

Summary
Telomerase is an attractive target for anti-tumor therapy as it is almost universally expressed in cancer cells. Here we show that treatment with a telomere-targeting drug, 6-thio-2’-deoxyguanosine (6-thio-dG), leads to tumor regression through innate and adaptive immune-dependent responses in syngeneic and humanized mouse models of telomerase-expressing cancers. 6-thio-dG treatment causes telomere-associated DNA damages that are sensed by dendritic cells (DCs) and activates the host cytosolic DNA sensing STING/IFN-I pathway, resulting in enhanced cross-priming capacity of DCs and tumor-specific CD8+ T cell activation. Moreover, 6-thio-dG overcomes resistance to checkpoint blockade in advanced cancer models. Our results unveil how telomere stress increases innate sensing and adaptive anti-tumor immunity and provide strong rationales for combining telomere-targeted therapy with immunotherapy.
Keywords: Telomerase, telomere-targeted therapy, immunotherapy, 6-thio-dG, innate sensing, PD-1/PD-L1, DNA damage, STING, anti-tumor immunity, checkpoint blockade
Graphical Abstract
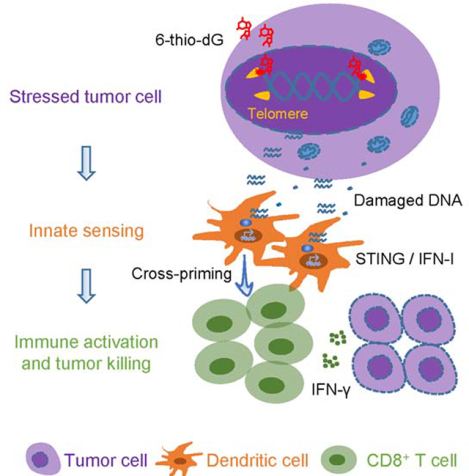
In Brief:
Mender et al. show that cancer cells treated with the nucleoside analog 6-thio-2’-deoxyguanine (6-thio-dG) undergo telomere stress and release DNA that is sensed by dendritic cells via STING-interferon signaling, which in turn activate CD8+ T cells. 6-thio-dG synergizes with immune checkpoint inhibitors.
Introduction
Immunotherapies have revolutionized the treatment of many cancers in the immuno-oncology field (Brahmer et al., 2012; Hodi et al., 2010; Ribas and Wolchok, 2018; Topalian et al., 2012). The most commonly used immunotherapies are PD-L1/PD-1 checkpoint blockades that have been approved by the FDA for advanced cancers such as melanoma and non-small cell lung cancer (Garon et al., 2015; Ribas et al., 2016; Rizvi et al., 2015b; Socinski et al., 2018). Despite the success of immunotherapies, many patients do not respond well to these therapies due to the immune suppressive tumor microenvironment, tumor immunogenicity and the emergence of primary and adaptive resistance (Chen and Han, 2015; Gide et al., 2018). Although recent studies show that the abundance of tumor mutations and neoantigens partially dictate cancer patient responses to checkpoint blockade, there are still considerable numbers of patients with high mutations and neoantigens that do not respond well (Le et al., 2017; Mandal et al., 2019; Rizvi et al., 2015a), suggesting neoantigens are not sufficient for provoking anti-tumor immune responses. Therefore, there is an urgent need to identify other factors for better immune responses and to develop new approaches to improve patient overall survival.
The generation of effective anti-tumor adaptive immune responses require tumor antigen presentation by antigen presenting cells, whose activation heavily rely on adequate innate sensing. Innate sensing is often provided by danger signals such as high mobility group box 1 protein, extracellular ATP and tumor DNAs released from stressed tumor cells (Kroemer et al., 2013; Pitt et al., 2017). Recent studies highlight the importance of cytosolic DNA sensing in radiation and DNA damaging therapies (Deng et al., 2014; Sen et al., 2019). The presence of DNA in the cytoplasm, for example, in the form of micronuclei (small DNA containing organelles) that lose nuclear envelop membranes can trigger immune responses. Micronuclei are the products of chromosome damage as a result of genotoxic stress and chromosome missegregation during cell division (Fenech et al., 2011). The cytosolic DNA sensor cGAS recognizes micronuclei and converts GTP (guanosine triphosphate) and ATP (adenosine triphosphate) into second messenger cGAMP (cyclic GMP-AMP) (Wu et al., 2013). Then the adaptor protein Stimulator of IFN Gene (STING) binds to cGAMP (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013; Zhang et al., 2013). This complex process activate TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3) (Liu et al., 2015; Tanaka and Chen, 2012) and further activate the downstream transcription of type I IFNs and other cytokines (reviewed in (Li and Chen, 2018)), ultimately increases innate sensing.
Eukaryotic linear chromosomes are capped by special structures called telomeres (TTAGGG), which are essential to maintain chromosomal stability (reviewed in (Blackburn, 1991)). Telomeres constitute the final ~10 kb of all human chromosomes and the final 12–80 kb of all mouse chromosomes (Lansdorp et al., 1996; Zijlmans et al., 1997). In all somatic human cells, telomeres shorten with each cell division due to the end replication problem and the absence of a telomere maintenance mechanism (reviewed in (Greider, 1996)). However, unicellular eukaryotes, germline cells and immortal cancer cells maintain their telomeres at a constant length almost always by activating the enzyme telomerase (Greider and Blackburn, 1985; McEachern and Blackburn, 1996; Morin, 1989; Nakamura et al., 1997; Singer and Gottschling, 1994; Yu et al., 1990). Telomerase is a reverse transcriptase enzyme that elongates telomeres by adding TTAGGG repeats to the ends of chromosomes and is expressed in ~90% of human tumors, but not in most normal cells (Shay and Bacchetti, 1997). Therefore, telomerase is an attractive target to develop anti-cancer therapies.
The nucleoside analogue, 6-thio-2’-deoxyguanosine (6-thio-dG), is an effective therapeutic approach in the cancer field. Its incorporation into de novo synthesized telomeres by telomerase is known to induce damage on telomeric DNA (Mender et al., 2015a). This results in rapid tumor shrinkage or growth arrest in many tumor-derived xenograft models with minimal side effects (Mender et al., 2018; Sengupta et al., 2018; Zhang et al., 2018). The most important advantage of this telomere-targeted therapy over direct telomerase inhibitors is that 6-thio-dG does not have a long lag period for tumor killing effects. Additionally, it does not directly inhibit telomerase but is preferentially recognized by telomerase over other polymerases and incorporated into the telomeres resulting in an immediate DNA chain termination. Importantly, its effect is independent of initial telomere length by hijacking tumor telomerase to make unstable telomeres (Mender et al., 2015b).
In this study, we aimed to explore whether 6-thio-dG that induces telomere stress in telomerase positive cancer cells could initiate rapid DNA damage for innate sensing. We used syngeneic wild type and genetic deficient mice to evaluate how 6-thio-dG triggers innate sensing and how it contributes to host anti-tumor immunity. Importantly, we demonstrate that 6-thio-dG overcomes PD-L1 blockade resistance in advanced tumors.
Results
The therapeutic effect of 6-thio-dG depends on CD8+ T cells.
All previous studies with xenograft models showed that intensive daily treatment with 6-thio-dG over 10 days could partially control tumor growth in many tumor models (Mender et al., 2015a; Mender et al., 2018; Zhang et al., 2018). However, the potential role of this drug on interaction between tumors and the adaptive immune system is unknown. In order to explore whether 6-thio-dG induces telomere-based DNA sensing for T cell responses, we first determined the inhibition of cell viability by 6-thio-dG on telomerase-positive murine colon cancer cells (MC38) in immunocompetent host. MC38 tumor cells are sensitive to 6-thio-dG with an IC50 concentration of 370 nM (Figure 1A). We also confirmed 6-thio-dG sensitivity in MC38 cells by a separate colony formation assay. MC38 cells treated with 6-thio-dG every three days for 13 days, resulted in less than 50% of the cells forming colonies with 0.5 μM 6-thio-dG treatment (Figures 1B and 1C). To evaluate whether 6-thio-dG reduces tumor burden in syngeneic mouse models in vivo, we subcutaneously inoculated MC38 cell into immunocompetent wild-type (WT) C57BL/6 mice. Seven days after tumor inoculation (when the tumor volume was ~ 100 mm3), 3 mg/kg 6-thio-dG was administrated daily for only three days and tumor growth was significantly reduced (Figure 1D) compared to the control tumor. This was not a unique response to MC38 tumor model as we also observed cell viability inhibition in vitro and significant tumor growth delay in vivo in telomerase-positive LLC (Lewis lung murine carcinoma derived from the C57BL/6 mouse) and CT26 (Colon murine carcinoma derived from the BALB/C mouse) tumor models with only three days treatment (Figures S1A–S1D).
Figure 1. The therapeutic effect of 6-thio-dG depends on CD8+ T cells.
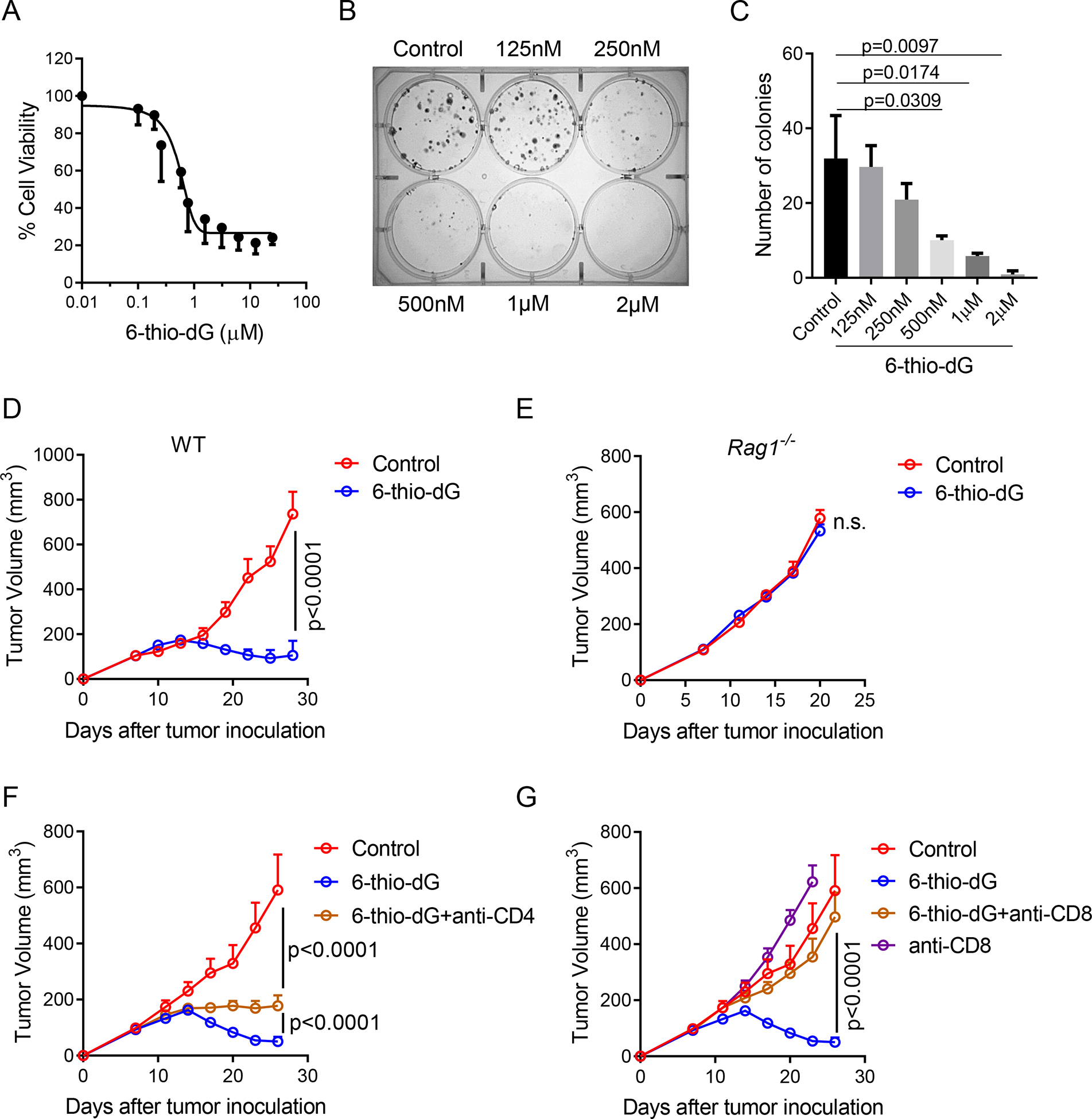
(A) Cell viability (IC50) of 6-thio-dG in MC38 cells. Cells were treated with 6-thio-dG for 5 days.
(B and C) Colony formation assay of 6-thio-dG in MC38 cells at indicated doses for 13 days. Cells were treated with 6-thio-dG every 3 days, then fixed and stained with crystal violet. Representative image of three biological replicates were shown in (B) and the quantification data was shown in (C).
(D and E) WT (D) or Rag1−/− (E) C57BL/6 mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9).
(F and G) C57BL/6 mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 200 μg of anti-CD4 (F) or anti-CD8 (G) was administrated one day before treatment initiation and then twice a week for 3 weeks. Tumor growth was measured every 3 days.
Data were shown as mean ± SEM from two to three independent experiments. P value was determined by two-tailed unpaired t test (C) or two-way ANOVA (D-G). See also Figure S1.
Because we administered 6-thio-dG for such a short duration compared to the intensive dosing strategy in xenograft models (5 mg/kg, daily for two weeks) and achieved better anti-tumor effect in the syngeneic mouse models, we speculated that 6-thio-dG might have an immune stimulatory role in vivo. Therefore, we inoculated tumors on Rag1 KO mice that cannot generate mature T and B cells. Indeed, the therapeutic effect of 6-thio-dG was completely diminished (Figure 1E), indicating adaptive immune cells are largely required for tumor control in vivo. To find out which subset of T cells contributes to the 6-thio-dG-mediated anti-tumor effect, we depleted CD4+ or CD8+ T cells while giving 6-thio-dG treatment and observed a marginal influence of CD4+ T cell depletion (Figure 1F). However, depletion of CD8+ T cells completely abolished the therapeutic effect of 6-thio-dG (Figure 1G). Together the data can be interpreted to suggest an essential role of CD8+ T cells in 6-thio-dG treatment.
6-thio-dG treatment increases tumor-specific T cell response.
As the therapeutic effect of 6-thio-dG depends on T cells, we reasoned that 6-thio-dG treatment might change immune cell expansion in the tumor microenvironment. To test this, we analyzed the number of tumor infiltrating lymphocytes (TILs) 6 days after the last of three daily doses of 6-thio-dG treatment. We found an increase in the frequency of CD3+ T cells and CD8+ T cells in TILs after 6-thio-dG treatment (Figures 2A, S2A and S2B). We also observed a significant upregulation of CD8+ T cell proliferation indicated by elevated Ki67 expression (Figure 2B), but no significant changes of Treg cells (Figure S2C). Although tumor infiltrating NK cells were also increased, we did not see an impact of NK cell depletion on the therapeutic effect of 6-thiodG (Figures S2D and S2E). Together with our CD8 depletion experiments, this suggests that NK cells are not essential but CD8+ T cell responses are required in 6-thio-dG mediated anti-tumor effects.
Figure 2. 6-thio-dG treatment increases tumor-specific T cell response.
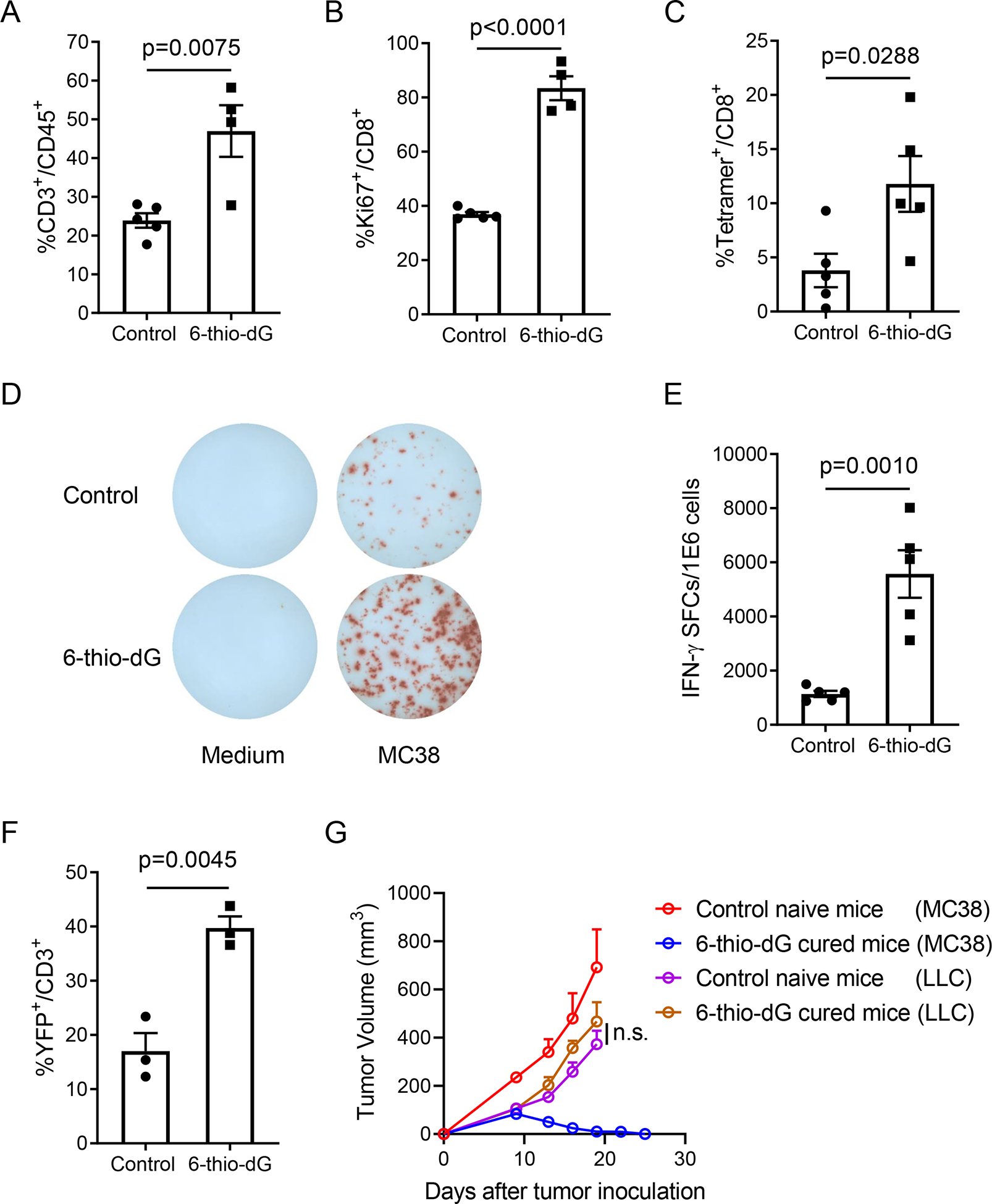
(A and B) C57BL/6 mice (n=4–5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Six days after last treatment, tumor infiltrating T cells were analyzed for the frequency of total T cells (A) and Ki67+CD8+ T cells (B).
(C) C57BL/6 mice (n=5) bearing MC38-OVA tumor were treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Three days after last treatment, tumor infiltrating T cells were analyzed for OVA specific CD8+ T cells with H-2Kb-OVA257–264 tetramer.
(D and E) Same experiment scheme as in (A), splenocytes were collected and re-stimulated with irradiated MC38 tumor cells for 48 h. IFN-γ producing cells were determined by ELISPOT assay. Representative spots were shown in (D) and the quantification data (n=5) was shown in (E).
(F) IFN-γ reporter mice (n=3) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Eleven days after the last treatment, tumors were minced and digested for flow cytometric detecting of YFP+ T cells.
(G) C57BL/6 mice were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 5 weeks later, tumor-free mice from 6-thio-dG treated group (n=5) and control naïve mice (n=5) were re-challenged with 5×106 MC38 tumor cells on the opposite flank (left), and 5×105 LLC tumor cells were injected on the right flank. Tumor growth was measured every 3 days.
Data were shown as mean ± SEM from two to three independent experiments. P value was determined by two-tailed unpaired t test (A-C, E and F) or two-way ANOVA (G). See also Figure S2.
We further tested the antigen-specific T cell response after 6-thio-dG treatment by using the MC38-OVA tumor model, which allows tracking of antigen specific T cells in the tumor tissue. Indeed, we observed increased tumor-specific CD8+ T cells in tumors 6 days after 6-thiodG treatment (Figure 2C). We also observed enhanced tumor-specific cytotoxic T cell responses in the MC38 tumor model by measuring IFN-γ producing T cells after 6-thio-dG treatment (Figures 2D and 2E). To directly assess the capacity of T cells to produce IFN-γ in vivo, we utilized IFN-γ YFP reporter mice that allow tracking of IFN-γ producing T cells with YFP expression (Reinhardt et al., 2009). 6-thio-dG treatment significantly increased YFP+ T cells in the tumor, suggesting enhanced IFN-γ production ability of T cells (Figures 2F and S2F). The hallmark of an adaptive immune response is the formation of memory that initiates a rapid recall response when the same antigen appears. To determine if 6-thio-dG treatment induces a memory response, mice with completely regressed tumors after 6-thio-dG treatment were rested for 5 weeks and re-challenged with the same MC38 tumor but with 10 times more tumor cells on the opposite flank (left flank), and LLC tumor cells were inoculated as control on the right flank. When naïve mice (never exposed to MC38 cells or 6-thio-dG) were injected with the same number of MC38 cells, the tumors grew aggressively. Remarkably, all tumor-free mice by 6-thio-dG treatment spontaneously rejected re-challenged MC38 tumors. In contrast, the control LLC tumors grew at a similar rate in naïve versus 6-thio-dG cured mice (Figure 2G), demonstrating the induction of antigen-specific immune memory protection after 6-thio-dG treatment.
6-thio-dG treatment enhances the cross-priming capacity of dendritic cells.
Antigen cross-presentation by antigen presenting cells (APCs) such as DCs or macrophages accounts for the tumor-specific CD8+ T cell activation. To explore which APC subset contributes to 6-thio-dG induced T cell activation, we first used anti-CSF1R antibody to deplete macrophages. We found that 6-thio-dG worked even better in macrophage depleted group (Figure 3A), which can be explained by the additive effect of the removal of immune suppressive tumor associated macrophages. BATF3 (basic leucine zipper ATF-like transcription factor 3)-dependent DCs are critical for the priming of antigen-specific CD8+ T cells (Broz et al., 2014; Edelson et al., 2010). 6-thio-dG treatment in Batf3 deficient mice partially delayed tumor growth but was significantly less effective compared with WT mice (Figure 3B). Noticeably, 60% of WT mice were completely tumor free but none of mice were tumor free in Batf3−/− mice (Figure 3C), suggesting an important role of BATF3-dependent DCs in the therapeutic effect of 6-thio-dG.
Figure 3. 6-thio-dG treatment enhances the cross-priming capacity of dendritic cells.
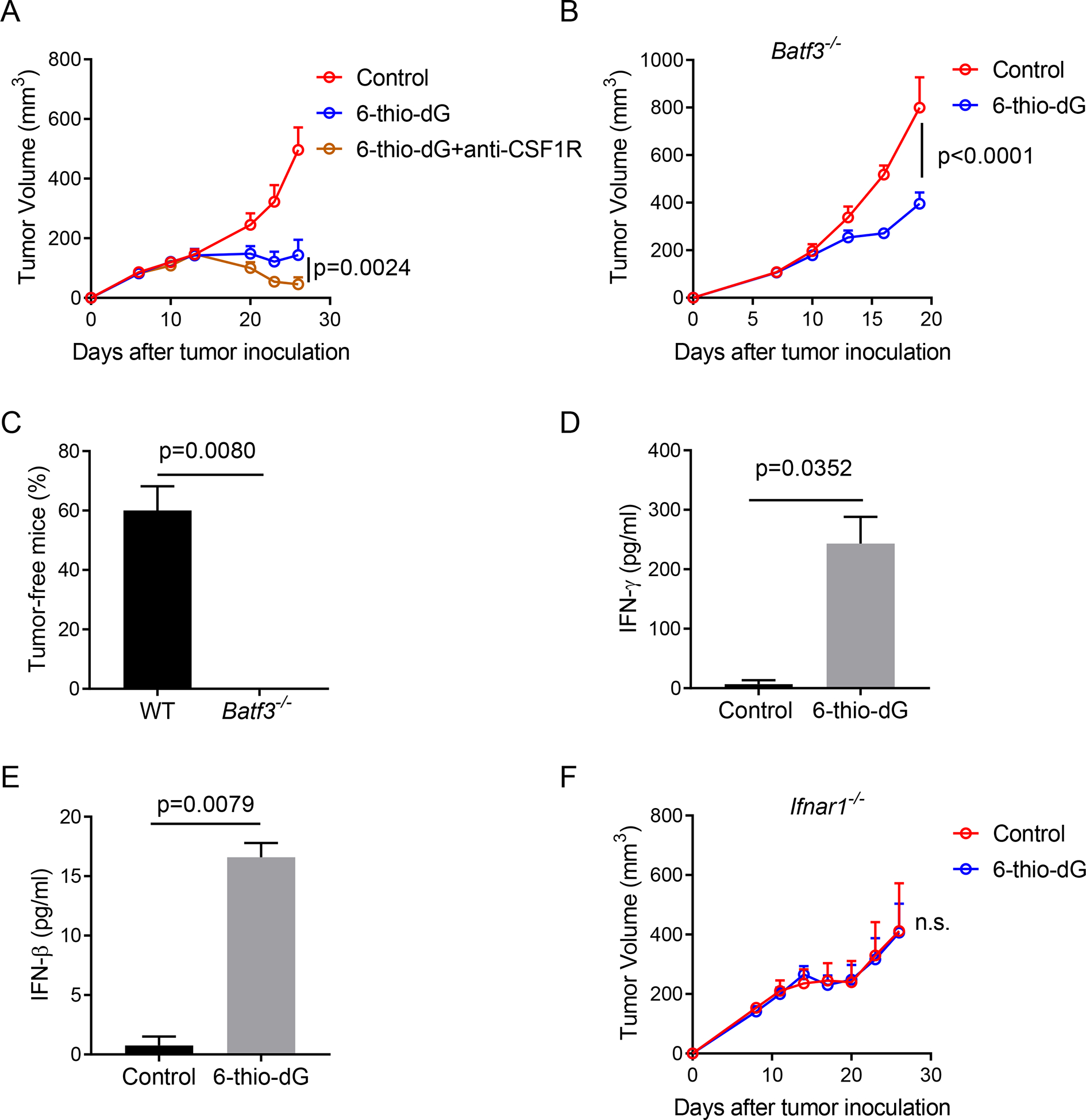
(A) C57BL/6 mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thiodG (3 mg/kg, days 7, 8, 9). 200 μg anti-CSF1R was administrated one day before treatment initiation and then twice a week for 3 weeks.
(B) Batf3−/− mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days.
(C) Percentage of tumor-free mice in WT and Batf3−/− mice (n=5) after 6-thio-dG treatment.
(D) BMDCs were cultured with MC38 tumor cells that were pretreated with 200 nM 6-thio-dG or vehicle for overnight, and then DCs were purified and co-cultured with naïve OT-1 T cells. 48 h later, supernatant was collected and tested for IFN-γ production by cytometric bead array (CBA).
(E) BMDC were cultured with MC38 tumor cells that were pretreated with 200 nM 6-thio-dG or vehicle for 18 h, supernatant was collected for IFN-β ELISA.
(F) Ifnar1−/−mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thiodG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days.
Data were shown as mean ± SEM from two to three independent experiments. P value was determined by two-way ANOVA (A, B and F) or two-tailed unpaired t test (C-E).
To directly demonstrate that 6-thio-dG treatment enhances cross-priming capacity of DCs, we co-cultured 6-thio-dG pretreated MC38-OVA tumor cells with bone marrow derived DCs (BMDCs) overnight. Then the DCs were purified and co-cultured with naïve OT-1 transgenic CD8+ T cells that express the TCR with the specificity to recognize the OVA257–264 epitope. We observed a significant increase of IFN-γ production by CD8+ T cells in the 6-thio-dG treatment group (Figure 3D), which indicates an increased cross-priming capacity of DCs after 6-thio-dG treatment. Because IFN-I signaling promotes the cross-priming capacity of DCs (Diamond et al., 2011; Le Bon et al., 2003; Sanchez-Paulete et al., 2017), we tested the production of IFN-β by DCs after co-culturing them with 6-thio-dG treated tumor cells. Indeed, IFN-β production significantly increased in the 6-thio-dG treatment group, indicating increased innate sensing of DCs (Figure 3E). We further investigated whether the IFN-I pathway is essential for 6-thio-dG-mediated anti-tumor effect. Using Ifnar1−/− mice, we showed that the loss of IFN-I signaling in the host abolished the anti-tumor effect of 6-thio-dG (Figure 3F), indicating the indispensable role of IFN-I signaling in 6-thio-dG treatment.
STING signaling in the host is required for 6-thio-dG induced innate sensing.
Tumor cells under stress might release danger-associated molecular patterns (DAMPs) to engage TLR/Myd88 pathways in APCs and initiate IFN-I signaling. Tumor-derived DNAs can also trigger the cytosolic DNA sensing cGAS/STING pathway and activate IFN-I pathways (Deng et al., 2014; Li et al., 2019). To further delineate which upstream pathway is essential in 6-thio-dG triggered IFN-I signaling activation in host cells, we inoculated MC38 tumors into Myd88−/− and Tmem173−/− (Tmem173 encodes STING) mice. 6-thio-dG treatment controlled tumor growth well in Myd88−/− mice but completely lost efficacy in Tmem173−/− mice (Figures 4A and 4B), suggesting an essential role of host STING signaling in 6-thio-dG triggered innate sensing. We further investigated whether 6-thio-dG treatment activates the host STING / IFN-I pathway. We observed an increase of TBK1 phosphorylation in DCs after co-culture with 6-thio-dG pretreated tumor cells and the phosphorylation was completely diminished in Tmem173 KO DCs (Figure S3A). 6-thio-dG treatment induced IFN-β production in DCs in a STING-dependent manner (Figure S3B). As previous studies reported that tumor-intrinsic STING signaling is critical in innate-sensing inducing cancer therapies (Sen et al., 2019; Vanpouille-Box et al., 2017), we tested whether tumor-intrinsic STING signaling also contributes to 6-thio-dG treatment efficacy. We used CRISPR/Cas9 to knock out Tmem173 and Mb21d1 (Mb21d1 encodes cGAS) in MC38 tumor cells. In contrast to other studies, tumor-intrinsic STING signaling played a non-essential role, as 6-thio-dG treatment still controlled tumor growth in mice bearing Tmem173 KO and Mb21d1 KO tumor cells (Figures 4C and 4D).
Figure 4. STING signaling in host is required for 6-thio-dG induced innate sensing.
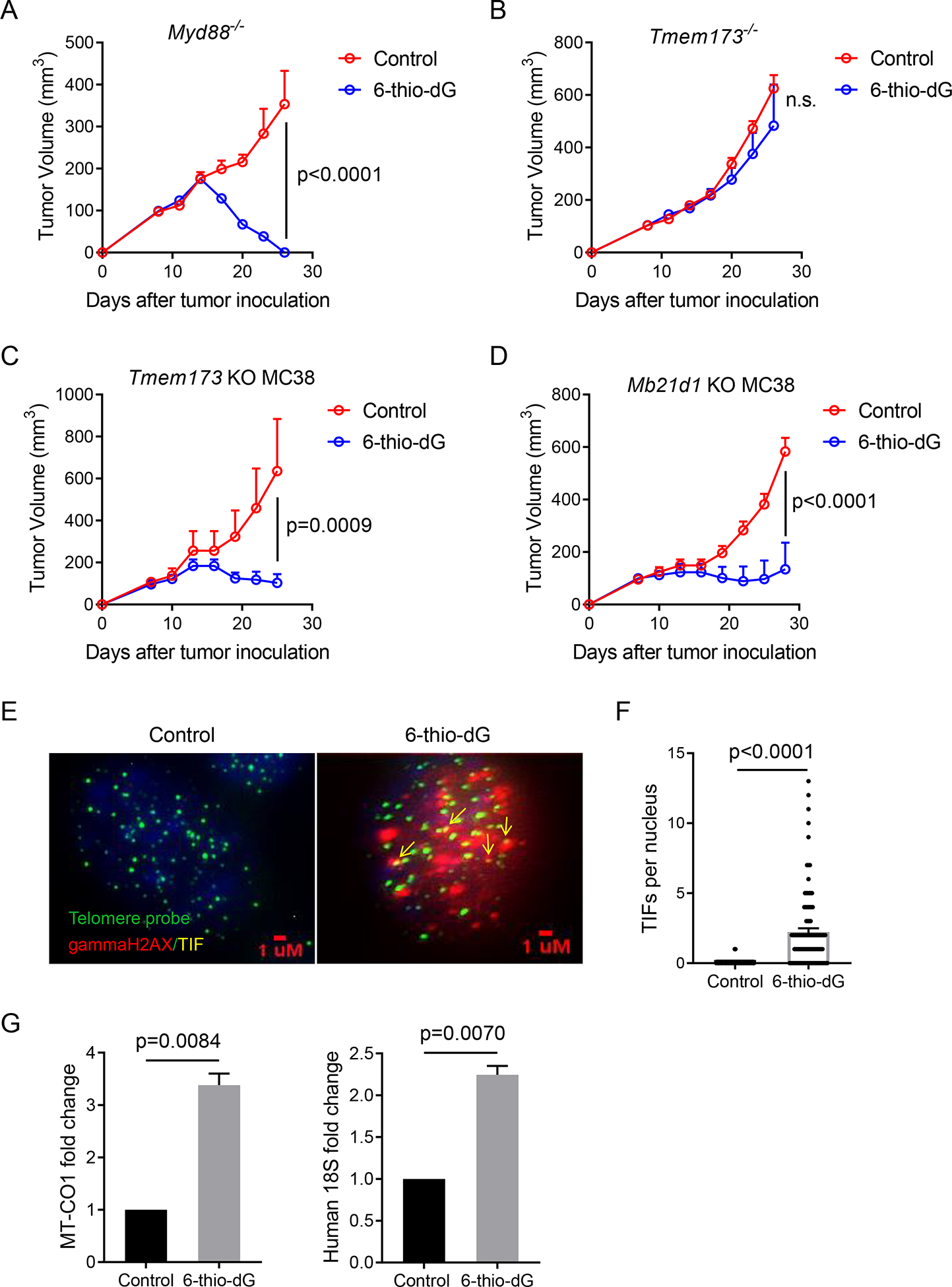
(A and B) Myd88−/− (A) or Tmem173−/− (B) mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days.
(C and D) C57BL/6 mice (n=5) were inoculated with 5×105 Tmem173 KO (C) or Mb21d1 KO (D) MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days.
(E and F) MC38 tumor cells were treated with 1 μM 6-thio-dG for 24 h. TIF (Telomere dysfunction Induced Foci) assay confirms induction of TIFs with 6-thio-dG treatment in MC38 cells. n=100 (control), n=100 (6-thio-dG). Scale bar: 1 μM..
(G) BMDCs were cultured with HCT116 human colon cancer cells that were pretreated with 500 nM 6-thio-dG or vehicle for 4 h, then DCs were purified and cytosolic DNAs were extracted. Relative abundance of MT-CO1 and human 18S in the cytosol of DC were detected by qPCR. Data were shown as mean ± SEM from two to three independent experiments. P value was determined by two-way ANOVA (A-D) or two-tailed unpaired t test (F and G). See also Figure S3.
We then sought to determine how 6-thio-dG treated tumor cells trigger innate sensing in DCs. Since 6-thio-dG is a telomere-targeting drug, 6-thio-dG induced telomere stress might contribute to innate sensing of DCs by releasing DNAs. Therefore, we first analyzed telomere stress by the TIF (Telomere dysfunction Induced Foci) assay and showed that 6-thio-dG induced telomere damages in MC38 cells (Figures 4E and 4F). Since telomeres are only a small fraction of genomic DNA (~1/6000) any co-localization of telomeres with DNA damage is significant. We also observed similar increases of TIFs in 6-thio-dG treated tumor tissues from MC38-tumor bearing mice (Figures S3C and S3D). 6-thio-dG also induced interphase bridges between the two daughter cells during telophase and since many contained telomere sequences, this may explain why many micronuclei containing telomere signals when cells re-entered interphase after mitosis (Figure S3E). These cytosolic fragments formed micronuclei with fragile nuclear envelopes (Figures S3E and S3F), which can be eventually recognized as a danger signal. These DNA fragments are released from the cells and can be taken up by DCs.
To substantiate this hypothesis, we treated HCT116, a human colon cancer cell line, with 6-thio-dG and co-cultured them with mouse BMDC for 4 h, and then we isolated DCs and extracted cytosolic DNA. The short-time co-culture of a human tumor cell line with mouse BMDCs allowed us to distinguish DNAs from different origins. We found an increase of human DNAs (MT-CO1 and human 18S) in the cytosol of mouse DCs after 6-thio-dG treatment, which suggests that DNAs from the tumors enter the host DCs (Figure 4G). To determine if 6-thio-dG treatment increases the uptake of unique telomeric DNAs by DCs, we labeled tumor cells with EdU, then washed the cells. Next, we treated with 6-thio-dG, then washed cells again. Finally, we co-cultured tumor cells with DCs and then isolated the DC for analysis. Among DCs had the uptake of tumor DNAs (EdU+ DC) in the cytosol, we observed an increase of telomere co-localization with EdU after 6-thio-dG treatment, suggesting a significant uptake of tumor derived telomeric DNAs (Figures S3G and S3H). Together, we demonstrated that 6-thio-dG triggers innate sensing through the activation of the host cytosolic DNA sensing STING/IFN-I pathway.
6-thio-dG overcomes PD-L1 blockade resistance in advanced tumors.
While 6-thio-dG treatment activated CD8+ T cells, it also upregulated PD-1 expression in the frequency of total CD8+ T cells and on per cell basis (Figure 5A). PD-1 is a co-inhibitory molecule that limits T cell activation. The elevated PD-1 expression might eventually inhibit the cytotoxic CD8+ T cell function after 6-thio-dG treatment. Therefore, we reasoned that combination of 6-thio-dG with PD-1/PD-L1 blockade might augment the overall anti-tumor immune response, especially in the advanced tumor setting which harbors a more immune suppressive microenvironment containing multiple resistance mechanisms that limit single treatment efficacy. Since 6-thio-dG single treatment was only effective in relatively small tumor sizes ~ 100 mm3, for advanced tumor treatment we let the tumor sizes reach to 150–200 mm3 and then treated with 6-thio-dG and/or anti-PD-L1 treatment. In such advanced cancers, tumor volume is difficult to control with two daily treatments with 6-thio-dG or by two treatments with anti-PD-L1 (Figure 5B). However, sequential administration of 6-thio-dG and anti-PD-L1 completely inhibited the tumor growth (Figure 5B). Remarkably, only mice in the combination treatment group achieved a 100% survival rate (Figure 5C), showing a synergistic effect of 6-thio-dG treatment with PD-L1 blockade. In addition, we did not observe any body weight loss of mice in the combination treatment group (Figure S4A). We further analyzed the tumor-specific T cell response in draining lymph nodes (dLNs) and found that the anti-PD-L1 treatment had little effect on T cell activation in advanced tumors. In contrast, combination therapy significantly increased IFN-γ production compared to other groups. The immune response was MC38 tumor specific as there were almost no IFN-γ spots in the control LLC tumor stimulation group (Figure 5D).
Figure 5. 6-thio-dG overcomes PD-L1 blockade resistance in advanced tumor models.
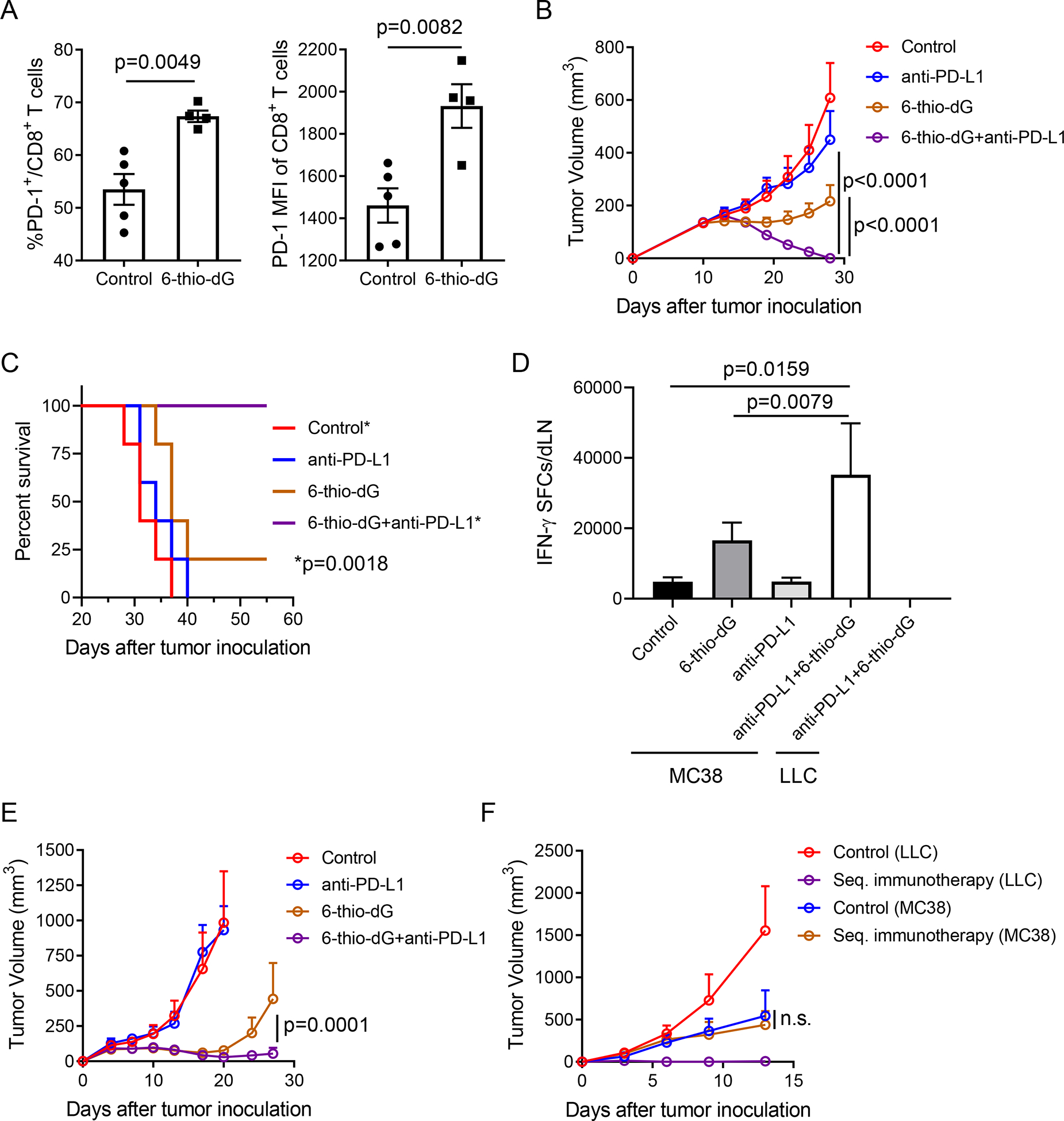
(A) C57BL/6 mice bearing MC38 tumor (n=4–5) were treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 7 days after first treatment, PD-1+CD8+ T cell frequency (left) and PD-1 MFI (right) were tested.
(B and C) C57BL/6 mice (n=5) were inoculated with 5×105 MC38 tumor cells and treated with 6-thio-dG (3 mg/kg, days 10, 11). 50 μg anti-PD-L1 antibody was administrated on days 13 and 17. Tumor growth (B) and survival capacity (C) were shown.
(D) C57BL/6 mice (n=5) bearing MC38 tumor were treated with 6-thio-dG (3 mg/kg, days 10 and 11) or anti-PD-L1 (2.5 mg/kg, day 10) or combination both treatment. 7 days after first treatment, draining lymphoid was harvested and stimulated with irradiated MC38 tumor cells or LLC tumor cells for IFN-γ ELISPOT.
(E and F) C57BL/6 mice (n=5) were inoculated with 1×106 LLC murine lung tumor cells and treated with 6-thio-dG (3 mg/kg, days 4, 5, 6 and 10, 11). 200 μg anti-PD-L1 antibody was administrated on days 8 and 13. Tumor growth was measured every 3–4 days (E). Six weeks later, tumor free mice (n=4) in sequential treatment group and control mice were re-challenged with 5×106 LLC (right flank) and 5×106 MC38 (left flank) tumor cells. Tumor growth was measured every 3–4 days (F).
Data were shown as mean ± SEM from two independent experiments. P value was determined by two-tailed unpaired t test (A, D) or two-way ANOVA (B, E and F) or Log-rank test (C). See also Figure S4.
MC38 is known to be an immunogenic tumor model. To test if the combination therapy can also overcome PD-L1 blockade resistance in less immunogenic tumor models, we employed the mouse LLC tumor model that has been reported to be resistant to PD-L1 blockade (Bullock et al., 2019; Li et al., 2017). Consistent with previous reports, single treatment with anti-PD-L1 had no therapeutic effect (Figure 5E). Notably, combination of 6-thio-dG with anti-PD-L1 significantly reduced mouse tumor burden and 40% of mice eventually completely rejected tumors (Figure 5E). We re-challenged tumor-free mice 6 weeks after tumor regression to check the memory response. All combination treated mice spontaneously rejected LLC tumors but not MC38 tumors, suggesting a long lasting tumor-specific immune memory (Figure 5F). Based on these results, 6-thio-dG treatment overcomes PD-L1 blockade resistance in advanced tumors. This will potentially benefit PD-1/PD-L1 blockade resistant patients in the clinic.
6-thio-dG reduces human colon cancer burden in a humanized mouse model.
Previous studies showed high TERT (the catalytic subunit of the telomerase) expression patients have poor clinical outcomes in various cancers such as non-small cell lung cancer and B cell chronic lymphocytic leukemia (Terrin et al., 2007; Wang et al., 2002). We thus analyzed colorectal adenocarcinoma patients from the TCGA database and found patients with abnormal high expression of TERT had significantly worse overall survival rates compared to the colon cancer patients with low TERT expression (Figure 6A). To directly demonstrate whether 6-thiodG induced telomere stress can benefit cancer patients in a more clinically relevant model, we developed a humanized mouse model with NSG-SGM3 mouse which has human SCF-1, GM-CSF and IL-3 transgenic expression that support the better development of human myeloid cells. We reconstituted the human immune system in NSG-SGM3 mice with human CD34+ hematopoietic stem cells (HSCs). 12 weeks after HSCs transfer, the humanized mice had an average of over 60% human CD45+ cells and over 20% human T cells among human CD45+ cells in circulation (Figures S5A–S5C). Then we inoculated HCT116, a human colon cancer cell line that is sensitive to 6-thio-dG treatment with an IC50 of 0.73 μM (Figure 6B), into NSG-SGM3 control mice and humanized NSG-SGM3 mice. The control group of humanized mice had similar constitution of human immune cells with 6-thio-dG treated group before treatment started (Figures S5B and S5C). After three doses of 6-thio-dG treatment, immunocompromised mice did not have significant difference compared to control group (Figure 6D). Remarkably, the humanized mice significantly delayed tumor growth with 6-thio-dG treatment (Figures 6C and 6E). We then tested a human melanoma cell line A375 that is sensitive to 6-thio-dG in vitro (Figure S5D). We did not observe any effect in immunocompromised NSG-SGM3 mice since we provided a relatively short-time 6-thio-dG treatment (Figure S5E). Notably, we found a treatment with two doses of 6-thio-dG partially delayed tumor growth in humanized mice. In addition, combination with checkpoint blockades further reduced tumor burden, suggesting pretreatment with 6-thio-dG sensitizes human tumors to checkpoint blockades (Figure S5F). Given that humanized mice only partially restore human immunity due to missing some immune cells and limited number of human T cells, it is not surprising that we did not observe complete tumor regression.
Figure 6. 6-thio-dG reduces human colon cancer burden in a humanized mouse model.
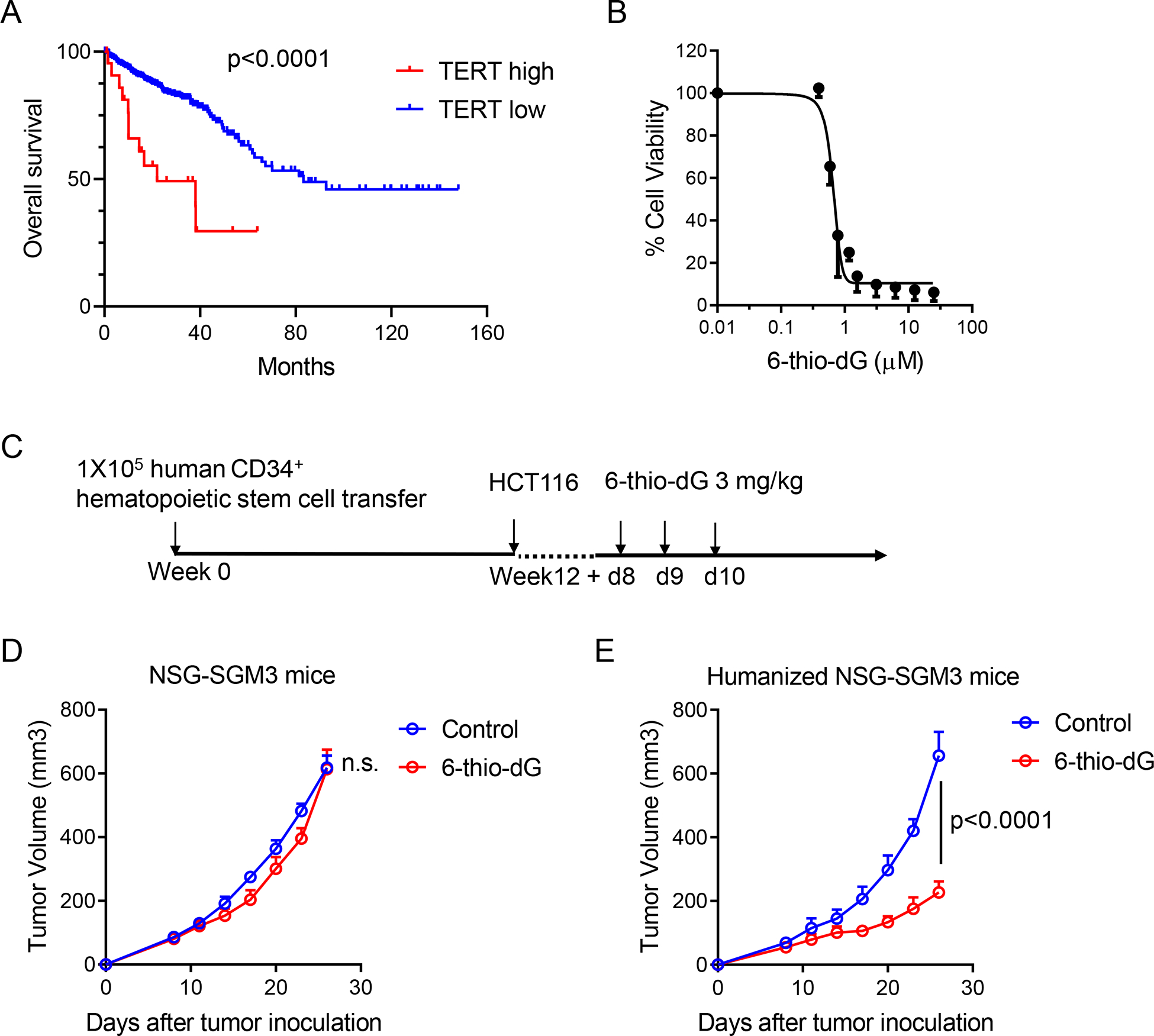
(A) Overall survival in high and low TERT (Telomerase reverse transcriptase, the catalytic subunit of the telomerase) expression colorectal adenocarcinoma patients from TCGA database.
(B) Cell viability (IC50) of 6-thio-dG in HCT116 human colon cancer cells. Cells were treated with 6-thio-dG for 5 days.
(C) The schema for humanized mouse tumor model.
(D and E) NSG-SGM3 mice (n=5) (D) or humanized NSG-SGM3 mice (n=4) (E) were inoculated with 1×106 HCT116 tumor cells and treated with 6-thio-dG (3 mg/kg, days 8, 9, 10). Tumor growth was measured every 3 days.
Data were shown as mean ± SEM from two independent experiments. P value was determined by Log-rank test (A) or two-way ANOVA (D and E). See also Figure S5.
Overall, our data can be interpreted to support that 6-thio-dG induces telomerase-dependent DNA damage and increases tumor DNAs taken up by DCs. The increased cytosolic DNAs trigger the DC-intrinsic STING/IFN-I pathway, resulting in enhanced cross-priming capacity of DCs and subsequent tumor-specific T cell activation. Moreover, 6-thio-dG overcomes PD-L1 blockade resistance in advanced tumors. Our study identifies 6-thio-dG as a unique immune stimulatory drug that will potentially benefit a wide population of cancer patients in the clinic.
Discussion
High telomerase expression in tumor cells is recognized as a poor prognostic factor for cancer development (Zhang et al., 2018). Here, we report a previously undefined role of a telomerase dependent telomere targeting therapy (6-thio-dG) in inducing anti-tumor immune responses in syngeneic colon and lung mouse models and humanized mouse cancer models. This effect is mediated through triggering the cytosolic DNA sensing STING/IFN-I pathway in DCs, which ultimately enhances the cross-priming capacity of DCs and subsequent tumor specific T cell activation. This is a remarkable finding since telomerase is a universal tumor marker and it can potentially be applied to many other telomerase positive cancers. Moreover, sequential administration of 6-thio-dG and anti-PD-L1 overcomes PD-L1 resistance in PD-L1 blockade resistant tumors, suggesting the combination therapy can benefit PD-L1 resistant patients in the clinic.
Current dogma is that 6-thio-dG treatment kill tumor cells mainly by impairing telomeres and inducing DNA damage. Our study demonstrates that this drug also controls tumors largely depending on DNA sensing and T cell responses. Most previous studies use xenograft models without an intact immune system. In these models, they can only study tumor intrinsic effects or part of the innate immune responses. Even though these might be important factors, T cells are essential for long-term tumor control. In addition, most previous studies tend to use high dose or intensive dosing strategies that directly kill tumor cells more efficiently but actually dampen immune responses, either due to the toxicity to immune cells or the non-immunogenic death of tumor cells (Galluzzi et al., 2017; Kroemer et al., 2013). Also, these intensive dosing strategies often lead to the emergence of tumor resistance mechanisms. In the present study, we took advantage of syngeneic mouse models with intact immune systems and humanized mouse model with more clinical relevance to fully evaluate the impact of lower doses and shorter treatment regimens with 6-thio-dG on host immune responses in tumor bearing mice. This discovery that 6-thio-dG is an immune stimulatory drug might allow the design of better combinational treatments including immunotherapy to amplify initial immunity.
Accumulating studies show that tumor DNA mediated innate sensing is critical for the induction of anti-tumor immune responses and the STING/IFN-I pathway is primarily involved in initiation of anti-tumor immune response, but whether host or tumor autonomous STING is more essential depends on different treatment regimens (Deng et al., 2014; Li et al., 2019; Qiao et al., 2017; Sen et al., 2019; Vanpouille-Box et al., 2017; Woo et al., 2014). This discrepancy is likely to be explained by the relative STING activation strength of hosts versus tumor cells, for example, tumor cells might have STING pathway suppression or low activity (Xia et al., 2016). We demonstrated that the 6-thio-dG treatment triggered innate sensing is host STING signaling dependent as 6-thio-dG completely lost its efficacy in Tmem173 deficient mice but not in Tmem173 deficient tumors. Since STING signaling in MC38 tumors is active, one explanation is after 6-thio-dG treatment, tumor intrinsic STING was activated but most tumor cells died, so little type I IFN can be produced. Another possibility is that there might be an intrinsic mechanism that limits STING activation in tumor cells, which still remains poorly defined. Recent reports showed STING signaling can also be involved in autophagy activation, which is less likely to contribute to the therapeutic effect of 6-thio-dG (Gui et al., 2019; Nassour et al., 2019) since we did not see activation of autophagy in tumor cells after 6-thio-dG treatment (data not shown). Also, 6-thio-dG lost efficacy in Ifnar1 deficient mice, suggesting the involvement of IFN I signaling. However, STING activation of autophagy is IFN I signaling independent.
Compared to the general DNA damage induction approaches, for example, radiation therapy or chemotherapy that non-selectively induce DNA damage in all proliferating cells, one unique feature of 6-thio-dG is the specific induction of telomere-associated DNA damage in telomerase expressing cells, primarily tumor cells, but not affecting immune cells and other telomerase-silent somatic cells. Importantly, 6-thio-dG can be preferentially incorporated into de novo-synthesized telomeres and causes rapid tumor shrinkage. However, direct telomerase inhibitors function through the inhibition of telomerase activity and rely on the progressive shortening of telomeres. In contrast, 6-thio-dG takes effect rapidly regardless of the initial telomere length. This is critical in reducing toxicity compared with a direct telomerase inhibitor (Gryaznov et al., 2007; Mender et al., 2015b). We show that 6-thio-dG induced DNA damage is significantly co-localized with telomeres, indicating the formation of telomere dysfunction induced foci (TIF). Telomeres are only ~1/6000th of genomic DNA so any TIF is highly significant. Moreover, some TIFs are taken up by DCs and further trigger STING-dependent IFN I signaling.
Despite the overwhelming success of checkpoint blockade, especially PD-1/PD-L1 blockade, in the clinic, only a minority of patients respond well. Both primary and adaptive resistances limit clinical benefit of PD-1/PD-L1 therapy (Chen and Han, 2015; Gide et al., 2018; Zaretsky et al., 2016; Zou et al., 2016). We believe that lack of proper innate sensing might limit T cell activation inside the tumor microenvironment, therefore combination therapy of targeting both innate and adaptive immune cells is urgently needed. PD-L1 blockade reinvigorates adaptive immune responses by “releasing the brake”, while 6-thio-dG induced innate sensing by “adding fuel”. We hypothesized combination of 6-thio-dG with PD-L1 blockade should augment overall anti-tumor immunity responses. Indeed, our study showed sequential administration of 6-thio-dG and anti-PD-L1 have synergistic effect in advanced tumors and in PD-L1 blockade resistant tumors. Further studies should be carried out regarding optimal combination regimens.
Overall, our results reveal a previously undefined role of 6-thio-dG, a telomerase-dependent telomere targeting small molecule drug, in potentiating anti-tumor immune responses. Mechanistically, 6-thio-dG induces telomere dysfunction and increases cytosolic DNA release. Importantly, these telomeric DNA fragments are taken up by DCs and activate the DC intrinsic STING/IFN-I pathway, resulting in enhanced cross-priming capacity of DCs and subsequent tumor specific T cell activation. Moreover, our study showing the remarkable efficacy of sequential administration of 6-thio-dG and anti-PD-L1 in advanced tumors and PD-L1 blockade resistant tumors provides a strong scientific rationale for propelling combination therapy into clinical trials. We expect that these findings will be translated in the near future and benefit more patients in the clinic.
STAR★Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Anli Zhang (Anli.Zhang@UTSouthwestern.edu).
Materials Availability
Unique reagents generated in this study are available with a MTA.
Data and Code Availability
The TERT gene expression data of colorectal adenocarcinoma was downloaded from TCGA database (https://www.cbioportal.org/) for correlation analysis.
Experimental Model and Subject Details
Mice
Female C57BL/6J, BALB/cJ, Myd88−/−, Tmem173−/−, Batf3−/− and OT-1 CD8+ T cell receptor transgenic mice in the C57BL/6J background and NSG-SMG3 mice were purchased from The Jackson Laboratory. Rag1−/− mice and IFN-γ reporter mice (Ifngtm3.1Lky/J) on C57BL/6 background were purchased from UT southwestern mice breeding core. Ifnαr1−/− mice were provided by Dr. Anita Chong from the University of Chicago. All mice were maintained under specific pathogen-free conditions. Animal care and experiments were carried out under institutional and National Institutes of Health protocol and guidelines. This study has been approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
Cell lines and reagents
MC38, CT26, LLC A375 and HCT116 cells were purchased from ATCC. MC38-OVA cells were made by lentiviral transduction of OVA gene. All cell lines were routinely tested using mycoplasma contamination kit (R&D) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin under 5% CO2 at 37 °C.
Anti-CD4 (GK1.5), anti-NK1.1 (PK136), anti-CD8 (53–5.8) and anti-CSF1R (AFS98) mAbs were purchased from BioXCell. Anti-PD-L1 (atezolizumab) and anti-CTLA-4 (Ipilimumab) were kindly provided by UT Southwestern Simmons Cancer Center Pharmacy. 6-thio-dG was purchased from Metkinen Oy. For in vitro studies, 6-thio-dG was dissolved in DMSO/water (1:1) to prepare 10mM stock solutions. For in vivo studies, 3 mg/kg 6-thio-dG was prepared in 5% DMSO (in 1xPBS) for intraperitoneal injection. Drugs were kept frozen at −20°C until use.
Method Details
Cell viability assay
For determination of IC50 with cell proliferation assays, murine and human cancer cell lines were screened with 6-thio-dG with a 2 fold dilution series in 8 different points in 96-well plates. Cells were plated 24 h prior to the addition of drug, incubated for 4–5 days, and assayed using CellTiter 96® Aqueous One Solution Cell Proliferation Assay according to the manufacturer’s instructions (Promega). Cell number per well ranged from 1,000 to 10,000 cells per well inversely proportional to doubling times. Dose response curves were generated and IC50s calculated using Graphpad Prism. All samples were analyzed in triplicate and standard deviations are from 2–3 independent experiments.
Colony formation assay
MC38 cells were seeded in three different concentrations on six well plates (1000–4000 cells/ well) and treated with various drug concentrations every 3–4 days. Following 13 days treatment, cells were fixed and stained with 6% glutaraldehyde (Fisher Scientific) plus 0.5% crystal violet (Sigma) solution. After washing with tap water, cells were air-dried and images captured using a G-BOX (Syngene, model: G-BOX F3).
Telomere dysfunction Induced Foci (TIF) and micronuclei assays
The TIF assay was conducted as described (Mender and Shay, 2015). Briefly, cells were seeded into 4-well chamber slides. Next day, cells were treated with 1 μM 6-thio-dG for 24 h (for TIF assay) or 1 μM 6-thio-dG for 48 h (for micronuclei assay). Slides were then rinsed twice with PBS and fixed in 4% formaldehyde (Thermo Fisher) in PBS for 10 min. Then, cells were washed twice with PBS and permeabilized in 0.5% PBST for 10 min. Following permeabilization, cells were washed and blocked with 10% goat serum in 0.1% PBST for 1 h. γ-H2AX (TIF assay) (Millipore) or lamin A/C (micronuclei assay) (Santa Cruz) was diluted in blocking solution and incubated on cells for 2 h. Following washes with 0.1% PBST and PBS, cells were incubated with Alexaflour 568 conjugated goat anti-mouse antibody (Invitrogen) for 40 min, then washed five times with 0.1% PBST. Cells were fixed in 4% formaldehyde in PBS for 20 min at RT. The slides were sequentially dehydrated with 70%, 90%, 100% ethanol followed by denaturation with hybridization buffer containing FAM-conjugated telomere sequence-specific peptide nucleic acid (PNA) probe, 70 % formamide, 30% 2 × SSC, 10% (w/v) MgCl2.6H2O (Fisher Sci), 0.25% (w/v) blocking reagent for nucleic acid hybridization and detection (Roche) for 7 min at 80 °C on a heat block, followed by overnight incubation at RT. Slides were washed sequentially with 70% formamide (Ambion) / 0.6 × SSC (Invitrogen), 2 × SSC, PBS and sequentially dehydrated with 70%, 90%, 100% ethanol, then mounted with Vectashield mounting medium with DAPI (Vector Laboratories). TIF images were captured with a fluorescence microscope using the 100X magnification and quantified using Image J. Micronuclei images were captured at 63X magnification with an Axio Imager Z2 equipped with an automatic metaphase capture system (Coolcube1 camera) and analyzed with ISIS software (Metasystems).
Detection of DNA in Bone Marrow Derived Dendritic Cells
Cells were labeled with EdU as described previously (Min et al., 2019). Briefly, 1× 105 MC38 cells were seeded to 6-well plate and labeled with 25 μM EdU. Two days later, cells were washed out and treated with 1 μM 6-thio-dG for 24 h. Cells were washed out again and co-cultured with BMDCs for overnight. Next day, DCs were sorted out with magnetic beads, washed, fixed and cytospun. Slides were then stained with 6-carboxytetramethlyrhodamine fluorescent azide (Invitrogen) in fresh homemade EdU-staining solution (PBS containing 1mM CuSO4, 2mM ascorbic acid) for 30min. Slides were then washed vigorously with PBS for at least 1 h, and then telomere FISH steps using a FAM-TelG probe were followed as described in “Telomere dysfunction Induced Foci (TIF) and micronuclei assays” method parts. Images were captured at 63X magnification with an Axio Imager Z2 equipped with an automatic metaphase capture system (Coolcube1 camera) and analyzed with ISIS software (Metasystems).
ImmunoFISH
Deparaffinized tissue sections (5 μM) were incubated in sodium citrate buffer (10 mM Na-citrate, 0.05 % Tween 20, pH=6.0) at microwave for 20 min to retrieve antigens. Then tissue sections were rinsed with PBS and dehydrated in 95% ethanol. Denaturation was conducted with hybridization buffer (as in TIF assay) containing FITC-conjugated telomere sequence (TTAGGG)3-specific PNA probe for 7 min at 80˚C on a heat block. Slides were washed sequentially with 70% formamide / 0.6 × SSC, 2 × SSC, PBS, 0.1% PBST and incubated with blocking buffer (4 % BSA in PBST) for 30 min. Sections were incubated with phospho-histone H2AX antibody (Cell Signaling) in blocking buffer at RT for 1 h and washed with PBST and incubated with Alexaflour 568 conjugated goat anti-Rabbit antibody in blocking buffer at RT for 1 h. Sections were washed sequentially with 0.1% PBST and PBS. The slides were mounted with Vectashield mounting medium with DAPI. Images were captured with a fluorescence microscope using a 100X objective. TIFs were quantified using Image J.
Tumor growth and treatment
A total of 5×105 MC38, 5×105 CT26 or 1×106 LLC cells were inoculated subcutaneously into right dorsal flanks of the mice in 100 μL phosphate buffered saline (PBS). Tumor-bearing mice were randomly grouped into treatment groups when tumors grew to around 100 mm3. For 6-thio-dG single treatment, 3 mg/kg 6-thio-dG was intraperitoneally given on days 7, 8 and 9 in MC38 tumor and LLC tumor and on days 5, 6, 7 for CT26 tumor. For CSF1R, NK1.1, CD4+ and CD8+ T cell depletion, 200 μg of antibodies were intraperitoneally injected 1 day before treatment initiation and then twice a week for 2 weeks. For PD-L1 blockade combination therapy in MC38 model, 6-thio-dG was given on days 10 and 11, 50 μg PD-L1 was intraperitoneally injected on days 13 and 17. For PD-L1 blockade combination therapy in LLC model, 6-thio-dG was given on days 4, 5, 6, 10 and 11, 200 μg PD-L1 was intraperitoneally injected on days 8 and 13. Tumor volumes were measured by the length (a), width (b) and height (h) and calculated as tumor volume = abh/2.
Humanized mouse tumor models
Humanized mouse reconstitution was previously described (Qiao et al., 2019). Briefly, four-week-old NSG-SGM3 female mice were irradiated with 100 cGy (X-ray irradiation with X-RAD 320 irradiator) one day prior to human CD34+ cells transfer. Cord blood was obtained from UT Southwestern Parkland Hospital. Human CD34+ cells were purified from cord blood by density gradient centrifugation (Ficoll® Paque Plus, GE healthcare) followed by positive immunomagnetic selection with anti-human CD34 microbeads (Stemcell). 1 × 105 CD34+ cells were intravenously injected into each recipient mouse. 12 weeks after engraftment, humanized mice with over 50 % human CD45+ cells reconstitution and age and sex matched non-humanized mice were inoculated with 1 × 106 HCT116 tumor cells subcutaneously on the right flank. 3 mg/kg 6-thio-dG was intraperitoneally given on days 7, 8 and 9. Tumor volumes were measured twice a week. Experiments were performed in compliance with UTSW Human Investigation Committee protocol and UTSW Institutional Animal Care and Use Committee.
Tmem173 and Mb21d1 KO MC38 cell line
Tmem173 and Mb21d1 genes in MC38 cells were knocked out by CRISPR/Cas9 technology. The guide sequence 5’-CACCTAGCCTCGCACGAACT-3’for Tmem173 and 5’-CGCAAAGGGGGGCTCGATCG-3’for Mb21d1 were cloned into px458 plasmids(non-integrating plasmid with GFP selecting marker), and then were transiently transfected into tumor cells using lipofectamine 2000 (Thermo Fisher). 24 h later, GFP positive cells were sorted and cultured for another one week. Then sorted cells were seeded into 96-well plates. Another week later, GFP negative clones were passed into 12 well plates, and western blot was performed to identify the KO clones. Finally, all KO clones were pooled together for experiments.
IFN-γ enzyme-linked immunosorbent spot assay (ELISPOT)
MC38 tumors were injected subcutaneously on the right flank of C57BL/6. For 6-thio-dG single treatment, 3 mg/kg 6-thio-dG was intraperitoneally given on days 7, 8 and 9; for PD-L1 blockade combination therapy in MC38 model, 3 mg/kg 6-thio-dG was given on days 10 and 11, 50 μg PD-L1 was intraperitoneally injected on day 11. 7 days after last treatment, tumor draining lymphoid and spleen from tumor-bearing mice were collected and single-cell suspension was prepared. Irradiated MC38 tumor cells and control LLC tumor cells were used to re-stimulate the tumor-specific T cells. 1.5 × 105 draining lymph nodes cells or splenocytes and 7.5 × 104 irradiated tumor cells were co-cultured for 48 h, and ELISPOT assay was performed using the IFN-γ ELISPOT kit (BD Bioscience) according to the manufacturer’s instructions. IFN-γ spots were enumerated with the CTL-ImmunoSpot® S6 Analyzer (Cellular Technology Limited).
In vitro co-culture of bone marrow dendritic cells (BMDC) and T cells
Single-cell suspensions of bone marrow (BM) cells were collected from tibias and femurs of C57BL/6 mice. The BM cells were placed in 10cm dish and cultured with complete RPMI 1640 medium containing 20 ng/mL recombinant mouse GM-CSF (BioLegend). Fresh medium with was added into the culture on days 3 and 6. The BMDCs were harvested Day 7. CD8+ T cells were isolated from lymph nodes and spleens of OT-1 transgenic mice with a negative CD8+ T cell isolation kit (Stemcell). MC38-OVA cells pretreated with 200 nM 6-thio-dG for 4 h. Then the drug was washed out, tumor cells were continued to culture for 72 h and were harvested on the same day as BMDC harvest. Then MC38-OVA cells were co-cultured with BMDC for overnight. Supernatant was collected for IFN-β ELISA test (PBL). BMDCs were sorted with CD11c positive selection kit (Stemcell) and co-cultured with OT-1 CD8+ T cells for 48 h. Supernatants were collected and IFN-γ was measured by cytometric bead array assay (BD Biosciences).
Cytosolic DNA extraction and quantitative real-time PCR
HCT116 cells were pretreated with 500 nM 6-thio-dG for 4 h. Then drug was washed out, tumor cells were continued to culture for 72 h and harvest on the same day as BMDC harvest. Then HCT116 cells were mixed 1:1 with 1 × 106 BMDC for 4 h. BMDC was purified and divided into two equal aliquots. One aliquot was extracted for total genomic DNA with Purelink Genomic DNA kit (Invitrogen) and served as normalization control. The other aliquot was resuspended in 100 μL cytosolic extract buffer containing 150 mM NaCl, 50 mM HEPES and 25 mg/mL digitonin (Sigma) and incubated for 10 min at RT for plasma membrane permeabilization(West et al., 2015). Then cells were centrifuged to pellet intact cells. The cytosolic supernatants were collected and centrifuged at 12000g for 10 min to pellet the remaining cellular debris. Then cytosolic DNA was extracted with Purelink Genomic DNA kit (Invitrogen). Quantitative PCR was performed on both whole-cell extracts and cytosolic fractions using human DNA primers and mouse DNA primers (Xu et al., 2017).
Tumor digestion
Tumor tissues were excised and digested with 1 mg/mL Collagenase I (Sigma) and 0.5 mg/mL DNase I (Roche) in the 37°C for 30 min, tumor was then passed through a 70 μm cell strainer to remove large pieces of undigested tumor. Tumor infiltrating cells were washed twice with PBS containing 2 mM EDTA.
Flow cytometry analysis
Single cell suspensions of cells were incubated with anti-FcγIII/II receptor (clone 2.4G2) for 15 minutes to block the non-specific binding before staining with the conjugated antibodies, and then incubated with indicated antibody for 30 min at 4 °C in the dark. Fixable viability Dye eFlour 506 or eFlour780 (eBioscience) was used to exclude the dead cells. Foxp3 and Ki67 were stained intracellularly by using True-Nuclear transcription factor buffer set (BioLegend) following the manufacturer’s instructions. Data were collected on CytoFLEX flow cytometer (Beckman Coulter, Inc) and analyzed by using FlowJo (Tree Star Inc., Ashland, OR) software.
Quantitative real-time PCR
Real-time PCR was performed with SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) according to the manufacturer’s instructions with different primer sets (human MT-CO1, forward primer 5’- CGCCACACTCCACGGAAGCA-3’, reverse primer 5’- CGGGGCATTCCGGATAGGCC-3’; human18s rRNA, forward primer 5’- ACCGATTGGATGGTTTAGTGAG-3’, reverse primer 5’- CCTACGGAAACCTTGTTACGAC-3’; mouse IFN-β, forward primer 5’-ATGAGTGGTGGTTGCAGGC-3’, reverse primer 5’-TGACCTTTCAAATGCAGTAGATTCA-3’; mouse GAPDH, forward primer 5’-CATCAAGAAGGTGGTGAAGC-3’, reverse primer 5’- CCTGTTGCTGTAGCCGTATT-3’) mouse GAPDH was used as the internal control. 2−ΔΔCt method was used to calculate relative expression changes.
Immunoblotting
BMDC and MC38 treatment were same as “In vitro co-culture of bone marrow dendritic cells”. 6 h after co-culture, DC was isolated with CD11c positive selection kit (Stemcell). Protein sample preparation and immunoblot procedures were performed as previously described (Liu et al., 2019). Proteins were detected with rabbit monoclonal antibodies for pSTING (Cell signaling, 72971), STING (Cell signaling, 50494), pTBK1 (Cell signaling, 5483), TBK1 (Cell signaling, 3504). Protein loading was determined with antibodies against with Cyclophilin A (Cell signaling, 2175). Anti-rabbit was used for secondary antibody (Cell signaling, 7074). X-ray film (GeneMate, F-9024–8X10) was used to develop the membranes. Clarity Max Western ECL Substrate (Biorad, 1705062) or Supersignal West PicoPlus Chemiluminescent Substrate (Thermoscientific, 34577) was used for chemiluminescent western blot.
Quantification and Statistical Analysis
All the data analyses were performed with GraphPad Prism statistical software and shown as mean ± SEM. P value determined by two-way ANOVA for tumor growth or Log-rank test for survival or unpaired two-tailed t-tests for other analysis. A value of p < 0.05 was considered statistically significant.
Supplementary Material
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse CD4 (GK1.5) | BioXcell | Cat# BE0003–1 |
| InVivoMAb anti-mouse CD8 (53–5.8) | BioXcell | Cat# BE0223 |
| InVivoMAb anti-mouse NK1.1 (PK136) | BioXcell | Cat# BE0036 |
| InVivoMAb anti-mouse CSF1R (AFS98) | BioXcell | Cat# BE0213 |
| anti-PD-L1 (atezolizumab) | Genentech | |
| anti-human CTLA-4 (Ipilimumab) | Bristol-Myers Squibb | |
| Anti-mCD45 (Flow cytometry, 30-F11) | BioLegend | Cat# 103126 |
| Anti-mCD3 (Flow cytometry, 145–2C11) | BD Biosciences | Cat#564379 |
| Anti-mCD4(Flow cytometry, RM4–5) | BioLegend | Cat# 100526 |
| Anti-mCD8 (Flow cytometry, 53–6.7) | BioLegend | Cat# 100730 |
| Anti-mCD8a (Flow cytometry, KT15) | Invitrogen | Cat# MA5–16759 |
| Anti-mNK1.1 (Flow cytometry, PK136) | BD Biosciences | Cat#552878 |
| Anti-mFoxp3(Flow cytometry, MF-14) | Biolegend | Cat#126408 |
| Anti-mKi67(Flow cytometry, 16A8) | Biolegend | Cat#652413 |
| Anti-mPD-1(Flow cytometry, 29F.1A12) | Biolegend | Cat#135224 |
| Anti-hCD45(Flow cytometry, HI30) | Biolegend | Cat#304021 |
| Anti-hCD3(Flow cytometry, HIT3a) | Biolegend | Cat#300327 |
| Anti-hCD8(Flow cytometry, HIT8a) | Biolegend | Cat#300905 |
| Anti-hCD4(Flow cytometry, A161A1) | Biolegend | Cat#357417 |
| Fixable Viability Dye eFluor™ 506 | Thermo Fisher | Cat# 65-0866-18 |
| iTAg Tetramer/PE - H-2 Kb OVA (SIINFEKL) | MBL | Cat# TB-5001–1 |
| Anti-FcyIII/II receptor (clone 2.4G2) | BD Biosciences | Cat# 553141 |
| Phospho-Histone H2AX (Ser139) Rabbit mAb | Cell Signaling | Cat# 9718 |
| STING (D2P2F) Rabbit mAb | Cell Signaling | Cat# 50494 |
| Phospho-STING (Ser365) (D8F4W) Rabbit mAb | Cell Signaling | Cat# 72971 |
| TBK1/NAK Antibody | Cell Signaling | Cat# 3504 |
| Phospho-TBK1/NAK (Ser172) (D52C2) XP® Rabbit mAb | Cell Signaling | Cat# 5483 |
| Phospho-STING (Ser366) (D7C3S) Rabbit mAb | Cell Signaling | Cat# 19781 |
| Cyclophilin A Antibody | Cell Signaling | Cat# 2175 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat# 7074 |
| Lamin A/C Ab (E-1) | SantraCruz | Cat# 376248 |
| γ-H2AX | Millipore | Cat# 05–636 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TMB Solution (1X) | eBioscience | Cat# 00-4201-56 |
| Sulfadiazine/ Trimethoprim (Aurora Pharmaceutical LLC) | UTSW- Veterinary Drug Services | Cat# 302 |
| GE Healthcare Ficoll-Paque™ PLUS Media | Fisher | Cat# 45-001-750 |
| Collagenase type I | Sigma | Cat# C0130 |
| DNAse I | Roche | Cat# 11284932001 |
| Recombinant mouse GM-CSF | BioLegend | Cat# 576306 |
| 6-carboxytetramethlyrhodamine tetramethlyrhodamine fluorescent azide | Invitrogen | Cat# T10182 |
| Critical Commercial Assays | ||
| BD™ Cytometric Bead Array (CBA) Mouse Inflammation Kit | BD Biosciences | Cat# 552364 |
| BD Mouse IFN-γ ELISPOT Sets | BD Biosciences | Cat# 551083 |
| Mouse IFN Beta ELISA Kit | Cat# 42410 | |
| SsoAdvanced Uni SYBR Grn Supmix | Bio-Rad | Cat# 1725272 |
| True-Nuclear™ Transcription Factor Buffer Set | BioLegend | Cat# 424401 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | STEMCELL | Cat# 19853 |
| EasySep™ Human CD34 Positive Selection Kit II | STEMCELL | Cat# 17856 |
| EasySep™ Mouse CD11 c Positive Selection Kit II | STEMCELL | Cat# 18780 |
| PureLink™ Genomic DNA Mini Kit | Thermo Fisher | Cat# K182002 |
| Reasy Mini Kit | Qiagen | Cat# 74104 |
| Cell Titer-Glo Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Blue Lite Autorad Film | GeneMate | Cat# 9024 |
| Clarity Max Western ECL Substrate | Bio-Rad | Cat# 1705062 |
| Supersignal West PicoPlus Chemiluminescent Substrate | Thermo Fisher | Cat# 34577 |
| Deposited data | ||
| TERT gene expression | TCGA | https://www.cbioportal.org/ |
| Experimental Models: Cell Lines | ||
| MC38 | ATCC | |
| LLC | ATCC | |
| CT26 | ATCC | |
| HCT116 | ATCC | |
| A375 | ATCC | |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | Cat# 000664 |
| BALB/cJ | Jackson Laboratory | Cat# 000651 |
| Ragi KO mice | UTSW breeding Core | |
| NSG-SGM3 mice | Jackson Laboratory | Cat# 013062 |
| Batf3−/− mice | Jackson Laboratory | Cat# 013755 |
| OT-1 mice | Jackson Laboratory | Cat# 003831 |
| Tmeml73−/− mice | Jackson Laboratory | Cat# 25805 |
| Myd88−/− mice | Jackson Laboratory | Cat# 009088 |
| Ifngtm3.1Lky/J mice | UTSW breeding Core | |
| Ifnar1−/− mice | Dr.Anita Chong, U Chicago | |
| Oligonucleotides | ||
| human Co1, forward primer 5’- CGCCACACTCCACGGAAGCA-3 ‘ | Sigma | |
| human Co1, reverse primer 5’- CGGGGCATTCCGGATAGGCC-3 ‘ | Sigma | |
| human18s rRNA, forward primer 5’- ACCGATT GGAT GGTTT AGT GAG-3 ‘ | Sigma | |
| human18s rRNA, reverse primer 5’- CCTACGGAAACCTTGTTACGAC-3 ‘ | Sigma | |
| mouse IFN-β, forward primer 5’- ATGAGTGGTGGTTGCAGGC-3 ‘ | Sigma | |
| mouse IFN-β, reverse primer 5’- T GACCTTT CAAAT GCAGTAGATT C A-3’ | Sigma | |
| mouse GAPDH, forward primer 5’- CATCAAGAAGGTGGTGAAGC-3 ‘ | Sigma | |
| mouse GAPDH, reverse primer 5’- CCTGTTGCTGTAGCCGTATT-3 ‘ | Sigma | |
| Software and Algorithms | ||
| GraphPad Prism software 7.0 | GraphPad Software, Inc. | https://graphpad.com/scientific-software/prism/ |
| CTL-ImmunoSpot® S6 Analyzer | Cellular Technology Limited | http://www.immunospot.com/ImmunoSpot-analyzers |
| CytExpert | Beckman Coulter, Inc | https://www.beckman.com/coulter-flow-cytometers/cytoflex/cytexpert |
| BD FACSAria™ III System | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-sorters/facsaria-iii |
| FlowJo | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
Highlights.
6-thio-dG induces telomere-associated DNA damage in telomerase positive tumor cells
6-thio-dG reduces tumor burden in T cell dependent manner in syngeneic tumor models
Host STING signaling is required for 6-thio-dG mediated anti-tumor effects
6-thio-dG overcomes PD-L1 blockade resistance in advanced tumors
Significance.
Telomerase is almost universally expressed in tumor cells but not in normal cells. The nucleoside analog 6-thio-dG is directly incorporated into telomeres by telomerase, where it rapidly induces telomere damage in telomerase-positive cancer cells but not in normal telomerase silent cells. Unexpectedly, 6-thio-dG induced DNA mediated innate sensing and activation of immune responses in a host STING-dependent manner, leading to improved anti-tumor efficacy. Moreover, 6-thio-dG sequentially followed by anti-PD-L1 therapy can completely eliminate advanced tumors in mouse models. Thus, 6-thio-dG is a unique tumor-targeting and immune-stimulating drug that can benefit telomerase-positive and PD-L1 resistant cancer patients in the clinic.
Acknowledgments
This work was in part supported by Texas CPRIT grant RR150072 and RP180725 to Y.-X.F. NCI SPORE P50CA70907, NIH grant C06 RR30414 to J.W.S. Y.-X.F. holds the Mary Nell and Ralph B. Rogers Professorship in Immunology. J.W.S. holds the distinguished Southland Financial Corporation Distinguished Chair in Geriatrics Research. We acknowledge Jaewon Min and Peter Ly for technical help and the Cancer Biology Training Grant, NCI T32CA124334 (IM), Cancer Intervention and Prevention Discovery Training Program Fellowship, RP160157 (SS). We also thank Zhida Liu, Longchao Liu, Xuezhi Cao, Changzheng Lu and Jian Qiao for providing experiment materials and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Jerry Shay is a founding scientist of Maia Biotechnology who is progressing 6-thio-dG into clinical development. In addition, Jerry Shay, Anli Zhang, Ilgen Mender and Yang-Xin Fu are named co-inventors on pending patents related to this work.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, and Hornung V (2013). cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH (1991). Structure and function of telomeres. Nature 350, 569–573. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 938. [DOI] [PubMed] [Google Scholar]
- Bullock BL, Kimball AK, Poczobutt JM, Neuwelt AJ, Li HY, Johnson AM, Kwak JW, Kleczko EK, Kaspar RE, Wagner EK, et al. (2019). Tumor-intrinsic response to IFNgamma shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci Alliance 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Han X (2015). Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 125, 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208, 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, and Vance RE (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. (2010). Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 207, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, and Thomas P (2011). Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125–132. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp O, Zitvogel L, and Kroemer G (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97–111. [DOI] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013). Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Gide TN, Wilmott JS, Scolyer RA, and Long GV (2018). Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin Cancer Res 24, 1260–1270. [DOI] [PubMed] [Google Scholar]
- Greider CW (1996). Telomere length regulation. Annual review of biochemistry 65, 337–365. [DOI] [PubMed] [Google Scholar]
- Greider CW, and Blackburn EH (1985). Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405–413. [DOI] [PubMed] [Google Scholar]
- Gryaznov SM, Jackson S, Dikmen G, Harley C, Herbert BS, Wright WE, and Shay JW (2007). Oligonucleotide conjugate GRN163L targeting human telomerase as potential anticancer and antimetastatic agent. Nucleosides Nucleotides Nucleic Acids 26, 1577–1579. [DOI] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, and Chen ZJ (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Kepp O, and Zitvogel L (2013). Immunogenic Cell Death in Cancer Therapy. Annual Review of Immunology 31, 51–72. [DOI] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap AK, and Tanke HJ (1996). Heterogeneity in telomere length of human chromosomes. Human molecular genetics 5, 685–691. [DOI] [PubMed] [Google Scholar]
- Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, and Tough DF (2003). Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 4, 1009–1015. [DOI] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC, Clambey ET, and Nemenoff RA (2017). The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res 5, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. The Journal of experimental medicine 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu Z, Zhang A, Han C, Shen A, Jiang L, Boothman DA, Qiao J, Wang Y, Huang X, and Fu YX (2019). NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat Commun 10, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Liu Z, Han C, Dong C, Shen A, Hsu E, Ren Z, Lu C, Liu L, Zhang A, Timmerman C, et al. (2019). Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Sci Immunol 4. [DOI] [PubMed] [Google Scholar]
- Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, and Blackburn EH (1996). Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes & development 10, 1822–1834. [DOI] [PubMed] [Google Scholar]
- Mender I, Gryaznov S, Dikmen ZG, Wright WE, and Shay JW (2015a). Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2’-deoxyguanosine. Cancer Discov 5, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, Gryaznov S, and Shay JW (2015b). A novel telomerase substrate precursor rapidly induces telomere dysfunction in telomerase positive cancer cells but not telomerase silent normal cells. Oncoscience 2, 693–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, LaRanger R, Luitel K, Peyton M, Girard L, Lai TP, Batten K, Cornelius C, Dalvi MP, Ramirez M, et al. (2018). Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance. Neoplasia 20, 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, and Shay JW (2015). Telomere Dysfunction Induced Foci (TIF) Analysis. Bio-protocol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Wright WE, and Shay JW (2019). Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev 33, 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin GB (1989). The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, and Cech TR (1997). Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959. [DOI] [PubMed] [Google Scholar]
- Nassour J, Radford R, Correia A, Fuste JM, Schoell B, Jauch A, Shaw RJ, and Karlseder J (2019). Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JM, Kroemer G, and Zitvogel L (2017). Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv Exp Med Biol 1036, 65–79. [DOI] [PubMed] [Google Scholar]
- Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, Fu K, Peng J, Wang Y, Ren Z, et al. (2019). Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+) T Cell Apoptosis. Cancer Cell 35, 901–915 e904. [DOI] [PubMed] [Google Scholar]
- Qiao J, Tang H, and Fu YX (2017). DNA sensing and immune responses in cancer therapy. Curr Opin Immunol 45, 16–20. [DOI] [PubMed] [Google Scholar]
- Reinhardt RL, Liang HE, and Locksley RM (2009). Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, et al. (2016). Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 315, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015a). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, et al. (2015b). Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol 16, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, Sancho D, and Melero I (2017). Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol 28, xii44–xii55. [DOI] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. (2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Sobo M, Lee K, Senthil Kumar S, White AR, Mender I, Fuller C, Chow LML, Fouladi M, Shay JW, and Drissi R (2018). Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Molecular cancer therapeutics 17, 1504–1514. [DOI] [PubMed] [Google Scholar]
- Shay JW, and Bacchetti S (1997). A survey of telomerase activity in human cancer. European journal of cancer 33, 787–791. [DOI] [PubMed] [Google Scholar]
- Singer MS, and Gottschling DE (1994). TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science 266, 404–409. [DOI] [PubMed] [Google Scholar]
- Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodriguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. (2018). Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378, 2288–2301. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Science signaling 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrin L, Trentin L, Degan M, Corradini I, Bertorelle R, Carli P, Maschio N, Bo MD, Noventa F, Gattei V, et al. (2007). Telomerase expression in B-cell chronic lymphocytic leukemia predicts survival and delineates subgroups of patients with the same igVH mutation status and different outcome. Leukemia 21, 965–972. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Soria JC, Kemp BL, Liu DD, Mao L, and Khuri FR (2002). hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin Cancer Res 8, 2883–2889. [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, and Barber GN (2016). Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li XD, Deng L, Chen ZJ, et al. (2017). Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363–373 e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GL, Bradley JD, Attardi LD, and Blackburn EH (1990). In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature 344, 126–132. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Wu LW, Mender I, Barzily-Rokni M, Hammond MR, Ope O, Cheng C, Vasilopoulos T, Randell S, Sadek N, et al. (2018). Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma. Clin Cancer Res 24, 4771–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Molecular cell 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK, and Lansdorp PM (1997). Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proceedings of the National Academy of Sciences of the United States of America 94, 7423–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TERT gene expression data of colorectal adenocarcinoma was downloaded from TCGA database (https://www.cbioportal.org/) for correlation analysis.