

Abstract
High fatty acid oxidation (FAO) is associated with lipotoxicity, but whether it causes lipotoxic cardiomyopathy remains controversial. Molecular mechanisms that may be responsible for FAO-induced lipotoxic cardiomyopathy are also elusive. In this study, increasing FAO by genetic deletion of acetyl-CoA carboxylase 2 (ACC2) did not induce cardiac dysfunction after 16 weeks of high fat diet (HFD) feeding. This suggests that increasing FAO, per se, does not cause metabolic cardiomyopathy in obese mice. We compared transcriptomes of control and ACC2 deficient mouse hearts under chow- or HFD-fed conditions. ACC2 deletion had a significant impact on the global transcriptome including downregulation of the peroxisome proliferator-activated receptors (PPARs) signaling and fatty acid degradation pathways. Increasing fatty acids by HFD feeding normalized expression of fatty acid degradation genes in ACC2 deficient mouse hearts to the same level as the control mice. In contrast, cardiac transcriptome analysis of the lipotoxic mouse model (db/db) showed an upregulation of PPARs signaling and fatty acid degradation pathways. Our results suggest that enhancing FAO by genetic deletion of ACC2 negatively regulates PPARs signaling through depleting endogenous PPAR ligands, which can serve as a negative feedback mechanism to prevent excess activation of PPAR signaling under non-obese condition. In obesity, excessive lipid availability negates the feedback mechanism resulting in over activation of PPAR cascade, thus contributes to the development of cardiac lipotoxicity.
Keywords: Fatty Acid Oxidation, PPAR, Lipotoxicity, Transcriptomics
Graphical Abstract
INTRODUCTION
Obesity and Type 2 diabetes mellitus are the major risk factors for the development of cardiovascular disease. Obesity and ectopic fat accumulation induce a variety of remodeling in the cardiovascular system leading to manifestations ranging from subclinical myocardial diastolic dysfunction to severe end-stage heart failure phenotype [1]. However, the mechanisms that lead to obesity/diabetes induced cardiomyopathy are complex and remain largely unknown. Abnormalities in cardiac metabolism and energy production have been proposed as a contributor to the development of obesity and diabetes related cardiovascular disease [2, 3]. Under obese condition, chronic systemic hyperlipidemia and intracellular lipid accumulation are the major causes for the dysregulated glucose and fatty acid metabolism, which in turn leads to the cardiac metabolic inflexibility, and thus contributes to impaired cardiac energy homeostasis [3]. Moreover, increased circulating free fatty acid plus a shift in substrate utilization towards fatty acid metabolism in obesity also contributes to intracellular accumulation of toxic lipid species, which leads to morphological and functional impairments in the heart. Since high fatty acid oxidation (FAO) is always associated with lipotoxicity, it has been proposed to be responsible for metabolic cardiomyopathy in obesity and/or diabetes [4, 5].
It has been suggested that over activation of peroxisome proliferator-activated receptors (PPARs) and its downstream transcriptional program is responsible for the exaggerated reliance on fatty acid utilization and the development of cardiac lipotoxicity in obese and diabetic animal models [6–9]. PPARs belong to the nuclear receptor superfamily of ligand-activated transcription factors and include three member isoforms, PPAR-α, PPAR-β/δ, and PPAR-γ. PPARs govern the expression of key molecules involved in fatty acid metabolic pathway, including the uptake, oxidation and storage of FAs [10–12]. The transcriptional activity of PPARs is regulated by their endogenous ligands and/or metabolites produced from fatty acids metabolic pathways [12–14]. Therefore, increased lipid load in the setting of obesity has been proposed to be responsible for the excess activation of PPAR-α and/or PPAR-γ, leading to the enhanced fatty acid utilization and the development of cardiac lipotoxicity.
We previously have demonstrated that deletion of Acetyl-CoA Carboxylase 2 (ACC2) in the mouse heart decreases malonyl-CoA level and enhanced FAO by relieving the inhibition of carnitine-palmityl-transferase 1 (CPT1). The ACC2 deficient heart maintains normal function in the long-term indicating that increasing FAO per se does not cause cardiac dysfunction [15]. However, increases of FAO in the setting of PPARs activation or obesity are associated with changes in fatty acid uptake or activation. It is unknown whether similar changes occur in ACC2 deficient hearts. More importantly, it has not been determined whether ACC2 deficient hearts will succumb to conditions of increased fatty acid supply, such as obesity.
In order to address the above questions, we compared cardiac transcriptome of ACC2 inducible knock out (iKO) mice after 16 weeks of chow or high fat diet (HFD) feeding with that of control mice fed a matched diet. We found that the PPARs signaling and fatty acid degradation pathway were downregulated in ACC2 iKO hearts under chow-fed condition, which was normalized by HFD feeding. On the contrary, activation of the PPARs signaling was observed in the heart of lipotoxicity models, e.g. HFD-fed and db/db mouse hearts. Consistent with the notion that enhanced FAO depleted the availability of PPARs ligands, global metabolomics analysis showed decreases of active lipids species in chow-fed ACC2 iKO mouse hearts, which was normalized by HFD. Collectively, these results identified a novel regulatory circuit between fatty acid oxidation and PPAR signaling, which served as a negative feedback mechanism for preventing the development of cardiac lipotoxicity.
RESULT
Four-month HFD feeding did not alter cardiac function in ACC2 iKO mice
We induced high FAO in adult mouse hearts by cardiac-specific deletion of ACC2 using a tamoxifen inducible model (iKO) as previously reported [16]. In order to examine the effect of increasing FAO on the regulation of gene expression in iKO heart under normal or obese conditions, both control (Con) and iKO mice were subject to chow or high fat diet (60 kcal% fat) feeding and the hearts were subsequently harvested for the transcriptome analysis (Figure 1A). From here on, we will use Con-chow, iKO-chow, Con-HFD and iKO-HFD to represent the four groups of mice in this study. After 16 weeks of HFD feeding, both Con and iKO mice showed similar obese phenotype with comparable body weight gain (Figure 1B). Since deletion of ACC2 shifted cardiac metabolism towards FAO in normal mice, we then asked whether increasing fatty acid supply with HFD feeding would further alter cardiac substrate metabolism in iKO mice. In isolated perfused hearts, iKO-chow exhibited higher FAO as compared to Con-chow, which was accompanied by a reduction in the contribution of glucose oxidation (Figure 1C). A similar increase in FAO was also observed in Con-HFD (Figure 1C). Interestingly, HFD feeding further increased FAO in iKO compared with Con-HFD (Figure 1C). During the perfusion, we found that cardiac systolic function was maintained in both Con-HFD and iKO-HFD mice compared with chow-fed mice (Supplementary Table 1). Since cardiac diastolic dysfunction was often observed in obese mice, we further examined the Pressure-Volume relationship for left ventricular end-diastolic pressure to determine whether cardiac diastolic function was altered in these mice. We found that the P-V relationship was indistinguishable in Con and ACC2 iKO mice under both chow- and HFD-fed conditions (Figure 1D). Consistently, echocardiography analysis revealed that four months of HFD feeding did not cause any cardiac contractile dysfunction, evidenced by the preserved ejection fraction in Con-HFD mice compared with Con-chow mice (Figure 1E, Supplementary Table 2) [17]. More importantly, iKO-HFD mice also maintained normal cardiac function (Figure 1E, Supplementary Table 2). There is no obvious cardiac hypertrophy induced by this short-term HFD feeding in both Con and ACC2 iKO mice (Supplementary Figure 1). At cellular level, HFD feeding increased the MDA level in Con heart, indicative of oxidative stress, which was prevented in iKO-HFD hearts (Figure 1F). Taken together, these data indicated that increasing myocardial FAO per se did not cause the development of cardiac dysfunction in obese mice.
Figure 1. Four months’ HFD feeding did not alter cardiac function in ACC2 iKO mice.

(A) Schematic demonstration of the experimental procedure. (B-F) Con and ACC2 iKO mice were subjected to HFD feeding for 16 weeks. (B) Body weight was measured. (C-D) Hearts were perfused with a buffer containing 13C labeled fatty acids, 13C labeled glucose, lactate, and insulin (mixed substrates). (C) Contribution of 13C labeled substrates to tricarboxylic acid (TCA) cycle was determined by 13C NMR spectroscopy in heart extracts. Relative contribution of fatty acids, glucose, and other unlabeled substrates (lactate, endogenous) is reported as fold changes over Con-chow (dotted line) (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=3–6). (D) Left ventricular pressure–volume relationship for end diastolic pressure is shown (n=3–6). (E) Left ventricular ejection fraction (LVEF %) was measured by echocardiography (n=4–6). (F) Evaluation of ROS generation by TBARS assay. Malondialdehyde (MDA) was quantified colorimetrically to monitor lipid peroxidation (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=4).
Global cardiac transcriptome analysis in ACC2 iKO mice
In order to explore the molecular mechanisms by which iKO mice adapted to high cardiac FAO, RNA deep-sequencing and transcriptome analysis was conducted using heart samples from Con and iKO mice under chow- or HFD-fed conditions. Principal component analysis (PCA) demonstrated that differentially expressed genes (DEGs) were clustered into four groups by two preselected factors, genotype and diet, suggesting that these two factors impacted the global transcriptome in the heart (Figure 2A). We then performed the pairwise comparison of gene expression in Con and iKO hearts under chow- or HFD-fed conditions, respectively. The volcano plot demonstrated that 50 transcripts were upregulated and 182 transcripts were downregulated in iKO-chow hearts compared with Con-chow hearts (Figure 2B). This accounts for 0.92% of the 25053 mouse transcripts covered by the global transcriptome. On the other hand, in response to HFD feeding, 34 transcripts were upregulated and 72 transcripts were downregulated in iKO-HFD hearts compared with Con-HFD hearts (Figure 2C), which accounts for 0.42% of the global transcriptome. These observations suggested that selectively enhancing mitochondrial FAO, via deletion of ACC2 in adult hearts, affected the transcription of a very limited number of genes, and interestingly, the effect was minimized by HFD. Notably, the numbers of DEGs, especially the downregulated genes, were much lower (by 2–3 folds) in HFD than that of chow-fed conditions (Figure 2D). Furthermore, we found that less than 10% of differentially regulated genes, i.e. 19 downregulated transcripts and 4 upregulated transcripts, were shared between chow- and HFD-fed conditions (Figure 2D). These observations collectively suggest that ACC2 iKO causes specific changes of gene transcription that is likely dependent on dietary fat.
Figure Figure . Global transcriptome analysis in Con and ACC2 iKO mouse hearts under chow or HFD feeding.

(A) Principle component analysis (PCA) of RNA-seq data sets from Con and ACC2 iKO mouse hearts after 16-weeks chow and HFD feeding. PCA is based upon the abundance of all the transcripts detected in RNA-seq analysis. (B-C) Patterns of differential gene expression in ACC2 iKO mouse hearts compared with Con under chow-fed (B) and HFD-fed (C) conditions were analyzed by Enhanced Volcano Package. Red dots represent genes with p value < 0.001. (D) The number of shared DEGs between ACC2 iKO and Con mouse hearts under chow-fed and HFD-fed conditions was shown by Venn diagrams.
The downregulated DEGs revealed that the PPARs signaling pathway was suppressed in ACC2 iKO hearts
In order to examine the mechanisms by which enhancing cardiac FAO regulated gene expression, we performed the pathway enrichment analysis using the DEGs in iKO-chow hearts. Among upregulated transcripts, ribosome was the only pathway enriched in iKO-chow hearts compared with Con-chow hearts (p< 0.05, Supplementary Table 3). On the other hand, in the group of downregulated DEGs, twelve pathways were significantly enriched in iKO-chow group (p< 0.05, Supplementary Table 3). In the top ten pathways, ranked by p. value, relative enrichment and the number of genes involved, fatty acid degradation is the most significantly enriched pathway (Figure 3A). Fifteen out of 50 genes in the fatty acid degradation pathway were significantly downregulated in iKO-chow hearts (Supplementary Table 3). This is somewhat unexpected as iKO hearts exhibited ~50% higher FAO compared with Con hearts [16]. The association of a greater FAO with a downregulation of gene expressions in the fatty acid degradation pathway suggested that a negative feedback mechanism existed between the metabolic flux and the transcriptional regulation of the pathway in iKO hearts. Furthermore, a large fraction of downregulated DEGs in iKO-chow hearts were enriched in pathways related to fatty acid metabolism, i.e. peroxisome pathway, peroxisome proliferator-activated receptor (PPAR) signaling pathway, and fatty acid elongation or biosynthetic pathways (Figure 3A).
Figure 3. Pathways enrichment and TF binding analysis of the downregulated DEGs in ACC2 iKO mouse heart under chow-fed condition.

(A) Pathway enrichment analysis of the downregulated genes in ACC2 iKO hearts under chow-fed condition was conducted using Cytoscape with ClueGo and CluePedia plugins. Top 10 clusters are shown. Adjusted p value was represented by dot color. Gene numbers were represented by dot size. Rich factor indicated the percentage of genes in the pathway. (B) Transfac Positional Weight Matrix for PPARα (upper panel) and PPARγ (lower panel). Motif name is attached. PPARs and RXR binds to direct repeat of a nuclear receptor-binding site (AGGTCA) spaced by one nucleotide. (C–D) The downregulated DEGs in ACC2 iKO hearts were subjected to transcriptional factor binding analysis. A network representing the selected targets (oval yellow nodes) regulated by the top potential transcriptional factor PPARα (C) and PPARγ (D) were shown. (E-F) Con and ACC2 iKO mice were subjected to HFD feeding for 16 weeks. ChIP-qPCR assays were performed with anti-PPARα (E) and anti-PPARγ (F) antibodies, respectively. The enrichment of individual PPARs’ targets was calculated as % of input DNA. (*p<0.05 vs. Con/chow, n=4–6).
To search for transcriptional regulatory mechanisms responsible for the global suppression of fatty acid degradation/metabolism in iKO hearts, we performed “regulons” analysis using the downregulated DEGs in iKO hearts. A regulon is defined as a group of genes which share one or more transcription factor binding motifs [18]. Motifs identified this way were clustered by similarity, ranked by NES (normalized enrichment score) and named by M1, M2 and etc. (Supplementary Table 4). Four out of the top five transcription factor binding motifs found among the downregulated DEGs in iKO hearts were categorized as M1 cluster and predicted as ones that occupied by RXRα, PPARα and PPARγ (Supplementary Table 4). We first selected several predicted PPARα and PPARγ target genes, which have shown the most significant changes in iKO mice and validated their expression by RT-PCR. The gene expression of PPARs’ targets examined by RT-PCR in Con and iKO mice under both chow-and HFD-fed conditions showed similar expression patterns compared with RNA sequencing results (Supplementary Figure 2A–2D). From this analysis, we have identified 29 DEGs (16% of the total downregulated DEGs) as predicted targets of PPARα, and 42 DEGs (23% of the total downregulated DEGs) as predicted targets of PPARγ (Figure 3B–3D). Notably, multiple downregulated DEGs that had been grouped in the fatty acid degradation pathway (Figure 3A) were also predicted as targets of PPARα and PPARγ. To examine whether PPARα and PPARγ directly bind to the promoters of these genes in vivo as predicted, we conducted chromatin immunoprecipitation (ChIP) experiments using PPARα and PPARγ antibodies. We found that the enrichment of PPARα or PPARγ on the promoters of the selected target genes were significantly reduced in ACC2 iKO mice, which is consistent with the gene expression profiles (Figure 3E–3F, Supplementary Figure 2B and 2D). On the other hand, ChIP analysis also confirmed that HFD treatment increased PPARs transcriptional activity in Con hearts and normalized the promoter binding occupancy in ACC2 iKO mice (Figure 3E–3F, Supplementary Figure 2B and 2D). However, the downregulated DEGs did not include any isoforms of PPAR (data not shown), suggesting a change of transcriptional activity rather than the expression level of the transcription factors in iKO hearts. Taken together, these analyses suggested that iKO hearts exhibited a deactivation of PPARα and/or PPARγ transcriptional program, which likely resulted in the suppression of gene expression for fatty acid degradation.
Cardiac transcriptome of lipotoxicity mouse models was distinct from ACC2 iKO and showed upregulation of the PPARs signaling and fatty acid degradation pathways
Cardiac dysfunction observed in several lipotoxic animal models was associated with increased FAO [19–21]. Despite higher FAO in iKO hearts, no obvious adverse effect on cardiac function or morphology has been observed even under chronic conditions [15, 16]. These observations suggest that high FAO is unlikely the culprit of lipotoxic cardiomyopathy. Thus, we reasoned that a comparison of the transcriptomes of these two types of high FAO hearts might generate mechanistic insight into cardiac lipotoxicity. Accordingly, we performed similar pathway analysis using transcriptome data from two well-established lipotoxic models: HFD induced obesity (the present study) and db/db mouse models (previously published database) [22], in which higher FAO has been observed [6, 20, 23]. We found that there was a total of 1736 DEGs in Con-HFD hearts compared with the Con-chow group (Supplementary Figure 3). We then compared the downregulated DEGs or the upregulated DEGs generated from iKO-chow hearts with those in HFD-fed or db/db mouse hearts. Venn diagram indicated that there are total 84 downregulated transcripts overlapping between HFD and db/db mouse hearts, whereas iKO-chow shares only 2 transcripts with db/db and 32 transcripts with HFD-fed mice (Supplementary Figure 4A). We further conducted the pathway analysis on these downregulated and overlapping genes, and found that the p53 signaling pathway was significantly enriched among the shared DEGs between HFD and db/db mouse hearts (Supplementary Figure 4B). Due to the small number of overlapping genes between iKO-chow and HFD-fed mouse hearts or between iKO-chow and db/db mouse hearts, we were unable to perform the pathway analysis. For upregulated genes, there were total 37 transcripts overlapping between HFD and db/db mouse hearts, whereas iKO-chow mice shared only 3 transcripts with HFD-fed mouse hearts and 0 transcripts with db/db mouse hearts (Supplementary Figure 5A). Overall, there were minimal DEGs in iKO mouse hearts that overlapped with that in HFD-fed or db/db mouse hearts. These findings suggest that despite high FAO, the transcriptional changes in iKO mouse hearts are distinct from that of lipotoxic hearts.
In the subsequent pathway analysis, we found that HFD-fed and db/db mouse hearts did not share any pathways in the downregulated DEGs (Figure 4A and 4C). On the other hand, the two models shared four pathways that were significantly enriched among upregulated DEGs, including fatty acid degradation, PPAR signaling pathway, peroxisome and fatty acid synthesis (Figure 4B and 4D, Supplementary Figure 5B). Surprisingly, all of the four pathways were significantly downregulated in iKO-chow mouse hearts (Figure 3A). Taken together, these data suggested that despite the elevated cardiac FAO in all three groups, expression of genes for fatty acid degradation as well as PPARs signaling pathway were changed in the opposite direction in iKO hearts compared to that in HFD-fed or db/db mice. Thus, the lipotoxic phenotype does not correlate with the level of FAO but closely associated with gene expression profiles for the PPARs signaling and its targets involved in fatty acid metabolism.
Figure 4. PPAR signaling and fatty acid degradation genes are upregulated in lipotoxic hearts but not in ACC2 iKO mouse hearts.

Pathway enrichment analysis of the downregulated DEGs in HFD-fed mice (A) and db/db mice (C) (GEO accession: GSE36875, p-value < 0.01) compared to their respective control. Pathway enrichment analysis of the upregulated DEGs in HFD-fed mice (B) and db/db mice (D) compared to their respective control.
Fatty acid degradation gene expressions were normalized in ACC2 iKO-HFD mouse hearts
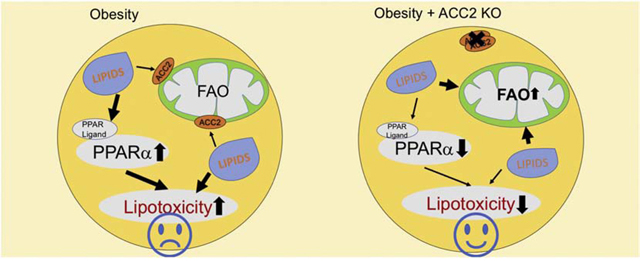
PPARs activation in the heart of HFD-fed and db/db mice is considered to be driven by increased availability of lipid ligands [24, 25]. We speculated that the deactivation of PPARs signaling in iKO-chow hearts was due to the depletion of lipid ligands secondary to increased FAO. This appeared to be consistent with the observation of no change in PPAR transcripts in the iKO hearts (data not shown). If our hypothesis is correct, increasing lipid supply, such as HFD feeding, should restore PPARs signaling in iKO hearts. We tested the hypothesis by using the top twenty genes from the pool of downregulated DEGs in iKO hearts that were involved in fatty acid degradation and PPARs signaling pathway. Consistent with our hypothesis, majority of the twenty genes was upregulated in the heart of HFD-fed or db/db mice (Figure 5A). Furthermore, when the iKO mice were subjected to HFD feeding, log2 fold changes for these genes were near 0, indicating that the expression of these genes was normalized to the level of control mice under chow-fed condition (Figure 5A). Independent metabolomics analysis showed that a group of active lipid species were downregulated in iKO-chow hearts, but upregulated in Con-HFD hearts, consistent with the expression pattern of PPAR target genes (Figure 5B, Dataset S1). Furthermore, HFD feeding normalized the amount of those lipids in iKO hearts to the similar level as Con-chow hearts (Figure 5B, Dataset S1). Among the altered active lipid metabolites, eicosapentaenoic acid (EPA), one of the eicosanoids/docosanoids species, has been shown to directly bind to PPARs and activate PPARs transcriptional activity [26, 27]. Arachidonic acid and its metabolites are also considered as biologically active mediators for PPARs activation [28]. These data suggested that downregulation of fatty acid degradation and PPAR signaling pathway in iKO mice was likely due to reduced PPAR ligands under chow-fed condition, which could be restored to the normal level through increasing lipid supply, such as HFD feeding. Activation of PPARs leads to cardiac lipid accumulation, an indication that lipid uptake exceeds FAO [29, 30]. Consistently, we found that HFD treatment induced cardiac triglyceride (TG) accumulation in Con hearts (Figure 5C). On the other hand, iKO mice attenuated HFD induced TG accumulation with no activation of PPAR signaling (Figure 5B and 5C). Taken together, the results suggest that a positive loop between ligands mediated PPAR activation and enhanced FAO in HFD-fed or db/db mice may contribute to the development of intracellular lipotoxicity and cardiac remodeling. On the contrary, enhancing FAO via a PPAR independent mechanism, e.g. deletion of ACC2, consumes intracellular lipid ligands and triggers a negative feedback to prevent over activation of the PPAR signaling and the development of lipotoxicity (Figure 5D).
Figure 5. Differential transcriptional regulation of fatty acid degradation genes by FAO and fatty acid supply.

(A) Heatmap of FAO related gene transcription levels in ACC2 iKO/chow, Con/HFD, db/db and ACC2 iKO/HFD hearts. Color coding for each gene was assigned using a log2 fold change versus the mean value of Con/chow. (B) Heatmap of bioactive lipids in Con and ACC2 iKO hearts with chow or HFD feeding. Color coding for each gene was assigned using a log2 fold change versus the mean value of Con/chow. (C) Cardiac triglyceride (TG) content normalized to tissue weight (*p<0.05 vs. Con/chow, #p<0.05 vs. Con/HFD, n=5). (D) Schematic working hypothesis. Ligands mediated PPAR activation and enhanced FAO in HFD induced obesity or db/db mice may contribute to the development of lipotoxicity. On the other hand, enhancing FAO by ACC2 deletion may deplete intracellular fatty acids, which can serve as a negative feedback to prevent over activation of PPAR pathway.
DISCUSSION
In this study, we have compared cardiac transcriptional profiles in mouse models with elevated cardiac FAO but demonstrated opposite susceptibility to lipotoxic cardiomyopathy. Increased FAO in ACC2 deficient hearts transcriptionally suppresses the PPARs signaling and fatty acid oxidation pathway under normal condition, while HFD feeding restores those pathways to the normal level. The negative feedback between FAO and PPARs signaling in these hearts maintains the homeostasis of lipid metabolism and prevents the development of cardiomyopathy during HFD induced obesity. On the other hand, upregulation of PPARs signaling and fatty acid oxidation pathways is observed in HFD-fed and db/db mouse hearts, two well-established lipotoxic cardiomyopathy models, suggesting that lipid overload in these hearts triggers a positive coupling of PPARs signaling and FAO that leads to the detrimental outcome. Our observations suggest that appropriate control of PPARs signaling to maintain the balance between fatty acid supply and oxidation is essential for preventing the development of lipotoxicity in the heart.
Although we have observed increased cardiac FAO in ACC2 deficient mouse hearts [15, 16], RNA sequencing data revealed that ACC2 iKO mouse hearts exhibited the coordinated decrease in the expression of genes involved in fatty acid degradation pathway. Consistent with our observation, downregulation of several fatty acid degradation genes, e.g. medium-chain acyl-CoA dehydrogenase (MCAD) and CPT1, was reported in the heart of the whole body ACC2 KO mice [31]. Therefore, we speculate that there is a negative feedback mechanism between FAO and transcriptional program of fatty acid degradation under normal conditions. In support of this notion, another study reported decreased PPAR signaling in the mouse heart with increased FAO due to an active mutation in Cpt1b (Cpt1bE3A) [32]. On the other hand, reducing FAO by inhibiting CPT1 via pharmacological or genetic approaches led increased mRNA levels of PPAR downstream targets, e.g. citrate synthase, Cpt1, PDK-4 or CD36 [33–35]. Our unbiased search using Iregulon motif analysis clearly show that a large pool of downregulated DEGs in ACC2 iKO mouse heart contains PPAR binding elements, thus identifying that FAO suppresses the transcription of fatty acid degradation genes via regulating the PPAR signaling pathway.
Previously, we found that ACC2 deletion did not change myocardial triglyceride content or acylcarnitine levels in the heart despite higher FAO [15]. Results here suggest that these hearts maintain lipid homeostasis through regulating the level of PPAR ligands. Previous report suggest that mRNA levels of PPARs target genes are upregulated by exposure of the heart to fatty acids [36, 37]. In hearts examined in this study, the expression of PPAR targeted genes closely correlates with a group of bioactive lipid species while the transcripts of PPAR remain unaltered. Among identified lipid species, long-chain fatty acids such as eicosapentaenoic acid, has been shown as a PPAR agonist [14]. Our metabolomics analysis confirms that cardiac level of EPA in ACC2 iKO mice is reduced while its level is increased in HFD-fed control mice. More importantly, increasing fatty acid supply by HFD feeding normalized the cardiac EPA level and restored the expression of genes involved in PPARs signaling in iKO hearts.
In the present study, 16 weeks of HFD feeding significantly increased body weight of the mice without causing systolic or diastolic dysfunction of the heart. Our previous findings demonstrated that HFD feeding for a similar duration increased blood glucose level and caused cardiac lipid accumulation in mice [17]. Therefore, analysis of cardiac transcriptome at this time point allow us to decipher the impact of metabolic changes independent of the confounding effects due to contractile dysfunction. In the absence of measurable changes of cardiac function, perturbing the lipid availability and/or FAO in the heart leading to distinct transcriptional responses that is not predicted by FAO rate but is concordant with the balance between supply and consumption.
The observation is consistent with the emerging concept that the association between high FAO and cardiac lipotoxicity, though often observed, does not indicate a causal relationship. Instead, the balance between fatty acid supply and oxidation is essential for the maintenance of lipid homeostasis and prevention of lipotoxicity in the heart. Promoting fatty acid oxidation under conditions of normal lipid supply will reduce PPAR ligands and thus, limit excessive oxidation via transcriptional suppression of fatty acid degradation pathway. Under conditions of increased lipid load, enhancing fatty acid oxidation will sustain the balance of supply and consumption. This not only prevents the accumulation of toxic lipid species but also limits further activation of PPAR signaling which would otherwise promote fatty acids import and activation. In support of this concept, reducing fatty acid uptake or promoting neutral lipid storage have also been shown to prevent cardiac lipotoxicity [38–40]. On the other hand, worsened cardiac toxicity and adverse cardiovascular events have been observed in rodents and human with diabetic complications after treatment with PPARs agonists (e.g. tesaglitazar and muraglitazar) [41–43]. Although the detailed mechanisms are not completely understood, observations from the current study suggest that excessive activation of PPAR signaling and the consequent imbalance between fatty acid uptake and utilization contributes to excess lipid accumulation and cardiac lipotoxicity. Consistently, PPARs activation on top of high FAO in the heart has been shown to mediate severe cardiomyopathy through the downregulation of Sirt1-PGC1 alpha pathway in obese mice [43]. It is worth to test whether the activity of Sirt1-PGC1 alpha axis was altered in our animal models in the future. Apparently, complete blocking PPARs activity in the heart is detrimental and not ideal under the obese conditions. Possible interventions which prevent excessive PPARs activation or increasing FAO through PPAR independent mechanisms may be effective approaches to prevent the development of obesity induced cardiomyopathy.
In conclusion, we report that high FAO causes downregulation of the PPAR signaling and fatty acid degradation pathways in ACC2 deficient hearts. The suppression of PPARs signaling may serve as a compensatory mechanism to prevent excessively high FAO under normal conditions and the development of cardiac lipotoxicity during lipid overload conditions.
MATERIALS AND METHODS
Animal Model
ACC2 flox/flox-MerCreMer+ (ACC2−f/f−MCM+) mice were mated with ACC2f/f to produce both study and control littermates. All the mice were housed at 22°C with a 12-hour light, 12-hour dark cycle with free access to water and standard chow. At 8 weeks of age, both ACC2f/f−MCM+ and ACC2f/f mice received an intraperitoneal injection of tamoxifen (20mg/kg) for 5 days, which was sufficient to cause ACC2 deletion in ACC2f/f−MCM+. Four weeks after the last injection of tamoxifen, ACC2f/f (designated as Con) and ACC2f/f−MCM+ (designated as iKO) mice were subjected to high fat diet (Research Diets, D12492) feeding for 16 weeks. The experiments included in this study were performed with male mice maintained on a C57BL/6 background. All protocols concerning animal use were approved by the Institutional Animal Care and Use Committee at University of Washington.
Transthoracic Echocardiography
The mice were anesthetized and maintained with 1–2% isofluorane in 95% oxygen. Transthoracic echocardiography was conducted at 16 weeks post high fat diet feeding with Vevo 2100 high-frequency, high-resolution digital imaging system (VisualSonics) equipped with a MS400 MicroScan Transducer. A parasternal short axis view was used to obtain M-mode images for analysis of fractional shortening, ejection fraction, and other cardiac functional parameters.
Isolated Heart Perfusion Experiments and NMR Spectroscopy
Mouse hearts were excised and perfused in Langendorff-mode at a constant pressure of 80 mmHg at 37°C. The equilibration perfusate contained (in mmol/L): NaCl 118, NaHCO3 25, KCl 5.3, CaCl2 2, MgSO4 1.2, EDTA 0.5, glucose 5.5, mixed long-chain fatty acids (LCFA) 0.4 (bound to 1.2% albumin), lactate 1.2, and insulin 50 μU/mL, equilibrated with 95% O2 and 5% CO2 (pH 7.4). After a 20-minute period, hearts were then perfused with a buffer in which the unlabeled LCFA and glucose were replaced with uniformly labeled 13C long-chain fatty acids and 1,6-13C glucose to determine the relative contribution of each substrate to oxidative metabolism. At the end of the 40-minute perfusion period, hearts were freeze-clamped. The frozen tissue was pulverized under liquid nitrogen, extracted with perchloric acid, neutralized, and lyophilized. The sample was resuspended in deuterium oxide and analyzed via 13C NMR spectroscopy. The contributions of glucose and fatty acids was determined by isotopomer analysis of the C3 and C4 glutamate multiplets from the NMR spectra. Left ventricular (LV) function was monitored via a water-filled balloon inserted into the LV and connected to a pressure transducer with the data acquisition system (PowerLab, ADInstruments). After 5 min of stabilization, hearts were equilibrated for 10 min at spontaneous HRs and then fixed at a HR of ~450 beats/min with an electrical stimulator (Grass Technologies). Pressure-volume relationships (i.e., Frank-Starling curves) were assessed by gradually increasing the volume of the LV balloon by 5μl increments [16, 44].
RNA Extraction and Deep Sequencing
Total RNA was extracted from frozen LV tissue using the RNeasy Fibrous Tissue Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA samples (RNA integrity number (RIN) >8) were used for library preparation and sequencing. Poly(A)- enriched cDNA libraries were prepared using the Illumina TruSeq RNA Library Preparation kit according to the manufacturer’s instructions. Then, library templates were sequenced on the HiSeq 4000 sequencing system at a read length of 100nt single end, and a depth of about 50 million per sample (Illumina, USA). Five mice per genotype or diet condition are used for subsequent data analysis.
RNA Sequencing Data Processing
The low quality reads, reads with adaptors and reads with unknown bases were filtered with the software SOAPnuke to get the clean reads [45]. Clean reads were mapped to the reference using Bowtie 2 [46], and then calculate gene expression level using RSEM [47]. Differential gene expression analysis was conducted with R packages DESeq2 [48]. Cut off for differential expression was chosen at an adjusted p value <0.05.
Pathway Enrichment and Transcription Factor Binding Analysis
Pathway enrichment analysis was performed using the ClueGO v2.5.1 plugins in Cytoscape (v3.6.1) with the KEGG pathway and Reactome database (adjusted P value (Bonferroni) <0.01) [49]. The analysis was visualized by CluePedia v1.5.1. Iregulon v1.3 was used to predict transcription factor binding motif [18]. The following options, Motif collection: 10K (9713 PWMs), putative regulatory region: 20kb centered around TSS (7species) alongside the program default settings for Recovery and Transcription factor prediction options were selected for the analysis.
Biochemical assays
Cardiac TG level was measured using Triglyceride Colorimetric Assay (Cayman Chemical) according to the supplier’s protocol. To assess the extent of oxidative stress, the levels of malondialdehyde (MDA) in heart tissue were measured using Lipid Peroxidation (MDA) Assay (Abcam) according to the supplier’s protocol. Values were normalized to frozen tissue weight.
Lipidomics Analysis
Pre-weighed frozen tissue was transferred to a pre-chilled 2 mL homegenization tube containing 300 mg of 1 mm zirconium disruption beads. Ethanol:water (80:20) extraction solvent, containing deuterated internal standards, was added at volumes to achieve a normalized concentration of 50 mg of tissue per 1 μL of solvent for each sample. The samples were then homogenized for 3 cycles of 10 seconds on followed by 10 seconds of dwell time at a speed of 8 m/s using an Omni International Bead Ruptor Elite homogenizer. The samples were then placed on dry ice for 1 minute to cool. The homogenization and cooling step were then repeated 2 more times. The homogenate was then transferred to a clean 1.5 mL microfuge tube and allowed to deproteinize for 30 minutes at −20 °C. To aide in more efficient SPE extraction, each sample was transferred to an Axygen 500 μL V-bottom 96-well plate (P/N P-DW-500-C) with 300 μL of water in each well. A Phenomenex Strata-X SPE plate was then washed with subsequent additions of 600 μL of methanol, 600 μL of ethanol, and 900 μL of water per well. Each addition of solvent was pulled through using a vacuum manifold taking care to not let the wells dry out between additions of solvents. The entire volume of the diluted samples were then added to the SPE wells and allowed to drain fully using gravity. Each well was then washed with 600 μL of a mixture of water:methanol (90:10) which was then slowly pulled through (5 mmHg vacuum) until no liquid was showing in the wells. The wells were then dried for an additional 60 seconds under 20 mmHg of vacuum. Metabolites were then eluted into an Axygen 500 μL V-bottom 96-well plate with the addition of 450 μL of room temperature ethanol and allowed to sit for 120 seconds. The eluent was then pulled through with the aid of 5 mmHg of vacuum. The eluent was then immediately dried in vacuo using a Thermo Savant centrifugal vacuum concentrator at a temperature of 40 °C. Once dried, each well was resuspended with 50 μL of a solution containing methanol:water (40:60) contain 0.1% acetic acid as well as CUDA, Resolvin D1-d5, and LTB4-d4 internal standards. The plate was then immediately sealed and vortexed at 500 rpm at 4 °C for 10 minutes. Samples were then immediately transferred into a Greiner deep-well 96-well plate (P/N 780215) where each well contains a Wheaton 300 μL glass insert (P/N 11–0000-101B). The sample plate was then immediately sealed and centrifuged at 500 rpm at 4 °C for 5 minutes to remove any air bubbles. Samples were then placed into a Thermofisher Vanquish UHPLC autosampler and maintained at 4 °C until 20 μL of each sample was injected for analysis by LC-MS/MS [50, 51].
Chromatographic separation was performed on a Thermofisher Vanquish UHPLC equipped with a Phenomenex Kinetex C18 column (1.7 μm particle size, 100Å, 100 × 2.1 mm. P/N 00D-4475-AN) maintained at a temperature of 50 °C. Elution was performed with mobile phases A (70:30 water:acetonitrile and 0.1% acetic acid) and B (50:50 acetonitrile:isopropanol and 0.02% Acetic Acid), flowing at 0.375 mL/min with following gradient was used: 1% B from 0 to 0.25min, 1% to 55%B from 0.25 to 5.00 minutes, 55% to 99%B from 5 to 5.50 minutes, and 99% B from 5.50 to 7 minutes. Mass detection was performed with a Thermo Scientific Q Exactive HF orbitrap mass spectrometer equipped with a heated electrospray injection (HESI) source. Mass detection was performed in negative ionization mode using the following source conditions: spray voltage 3.55 kV; sweep gas flow rate 2 arbitrary units; AUX gas flow rate 10 arbitrary units; sheath gas flow rate 40 arbitrary units; capillary temperature 265 °C; S-lens RF level 45; AUX gas heater temperature 350 °C. MS1 acquisition was performed under the following settings: Resolution 15,000; AGC volume 1×106; Maximum IT 50 ms; scan range 250 to 650 m/z. Data Dependent MS/MS acquisition was performed under the following settings: Resolution 15,000; AGC volume 1×105; Loop count 8; Isolation Window 1.0 m/z; Normalized collision energy 35 eV; minimum AGC volume 2×103; Dynamic Exclusion 5.0 seconds [50, 51].
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted from frozen LV tissue using the RNeasy Fibrous Tissue Mini Kit (Qiagen) according to the manufacturer’s instructions. Total RNA was reverse-transcribed into the first-strand cDNA using the Superscript First-Strand Synthesis Kit (Invitrogen). cDNA transcripts were quantified by Rotor Gene Real-Time PCR System (Qiagen) using SYBR Green (Biorad). Results of mRNA levels were normalized to 36B4 rRNA levels and reported as fold-change over Control. Primer information is listed below:
| Gene | Species | Forward sequence | Reverse sequence |
|---|---|---|---|
| Agt | Mouse | GACCTCCTGACTTGGATAGAGA | GAGTTCGAGGAGGATGCTATTG |
| Fbp2 | Mouse | GCTCGTCTCCGAAGAGAATAAA | CAGGGTTGCACTACCATACA |
| Acsf2 | Mouse | TTCAGTCCTCCCACACAAAG | GCAAAGTGCTGGGAAATTAGG |
| Decr1 | Mouse | CACCATTGTCCTCTGTGCTATAA | GCCTTGTTCCAAACCCAAAC |
| Slc25a20 | Mouse | CTGACAGACAGACAGACAGAAG | TCCAAGGTCCCAGAGTACATA |
| Acot2 | Mouse | ACAGTTGGGTAGCAAGAGTTAG | GCCAAGGGTACACAGAGAAA |
| Scn4b | Mouse | CAAGGCATAGAGCAGAAGAGAG | CCAGAGATGAGGACAGGATAGA |
| Slc25a34 | Mouse | GACAAGGCTACATCAGGACTAC | CTACCACAGCACCACAGATAA |
| 36B4 | Mouse | AGATTCGGGATATGCTGTTGG | AAAGCCTGGAAGAAGGAGGTC |
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the ChIP-IT® Express Chromatin Immunoprecipitation Kit (Active motif) with some modifications. Hearts were dissected from mice and atria were removed. Ventricles corresponding to 70–75 mg were minced to 1 mm block and homogenized with douce homogenizer (1400 rpm, 20 strokes) with 5 mL cold PBS until no big tissue chunk can be seen. The samples were centrifuged at 600 g for 5 minutes and resuspended in 1% Formalin/PBS. After incubation and rotation at RT for 10 minutes, 10× Glycine solution was added, followed by incubation for 5 minutes with rotation. After centrifugation, the pellets were washed 10 ml ice cold PBS with 10 mM PMSF (Phenylmethanesulfonyl fluoride) twice. The pellets then were resuspended in 1mL ChIP lysis buffer (20mM Tris-HCl pH 8.0, 85mM KCl, 0.5% NP-40) for 20 min at 4 °C and centrifuge at 5,000 rpm to pellet the nuclei. The cell nuclei were resuspended in 400 μL nuclei lysis buffer (50mM Tris-HCl pH8.0, 10mM EDTA, 1% SDS) and then subjected for sonication for 15 min with 30s intervals. Purified chromatin was analyzed by 1% agarose gel to determine the shearing efficiency and concentration. Chromatin solution corresponding to 3 μg DNA dissolved with ChIP buffer 1, 1 g antibodies and 20 μL protein G magnetic beads were used for each immunoprecipitation. Antibodies used for ChIP assays were PPARα (Santa Cruz) and PPARγ (Cell Signaling). As a control, normal IgG was used as a replacement of PPARα and PPARγ (Santa Cruz). Procedures for immunoprecipitation, chromatin elution and reverse crosslinking were performed by following the manufacturer’s instructions. Collected chromatin fragments were measured by quantitative PCR using the same conditions used for determination of mRNA expression levels. The mean value of the control mice was expressed as 1. PCRs were carried out using the following oligonucleotide primers:
PPARα targets:
| Gene | Species | Forward sequence | Reverse sequence |
|---|---|---|---|
| Agt | Mouse | CAGGCTTGACCAAGATGGAT | CCCAGAGAGGCTTACGAGTG |
| Fbp2 | Mouse | TCATTCTGCACGGTTGAGAC | AACTGGCAAATCAACCCTGA |
| Decr1 | Mouse | CTGCCTTACCCTCTGAGGAC | GAGAGGGAGCGGAGAACTG |
PPARγ targets:
| Gene | Species | Forward sequence | Reverse sequence |
|---|---|---|---|
| Agt | Mouse | GGAAAGCAGCAGTTTTGGAG | GTCCACATGGCCAAGAGATT |
| Scn4b | Mouse | GGTTCCAGGCCACAAAATAA | CTTGGAGCTGGAGACAAAGG |
| Acot2 | Mouse | TTTGTGTGGGAGGACCTAGC | GGAAATGAGTTCAAGTCCTTCG |
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical comparisons between groups were conducted by one-way or two-way ANOVA followed by a Newman-Keuls comparison test by Prism (GraphPad). The value of p<0.05 was considered to be significant.
Data Availability
RNA-seq data displayed are deposited in NCBI Gene Expression Omnibus under accession code GSE131122. All other remaining data are available within the article and Supplementary Files, or available from the authors upon request.
Supplementary Material
Supplementary Figure 1
The heart weight to tibia length (HW/TL) ratio of ACC2 iKO and Con mice under chow- or HFD-fed conditions (n=3–6).
Supplementary Figure 2
Con and ACC2 iKO mice were subjected to HFD feeding for 16 weeks. (A and C) RNA sequencing analysis of the selected PPAR⍺ (A) and PPAR γ (C) targeted genes. The expression was reported as fold changes over Con. (B and D) qRT-PCR validation of the selected PPAR⍺ and PPAR γ targeted genes shown in (A) and (C). The expression was normalized to 36B4 rRNA and reported as fold change over Con (*p<0.05 vs. Con/chow, #p<0.05 vs. iKO/chow, n=5).
Supplementary Figure 3
Pattern of the differential gene expression in mouse hearts under HFD-fed condition compared with chow-fed condition was analyzed by Enhanced Volcano Package. Red dots represent genes with p value < 0.001.
Supplementary Figure 4
(A) Total number and shared numbers of the downregulated DEGs in ACC2 iKO, db/db, and HFD-fed mice hearts with each other are shown by Venn diagram. (B) The shared downregulated DEGs between db/db and HFD-fed mouse hearts were subjected to pathway enrichment analysis. GOTerm, gene number and associated gene name are shown.
Supplementary Figure 5
(A) Total number and shared numbers of the upregulated DEGs in ACC2 iKO, db/db, and HFD-fed mouse hearts with each other are shown by Venn diagram. (B) The shared upregulated DEGs between db/db and HFD-fed mouse hearts were subjected to pathway enrichment analysis. GOTerm, gene number and associated gene name are shown.
Supplementary Table 1
Con and ACC2 iKO mice were subjected to chow or HFD feeding for 16 weeks. Ex vivo cardiac functions were assessed in isolated perfused hearts. LVDevP, left ventricular developed pressure; RPP, rate pressure product (the product of LVDevP and heart rate).
Supplementary Table 2
Echocardiography examination of Con and ACC2 iKO mice under chow or HFD feeding conditions.
Supplementary Table 3
The top 21 KEGG pathways identified in ACC2 iKO hearts.
Supplementary Table 4
Summary of Iregulon analysis of the downregulated DEGs in ACC2 iKO mouse hearts under chow-fed condition. Total 179 downregulated DEGs from ACC2 iKO mouse hearts were subjected to Iregulon analysis. Top transcription binding motifs and their associated transcription factors enriched in the cis-regulatory regions of the DEGs are shown. NES (normalized enrichment score) corresponds to the enrichment score of the motif or the maximal enrichment score for a given TF. The enriched motifs are clustered by similarity and similar motifs that belong to the same cluster will have the same cluster code. The cluster code is given by the order of maximal NES per motif cluster.
Highlight.
Increasing FAO by ACC2 deletion does not cause cardiac dysfunction in response to HFD feeding.
ACC2 deletion increases FAO and negatively regulates PPAR signaling likely via reduction of endogenous PPAR ligands in non-obese mice.
Activation of PPARs signaling and downstream lipid metabolism pathways is observed in the heart of lipotoxic mouse models.
Enhancing FAO by ACC2 deletion in obese mice consumes intracellular lipid ligands and triggers a negative feedback to prevent over activation of the PPAR signaling and the development of lipotoxicity.
ACKNOWLEDGEMENTS
We thank Dr. Stephen C. Kolwicz Jr for his help of tissue collection. This work was supported in part by U.S. National Institutes of Health Grants HL-110349 and HL-129510 (to R. Tian), the American Heart Association Career Development Award 18CDA34080486 (to D. Shao), UCSD Chancellor’s Research Excellence Scholarship (CRES) award (to J. Ding) and Ledell Family Research Scholarship for Science and Engineering (to J. Ding). D. Shao is also supported by the pilot and feasibility program from the Diabetes Research Center at the University of Washington (P30 DK017047).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference:
- 1.Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36(27):1718–27, 27a-27c. [DOI] [PubMed] [Google Scholar]
- 2.Isfort M, Stevens SC, Schaffer S, Jong CJ, Wold LE. Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail Rev. 2014;19(1):35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101(4):335–47. [DOI] [PubMed] [Google Scholar]
- 4.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–23. [DOI] [PubMed] [Google Scholar]
- 5.Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. 2014;171(8):2080–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146(12):5341–9. [DOI] [PubMed] [Google Scholar]
- 7.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100(3):1226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borradaile NM, Schaffer JE. Lipotoxicity in the heart. Curr Hypertens Rep. 2005;7(6):412–7. [DOI] [PubMed] [Google Scholar]
- 9.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801(3):311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Q, Li Y. Roles of PPARs on regulating myocardial energy and lipid homeostasis. J Mol Med (Berl). 2007;85(7):697–706. [DOI] [PubMed] [Google Scholar]
- 11.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20(5):649–88. [DOI] [PubMed] [Google Scholar]
- 12.Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000;10(6):238–45. [DOI] [PubMed] [Google Scholar]
- 13.Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11(6):779–91. [DOI] [PubMed] [Google Scholar]
- 14.Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications--a review. Nutr J. 2014;13:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111(6):728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. 2016;100:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen S, Shao D, Tomasi LC, Braun A, de Mattos ABM, Choi YS, et al. The effects of fatty acid composition on cardiac hypertrophy and function in mouse models of diet-induced obesity. J Nutr Biochem. 2017;46:137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014;10(7):e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, et al. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56(8):2085–92. [DOI] [PubMed] [Google Scholar]
- 20.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52(2):434–41. [DOI] [PubMed] [Google Scholar]
- 21.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53(9):2366–74. [DOI] [PubMed] [Google Scholar]
- 22.Wilson KD, Li Z, Wagner R, Yue P, Tsao P, Nestorova G, et al. Transcriptome alteration in the diabetic heart by rosiglitazone: implications for cardiovascular mortality. PLoS One. 2008;3(7):e2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, et al. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106(3):447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54(12):3496–502. [DOI] [PubMed] [Google Scholar]
- 25.Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab. 2004;89(2):463–78. [DOI] [PubMed] [Google Scholar]
- 26.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3(3):397–403. [DOI] [PubMed] [Google Scholar]
- 27.Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci U S A. 1993;90(6):2160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol. 2000;129(5):823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117(10):2791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essop MF, Camp HS, Choi CS, Sharma S, Fryer RM, Reinhart GA, et al. Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am J Physiol Heart Circ Physiol. 2008;295(1):H256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Weeghel M, Abdurrachim D, Nederlof R, Argmann CA, Houtkooper RH, Hagen J, et al. Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B. Cardiovascular research. 2018;114(10):1324–34. [DOI] [PubMed] [Google Scholar]
- 33.Bristow M. Etomoxir: a new approach to treatment of chronic heart failure. Lancet. 2000;356(9242):1621–2. [DOI] [PubMed] [Google Scholar]
- 34.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Kung TA, Hoit BD, et al. Carnitine palmitoyl transferase-I inhibition is not associated with cardiac hypertrophy in rats fed a high-fat diet. Clin Exp Pharmacol Physiol. 2007;34(1–2):113–9. [DOI] [PubMed] [Google Scholar]
- 35.Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, et al. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006;114(16):1721–8. [DOI] [PubMed] [Google Scholar]
- 36.Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92(5):518–24. [DOI] [PubMed] [Google Scholar]
- 37.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95(6):568–78. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, et al. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100(8):1208–17. [DOI] [PubMed] [Google Scholar]
- 39.Steinbusch LK, Luiken JJ, Vlasblom R, Chabowski A, Hoebers NT, Coumans WA, et al. Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice. Am J Physiol Endocrinol Metab. 2011;301(4):E618–27. [DOI] [PubMed] [Google Scholar]
- 40.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr., Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100(6):3077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294(20):2581–6. [DOI] [PubMed] [Google Scholar]
- 42.Goldstein BJ, Rosenstock J, Anzalone D, Tou C, Ohman KP. Effect of tesaglitazar, a dual PPAR alpha/gamma agonist, on glucose and lipid abnormalities in patients with type 2 diabetes: a 12-week dose-ranging trial. Curr Med Res Opin. 2006;22(12):2575–90. [DOI] [PubMed] [Google Scholar]
- 43.Kalliora C, Kyriazis ID, Oka SI, Lieu MJ, Yue Y, Area-Gomez E, et al. Dual peroxisome-proliferator-activated-receptor-alpha/gamma activation inhibits SIRT1-PGC1alpha axis and causes cardiac dysfunction. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116(8):901–9. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 2018;7(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watrous JD, Niiranen TJ, Lagerborg KA, Henglin M, Xu YJ, Rong J, et al. Directed Non-targeted Mass Spectrometry and Chemical Networking for Discovery of Eicosanoids and Related Oxylipins. Cell Chem Biol. 2019;26(3):433–42 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lagerborg KA, Watrous JD, Cheng S, Jain M. High-Throughput Measure of Bioactive Lipids Using Non-targeted Mass Spectrometry. Methods Mol Biol. 2019;1862:17–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
The heart weight to tibia length (HW/TL) ratio of ACC2 iKO and Con mice under chow- or HFD-fed conditions (n=3–6).
Supplementary Figure 2
Con and ACC2 iKO mice were subjected to HFD feeding for 16 weeks. (A and C) RNA sequencing analysis of the selected PPAR⍺ (A) and PPAR γ (C) targeted genes. The expression was reported as fold changes over Con. (B and D) qRT-PCR validation of the selected PPAR⍺ and PPAR γ targeted genes shown in (A) and (C). The expression was normalized to 36B4 rRNA and reported as fold change over Con (*p<0.05 vs. Con/chow, #p<0.05 vs. iKO/chow, n=5).
Supplementary Figure 3
Pattern of the differential gene expression in mouse hearts under HFD-fed condition compared with chow-fed condition was analyzed by Enhanced Volcano Package. Red dots represent genes with p value < 0.001.
Supplementary Figure 4
(A) Total number and shared numbers of the downregulated DEGs in ACC2 iKO, db/db, and HFD-fed mice hearts with each other are shown by Venn diagram. (B) The shared downregulated DEGs between db/db and HFD-fed mouse hearts were subjected to pathway enrichment analysis. GOTerm, gene number and associated gene name are shown.
Supplementary Figure 5
(A) Total number and shared numbers of the upregulated DEGs in ACC2 iKO, db/db, and HFD-fed mouse hearts with each other are shown by Venn diagram. (B) The shared upregulated DEGs between db/db and HFD-fed mouse hearts were subjected to pathway enrichment analysis. GOTerm, gene number and associated gene name are shown.
Supplementary Table 1
Con and ACC2 iKO mice were subjected to chow or HFD feeding for 16 weeks. Ex vivo cardiac functions were assessed in isolated perfused hearts. LVDevP, left ventricular developed pressure; RPP, rate pressure product (the product of LVDevP and heart rate).
Supplementary Table 2
Echocardiography examination of Con and ACC2 iKO mice under chow or HFD feeding conditions.
Supplementary Table 3
The top 21 KEGG pathways identified in ACC2 iKO hearts.
Supplementary Table 4
Summary of Iregulon analysis of the downregulated DEGs in ACC2 iKO mouse hearts under chow-fed condition. Total 179 downregulated DEGs from ACC2 iKO mouse hearts were subjected to Iregulon analysis. Top transcription binding motifs and their associated transcription factors enriched in the cis-regulatory regions of the DEGs are shown. NES (normalized enrichment score) corresponds to the enrichment score of the motif or the maximal enrichment score for a given TF. The enriched motifs are clustered by similarity and similar motifs that belong to the same cluster will have the same cluster code. The cluster code is given by the order of maximal NES per motif cluster.
Data Availability Statement
RNA-seq data displayed are deposited in NCBI Gene Expression Omnibus under accession code GSE131122. All other remaining data are available within the article and Supplementary Files, or available from the authors upon request.