

Protein-mediated tethering of vesicles in the extracellular matrix contributes to microcalcification formation.
Abstract
Extracellular vesicles (EVs) including plasma membrane–derived microvesicles and endosomal-derived exosomes aggregate by unknown mechanisms, forming microcalcifications that promote cardiovascular disease, the leading cause of death worldwide. Here, we show a framework for assessing cell-independent EV mechanisms in disease by suggesting that annexin A1 (ANXA1)–dependent tethering induces EV aggregation and microcalcification. We present single-EV microarray, a method to distinguish microvesicles from exosomes and assess heterogeneity at a single-EV level. Single-EV microarray and proteomics revealed increased ANXA1 primarily on aggregating and calcifying microvesicles. ANXA1 vesicle aggregation was suppressed by calcium chelation, altering pH, or ANXA1 neutralizing antibody. ANXA1 knockdown attenuated EV aggregation and microcalcification formation in human cardiovascular cells and acellular three-dimensional collagen hydrogels. Our findings explain why microcalcifications are more prone to form in vulnerable regions of plaque, regulating critical cardiovascular pathology, and likely extend to other EV-associated diseases, including autoimmune and neurodegenerative diseases and cancer.
INTRODUCTION
Considerable molecular understanding of membrane vesicle trafficking within and between cells related to cell growth and maintenance, neurotransmission, and regulated insulin secretion has been achieved (1–4). Yet, pathophysiological roles of vesicles outside of cells, termed extracellular vesicles (EVs), are less defined. Intracellularly, vesicles serve as transporters with vesicle and cell membrane proteins mediating tethering at membrane contact sites by serving as a bridge between membranes (5), a process often followed by SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor)–driven membrane fusion. Membrane tethering also occurs in the absence of SNARE-driven fusion, for example, in mitochondrial dynamics and endoplasmic reticulum tethering to mitochondria (6). Whether vesicle tethering occurs outside of cells in acellular areas of extracellular matrix (ECM) is unknown.
EVs, including plasma membrane–derived microvesicles and endosomal-derived exosomes, are well-established mediators of mineralization in bone (7). Smooth muscle cell (SMC)–and valvular interstitial cell (VIC)–derived EVs serve as vascular and valvular calcification nucleation sites (8–11). Mechanisms by which EVs generate microcalcifications hold great clinical interest as they contribute to atherosclerotic plaque rupture and heart failure (12, 13), components of cardiovascular disease, the leading cause of death worldwide. Cardiovascular cells secrete EVs that aggregate and promote mineralization (8), but how EVs aggregate and generate ectopic calcification is unknown. Established EV cargos involved in ECM mineralization include the phosphate metabolizing enzyme tissue nonspecific alkaline phosphatase (TNAP), annexin A5 (ANXA5), and ANXA6 that may act in calcium-binding functions involved in formation of hydroxyapatite mineral (9–11, 14–16). However, mechanistic functions of annexins in calcification remain largely unclear. Whether annexins are involved in EV aggregation is unknown, although ANXA5 is unlikely to function in this manner as human ANXA5 does not aggregate lipid membranes (17). In addition to cardiovascular diseases, EV and ectopic calcifications associate with autoimmune and neurodegenerative diseases and cancer. Intracranial calcifications occur in oligodendroglioma brain cancer, and oligodendroglioma cells release EVs containing ECM-degrading proteins that can contribute to malignant invasion (18). In neurodegenerative diseases, including Alzheimer’s disease, ECM alterations also occur. Vascular calcification is associated with increased risk of dementia and Alzheimer’s disease (19). Chemically inhibiting EV secretion reduces amyloid plaques that are associated with Alzheimer’s disease, whereas injection of EVs into mouse brains promotes amyloid beta aggregation and plaque formation (20). Systemic sclerosis autoimmune disease is also associated with ectopic calcification, and human systemic sclerosis platelet–derived EVs induce inflammation, ECM changes, and fibrosis in mice (21). These reports support a role of EVs in mediating disease pathology, particularly in conditions associated with ectopic calcification. Therefore, we hypothesized that EVs contain tethering proteins that promote EV aggregation in ECM, driving critical pathology in cardiovascular and other EV-associated diseases. Here, we present a method to distinguish microvesicles and exosomes at a single-EV level and use cardiovascular disease as a model to demonstrate a new functional role of annexin in tethering EVs, primarily microvesicles, which promotes EV aggregation and microcalcification formation.
RESULTS
Human cardiovascular tissue SMC EVs and VIC EVs aggregate and calcify
To determine cellular origins of calcifying EVs, we examined EVs in examples of common cardiovascular pathology, calcified human carotid artery, and aortic valve tissues. Using transmission electron microscopy, we observed EVs originating from SMCs in calcified artery and VICs in calcified valve tissues (Fig. 1, A and B). SMCs in arteries and VICs in aortic valve are predominant cell types in these tissues and both have been associated with EV-mediated cardiovascular calcification (8–11). However, our electron microscopy analysis does not exclude likely involvement of other cell types, including macrophages (15), contributing to observed EVs in cardiovascular tissues. In agreement with EVs being derived from plasma membrane and endosomal pathways (9, 10, 16), we observed microvesicles originating by plasma membrane budding and, to a much lesser extent, multivesicular body release of exosomes in human artery tissue SMCs and aortic valve tissue VICs (Fig. 1, A and B). Multivesicular bodies were observed extremely rarely in comparison to EV budding from plasma membrane, which were very frequently observed on cells. As such, microvesicle release may be a predominant means through which EV aggregation occurs in cardiovascular tissue. Notably, we observed EVs in acellular ECM regions tethered to each other and aggregated in calcified human artery and valve tissues (Fig. 1, A and B). Supporting an EV origin of cardiovascular microcalcification, aggregated and calcifying EVs localized within acellular collagen ECM of calcified human artery and valve tissues (Fig. 1C). Using density-dependent scanning electron microscopy, we observed calcifying EV aggregates in diseased human artery and valve tissues (Fig. 1D), further supporting an EV origin of cardiovascular microcalcifications. Together, these data suggest that SMCs in human artery and VICs in human aortic valve tissues release EVs that may aggregate in acellular collagen ECM by binding to each other and then mineralize to form microcalcifications.
Fig. 1. Human cardiovascular tissue SMCs and VICs released EVs that aggregated and calcified in acellular ECM.
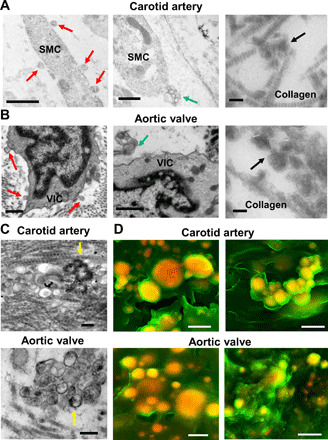
(A) SMCs in calcified human carotid artery and (B) VICs in calcified human aortic valve tissue transmission electron microscopy images (n = 5 donors, with representative images shown). Red arrows indicate EVs that likely budded from plasma membrane (scale bars, 500 nm), blue arrows indicate multivesicular bodies likely being released (scale bars, 500 nm), and black arrows indicate aggregated EVs in acellular collagen ECM (scale bars, 100 nm). (C) Transmission electron microscopy images of aggregated and calcifying EVs (yellow arrows indicate EVs with membrane hydroxyapatite formation) in collagen ECM in human carotid artery and aortic valve tissues (n = 5 donors, with representative images shown; scale bars, 200 nm). (D) Density-dependent scanning electron microscopy images of aggregated microcalcifications (yellow/orange color) in human carotid artery and aortic valve tissue ECM (green color); scale bars, 1 μm (n = 5 donors with two representative images shown).
Osteogenic conditions alter human cardiovascular EV protein composition
To investigate EV protein mechanistic contributions to calcification, we assessed whether human vascular and valvular EV protein composition changed under calcifying conditions. While other cell types, including macrophages, contribute to calcification pathology (15), we focused on SMC- and VIC-derived EVs obtained from both normal media (NM) and calcifying osteogenic media (OM) cultured cells. To do this, we used label-free quantitative proteomics to assess human vascular and valvular cell EV protein composition in calcification-promoting OM. EVs were isolated in a serial centrifugation method, first centrifuging conditioned media at 2500 times gravity to exclude cell debris and apoptotic bodies and then at 100,000 times gravity to pellet EVs. We characterized our proteomics starting material by three distinct methods (Western blot, nanoparticle tracking analysis, and electron microscopy) that together validated our proteomics starting material, which is purified EV. We first assessed EV purity by comparing abundances of EV marker proteins and cellular organelle marker proteins. Western blot of equal loaded (total protein content) EVs and cell lysates showed a clear separation of EVs with our isolation method (fig. S1A). To further exclude a possibility of apoptotic vesicle contamination in our isolation, we also assessed cells for apoptosis markers cleaved caspase 3 and cleaved poly(adenosine diphosphate–ribose) polymerase (PARP), which were not detected by Western blot in NM- or OM-incubated cells (fig. S1B). Next, we used nanoparticle tracking analysis to confirm that our starting material was a correct size for EVs. No significant differences were measured by nanoparticle tracking analysis for human coronary artery SMC– and aortic VIC–derived EV quantity or size (ranging from about 50 nm to around 1 μm, with a majority of EVs in a 50 to 200 nm range, a correct size range of microvesicles and exosomes) in OM compared to NM, with each cell type analyzed separately (Fig. 2A). Last, we used electron microscopy to validate that our SMC and VIC EV proteomics starting material was composed of EVs, which revealed EVs distinguishable with lipid bilayers (Fig. 2B). Of 863 detected proteins, 103 were increased and 9 were decreased in SMC EVs in OM (data file S1, Fig. 2C). Of 549 detected proteins, 53 were increased and 29 were decreased in VIC EVs in OM (data file S1, Fig. 2C). Proteomics further validated an enrichment of EVs in our samples through identification of EV markers, including flotillin-1 and tetraspanins (data file S1). Our quantitative proteomics data demonstrated that protein composition changes occur in primary human SMC EVs and VIC EVs under osteogenic conditions.
Fig. 2. Human cardiovascular EV protein composition was altered under osteogenic conditions.
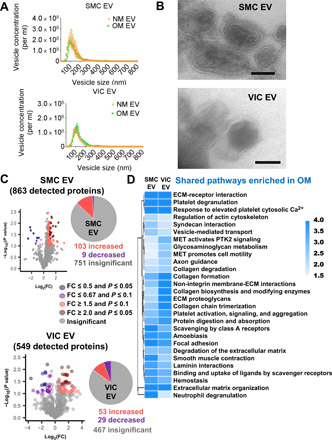
(A) Nanoparticle tracking analysis of size and abundance of human SMC EV and VIC EV in conditioned media from cells cultured in NM or OM and used in proteomics analysis; error bars are means ± SD from three donors. (B) Transmission electron microscopy images of EVs isolated from SMC and VIC conditioned media and used in proteomics analysis (n = 3 pooled donors, with representative images shown); scale bars, 100 nm. (C) Proteomics protein volcano plot analysis for EVs derived from human SMC (n = 9 donors) and VIC (n = 7 donors) conditioned media. Plots show increased, insignificant, and decreased EV protein abundances, along with pie charts of total detected protein distribution in OM relative to control NM. SMC EV and VIC EV full proteomics datasets and statistical analysis included in data file S1. (D) Heatmap of shared enriched pathways based on significantly changed proteins in OM from EVs obtained from both SMCs (n = 9 donors) and VICs (n = 7 donors). SMC EV and VIC EV full pathway datasets and statistical analysis included in data file S1, and full labeled pathway networks included in figs. S2 and S3. PTK2, protein tyrosine kinase 2.
Human SMC EVs and VIC EVs contain tethering proteins
Next, we examined whether EVs isolated from human SMCs and VICs had tethering proteins that could generate calcified EV aggregates aggregates such as those that we observed in human cardiovascular tissues. We performed quantitative pathway analysis (22) to triage candidate tethering proteins from our human EV proteomics datasets. Human SMC EVs and VIC EVs in OM showed enrichment in several pathways compared to EVs in control NM (data file S1 and figs. S2 and S3). Comparative analysis revealed that human SMC EVs and VIC EVs had shared enriched pathways in OM (Fig. 2D). Annexins associate with several of these shared enriched pathways. ANXA1 was identified in an enriched “smooth muscle contraction” pathway in our EV datasets (data file S1 and Fig. 2D). “Vesicle-mediated transport” pathway was enriched in SMC EVs and VIC EVs in OM (data file S1 and Fig. 2D). Linking annexins to this pathway, ANXA1 mediates intracellular endosomal transport (23). Response to elevated cytosolic calcium pathway was enriched in both SMC EVs and VIC EVs in OM (data file S1 and Fig. 2D). We previously reported that mitochondrial dynamics and intracellular calcium signaling are altered in cardiovascular calcification (24). Annexins associate with plasma membrane in response to cytosolic calcium (25), where they can be loaded onto EVs, supporting the role of altered calcium signaling in annexin EV loading in OM. Together, our proteomics and pathway analysis revealed that human SMC EVs and VIC EVs contain proteins, including ANXA1 that could tether EVs.
To assess the role of annexin in mediating EV tethering that leads to aggregation, we compared abundances of ANXA1 and related binding proteins in human SMC EVs and VIC EVs under calcifying conditions. ANXA1 was increased in SMC EVs and was detected in VIC EVs in OM (data file S1 and Fig. 3A). Comparative proteomics analysis revealed 16 up-regulated proteins out of 529 detected in both SMC EVs and VIC EVs in OM, including an ANXA1 calcium-dependent binding partner, S100 calcium-binding protein A11 (S100A11) (data file S1 and Fig. 3A). Supporting the role of S100A11 in promoting annexin activity, formation of an S100A11-ANXA1 complex is required for proper ANXA1-mediated endosomal trafficking in HeLa cells (23). Our data support the idea that annexin tethering–mediated EV aggregation could occur both with and without increased annexin content, as increased S100 calcium and annexin-binding proteins in EVs could also induce annexin tethering activity. As ANXA1 was highlighted by our pathway analysis and has not been previously demonstrated to regulate vascular or valvular calcification, we focused on assessing the mechanistic roles of ANXA1 in EV tethering, aggregation, and microcalcification formation. Further supporting the rationale for potential ANXA1 mechanistic involvement in cardiovascular calcification is the fact that in bone osteosarcoma Soas-2 cells, ANXA1, ANXA2, ANXA6, and ANXA7 are enriched in mineralizing EVs (7). ANXA6 is an established regulator of cardiovascular calcification (9, 10), which additionally supports our rationale and focus on determining unknown mechanistic involvement of ANXA1 in this disease.
Fig. 3. Human SMC EV and VIC EV contained tethering proteins.
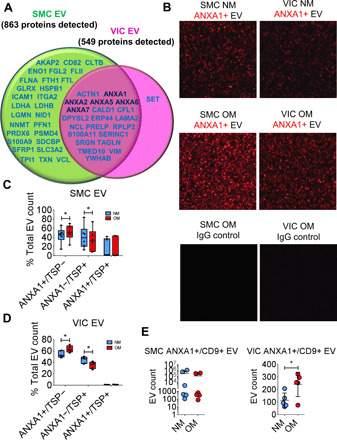
(A) Venn diagram of detected EV proteins increased in OM, with the green circle including proteins detected in SMC EV (n = 9 donors) and pink circle including proteins detected in VIC EV (n = 7 donors). Annexin proteins (ANXA1, ANXA2, ANXA5, ANXA6, and ANXA7) detected in both SMC EV and VIC EV and increased in OM in either or both are indicated in dark blue. Additional proteins detected and increased in OM in SMC EV (green circle), VIC EV (pink circle), or both SMC EV and VIC EV (overlapping region) are indicated in light blue. Full comparative EV proteomics dataset and statistical analysis included in data file S1. (B) Representative single-EV microarray images obtained using the ExoView R100 platform with SMC EV (n = 6 donors) and VIC EV (n = 5 donors). Top and middle panels show ANXA1+ EV in NM and OM with ANXA1+ capture, and bottom panels show lack of ANXA1+ EV with IgG negative control capture. (C) Box and whisker plots for single-EV microarray assessment of ANXA1+ EV, tetraspanin+ EV (TSP+; CD9/CD63/CD81), and ANXA1+/TSP+ EV from SMCs (n = 6 donors, *P < 0.05, analyzed by Wilcoxon matched pairs test) and (D) VICs (n = 5 donors, *P < 0.05 analyzed by Welch’s t test) cultured in NM or OM. Data presented as a percentage of the total EV count (ANXA1+, TSP+, and ANXA1+/TSP+ EV combined) for each donor. (E) Single-EV microarray EV count for ANXA1+/CD9+ EV in SMCs (n = 6 donors, error bars are means ± SD) and VICs (n = 5 donors, error bars are means ± SD, *P < 0.05 analyzed by Welch’s t test) cultured in NM and OM.
Single-EV microarray detected ANXA1 on microvesicles
EVs are a heterogeneous population composed of both microvesicles and exosomes. To assess which EV subtypes ANXA1 localized within our study, we tailored a customized method to detect microvesicles and exosomes by performing multicolor single-EV analysis, which we term “single-EV microarray.” This state-of-the-art method allowed us to assess EV protein content and largely distinguish microvesicles from exosomes at a single-EV level. Beyond an initial 2500 times gravity spin to get rid of any cell debris in conditioned media, no other purification steps were required, which increased our overall yield and maintained vesicles in their natural state compared to traditional bulk EV isolation methods using ultracentrifugation or density gradients. Microarray chips were custom-printed with validated ANXA1 and tetraspanin (CD9, CD63, and CD81) capture antibodies, incubated with SMC and VIC conditioned media containing EVs, then incubated with tetraspanin and ANXA1 detection antibodies, and imaged using the ExoView R100 platform (Fig. 3B). ANXA1 knockdown samples and immunoglobulin G (IgG) control antibodies were used to demonstrate signal specificity. With single-EV microarray, we found that ANXA1 small interfering RNA (siRNA) treatment resulted in a strong reduction of ANXA1+ signal on SMC EV and VIC EV (fig. S4A). Demonstrating a calcium dependency of ANXA1 on EV, ANXA1+ EV signal was reduced by chelating calcium either before or after binding of EVs on microarray chips (fig. S4B). Assessing vesicles for tetraspanins and ANXA1 allowed us to quantitatively distinguish likely microvesicles (ANXA1+/tetraspanin−) from likely exosomes (ANXA1−/tetraspanin+) at a single-EV level, although we cannot exclude the existence of subpopulations of tetraspanin+ microvesicles or ANXA1+ exosomes. As expected, ANXA1 was observed mostly on microvesicles (Fig. 3, C and D). ANXA1 was also on a small fraction (about 1 to 2% of the total EV population in VIC donors and about 1 to 2% in two-thirds of the assessed SMC donors, with a higher portion observed in one-third of SMC donors) of a putative exosome (tetraspanin+ EVs) subpopulation (Fig. 3, C and D, and fig. S4C). In addition to detecting increased ANXA1+ EVs from SMCs (Fig. 3C) and VICs (Fig. 3D) in OM, single-EV microarray detected increased ANXA1+/CD9+ EVs from VICs in OM (Fig. 3E and fig. S4D). Together, these data suggest that annexin was predominantly on microvesicles and potentially a putative subpopulation of ANXA1+/tetraspanin+ exosomes or ANXA1+/tetraspanin+ microvesicles under osteogenic conditions.
ANXA1 localized to tethered EVs in calcified human cardiovascular tissues
To localize ANXA1 ex vivo, we performed immunostaining with a knockout validated monoclonal antibody on noncalcified and calcified human carotid artery and aortic valve tissues. Calcified human carotid artery and aortic valve tissues contained immunoreactive ANXA1 (Fig. 4A). ANXA1 presence near aggregated microcalcifications in human carotid artery and aortic valve ECM was confirmed using ANXA1 immunofluorescence with a near-infrared calcium tracer (OsteoSense 680) (26) and the collagen probe CNA35-OG488 (Fig. 4B) (27). To evaluate ANXA1 presence on aggregated EVs in calcified human cardiovascular tissue, we used immunogold electron microscopy with a knockout-validated ANXA1 monoclonal antibody. Both calcified human carotid artery and aortic valve tissues showed ANXA1 on the surface of EVs in close proximity (Fig. 4C and fig. S5A). As ANXA1 is a putative marker of microvesicles (28), these data further support the idea that microvesicles contribute to aggregated EV formation. ANXA1 protein was also observed in noncalcified human artery and valve tissues (fig. S5B). We validated that total abundance of annexins was not changed between calcified and noncalcified human cardiovascular tissues using our previously reported untargeted proteomics dataset (22), which showed no statistical differences in total abundance of ANXA1, ANXA2, ANXA5, ANXA6, or S100A11 in calcified human valve tissue compared to noncalcified valve tissue (fig. S6A). In addition, we assessed annexin mRNA levels in calcified and noncalcified human carotid artery and aortic valve tissues, which revealed no significant changes in annexin mRNA expression in calcified tissues (fig. S6B). In agreement with our ex vivo data, ANXA1, ANXA2, and ANXA6 mRNA levels were not significantly altered in SMCs or VICs cultured in OM compared to NM (fig. S6C). These data suggested that trafficking changes of calcium-binding proteins most likely account for increased protein abundance of annexins and related binding partners on EVs under osteogenic conditions rather than transcriptional induction.
Fig. 4. ANXA1 knockdown attenuated human SMC and VIC calcification.
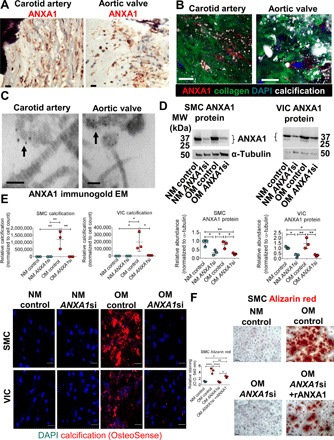
(A) ANXA1 immunohistochemistry (red color) in calcified (hematoxylin stain, purple color) human carotid artery and aortic valve tissues. Scale bars, 20 μm; n = 5 donors, with representative images shown and additional images included in fig. S5. (B) Human carotid artery and aortic valve tissue ANXA1 immunofluorescence (red color) near aggregated microcalcifications (OsteoSense 680, white color) in collagen ECM (CNA35-OG488, green color). Scale bars, 20 μm; n = 5 donors, with representative images shown. (C) Calcified human carotid artery and aortic valve tissue transmission electron microscopy (EM) of ANXA1 immunogold labeling (black dots) on EV in close proximity (arrow) in collagen ECM. Scale bars, 100 nm; n = 5 donors, with representative images shown, and additional images included in fig. S5. (D) Human SMC and VIC ANXA1 protein from cells cultured in control NM or OM and incubated with control siRNA (control) or ANXA1 siRNA (ANXA1si); quantification is the sum of both bands. Error bars are means ± SD from three donors. (E) Confocal microscopy images of microcalcifications (red color, OsteoSense 680 staining; blue color, DAPI) in SMCs and VICs cultured in NM or OM with control siRNA or ANXA1si; n = 3 donors, error bars are means ± SD; scale bars, 20 μm. (F) Alizarin red calcification stain and quantification for SMCs cultured in NM or OM with control, ANXA1si, or ANXA1si and recombinant human ANXA1 (rANXA1). Scale bars, 100 μm; error bars are means ± SD from three donors. ****P < 0.0001, **P < 0.01, and *P < 0.05, analyzed by analysis of variance (ANOVA).
ANXA1 knockdown attenuated calcification of human SMCs and VICs
To assess the roles of ANXA1 in cardiovascular cell calcification, we knocked down ANXA1 in human cardiovascular cells undergoing osteogenic differentiation. ANXA1 siRNA-incubated primary human SMCs and VICs had significantly reduced ANXA1 mRNA levels and ANXA1 protein (fig. S6C and Fig. 4D), without altering other annexin mRNA levels (fig. S6C). ANXA1 siRNA suppressed OM-induced TNAP activation in human SMCs and VICs (fig. S7A). Several annexins have been reported to act as calcium channels, with this action of ANXA5 suggested to induce TNAP activity (14). Whether annexins truly act as calcium channels is still a matter of debate. We therefore looked at an additional inducer of TNAP, p38, which is activated by ANXA1 in human lung fibroblasts (29). ANXA1 siRNA suppressed p38 protein induction by OM in human SMCs, supporting a role of an ANXA1-p38-TNAP pathway in cardiovascular cells (fig. S7B). In addition, ANXA1 siRNA attenuated microcalcification formation in OM, visualized with a near-infrared calcium tracer in cultured human SMCs and VICs (Fig. 4E), without altering EV size or quantity (fig. S7C). Addition of recombinant human ANXA1 to culture media partially reversed a low OM calcification response of human SMCs incubated with ANXA1 siRNA, visualized with Alizarin red stain (Fig. 4F). Together, our data demonstrate that ANXA1 is a previously unidentified regulator of human cardiovascular calcification.
ANXA1 is a calcium-dependent human cardiovascular EV tethering protein
To demonstrate ANXA1 as an EV tethering protein, we used several in vitro biochemical approaches. We first validated that ANXA1 localized on surface of both SMC EVs and VIC EVs and could therefore tether EVs, by incubating EVs with EDTA to chelate calcium and release surface bound ANXA1. Addition of EDTA to human SMC EVs and VIC EVs led to increased ANXA1 release from EVs (fig. S7D). These data agreed with our single-EV microarray data that suggested that ANXA1 is on EV surfaces and interacts with EV membrane via calcium-dependent binding.
Next, we validated that ANXA1 vesicle tethering can induce vesicle aggregation by two different methods that assess tethering and aggregation activity. Annexins bind phosphatidylserine, a phospholipid enriched in the membranes of calcifying EVs (10, 15). We first generated single vesicles that could be visualized using confocal microscopy by swelling phosphatidylserine on polyvinyl alcohol–coated coverslips. We immunopurified green fluorescent protein (GFP)–tagged human ANXA1 to incubate with these swelling-generated vesicles (fig. S8A). Incubating GFP-tagged human ANXA1 with phosphatidylserine vesicles revealed enriched ANXA1 at tethered membrane contact sites of single vesicles (Fig. 5A and movie S1). We next confirmed that recombinant human ANXA1 tethering promoted EV aggregation using phosphatidylserine vesicles of the same size as the majority of cardiovascular EVs (around 50 to 200 nm in diameter). In this second assay, phosphatidylserine vesicles were generated by filter extrusion and verified to be around 50 to 200 nm in diameter by nanoparticle tracking analysis (fig. S8B). Addition of recombinant human ANXA1 protein to extrusion generated vesicles led to both time- and concentration-dependent vesicle aggregation, assessed by measuring turbidity with a 96-well plate reader (Fig. 5B). As annexins bind calcium, calcium regulation of annexin membrane interaction and calcium-altered EV charge could affect annexin-mediated EV tethering that drives aggregation. Physiologic levels of calcium at 1 mM (30) in vesicle buffer without addition of ANXA1 did not induce vesicle aggregation (Fig. 5B). We also confirmed that vesicle aggregation was calcium dependent using GFP-tagged human ANXA1 (fig. S8C). These data demonstrate that factors in addition to physiologic levels of calcium, such as annexins, are required for vesicle tethering that induce EV aggregation. Next, we assessed pH regulation of annexin-mediated EV aggregation, as microcalcifications are more prone to form in artery and valve tissue areas with slightly acidic pH. ANXA1 EV aggregation was highest in slightly acidic pH (6.75) compared to more basic (8.25) or more acidic (5.25) pH (Fig. 5C). Demonstrating calcium dependence for ANXA1-mediated vesicle aggregation, addition of a calcium chelator, EDTA, attenuated ANXA1-mediated vesicle aggregation (Fig. 5D). Demonstrating ANXA1 specificity, we also suppressed vesicle aggregation with the addition of a knockdown-validated N-terminal ANXA1 neutralizing antibody (Fig. 5E). Together, these data show that ANXA1 is a calcium-dependent tethering protein for human cardiovascular EVs and that tethering of single EVs together by annexin promotes EV aggregation.
Fig. 5. ANXA1 tethered vesicles and its inhibition suppressed vesicle aggregation.
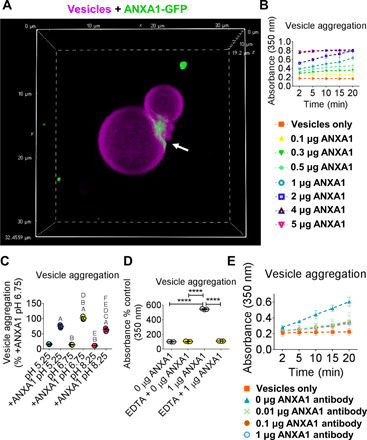
(A) Confocal Z stack image of DiR′ [DiIC18(7) 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide]–labeled (purple color) swelling produced vesicles with ANXA1-GFP (green color) binding at vesicle tethered sites (white arrow) (x and y planes, 32.4559 μm, and z, 19.2 μm); n = 3 experiments, with representative image shown. Movie of ANXA1-GFP vesicle binding included as movie S1. (B) ANXA1 time- and concentration-dependent vesicle aggregation with extrusion produced vesicles; error bars are means ± SD from three experiments. (C) pH-dependent vesicle aggregation with extrusion produced vesicles with 1 μg of ANXA1 (+ANXA1) or without ANXA1; data are means ± SD from three experiments. (D) EDTA and (E) N-terminal ANXA1 neutralizing antibody aggregation inhibition of extrusion produced vesicles. Error bars are means ± SD from three experiments. ****P < 0.0001, A < 0.0001 versus pH 5.25, B < 0.0001 versus +ANXA1 pH 5.25, C < 0.05 versus +ANXA1 pH 5.25, D < 0.0001 versus pH 6.75, E < 0.0001 versus +ANXA1 pH 6.75, F < 0.0001 versus pH 8.25, analyzed by ANOVA.
ANXA1 knockdown attenuated EV microcalcification in three-dimensional collagen hydrogels, a cardiovascular calcification model
Last, we tested whether ANXA1 knockdown could suppress human EV-mediated microcalcification formation. As ANXA1 siRNA did not alter quantity or size of human SMC EVs and VIC EVs released into conditioned media (fig. S7C), collagen was likely involved in first trapping secreted EVs that go on to tether, aggregate, and form microcalcifications. To assess aggregation and calcification potential of EVs following ANXA1 knockdown, we incubated conditioned media containing EVs in cell-free three-dimensional (3D) collagen hydrogels. Conditioned media containing EVs were added after a 2500 times gravity spin to eliminate cell debris, but without ultracentrifugation to avoid artifact aggregates formed by ultracentrifugation. We previously established this acellular model to study formation and growth of EV-derived microcalcifications (8). We assessed OM conditioned media SMC EV and VIC EV aggregation by labeling EVs with a lipophilic tracer and analyzing size of EV aggregates in 3D collagen hydrogels with confocal microscopy (Fig. 6A). Single EVs are below the imaging resolution of confocal microscopy, which allowed us to specifically analyze aggregated EV size. ANXA1 siRNA in OM decreased the size of both SMC EV and VIC EV aggregates in 3D collagen hydrogels (Fig. 6B). EVs in conditioned media from human SMCs and VICs cultured in OM generated microcalcifications in 3D collagen hydrogels that were attenuated by ANXA1 siRNA, visualized by confocal microscopy with a near-infrared calcium tracer (Fig. 6C). We further demonstrated that EV tethering led to aggregated EV and microcalcification formation by using super-resolution microscopy. Calcified EV aggregates were observed with EVs from both human SMCs and VICs in OM (Fig. 6D). Together, these data demonstrate that ANXA1 promotes EV tethering, leading to EV aggregation and microcalcification formation.
Fig. 6. ANXA1 inhibition suppressed EV microcalcification in 3D collagen hydrogels.
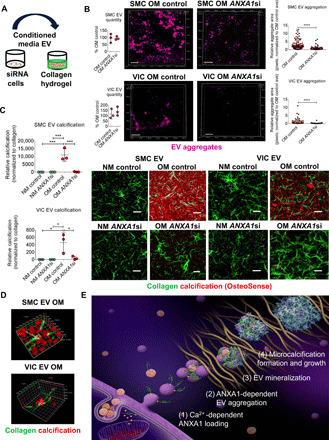
(A) Illustration showing 3D collagen hydrogel EV microcalcification experiment design. (B) Nanoparticle tracking analysis of single-EV quantity in conditioned media used for 3D collagen hydrogel EV experiments (n = 3 donors; analyzed by Welch’s t test). Confocal Z stack images and aggregate size quantification for DiR′-labeled (purple color) OM EV aggregates from SMCs [n = 3 donors with 352 control siRNA (control) and 225 ANXA1 siRNA (ANXA1si) EV aggregate areas quantified] and VICs (n = 3 donors with 65 control and 56 ANXA1si EV aggregate areas quantified). Scale bars, 20 μm; error bars are means ± SD; analyzed by Mann-Whitney test, ****P < 0.0001. (C) EV-generated microcalcifications (OsteoSense 680, red color) in 3D collagen hydrogels (CNA35-OG488, green color) using human SMC EV and VIC EV in conditioned NM or OM with control or ANXA1si; scale bars, 20 μm; n = 3 donors; error bars are means ± SD; ***P < 0.001 and *P < 0.05, analyzed by ANOVA. (D) Super-resolution microscopy images of OM conditioned media SMC EV and VIC EV calcification (OsteoSense 680) aggregates in 3D collagen hydrogels (CNA35-OG488). SMC EV x and y planes, 3200 nm; z, 800 nm; and VIC EV x, 10,800 nm, y, 9600 nm, z, 4800 nm; n = 3 donors, with representative images shown. (E) Working model in which altered calcium signaling may lead to increased calcium-binding proteins, including ANXA1 on EVs that promotes tethering of EVs trapped in ECM, leading to EV aggregation and formation and growth of microcalcifications.
We present a working model for a role of tethering proteins in promoting critical cardiovascular pathology (Fig. 6E): Osteogenic conditions induce TNAP activation and loading onto EVs, producing mineralization-capable EVs (16). Reduced calcium buffering (24) may lead to increased cytosolic calcium that induces annexins and annexin-interacting proteins trafficking to cell membranes and incorporation on EVs, mostly microvesicles but perhaps also a subpopulation of endosomal-derived exosomes. Secreted EVs get trapped in collagen fibers and other ECM components (8), which increases single-EV interactions. Increased calcium-binding and annexin-interacting proteins on EVs trapped in ECM induce ANXA1-dependent EV tethering. EV tethering promotes EV aggregation, mineralization, and the formation and growth of microcalcifications that might lead to plaque instability or heart valve failure.
DISCUSSION
EVs act as mediators of cell-cell communication via transporting protein, lipid, and nucleic acid cargos that are important for maintaining proper cell and organ homeostasis. EVs contribute to regulation of inflammation, tissue repair, and regeneration processes (31). How this goes awry in pathological conditions like cardiovascular calcification leading to changes in EV cargos and aggregation is not clear. We and others have demonstrated that EVs serve as a precursor for microcalcification formation (8–11). Changes in protein transporters, plasma membrane microdomains, and lipid metabolism occur in cardiovascular calcification that can alter EV formation and cargos (9, 16). Changes in mitochondria, calcium buffering, and stress also occur in calcification (16, 24), which could promote ANXA1 EV loading and contribute to slightly acidifying the extracellular environment further inducing EV aggregation, but this remains to be determined. How ANXA1 localizes to EV surfaces is unclear. Calcium allows ANXA1 to interact with membranes including plasma membrane (25), where it may be loaded on budding microvesicles or secreted and then associate with EVs. We have previously shown that cytoskeleton disruption in calcifying vascular cells is corrected by increasing mitochondrial calcium buffering (24). As cytosolic annexins reversibly associate with cytoskeleton and cytoskeleton regulators like S100a11, it is probable that cytoskeleton alterations and reduced calcium buffering contribute to increased ANXA1 EV localization. Calcium signaling alters cytoskeleton involved in budding of microvesicles from plasma membrane and promotes phosphatidylserine exposure on the outer leaflet by promoting flippase, floppase, and scambalase activity (31), which likely results in phosphatidylserine-bound ANXA1 on the surface of budding microvesicles.
Understanding how aggregates form is a fundamental area of research extending from physical to biological science. In biological science, aggregation of nanometer-sized EVs explains the origin of micrometer-sized spherical calcifications that we observed in diseased human artery and valve tissues. By identifying tethering proteins on calcifying EVs, we provide a mechanistic explanation of how EV aggregation occurs. In addition to protein-driven tethering, physical changes including EV charge state may play a role in aggregation. EVs are generally negatively charged, which suppresses EV aggregation (32). Calcifying cardiovascular EVs show enrichment in phosphatidylserine (10, 15), which bears a negative charge. Demonstrating a role of ANXA1 in physiologic EV tethering, we did not observe phosphatidylserine vesicle aggregation without addition of ANXA1 in 1 mM calcium, a physiologic concentration of free ionized calcium in human blood (30). Annexins and associated S100 calcium-binding proteins could potentially further act to neutralize EV negative charge by binding and taking up supraphysiological amounts of positively charged calcium ions to increase EV aggregation potential.
We demonstrate the functional roles of annexins, namely, ANXA1, are more complex than formerly thought by adding a previously undescribed functional role of ANXA1 in EV tethering, leading to aggregation and ectopic calcification. A previous study found that aortic SMC EVs contain ANXA1 among other annexins (10); however, ANXA1 protein quantification, location of ANXA1 on EV surface, and inhibition and function of ANXA1 in cardiovascular calcification was not assessed. The same study suggested that ANXA6 but not ANXA2 or ANXA5 calcium binding may promote hydroxyapatite nucleation in a model that rapidly (16 to 24 hours) induced SMC calcification without altering TNAP levels by incubating cells with high inorganic phosphate (2.5 mM) and calcium (2.7 mM) (10). Culturing coronary artery SMCs and aortic VICs with organic phosphate without adding high calcium, we found that ANXA1 actively mediated calcification via regulation of TNAP activity, ANXA1 EV surface localization, and EV aggregation that promoted formation of microcalcification. Intracellularly, annexins physiologically act in membrane contact functions (23). We have previously shown that normal vascular EVs do not aggregate in collagen hydrogels (8), suggesting that EV aggregation could be more of a pathological response than a normal physiological function. Physiological intracellular membrane contact functions may occur extracellularly in disease when EV cargos and ECM alterations co-occur, inducing aggregation. Occurrence of a related membrane-contact mechanism in acellular ECM could not be previously assumed given lack of cellular membranes in acellular ECM where EV aggregates occur. We demonstrated greater ANXA1-mediated vesicle aggregation in slightly acidic pH (6.75) compared to more acidic (5.25) or basic (8.25) pH. This mechanistic finding explains why spotty microcalcifications are more prone to form nearby vulnerable lipid pool/necrotic core regions of human plaque (13), which are slightly more acidic than regions with sheet-like calcification (33). In addition, our findings help to explain the formation of calcification in valves, which is also more prone to form in a slightly acidic pH around 6.75 than more acidic pH, as demonstrated in a transplanted valve tissue in vivo model (34).
Before our present study, the role of annexins or any other proteins in promoting microvesicle tethering in acellular space and the role of ANXA1 in mediating cardiovascular calcification had not been demonstrated. Tetherin (also called BST2) protein tethers exosomes to each other in conjunction with tethering aggregated exosomes to plasma membrane of HeLa cells incubated with a vacuolar acidification and autophagy inhibitor, bafilomycin (35). We did not detect a change in tetherin protein abundance in SMC- and VIC-derived EVs in OM (data file S1). With single-EV microarray, we found the majority of ANXA1 on microvesicles, whereas exosomes were the EV subset suggested to be tethered by tetherin (35). ANXA1 has been suggested to distinguish microvesicles from tetraspanin-containing exosomes (28). While our findings largely agree with this claim, we found the likely existence of a putative ANXA1+ exosome subpopulation that was missed by this prior study that used a bulk EV isolation method. We cannot exclude the alternative possibility of an ANXA1+/tetraspanin+ microvesicle subpopulation. In either case, we show the existence of an ANXA1+/tetraspanin+ EV subpopulation, strongly supporting a need for analysis of EVs at a single-EV level when making definitive marker claims. Developing tools that assess single EVs is a high-impact area of research that has only recently been obtainable. By using both ANXA1 and several tetraspanins as markers, we further developed a method from which to quantitatively distinguish between microvesicles (ANXA1+/tetraspanin−) and exosomes (ANXA1−/tetraspanin+) on a single-EV level. In all of our VIC donors and two-thirds of our SMC donors, ANXA1+/tetraspanin+ EV made up about 2% of the EV population; however, in one-third of the SMC donors, this population made up a little less than 50% of the EV population. Given the low abundance of this EV subpopulation in a majority of our donors and the prior claim of ANXA1 EV being distinct from tetraspanin-containing EVs (28), it is unclear whether this variation exists because of potentially increased tetraspanin localization to the plasma membrane and incorporation on microvesicles in some SMC donors or some other technical reason. Ongoing methodological development that further incorporates assessment of additional EV surface markers and protein cargos trapped within ANXA1+ EV and tetraspanin+ EV will help to further distinguish EV subpopulations and origin at a single-EV level.
Our findings can be extrapolated to other EV- and annexin-associated diseases. Copy number variants with a small recurrent duplication in the ANXA1 gene have been identified in humans that associate with autism spectrum disorders (36), but a genetic association of ANXA1 to human cardiovascular disease has not been established. Elevated annexin levels, including ANXA1, have been associated with a wide range of human diseases, such as rheumatoid arthritis (37) and cancer (38). Additional work is needed to assess the role of EV tethering in a diverse range of pathophysiologic processes, including in cell-cell communication, EV-collagen association, ECM degradation, and extracellular protein aggregate seeding. All of which would be aided by methods developed here to assess contributions of EVs on a single-EV level. We detected ECM-degrading enzymes in our cardiovascular EV proteomics dataset (data file S1) that could be important in cancer metastasis. Microcalcifications are observed in patients with breast cancer (39). ANXA1 is increased in patients with triple-negative breast cancer, and ANXA1 knockdown suppresses invasion of these cancer cells in vitro (38). Whether aggregated EVs release proteases as EV membranes break down, which promotes ECM remodeling and leads to cell invasion, requires further study. Microcalcifications are observed in vascular structures of human hippocampus (40), a vulnerable brain region in the development of Alzheimer’s disease. Brains of patients with Alzheimer’s disease have increased ANXA1 (41). While beyond the scope of our present study, we detected amyloid beta in our human SMC-derived EV proteomics dataset (data file S1). Tethered EVs could be involved in amyloid seeding and aggregation by serving as a scaffold that acts as an early Alzheimer’s disease pathology step accelerating protein aggregate formation.
EV aggregation is a key step in forming microcalcifications (8). Understanding mechanisms by which this aggregation process occurs could lead to development of early anticalcification therapies. Future in vivo studies inhibiting ANXA1-mediated EV aggregation could demonstrate the therapeutic potential of our findings that help mechanistically explain ectopic calcification formation. Translation of tethering inhibition is not trivial, as annexins have beneficial functions, including ANXA1 inflammation resolution demonstrated in atherosclerotic mice (42). While outside the scope of our present study, correcting dysfunctional calcium buffering that may increase ANXA1 binding to EV membranes could be tested in vivo to inhibit ANXA1 action on EV aggregation without impairing inflammation resolution. In addition, we found that treatment with an N-terminal ANXA1 antibody was able to suppress ANXA1-mediated vesicle tethering. As such, another option might be to increase cleavage and release of ANXA1 N-terminal signaling peptide to inhibit EV aggregation while simultaneously promoting inflammation resolution. The N terminus of ANXA1 mediates ANXA1 anti-atherosclerosis effects in hypercholesterolemic mice through the A lipoxin and formyl peptide receptor 2 (42). Complicating potential therapeutic interpretation of this pathway is that while genetic deletion of A lipoxin and formyl peptide receptor 2 increased atherosclerosis in one study using apolipoprotein E–deficient mice, it reduced or had no effect on atherosclerosis in separate studies also using apolipoprotein E–deficient mice or alternatively using low-density lipoprotein receptor-deficient mice (42). Further complicating matters is that inflammation regulation by ANXA1 may be cell type dependent. While ANXA1 generally associates with inflammation resolution, circulating ANXA1 was increased in a small sample of patients with chronic inflammatory lung disease (29), a disease with increased cardiovascular calcification risk (43). In addition, in human lung fibroblasts, ANXA1 activated p38 and increased inflammatory cytokine mRNA levels (29). In agreement, we found that ANXA1 siRNA reduced p38 induced by OM in human SMCs, supporting a likely ANXA1-p38-TNAP calcification pathway in addition to a novel mechanistic role of ANXA1 in aggregating EVs, leading to microcalcification formation and growth.
Our findings open an unexplored area of research involving EV tethering that could pave the way for development of therapeutic strategies in cardiovascular diseases and likely a wide range of other human pathologies including autoimmune and neurodegenerative diseases and cancer. In addition, implementing a single-EV microarray method further developed here to assess both microvesicles and exosomes may help identify novel disease drivers and low-abundance biomarkers in EV-associated diseases.
MATERIALS AND METHODS
Human tissue
Human carotid artery specimens derived from endarterectomy (Brigham and Women’s Hospital Institutional Review Board protocol #1999P001348) and autopsy artery samples were obtained from Brigham and Women’s Hospital (Institutional Review Board protocol #2013P002517/BWH). Human aortic valve tissue was obtained from patients undergoing valve replacement (Institutional Review Board protocol #2011P001703; written informed consent was obtained). Samples were transferred promptly from the operating room on ice and further processed within 30 min of surgical extraction. Samples were visually assessed as calcified or noncalcified tissue. Calcified samples were further validated by incubating sections with OsteoSense 680 (PerkinElmer, Waltham, MA; 1:100), a near-infrared based bisphosphonate calcium tracer, for 1 hour at room temperature.
Transmission electron microscopy
Transmission electron microscopy was performed at the Massachusetts General Hospital Program in Membrane Biology Electron Microscopy Core using a JEOL 1011 electron microscope. For morphology analysis, freshly isolated human tissue was collected directly after surgical removal and was fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer. Tissues were fixed for 24 hours at room temperature. Fixative was decanted, and tissues were rinsed three times with 0.1 M sodium cacodylate buffer. For immunogold electron microscopy, tissues were fixed in 4.0% paraformaldehyde with 0.2% glutaraldehyde in 0.1 M sodium cacodylate buffer for 24 hours at room temperature and then incubated with a knockout-validated ANXA1 antibody (Abcam, Cambridge, MA; #ab214486). For EV analysis, conditioned cell culture media were collected and centrifuged at 2500 rpm for 5 min to remove cell debris. Supernatant was collected and EVs were pelleted by ultracentrifugation (Optima MAX-XP, Beckman Coulter, Brae, CA) at 100,000g for 60 min at 4°C (TLA 120.2 rotor, Beckman Coulter). EV pellets were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer for 2 hours, washed with 0.1 M sodium cacodylate buffer, and processed similarly to human tissues.
Density-dependent scanning electron microscopy
Density-dependent scanning electron microscopy was performed at University College London as previously described (8). Briefly, human carotid artery or aortic valve sections on glass slides were secured to aluminum sample holders with carbon tape, and silver paint was applied to the area immediately surrounding each sample. Samples were then coated with 5-nm carbon (Quorum Technologies Turbo-Pumped Thermal Evaporators model K975X, Lewes, UK). Following coating, samples were imaged on a scanning electron microscope (SEM; Zeiss VP), operated at 10 kV, and equipped with both an inlens detector that recorded secondary electrons and a backscatter electron detector. Images were obtained by imaging a region in inlens mode and subsequently imaging the same region in backscatter mode. Adobe Photoshop CC 2018 was used on stacked images, and the inlens image was assigned to the green channel whereas the backscatter image was assigned to the red channel.
Primary cell culture
Primary human coronary artery SMCs were obtained from Promocell (Heidelberg, Germany) and expanded in SMC growth medium 2 (Promocell) supplemented with epidermal growth factor (0.5 ng/ml), insulin (5 μg/ml), basic fibroblast growth factor-B (2 ng/ml), and 5% fetal bovine serum (FBS). Primary human VICs were obtained from human aortic valve tissue using collagenase digestion (22, 24). Tissue-derived SMCs were confirmed as alpha smooth muscle actin–positive by fluorescence-activated cell sorting (24). Tissue-derived VICs were confirmed as vimentin positive by immunohistochemistry (22). Cells were cultured at 37°C (5% CO2, 90% humidity) and used between passages 1 and 5. Fully confluent control SMCs or VICs were cultured for a total of 14 to 21 days in Dulbecco’s modified Eagle’s medium containing glucose (4.5 g/liter) and l-glutamine (Lonza, Walkersville, MD), supplemented with 10% FBS and 1% penicillin-streptomycin (control condition: NM). Osteogenic differentiation of SMCs and VICs was induced in fully confluent cells cultured for 14 to 21 days in OM, which is NM with the following additions: 10 nM dexamethasone, 10 mM β-glycerol phosphate, and 100 μM l–ascorbic acid 2-phosphate (OM). Media were changed every 3 to 4 days. For EV data collection and assays, the last media change occurred 3 days before using media that were first centrifuged overnight at 100,000 times gravity and 4°C to minimize FBS EV contamination and then sterile-filtered. For recombinant human ANXA1 (R&D Systems, Minneapolis, MN) treatment, SMCs were incubated at each media change with the same 100 ng/ml dose used in a study assessing ANXA1 effects in human lung fibroblasts (29). TNAP activity was analyzed after 14 days in culture using whole-cell lysates and the TNAP enzyme activity assay (BioVision Inc., Milpitas, CA), according to the manufacturer’s protocol. Calcification was analyzed in cells after 21 days in culture by incubating culture wells with OsteoSense 680 (1:100), a near-infrared based bisphosphonate calcium tracer, for 24 hours at 37°C (5% CO2, 90% humidity). Cells were then fixed in 4% paraformaldehyde for 15 min at room temperature. Nuclear staining for cell count analysis was performed with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific), according to the manufacturer’s protocol. Calcification was also analyzed by Alizarin red in cells after 21 days in culture by fixing cells in 4% paraformaldehyde for 15 min at room temperature, washed with distilled water, stained with Alizarin red (Sigma-Aldrich), and washed three times with distilled water. Alizarin red stain was quantified by extracting the stain in 100 mM cetylpyridinium chloride (Thermo Fisher Scientific) with gentle shaking at room temperature for 30 min, and then the absorbance was measured at 540 nm on a SpectraMax M5 96-well plate reader (Molecular Devices, San Jose, CA). RNA knockdown was performed by using 20 nM silencer select validated ANXA1 siRNA (Thermo Fisher Scientific, #4390825 assay ID s1382) or negative control silencer select siRNA (Thermo Fisher Scientific #4390843), and transfection was performed using Lipofectamine RNAi MAX (Thermo Fisher Scientific), according to the manufacturer’s protocol. The siRNA was added at the beginning of experiments and, with each media change, every 3 days until sample collection.
Mass spectrometry
Conditioned cell culture media were collected after cells were incubated in NM or OM for 14 days. Media were changed every 3 to 4 days. Conditioned media were collected on day 14 with the last media change occurring 3 days prior. Conditioned media were centrifuged at 2500g for 5 min to remove cell debris. Supernatant was collected and EVs were pelleted by ultracentrifugation (Optima MAX-XP, Beckman Coulter, Brae, CA) at 100,000 times gravity for 60 min at 4°C (TLA 120.2 rotor, Beckman Coulter). Pellets were suspended in 10 μl of lyse buffer of the PreOmics iST kit for subsequent proteolysis steps, according to the manufacturer’s protocol (PreOmics GmbH, Planegg/Martinsried, Germany). Peptide samples were analyzed on an Orbitrap Fusion Lumos mass spectrometer fronted with an EASY-Spray Source (heated at 45°C) and coupled to an Easy-nLC1000 HPLC pump (Thermo Fisher Scientific). Peptides were subjected to a dual-column setup: an Acclaim PepMap RSLC C18 trap analytical column, 75 μm × 20 mm (precolumn), and an EASY-Spray LC column, 75 μm × 250 mm (Thermo Fisher Scientific). The analytical gradient was run at 300 nl/min from 5 to 21% solvent B (acetonitrile/0.1% formic acid) for 75 min, 21 to 30% solvent B for 10 min, followed by 10 min of 95% solvent B. Solvent A was water/0.1% formic acid. Acetonitrile and water were LC-MS–grade. The Orbitrap analyzer was set to 120 K resolution, and the top N precursor ions in 3 s cycle time within a scan range of 375 to 1500 m/z (mass/charge ratio) (60 s dynamic exclusion enabled) were subjected to collision-induced dissociation [CID; collision energy, 30%; isolation window, 1.6 m/z; AGC (Automatic Gain Control) target, 1.0 × 104]. The ion trap analyzer was set to a rapid scan rate for peptide sequencing (MS/MS). MS/MS data were queried against the Human UniProt database (downloaded on 1 August 2014) and Bovine UniProt database simultaneously (downloaded on 1 August 2014), using the SEQUEST-HT search algorithm, via the Proteome Discoverer Package (version 2.2, Thermo Fisher Scientific). VIC and SMC EV data were searched separately. Any residual bovine peptides coming from the media bovine serum in the culture media were excluded in the final EV protein analysis. Trypsin was set as the digestion enzyme in the software allowing up to four missed cleavages, using a 10 ppm (parts per million) precursor tolerance window and a 0.6-Da fragment tolerance window. Oxidation of methionine was set as variable modifications, and carbamidomethylation of cysteine was set as a fixed modification. The peptide false discovery rate was calculated using a percolator provided by Proteome Discoverer, and peptides were filtered based on a 1.0% false discovery rate. Peptides assigned to a given protein group (master protein), and not present in any other protein group, were considered as unique and used for quantification. A minimum of two unique peptides were included for each dataset. To quantify peptide precursors detected in the MS1, but not sequenced from sample to sample, we enabled the “Feature Mapper” node. Chromatographic alignment was done with a maximum retention time shift of 10 min and a mass tolerance of 10 ppm. Feature linking and mapping settings were as follows: a minimum retention time tolerance of 0 min, mass tolerance of 10 ppm, and a minimum signal-to-noise ratio of 5. Precursor peptide abundances were based on their chromatographic intensities, and total peptide amount was used for normalization. Assessment of calcified and noncalcified human valve tissue proteomics for annexins and S100A11 was performed using our previous reported nontargeted proteomics dataset (22).
Network analysis
Pathway enrichment analysis was performed by testing the statistically significant proteins from proteomics datasets for overrepresentation in biological pathways by a hypergeometric test. The resulting P values were adjusted for multiple comparisons using the Benjamini and Hochberg method for controlling false discovery rate to obtain q values. In the network representation of significantly enriched pathways (q < 0.05), nodes represent pathways and connections (edges) between nodes represent the shared proteins between the pathways. Node size was made proportional to the significance of enrichment measured in units of −log(q value). Edge thickness was made proportional to the number of overlapping proteins between the two connected pathways in units of the Jaccard index, which is defined as
where sA and sB are the set of proteins detected in proteomics that belong to pathways A and B, respectively. Edges with a Jaccard index < 0.1 were discarded in the visualization for clarity. For the pathway enrichment analysis, the canonical pathways from KEGG, Biocarta, and Reactome were considered.
Immunohistochemistry and immunofluorescence
Tissues were cut into slices with 7-μm thickness, and cryosections were fixed in acetone. Two sections were analyzed per donor. Following blocking in 4% serum, sections were incubated with ANXA1 antibody (Abcam #ab214486; 1:4000) or IgG negative control antibody (Abcam #ab172730; 1:4000) in place of the primary antibody. For immunohistochemistry, sections were then incubated with biotin-labeled secondary antibody (Vector Laboratories, Burlingame, CA), followed by incubation with streptavidin-peroxidase (KPL, Gaithersburg, MD), and incubated in AEC solution (Dako, Lexington, MA). For immunofluorescence, sections were incubated with OsteoSense 680 (1:100), a near-infrared based bisphosphonate calcium tracer, for 1 hour at room temperature, and CNA35-OG488 (1:50), a collagen probe, for 1 hour at room temperature before fixation. ANXA1 antibody incubated slides were incubated with Alexa Fluor 594–labeled secondary antibody (Life Technologies, Carlsbad, CA; 1:200). Immunofluorescence slides were examined using a confocal microscope A1 (Nikon Instruments Inc., Melville, NY), and all images were processed with Elements 3.20 software (Nikon Instruments Inc.).
ANXA1 vesicle binding and tethering assay
Giant unilamellar vesicles were generated by swelling phosphatidylserine on polyvinyl alcohol. For the confocal-based ANXA1 vesicle binding assay, 12-mm-diameter cover glass circles were coated with 5% polyvinyl alcohol (Millipore, Burlington, MA) in water and left to dry overnight. Dried slides were then coated with 20 μl of phosphatidylserine (10 mg/ml; Avanti Polar Lipids, Alabaster, AL) in chloroform containing 0.1% DiR′ [DiIC18(7) 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide] (Thermo Fisher Scientific) and dried under vacuum. One milliliter of tris buffer (pH 6) was added to cover the coated cover glass circle in a 24-well plate, and giant unilaminar vesicles were formed by swelling at room temperature for 1 hour. Vesicles were detached from the cover glass by gently tapping the plate directly beneath the slide. ANXA1 C terminus–tagged GFP (ANXA1-GFP) was obtained by transfecting human embryonic kidney (HEK) 293 cells (ATCC, Manassas, VA) with human ANXA1-GFP plasmid (OriGene, Rockville, MD, #RG201569). Transfected HEK-293 cells were lysed in Pierce IP lysis buffer (Thermo Fisher Scientific), and 3 mg of total protein lysate was incubated overnight with 10 μl of ANXA1 antibody (Abcam #ab214486) and 50 μl of Dynabeads protein A (Thermo Fisher Scientific) under rotation at 4°C. Dynabeads were washed three times in lysis buffer, and then ANXA1-GFP was eluted by adding 0.2 mM glycine. Eluted proteins were immediately neutralized and diluted in tris buffer (pH 8) and stored at −80°C until use. One-hundred-microliter vesicles were incubated with 2 μg of ANXA1-GFP and 1 mM CaCl2 (Sigma-Aldrich) for 5 min and then imaged on a Nikon confocal microscope. GFP laser settings were adjusted such that no GFP signal was detected in samples without ANXA1-GFP added.
Vesicle aggregation assay
Vesicle aggregation was assessed by using a 96-well plate reader format. Large unilaminar vesicles of ~150 nm in diameter on average were generated by filter extrusion. Briefly, 2.5 mg of phosphatidylserine (Avanti Polar Lipids) was solubilized in 1 ml of buffer containing 10 mM Hepes (Sigma-Aldrich, St. Louis, MO), 100 mM NaCl (Sigma-Aldrich), and 100 μM EDTA (Boston BioProducts, Ashland, MA) (pH 6). The lipid mixture was run through an extruder containing 0.1-μm polycarbonate membranes (Avanti Polar Lipids) 10 times, during which point the lipid mixture went from cloudy to clear in appearance. Vesicles were assessed for correct size and abundance before use by NanoSight nanoparticle tracking analysis. Fifty micrograms of recombinant human ANXA1 (R&D Systems) was resuspended in 500 μl of buffer made the same day as the assays were run: 300 mM sucrose (Sigma-Aldrich), 40 mM l-histidine monohydrochloride (Sigma-Aldrich), 1 mM CaCl2 (Sigma-Aldrich), and 0.5 mM MgCl2 (Sigma-Aldrich) (pH 6). For variable pH vesicle aggregation assessment, all buffers used in the assay were adjusted to the indicated pH (5.25, 6.75, and 8.25). In a 96-well plate, 0 μg (referred to as “vesicles only”) to 5 μg of recombinant ANXA1 was added (1 μg was used for assays unless otherwise stated), and the volume was brought to 73 μl using the same buffer that was used to resuspend the ANXA1 protein. Twenty-seven microliters of extrusion generated vesicles was added to each well and vesicle aggregation was measured at wavelength 350 on a SpectraMax M5 96-well plate reader. For calcium chelation, 0.48 mM EDTA was incubated with 1 μg of human ANXA1 protein for 5 min at room temperature before adding the extrusion generated vesicles, after which the reaction was carried out for 20 min and then aggregation was determined on a plate reader. For ANXA1 neutralizing antibody assessment, 0.01 to 1 μg of N-terminal ANXA1 antibody (Abcam, #ab33061) designed against a synthetic peptide, MAMVSEFLKQAWFIENEEQEYVQTVKSSKGGPGSAVSPYPTFNPSSDVAA, corresponding to the N-terminal amino acid 1-50 region of human ANXA1, was incubated with 1 μg of human ANXA1 protein for 5 min before adding the extrusion generated vesicles. For ANXA1-GFP analysis, 1 μg of protein was added to the aggregation buffer with or without 1 mM CaCl2 and absorbances were assessed at 10 min and compared as a percentage of the control (0 μg of protein) absorbance.
Nanoparticle tracking analysis
Vesicle diameter and particle abundance were determined by NanoSight LM10 (Malvern Instruments, Malvern, UK) nanoparticle tracking analysis. Conditioned cell culture media were collected after cells were incubated in NM or OM for 14 days. Media were changed every 3 to 4 days. Conditioned media were collected on day 14 with the last media change occurring 4 days prior. Conditioned media were centrifuged at 2500 times gravity for 5 min to remove cell debris. Supernatant was then diluted 1:10 in phosphate-buffered saline (PBS) and injected continuously via a syringe pump, with five runs used to calculate the mean and SD for each sample. NanoSight camera gain was set at a constant value of 9, and the threshold value for vesicle detection was set to 2. The size and concentration from the collected data were averaged to obtain the distribution in each sample.
Single-EV microarray
SMC and VIC conditioned media were collected and stored at −80°C after culturing cells in NM or OM for 14 days, changing media every 3 to 4 days, with the last media change occurring 3 days prior. Samples were centrifuged at 2500 times gravity for 15 min to exclude any cell debris and diluted in annexin EV binding buffer (pH 7.4) (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2, and 0.04% NaN3) with 1:2, 1:3, 1:5, 1:10, 1:25, 1:200, 1:250, 1:400, and 1:500 dilutions. Thirty-five microliters of diluted sample was incubated overnight on microarray chips (NanoView Biosciences, Brighton, MA) coated with capture antibodies for CD81 (clone JS-81, NanoView Biosciences), CD63 (clone H5C6, NanoView Biosciences), CD9 (clone H19a, BioLegend), and ANXA1 (BioLegend, #831601; Abcam, #ab214486), or negative control IgG (clone MOPC-21, NanoView Biosciences). Microarray chips were washed three times with annexin binding buffer to remove unbound material, and bound EVs were fixed for 10 min at room temperature. After fixation, microarray chips were washed three more times for 5 min each with annexin EV binding buffer to remove fixative and incubated with fluorescently labeled CD9 (0.1 μg/ml, 488-labeled, clone H19a, BioLegend) and ANXA1 antibodies (0.5 μg/ml, 555-labeled, Abcam, #ab214486, and 647-labeled, BioLegend, #831601) for 1 hour. Last, microarray chips were washed three times with annexin EV binding buffer, rinsed, dried, and then imaged and quantified using the ExoView R100 platform (NanoView Biosciences). ANXA1 detection specificity was assessed using EVs from donors incubated with ANXA1 siRNA for 14 days. Assessment of calcium chelation effects on the release of ANXA1 from EVs was carried out as described with the slight modification of adding EDTA calcium chelator in an incubation solution (NanoView Biosciences) during the overnight EV binding step or during the wash step after overnight binding but before EV fixation. ANXA1+ detection on captured EVs was compared to EVs not incubated with calcium chelator during the EV binding or the postbinding wash step.
EV calcification assay
3D collagen hydrogels were cast as previously described (8). Rat tail collagen type I (Corning, Corning, NY) at a pH 7 to 8 was used, as the collagen forms a hydrogel network in this pH range. Collagen solution (150 μl) was added to chambered cover glass wells (Thermo Fisher Scientific, LAB-TEK, #1.5 borosilicate) and allowed to solidify in a cell culture incubator for 2 hours. Conditioned cell culture media were collected after SMCs and VICs were incubated in NM or OM for 14 days. Media were changed every 3 to 4 days. Conditioned media were collected on day 14 with the last media change occurring 3 days prior. Conditioned media were centrifuged at 2500g for 5 min to remove cell debris. EVs were not further isolated by ultracentrifugation to avoid artifact aggregation. One hundred microliters of EVs in day 14 SMC or VIC conditioned media was added to the collagen hydrogels and incubated at 37°C, replacing media with new day 14 conditioned media containing EV every 3 to 4 days for a total of 21 days. For calcification assessment, on the day before imaging, OsteoSense 680 (PerkinElmer; 1:100) was added to the collagen hydrogels and incubated overnight at 37°C. On the day of imaging, CNA35-OG488 collagen probe (1:400) was added and incubated for 1 hour at 37°C, followed by washing gels in PBS. For EV aggregation assessment, day 14 OM conditioned media were first mixed with the lipophilic tracer 0.1% DiR′ and then added to the collagen hydrogels. Hydrogels with the lipophilic tracer-labeled EV were incubated at 37°C for 3 days and then imaged on a Nikon confocal microscope A1, and the size of EV aggregates was quantified using National Institutes of Health (NIH) ImageJ software.
Calcification quantification
EV aggregation-mediated calcification was imaged by confocal microscopy on a confocal microscope A1 (Nikon Instruments Inc.), and all images were processed with Elements 3.20 software (Nikon Instruments Inc.). Structure illumination microscopy (Zeiss ELYRA Super-Resolution) was performed at the Harvard Center for Biological Imaging to image calcification aggregates in the collagen hydrogels. Assessments of calcification and collagen staining were performed using ImageJ (NIH) analysis of confocal-obtained images in both normal and osteogenic conditioned media treatments. First, the image channels were separated by color (red and green), and the intensities of red staining (calcification) or green staining (collagen) were computed by ImageJ as the integral of the red or green channel histogram. Calcification was normalized to the amount of collagen stain in each well for quantification. For SMC and VIC calcification quantification, calcification was imaged and quantified in a similar manner to EVs, except that normalization was made using total cell count quantified by DAPI staining. Investigators were blinded to the treatment group for quantification.
RNA and protein analysis
RNA was extracted from human tissues and cells using TRIzol (Thermo Fisher Scientific), and cDNA was made using the qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD). Quantitative PCR was performed using the following Taqman probes (Thermo Fisher Scientific): Hs02758991_g1 (GAPDH), Hs00167549_m1 (ANXA1), Hs00743063_s1 (ANXA2), Hs00996187_m1 (ANXA5), and Hs01049082_m1 (ANXA6). Fold changes were normalized to GAPDH and determined by the ∆∆Ct method. Western blot analysis was performed by lysing cells or isolated EVs in RIPA buffer (Thermo Fisher Scientific) containing EDTA-free protease inhibitor (Sigma-Aldrich), loading 20 μg of total protein lysate, and using the following antibodies: α-tubulin (Abcam #ab15246; 1:1000), ANXA1 (Abcam #ab214486; 1:1000), β-actin (Novus Biologicals LLC, Centennial, CO, #NB600-501; 1:5000), CALNEXIN (Cell Signaling, Danvers, MA, #2679S; 1:1000), caspase 3 (Cell Signaling Technology #9662; 1:1000), CD9 (Cell Signaling Technology #13403S; 1:1000), CD63 (Abcam #ab68418; 1:1000), GAPDH (Santa Cruz Biotechnology, Dallas, TX, #sc-47724: 1:200), GM130 (Abcam #ab52649; 1:1000), Histone H3 (Abcam #ab1791; 1:1000), LAMP1 (Abcam #ab25630; 1:1000), p38 (Cell Signaling Technology #8690; 1:1000), phospho-p38 (Cell Signaling Technology #4511; 1:1000), PARP (Cell Signaling Technology #9542; 1:1000), and TOMM20 (Abcam #ab210665; 1:1000). Antibody-positive control for cleaved caspase 3 and cleaved PARP was obtained from Cell Signaling Technology (Jurkat cells and Jurkat cells incubated with cytochrome c control cell extracts, #9663; 10 μl loaded per well). ANXA1 EV surface localization was assessed by isolating human SMC and VIC EVs. Ten milliliters of conditioned media was centrifuged at 2500 times gravity for 5 min to remove any debris and then at 100,000 times gravity for 40 min, and the resulting EV pellet was resuspended in PBS or PBS containing 0.48 mM EDTA and incubated for 30 min at 37°C. EVs were pelleted a second time at 100,000 times gravity for 40 min. The supernatant was collected, and the pellet was resuspended in 100 μl of PBS. Samples were incubated with 6× loading buffer (Boston BioProducts) and boiled for 10 min, before analysis by Western blot. ANXA1 released into the supernatant by EDTA treatment was assessed as being on the EV surface.
Statistical analysis
PRISM software (GraphPad, San Diego, CA) was used to analyze data by analysis of variance (ANOVA) with Tukey’s multiple comparisons test, Welch’s t test, Mann-Whitney test, or Wilcoxon matched pairs test when appropriate. Data were analyzed for normality and equal variance to determine whether applied parametric tests were appropriate using Prism software. Data were plotted as means ± SD, and individual data points were included as dot points. For mass spectrometry analysis, P values were adjusted using the Benjamini and Hochberg method (false discovery rate), and the results were visualized using volcano plots.
Supplementary Material
Acknowledgments
We thank J. Hutcheson and C. Goettsch for helpful discussion with the project rationale and experimental design, and H. Muller-Kahle and NanoView Biosciences (Brighton, MA) for assistance with the single-EV microarray method development and data collection. Funding: This study was supported by a research grant from Kowa Company Ltd. (Tokyo, Japan, to M.A.) and NIH grants R01 HL136431, R01 HL141917, and R01 HL147095, and a Harvard Catalyst Advanced Microscopy Pilot grant (to E.A.). We thank the Harvard Center for Biological Imaging for infrastructure and support and acknowledge NIH SIG award (1S10RR029237-01), which was used to acquire the ELYRA microscope. Author contributions: M.A.R., F.B., F.S., S.K.A., L.H.L., A.H., M.C.B., E.T., K.L., G.D., H.H., S.C.B., S.A.S., S.B., P.L., M.A., and E.A. contributed to the collection and analysis of the data. C.V.C.B. provided the CNA35-OG488 collagen probe. M.A.R. wrote the manuscript. M.A. and E.A. contributed to overall project supervision and funding. All authors contributed to revising and editing of the manuscript. Competing interests: K.L. and G.D. are employed by NanoView Biosciences. Portions of this publication are covered in a pending patent application (no. 62/715,428) submitted by Brigham and Women’s Hospital (Boston, MA) with M.A.R., M.A., and E.A. as listed inventors. P.L. is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Esperion Therapeutics, Ionis Pharmaceuticals, Kowa Pharmaceuticals, Novartis, Pfizer, Sanofi-Regeneron, and XBiotech Inc. P.L. is a member of the scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, IFM Therapeutics, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech Inc. P.L. serves on the Board of XBiotech Inc. P.L.’s laboratory has received research funding in the last 2 years from Novartis. P.L. has a financial interest in XBiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabb1244/DC1
REFERENCES AND NOTES
- 1.Caro L. G., Palade G. E., Protein synthesis, storage, and discharge in the pancreatic exocrine cell: An autoradiographic study. J. Cell Biol. 20, 473–495 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block M. R., Glick B. S., Wilcox C. A., Wieland F. T., Rothman J. E., Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. U.S.A. 85, 7852–7856 (1988ss). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., Schekman R., COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Luo F., Liu X., Südhof T. C., Acuna C., Efficient stimulus-secretion coupling at ribbon synapses requires RIM-binding protein tethering of L-type Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 114, E8081–E8090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao X., Ballew N., Barlowe C., Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 17, 2156–2165 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Brito O. M., Scorrano L., Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Cmoch A., Strzelecka-Kiliszek A., Palczewska M., Groves P., Pikula S., Matrix vesicles isolated from mineralization-competent Saos-2 cells are selectively enriched with annexins and s100 proteins. Biochem. Biophys. Res. Commun. 412, 683–687 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Hutcheson J. D., Goettsch C., Betazzo S., Maldonado N., Ruiz J. L., Goh W., Yabusaki K., Faits T., Bouten C., Franck G., Quillard T., Libby P., Aikawa M., Weinbaum S., Aikawa E., Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 15, 335–343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapustin A. N., Chatrou M. L., Drozdov I., Zheng Y., Davidson S. M., Soong D., Furmanik M., Sanchis P., De Rosales R. T., Alvarez-Hernandez D., Shroff R., Yin X., Muller K., Skepper J. N., Mayr M., Reutelingsperger C. P., Chester A., Bertazzo S., Schurgers L. J., Shanahan C. M., Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 116, 1312–1323 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Kapustin A. N., Davies J. D., Reynolds J. L., McNair R., Jones G. T., Sidibe A., Schurgers L. J., Skepper J. N., Proudfoot D., Mayr M., Shanahan C. M., Calcium regulates key components of vascular smooth muscle cell–derived matrix vesicles to enhance mineralization. Circ. Res. 109, e1–e12 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Cui L., Rashdan N. A., Zhu D., Milne E. M., Ajuh P., Milne G., Helfrich M. H., Lim K., Prasad S., Lerman D. A., Vesey A. T., Dweck M. R., Jenkins W. S., Newby D. E., Farquharson C., Macrae V. E., End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J. Cell. Physiol. 232, 2985–2995 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers M. A., Aikawa E., Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nat. Rev. Cardiol. 16, 261–274 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Kelly-Arnold A., Maldonado N., Laudier D., Aikawa E., Cardoso L., Weinbaum S., Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. U.S.A. 110, 10741–10746 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen N. X., O’Neill K. D., Chen X., Moe S. M., Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J. Bone Miner. Res. 23, 1798–1805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.New S. E., Goettsch C., Aikawa M., Marchini J. F., Shibasaki M., Yabusaki K., Libby P., Shanahan C. M., Croce K., Aikawa E., Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 113, 72–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goettsch C., Hutcheson J. D., Aikawa M., Iwata H., Pham T., Nykjaer A., Kjolby M., Rogers M., Michel T., Shibasaki M., Hagita S., Kramann R., Rader D. J., Libby P., Singh S. A., Aikawa E., Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 126, 1323–1336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee G., Pollard H. B., Highly sensitive and stable phosphatidylserine liposome aggregation assay for annexins. Anal. Biochem. 252, 160–164 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Lo Cicero A., Maikowska I., Nagase H., Di Liegro I., Troeberg L., Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol. 31, 229–233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bos D., Vernooij M. W., de Bruijn R. F., Koudstaal P. J., Hofman A., Franco O. H., van der Lugt A., Ikram M. A., Atherosclerotic calcification is related to a higher risk of dementia and cognitive decline. Alzheimers Dement. 11, 639–647.e1 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Dinkins M. B., Dasgupta S., Wang G., Zhu G., Bieberich E., Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 1792–1800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maugeri N., Capobianco A., Rovere-Querini P., Ramirez G. A., Tombetti E., Valle P. D., Monno A., D’Alberti V., Gasparri A. M., Franchini S., D’Angelo A., Bianchi M. E., Manfredi A. A., Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 10, eaao3089 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Schlotter F., Halu A., Goto S., Blaser M. C., Body S. C., Lee L. H., Higashi H., Delaughter D. M., Hutcheson J. D., Vyas P., Pham T., Rogers M. A., Sharma A., Seidman C. E., Loscalzo J., Seidman J. G., Aikawa M., Singh S. A., Aikawa E., Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation 138, 377–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eden E. R., Sanchez-Heras E., Tsapara A., Sobota A., Levine T. P., Futter C. E., Annexin A1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev. Cell 37, 473–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers M. A., Maldonado N., Hutcheson J. D., Goettsch C., Goto S., Yamada I., Faits T., Sesaki H., Aikawa M., Aikawa E., Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ. Res. 121, 220–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Draeger A., Wray S., Babiychuk E. B., Domain architecture of the smooth-muscle plasma membrane: Regulation by annexins. Biochem. J. 387, 309–314 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aikawa E., Nahrendorf M., Figueiredo J.-L., Swirski F. K., Shtatland T., Kohler R. H., Jaffer F. A., Aikawa M., Weissleder R., Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 116, 2841–2850 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Aper S. J. A., van Spreeuwel A. C., van Turnhout M. C., van der Linden A. J., Pieters P. A., van der Zon N. L., de la Rambelje S. L., Bouten C. V., Merkx M., Colorful protein-based fluorescent probes for collagen imaging. PLOS ONE 9, e114983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeppesen D. K., Fenix A. M., Franklin J. L., Higginbotham J. N., Zhang Q., Zimmerman L. J., Liebler D. C., Ping J., Liu Q., Evans R., Fissell W. H., Patton J. G., Rome L. H., Burnette D. T., Coffey R. J., Reassessment of exosome composition. Cell 177, 428–445.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai T., Li Y., Mai Z., Wen X., Lv Y., Xie Z., Lv Q., Chen M., Wu D., Wu B., Annexin A1 is elevated in patients with COPD and affects lung fibroblast function. Int. J. Chron. Obstruct. Pulmon. Dis. 13, 473–486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eqi M., Kim I., Nichol A., Stachowski E., French C. J., Hart G. K., Hegarty C., Bailey M., Bellomo R., Ionized calcium concentration and outcome in critical illness. Crit. Care Med. 39, 314–321 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Oggero S., Austin-Williams S., Norling L. V., The contrasting role of extracellular vesicles in vascular inflammation and tissue repair. Front. Pharmacol. 10, 1479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martins T. S., Catita J., Rosa I. M., da Cruz E Silva O. A. B., Henriques A. G., Exosome isolation from distinct biofluids using precipitation and column-based approaches. PLOS ONE 13, e0198820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naghavi M., John R., Naquib S., Siadaty M. S., Grasu R., Kurian K. C., van Winkle W. B., Soller B., Litovsky S., Madjid M., Willerson J. T., Casscells W., pH heterogeneity of human and rabbit atherosclerotic plaques; a new insight into detection of vulnerable plaque. Atherosclerosis 164, 27–35 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Steitz S. A., Speer M. Y., McKee M. D., Liaw L., Almeida M., Yang H., Giachelli C. M., Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 161, 2035–2046 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar J. R., Manna P. T., Nishimura S., Banting G., Robinson M. S., Tetherin is an exosomal tether. eLife 5, e17180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Catarina T. C., Conceição I. C., Oliveira B., Coelho J., Sousa I., Sequeira A. F., Almeida J., Café C., Duque F., Mouga S., Rogerts W., Gao K., Lowe J. K., Thiruvahindrapuram B., Walker S., Marshall C. R., Pinto D., Nurnberger J. I., Scherer S. W., Geschwind D. H., Oliveira G., Vicente A. M., Recurrent duplications of the annexin A1 gene (ANXA1) in autism spectrum disorders. Mol. Autism 5, 28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodríguez-García M. I., Fernández J. A., Rodríguez A., Fernández M. P., Gutierrez C., Torre-Alonso J. C., Annexin V autoantibodies in rheumatoid arthritis. Ann. Rheum. Dis. 55, 895–900 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okano M., Kumamoto K., Saito M., Onozawa H., Saito K., Abe N., Ohtake T., Takenoshita S., Upregulated annexin A1 promotes cellular invasion in triple-negative breast cancer. Oncol. Rep. 33, 1064–1070 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Boisserie-Lacroix M., Mac Grogan G., Debled M., Ferron S., Asad-Syed M., Brouste V., Mathoulin-Pelissier S., Hurtevent-Labrot G., Radiological features of triple-negative breast cancers (73 cases). Diagn. Interv. Imaging 93, 183–190 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Peters M. E. M., Kochelkoren R., de Brouwer E. J. M., Koek H. L., Bleys R. L. A. W., Mali W. P. T., Hendrikse J., Rozemuller A. M., de Jong P. A., Histological validation of calcifications in the human hippocampus as seen on computed tomography. PLOS ONE 13, e0197073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ries M., Loiola R., Shah U. N., Gentleman S. M., Solito E., Sastre M., The anti-inflammatory annexin A1 induces the clearance and degradation of the amyloid-β peptide. J. Neuroinflammation 13, 234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirault J., Bäck M., Lipoxin and Resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front. Pharmacol. 9, 1273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams M. C., Murchison J. T., Edwards L. D., Agusti A., Bakke P., Calverley P. M. A., Celli B., Coxson H. O., Crim C., Lomas D. A., Miller B. E., Rennard S., Silverman E. K., Tal-Singer R., Vestbo J., Wouters E., Yates J. C., van Beek E. J. R., Newby D. E., MacNee W.; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators , Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax 69, 718–723 (2014).24473329 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabb1244/DC1