

Abstract
Abasic (AP) sites are one of the most frequently occurring types of DNA damage. They lead to DNA strand breaks, interstrand DNA crosslinks, and block transcription and replication. Mutagenicity of AP sites arises from translesion synthesis (TLS) by error-prone bypass polymerases. Recently, a new cellular response to AP sites was discovered, in which the protein HMCES (5-hydroxymethlycytosine (5hmC) binding, embryonic stem cell-specific) forms a stable, covalent DNA-protein crosslink (DPC) to AP sites at stalled replication forks. The stability of the HMCES-DPC prevents strand cleavage by endonucleases and mutagenic bypass by TLS polymerases. Crosslinking is carried out by a unique, SRAP (SOS Response Associated Peptidase) domain conserved across all domains of life. Here, we review the collection of recently reported SRAP crystal structures from human HMCES and E. coli YedK, which provide a unified basis for SRAP specificity and a putative chemical mechanism of AP site crosslinking. We discuss the structural and chemical basis for the stability of the SRAP DPC and how it differs from covalent protein-DNA intermediates in DNA lyase catalysis of strand scission.
Keywords: DNA-protein crosslink, abasic site, thiazolidine, DNA lyase, HMCES, SRAP
1. Introduction
1.1. AP site formation and outcomes
Apurinic/apyrimidinic (AP, or abasic) sites are one of the most common forms of DNA damage, occurring at rates of 10,000 to 30,000 per cell per day (1–3). AP sites are generated directly by reactive oxygen species or ionizing radiation, and as intermediates during base excision repair of aberrant nucleotides. Frequent exposure of DNA to environmental toxins, UV radiation, and reactive cellular metabolites generates chemically modified nucleobases (4–6). AP sites arise from either spontaneous or DNA glycosylase catalyzed hydrolysis of the N-glycosidic bond that links the modified base to the deoxyribose (7,8) (Fig. 1A). DNA glycosylases remove a large number of alkylated, oxidized, and deaminated bases as the first step of the base excision repair (BER) pathway and are thus primarily responsible for AP sites (9,10).
Fig. 1. Consequences of abasic sites.
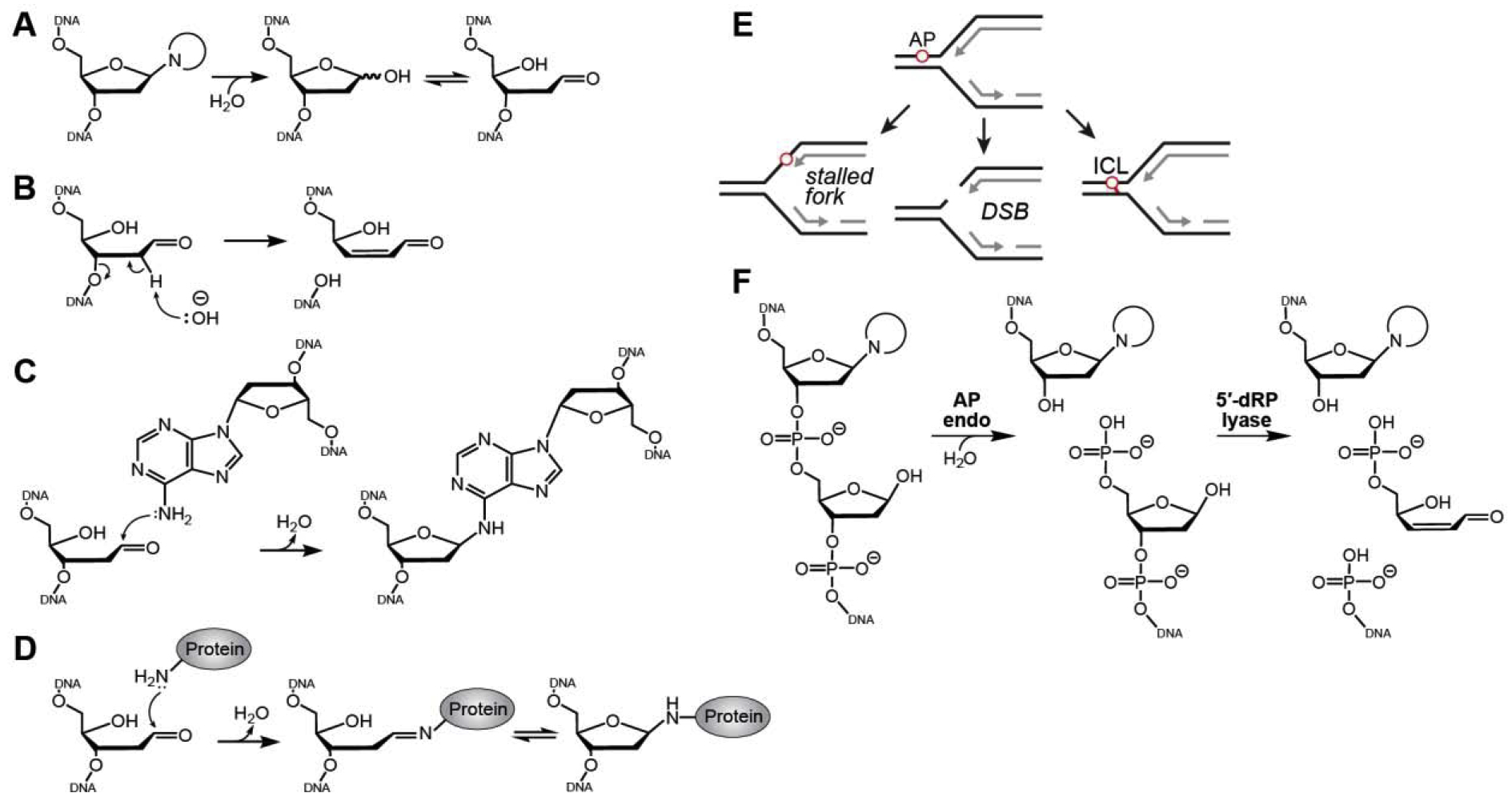
A. AP sites arise from enzymatic and spontaneous hydrolysis of the N-glycosidic bond and exist in either furanose or aldehyde forms. B. Base-catalyzed β-elimination of an AP site generates a strand break. C,D. Formation of an ICL (C) and a DPC (D) by nucleophilic attack of AP site C1ʹ by primary amines in DNA or proteins . E. Consequences of AP sites in the template strand during DNA replication. F. Incision of DNA by AP endonuclease and DNA lyases.
AP sites are unstable and reactive, and can lead to DNA strand breaks, interstrand DNA crosslinks (ICLs), and DNA-protein crosslinks (DPCs) (11–13). In solution, an AP site in DNA is at equilibrium between a ring-closed 2′-deoxy-D-erythro-pentofuranose and a ring-opened aldehyde (Fig. 1A). The AP site exists primarily in the cyclic furanose form as a mixture of α-and β-hemiacetals, with approximately 1% of the sugar in the ring-opened aldehyde form (14,15). This electrophilic aldehyde is susceptible to base-catalyzed β-elimination of the 3′ phosphoryl group, generating a single-strand break (16) (Fig. 1B). AP sites can also react with exocyclic groups of nucleobases on the complimentary strand to generate ICLs (12,17) (Fig. 1C), and with primary amines in proteins to generate DPCs (11) (Fig. 1D). In addition to their reactivity, AP sites lead to stalled replication forks by inhibiting replicative polymerases (18) (Fig. 1E). Moreover, replication forks that encounter an AP site on the template strand can lead to a double-strand break (DSB) (Fig. 1E). For a recent review of biological outcomes of AP sites, we direct the reader to an accompanying review in this special issue (19).
1.2. AP site repair and tolerance
Despite the fact that AP sites form more readily in single-stranded DNA (ssDNA) (7,20), AP site repair occurs within the context of double-stranded DNA (dsDNA) as part of BER (8,21,22). In the second step of BER, AP endonuclease I (APE1) incises DNA 5′ to the AP nucleotide to generate a 3′-OH and 5′-deoxyribose phosphate (5′-dRP) residue (Fig. 1F) (23). The 3′-OH serves as a substrate for long patch repair DNA synthesis by DNA pol δ or short-patch repair synthesis by DNA polymerase β (DNA pol β). DNA pol β also contains 5′-dRP lyase activity that cleaves the 3′ side of the AP site (Fig. 1F) (24).
AP sites that occur in ssDNA, such as those encountered during replication, are not removed by BER. APE1 has much weaker activity for AP sites in ssDNA than in dsDNA (25,26), and indeed, APE1 incision at an AP site in ssDNA would generate a strand break. Until recently, the only known fate of AP sites during DNA replication was lesion bypass by low-fidelity translesion synthesis (TLS) polymerases (18,27). This error-prone damage-tolerance pathway allows replication to continue at the expense of potentially introducing mutations (28,29).
An error-free pathway for repair of replication-associated AP sites was discovered recently that depends on the protein HMCES [5-hydroxymethlycytosine (5hmC) binding, embryonic stem cell-specific (originally C3Orf37)] (30). Cells lacking HMCES exhibit elevated levels and delayed repair of AP sites, as well as increased double-strand breaks and mutation frequency from TLS. HMCES is recruited to replication forks via a direct interaction with proliferating cell nuclear antigen (PCNA), the processivity factor for replicative polymerases and a hub for replication-associated processes (31). Importantly, HMCES forms a covalent DPC with AP sites present in ssDNA, but not dsDNA (30). The HMCES DPC is highly stable and persists in cells (30,32). Subsequent repair of the DPC is unknown, although the protein is eventually degraded via the proteasome (30). The current model is that the highly stable HMCES DPC protects AP sites from nuclease cleavage and mutagenic TLS polymerases (30,33).
2. Function of the SRAP domain in AP site repair
HMCES cellular function and AP-site crosslinking activity depends on a highly conserved SRAP (SOS Response Associated Peptidase) domain, which constitutes the majority of the protein. The SRAP domain is conserved across all domains of life, with a SRAP-containing protein found in most bacteria and eukaryotes, some archaea, as well as viruses and bacteriophages (30,32,34). HMCES is the only SRAP-containing protein present in humans. HMCES and other eukaryotic SRAP proteins have a C-terminal disordered region containing at least one non-canonical PCNA-interacting protein (PIP) box that mediates the direct interaction with PCNA (30), whereas the SRAP domain constitutes the entirety of the bacterial proteins (34). The bacterial SRAP proteins do not appear to have an interacting motif (QL(S/D)LF) for the DNA polymerase III β-subunit (β-clamp) (35)—the prokaryotic ortholog of PCNA (36)—and their potential to interact with the sliding β-clamp is unknown. Crosslinking to AP sites in ssDNA has been demonstrated for the SRAP domain of human HMCES and the E. coli ortholog YedK (30).
SRAP was named for the proximity of the gene to other prokaryotic DNA repair (SOS response) genes, and for its highly conserved triad of cysteine, glutamate, and histidine residues (Cys2, Glu127, and His210 in HMCES) reminiscent of cysteine proteases (34). Cys2 is invariant and required for HMCES function in cells and for the crosslinking activity of HMCES and YedK (30). AP site crosslinking by Cys2 depends on removal of the N-terminal methionine by an aminopeptidase activity that exposes Cys2 at the N-terminus (30,34,37). Cleavage of Met1 by HMCES autoproteolysis has been reported (37), although all organisms contain essential methionyl aminopeptidases that are capable of removing this residue. Crosslinking activity also depends on natural AP sites, as a tetrahydrofuran (THF) analog lacking the hydroxyl group at C1′ does not react with SRAP (30).
The native DPC formed between SRAP and AP sites is a unique DNA repair mechanism. Typically, proteins covalently conjugated to DNA occur either as deleterious lesions (38–40) or as a catalytic intermediate in DNA strand cleavage (lyase) reactions (41–43). For example, the AP lyase activity of a number of DNA repair proteins depends on formation of a transient Schiff base intermediate between protein amino and DNA carbonyl groups (44,45). In contrast, SRAP DPCs are highly stable—on the order of hours in cells and days in vitro at physiological temperature, and resistant to boiling for up to 10 minutes (30,32). Consequently, the SRAP DPC blocks nuclease activity by APE1, even when SRAP has been proteolyzed to leave a DNA-peptide crosslink (30,32). Thus, the high degree of stability of the SRAP DPC is likely important for shielding AP sites from cleavage and explains the HMCES-dependent reduction of spontaneous DNA strand breaks in cells (30).
3. Structural basis for SRAP interaction with DNA
Over the past 14 years, a number of SRAP protein structures have been deposited in the Protein Data Bank (PDB) (Table 1). The first of these were unpublished entries of bacterial SRAP proteins, including E. coli YedK, from structural genomics groups. More recently, structures of the HMCES-SRAP domain (residues 1–270 and lacking the 84-residue C-terminal tail) appeared (Fig. 2). Despite the handful of structures and the high conservation of this protein domain, the function of SRAP was unknown at their time of deposition. In 2019, 12 crystal structures from HMCES-SRAP and YedK were published, 11 of which were in complex with DNA (Table 1). This wealth of new structural information provided insights into the mechanism of these fascinating proteins, as well as a more detailed understanding of their interaction with DNA.
Table 1.
SRAP protein crystal structures
| Protein | DNAa | PDBID | Reference |
|---|---|---|---|
| H. sapiens HMCES SRAP domain | None | 5KO9 | (48) |
| H. sapiens HMCES SRAP domain | Non-covalent overhang 5′-CCAGACGTTG-3′ 3′-GGTCTG-5′ | 6OEA | (48) |
| H. sapiens HMCES SRAP domain | Non-covalent overhang 5′-CCAGACGTT-3′ 3′-GGTCTG-5′ | 6OEB | (48) |
| H. sapiens HMCES SRAP domain | Covalent overhang 5′-CCAGACGT(AP)-3′ 3′-GGTCTG-5′ | 6OE7 | (48) |
| H. sapiens HMCES SRAP domain | Non-covalent palindromic 5′-CAACGTTGTTTT-3′ 3′-TTTTGTTGCAAC-5′ | 6OOV | (49) |
| E. coli YedK | Covalent ssDNA 5′-GTC(AP)GGA-3′ | 6NUA | (32) |
| E. coli YedK | Non-covalent ssDNA 5′-GTC(C3)GGA-3′ | 6NUH | (32) |
| E. coli YedK | Covalent ssDNA 5′-AAA(AP)AA-3′ | 6KCQ | (47) |
| E. coli YedK | Covalent ssDNA 5′-TTC(AP)-3′ | 6KIJ | (47) |
| E. coliYedK | Covalent ssDNA 5′-CGGT(AP)-3′ | 6KBX | (47) |
| E. coli YedK | Non-covalent ssDNA 5′-GGT(THF)GATTC-3′ | 6KBZ | (47) |
| E. coliYedK | Non-covalent ssDNA 5′-GGTCGATTC-3′ | 6KBS | (47) |
| E. coli YedK | None | 6KBU | (47) |
| E coli YedK | None | 2ICU | SECSG, RSGIb |
| Bordetella bronchiseptica Q7WLM8 | None | 1ZN6 | NESGc |
| Bordetella bronchiseptica BB2244 | None | 2BDV | NESGc |
| Agrobacterium tumefaciens Atu5096 | None | 2AEG | NESGc |
| Bacteroides thetaiotaomicron BT1218 | None | 2F20 | NESGc |
AP, abasic site; C3, C3-spacer, THF, tetrahydrofuran
SECSG, Southeast Collaboratory for Structural Genomics; RSGI RIKEN Structural Genomics/Proteomics Initiative
NESG, Northeast Structural Genomics Consortium
Fig. 2. SRAP-DNA structure.
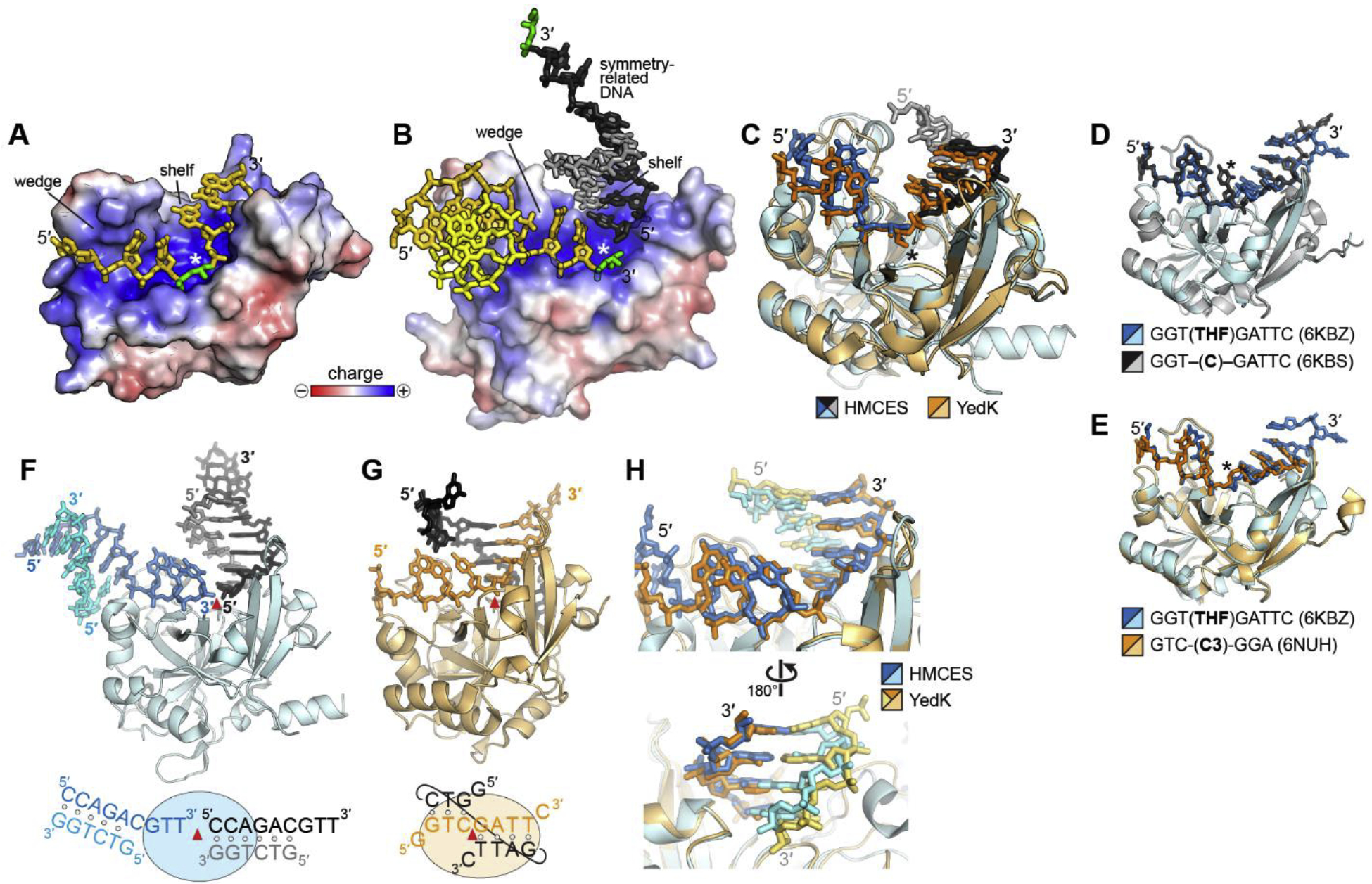
A,B. Electrostatic surface potential of (A) YedK-DPC (PDB ID 6NUA and (B) HMCES-SRAP DPC (PDB ID 6OE7) structures. The AP sites are green, and the symmetry-related DNA molecule in HMCES is shown in black and grey. The white asterisk denotes the position of the active site. C. Superposition of YedK-DPC (PDB ID 6NUA) and HMCES-SRAP DPC (PDB ID 6OE7). The DPC is marked with an asterisk. The ends of the DNA in the HMCES structure have been removed for clarity. D,E. Superposition of YedK bound noncovalently to ssDNA containing (D) a cytosine or an abasic site analog, and (E) two different abasic site analogs. The position of the nucleotide at the active site is marked with an asterisk. F,G. Similarity of dsDNA bound to (F) HMCES-SRAP (PDB ID 6OEB) and (G) YedK (PDB ID 6KBS). The asymmetric unit is colored blue or orange, and symmetry related DNA is shown in black and grey. The red triangle marks the position of the active site. In the schematic at the bottom, base pairs are denoted by open circles. H. Superposition of the two structures in panels F and G. DNA is colored blue/cyan in HMCES and orange/gold in YedK.
3.1. SRAP architecture
The HMCES SRAP domain and YedK, which share 29% sequence identity and 43% similarity, are highly similar in structure, with an RMSD of 1.2 Å for all Cα atoms (PDB IDs 5KO9 and 2ICU). The only noticeable differences between the HMCES and YedK structures are in the loop regions, which have weaker sequence similarity and in HMCES are noticeably longer. HMCES residues 149–159, found only in sequences from higher eukaryotes, are disordered in all available HMCES-SRAP structures. According to the SCOP (Structural Classification of Proteins) database (46), SRAP domains possess a unique BB1717-like fold containing a bifurcated β-sheet surrounded by several α-helices. The β-barrel core forms a positively charged DNA binding channel, with the Cys2 active site in the middle (Fig. 2A). Not surprisingly, this channel is the most highly-conserved region of the SRAP domain (32,47). The channel cradles the phosphoribose backbone in all available SRAP-DNA structures. Two highly conserved surface-exposed arginine residues, Arg98 and Arg212 in HMCES (Arg77 and Arg162 in YedK) interact with phosphates of the DNA backbone and are required for DNA binding (30,32,47,48).
Despite the differences in the DNA ligands used in the HMCES and YedK structures (Table 1), the observed confirmations of DNA and the protein-DNA interactions were remarkably similar, even between non-covalent and DPC complexes, indicating that SRAP interaction with DNA is likely not sequence-dependent. Several of the YedK-DNA structures (PDB IDs 6NUA, 6NUH, 6KBS, 6KBZ, 6KCQ) contained a continuous ssDNA molecule bound across the entire channel, which showed the details for how the DNA backbone is highly kinked and twisted at the position of the active site (Fig. 2A) (32,47). The HMCES-SRAP structures (PDB IDs 6OEA, 6OEB, and 6OE7) contained duplex DNA with a 3′-ssDNA overhang tail that bound to one side of the channel up to the active site (Fig. 2B) (48). The duplex portion of a symmetry-related molecule bound to the other side, creating a semi-continuous strand that overlays with the continuous ssDNA in the YedK structures (Fig. 2B,C). A more recent HMCES-SRAP structure (PDB ID 6OOV) crystallized with a palindromic DNA containing 3′ overhangs on each end. The overhangs link two symmetry related protomers by binding to one side of their positive channels in the same manner as the previous HMCES structures, but neither protomer contains DNA on the other side of the channel (49).
Importantly, all of the structures showed that a conserved “wedge” motif protrudes into one side of the positive channel and disrupts nucleobase stacking, precluding a complimentary second strand from pairing immediately adjacent to the 5′ side the AP site (Fig. 2A,B). This structural feature, together with the sharp kink in the DNA at the active site, explains SRAP’s preference for ssDNA (30,32,47,48). Comparison of DNA-bound and free forms of HMCES-SRAP and YedK show that there is very little change in the SRAP DNA binding site upon binding DNA (Cα RMSD of 1.16 Å for YedK and 1.00 Å for HMCES-SRAP), indicating that restructuring of the DNA in this particular way is a key feature of the protein. Furthermore, structures of YedK bound non-covalently to DNA containing either a full nucleotide or different types of abasic sites showed almost no difference in the DNA conformation, despite their different crystal packing arrangements (Fig. 2D,E).
3.2. SRAP accommodates 3′-junction structures
In contrast to the kinked DNA 5’ to the AP site by the wedge motif, all of the structures revealed that the DNA on the other side of the active site adopts a B-form conformation, and that duplex DNA can be accommodated immediately adjacent to the 3’-side of the AP site (32,47,48). Such a dsDNA-ssDNA junction would be formed by a stalled replicative polymerase during DNA synthesis. Four structures explicitly showed dsDNA bound to this side of the positively charged channel via crystal lattice interactions. Two HMCES-SRAP structures (PDB IDs 6OE7, 6OEB) contained the dsDNA portion of a symmetry-related molecule bound to the channel such that the blunt end of the duplex stacked against a highly conserved protein surface immediately adjacent to the active site, which we refer to as the DNA “shelf” (Fig. 2F). Similarly, in two YedK structures crystalized with ssDNA (PDB IDs 6KBS, 6KBZ), the DNA from one complex partially hybridizes to the DNA from a symmetry-related molecule, effectively establishing a dsDNA-ssDNA junction on the 3′ side of the AP site (Fig. 2G). The kinking of the continuous ssDNA along the channel allows for the nested 3’-end of the duplex to stack against the DNA shelf. The structures of the dsDNA and nested 3’-ends in HMCES and YedK are remarkably similar (Fig. 2H). An additional YedK-ssDNA structure (PDB ID 6NUA) did not contain dsDNA, but the ssDNA 3’ to the AP site adopted a B-form conformation such that duplex DNA could be easily modeled to create a dsDNA-ssDNA junction with a nested 3′-end (32). The structure of this modeled 3’-junction is virtually identical to both the human and bacterial SRAP structures that contain duplex from different crystal packing interactions, indicating that this interaction is an inherent property of SRAP proteins. Interestingly, ssDNA 3′ to the AP site in two additional YedK-DPC structures (PDB IDs 6KIJ, 6KBX) and in a non-covalent YedK-DNA complex (PDB ID 6NUH) either did not exhibit sufficient density to be modeled or showed high B-factors, suggesting that ssDNA 3’ to the AP site is relatively mobile in the absence of a base-paired strand to stabilize that region of DNA. The preferential binding to 3’-junctions were verified biochemically in both HMCES and YedK (32,47,48).
4. The unique SRAP DNA-protein crosslink
The molecular basis for the stability of the SRAP DPC was elucidated by a 1.6-Å crystal structure of YedK crosslinked to AP-DNA (6NUA), which revealed a thiazolidine linkage between the ring-opened AP deoxyribose and the α-amino and sulfhydryl groups of the N-terminal Cys2 residue (32) (Fig. 3A). This specific DPC linkage was also observed in subsequent high-resolution (1.2 Å) YedK-DPC structures (47) and in a 2.2-Å DPC structure of HMCES-SRAP (PDB ID 6OE7, Fig. 3B), which revealed the conserved mode of SRAP crosslinking across domains of life (48). Thiazolidine adducts between proteins and formaldehyde have been identified (50,51), but to our knowledge a thiazolidine linkage between protein and DNA has not been described previously.
Fig. 3. SRAP active site and mechanism of crosslinking.
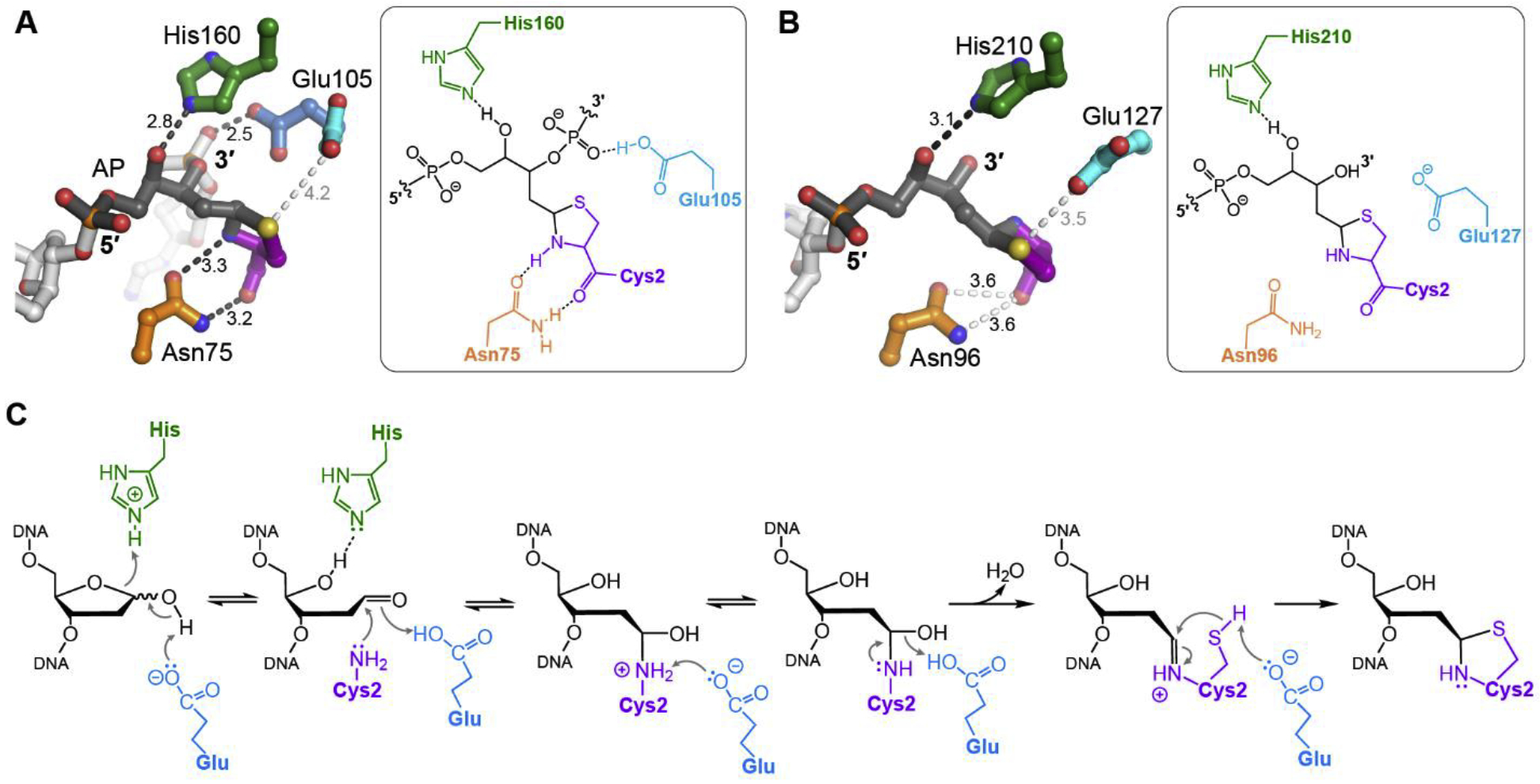
A,B. Atomic details of the thiazolidine DPC in (A) YedK (PDB ID 6NUA) and (B) HMCES-SRAP (PDB ID 6OE7). DNA is greyscale with AP site dark grey, and SRAP is colored by amino acid. Interatomic distances (Å) are labeled, with hydrogen bonds indicated by dark dashes and close contacts with light dashes. C. Proposed catalytic mechanism of crosslinking.
4.1. The SRAP active site
The residues responsible for DPC formation lie at the center of the DNA binding channel and include the invariant Cys2 and a highly conserved glutamate (Glu127 in HMCES; Glu105 in YedK), histidine (His210 in HMCES, His160 in YedK), and asparagine (Asn96 in HMCES; Asn75 in YedK). These residues are critical for crosslinking as substitution to alanine either abrogates or significantly reduces activity (30,32,47). Their interactions with the AP site are conserved between HMCES and YedK structures (Fig. 3A,B). In the absence of DNA, the thiol side chain of Cys2 points down into the protein, with the N-terminal amine position varying (HMCES, PDB ID 5KO9; YedK, PDB ID 6KBU). In one structure (YedK, PDB ID 2ICU) Cys2 was not observed in the crystallographic data, suggesting a degree of disorder potentially due to the presence of an N-terminal tag. When either HMCES or YedK is bound to DNA containing a natural base (PDB IDs 60EA, 60EB, 600V, 6KBS), Cys2 is observed in multiple positions and the nucleobase is facing away from the active site (47,48). In non-covalent complexes of YedK with an AP site analog, THF or C3-spacer (PDB IDs 6KBZ, 6NUH), Cys2 is rotated 180° relative to its buried position in the unbound structures, restructured into a pre-catalytic, crosslinkable position (32,47). Notably, the backbone of the THF structure is twisted by 90° such that the THF is positioned towards the Cys2, similar to the DNA conformation seen in the DPC structures.
The histidine side chain hydrogen bonds to the O4′ hydroxyl of the ring-opened AP site in the DPC structure but not in the non-covalent THF structure. The structure of YedK noncovalently bound to DNA with a nucleobase in the active site position shows the nucleotide rotated such that His160 now forms a hydrogen bond with O3′ as opposed to O4′ in the DPC structure (47). The structure of HMCES-SRAP bound to palindromic DNA containing 3′ overhangs (PDB ID 6OOV) shows the 3′-OH of each overhang hydrogen bonded to His210 (49). In the various DPC structures, the glutamate side chain is in close contact to the thiazolidine ring, anywhere from 2.6–4.2 Å observed in the various DPC structures. In the YedK DPC structures, a second glutamate conformer is observed within hydrogen bonding distance of the phosphate 3′ to the AP site, and is thus likely protonated to avoid repulsion with the negative backbone (32,47). On the other side of the thiazolidine crosslink, the asparagine hydrogen bonds with the carbonyl oxygen and the backbone amide nitrogen of Cys2.
4.2. Catalytic mechanism of SRAP DPC formation
Based on the SRAP-DNA structures and previous work on thiazolidine chemistry (52–55), we propose the following mechanism for SRAP DPC formation at AP sites. Thiazolidine formation would proceed by nucleophilic attack of the AP site C1′ carbon by the N-terminal Cys2 α-amino group to form a Schiff base intermediate, followed by a second nucleophilic attack of the imino carbon by the Cys2 thiolate side chain (Fig. 3C, Fig. 4A). Because of the low abundance of the aldehyde form of an AP site (14), it is possible that SRAP first catalyzes AP site ring opening, although the alternative in which SRAP traps a spontaneously formed aldehyde is also possible. Based on their positions in the crystal structures, Glu127 and His210 could catalyze ring opening by providing a general base to deprotonate the O1′ hydroxyl and a general acid to protonate O4′, both of which are necessary for aldehyde formation. The glutamate would then facilitate Schiff base formation by acid-base cycling to deprotonate Cys2 α-NH2 and possibly protonate the AP site O1′. Glu127 would also be positioned to deprotonate the Cys2 sulfhydryl to create the thiolate nucleophile required to complete formation of the thiazolidine (Fig. 3C). A cycle of Glu127 ionization is supported by the two observed conformations—an ionized conformer contacting the thiazolidine nitrogen and a protonated conformer contacting the phosphate 3′ to the AP site (32). The hydrophobic residues around the active site would raise the pKa of the glutamate, increasing the likelihood that it acts as both a proton donor and acceptor in the acid-base cycle. Finally, based on its position in the structures, the highly conserved asparagine likely positions the N-terminal Cys2 for nucleophilic attack.
Fig. 4. Comparison of DPCs formed by SRAP and AP lyases.
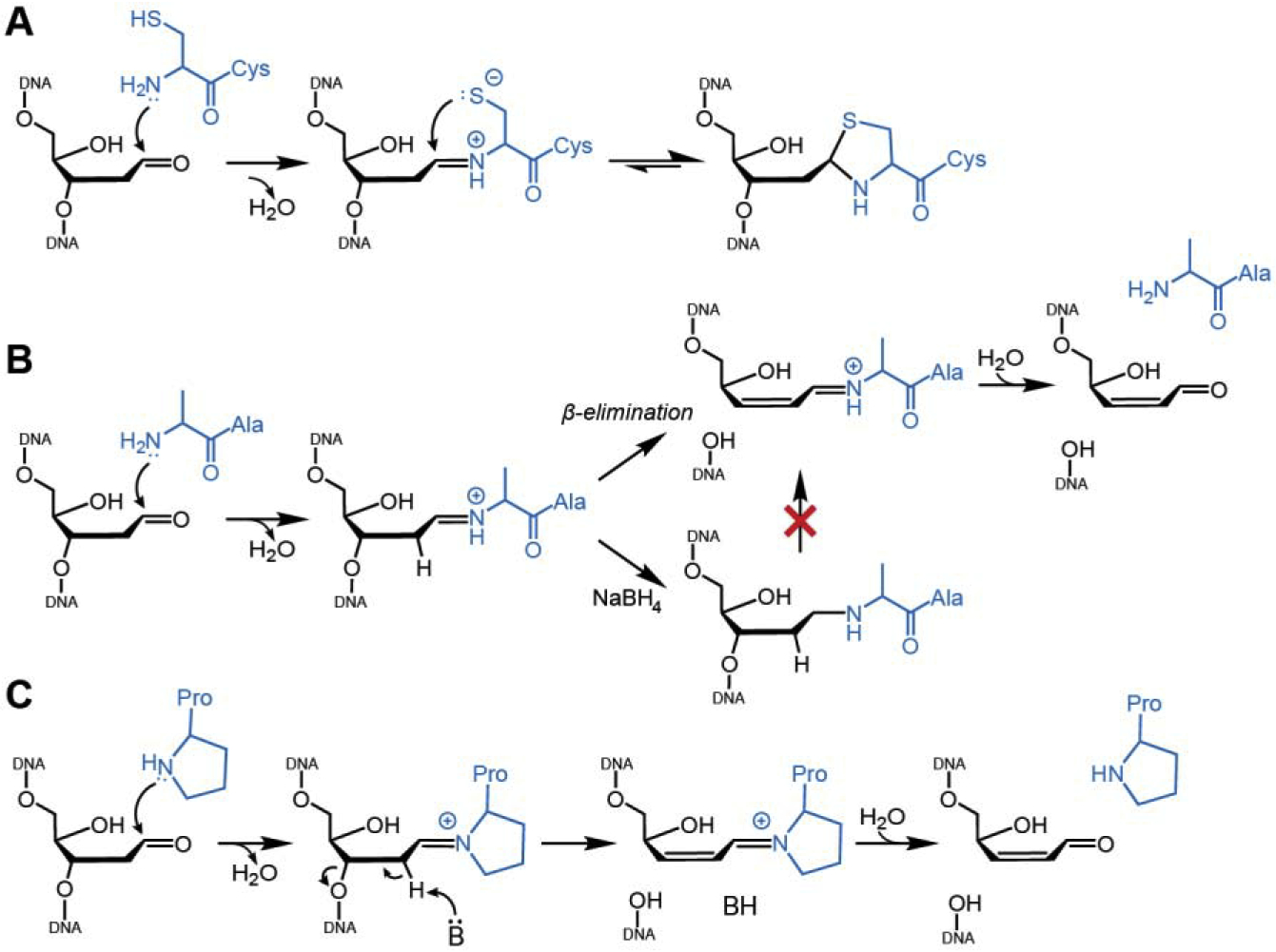
A. Wild-type SRAP forms a thiazolidine DPC via a Schiff base intermediate. B. The Schiff base formed by a SRAP C2A mutant is prone to β-elimination and can be reduced to a stable DPC via borohydride treatment. C. Catalysis of DNA lyase activity by bifunctional DNA glycosylases.
4.3. Thiazolidine stability
The Cys2 active site is surrounded by a pocket of strongly conserved hydrophobic residues. The solvent inaccessibility of this conserved DNA binding pocket likely provides an optimal environment for crosslink formation and helps to shield it from hydrolysis. However, shielding is not the sole basis for crosslink stability as proteolysis of the DPC down to a peptide-DNA crosslink is also persistent (32). Indeed, the chemical nature of the thiazolidine linkage is inherently stable and its formation proceeds efficiently under acidic conditions, as revealed by reactions between free cysteine and various aldehydes (52,53,56,57). The thiazolidine ring is stable up to at least 3M HCl, and reversal back to aldehyde and free cysteine is only observed under basic conditions greater than 1 M NaOH (54). This stability has also been exploited in conjugation reactions to generate peptide-peptide linkages or to attach site-specific labels (58,59).
The stability of the thiazolidine ring explains how SRAP protects AP sites from spontaneous strand breakage. Both AP sites and their Schiff base conjugates to proteins are susceptible to β-elimination, leading to cleavage of the DNA backbone (Fig 1B, Fig 4). Thiazolidines exist in equilibrium with the Schiff base, but the ring-closed thiazolidine is favored over the ring-opened iminium ion by 5 orders of magnitude (52,53,55). Thus, the closed thiazolidine ring draws the equilibrium away from Schiff base degradation (Fig. 4A). Consistent with this premise, removal or substitution of the thiolate by a YedK C2A or C2S mutant not only abrogated crosslink formation but also led to cleavage of the DNA to form a product consistent with β-elimination at the AP site (Fig. 4B) (32). Evidence for the Schiff base intermediate in SRAP was provided by borohydride trapping (60) of YedK C2A and C2S DPC intermediates (Fig. 4B), which dramatically reduced DNA strand cleavage (32). Formation of a Schiff base also indicates that the N-terminal amine—not the Cys2 thiolate—initiates crosslink formation. Indeed, the hydrophobic environment of this residue would raise the pKa of the thiol and lower the pKa of the amine, favoring the amine as the nucleophile.
A C2S mutant could theoretically form an oxazolidine ring by the same mechanism as thiazolidine formation by Cys2. To our knowledge, however, there is no evidence of a SRAP C2S variant in nature. We did not observe any DPC formation in our biochemical experiments with a C2S mutant. An oxazolidine is less stable than a thiazolidine (55,61) likely because sulfur is 0.4 Å larger than oxygen, and can form longer bonds that could aid in the stability of the thiazolidine (62,63). Moreover, the side chain of cysteine has a lower pKa than that of serine, making cysteine a better nucleophile. Thus, an oxazolidine is unlikely to form, and if it did form, it would be unable to shift the equilibrium away from the competing β-elimination reaction.
4.4. Comparison to DPC formation in DNA lyases
The N-terminal cysteine in SRAP is unique among DNA repair proteins. A number of repair enzymes interact with AP sites via Schiff base intermediates, but typically do so to catalyze strand scission (64–67). Bifunctional DNA glycosylases contain AP lyase activity that cleaves the DNA backbone 3′ to the AP site (68). Pol β and Ku antigen contain 5′-deoxyribophosphate (5′-dRP) lyase activities important for single-strand break processing during BER and non-homologous end joining (NHEJ), respectively (24,69,70). PARP1 and PARP2 have been shown to exhibit AP lyase and 5’-dRP lyase activity in vitro (71,72). Similar to the SRAP crosslinking mechanism, DNA lyases use an amine nucleophile—either an internal lysine or an N-terminal proline or valine—to form a Schiff base intermediate that can be trapped by borohydride reduction (44,60,73). Lyases lack a second nucleophile to stabilize the DPC, and instead the Schiff base increases the basicity of C2′ to promote catalysis of β-elimination and cleavage of the DNA (Fig. 4C). Thus, the main distinction between AP lyases and SRAP is the ability of the SRAP crosslinking nucleophile to exhibit a second nucleophilic attack of the imino C1′ carbon to form a stable linkage, thereby acting as an AP-site sink to inhibit the competing elimination reaction. Wild type SRAP does not lead to, but rather prevents strand cleavage.
Interestingly, some AP lyases form stable DPCs with oxidized AP sites in mammalian cells, exhibiting half-lives of 15–60 min before proteasome dependent resolution (40,74,75). These AP lyase DPCs were associated with an increase in double-strand breaks in cells, in contrast to the HMCES dependent reduction in double-strand breaks (30,69). Thus, whereas HMCES DPCs play a protective role, the oxidative AP lyase DPCs are thought to be toxic byproducts of DNA repair proteins trapped in unproductive complexes (40). In contrast, PARP1 has been shown to form a DPC with AP sites by reduction of the Schiff base via an intrinsic redox activity of the protein, similar to the reaction in borohydride trapping experiments. Like HMCES, the PARP1 DPC is proposed to protect AP sites and recruit BER factors for repair (76).
5. Concluding remarks
This review has focused primarily on the mechanics of SRAP crosslinking to AP sites. However, in addition to its role in the replication stress response (30,77), HMCES is implicated in a number of other DNA related transactions across species, including viral replication (78), aging (79), alternative end-joining during B-cell class switch recombination (49), and long interspersed element-1 (LINE-1) retrotransposition (80–82). Moreover, HMCES expression is significantly down regulated in both B-cell lymphoma and multidrug resistant osteosarcoma cells (83,84). HMCES was named for its identification in a screen for readers of modified bases in embryonic stem cells (85), although this name is likely a misnomer as HMCES is expressed in all cell lineages (86) and its binding to oxidatively modified bases has not been reproduced. Moreover, global DNA methylation patterns are not significantly altered in HMCES knockouts (30,49), and the catalytic SRAP domain is conserved in prokaryotes, which do not utilize 5hmC (87).
An important open question relates to the fate of the HMCES DPC in cells. HMCES is ultimately targeted for ubiquitin-dependent destruction by the proteasome (30). Is the resulting peptide-DNA crosslink repaired by nucleotide excision or some other repair pathway? In HMCES, several lysine residues located in the C-terminal tail and on the surface of the protein opposite the DNA binding channel have been found to be ubiquitylated via mass spectrometry (88,89). The majority of these lysines are highly conserved in eukaryotic SRAP proteins. These lysines and other predicted phosphorylation and SUMOylation sites are mutated in some human tumors (88,90). Other cancer associated mutations appear to be destabilizing mutations as they reside in the protein interior and likely result in improper folding and/or subsequent degradation. HMCES regulation and the fate of SRAP-AP DPCs in cells represent the next frontier in understanding this important class of protein.
ACKNOWLEDGEMENTS
Research on abasic site repair in the Eichman lab is funded by National Institutes of Health (USA) grants R35GM136401 and R01GM131071. K.M.A. is supported by National Institutes of Health (USA) grant T32GM008320.
Abbreviations:
- AP
Apurinic/apyrimidinic
- ssDNA
single-stranded DNA
- dsDNA
double-stranded DNA
- DSB
double-strand break
- DPC
DNA-protein crosslink
- ICL
interstrand DNA crosslink
- THF
tetrahydrofuran
- UDG
uracil DNA glycosylase
- 5′-dRP
5′-deoxyribose phosphate
- BER
base excision repair
- NER
nucleotide excision repair
- PARP
poly(ADP-ribose) polymerase
- PCNA
proliferating cell nuclear antigen
- PDB
Protein Data Bank
- PIP
PCNA-interacting protein
- TLS
translesion synthesis
- HMCES
5-hydroxymethylcytosine embryonic stem cells specific
- 5hmC
5-hydroxymethylcytosine
- pol β
DNA polymerase β
- SRAP
SOS response associated peptidase
- NHEJ
non-homologous end joining
Footnotes
Publisher's Disclaimer: This is a PDF filele of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF COMPETING INTEREST
There are no conflicts of interest to declare.
REFERENCES
- 1.Nakamura J, Walker VE, Upton PB, Chiang S-Y, Kow YW and Swenberg JA (1998) Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer research, 58, 222–225. [PubMed] [Google Scholar]
- 2.Nakamura J and Swenberg JA (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer research, 59, 2522–2526. [PubMed] [Google Scholar]
- 3.Lindahl T (1993) Instability and decay of the primary structure of DNA. nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 4.Barnes DE and Lindahl T (2004) Repair and Genetic Consequences of Endogenous DNA Base Damage in Mammalian Cells. Annual Review of Genetics, 38, 445–476. [DOI] [PubMed] [Google Scholar]
- 5.Cadet J and Wagner JR (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Bont R and van Larebeke N (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis, 19, 169–185. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl T and Nyberg B (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry, 11, 3610–3618. [DOI] [PubMed] [Google Scholar]
- 8.Krokan HE and Bjørås M (2013) Base excision repair. Cold Spring Harbor perspectives in biology, 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullins EA, Rodriguez AA, Bradley NP and Eichman BF (2019) Emerging roles of DNA glycosylases and the base excision repair pathway. Trends in biochemical sciences. [DOI] [PMC free article] [PubMed]
- 10.Hitomi K, Iwai S and Tainer JA (2007) The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA repair, 6, 410–428. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura J and Nakamura M (2020) DNA-protein crosslink formation by endogenous aldehydes and AP sites. DNA repair, 102806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z, Price NE, Johnson KM, Wang Y and Gates KS (2017) Interstrand cross-links arising from strand breaks at true abasic sites in duplex DNA. Nucleic acids research, 45, 6275–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindahl T and Andersson A (1972) Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry, 11, 3618–3623. [DOI] [PubMed] [Google Scholar]
- 14.Overend WG (1950) 533. Deoxy-sugars. Part XIII. Some observations on the Feulgen nucleal reaction. Journal of the Chemical Society (Resumed), 2769–2774. [Google Scholar]
- 15.Manoharan M, Ransom SC, Mazumder A, Gerlt JA, Wilde JA, Withka JA and Bolton PH (1988) The characterization of abasic sites in DNA heteroduplexes by site specific labeling with carbon-13. Journal of the American Chemical Society, 110, 1620–1622. [Google Scholar]
- 16.Lhomme J, Constant JF and Demeunynck M (1999) Abasic DNA structure, reactivity, and recognition. Biopolymers: Original Research on Biomolecules, 52, 65–83. [DOI] [PubMed] [Google Scholar]
- 17.Price NE, Johnson KM, Wang J, Fekry MI, Wang Y and Gates KS (2014) Interstrand DNA–DNA cross-link formation between adenine residues and abasic sites in duplex DNA. Journal of the American Chemical Society, 136, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S and Prakash L (2001) Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes & development, 15, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson PS and Cortez D (2020) New insights into abasic site repair and tolerance. DNA Repair (Amst), 90, 102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kavli B, Otterlei M, Slupphaug G and Krokan HE (2007) Uracil in DNA—general mutagen, but normal intermediate in acquired immunity. DNA repair, 6, 505–516. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava DK, Berg BJV, Prasad R, Molina JT, Beard WA, Tomkinson AE and Wilson SH (1998) Mammalian abasic site base excision repair Identification of the reaction sequence and rate-determining steps. Journal of Biological Chemistry, 273, 21203–21209. [DOI] [PubMed] [Google Scholar]
- 22.Krokan HE, Standal R and Slupphaug G (1997) DNA glycosylases in the base excision repair of DNA. Biochemical Journal, 325, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiering A and Deutsch W (1986) Drosophila apurinic/apyrimidinic DNA endonucleases. Characterization of mechanism of action and demonstration of a novel type of enzyme activity. Journal of Biological Chemistry, 261, 3222–3228. [PubMed] [Google Scholar]
- 24.Allinson SL, Dianova II and Dianov GL (2001) DNA polymerase β is the major dRP lyase involved in repair of oxidative base lesions in DNA by mammalian cell extracts. The EMBO journal, 20, 6919–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erzberger JP, Barsky D, Schärer OD, Colvin ME and Wilson III DM (1998) Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic acids research, 26, 2771–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson DM, Takeshita M, Grollman AP and Demple B (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. Journal of Biological Chemistry, 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- 27.Schaaper RM, Kunkel TA and Loeb LA (1983) Infidelity of DNA synthesis associated with bypass of apurinic sites. Proceedings of the National Academy of Sciences, 80, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen PL, Xu F and Xiao W (2008) Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell research, 18, 162. [DOI] [PubMed] [Google Scholar]
- 29.Powers KT and Washington MT (2018) Eukaryotic translesion synthesis: Choosing the right tool for the job. DNA repair, 71, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KP and Cortez D (2019) HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell, 176, 144–153. e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boehm EM, Gildenberg MS and Washington MT (2016) The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes, 39, 231–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson PS, Amidon KM, Mohni KN, Cortez D and Eichman BF (2019) Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nature Structural & Molecular Biology, 26, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR and Cortez D (2020) HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell Reports, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aravind L, Anand S and Iyer LM (2013) Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems. Biology direct, 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duzen JM, Walker GC and Sutton MD (2004) Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD’. DNA Repair (Amst), 3, 301–312. [DOI] [PubMed] [Google Scholar]
- 36.Tomer G, Reuven NB and Livneh Z (1998) The beta subunit sliding DNA clamp is responsible for unassisted mutagenic translesion replication by DNA polymerase III holoenzyme. Proc Natl Acad Sci U S A, 95, 14106–14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kweon S-M, Zhu B, Chen Y, Aravind L, Xu S-Y and Feldman DE (2017) Erasure of Tet-Oxidized 5-Methylcytosine by a SRAP Nuclease. Cell reports, 21, 482–494. [DOI] [PubMed] [Google Scholar]
- 38.Covey JM, Jaxel C, Kohn KW and Pommier Y (1989) Protein-linked DNA strand breaks induced in mammalian cells by camptothecin, an inhibitor of topoisomerase I. Cancer Res, 49, 5016–5022. [PubMed] [Google Scholar]
- 39.Ide H, Nakano T, Salem AMH and Shoulkamy MI (2018) DNA-protein cross-links: Formidable challenges to maintaining genome integrity. DNA Repair (Amst), 71, 190–197. [DOI] [PubMed] [Google Scholar]
- 40.Quiñones JL, Thapar U, Wilson SH, Ramsden DA and Demple B (2020) Oxidative DNA-protein Crosslinks Formed in Mammalian Cells by Abasic Site Lyases Involved in DNA Repair. DNA Repair, 102773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zharkov DO, Rieger RA, Iden CR and Grollman AP (1997) NH2-terminal proline acts as a nucleophile in the glycosylase/AP-lyase reaction catalyzed by Escherichia coli formamidopyrimidine-DNA glycosylase (Fpg) protein. Journal of Biological Chemistry, 272, 5335–5341. [DOI] [PubMed] [Google Scholar]
- 42.Gilboa R, Zharkov DO, Golan G, Fernandes AS, Gerchman SE, Matz E, Kycia JH, Grollman AP and Shoham G (2002) Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. Journal of Biological Chemistry, 277, 19811–19816. [DOI] [PubMed] [Google Scholar]
- 43.Pommier Y, Pourquier P, Urasaki Y, Wu J and Laco GS (1999) Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resistance Updates, 2, 307–318. [DOI] [PubMed] [Google Scholar]
- 44.McCullough AK, Sanchez A, Dodson M, Marapaka P, Taylor J-S and Lloyd RS (2001) The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry, 40, 561–568. [DOI] [PubMed] [Google Scholar]
- 45.Cordes EH and Jencks WP (1962) On the Mechanism of Schiff Base Formation and Hydrolysis. Journal of the American Chemical Society, 84, 832–837. [Google Scholar]
- 46.Andreeva A, Kulesha E, Gough J and Murzin AG (2019) The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Research, 48, D376–D382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang N, Bao H, Chen L, Liu Y, Li Y, Wu B and Huang H (2019) Molecular basis of abasic site sensing in single-stranded DNA by the SRAP domain of E. coli yedK. Nucleic Acids Research, 47, 10388–10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L and Arrowsmith CH (2019) Structural basis of HMCES interactions with abasic DNA and multivalent substrate recognition. Nature Structural & Molecular Biology, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shukla V, Halabelian L, Balagere S, Samaniego-Castruita D, Feldman DE, Arrowsmith CH, Rao A and Aravind L (2019) HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Molecular Cell, 77, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Higgins KA and Giedroc D (2014) Insights into Protein Allostery in the CsoR/RcnR Family of Transcriptional Repressors. Chem Lett, 43, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJ et al. (2004) Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J Biol Chem, 279, 6235–6243. [DOI] [PubMed] [Google Scholar]
- 52.Ratner S and Clarke HT (1937) The Action of Formaldehyde upon Cysteine. Journal of the American Chemical Society, 59, 200–206. [Google Scholar]
- 53.Kallen RG (1971) Mechanism of reactions involving Schiff base intermediates. Thiazolidine formation from L-cysteine and formaldehyde. Journal of the American Chemical Society, 93, 6236–6248. [DOI] [PubMed] [Google Scholar]
- 54.Pesek J and Frost J (1975) Decomposition of thiazolidines in acidic and basic solution: Spectroscopic evidence for Schiff base intermediates. Tetrahedron, 31, 907–913. [Google Scholar]
- 55.Canle M, Lawley A, McManus EC and O’Ferrall RAM (1996), Pure and Applied Chemistry, Vol. 68, pp. 813. [Google Scholar]
- 56.Schubert MP (1936) COMPOUNDS OF THIOL ACIDS WITH ALDEHYDES. Journal of Biological Chemistry, 114, 341–350. [Google Scholar]
- 57.Cook AH and Heilbron IM (1949) In Clarke HT, Johnson JR and Robinson R (eds.), Chemistry of Penicillin. Princeton University Press, pp. 921–972. [Google Scholar]
- 58.Gentle IE, De Souza DP and Baca M (2004) Direct production of proteins with N-terminal cysteine for site-specific conjugation. Bioconjugate chemistry, 15, 658–663. [DOI] [PubMed] [Google Scholar]
- 59.Zhang L and Tam JP (1996) Thiazolidine formation as a general and site-specific conjugation method for synthetic peptides and proteins. Analytical biochemistry, 233, 87–93. [DOI] [PubMed] [Google Scholar]
- 60.Billman JH and Diesing AC (1957) Reduction of Schiff bases with sodium borohydride. The Journal of Organic Chemistry, 22, 1068–1070. [Google Scholar]
- 61.Just G, Chung BY, Kim S, Rosebery G and Rossy P (1976) Reactions of oxygen and sulphur anions with oxazolidine and thiazolidine derivatives of 2-mesyloxymethylglyceraldehyde acetonide. Canadian Journal of Chemistry, 54, 2089–2093. [Google Scholar]
- 62.Bondi A.v. (1964) van der Waals volumes and radii. The Journal of physical chemistry, 68, 441–451. [Google Scholar]
- 63.Eramian H, Tian Y-H, Fox Z, Beneberu HZ and Kertesz M (2013) On the anisotropy of van der Waals atomic radii of O, S, Se, F, Cl, Br, and I. The Journal of Physical Chemistry A, 117, 14184–14190. [DOI] [PubMed] [Google Scholar]
- 64.Piersen CE, Prasad R, Wilson SH and Lloyd RS (1996) Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. Journal of Biological Chemistry, 271, 17811–17815. [DOI] [PubMed] [Google Scholar]
- 65.Bailly V, Verly W, O’Connor T and Laval J (1989) Mechanism of DNA strand nicking at apurinic/apyrimidinic sites by Escherichia coli [formamidopyrimidine] DNA glycosylase. Biochemical Journal, 262, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun B, Latham KA, Dodson M and Lloyd RS (1995) Studies on the Catalytic Mechanism of Five DNA Glycosylases PROBING FOR ENZYME-DNA IMINO INTERMEDIATES. Journal of Biological Chemistry, 270, 19501–19508. [DOI] [PubMed] [Google Scholar]
- 67.Golan G, Zharkov DO, Grollman AP, Dodson M, McCullough AK, Lloyd RS and Shoham G (2006) Structure of T4 pyrimidine dimer glycosylase in a reduced imine covalent complex with abasic site-containing DNA. Journal of molecular biology, 362, 241–258. [DOI] [PubMed] [Google Scholar]
- 68.Purmal AA, Rabow LE, Lampman GW, Cunningham RP and Kow YW (1996) A common mechanism of action for the N-glycosylase activity of DNA N-glycosylase/AP lyases from E. coli and T4. Mutat Res, 364, 193–207. [DOI] [PubMed] [Google Scholar]
- 69.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P and Ramsden DA (2010) Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature, 464, 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strande N, Roberts SA, Oh S, Hendrickson EA and Ramsden DA (2012) Specificity of the dRP/AP lyase of Ku promotes nonhomologous end joining (NHEJ) fidelity at damaged ends. Journal of Biological Chemistry, 287, 13686–13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kutuzov MM, Khodyreva SN, Ilina ES, Sukhanova MV, Amé JC and Lavrik OI (2015) Interaction of PARP-2 with AP site containing DNA. Biochimie, 112, 10–19. [DOI] [PubMed] [Google Scholar]
- 72.Khodyreva S, Prasad R, Ilina E, Sukhanova M, Kutuzov M, Liu Y, Hou E, Wilson S and Lavrik O (2010) Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly (ADP-ribose) polymerase-1 (PARP-1). Proceedings of the National Academy of Sciences, 107, 22090–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prakash A, Eckenroth BE, Averill AM, Imamura K, Wallace SS and Doublié S (2013) Structural investigation of a viral ortholog of human NEIL2/3 DNA glycosylases. DNA Repair (Amst), 12, 1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW and Demple B (2015) Enzyme mechanism-based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proceedings of the National Academy of Sciences, 112, 8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeMott MS, Beyret E, Wong D, Bales BC, Hwang J-T, Greenberg MM and Demple B (2002) Covalent trapping of human DNA polymerase β by the oxidative DNA lesion 2-deoxyribonolactone. Journal of Biological Chemistry, 277, 7637–7640. [DOI] [PubMed] [Google Scholar]
- 76.Prasad R, Horton JK and Wilson SH (2020) Requirements for PARP-1 covalent crosslinking to DNA (PARP-1 DPC). DNA Repair (Amst), 89, 102824. [DOI] [PubMed] [Google Scholar]
- 77.Srivastava M, Su D, Zhang H, Chen Z, Tang M, Nie L and Chen J (2020) HMCES safeguards replication from oxidative stress and ensures error-free repair. EMBO Rep, e49123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Viktorovskaya OV, Greco TM, Cristea IM and Thompson SR (2016) Identification of RNA Binding Proteins Associated with Dengue Virus RNA in Infected Cells Reveals Temporally Distinct Host Factor Requirements. PLoS Negl Trop Dis, 10, e0004921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Horikoshi M, Day FR, Akiyama M, Hirata M, Kamatani Y, Matsuda K, Ishigaki K, Kanai M, Wright H and Toro CA (2018) Elucidating the genetic architecture of reproductive ageing in the Japanese population. Nature communications, 9, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor MS, LaCava J, Mita P, Molloy KR, Huang CRL, Li D, Adney EM, Jiang H, Burns KH and Chait BT (2013) Affinity proteomics reveals human host factors implicated in discrete stages of LINE-1 retrotransposition. Cell, 155, 1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor MS, Altukhov I, Molloy KR, Mita P, Jiang H, Adney EM, Wudzinska A, Badri S, Ischenko D and Eng G (2018) Dissection of affinity captured LINE-1 macromolecular complexes. Elife, 7, e30094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyoshi T, Makino T and Moran JV (2019) Poly (ADP-Ribose) Polymerase 2 Recruits Replication Protein A to Sites of LINE-1 Integration to Facilitate Retrotransposition. Molecular cell, 75, 1286–1298. e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Zeng L, Liang C, Zan R, Ji W, Zhang Z, Wei Y, Tu S and Dong Y (2019) Integrated analysis of transcriptome-wide m6A methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics, 11, 1693–1715. [DOI] [PubMed] [Google Scholar]
- 84.Gao H-X, Nuerlan A, Abulajiang G, Cui W-L, Xue J, Sang W, Li S-J, Niu J, Ma Z-P and Zhang W (2019) Quantitative proteomics analysis of differentially expressed proteins in activated B-cell-like diffuse large B-cell lymphoma using quantitative proteomics. Pathology-Research and Practice, 215, 152528. [DOI] [PubMed] [Google Scholar]
- 85.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Münzel M, Wagner M, Müller M and Khan F (2013) Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell, 152, 1146–1159. [DOI] [PubMed] [Google Scholar]
- 86.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A and Edlund K (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & Cellular Proteomics, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pfeifer GP, Szabó PE and Song J (2019) Protein Interactions at Oxidized 5-Methylcytosine Bases. Journal of molecular biology. [DOI] [PMC free article] [PubMed]
- 88.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E and Zhang B (2004) PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics, 4, 1551–1561. [DOI] [PubMed] [Google Scholar]
- 89.Oughtred R, Stark C, Breitkreutz B-J, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G and McAdam R (2019) The BioGRID interaction database: 2019 update. Nucleic acids research, 47, D529–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM and Network CGAR (2013) The cancer genome atlas pan-cancer analysis project. Nature genetics, 45, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]