

Abstract
The APOBEC family of cytidine deaminases has been proposed to represent a major enzymatic source of mutations in cancer. Here, we summarize available evidence that links APOBEC deaminases to cancer mutagenesis. We also highlight newly identified human cell models of APOBEC mutagenesis, including cancer cell lines with suspected endogenous APOBEC activity and a cell system of telomere crisis-associated mutations. Finally, we draw on recent data to propose potential causes of APOBEC misregulation in cancer, including the instigating factors, the relevant mutator(s), and the mechanisms underlying generation of the genome-dispersed and clustered APOBEC-induced mutations.
Keywords: APOBEC mutations, Cancer mutagenesis, Mutational signature, Kataegis, Chromothripsis, Cancer cell lines
1. Introduction
Over the past decade the apolipoprotein B mRNA-editing enzyme catalytic polypeptidelike (APOBEC) family of single-stranded DNA (ssDNA) cytidine deaminases has emerged as a prominent putative enzymatic source of mutations in cancer. APOBEC1 was the first member identified for its role in plasma metabolism by apoprotein B pre-mRNA editing [1,2]. It was later recognized that APOBEC1 can also deaminate cytidine in DNA [3,4]. APOBEC2 and APOBEC4 have no confirmed DNA deamination activities [7,8]. The APOBEC3 subfamily, comprised of seven APOBEC members, restricts viral replication through base-editing of viral intermediates during reverse transcription [9]. Importantly, a selection of APOBEC3 enzymes is currently believed to underlie most of the genome-dispersed and clustered APOBEC-associated mutations. Notably, another APOBEC family member, Activation-induced cytidine deaminase (AID) with a role in initiating the hypermutation and recombination of immunoglobulin genes, has been tied to chromosomal translocations and the development of specifically B cell malignancies, reviewed previously [10].
APOBEC-associated mutations represent traces of some of the most common mutational processes in cancer, detected in approximately 75% of cancer types and >50% of all cancers analyzed [11]. Independent of base editing, APOBEC3-mediated cytosine deamination can also elicit a DNA damage response, if DNA double-strand breaks form upon excision of the resulting uracil bases by uracil DNA glycosylase (UNG) [12-14]. This APOBEC3-dependent DNA damage may place unique pressures on the cell and offer opportunities for therapeutic intervention via induction of synthetic lethality [15-18].
The clinical impact of APOBEC-associated mutations appears to be significant [19,20]. APOBEC-associated mutations likely influence tumor evolution through activation of oncogenes and loss of tumor suppressors [21]. Expression levels of some APOBECs also influence the response to therapies: high APOBEC3 expression levels are associated with reduced clinical benefit from tamoxifen in estrogen receptor (ER)-positive breast cancer [22]. These correlative results are corroborated by mouse xenograft models where induced APOBEC3 overexpression abrogates the benefits of tamoxifen [23]. Consistent with activation late in disease progression, APOBEC activity is also associated with tumor subclonal diversification and heterogeneity [22,24]. Finally, overexpression of APOBEC3B can also improve sensitivity to immune checkpoint blockade in a mouse model of melanoma [25]. It is thus possible that APOBEC enzymes may play a role in generating neoepitopes and promote tumor immunogenicity, thus calling for future studies in this direction.
2. Links between APOBEC enzymes and mutations in cancer
Accumulating evidence from yeast and human cell models, transgenic animals, and human cancer genomics point towards a role for APOBEC deaminases in cancer mutagenesis. The first data connecting APOBEC enzymes to cancer was the discovery that expression of APOBEC1 promotes hepatocellular carcinoma in transgenic animals [26]. The subsequent demonstration that overexpression of APOBECs can induce DNA damage and mutagenesis nominated APOBECs as putative DNA mutators in cancer [3,12], in addition to their known roles as RNA editors in viral restriction. The identification of mutations at motifs characteristic of APOBEC activity in viral restriction and cell models (cytosine bases in 5’-TCN-3’ (T = thymine, C = cytosine, N = any base) in cancer genes [27] further suggested that APOBECs are also active in cancer.
2.1. Mutational signature analysis implicates APOBECs in cancer mutagenesis
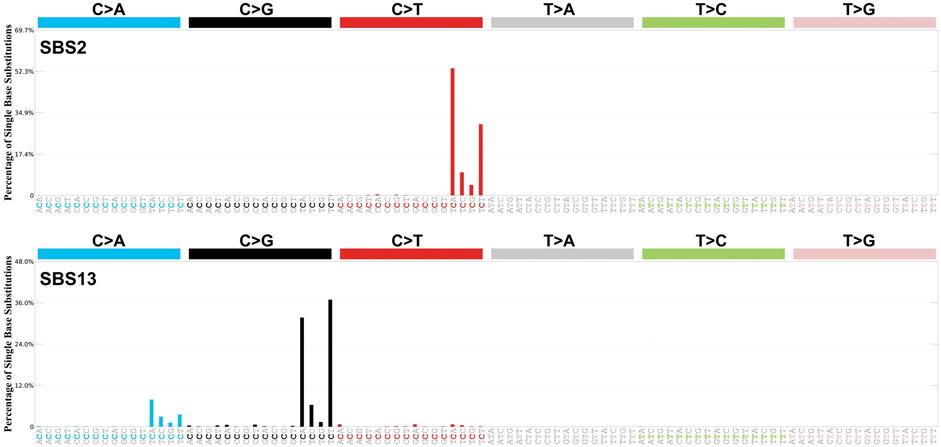
With the increase in numbers of cancer genomes sequenced and analyzed, a growing number of studies have started to identify mutations resembling APOBEC cytosine deamination in cancer [28,29]. Mathematical deconvolution of somatic mutations from individual cancer genomes identified two separate signatures of genome-dispersed mutations associated with APOBEC deamination, first in breast cancer [29] and later across multiple cancer types [30]. The two signatures, termed ‘Signature 2’ and ‘Signature 13’ (also commonly abbreviated as SBS2 and SBS13 signatures; where SBS stands for ‘single base substitution’), are respectively characterized by C>T transitions, and C>G and C>A transversions, at TCN trinucleotides [11,30] (Fig. 1). These are likely initiated by APOBEC cytosine deamination, as the mutations occur within APOBEC-favored TCN motifs, whereas the distinct mutation types that characterize these signatures may arise from different DNA replication and repair mechanisms [31]. The transversion mutations that characterize Signature 13 are thought to arise via base excision repair (BER), which requires UNG-mediated removal of uracils and insertion of cytosines opposite abasic sites by error-prone translesion polymerases, such as REV1 [31]. The C>T transition mutations that characterize Signature 2 are thought to arise predominantly by replication across the uracil bases [31], but they could also reflect activities of other polymerases with translesion potential, such as POLD, following BER. Further supporting a shared etiology, Signatures 2 and 13 are almost always present together, albeit in different proportions, in individual cancers [11,30].
Figure 1. APOBEC-associated genome wide signatures SBS2 and SBS13.

SBS2 and SBS13 identified from whole-genome sequences of 2,780 primary cancer from various types, as part of the Pan Cancer Analysis of Whole Genomes (PCAWG) effort [9]. Each signature is displayed according to the alphabetical 96-substitution classification on horizontal axes, defined by the six color-coded substitution types (C>A, C>G, C>T, T>A, T>C, and T>G; whereby all substitutions are referred to by the pyrimidine of the mutated Watson—Crick base pair) and sequence context immediately 5' and 3' to each mutated base. Vertical axes indicate the percentage of mutations attributed to specific mutation types, adjusted to the actual frequencies of each trinucleotide in the reference human genome version GRCh37. Images acquired from COSMIC (Mutational Signatures v3, May 2019).
In addition to the genome-dispersed signatures, mutations in APOBEC-characteristic TCN motifs also occur in patterns of localized hypermutation, where multiple same-strand base substitutions at C:G pairs cluster over kilobase-sized regions [29,32-34]. This regional signature of APOBEC-associated hypermutation has been termed kataegis (from Greek for shower or thunderstorm, i.e. mutational showers) and often colocalizes with sites of DNA rearrangements in cancer [29]. The different densities of mutations that form genome-dispersed and clustered APOBEC-associated signatures are thought to be governed by the lengths of the target single-stranded DNA (ssDNA) substrates, required for APOBEC-mediated deamination [35,36].
Since these initial discoveries, researchers have further dissected APOBEC-associated mutations in cancer at extended sequence contexts (i.e. pentanucleotide motifs) [37], at genomic meso-scales (e.g. at longer sequences predicted to form hairpin loops) [38] and at macro-scales (e.g. at regions related to transcription, DNA replication and chromatin organization) [39-42]. Such refinements were often combined with overexpression of individual APOBEC enzymes in model systems to investigate which family member causes mutations exhibiting genomic features that are most similar to the signatures observed in cancer (discussed in the following section) [3,37,43-45]. These studies collectively demonstrated that some APOBEC deaminases, when overexpressed in experimental model systems, can recapitulate mutational patterns seen in cancer.
2.2. APOBEC gene locus polymorphisms associate with mutation burdens
The second collection of links between APOBEC deaminases and mutations in cancer stems from genetic associations between polymorphisms in the APOBEC gene cluster on human chromosome 22 and burdens of the relevant signatures in cancer genomes. A germline polymorphism that results in the loss of the coding sequence of APOBEC3B, prevalent in East Asian populations (> 30%) [46], has been associated with an elevated risk of breast cancer in Asian populations [47,48] and the higher overall burden of APOBEC-association mutational signatures in breast cancers [49]. The results on the association between this polymorphism and cancer risk in European populations, where it is less common (~6%) [46], have so far been conflicting [50-52]. Whereas the association was not replicated in bladder cancers [53], it was found in lung and prostate cancers in individuals under 50 years of age [50]. In addition to the deletion polymorphism, a single nucleotide polymorphism (SNP) [50] rs1014971, located upstream of the APOBEC3 cluster, was associated with bladder cancer risk, increased APOBEC3B expression, and enrichment of APOBEC-signature mutations in bladder tumors [53]. Finally, APOBEC3H haplotype I has been implicated as another putative source of mutation in cancers, but its role remains elusive [54]. These studies provide important genetic associations between the APOBEC gene cluster, cancer risk and mutational burdens in human cancer. The impact of individual polymorphisms in different populations and cancer types, as well as their underlying mechanistic bases, require further clarification.
2.3. APOBEC expression correlates weakly, but positively, with mutation burden
The third collection of studies linking mutations in cancer and APOBEC deaminases identified correlations between upregulation of expression of individual APOBEC genes and burdens of APOBEC-associated signatures [14,28,38,55-57]. Notably, the correlations have been weak in comparison to the often-striking mutational burdens attributed to APOBEC-associated signatures in cancer and have generated opposing results to experimental overexpression systems with respect to the relevant mutator APOBEC enzyme [37,38,56] (discussed later). The major factor limiting comparisons between mutational burdens and RNA abundance are potential differences in APOBEC expression levels at the time of mutagenesis and at the time of RNA sampling. Mutations captured in cancer genomes could have been generated by APOBEC deaminases over the lifetime of a cell lineage, whereas RNA captures expression at the single time point of sample acquisition and not necessarily at the time of active mutagenesis [58]. It is also possible that signals of APOBEC expression in tumors originate from immune cells with naturally higher APOBEC levels [59].
3. APOBEC activity in cancer and non-malignant cells
The following section summarizes the prevalence, timing and activity of APOBEC-associated mutagenesis using mutational readouts from cancer and healthy tissues to provide insights into the features of APOBEC misregulation.
3.1. Prevalence of APOBEC mutagenesis in cancer and non-malignant cells
APOBEC-associated mutational signatures are among the most widespread in cancer and contribute particularly prominent mutational loads (median >0.25 mutations per Mb) in cancers of the cervix, head and neck, bladder, lung, and esophagus [11,28,30]. However, they have not been reported in cancers of the colon, liver, brain, chronic lymphocytic leukemia and acute myeloid leukemias (AML) [11,28,30,60]. Thus, APOBEC-associated mutagenesis is generally prevalent, but absent or rare in some cancer types. Furthermore, APOBEC-associated mutations are rare in childhood cancers [11], suggesting that the underlying misregulation may be age-related.
APOBEC-associated signatures have not been systematically examined in non-malignant cells, due to challenges associated with detection of somatic mutations among non-clonal cell populations. Strategies have recently been developed to circumvent this, including sequencing of clonal cell colonies [61-65] or clonal microscopic biopsies [66,67] and structures from healthy tissues, such as colonic crypts and endometrial glands [68-71]. Specialized methods [72,73] and single-cell sequencing [74,75] have also been employed to identify somatic mutations. These initial data sets have started to reveal the prevalence and patterns of APOBEC activity across healthy tissues. APOBEC-associated signatures have been detected in a small number of healthy colonic crypts [68] and colonies derived from single bronchial epithelial cells [65], indicating that APOBEC mutagenesis likely occurs in healthy tissues.
APOBEC-associated signatures were not detected in clones from normal esophageal epithelium [66] nor in healthy endometrial glands [71], despite their presence in esophageal and endometrial cancers [11,30], implying that they may be acquired later in cancer evolution, as suggested before [76,77], or that such cancers are more likely to evolve from clones with APOBEC mutagenesis [66]. APOBEC-associated signatures have also not been detected in healthy and cirrhotic liver clones, nor in healthy hematopoietic stem cells and myeloid progenitor cells [63,70]. Over the following years, systematic examination of mutational burdens in healthy tissues will shed further light onto the prevalence and emerging cell-type-specificity of APOBEC activity.
3.2. Episodic APOBEC mutagenesis in cancer cell lines
Generation of mutational signatures was recently assessed during in vitro culture of human cancer cell lines [58] (Fig. 2A). Whereas mutations detected in cancer genomes sampled at a single time point reflect readouts of historic mutagenic activities, cell lines are amenable to tracking ongoing mutation acquisition over serial and defined timeframes. Cell lines that carry APOBEC-associated mutational signatures in their genomes, and were therefore exposed to APOBEC mutagenesis in their past, often continued to generate APOBEC-associated signatures over time in culture. This indicates that APOBEC-associated signatures reflect traces of active rather than a historic mutational processes. Given the natural roles of APOBEC deaminases in innate immunity, induction of APOBEC misregulation by immune cells was foreseeable. Identification of cancer cell lines which generate APOBEC-associated signatures over time is the first demonstration that underlying mutagenesis can occur in the absence of the immune system [58].
Figure 2. Cell models of APOBEC mutagenesis.
A. Human cancer cell lines which generate APOBEC-associated mutations episodically in vitro represent genetically amenable models to experimentally explore origins of such mutations in cancer cells. CRISPR-Cas9 modifications of the candidate genes, such as individual APOBEC enzymes or other putative modulators of mutagenesis, can be generated in such cell lines to examine changes in mutation acquisition on various genetic backgrounds and during the defined in vitro timeframes (t). Genetically modified single-cell derived clones (‘parent clones’) are cultured for specified periods of time, e.g. depending on the mutation rates previously determined in cell lines [58]. Multiple ‘daughter clones’ are derived subsequently from single cells of individual parent clones. DNA isolated from parent clones and their corresponding daughters are subjected to WGS. Clonal mutations in daughter clones absent from their respective parent clones represent de novo mutations acquired during the specified in vitro timeframes spanning the two single cell bottlenecks. APOBEC-associated mutational burdens can be examined among de novo mutations acquired in different genetic backgrounds. B. Dicentric chromosomes formed by telomere fusion persist through mitosis intact and develop into cell-connecting DNA bridges [33]. Nuclear membrane rupture at DNA bridges enables the cytoplasmic exonuclease TREX1 to access and resect DNA. The resulting single-stranded DNA is targeted by APOBEC3B when broken DNA bridges are re-integrated into the nucleus. Repair of the fragmented DNA generates chromothripsis chromosomes frequently associated with kataegis. Uracil excision by UNG2 and abasic site cleavage by APE1 at single-stranded DNA may further enhance chromothriptic fragmentation, but APOBEC3B is not required for chromosome fragmentation.
Active APOBEC-associated mutagenesis was estimated to persist in at least 75% of cancer cell lines, with no ongoing APOBEC-associated mutagenesis detectable across only a smaller proportion of cell lines [58]. However, cancer cell models of APOBEC mutagenesis should be selected with caution, because the mutagenesis may not remain strongly or at all active in all cell lines that were previously exposed to it and carry the relevant signatures. The amenability of cell lines to serial single-cell cloning, followed by short propagation timeframes and whole-genome sequencing, revealed that unlike other examined mutational signatures, APOBEC-associated signatures exhibit substantial fluctuations in mutation rates over time [58]. In some cases, acquisition of signatures virtually completely ceased before reactivating and then discontinuing again. This phenomenon was termed ‘episodic mutagenesis’ [58]. The variability of APOBEC-associated mutational burdens was also detected between individual branches of phylogenetic trees from primary tumors [78,79] and healthy bronchial epithelial cells [65]. Collectively, these data indicate that some or all aberrant APOBEC activities in vivo may be unleashed onto the genome in mutagenic bursts. Genetically amenable cancer cell lines that exhibit active accumulation of APOBEC-associated mutations during propagation in vitro represent useful tools to probe the mechanisms of APOBEC mutagenesis (Fig. 2A).
Whereas the origins of APOBEC dysfunction remain unknown, these data imply that the instigating factors can be episodic in nature and possibly endogenous to affected cells. In addition, APOBEC-associated mutations seem to present rarely in healthy cells and/or may be more damaging to certain cell types. Collectively, these are some of the fundamental features to be considered in future investigations on the mechanisms underlying APOBEC misregulation in cancer.
4. Prime candidate mutators underlying APOBEC-associated mutagenesis
The identity of the APOBEC deaminase that is primarily responsible for mutagenesis in cancer is a persistent question in the field. Recent efforts have employed correlative analyses of APOBEC3 expression with mutational burden and developed transgenic mouse and cell overexpression models to assess the mutagenic potential of specific APOBEC3 paralogs. These studies nominated two APOBEC3 family members, APOBEC3A and APOBEC3B, which preferentially operate in a TCN context which brands the associated signatures in cancer, as the most likely candidates. Current focus has shifted away from the other nine AID/APOBEC members as potential mutators, due to lack of known DNA deaminase activity, distinct sequence preferences and/or apparent limited access to genomic DNA (reviewed in [80,81]), although their contributions cannot be in entirety dismissed.
4.1. Studies supporting a role for APOBEC3B in carcinogenesis
APOBEC3B mRNA is upregulated in at least 6 distinct cancers (bladder, cervix, lung adenocarcinoma, lung squamous cell carcinoma, head and neck, and breast) relative to healthy tissue [14,55]. The other APOBEC3 paralogs are generally expressed at significantly lower levels than APOBEC3B, aside from a few limited examples (e.g. APOBEC3A overexpression in AML [15]). Several lines of evidence implicate the observed APOBEC3B upregulation with carcinogenesis.
First, APOBEC3B expression correlates with specific clinical outcomes indicating a role in disease progression. APOBEC3B expression is positively correlated with adverse outcomes in estrogen receptor-positive breast cancers [22]. High APOBEC3B expression predicts worse outcomes in lung cancer and recurrence of clear cell renal cell carcinomas [82,83]. However, in a subset of clear cell ovarian carcinomas, high APOBEC3B expression is associated with improved progression-free survival [84]. Improved progression-free survival in this subset of patients may be explained by enhanced sensitivity to cisplatin from increased DNA damage resulting from APOBEC3B-mediated deamination [84].
Second, APOBEC3B expression levels were reported to weakly, albeit positively correlate with overall mutation burden [14]. Breast cancers with high levels of APOBEC3B expression exhibit nearly twice as many C-to-T mutations [14]. This correlation extends to distinct cancer types including cervical, bladder, and other cancers [55]. In line with APOBEC3B being a putative mutator in cancer, it is the only APOBEC3 that is constitutively nuclear as it exhibits a nuclear localization in all cell types examined thus far [14,85]. However, another widely considered mutator, APOBEC3A, also appears to access the nucleus and displays a pan-cellular localization in some cell types, but restriction to the cytosol in others [12,85].
Third, APOBEC3B expression levels correlate with the course of disease. Consistent with clinical data showing that high APOBEC3B levels inversely correlate with response to tamoxifen in estrogen receptor-positive disease, APOBEC3B overexpression promotes tamoxifen resistance in murine xenografts [23] and increases sensitivity to immune checkpoint blockade in murine models of melanoma [25].
Collectively these studies suggest an important role for APOBEC3B in cancer. However, most experiments have used correlations to implicate APOBEC3B as the relevant mutator. In fact, recent refinement of the expression-based studies, suggests that APOBEC3A expression correlates more strongly with APOBEC-associated mutation load in breast cancer than APOBEC3B expression [56]. It is thus conceivable that high APOBEC3B expression influences disease progression through independent means, such as the generation of DNA breaks and induction of chromosome rearrangements. In the future it will be important to experimentally connect endogenous APOBEC3B activity to mutations in cancer to assess its role as a mutator. Thus far, endogenous APOBEC3B has also been implicated in generation of kataegis in a cell model of telomere crisis (more below) [34]. This type of mechanistic clarity is especially important because several studies have suggested that APOBEC3B plays a limited role in cancer mutagenesis [37,56].
4.2. Studies supporting a role for APOBEC3A in cancer mutagenesis
Independent lines of evidence point towards the APOBEC3A paralog as a more prominent source of mutations in cancer. The first piece of evidence to suggest the presence of APOBEC3B-independent sources of mutagenesis comes from the APOBEC3B deletion polymorphism discussed above, associated with increased APOBEC-associated mutational burdens in some populations [46,49]. This deletion effectively removes APOBEC3B and fuses the APOBEC3A coding sequence with the 3’UTR of APOBEC3B. This alteration is proposed to stabilize the APOBEC3A mRNA thus resulting in higher mutation burdens [49,86]. An alternative proposal is that APOBEC3H may be a putative mutator in cancers with the APOBEC3A/B deletion polymorphism [54], but conclusive experimental evidence is lacking.
Data from yeast suggest that human APOBEC3A and APOBEC3B preferentially operate at distinct motifs and that the APOBEC3A-preferred motif is more common in cancer [37,43]. This study used overexpression of human APOBEC3A and APOBEC3B in yeast models engineered to accumulate long, ssDNA substrates to generate sufficient mutations to distinguish sequence motifs preferred by each paralog [37]. This analysis revealed that APOBEC3A favors YTCA sites, while APOBEC3B favors RTCA sites (Y = pyrimidine, R = purine) [37]. The structural basis for this sequence preference is not clear as APOBEC3A and a chimeric version of APOBEC3B have limited contact with ssDNA aside from the central 5′-TC motif [87]. The APOBEC3A-preferred YTCA site is a more prominent source of mutations in cancer genomes and in cell lines with ongoing mutagenesis [37,58]. This correlation suggests that APOBEC3A may be a prime mutator in human cancer, but the substrate preferences of other APOBEC3s expressed in yeast and human systems have not been rigorously assessed. More recently, APOBEC3A was found to prefer DNA substrates predicted to form stem-loops and multiple recurring APOBEC-resembling mutations were found at such APOBEC3A-favored sequences in cancer genomes [38]. It thus seems likely substrate preferences of APOBEC3A, rather than selection, drive recurring occurrence of mutations at such hotspot sequences in cancer, highlighting how understanding the substrate preferences at which mutational processes operate may be helpful in distinguishing ‘driver’ mutations with functional consequences from ‘passenger’ mutations in cancer genomes that not affected by selection [38]. Importantly, although APOBEC3A-may prefer targeting sequences predicted to form stem-loops, the majority of the APOBEC-associated mutations is expected to present outside of such sequences, that are otherwise rare across the human genome.
Unlike APOBEC3B, APOBEC3A, is expressed at a low level in most cancers [28] posing a significant challenge to accurately detect its activities. A lack of sensitive, APOBEC3A-specific antibodies has further compounded the challenge of detecting APOBEC3A protein in cancer. Recently, RNA stem-loops were found to be commonly edited in cancer, within the sequence motifs characteristic of APOBEC mutagenesis on DNA [88]. Although the specific RNA loci were not mutated in their DNA templates of analyzed tumors, these data indicated that structural preferences of the APOBEC3A activities on RNA, mimic its ability to also mutate DNA within the similar hairpin-like structures [38] and as such differ from the preferences of APOBEC3B [88]. A selection of the RNA hotspots was subsequently leveraged in a digital droplet PCR assay to quantify the RNA-editing activities associated with APOBEC3A in individual tumors. In the future, it will be critical to determine whether detection of APOBEC3A-associated RNA editing also translates to active APOBEC3A mutagenesis in cancer genomes. APOBEC3A activity, as inferred from the active RNA editing, was frequently observed in the panel of AML and myeloproliferative neoplasms (MPN), in keeping with such activities previously reported in monocytes and macrophages during M1 polarization [89]. However, APOBEC-associated mutational signatures have not been detected in AML and are rare in MPN [11]. It is thus possible that APOBEC3A activities on RNA may not represent an optimal proxy for an active mutagenesis in cancer.
Finally, transgenic expression of APOBEC3A also supports a role for this paralog in carcinogenesis. Transgenic APOBEC3A expression in cell models results in more cellular DNA damage than overexpression of other APOBEC3s [12,13]. This is consistent with observations that APOBEC3A exhibits the highest catalytic activity among the human APOBEC3 enzymes [90]. The structural basis of this high catalytic activity may reflect the relatively open conformation of APOBEC3A that is capable of binding to DNA in the absence of modification [87]. In contrast, APOBEC3B exhibits a closed active site that must be converted in order to bind DNA substrate [87]. Transgenic expression of APOBEC3A in mice has also recently been shown to induce APOBEC signature mutations and increase tumorigenesis in murine colon and liver tissues [45].
Overall, the discussed studies suggest that APOBEC3A likely generates APOBEC-associated mutations in cancer genomes. However, the experimental evidence and investigation into the extent of its mutagenic activities relative to APOBEC3B and possibly other APOBECs that may contribute additional mutations, are outstanding.
4.3. APOBEC3B generates clustered mutations during telomere crisis
In addition to mutations throughout the genome, APOBEC-induced mutations can also appear in kataegis, a pattern of localized hypermutation often associated with chromosome rearrangement breakpoints [29]. Recent work has identified kataegis in >60% of cancers and at high frequency in lung squamous cell carcinoma, bladder cancer, acral melanoma, and sarcomas [91]. APOBEC signatures account for the majority of kataegis events, but some may be driven by independent mechanisms including error-prone DNA synthesis [91,92]. APOBEC3B appears to be a prime driver because, along with structural variant burden and age of diagnosis, kataegis positively correlate with APOBEC3B expression levels [91].
Recent work implicates endogenous APOBEC3B in the generation of clustered mutations during telomere crisis [34] (Fig. 2B). Telomere crisis is a period of genome instability that occurs during tumorigenesis when depletion of the telomere reserve generates unstable dicentric chromosomes [93]. Dicentric chromosomes persist through mitosis to form DNA bridges that are generally broken hours after mitotic exit [33]. Rearranged clonal cell lines isolated after transit through telomere crisis exhibit frequent chromothripsis often associated with kataegis [33]. Reduction of kataegis in APOBEC3B knockout cells recovered after telomere crisis indicates that APOBEC3B is the major enzymatic source of kataegis in this telomere crisis model [34]. Kataegis-like clusters have also been generated in yeast by expression of AID/APOBEC family deaminases [32,43,94].
APOBEC3B base editing during telomere crisis occurs independently of APOBEC3B upregulation and instead depends on ssDNA formation at DNA bridges [34] (Fig. 2B). The ability of APOBECB to generate clustered mutations may derive from its ability to execute processive deamination [95]. However, it is not clear if APOBEC3B is uniquely capable of catalyzing kataegis during telomere crisis. RPE1-hTERT cells are non-tumorigenic and do not express APOBEC3A [34]. Therefore, a potential role for APOBEC3A in generating kataegis during telomere crisis is not clear from these experiments.
The identity of the prime mutator(s) may depend on unknown contexts specific a particular cell or tissue. Clustered and genome-dispersed mutations may also reflect the activities of distinct APOBEC3 mutators. The identity of the prime mutator is a fundamentally important problem because answers can guide the development of targeted therapies and studies on factors that instigate APOBEC mutagenesis. Cell line models that recapitulate APOBEC3 mutagenesis during propagation in vitro [33,34,58] will be useful tools to dissect the mutagenic potential of specific APOBEC3 paralogs.
5. Speculative mechanisms of episodic APOBEC misregulation in cancer
Mechanisms which drive APOBEC-mediated mutagenesis remain a major open question. We specifically focus on putative intrinsic sources of APOBEC misregulation in cancer cells, in the light of recent observations of endogenous and episodic APOBEC-associated mutagenesis in cancer cell lines and in a model of telomere crisis [34,58].
Thus far, recurrent mutations in APOBEC genes that are associated with the relevant mutational signatures in cancer have not been identified [49], implying a perturbation in activity or abundance. Expression of APOBEC genes in healthy tissues is considered low, with notable exceptions of APOBEC1 expression in small intestine, where it has a critical role in plasma metabolism by apoprotein B pre-mRNA editing [1,2], and expression of APOBEC3 members in immune cells [9]. Among the most prominent basal levels of APOBECs in healthy tissues is that of APOBEC3A in myeloid cells [96-98]. Upregulation of several APOBEC3 family members in cancer compared to healthy tissues has been reported [14,28,37,57]. However, correlations between individual APOBEC gene expressions and mutational burdens in cancer are weak [14,28,37,55-57], possibly due to aforementioned limitations or because other factors are involved. Prolonged APOBEC3 expression is cytotoxic in cell culture models [14]. Stable hyperexpression in cancer thus may be cytotoxic, but short and intermittent periods of APOBEC3 expression may limit genotoxicity in individual cancer cells.
5.1. Viruses and retrotransposons
APOBEC3 family members are believed to have evolved primarily as editors of viral and retrotransposon nucleic acid intermediates [80]. It was proposed that APOBEC-associated signatures in cancer may represent collateral damage on human DNA during APOBEC-mediated restriction of retrotransposons or exogenous viruses [30]. APOBEC-associated signatures are among the most prevalent in human papilloma virus (HPV)-associated cervix and head and neck cancers [11]. However, APOBEC-associated signatures are also found in many HPV-negative head and neck cancers [99], and are prevalent in many cancers types otherwise not linked to obvious infections (e.g. breast, lung, esophageal cancers) [11,30]. Furthermore, APOBEC-associated signatures have not been found in liver cancers [11,60], commonly infected with hepatitis viruses restricted by APOBEC deaminases [100] and active APOBEC-associated mutagenesis does not persist in cervical cancer cell lines where HPV sequences and related transcripts were detected [58]. Similarly, the burden of APOBEC-associated signatures did not correlate with somatic retrotransposon insertions in analysis across >2,300 primary cancers and the association was only weak in cell lines [58]. Whereas these data do not exclude the roles of viruses and retrotransposons in modulating APOBEC-associated mutagenesis, it indicates that there are cases in which they are neither required nor sufficient to drive it.
Although viruses and retrotransposons may not be the major activators of aberrant APOBEC activities, the same underlying modulators of APOBEC upregulation in the context of an anti-viral response may be involved in the context of cancer. APOBEC3 genes are interferon (IFN)-stimulated during the innate immune response, with APOBEC3A showing exceptional sensitivity to IFN stimulation [44]. Both IFN-dependent and IFN-independent mediators of APOBEC3 upregulation may play a role and have been extensively reviewed elsewhere [9], but are not likely to be thoroughly captured by correlations between mutation burdens and signals from RNA-sequencing [101] due to the potential for temporal disconnect between APOBEC mutation burden and sample acquisition discussed above.
Expression of some APOBEC3 enzymes can be upregulated by the cytosolic nucleic acid sensors, which detect single- and double-stranded RNA and DNA [98]. In myeloid cells, STING (stimulator of IFN genes) pathway can modulate APOBEC3A expression via IFN- β induction [102]. Multiple sources of cytosolic nucleic acid in cancer cells have been documented, including micronuclei [103-105] and expression of endogenous retroviruses [106]. Whereas APOBEC3A is induced by IFN to mediate clearance of foreign DNA from human cells, nuclear DNA remains intact and the restriction mechanisms are possibly compartmentalized to mitigate the risk to the genomic DNA [44]. Nuclear envelope rupture is one event that may break such barriers and that is common in cancer [107-111]. Transient upregulation of APOBEC expression and intermittent access to nuclear DNA are compatible with the episodic nature of underlying mutagenesis.
Multiple exogenous stimuli can activate APOBEC expression in experimental cell systems and have been reviewed elsewhere [9]. It is not clear whether these exogenous stimuli also induce mutagenesis and account for mutations observed in cancer as APOBEC upregulation may not always translate to active mutagenesis. However, it is likely that multiple environmental agents, including viruses, may contribute to misregulation. For example, lung adenocarcinomas from smokers carry more APOBEC-associated signatures compared to non-smokers [112], possibly because inflammatory responses associated with tobacco smoking also trigger APOBEC activity. Cancer cell lines with ongoing APOBEC-associated mutagenesis represent experimental models to investigate endogenous sources of episodic APOBEC misregulation [58]. Epidemiological associations may shed light further on the environmental modulators of APOBEC-associated mutagenesis.
Conclusions
Extensive data implicates APOBEC deaminases as some of the most prominent endogenous sources of mutations in human cancer. However, current links between APOBEC enzymes and mutations in cancer are based on associations, not causation. The critical evidence that APOBEC deaminases generate mutations observed in cancer cells is outstanding and the responsible mutator remains only speculative and is under debate [14,28,37,55-57]. These fundamental gaps remain because genetically amenable models that accurately recapitulate mutagenesis were not available until recently. Transgenic murine models of APOBEC mutagenesis should be cautiously interpreted. APOBEC3 sequences are rapidly evolving and exhibit significant divergence between rodent and primate species [113-115]. Given the mutagenic potential of aberrant APOBEC3 activity, it is possible that primates may have evolved mechanisms to protect against the risks associated with the expression of endogenous enzymatic mutators. Newly discovered negative regulators of APOBEC3 mutagenesis provide some experimental support for this view [116,117]. The absence of these potential regulatory mechanisms in rodents may compromise the utility of transgenic mouse models to fully understand APOBEC mutagenesis.
Recently described human cancer cell lines which generate APOBEC-associated mutational signatures endogenously over time were reported [58]. These cell lines also carry signatures of the historic exposure to mutagenesis, which is predicted to have at least in part occurred in the corresponding primary cancers. The in vitro mechanisms of APOBEC misregulation in cell lines thus likely operated in vivo and were inherited from their originating cancers [58]. Such cell lines with intrinsic APOBEC-associated mutagenesis represent suitable experimental systems for future investigations on its origins in cancer, to identify the relevant mutator member(s) and identify mechanisms which instigate the mutagenic behavior.
Acknowledgements
We thank Dr. Ludmil Alexandrov’s lab for sharing the high-resolution SBS plots and Allie Dananberg and members of the Maciejowski lab for critical reading of this manuscript. We are further grateful to Mike Stratton for his critical review and suggestions. Work in J.M.’s laboratory is supported by the NCI (R00CA212290), the Pew Charitable Trusts, the V Foundation, the Starr Cancer Consortium, the Emerald Foundation, the Geoffrey Beene and Ludwig Centers at MSKCC, and the NIH/NCI Cancer Center Support Grant P30 CA008748. M.P. is supported by the European Molecular Biology Organization (EMBO) Long-Term Fellowship (ALTF 760-2019).
Abbreviations:
- AID
activation induced cytidine deaminase
- AML
acute myeloid leukemia
- APOBEC
apolipoprotein B mRNA editing enzyme, catalytic polypeptide
- BER
base excision repair
- ER
estrogen receptor
- HPV
human papilloma virus
- MPN
myeloproliferative neoplasms
- SBS
single base substitution
- ssDNA
single-stranded DNA
- UNG
uracil DNA glycosylase
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Navaratnam N, Morrison JR, Bhattacharya S, Patel D, Funahashi T, Giannoni F, Teng BB, Davidson NO, Scott J, The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase, J. Biol. Chem 268 (1993) 20709–20712. [PubMed] [Google Scholar]
- [2].Teng B, Burant C, Davidson N, Molecular cloning of an apolipoprotein B messenger RNA editing protein, Science. 260 (1993) 1816–1819. 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- [3].Harris RS, Petersen-Mahrt SK, Neuberger MS, RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators, Mol. Cell 10 (2002) 1247–1253. 10.1016/S1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- [4].Petersen-Mahrt SK, Neuberger MS, In vitro deamination of cytosine to uracil in single-stranded DNA by apolipoprotein B editing complex catalytic subunit 1 (APOBEC1), J. Biol. Chem 278 (2003) 19583–19586. 10.1074/jbc.C300114200. [DOI] [PubMed] [Google Scholar]
- [5].Sowden M, Hamm JK, Smith HC, Overexpression of APOBEC-1 results in mooring sequence-dependent promiscuous RNA editing, J. Biol. Chem 271 (1996) 3011–3017. 10.1074/jbc.271.6.3011. [DOI] [PubMed] [Google Scholar]
- [6].Bishop KN, Holmes RK, Sheehy AM, Malim MH, APOBEC-mediated editing of viral RNA, Science. 305 (2004) 645 10.1126/science.1100658. [DOI] [PubMed] [Google Scholar]
- [7].Marino D, Perković M, Hain A, Vasudevan AAJ, Hofmann H, Hanschmann KM, Mühlebach MD, Schumann GG, König R, Cichutek K, Häussinger D, Münk C, APOBEC4 Enhances the Replication of HIV-1, PLoS One. 11 (2016) 1–23. 10.1371/journal.pone.0155422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Okuyama S, Marusawa H, Matsumoto T, Ueda Y, Matsumoto Y, Endo Y, Takai A, Chiba T, Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis, Int. J. Cancer 130 (2012) 1294–1301. 10.1002/ijc.26114. [DOI] [PubMed] [Google Scholar]
- [9].Stavrou S, Ross SR, Stavrou S, Ross SR, APOBEC3 Proteins in Viral Immunity, J. Cell Biol (2015) 4565–4570. 10.4049/jimmunol.1501504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM, Mutations, kataegis and translocations in B cells: Understanding AID promiscuous activity, Nat. Rev. Immunol 16 (2016) 164–176. 10.1038/nri.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, Alexandrov LB, Bergstrom EN, Boot A, Boutros P, Chan K, Covington KR, Fujimoto A, Getz G, Gordenin DA, Haradhvala NJ, Huang MN, Islam SMA, Kazanov M, Kim J, Klimczak LJ, Lopez-Bigas N, Lawrence M, Martincorena I, McPherson JR, Morganella S, Mustonen V, Nakagawa H, Tian Ng AW, Polak P, Prokopec S, Roberts SA, Rozen SG, Sabarinathan R, Saini N, Shibata T, Shiraishi Y, Stratton MR, Teh BT, Vázquez-García I, Wheeler DA, Wu Y, Yousif F, Yu W, Getz G, Rozen SG, Stratton MR, The repertoire of mutational signatures in human cancer, Nature. 578 (2020) 94–101. 10.1038/S41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Landry S, Narvaiza I, Linfesty DC, Weitzman MD, APOBEC3A can activate the DNA damage response and cause cell-cycle arrest, EMBO Rep. 12 (2011) 444–450. 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Green AM, Landry S, Budagyan K, Daphne C, Shalhout S, Bhagwat AS, Weitzman MD, Green AM, Landry S, Budagyan K, Daphne C, Shalhout S, Bhagwat AS, Apobeca MDW, APOBEC3A damages the cellular genome during DNA replication, CELL CYCLE. 15 (2016) 998–1008. 10.1080/15384101.2016.1152426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, Mcdougle RM, Brown WL, Law EK, Harris RS, APOBEC3B is an enzymatic source of mutation in breast cancer, Nature. 494 (2013) 366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, Weitzman MD, Cytosine deaminase APOBEC3A sensitizes leukemia cells to inhibition of the DNA replication checkpoint, Cancer Res. 77 (2017) 4579–4588. 10.1158/0008-5472.CAN-16-3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Buisson L, Rémi, Lawrence, Michael S, Benes Cyril H., and Zou, APOBEC3A and 3B Activities Render Cancer Cells Susceptible to ATR Inhibition, Cancer Res. 77 (2017) 4567–4578. 10.1158/0008-5472.CAN-16-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Serebrenik AA, Starrett GJ, Leenen S, Jarvis MC, Shaban NM, Salamango DJ, Nilsen H, Brown WL, Harris RS, The deaminase APOBEC3B triggers the death of cells lacking uracil DNA glycosylase, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 22158–22163. 10.1073/pnas.1904024116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Green AM, Weitzman MD, The spectrum of APOBEC3 activity: From anti-viral agents to anti-cancer opportunities, DNA Repair (Amst). 83 (2019) 102700 10.1016/j.dnarep.2019.102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vlachostergios PJ, Faltas BM, Treatment resistance in urothelial carcinoma: an evolutionary perspective, Nat. Rev. Clin. Oncol 15 (2018) 495–509. 10.1038/S41571-018-0026-y. [DOI] [PubMed] [Google Scholar]
- [20].Swanton C, McGranahan N, Starrett GJ, Harris RS, APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity, Cancer Discov. 5 (2015) 704–712. 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR, APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development, Cell Rep. 7 (2014) 1833–1841. 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- [22].Sieuwerts AM, Willis S, Burns MB, Look MP, Jacobs H, Wessels L, Leyland-jones B, Gray KP, Foekens JA, Harris RS, Martens JWM, Elevated APOBEC3B Correlates with Poor Outcomes for Estrogen-Receptor-Positive Breast Cancers, Horm. Cancer (2014) 405–413. 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Law EK, Sieuwerts AM, Lapara K, Leonard B, Starrett GJ, Molan AM, Temiz NA, Vogel RI, Gelder MEM, Sweep FCGJ, Span PN, Foekens JA, Martens JWM, Yee D, Harris RS, The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer, Sci. Adv (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Grönroos E, Muhammad MA, Horswell S, Gerlinger M, Varela I, Jones D, Marshall J, Voet T, Van Loo P, Rassl DM, Rintoul RC, Janes SM, Lee SM, Forster M, Ahmad T, Lawrence D, Falzon M, Capitanio A, Harkins TT, Lee CC, Tom W, Teefe E, Chen SC, Begum S, Rabinowitz A, Phillimore B, Spencer-Dene B, Stamp G, Szallasi Z, Matthews N, Stewart A, Campbell P, Swanton C, Spatial and temporal diversity in genomic instability processes defines lung cancer evolution, Science. 346 (2014) 251–256. 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Driscoll CB, Schuelke MR, Kottke T, Thompson JM, Wongthida P, Tonne JM, Huff AL, Miller A, Shim KG, Molan A, Wetmore C, Selby P, Samson A, Harrington K, Pandha H, Melcher A, Pulido JS, Harris R, Evgin L, Vile RG, APOBEC3B-mediated corruption of the tumor cell immunopeptidome induces heteroclitic neoepitopes for cancer immunotherapy, Nat. Commun 11 (2020) 1–14. 10.1038/s41467-020-14568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yamanaka S, Balestra ME, Ferrell LD, Fan J, Arnold KS, Taylor S, Taylor JM, Innerarity TL, Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals, Proc. Natl. Acad. Sci. U. S. A 92 (1995) 8483–8487. 10.1073/pnas.92.18.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beale RCL, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS, Comparison of the Differential Context-dependence of DNA Deamination by APOBEC Enzymes: Correlation with Mutation Spectra in Vivo, J. Mol. Biol 337 (2004) 585–596. 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- [28].Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA, An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers, Nat. Genet 45 (2013) 970–976. 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nik-zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Ranee R, Lau KW, Mudie LJ, Varela I, Mcbride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, Mclaren S, Garber JE, Silver D, Miron P, Fatima A, Butler AP, Teague JW, Boyault S, Langerød A, Tutt A, Martens JWM, Aparicio SAJR, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR, Cancer B, Mutational Processes Molding the Genomes of 21 Breast Cancers, Cell. (2012) 979–993. 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinsk M, Jäger N, Jones DTW, Jonas D, Knappskog S, Koo M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, Van Buuren MM, Van ’T Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Andrew Futreal P, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Signatures of mutational processes in human cancer, Nature. 500 (2013) 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Helleday T, Eshtad S, Nik-Zainal S, Mechanisms underlying mutational signatures in human cancers, Nat. Rev. Genet 15 (2014) 585–598. 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Male E, Mieczkowski PA, Resnick MA, Gordenin DA, Clustered Mutations in Yeast and in Human Cancers Can Arise from Damaged Long Single-Strand DNA Regions, Mol. Cell. 46 (2012) 424–435. 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maciejowski J, Li Y, Bosco N, Campbell PJ, De Lange T, Chromothripsis and Kataegis Induced by Telomere Crisis, Cell. 163 (2015) 1641–1654. 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maciejowski J, Chatzipli A, Dananberg A, de Lange T, Campbell P, APOBEC3B-dependent kataegis and TREX1-driven chromothripsis in telomere crisis, BioRxiv. (2019) 725366. 10.1101/725366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu Q, König R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR, Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome, Nat. Struct. Mol. Biol 11 (2004) 435–442. 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- [36].Harris RS, Liddament MT, Hall J, RETROVIRAL RESTRICTION BY APOBEC PROTEINS, Nat. Rev. Immunol 4 (2004) 868–877. 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- [37].Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, Getz G, Gordenin DA, An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers, Nat. Genet 47 (2015) 1067–1074. 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, Lawrence MS, Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features, Science (80-.). 364 (2019). 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, Butler A, Kim HY, Borg Å, Sotiriou C, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Stunnenberg HG, Van De Vijver MJ, Martens JWM, Børresen-Dale AL, Richardson AL, Kong G, Thomas G, Sale J, Rada C, Stratton MR, Birney E, Nik-Zainal S, The topography of mutational processes in breast cancer genomes, Nat. Commun 7 (2016) 1–11. 10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI, APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication, Genome Res. 26 (2016) 174–182. 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA, APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication, Cell Rep. 14 (2016) 1273–1282. 10.1016/j.celrep.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, Kamburov A, Hanawalt PC, Wheeler DA, Koren A, Lawrence MS, Getz G, Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair, Cell. 164 (2016) 538–549. 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Taylor BJM, Nik-zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS, DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis, Elife. (2013) 1–14. 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS, APOBEC3 proteins mediate the clearance of foreign DNA from human cells, Nat. Struct. Mol. Biol 17 (2010) 222–229. 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, Starrett GJ, Larson LK, Burns MB, Vogel RI, Stavrou S, Aguilera AN, Wagner S, Largaespada DA, Starr TK, Ross SR, Harris RS, APOBEC3A Catalyzes Mutation and Drives Carcinogenesis In Vivo, BioRxiv. (2019) 2019.12.27.889345. 10.1101/2019.12.27.889345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE, Population Stratification of a Common APOBEC Gene Deletion Polymorphism, PLoS Genet. 3 (2007) 0584–0592. 10.1371/journal.pgen.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Long J, Delahanty RJ, Li G, Gao YT, Lu W, Cai Q, Xiang YB, Li C, Ji BT, Zheng Y, Ali S, Shu XO, Zheng W, A common deletion in the APOBEC3 genes and breast cancer risk, J. Natl. Cancer Inst 105 (2013) 573–579. 10.1093/jnci/djt018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Komatsu A, Nagasaki K, Fujimori M, Amano J, Miki Y, Identification of novel deletion polymorphisms in breast cancer, Int. J. Oncol 33 (2008) 261–270. 10.3892/ijo. [DOI] [PubMed] [Google Scholar]
- [49].Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, Davies HR, Knappskog S, Martin S, Papaemmanuil E, Ramakrishna M, Shlien A, Simonic I, Xue Y, Tyler-Smith C, Campbell PJ, Stratton MR, Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer, Nat. Genet 46 (2014) 487–491. 10.1038/ng.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gansmo LB, Romundstad P, Hveem K, Vatten L, Nik-Zainal S, Lønning PE, Knappskog S, APOBEC3A/B deletion polymorphism and cancer risk, Carcinogenesis. 39 (2018) 118–124. 10.1093/carcin/bgx131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xuan D, Li G, Cai Q, Deming- S, Shrubsole MJ, Shu X, Kelley C, Zheng W, Long J, APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry, Carcinogenesis. 34 (2013) 2240–2243. 10.1093/carcin/bgt185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Klonowska K, Kluzniak W, Rusak B, Jakubowska A, Ratajska M, Krawczynska N, Vasilevska D, Czubak K, Wojciechowska M, Cybulski C, Lubinski J, Kozlowski P, The 30 kb deletion in the APOBEC3 cluster decreases APOBEC3A and APOBEC3B expression and creates a transcriptionally active hybrid gene but does not associate with breast cancer in the European population, Oncotarget. 8 (2017) 76357–76374. 10.18632/oncotarget.19400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, Figueroa JD, Zhu B, Koutros S, Kubo M, Shuin T, Freedman ND, Kogevinas M, Malats N, Chanock SJ, Garcia-Closas M, Silverman DT, Rothman N, Prokunina-Olsson L, Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors, Nat. Genet 48 (2016) 1330–1338. 10.1038/ng.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA, Harris RS, The DNA cytosine deaminase APOBEC3H haplotype i likely contributes to breast and lung cancer mutagenesis, Nat. Commun 7 (2016) 1–13. 10.1038/ncomms12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Burns MB, Temiz NA, Harris RS, Evidence for APOBEC3B mutagenesis in multiple human cancers, Nat. Publ. Gr 45 (2013) 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cortez LM, Brown AL, Dennis MA, Collins CD, Brown AJ, Mitchell D, Mertz TM, Roberts SA, APOBEC3A is a prominent cytidine deaminase in breast cancer, 2019. 10.1371/journal.pgen.1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, Oberg AL, Cunningham JM, Shridhar V, Bell DA, April C, Bentley D, Bibikova M, Cheetham RK, Fan JB, Grocock R, Humphray S, Kingsbury Z, Peden J, Chien J, Swisher EM, Hartmann LC, Kalli KR, Goode EL, Sicotte H, Kaufmann SH, Harris RS, APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma, Cancer Res. 73 (2013) 7222–7231. 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Petljak M, Alexandrov LB, Price S, Brammeld JS, Koh CC, Wedge DC, Ju YS, lorio F, Jose M, Tubio C, Dawson KJ, Meara SO, Butler AP, Menzies A, Characterization of mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis, Cell. accepted (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ng JCF, Quist J, Grigoriadis A, Malim MH, Fraternali F, Pan-cancer transcriptomic analysis dissects immune and proliferative functions of APOBEC3 cytidine deaminases, Nucleic Acids Res. 47 (2019) 1178–1194. 10.1093/nar/gky1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J, Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets, Nat. Genet 47 (2015) 505–511. 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, Riley-Gillis B, Sestan N, Urban AE, Abyzov A, Vaccarino FM, Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis, Science. 359 (2018) 550–555. 10.1126/science.aan8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Franco I, Johansson A, Olsson K, Vrtačnik P, Lundin P, Helgadottir HT, Larsson M, Revêchon G, Bosia C, Pagnani A, Provero P, Gustafsson T, Fischer H, Eriksson M, Somatic mutagenesis in satellite cells associates with human skeletal muscle aging, Nat. Commun 9 (2018). 10.1038/s41467-018-03244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lee-Six H, Øbro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly BJP, Martincorena I, Anderson E, O’Neill L, Stratton MR, Laurenti E, Green AR, Kent DG, Campbell PJ, Population dynamics of normal human blood inferred from somatic mutations, Nature. 561 (2018) 473–478. 10.1038/S41586-018-0497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Blokzijl F, De Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EES, Verstegen MMA, Van Der Laan LJW, De Jonge J, Ijzermans JNM, Vries RG, Van De Wetering M, Stratton MR, Clevers H, Cuppen E, Van Boxtel R, Tissue-specific mutation accumulation in human adult stem cells during life, Nature. 538 (2016) 260–264. 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T, Maughan EF, Beal K, Menzies A, Millar FR, Anderson E, Clarke SE, Pennycuick A, Thakrar RM, Butler CR, Kakiuchi N, Hirano T, Hynds RE, Stratton MR, Martincorena I, Janes SM, Campbell PJ, Tobacco smoking and somatic mutations in human bronchial epithelium, Nature. 578 (2020). 10.1038/s41586-020-1961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, Saeb-Parsy K, Jones PH, Somatic mutant clones colonize the human esophagus with age, Science (80-.). 362 (2018) 911–917. 10.1126/science.aau3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ, High burden and pervasive positive selection of somatic mutations in normal human skin, Science (80-. ). 348 (2015) 880–886. 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, Georgakopoulos N, Torrente F, Noorani A, Goddard M, Robinson P, Coorens THH, O’Neill L, Alder C, Wang J, Fitzgerald RC, Zilbauer M, Coleman N, Saeb-Parsy K, Martincorena I, Campbell PJ, Stratton MR, The landscape of somatic mutation in normal colorectal epithelial cells, Nature. 574 (2019) 532–537. 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- [69].Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H, Tanaka K, Yamamoto T, Motoyama T, Inoue I, Enomoto T, Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium, Cell Rep. 24 (2018) 1777–1789. 10.1016/j.celrep.2018.07.037. [DOI] [PubMed] [Google Scholar]
- [70].Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, Sanders MA, Ellis P, Alder C, Hooks Y, Abascal F, Stratton MR, Martincorena I, Hoare M, Campbell PJ, Somatic mutations and clonal dynamics in healthy and cirrhotic human liver, Nature. 574 (2019) 538–542. 10.1038/s41586-019-1670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, Dawson KJ, Butler T, Rahbari R, Mitchell TJ, Maura F, Nangalia J, Tarpey PS, Brunner SF, Lee-Six H, Hooks Y, Moody S, Mahbubani KT, Jimenez-Linan M, Brosens JJ, Iacobuzio-Donahue CA, Martincorena I, Saeb-Parsy K, Campbell PJ, Stratton MR, The mutational landscape of normal human endometrial epithelium, Nature. 580 (2020) 640–646. 10.1038/s41586-020-2214-z. [DOI] [PubMed] [Google Scholar]
- [72].Hoang ML, Kinde I, Tomasetti C, McMahon KW, Rosenquist TA, Grollman AP, Kinzler KW, Vogelstein B, Papadopoulos N, Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing, Proc. Natl. Acad. Sci 113 (2016) 9846–9851. 10.1073/pnas.1607794113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA, Detection of ultra-rare mutations by next-generation sequencing, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 14508–14513. 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, Luquette LJ, Lee E, Park PJ, Walsh CA, Somatic mutation in single human neurons tracks developmental and transcriptional history, Science. 350 (2015) 94–98. 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, Yang P, Chittenden TW, Hatem NE, Ryu SC, Woodworth MB, Park PJ, Walsh CA, Aging and neurodegeneration are associated with increased mutations in single human neurons, Science. 359 (2018) 555–559. 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C, deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution, Genome Biol. 17 (2016) 1–11. 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].McGranahan N, Favero F, De Bruin EC, Birkbak NJ, Szallasi Z, Swanton C, Clonal status of actionable driver events and the timing of mutational processes in cancer evolution, Sci. Transl. Med 7 (2015) 1–12. 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O’Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C, Tracking the evolution of non-small-cell lung cancer, N. Engl. J. Med 376 (2017) 2109–2121. 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- [79].Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, An Y, Keam B, Kim DW, Heo DS, Kim YT, Kim JS, Kim SH, Lee JS, Lee SH, Park K, Ku JL, Jeon YK, Chung DH, Park PJ, Kim J, Kim TM, Ju YS, ClonalHistory & genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas, J. Clin. Oncol 35 (2017) 3065–3074. 10.1200/JCO.2016.71.9096. [DOI] [PubMed] [Google Scholar]
- [80].Conticello SG, The AID/APOBEC family of nucleic acid mutators, Genome Biol. 9 (2008). 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Conticello SG, Langlois MA, Yang Z, Neuberger MS, DNA Deamination in Immunity: AID in the Context of Its APOBEC Relatives, Adv. Immunol 94 (2007) 37–73. 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- [82].Yan S, He F, Gao B, Wu H, Li M, Huang L, Liang J, Wu Q, Li Y, Increased APOBEC3B predicts worse outcomes in lung cancer: A comprehensive retrospective study, J. Cancer. 7 (2016) 618–625. 10.7150/jca.14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xu L, Chang Y, An H, Zhu Y, Yang Y, Xu J, High APOBEC3B expression is a predictor of recurrence in patients with low-risk clear cell renal cell carcinoma, Urol. Oncol. Semin. Orig. Investig 33 (2015) 340.e1–340.e8. 10.1016/j.urolonc.2015.05.009. [DOI] [PubMed] [Google Scholar]
- [84].Serebrenik AA, Argyris P, Jarvis MC, Brown WL, Bazzaro M, Vogel RI, Erickson BK, Lee S-H, Goergen KM, Maurer MJ, Heinzen EP, Oberg AL, Huang Y, Hou X, Weroha SJ, Kaufmann SH, Harris RS, The DNA cytosine deaminase APOBEC3B is a molecular determinant of platinum responsiveness in clear cell ovarian cancer, Clin. Cancer Res (2020) clincanres.2786.2019. 10.1158/1078-0432.ccr-19-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS, Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic, J. Biol. Chem 288 (2013) 17253–17260. 10.1074/jbc.M113.458661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Caval V, Suspene R, Shapira M, Vartanian J, Wain-hobson S, A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3’ UTR enhances chromosomal DNA damage, Nat. Commun (2014) 1–7. 10.1038/ncomms6129. [DOI] [PubMed] [Google Scholar]
- [87].Shi K, Carpenter MA, Banerjee S, Shaban NM, Kurahashi K, Salamango DJ, McCann JL, Starrett GJ, Duffy JV, Demir Ö, Amaro RE, Harki DA, Harris RS, Aihara H, Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B, Nat. Struct. Mol. Biol 24 (2017) 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jalili P, Bowen D, Langenbucher A, Park S, Aguirre K, Corcoran RB, Fleischman AG, Lawrence MS, Zou L, Buisson R, Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots, Nat. Commun 11 (2020) 1–13. 10.1038/S41467-020-16802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sharma S, Patnaik SK, Thomas Taggart R, Kannisto ED, Enriquez SM, Gollnick P, Baysal BE, APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages, Nat. Commun 6 (2015) 1–15. 10.1038/ncomms7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SMD, Bohn MF, Schiffer CA, Brown WL, Harris RS, Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A, J. Biol. Chem 287 (2012) 34801–34808. 10.1074/jbc.M112.385161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, Perry MD, Nahal-Bose HK, Ouellette BFF, Li CH, Rheinbay E, Nielsen GP, Sgroi DC, Wu CL, Faquin WC, Deshpande V, Boutros PC, Lazar AJ, Hoadley KA, Louis DN, Dursi LJ, Yung CK, Bailey MH, Saksena G, Raine KM, Buchhalter I, Kleinheinz K, Schlesner M, Zhang J, Wang W, Wheeler DA, Ding L, Simpson JT, O’Connor BD, Yakneen S, Ellrott K, Miyoshi N, Butler AP, Royo R, Shorser SI, Vazquez M, Rausch T, Tiao G, Waszak SM, Rodriguez-Martin B, Shringarpure S, Wu DY, Demidov GM, Delaneau O, Hayashi S, Imoto S, Habermann N, Segre AV, Garrison E, Cafferkey A, Alvarez EG, Heredia-Genestar JM, Muyas F, Drechsel O, Bruzos AL, Temes J, Zamora J, Baez-Ortega A, Kim HL, Mashl RJ, Ye K, DiBiase A, lin Huang K, Letunic I, McLellan MD, Newhouse SJ, Shmaya T, Kumar S, Wedge DC, Wright MH, Yellapantula VD, Gerstein M, Khurana E, Marques-Bonet T, Navarro A, Bustamante CD, Siebert R, Nakagawa H, Easton DF, Ossowski S, Tubio JMC, De La Vega FM, Estivill X, Yuen D, Mihaiescu GL, Omberg L, Ferretti V, Sabarinathan R, Pich O, Gonzalez-Perez A, Taylor-Weiner A, Fittall MW, Demeulemeester J, Tarabichi M, Roberts ND, Van Loo P, Cortés-Ciriano I, Urban L, Park P, Zhu B, Pitkänen E, Li Y, Saini N, Klimczak LJ, Weischenfeldt J, Sidiropoulos N, Alexandrov LB, Rabionet R, Escaramis G, Bosio M, Holik AZ, Susak H, Prasad A, Erkek S, Calabrese C, Raeder B, Harrington E, Mayes S, Turner D, Juul S, Roberts SA, Song L, Koster R, Mirabello L, Hua X, Tanskanen TJ, Tojo M, Chen J, Aaltonen LA, Rätsch G, Schwarz RF, Butte AJ, Brazma A, Chanock SJ, Chatterjee N, Stegle O, Harismendy O, Bova GS, Gordenin DA, Haan D, Sieverling L, Feuerbach L, Chalmers D, Joly Y, Knoppers B, Molnár-Gábor F, Phillips M, Thorogood A, Townend D, Goldman M, Fonseca NA, Xiang Q, Craft B, Piñeiro-Yáñez E, Muñoz A, Petryszak R, Füllgrabe A, Al-Shahrour F, Keays M, Haussler D, Weinstein J, Huber W, Valencia A, Papatheodorou I, Zhu J, Fan Y, Torrents D, Bieg M, Chen K, Chong Z, Cibulskis K, Eils R, Fulton RS, Gelpi JL, Gonzalez S, Gut IG, Hach F, Heinold M, Hu T, Huang V, Hutter B, Jäger N, Jung J, Kumar Y, Lalansingh C, Leshchiner I, Livitz D, Ma EZ, Maruvka YE, Milovanovic A, Nielsen MM, Paramasivam N, Pedersen JS, Puiggròs M, Sahinalp SC, Sarrafi I, Stewart C, Stobbe MD, Wala JA, Wang J, Wendl M, Werner J, Wu Z, Xue H, Yamaguchi TN, Yellapantula V, Davis-Dusenbery BN, Grossman RL, Kim Y, Heinold MC, Hinton J, Jones DR, Menzies A, Stebbings L, Hess JM, Rosenberg M, Dunford AJ, Gupta M, Imielinski M, Meyerson M, Beroukhim R, Reimand J, Dhingra P, Favero F, Dentro S, Wintersinger J, Rudneva V, Park JW, Hong EP, Heo SG, Kahles A, Van Lehmann K, Soulette CM, Shiraishi Y, Liu F, He Y, Demircioğlu D, Davidson NR, Greger L, Li S, Liu D, Stark SG, Zhang F, Amin SB, Bailey P, Chateigner A, Frenkel-Morgenstern M, Hou Y, Huska MR, Kilpinen H, Lamaze FC, Li C, Li X, Li X, Liu X, Marin MG, Markowski J, Nandi T, Ojesina AI, Pan-Hammarström Q, Park PJ, Pedamallu CS, Su H, Tan P, Teh BT, Wang J, Xiong H, Ye C, Yung C, Zhang X, Zheng L, Zhu S, Awadalla P, Creighton CJ, Wu K, Yang H, Göke J, Zhang Z, Brooks AN, Fittall MW, Martincorena I, Rubio-Perez C, Juul M, Schumacher S, Shapira O, Tamborero D, Mularoni L, Hornshøj H, Deu-Pons J, Muiños F, Bertl J, Guo Q, Gonzalez-Perez A, Xiang Q, Bazant W, Barrera E, Al-Sedairy ST, Aretz A, Bell C, Betancourt M, Buchholz C, Calvo F, Chomienne C, Dunn M, Edmonds S, Green E, Gupta S, Hutter CM, Jegalian K, Jones N, Lu Y, Nakagama H, Nettekoven G, Planko L, Scott D, Shibata T, Shimizu K, Stratton MR, Yugawa T, Tortora G, VijayRaghavan K, Zenklusen JC, Townend D, Knoppers BM, Aminou B, Bartolome J, Boroevich KA, Boyce R, Buchanan A, Byrne NJ, Chen Z, Cho S, Choi W, Clapham P, Dow MT, Dursi LJ, Eils J, Farcas C, Fayzullaev N, Flicek P, Heath AP, Hofmann O, Hong JH, Hudson TJ, Hübschmann D, Ivkovic S, Jeon SH, Jiao W, Kabbe R, Kahles A, Kerssemakers JNA, Kim H, Kim J, Koscher M, Koures A, Kovacevic M, Lawerenz C, Liu J, Mijalkovic S, Mijalkovic-Lazic AM, Miyano S, Nastic M, Nicholson J, Ocana D, Ohi K, Ohno-Machado L, Pihl TD, Prinz M, Radovic P, Short C, Sofia HJ, Spring J, Struck AJ, Tijanic N, Vicente D, Wang Z, Williams A, Woo Y, Wright AJ, Yang L, Hamilton MP, Johnson TA, Kahraman A, Kellis M, Polak P, Sallari R, Sinnott-Armstrong N, von Mering C, Beltran S, Gerhard DS, Gut M, Trotta JR, Whalley JP, Niu B, Espiritu SMG, Gao S, Huang Y, Lalansingh CM, Teague JW, Wendl MC, Abascal F, Bader GD, Bandopadhayay P, Barenboim J, Brunak S, Carlevaro-Fita J, Chakravarty D, Chan CWY, Choi JK, Diamanti K, Fink JL, Frigola J, Gambacorti-Passerini C, Garsed DW, Haradhvala NJ, Harmanci AO, Helmy M, Herrmann C, Hobolth A, Hodzic E, Hong C, Isaev K, Izarzugaza JMG, Johnson R, Juul RI, Kim J, Kim JK, Komorowski Jan, Lanzós A, Larsson E, Lee D, Li S, Li X, Lin Z, Liu EM, Lochovsky L, Lou S, Madsen T, Marchal K, Martinez-Fundichely A, McGillivray PD, Meyerson W, Paczkowska M, Park K, Park K, Pons T, Pulido-Tamayo S, Reyes-Salazar I, Reyna MA, Rubin MA, Salichos L, Sander C, Schumacher SE, Shackleton M, Shen C, Shrestha R, Shuai S, Tsunoda T, Umer HM, Uusküla-Reimand L, Verbeke LPC, Wadelius C, Wadi L, Warrell J, Wu G, Yu J, Zhang J, Zhang X, Zhang Y, Zhao Z, Zou L, Lawrence MS, Raphael BJ, Bailey PJ, Craft D, Goldman MJ, Aburatani H, Binder H, Dinh HQ, Heath SC, Hoffmann S, Imbusch CD, Kretzmer H, Laird PW, Martin-Subero JI, Nagae G, Shen H, Wang Q, Weichenhan D, Zhou W, Berman BP, Brors B, Plass C, Akdemir KC, Bowtell DDL, Burns KH, Busanovich J, Chan K, Dueso-Barroso A, Edwards PA, Etemadmoghadam D, Haber JE, Jones DTW, Ju YS, Kazanov MD, Koh Y, Kumar K, Lee EA, Lee JJK, Lynch AG, Macintyre G, Markowetz F, Navarro FCP, Pearson JV, Rippe K, Scully R, Villasante I, Waddell N, Yang L, Yao X, Yoon SS, Zhang CZ, Bergstrom EN, Boot A, Covington K, Fujimoto A, Huang MN, Islam SMA, McPherson JR, Morganella S, Mustonen V, Ng AWT, Prokopec SD, Vázquez-García I, Wu Y, Yousif F, Yu W, Rozen SG, Rudneva VA, Shringarpure SS, Turner DJ, Xia T, Atwal G, Chang DK, Cooke SL, Faltas BM, Haider S, Kaiser VB, Karlić R, Kato M, Kübler K, Margolin A, Martin S, Nik-Zainal S, P’ng C, Semple CA, Smith J, Sun RX, Thai K, Wright DW, Yuan K, Biankin AV, Garraway L, Grimmond SM, Adams DJ, Anur P, Cao S, Christie EL, Cmero M, Cun Y, Dawson KJ, Dentro SC, Deshwar AG, Donmez N, Drews RM, Gerstung M, Ha G, Haase K, Jerman L, Ji Y, Jolly C, Lee J, Lee-Six H, Malikic S, Mitchell TJ, Morris QD, Oesper L, Peifer M, Peto M, Rosebrock D, Rubanova Y, Salcedo A, Sengupta S, Shi R, Shin SJ, Spiro O, Vembu S, Wintersinger JA, Yang TP, Yu K, Zhu H, Spellman PT, Weinstein JN, Chen Y, Fujita M, Han L, Hasegawa T, Komura M, Li J, Mizuno S, Shimizu E, Wang Y, Xu Y, Yamaguchi R, Yang F, Yang Y, Yoon CJ, Yuan Y, Liang H, Alawi M, Borozan I, Brewer DS, Cooper CS, Desai N, Grundhoff A, Iskar M, Su X, Zapatka M, Lichter P, Alsop K, Bruxner TJC, Christ AN, Cordner SM, Cowin PA, Drapkin R, Fereday S, George J, Hamilton A, Holmes O, Hung JA, Kassahn KS, Kazakoff SH, Kennedy CJ, Leonard CR, Mileshkin L, Miller DK, Arnau GM, Mitchell C, Newell F, Nones K, Patch AM, Quinn MC, Taylor DF, Thorne H, Traficante N, Vedururu R, Waddell NM, Waring PM, Wood S, Xu Q, deFazio A, Anderson MJ, Antonello D, Barbour AP, Bassi C, Bersani S, Cataldo I, Chantrill LA, Chiew YE, Chou A, Cingarlini S, Cloonan N, Corbo V, Davi MV, Duthie FR, Gill AJ, Graham JS, Harliwong I, Jamieson NB, Johns AL, Kench JG, Landoni L, Lawlor RT, Mafficini A, Merrett ND, Miotto M, Musgrove EA, Nagrial AM, Oien KA, Pajic M, Pinese M, Robertson AJ, Rooman I, Rusev BC, Samra JS, Scardoni M, Scarlett CJ, Scarpa A, Sereni E, Sikora KO, Simbolo M, Taschuk ML, Toon CW, Vicentini C, Wu J, Zeps N, Behren A, Burke H, Cebon J, Dagg RA, De Paoli-Iseppi R, Dutton-Regester K, Field MA, Fitzgerald A, Hersey P, Jakrot V, Johansson PA, Kakavand H, Kefford RF, Lau LMS, Long GV, Pickett HA, Pritchard AL, Pupo GM, Saw RPM, Schramm SJ, Shang CA, Shang P, Spillane AJ, Stretch JR, Tembe V, Thompson JF, Vilain RE, Wilmott JS, Yang JY, Hayward NK, Mann GJ, Scolyer RA, Bartlett J, Bavi P, Chadwick DE, Chan-Seng-Yue M, Cleary S, Connor AA, Czajka K, Denroche RE, Dhani NC, Eagles J, Gallinger S, Grant RC, Hedley D, Hollingsworth MA, Jang GH, Johns J, Kalimuthu S, Ben Liang S, Lungu I, Luo X, Mbabaali F, McPherson TA, Miller JK, Moore MJ, Notta F, Pasternack D, Petersen GM, Roehrl MHA, Sam M, Selander I, Serra S, Shahabi S, Thayer SP, Timms LE, Wilson GW, Wilson JM, Wouters BG, McPherson JD, Beck TA, Bhandari V, Collins CC, Fleshner NE, Fox NS, Fraser M, Heisler LE, Lalonde E, Livingstone J, Meng A, Sabelnykova VY, Shiah YJ, Van der Kwast T, Bristow RG, Ding S, Fan D, Li L, Nie Y, Xiao X, Xing R, Yang S, Yu Y, Zhou Y, Banks RE, Bourque G, Brennan P, Letourneau L, Riazalhosseini Y, Scelo G, Vasudev N, Viksna J, Lathrop M, Tost J, Ahn SM, Aparicio S, Arnould L, Aure MR, Bhosle SG, Birney E, Borg A, Boyault S, Brinkman AB, Brock JE, Broeks A, Børresen-Dale AL, Caldas C, Chin SF, Davies H, Desmedt C, Dirix L, Dronov S, Ehinger A, Eyfjord JE, Fatima A, Foekens JA, Futreal PA, Garred Ø, Giri DD, Glodzik D, Grabau D, Hilmarsdottir H, Hooijer GK, Jacquemier J, Jang SJ, Jonasson JG, Jonkers J, Kim HY, King TA, Knappskog S, Kong G, Krishnamurthy S, Lakhani SR, Langerød A, Larsimont D, Lee HJ, Lee JY, Lee MTM, Lingjærde OC, MacGrogan G, Martens JWM, O’Meara S, Pauporté I, Pinder S, Pivot X, Provenzano E, Purdie CA, Ramakrishna M, Ramakrishnan K, Reis-Filho J, Richardson AL, Ringnér M, Rodriguez JB, Rodríguez-González FG, Romieu G, Salgado R, Sauer T, Shepherd R, Sieuwerts AM, Simpson PT, Smid M, Sotiriou C, Span PN, Stefánsson ÓA, Stenhouse A, Stunnenberg HG, Sweep F, Tan BKT, Thomas G, Thompson AM, Tommasi S, Treilleux I, Tutt A, Ueno NT, Van Laere S, Van den Eynden GG, Vermeulen P, Viari A, Vincent-Salomon A, Wong BH, Yates L, Zou X, van Deurzen CHM, van de Vijver MJ, van’t Veer L, Ammerpohl O, Aukema S, Bergmann AK, Bernhart SH, Borkhardt A, Borst C, Burkhardt B, Claviez A, Goebler ME, Haake A, Haas S, Hansmann M, Hoell JI, Hummel M, Karsch D, Klapper W, Kneba M, Kreuz M, Kube D, Küppers R, Lenze D, Loeffler M, López C, Mantovani-Löffler L, Möller P, Ott G, Radlwimmer B, Richter J, Rohde M, Rosenstiel PC, Rosenwald A, Schilhabel MB, Schreiber S, Stadler PF, Staib P, Stilgenbauer S, Sungalee S, Szczepanowski M, Toprak UH, Trümper LHP, Wagener R, Zenz T, Hovestadt V, von Kalle C, Kool M, Korshunov A, Landgraf P, Lehrach H, Northcott PA, Pfister SM, Reifenberger G, Warnatz HJ, Wolf S, Yaspo ML, Assenov Y, Gerhauser C, Minner S, Schlomm T, Simon R, Sauter G, Sültmann H, Biswas NK, Maitra A, Majumder PP, Sarin R, Barbi S, Bonizzato G, Cantù C, Dei Tos AP, Fassan M, Grimaldi S, Luchini C, Malleo G, Marchegiani G, Milella M, Paiella S, Pea A, Pederzoli P, Ruzzenente A, Salvia R, Sperandio N, Arai Y, Hama N, Hiraoka N, Hosoda F, Nakamura H, Ojima H, Okusaka T, Totoki Y, Urushidate T, Fukayama M, Ishikawa S, Katai H, Katoh H, Komura D, Rokutan H, Saito-Adachi M, Suzuki A, Taniguchi H, Tatsuno K, Ushiku T, Yachida S, Yamamoto S, Aikata H, Arihiro K, ichi Ariizumi S, Chayama K, Furuta M, Gotoh K, Hayami S, Hirano S, Kawakami Y, Maejima K, Nakamura T, Nakano K, Ohdan H, Sasaki-Oku A, Tanaka H, Ueno M, Yamamoto M, Yamaue H, Choo SP, Cutcutache I, Khuntikeo N, Ong CK, Pairojkul C, Popescu I, Ahn KS, Aymerich M, Lopez-Guillermo A, López-Otín C, Puente XS, Campo E, Amary F, Baumhoer D, Behjati S, Bjerkehagen B, Futreal PA, Myklebost O, Pillay N, Tarpey P, Tirabosco R, Zaikova O, Flanagan AM, Boultwood J, Bowen DT, Cazzola M, Green AR, Hellstrom-Lindberg E, Malcovati L, Nangalia J, Papaemmanuil E, Vyas P, Ang Y, Barr H, Beardsmore D, Eldridge M, Gossage J, Grehan N, Hanna GB, Hayes SJ, Hupp TR, Khoo D, Lagergren J, Lovat LB, MacRae S, O’Donovan M, O’Neill JR, Parsons SL, Preston SR, Puig S, Roques T, Sanders G, Sothi S, Tavaré S, Tucker O, Turkington R, Underwood TJ, Welch I, Fitzgerald RC, Berney DM, De Bono JS, Cahill D, Camacho N, Dennis NM, Dudderidge T, Edwards SE, Fisher C, Foster CS, Ghori M, Gill P, Gnanapragasam VJ, Gundem G, Hamdy FC, Hawkins S, Hazell S, Howat W, Isaacs WB, Karaszi K, Kay JD, Khoo V, Kote-Jarai Z, Kremeyer B, Kumar P, Lambert A, Leongamornlert DA, Livni N, Lu YJ, Luxton HJ, Marsden L, Massie CE, Matthews L, Mayer E, McDermott U, Merson S, Neal DE, Ng A, Nicol D, Ogden C, Rowe EW, Shah NC, Thomas S, Thompson A, Verrill C, Visakorpi T, Warren AY, Whitaker HC, Zhang H, van As N, Eeles RA, Abeshouse A, Agrawal N, Akbani R, Al-Ahmadie H, Albert M, Aldape K, Ally A, Appelbaum EL, Armenia J, Asa S, Auman JT, Balasundaram M, Balu S, Barnholtz-Sloan J, Bathe OF, Baylin SB, Benz C, Berchuck A, Berrios M, Bigner D, Birrer M, Bodenheimer T, Boice L, Bootwalla MS, Bosenberg M, Bowlby R, Boyd J, Broaddus RR, Brock M, Brooks D, Bullman S, Caesar-Johnson SJ, Carey TE, Carlsen R, Cerfolio R, Chandan VS, Chen HW, Cherniack AD, Chien J, Cho J, Chuah E, Cibulskis C, Cope L, Cordes MG, Curley E, Czerniak B, Danilova L, Davis IJ, Defreitas T, Demchok JA, Dhalla N, Dhir R, Doddapaneni HV, El-Naggar A, Felau I, Ferguson ML, Finocchiaro G, Fong KM, Frazer S, Friedman W, Fronick CC, Fulton LA, Gabriel SB, Gao J, Gehlenborg N, Gershenwald JE, Ghossein R, Giama NH, Gibbs RA, Gomez C, Govindan R, Hayes DN, Hegde AM, Heiman DI, Heins Z, Hepperla AJ, Holbrook A, Holt RA, Hoyle AP, Hruban RH, Hu J, Huang M, Huntsman D, Huse J, Iacobuzio-Donahue CA, Ittmann M, Jayaseelan JC, Jefferys SR, Jones CD, Jones SJM, Juhl H, Kang KJ, Karlan B, Kasaian K, Kebebew E, Kim HK, Korchina V, Kundra R, Lai PH, Lander E, Le X, Lee D, Levine DA, Lewis L, Ley T, Li HI, Lin P, Linehan WM, Liu FF, Lu Y, Lype L, Ma Y, Maglinte DT, Mardis ER, Marks J, Marra MA, Matthew TJ, Mayo M, McCune K, Meier SR, Meng S, Mieczkowski PA, Mikkelsen T, Miller CA, Mills GB, Moore RA, Morrison C, Mose LE, Moser CD, Mungall AJ, Mungall K, Mutch D, Muzny DM, Myers J, Newton Y, Noble MS, O’Donnell P, O’Neill BP, Ochoa A, Park JW, Parker JS, Pass H, Pastore A, Pennell NA, Perou CM, Petrelli N, Potapova O, Rader JS, Ramalingam S, Rathmell WK, Reuter V, Reynolds SM, Ringel M, Roach J, Roberts LR, Robertson AG, Sadeghi S, Saller C, Sanchez-Vega F, Schadendorf D, Schein JE, Schmidt HK, Schultz N, Seethala R, Senbabaoglu Y, Shelton T, Shi Y, Shih J, Shmulevich I, Shriver C, Signoretti S, Simons JV, Singer S, Sipahimalani P, Skelly TJ, Smith-McCune K, Socci ND, Soloway MG, Sood AK, Tam A, Tan D, Tarnuzzer R, Thiessen N, Thompson RH, Thorne LB, Tsao M, Umbricht C, Van Den Berg DJ, Van Meir EG, Veluvolu U, Voet D, Wang L, Weinberger P, Weisenberger DJ, Wigle D, Wilkerson MD, Wilson RK, Winterhoff B, Wiznerowicz M, Wong T, Wong W, Xi L, Yau C, Zhang H, Zhang H, Zhang J, Pan-cancer analysis of whole genomes, Nature. 578 (2020) 82–93. 10.1038/S41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Supek F, Lehner B, Clustered Mutation Signatures Reveal that Error-Prone DNA Repair Targets Mutations to Active Genes, Cell. 170 (2017) 534–547.e23. 10.1016/j.cell.2017.07.003. [DOI] [PubMed] [Google Scholar]
- [93].Maciejowski J, De Lange T, Telomeres in cancer: Tumour suppression and genome instability, Nat. Rev. Mol. Cell Biol 18 (2017) 175–186. 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sakofsky CJ, Saini N, Klimczak LJ, Chan K, Malc EP, Mieczkowski PA, Burkholder AB, Fargo D, Gordenin DA, Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation, 2019. 10.1371/journal.pbio.3000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Adolph MB, Love RP, Feng Y, Chelico L, Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B, Nucleic Acids Res. 45 (2017) 11925–11940. 10.1093/nar/gkx832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Koning FA, Newman ENC, Kim E-Y, Kunstman KJ, Wolinsky SM, Malim MH, Defining APOBEC3 Expression Patterns in Human Tissues and Hematopoietic Cell Subsets, J. Virol 83 (2009) 9474–9485. 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS, Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction, Nucleic Acids Res. 38 (2010) 4274–4284. 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Covino DA, Gauzzi MC, Fantuzzi L, Understanding the regulation of APOBEC3 expression: Current evidence and much to learn, J. Leukoc. Biol 103 (2018) 433–444. 10.1002/JLB.2MR0717-310R. [DOI] [PubMed] [Google Scholar]
- [99].Cannataro VL, Gaffney SG, Sasaki T, Issaeva N, Grewal NKS, Grandis JR, Yarbrough WG, Burtness B, Anderson KS, Townsend JP, APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma, Oncogene. 38 (2019) 3475–3487. 10.1038/s41388-018-0657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Janahi EM, McGarvey MJ, The inhibition of hepatitis B virus by APOBEC cytidine deaminases, J. Viral Hepat 20 (2013) 821–828. 10.1111/jvh.12192. [DOI] [PubMed] [Google Scholar]
- [101].Smid M, Rodríguez-González FG, Sieuwerts AM, Salgado R, Prager-Van Der Smissen WJC, Van Der Vlugt-Daane M, Van Galen A, Nik-Zainal S, Staaf J, Brinkman AB, Van De Vijver MJ, Richardson AL, Fatima A, Berentsen K, Butler A, Martin S, Davies HR, Debets R, Van Gelder MEM, Van Deurzen CHM, Macgrogan G, Van Den Eynden GGGM, Purdie C, Thompson AM, Caldas C, Span PN, Simpson PT, Lakhani SR, Van Laere S, Desmedt C, Ringnér M, Tommasi S, Eyford J, Broeks A, Vincent-Salomon A, Futreal PA, Knappskog S, King T, Thomas G, Viari A, Langerød A, Børresen-Dale AL, Birney E, Stunnenberg HG, Stratton M, Foekens JA, Martens JWM, Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration, Nat. Commun 7 (2016) 1–9. 10.1038/ncomms12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Suspène R, Mussil B, Laude H, Caval V, Berry N, Bouzidi MS, Thiers V, Wain-Hobson S, Vartanian JP, Self-cytoplasmic DNA upregulates the mutator enzyme APOBEC3A leading to chromosomal DNA damage, Nucleic Acids Res. 45 (2016) 3231–3241. 10.1093/nar/gkx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bakhoum SF, Ngo B, Laughney AM, Cavallo J, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, Duran M, Pauli C, Shaw C, Chadalavada K, Rajasekhar VK, Genovese G, Venkatesan S, Birkbak NJ, Mcgranahan N, Lundquist M, Laplant Q, Healey JH, Elemento O, Chung CH, Powell SN, Lammerding J, Lee NY, Imielenski M, Nanjangud G, Pe D, Don W, Chromosomal instability drives metastasis through a cytosolic DNA response, Nat. Publ. Gr 553 (2018) 467–472. 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA, Mitotic progression following DNA damage enables pattern recognition within micronuclei, Nature. 548 (2017) 466–470. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP, CGAS surveillance of micronuclei links genome instability to innate immunity, Nature. 548 (2017) 461–465. 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, Kuang Y, Zhang Y, Gjini E, Zhang G, Tian T, Sen DR, Miao D, Imamura Y, Thai T, Piel B, Terai H, Aref AR, Hagan T, Koyama S, Watanabe M, Baba H, Adeni AE, Lydon CA, Tamayo P, Wei Z, Herlyn M, Barbie TU, Uppaluri R, Sholl LM, Sicinska E, Sands J, Rodig S, Wong KK, Paweletz CP, Watanabe H, Barbie DA, Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses, Nat. Med 24 (2018) 1143–1150. 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Raab M, Gentili M, De Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil AM, Manel N, Piel M, ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death, Science (80-. ). 352 (2016) 359–362. 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- [108].Vargas JD, Hatch EM, Anderson DJ, Hetzer MW, Vargas JD, Hatch EM, Anderson DJ, Hetzer MW, Vargas JD, Hatch EM, Anderson DJ, Hetzer MW, Transient nuclear envelope rupture during interphase in human cancer cells, Nucleus. 3 (2012) 88–100. 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yang Z, Maciejowski J, De Lange T, Nuclear Envelope Rupture Is Enhanced by Loss of p53 or Rb, Mol. Cancer Res (2017) 1579–1587. 10.1158/1541-7786.MCR-17-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, Greenberg RA, Discher DE, DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration, Curr. Biol 27 (2017) 210–223. 10.1016/j.cub.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Denais CM, Gilbert RM, Isermann P, McGregor AL, Te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J, Nuclear envelope rupture and repair during cancer cell migration, Science (80-. ). 352 (2016) 353–358. 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR, Mutational signatures associated with tobacco smoking in human cancer, Science (80-.). 354 (2016) 618–622. 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Münk C, Willemsen A, Bravo IG, An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals, BMC Evol. Biol 12 (2012). 10.1186/1471-2148-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Duggal NK, Malik HS, Emerman M, The Breadth of Antiviral Activity of Apobec3DE in Chimpanzees Has Been Driven by Positive Selection, J. Virol 85 (2011) 11361–11371. 10.1128/jvi.05046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sawyer SL, Emerman M, Malik HS, Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G, PLoS Biol. 2 (2004). 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Matsumoto T, Shirakawa K, Yokoyama M, Fukuda H, Sarca AD, Koyabu S, Yamazaki H, Kazuma Y, Matsui H, Maruyama W, Nagata K, Tanabe F, Kobayashi M, Shindo K, Morishita R, Sato H, Takaori-Kondo A, Protein kinase A inhibits tumor mutator APOBEC3B through phosphorylation, Sci. Rep 9 (2019) 1–12. 10.1038/S41598-019-44407-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Aynaud MM, Suspène R, Vidalain PO, Mussil B, Guétard D, Tangy F, Wain-Hobson S, Vartanian JP, Human tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A, J. Biol. Chem 287 (2012) 39182–39192. 10.1074/jbc.M112.372722. [DOI] [PMC free article] [PubMed] [Google Scholar]