

Abstract
Cyclic dinucleotides (CDNs) trigger the innate immune response in eukaryotic cells through the STING (stimulator of interferon genes) signalling pathway. To decipher this complex cellular process, a better correlation between structure and downstream function is required. Here we report the design and immunostimulatory effect of a novel group of c-di-GMP analogues. By employing an “atomic mutagenesis” strategy, changing one atom at a time, a class of gradually modified CDNs was prepared. These c-di-GMP analogues induce type-I interferon (IFN) production, with some being more potent than c-di-GMP, their native archetype. To the best of our knowledge, this study demonstrates for the first time that CDN analogues bearing modified nucleobases can tune the innate immune response in eukaryotic cells.
Keywords: cyclic dinucleotide, innate immune response, modified nucleosides, STING agonist
Graphical Abstract
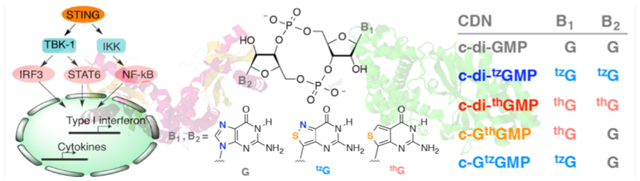
Cyclic dinucleotides (CDNs) play critical regulatory roles in bacteria and trigger the innate immune response in eukaryotic cells. Here we illustrate that a systematic modification of the nucleobases, rather than the phosphate or sugar moieties, can generate STING agonists that demonstrate strong immunostimulatory effects.
The innate immune system of eukaryotes possesses diverse mechanisms for detection of invading pathogens.[1] One of the most fundamental processes relies on cell surface or intracellular receptors that recognize molecular patterns unique to microorganisms.[2] Such pattern recognition receptors (PRRs) are capable of distinguishing pathogen-associated molecular patterns (PAMPs) from the molecular repertoire of endogenous host “self” patterns.[3] PRR engagement of PAMPs can then trigger the innate immune response to enhance antimicrobial activity, and further modulate the adaptive immune response.[2–3, 4] Cyclic dinucleotides (CDNs), which play critical roles in the cGAS-STING and RECON innate immune signaling pathways, have been recognized as PAMPs in recent years.[5] STING was the first mammalian receptor identified that directly binds CDNs,[6] most notably 2’,3’-cGAMP, the product of the cytosolic DNA sensor cGAS.[7] A conformational change upon ligand binding recruits and activates the kinase TBK1 (Figure 1),[8] and phosphorylation of STING by TBK1 facilitates recruitment of transcription factor IRF3. When IRF3 itself gets phosphorylated by TBK1, it forms an activated homo-dimer that induces expression of type-I interferon (IFN α/β) and other cytokines within the nucleus (Figure 1).[9] In addition to the TBK1–IRF3 pathway, STING can activate other signaling pathways, including NF-κB and STAT6.[9–10]
Figure 1.
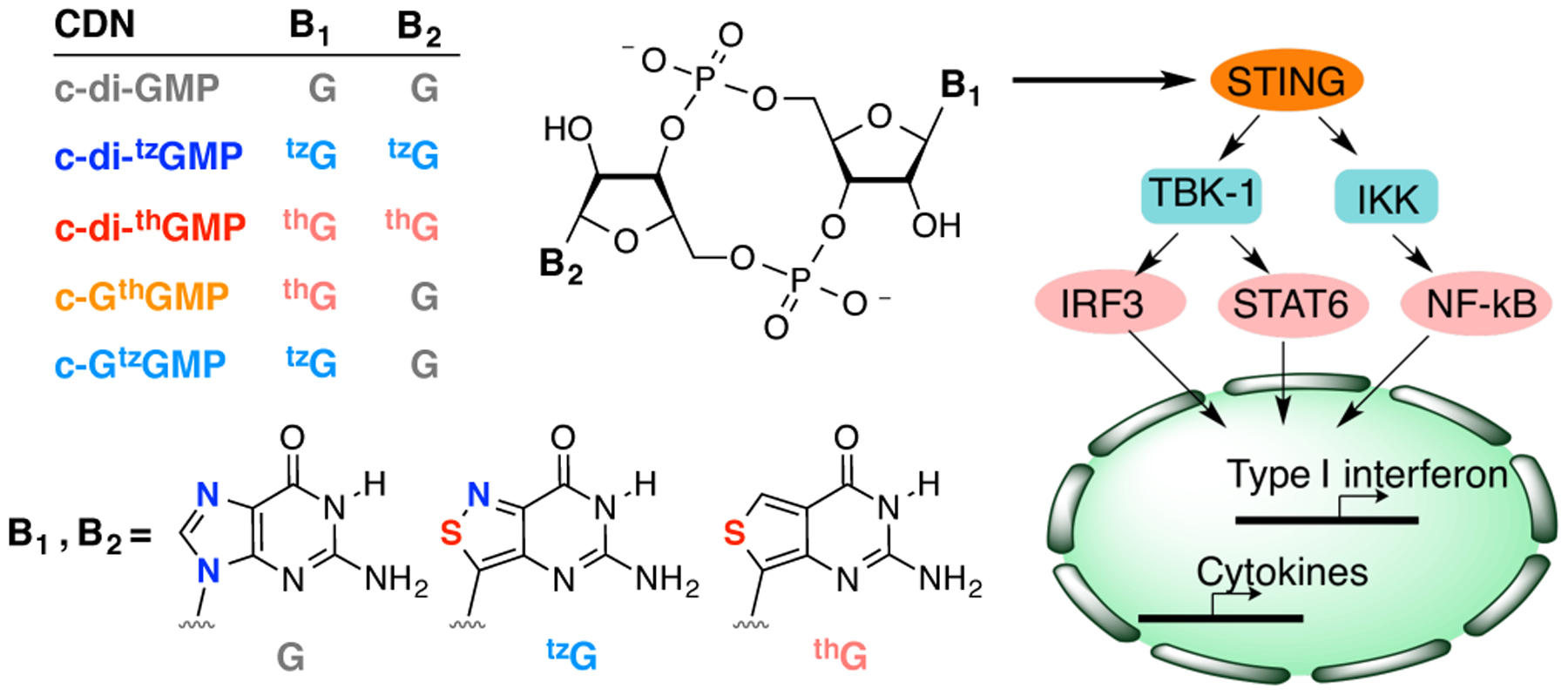
CDN analogues and their immunostimulatory effects.
Activation of STING by CDN analogues has shown pharmacological promise for improving the efficacy of cancer immunotherapies, including PD1 and CTLA-4 targeted drugs and CAR-T cell therapy.[11] Consequently, medicinal chemistry efforts have sought to develop hydrolysis-resistant CDNs with longer cellular residency time by altering the ribose and/or the phosphate moieties.[12] Chemical and chemo-enzymatic approaches have been taken for the preparation of CDN analogues bearing either backbone or nucleobase modification.[13] Analyses of the biological activities of such CDN analogues have provided valuable information on their binding properties to downstream sensors and augmented our knowledge regarding structure–activity relationships.[13–14]
In a recent publication, two fluorescent guanosine analogues developed in our laboratory have been used to prepare novel emissive CDNs, where atomic mutagenesis replaces the nucleobase’s imidazole ring with a thiophene or an isothiazole moiety (thG or tzG, respectively, Figure 1).[15] While this contribution primarily focused on the photophysical properties of the emissive CDNs,[15c] these compounds could also provide insight into CDNs and their biological recognition, as the G analogues differ by one atom, and along with native guanosine, thus present a gradually altered purine molecular architecture. To deepen our molecular level understanding of CDN signaling, we analyzed here the immunostimulatory effects of these systematically modified CDNs. We demonstrate that certain analogues can induce type-I IFN production more potently than their native archetype, highlighting potential new approaches to studying and manipulating the eukaryotic innate immune response.
The dimeric and mixed CDN analogues shown in Figure 1 were made from GTP, thGTP and tzGTP using DncV, a promiscuous dinucleotide cyclase from Vibrio cholerae (see Experimental Section and Figure S1 for data).[13b] The substrates and enzyme were incubated at 37 °C for 2–5 h, after which the reaction mixture was heat-inactivated and filtered before subjecting it to reverse-phase HPLC separation and purification. Pure fractions were collected, combined and lyophilized.[15c] The CDNs were re-dissolved in water for downstream experiments. To preliminarily determine whether the synthetic c-di-GMP analogues could activate the IFN response in eukaryotic cells, THP-1 cells were treated with 5 μM of c-di-GMP, c-GthGMP and c-di-thGMP. After 4 h incubation, induction of type-I IFN was measured with HEK-Blue IFN α/β reporter cells (see SI for experimental details). c-GthGMP induced type-I IFN production with comparable efficiency to c-di-GMP, while c-di-thGMP showed no activity (Figure 2).
Figure 2.
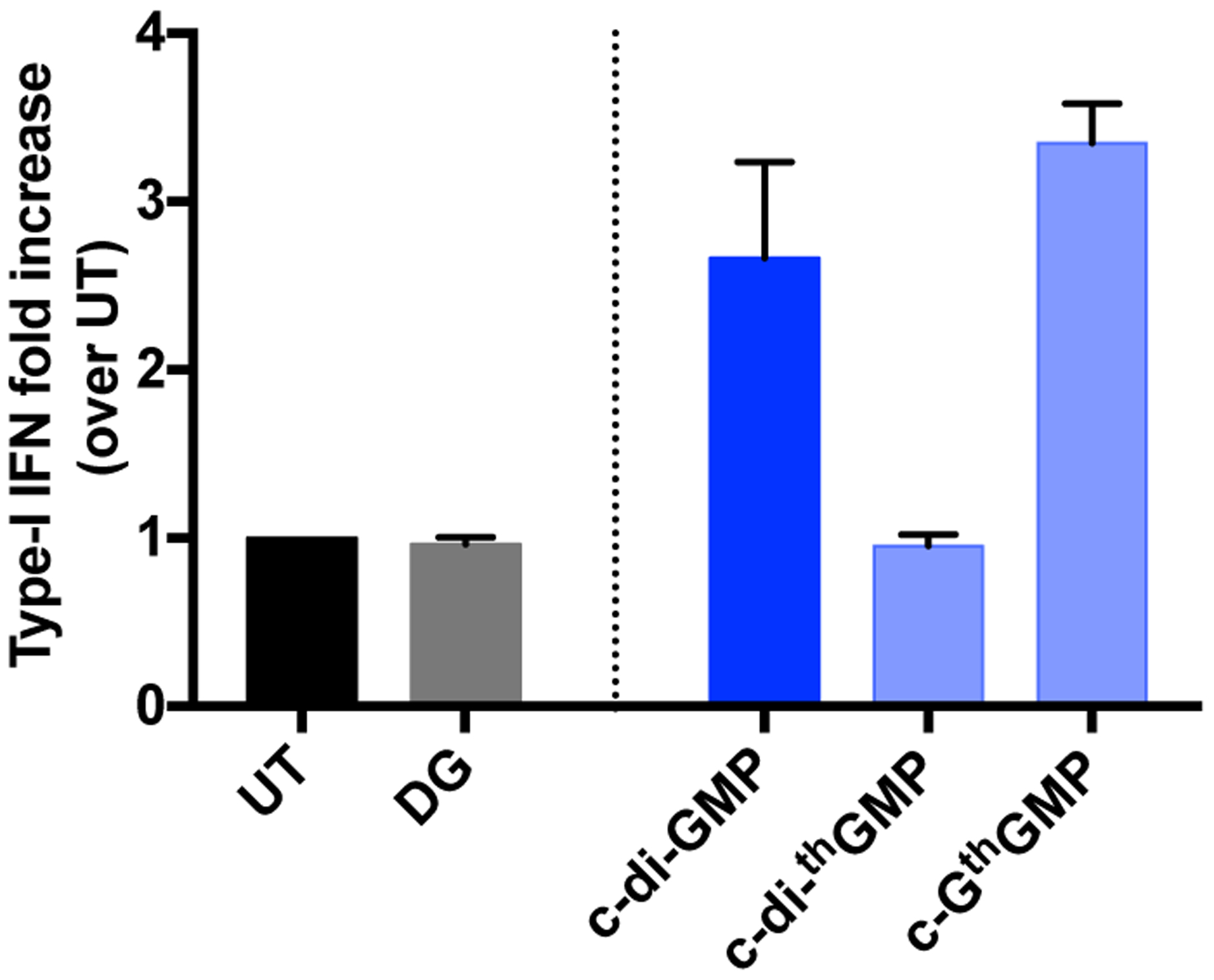
Type-I IFN induced by CDNs in THP-1 cells. THP-1 cells were seeded at a density of 100,000 cells/well in a 96-well cell culture plate and differentiated with 25 nM of PMA for approximately 20 h prior to treatment with CDNs. Cells were transfected with 5 μM of CDNs in a permeabilization buffer containing 5 μg/mL of digitonin, then washed and incubated in RPMI medium with 2% FBS at 37°C for 4 h. 50 μL of cell culture supernatant per well was transferred to 150 μL of HEK-Blue IFN α/β reporter cells seeded at 50,000 cells/well in a 96-well cell culture plate and incubated at 37 overnight. The reporter cells were spin down the next day, and 50 μL of cell culture supernatant per well was transferred to a 96-well plate and added with 150 μL of QUANTI-Blue™ SEAP detection medium (InvivoGen). The samples were then incubated at 37°C for 1 h 20 min before absorption was measured at 640 nm. The absorption signal of each sample was normalized to untreated samples. Two independent assays were done in duplicates or triplicates. Error bars indicate SD.
To analyze the immunostimulatory effects of all synthetic CDNs in greater detail, RAW 264.7 cells were treated with various concentrations of c-di-GMP, c-di-tzGMP, c-di-thGMP, c-GthGMP and c-GtzGMP and the phosphorylation of IRF3 to pIRF3 was evaluated. CDNs were thus transfected into RAW 264.7 murine cells with digitonin as described in previous studies.[7b, 16] Cells were then lysed with NP-40 buffer 2 h after transfection, and total protein collected for immunoblotting against phosphorylated IRF3 (pIRF3) and β-actin. No pIRF3 was observed for untreated cells (UT) nor digitonin permeabilized cells (DG) (Figure 3). Low concentrations (1 μM) of c-di-GMP did not induce obvious IRF3 activation, while 5 and 10 μM displayed comparable efficiency in inducing IRF3 phosphorylation. Increasing amounts of phosphorylated IRF3 were observed when cells were treated with higher concentrations of c-di-tzGMP and c-GtzGMP, while no clear dose-response was observed for c-GthGMP (Figure 3a–b). The least isomorphic analogue, c-di-thGMP, did not trigger observable IRF3 activation at any of the concentrations tested. Two other biological replicates produced similar trends (Figure S2).
Figure 3.
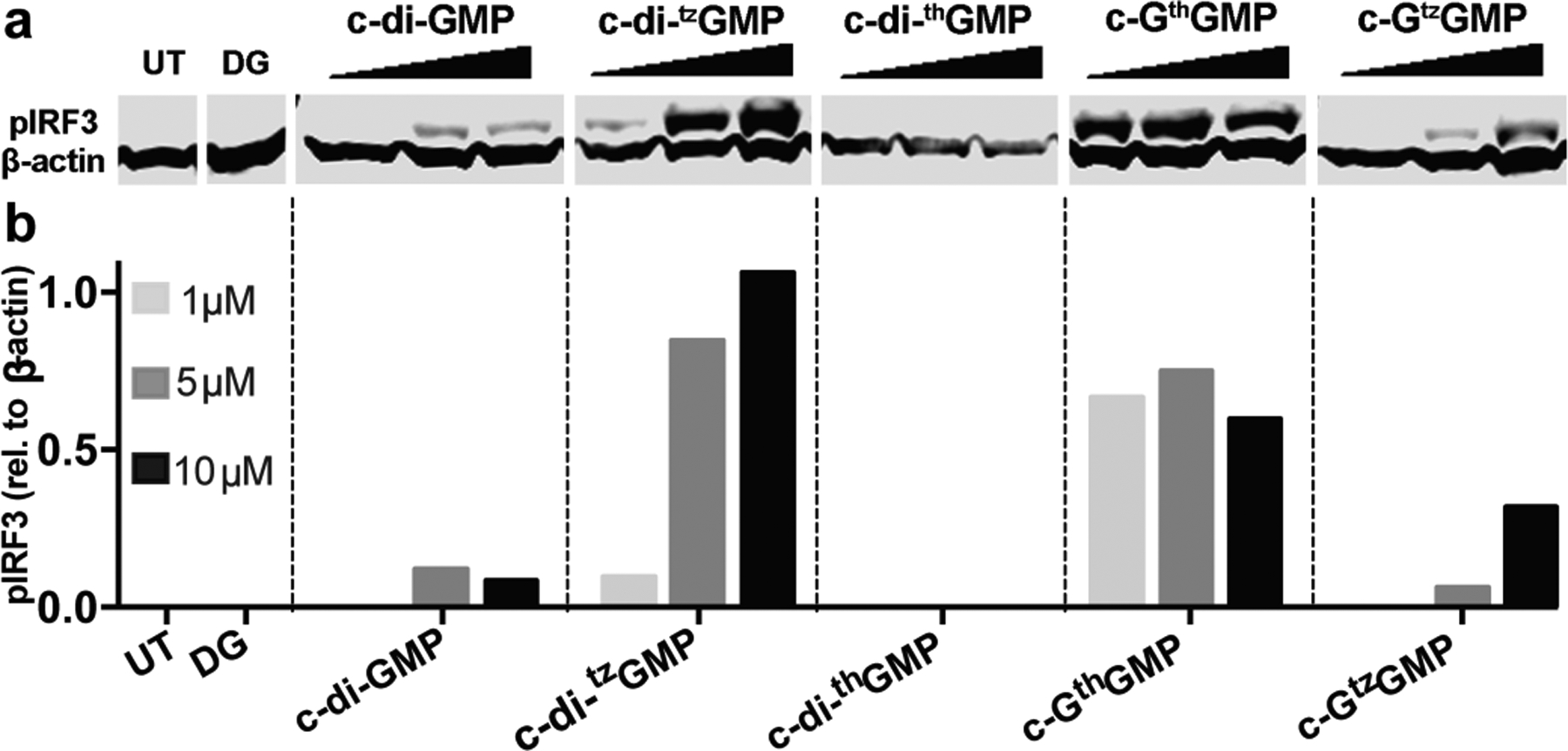
IRF3 phosphorylation induced by c-di-GMP and its analogues. (a) IRF3 phosphorylation induced by c-di-GMP analogues. 1, 5 and 10 μM of each CDN was used to transfect RAW 264.7 cells. Cells were lysed with NP-40 lysis buffer 2 h post transfection, 20 μg of total protein was loaded on SDS-polyacrylamide gel. Proteins were transferred to PVDF membrane after gel electrophoresis, and immunoblotted against pIRF3 and β-actin. (b) quantification of western blot. Y-axis indicates relative intensity of pIRF3 compare to β-actin.
Since most synthetic c-di-GMP analogues activated IRF3, we analyzed their dose- and time-dependency for inducing IFN-β mRNA production using RT-qPCR. RAW 264.7 cells were transfected with 1, 5 and 10 μM of CDNs as described above and incubated for 2, 4 and 6 h. Total RNA was isolated and used for RT-qPCR (see Experimental section in the SI). As shown in Figure 4a and 4b, c-di-GMP induced the most IFN-β mRNA production 4 h post transfection, while the highest response was observed after 2 h for c-di-tzGMP, c-GthGMP, and c-GtzGMP. The same trend was observed for all three concentrations of CDNs tested (Figure 4a, Figure S3a–b). The IFN response to c-di-thGMP was minimal, but c-GthGMP showed the highest potency in inducing IFN-β mRNA production (Figure 4a–c, Figure S3a–d) among all CDNs tested. After 2 h incubation, 5 μM of c-GthGMP induced 10-fold higher IFN-β mRNA production than c-di-GMP, the native messenger. The differences in activity displayed by the analogues and their dependency on the specific assay used are discussed below.
Figure 4.
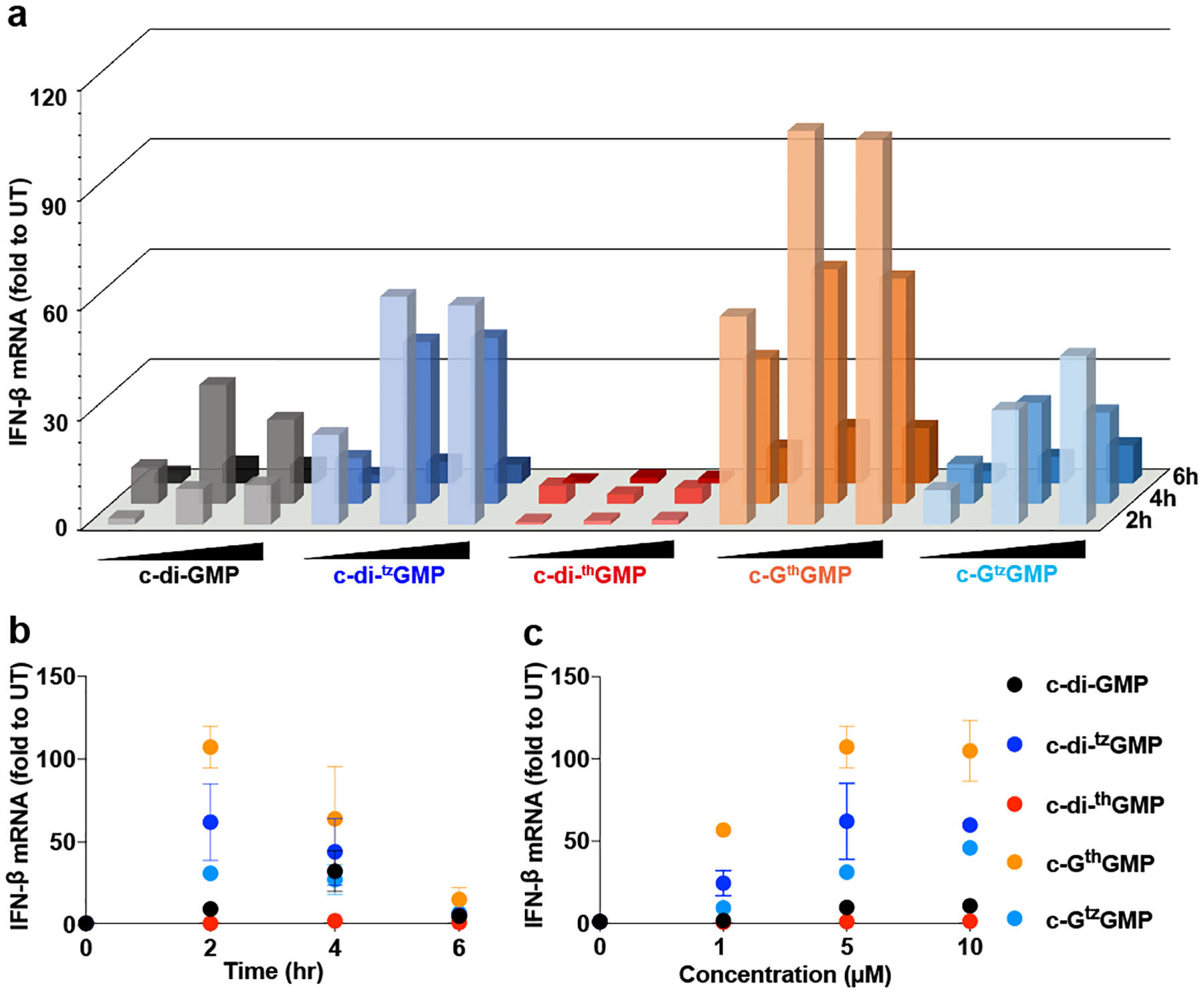
IFN-β mRNA induction triggered by c-di-GMP and its analogues. (a) IFN production induced by c-di-GMP and its analogues. RAW 264.7 cells were transfected with 1, 5, 10 μM of c-di-GMP, c-di-tzGMP, c-GthGMP, c-di-GthGMP and c-GtzGMP respectively, and incubated for 2, 4, 6 h respectively before lysed by TRIzol. RNA purification and RT-qPCR were conducted following the protocol described in the Methods section. (b) IFN response after 2, 4, 6 h incubation with 5 μM of CDNs. (c) IFN response to 1 μM, 5 μM and 10 μM of CDNs after 2 h incubation. Two independent assays were performed in triplicates (n=2). Error bars indicate SD.
Apparent STING activation by c-di-GMP analogues that contain unnatural isomorphic nucleobase was assessed here by three methods: type I IFN production measured by a reporter cell line, IRF3 phosphorylation measured by western blotting, and IFN-β mRNA production measured by RT-qPCR. The initial analysis was performed in THP-1, a human cell line, while more detailed analyses were performed in RAW 264.7, a murine cell line. Our results show that all analogues except for c-di-thGMP stimulated the STING pathway in RAW 264.7 cells. The other three analogues appear to stimulate IRF phosphorylation at comparable or higher levels than the parent c-di-GMP 2 h post transfection.
To quantitatively analyze activation of the STING pathway, CDN-induced, IFN-β production was measured by RT-qPCR in time- and dose-dependent manners. As seen in Figure 4, IFN-β induction drops in the order: c-GthGMP > c-di-tzGMP > c-GtzGMP > c-di-GMP > c-di-thGMP, although it is apparent the cellular processes show complex concentration/time dependency. The effect of CDN concentrations above 5 μM plateaued except for c-GtzGMP. Importantly, however, peak IFN-β responses occurred at different times for different analogues, with the synthetic analogues c-di-tzGMP, c-GtzGMP and c-GthGMP inducing earlier and stronger maximum IFN-β response compared to the native c-di-GMP (Figure 4, Figure S3 a–b). This pattern might result from negative feedback mechanisms of the CDNs-activated STING pathway and type-I IFN signalling.[17] We speculate that rapid and potent IFN induction might concomitantly activate early negative feedback responses, which ultimately result in down tuning IFN-β production.
Among the synthetic CDNs tested in RAW 264.7 cells, the two analogues containing thG, the least isomorphic G surrogate that lacks the basic N7 in the native purine scaffold, displayed dramatically different potency in activating the STING pathway, with c-di-thGMP appearing essentially inactive, while c-GthGMP exerting the strongest effect on IFN-β induction of all analogues tested. This stark difference was also observed in the THP-1 human cell line, although c-GthGMP showed comparable potency to c-di-GMP (Figure 2). Our findings could reflect differences in the assay themselves. RT-qPCR detects IFN- β mRNA levels and not necessarily the translated active protein levels, whereas the reporter assay detects secreted type I interferons, including both IFN α and β. Additionally, the difference between cell lines could be rationalized by the existence of multiple STING alleles in human cells compared to murine cells, which possess different sensitivity to CDNs.[7c, 18]
The observed intensity and duration of the cellular signalling response reflect both the affinity of the ligand to STING, as well as its resistance to intracellular degradation processes (assuming negligible differences in transfection efficiencies). It is perhaps not surprising that c-di-thGMP does not serve as a potent STING agonist, since it is the least isomorphic CDN analogue, with two altered purine cores. However, retaining one native G residue, as in c-GthGMP, restores STING activation. While speculative, this result is consistent with observations made for other asymmetric CDNs, 3’,3’-cGAMP and 2’,3’-cGAMP, that also induce more potent STING activation than c-di-GMP.[8a, 12a, 19] Either through enhanced binding affinity to STING or potentially increased resistance to hydrolytic degradation, c-GthGMP induces a faster and greater innate immune response relative to the native signal c-di-GMP in murine cells. It would be worthwhile to further investigate the binding affinity of these CDN analogues to different STING variants, to further build correlations with their biological activity. In this context, the intrinsic fluorescence of our modified CDNs could potentially provide an effective tool to facilitate such studies.
Modifying the phosphate and sugar moieties of CDNs has been explored as a strategy to alter the pharmacological potency of STING agonists. Most of the non-cognate base-modified CDNs have not been tested in immune response assays.[14f] Here we illustrate that a systematic modification of the nucleobases, rather than the phosphate or sugar moieties, can generate STING agonists that are more potent than c-di-GMP. Particularly intriguing is the high potency of the mixed analogues c-GthGMP and c-GtzGMP, where only one of the native guanosine residues is replaced by an unnatural synthetic C-nucleoside. Recognizing the complexity and intricacies of such cellular pathways, these observations put forth new approaches for the implementation of novel CDN analogues with altered recognition features, where the potency and duration of the triggered cellular immune response can potentially be tuned.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health for generous support (through grant GM 069773 to YT and grant GM 124589 to MCH) and the Chemistry & Biochemistry MS Facility. We also thank Professor Jeffery Esko for sharing his laboratory with us, Dr. Daniel Sandoval (Esko lab, UCSD) for technical assistance with cell culture and immunoblot, and Professor Phillip Gordts (UCSD) for discussion and suggestions regarding the immunogenicity assays.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Medzhitov R, Janeway CA Jr., Cell 1997, 91, 295–298. [DOI] [PubMed] [Google Scholar]
- [2].Akira S, Immunol. Rev 2009, 227, 5–8. [DOI] [PubMed] [Google Scholar]
- [3].a) Gordon S, Cell 2002, 111, 927–930; [DOI] [PubMed] [Google Scholar]; b) Takeuchi O, Akira S, Cell 2010, 140, 805–820. [DOI] [PubMed] [Google Scholar]
- [4].Iwasaki A, Medzhitov R, Science 2010, 327, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Motwani M, Pesiridis S, Fitzgerald KA, Nature Reviews Genetics 2019, 20, 657–674; [DOI] [PubMed] [Google Scholar]; b) McFarland AP, Luo S, Ahmed-Qadri F, Zuck M, Thayer EF, Goo YA, Hybiske K, Tong L, Woodward JJ, Immunity 2017, 46, 433–445; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ablasser A, Chen ZJ, Science 2019, 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- [6].Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, Nature 2011, 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Sun L, Wu J, Du F, Chen X, Chen ZJ, Science 2013, 339, 786–791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ, Science 2013, 339, 826–830; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE, Cell Rep. 2013, 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, Hartmann G, Barchet W, Tuschl T, Patel DJ, Cell 2013, 154, 748– 762; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Q, Sun L, Chen ZJ, Nat. Immunol 2016, 17, 1142–1149; [DOI] [PubMed] [Google Scholar]; c) Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, Chen ZJ, Science 2015, 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- [9].Margolis SR, C. Wilson S, Vance RE, Trends Immunol. 2017, 38, 733–743. [DOI] [PubMed] [Google Scholar]
- [10].a) Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, Xia W, Gu J, Ishikawa H, Gutman D, Barber G, Qin Z, Jiang Z, Cell 2011, 147, 436–446; [DOI] [PubMed] [Google Scholar]; b) Abe T, Barber GN, J. Virol 2014, 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW Jr., Kim Y, Sci. Transl. Med 2015, 7, 283ra252; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li A, Yi M, Qin S, Song Y, Chu Q, Wu K, J. Hematol. Oncol 2019, 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW Jr., Gajewski TF, Cell Rep. 2015, 11, 1018–1030; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, Mitchison TJ, Nat. Chem. Biol 2014, 10, 1043– 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Novotná B, Vaneková L, Zavřel M, Buděšínský M, Dejmek M, Smola M, Gutten O, Tehrani ZA, Polidarová M. Pimková, Brázdová A, Liboska R, Štěpánek I, Vavřina Z, Jandušík T, Nencka R, Rulíšek L, Bouřa E, Brynda J, Páv O, Birkuš G, J. Med. Chem 2019, 62, 10676–10690; [DOI] [PubMed] [Google Scholar]; b) Launer-Felty KD, Strobel SA, Nucleic Acids Res. 2018, 46, 2765–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Wang C, Sinn M, Stifel J, Heiler AC, Sommershof A, Hartig JS, J. Am. Chem. Soc 2017, 139, 16154–16160; [DOI] [PubMed] [Google Scholar]; b) Shanahan CA, Strobel SA, Org. Biomol. Chem 2012, 10, 9113– 9129; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Luo Y, Zhou J, Watt SK, Lee VT, Dayie TK, Sintim HO, Mol. Biosyst 2012, 8, 772–728; [DOI] [PubMed] [Google Scholar]; d) Zhou J, Zheng Y, Roembke BT, Robinson Sarah M., Opoku-Temeng C, Sayre DA, Sintim HO, RSC Adv . 2017, 7, 5421–5426; [Google Scholar]; e) Roembke BT, Zhou J, Zheng Y, Sayre D, Lizardo A, Bernard L, Sintim HO, Mol. Biosyst 2014, 10, 1568–1575; [DOI] [PubMed] [Google Scholar]; f) Clivio P, Coantic-Castex S, Guillaume D, Chem. Rev 2013, 113, 7354–7401. [DOI] [PubMed] [Google Scholar]
- [15].a) Shin D, Sinkeldam RW, Tor Y, J. Am. Chem. Soc 2011, 133, 14912–14915; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rovira AR, Fin A, Tor Y, J. Am. Chem. Soc 2015, 137, 14602–14605; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li Y, Ludford PT, Fin A, Rovira AR, Tor Y, Chem. Eur. J 2020, Accepted Author Manuscript, doi: 10.1002/chem.202001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Girardin SE, Boneca IG, Carneiro LAM, Antignac A, Jéhanno M, Viala J, Tedin K, Taha M, Labigne A, Zäthringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ, Science 2003, 300, 1584–1587. [DOI] [PubMed] [Google Scholar]
- [17].a) Konno H, Konno K, Barber Glen N., Cell 2013, 155, 688–698; [DOI] [PMC free article] [PubMed] [Google Scholar]; Porritt RA, Hertzog PJ, Trends Immunol. 2015, 36, 150–160. [DOI] [PubMed] [Google Scholar]
- [18].a) Gao P, Zillinger T, Wang W, Ascano M, Dai P, Hartmann G, Tuschl T, Deng L, Barchet W, Patel Dinshaw J., Cell Rep. 2014, 8, 1668–1676; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yi G, Brendel VP, Shu C, Li P, Palanathan S, Kao C. Cheng, PLoS One 2013, 8, e77846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ, Mol. Cell 2013, 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.