fibrosis was shown as the fraction of the light blue–stained area normalized to the total area (37).
Transmission electron microscopy
Small cubic pieces of left ventricles ≤1 mm3 were fixed with 2.5% glutaraldehyde in 0.1 M sodium phosphate (pH 7.4) overnight at 4°C before Epon Araldite embedding. Ultrathin sections (50 nm) were sliced using an ultramicrotome (Ultracut E, Leica) and were stained with uranyl acetate and lead citrate. The specimens were imaged through a Hitachi H-7000 Electron Microscope (Pleasanton, CA) equipped with a Gatan high-resolution digital camera (38).
Isolation of murine cardiomyocytes
After ketamine/xylazine sedation, hearts were removed and perfused with Krebs-Henseleit bicarbonate buffer containing 135 mM NaCl, 4.0 mM KCl, 1.0 mM MgCl2, 10 mM Hepes, 0.33 mM NaH2PO4, 10 mM glucose, and 10 mM butanedione monoxime, and the solution was gassed with 5% CO2 and 95% O2. Hearts were isolated using liberase enzymatic digestion for 20 min. Extracellular Ca2+ was added incrementally back to 1.20 mM over a period of 30 min. Isolated myocytes were used within 8 hours of isolation (8). To evaluate the role of FBXL2 and IP3R3 in HF diet intake– and FUNDC1 ablation–induced changes in cardiac function, a cohort of cardiomyocytes from WT or FUNDC1−/− mice was treated with palmitic acid (0.5 mM) (8) in the absence or presence of the FBXL2 stimulator BC-1258 (10 μg/ml) (35), the FBXL2 colocalization inhibitor GGTi-2418 (15 μM) (22), or the IP3R3 inhibitor 2-APB (30 μM) (39) for 8 hours before the assessment of cardiomyocyte mechanical and mitochondrial properties.
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using an IonOptix soft-edge system (IonOptix, Milton, MA). Cardiomyocytes were field stimulated at 0.5 Hz. Cell shortening and relengthening were assessed including PS, TPS, TR90, and maximal velocities of shortening/relengthening (±dL/dt) (8).
Measurement of intracellular and mitochondrial Ca2+
A cohort of myocytes was loaded with fura-2/acetoxymethyl ester (AM) (0.5 μM) for 15 min, and fluorescence intensity was recorded with a dual-excitation fluorescence photomultiplier system (IonOptix). Fluorescence emissions were detected between 480 and 520 nm; qualitative change in fura-2 fluorescence intensity (FFI) was inferred from the FFI ratio at the two wavelengths (360/380). Fluorescence decay time (single exponential) was calculated as an indicator of intracellular Ca2+ clearance (8). Mitochondrial Ca2+ transients were monitored by loading cells with Rhod-2 AM (1 μM, load for 1 hour), followed by de-esterification for another hour in Rhod-2–free Dulbecco’s modified Eagle’s medium (DMEM). Mitochondrial Ca2+ transients were presented as background-subtracted normalized fluorescence (F/F0) (40). Cells were exposed to light emitted by a 75-W lamp while being stimulated to contract at a frequency of 0.5 Hz. A cohort of Rhod-2–loaded cardiomyocytes was imaged via a confocal microscopy. Fluorescence intensity of Rhod-2 was normalized to that of the control group.
MitoSOX Red fluorescence measurement of mitochondrial O2•−
Cohorts of cells were loaded with MitoSOX Red (2 μM) for 10 min. Cells were rinsed with the perfusion buffer, and MitoSOX Red fluorescence intensity was captured at 510/580 nm using an Olympus BX51 microscope equipped with a digital cooled charged-coupled device camera (41).
Real-time quantitative polymerase chain reaction
Total mRNA was extracted from cardiac tissues using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and treated with deoxyribonuclease I (RNase-free) (Invitrogen). mRNA was then transcripted into complementary DNA (cDNA) with the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time polymerase chain reaction (RT-PCR) was performed on a CFX96TM RT-PCR detection system (Bio-Rad) using the SsoAdvanced Universal SYBR Green Supermix Kit (Bio-Rad). Primers were listed in table S1. Results are expressed relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using a ΔCT method (42).
Western blot analysis
Heart tissues or cardiomyocytes were homogenized and sonicated in a lysis buffer containing 20 mM tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, and a protease inhibitor cocktail. Protein samples were incubated with the anti-UCP2, anti-RIPK1, anti-RIPK3, anti-Bax, anti-Bcl2, anti–Caspase-1, anti-FBXL2, anti-IP3R1, anti-IP3R2, anti-IP3R3, anti-SERCA2a, anti-RyR2, anti–phosphorylated RyR2 (T2814), anti-LC3B, anti-Atg7, anti-Beclin1, anti-Atg5, anti-p62, anti-Pink1, anti-Parkin, anti-FUNDC1, anti-BNIP3, anti–cytochrome C, and anti-GAPDH, anti–voltage-dependent anion channel, or anti-COXIV [cytochrome c oxidase (complex IV)] (loading controls) antibodies. Horseradish peroxidase–coupled secondary antibodies were used. All antibodies were obtained from Cell Signaling Technology (Danvers, MA) or Santa Cruz Biotechnology (Santa Cruz, CA). After immunoblotting, films were scanned and detected with a Bio-Rad calibrated densitometer, and the intensity of bands was normalized with corresponding band intensity of GAPDH (10).
Immunofluorescence detection of inflammation
For immunofluorescent double staining, paraffin-embedded heart sections (3-μm thickness) were deparaffinized, rehydrated by serial immersion in ethanol, and pretreated with EDTA, followed by incubation in 50 mM NH4Cl, 0.1% Triton X-100 and 1% bovine serum albumin. Troponin T (Abcam, #ab8295) and IL-1β (Proteintech Group Inc., #10663-1-AP) were used as primary antibodies. Binding sites of the primary antibodies were revealed with Alexa Fluor 546 goat anti-rabbit and Alexa Fluor 488 goat anti-mouse secondary antibodies (Molecular Probes, Invitrogen, San Diego, CA). Nuclei were stained with 4′,6-diamidino-2-phenylindole (Life Technologies). Samples were visualized with a fluorescence microscope (Olympus, Berchem, Belgium) (43).
Structure-based protein interaction interface analysis between FUNDC1 and FBXL2
The protein structures of FUNDC1 were predicted by template-based homology structure modeling tool SWISS-MODEL (www.swissmodel.expasy.org), using Protein Data Bank (PDB) structure 2IP6, chain A (covering residues 82 to 131; sequence identity, 10.00%) and 3BK6, chain A (covering residues 74 to 256; sequence identity, 21.64%) as the template, respectively. The experimental structure of FBXL2 was downloaded from the PDB database (PDB ID: 6O60, chain C). Structures of proteins were submitted to the PRISM tool (http://cosbi.ku.edu.tr/prism) to predict their potential interaction interface. Prediction results were visualized by the PyMol tool (http://pymol.org).
Co-immunoprecipitation
The co-IP assay was performed using a Pierce Co-IP kit (Pierce, IL) as per the manufacturer’s instructions. One hundred micrograms of purified anti-FBXL2, anti-FUNDC1, or anti-flag antibodies were coupled with resin. Protein samples (1 mg) were then exposed to the antibody-coupled resin for 2 hours. Protein-antibody complexes were eluted in 50-μl elution buffer after mixing and washing. The eluted protein samples were subjected to immunoblotting with corresponding antibodies (38).
Plasmid constructs
FBXL2 (NM_178624.6) full-length, F-box domain deletion (FBXL2-Delta-F-box), and LRR1 domain deletion (FBXL2-Delta-LRR1) tagged with HA were cloned in pEGFP-C1 and purchased from Dongxuan Genes (Kunshan, China). FUNDC1 (NM_173794.4) tagged with Flag were cloned in pcDNA3.1+ and purchased from GenScript (Nanjing, China).
Immunopurification and MS
Human embryonic kidney–293T cells were transfected with FUNDC1-Flag plasmid, and 48 hours later, cell lysates were immunoprecipitated with the anti-Flag magnetic beads. Precipitates were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently stained with Coomassie blue. Stained protein bands were cut into small pieces and digested for peptide extraction. Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was applied for proteomic data analysis with the extracted peptides.
Neonatal cardiomyocyte isolation and viral transfection
Neonatal (1- to 2-day-old) WT mice were sterilized with 75% ethanol. Hearts were harvested and were cut into small pieces before trypsin digestion. Cardiomyocytes were centrifuged at 800g for 10 min at room temperature, and cell pellets were resuspended in DMEM containing fetal bovine serum (20%) with 1% penicillin and streptomycin before plating in an uncoated dish for 1 hour at 37°C (38). To evaluate the role of FBXL2 in HF diet intake–induced changes in mitochondrial integrity and cardiac function, a cohort of cardiomyocytes from WT or FUNDC1−/− mice was treated with palmitic acid (0.5 mM) (8) in the absence or presence of the FBXL2 stimulator BC-1258 (10 μg/ml) (35), the FBXL2 colocalization inhibitor GGTi-2418 (15 μM) (22), and the IP3R3 inhibitor 2-APB (30 μM) (39) for 8 hours before the assessment of cardiomyocyte mechanical and mitochondrial properties. To assess the role of FUNDC1 and FBXL2 in palmitic acid–induced responses, adenovirus overexpressing FUNDC1 (sequence, pAdeno-MCMV-Fundc1-3Flag-P2A-EGFP) or FBXL2.
Measurement of ΔΨm
ΔΨm was displayed by change in the ratio between red (aggregated JC-1) and green (monomeric JC-1) fluorescence. Briefly, cells were treated with JC-1 probe (2 μg/ml) for 30 min at 37°C. Samples were visualized using a fluorescence microscope (Olympus, Berchem, Belgium) or measured at an excitation wavelength of 490 nm and an emission wavelength of 530/590 nm using a spectrofluorimeter (SpectraMax Gemini XS, Spectra Max, Atlanta, Georgia). Fluorescence intensity was expressed as the 590- to 530-nm emission ratio (44).
Mitochondrial electron transport chain enzymatic activity (cytochrome c oxidase, succinate/cytochrome c reductase, and reduced nicotinamide adenine dinucleotide/succinate cytochrome c reductase)
Cytochrome c oxidase (CCO) activity was detected by monitoring the change in absorbance at 550 nm (∆A550) resulting from the oxidation of reduced cytochrome c. CCO activity was monitored until ∆A550 was ~0.6 U, at which point potassium cyanide (0.17 mM) was added to terminate the CCO reaction. Succinate (12.5 mM) was included to detect succinate–cytochrome c reductase activity by monitoring ∆A550. Next, malonate (17 mM) was added to terminate succinate–cytochrome c reductase. Reduced nicotinamide adenine dinucleotide (NADH) (2.8 nM) was added, and NADH–cytochrome c reductase activity was evaluated by monitoring ∆A550. The rate of cytochrome c oxidation or reduction (nanomole per minute) was derived using a molar extinction coefficient of 19,600 for cytochrome c (45).
Cellular viability detection
Necrosis was evaluated using MTT assay and LDH release assay with commercial kits (G020-1-1 and G020-2-2) from Jiancheng Biotech (Nanjing, China) (46).
Aconitase activity
Mitochondrial aconitase, an iron-sulfur enzyme that resides in citric acid cycle, is oxidized through removal of an iron from the [4Fe-4S] cluster. Mitochondrial fractions were resuspended in 0.2 mM sodium citrate. An aconitase activity assay kit (Aconitase-340 assay, OxisResearch, Portland, OR) was used as per the manufacturer’s instructions (47).
Assessment of cytochrome C
Cytochrome C levels were evaluated using immunofluorescent and immunoblotting. For immunofluorescence, cells were incubated with 4% paraformaldehyde for 30 min and were then permeabilized using 0.05% Triton X-100 for 30 min. Goat serum albumin (10%) was used to incubate cells for 1 hour at room temperature. Samples were incubated at 4°C overnight with an anti–cytochrome C antibody (1:1000; Abcam, 95029). Images were visualized using an Olympus IX81 inverted microscope using FV10-ASW 1.7 software. Relative fluorescence intensity of nuclear cytochrome C was analyzed using an Image-Pro Plus 4.5 software (NIH, Bethesda, MD, USA) (43).
Mitochondrial structure
To assess changes in mitochondrial structure, cells were fixed in 4% paraformaldehyde and were stained using a TOM20 antibody. Confocal microscope was used to capture single-cell images, which were analyzed using ImageJ 1.47v software to obtain the average of mitochondrial length. At least 30 cells were analyzed (15).
Pulse-chase analysis
Cells were radiolabeled with [35S]-Met (100 mCi) for the indicated durations in normal culture condition. Washed with phosphate-buffered saline, cells were chased in the complete DMEM (with 20% fetal bovine serum) for the indicated duration. Whole-cell lysates were made in tris-buffered saline (pH 7.6) as previously described. Lysates were immunoprecipitated with Protein G plus beads coated in the indicated antibodies. Precipitated proteins were eluted with SDS-PAGE loading buffer and analyzed through Western blots (43).
Statistical analysis
Data were expressed as means ± SEM. Statistical significance (P < 0.05) for all variables was estimated by one-way analysis of variance (ANOVA) followed by a Tukey’s test for post hoc analysis. All statistics was performed with GraphPad Prism 4.0 software (GraphPad, San Diego, CA).
Supplementary Material
Acknowledgment
We appreciate the excellent bioinformatics assistance from Q. Cui from the Department of Biomedical Informatics, Peking University, Beijing, China. Funding: Work in our laboratory has been supported by the National Key R&D Program of China (2017YFA0506000), Natural Science Foundation of China (91749128 and 81770261), Science and Technology Innovation Project of the Chinese Academy of Medical Sciences (Health and Longevity Pilot Special Project 2019-RC-HL-021), and the Training Program of Excellent Academic Leaders of Shanghai Health Mission (2018BR25). Author contributions: J.R., M.S., H.Z., A.A., Y. Zhou, J.T., and Y. Zhang performed the experiment. A.A. and J.R.S. participated in the discussion, graph drafting, and manuscript editing. J.R. and Y. Zhang conceived the study and drafted and proofed the manuscript. All authors approved the final submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The datasets used and/or analyzed supporting the findings of this study are available in this paper or the Supplementary Materials. Any other raw data that support the findings of this study are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc8561/DC1
REFERENCES AND NOTES
- 1.Emdin C. A., Khera A. V., Natarajan P., Klarin D., Zekavat S. M., Hsiao A. J., Kathiresan S., Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA 317, 626–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y., Sowers J. R., Ren J., Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 14, 356–376 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y., Ren J., Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol. Ther. 161, 52–66 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Lavie C. J., Ozemek C., Carbone S., Katzmarzyk P. T., Blair S. N., Sedentary behavior, exercise, and cardiovascular health. Circ. Res. 124, 799–815 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Reynolds A., Mann J., Cummings J., Winter N., Mete E., te Morenga L., Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 393, 434–445 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Alpert M. A., Karthikeyan K., Abdullah O., Ghadban R., Obesity and cardiac remodeling in adults: Mechanisms and clinical implications. Prog. Cardiovasc. Dis. 61, 114–123 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Cavalera M., Wang J., Frangogiannis N. G., Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 164, 323–335 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S., Wang C., Turdi S., Richmond K. L., Zhang Y., Ren J., ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: Role of CaM kinase II, histone H3K9 methyltransferase SUV39H, Sirt1, and PGC-1α deacetylation. Int. J. Obes. 42, 1073–1087 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Ren J., Sowers J. R., Zhang Y., Metabolic stress, autophagy, and cardiovascular aging: From pathophysiology to therapeutics. Trends Endocrinol. Metab. 29, 699–711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceylan A. F., Wang S., Kandadi M. R., Chen J., Hua Y., Pei Z., Nair S., Ren J., Cardiomyocyte-specific knockout of endothelin receptor a attenuates obesity cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3339–3352 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Li Z. L., Eirin A., Ebrahimi B., Pawar A. S., Zhu X. Y., Lerman A., Lerman L. O., Cardiac metabolic alterations in hypertensive obese pigs. Hypertension 66, 430–436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gustafsson A. B., Dorn G. W. II, Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 99, 853–892 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez-Resendiz S., Prunier F., Girao H., Dorn G., Hausenloy D. J.; EU-CARDIOPROTECTION COST Action (CA16225) , Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 24, 6571–6585 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H., Wang Y., Li W., Chen H., du L., Liu D., Wang X., Xu T., Liu L., Chen Q., Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 15, 1882–1898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H., Zhu P., Wang J., Zhu H., Ren J., Chen Y., Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 25, 1080–1093 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu S., Lu Q., Wang Q., Ding Y., Ma Z., Mao X., Huang K., Xie Z., Zou M. H., Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136, 2248–2266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao Y., Chen W., Zhong Z., Ding L., Bai H., Chen H., Zhang H., Gu Y., Lu S., Electroacupuncture preconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting mitophagy mediated by the mTORC1-ULK1-FUNDC1 pathway. Biomed. Pharmacother. 127, 110148 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Zhu P., Li R., Ren J., Zhou H., Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 30, 101415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S., Kandadi M. R., Ren J., Double knockout of Akt2 and AMPK predisposes cardiac aging without affecting lifespan: Role of autophagy and mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1865–1875 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S., Lu Q., Ding Y., Wu Y., Qiu Y., Wang P., Mao X., Huang K., Xie Z., Zou M. H., Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation 139, 1913–1936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamilton S., Terentyeva R., Kim T. Y., Bronk P., Clements R. T., O-Uchi J., Csordás G., Choi B. R., Terentyev D., Pharmacological modulation of mitochondrial Ca2+ content regulates sarcoplasmic reticulum Ca2+ release via oxidation of the ryanodine receptor by mitochondria-derived reactive oxygen species. Front. Physiol. 9, 1831 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuchay S., Giorgi C., Simoneschi D., Pagan J., Missiroli S., Saraf A., Florens L., Washburn M. P., Collazo-Lorduy A., Castillo-Martin M., Cordon-Cardo C., Sebti S. M., Pinton P., Pagano M., PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lock J. T., Smith I. F., Parker I., Spatial-temporal patterning of Ca2+ signals by the subcellular distribution of IP3 and IP3 receptors. Semin. Cell Dev. Biol. 94, 3–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C.-H., Wei Y.-H., Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 24, 70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Xu X., Ren J., MTOR overactivation and interrupted autophagy flux in obese hearts: A dicey assembly? Autophagy 9, 939–941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo R., Zhang Y., Turdi S., Ren J., Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: Role of autophagy. Biochim. Biophys. Acta 1832, 1136–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X., Hua Y., Sreejayan N., Zhang Y., Ren J., Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J. Mol. Cell Biol. 5, 61–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vannuvel K., Renard P., Raes M., Arnould T., Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 228, 1802–1818 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Lee J. S., Hou X., Bishop N., Wang S., Flack A., Cho W. J., Chen X., Mao G., Taatjes D. J., Sun F., Zhang K., Jena B. P., Aquaporin-assisted and ER-mediated mitochondrial fission: A hypothesis. Micron 47, 50–58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Yuan M., Bradley K. M., Dong F., Anversa P., Ren J., Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension 59, 680–693 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Gong Y., Li G., Tao J., Wu N. N., Kandadi M. R., Bi Y., Wang S., Pei Z., Ren J., Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165855 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Chernorudskiy A. L., Zito E., Regulation of calcium homeostasis by ER redox: A close-up of the ER/Mitochondria connection. J. Mol. Biol. 429, 620–632 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Miranda G., Romero-Garcia T., Barrera-Lechuga T. P., Mercado-Morales M., Rueda A., Impaired activity of ryanodine receptors contributes to calcium mishandling in cardiomyocytes of metabolic syndrome rats. Front. Physiol. 10, 520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arruda A. P., Pers B. M., Parlakgül G., Güney E., Inouye K., Hotamisligil G. S., Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen B. B., Glasser J. R., Coon T. A., Mallampalli R. K., Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell Death Dis. 4, e759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turdi S., Ge W., Hu N., Bradley K. M., Wang X., Ren J., Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J. Mol. Cell. Cardiol. 55, 117–129 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y., Hu N., Hua Y., Richmond K. L., Dong F., Ren J., Cardiac overexpression of metallothionein rescues cold exposure-induced myocardial contractile dysfunction through attenuation of cardiac fibrosis despite cardiomyocyte mechanical anomalies. Free Radic. Biol. Med. 53, 194–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S., Ge W., Harns C., Meng X., Zhang Y., Ren J., Ablation of toll-like receptor 4 attenuates aging-induced myocardial remodeling and contractile dysfunction through NCoRI-HDAC1-mediated regulation of autophagy. J. Mol. Cell. Cardiol. 119, 40–50 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Huang X., Jin M., Chen Y. X., Wang J., Chang Y., Yuan Q., Yao K. T., Ji G., ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 8, 1276–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie A., Zhou A., Liu H., Shi G., Liu M., Boheler K. R., Dudley S. C., Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes. PLOS ONE 13, e0200448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ceylan-Isik A. F., Guo K. K., Carlson E. C., Privratsky J. R., Liao S. J., Cai L., Chen A. F., Ren J., Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: Role of mitochondrial biogenesis. Hypertension 53, 1023–1031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q., Ren J., mTOR-Independent autophagy inducer trehalose rescues against insulin resistance-induced myocardial contractile anomalies: Role of p38 MAPK and Foxo1. Pharmacol. Res. 111, 357–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H., Toan S., Zhu P., Wang J., Ren J., Zhang Y., DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res. Cardiol. 115, 11 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Ma H., Li S. Y., Xu P., Babcock S. A., Dolence E. K., Brownlee M., Li J., Ren J., Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J. Cell. Mol. Med. 13, 1751–1764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Xu X., Bucala R., Ren J., Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J. Am. Heart Assoc. 2, e000439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Zhu P., Li R., Ren J., Zhang Y., Zhou H., Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics 10, 384–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren J., Yang L., Zhu L., Xu X., Ceylan A. F., Guo W., Yang J., Zhang Y., Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: Role of Sirt1-mediated autophagy regulation. Aging Cell 16, 976–987 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc8561/DC1