

Abstract
Recently, immunotherapeutic modalities with engineered cells and monoclonal antibodies have been effective in treating several malignancies. Nucleic acid aptamers can serve as alternative molecules to design immunotherapeutic agents with high functional diversity. Here we report a synthetic prototype consisting of DNA aptamers that can activate the T cell receptor cluster of differentiation 3 (TCR-CD3) complex in cultured T cells. We show that the activation potential is similar to that of a monoclonal antibody (mAb) against TCR-CD3, suggesting potential for aptamers in developing efficacious synthetic immunomodulators. The synthetic prototype of anti-TCR-CD3ε, as described here, was designed using aptamer ZUCH-1 against TCR-CD3ε, generated by ligand-guided selection (LIGS). Aptamer ZUCH-1 was truncated and modified with nuclease-resistant RNA analogs to enhance stability. Several dimeric analogs with truncated and modified variants were designed with variable linker lengths to investigate the activation potential of each construct. Among them, a dimeric aptamer with dimensions approximately similar to those of an antibody showed the highest T cell activation, suggesting the importance of optimizing linker lengths in engineering functional aptamers. The observed activation potential of dimeric aptamers shows the vast potential of aptamers in designing synthetically versatile immunomodulators with tunable pharmacokinetic properties, expanding immunotherapeutic designs by using nucleic acid-based ligands such as aptamers.
Keywords: immunotherapy, nucleic acids, aptamers, SELEX, LIGS, dimeric aptamer, T cells, TCR complex, cell binding, T-cell activation
Graphical Abstract
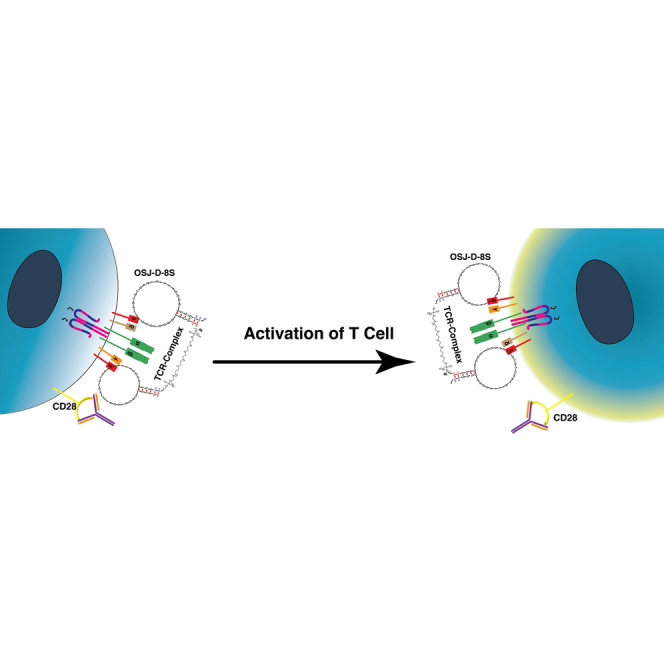
Freage et.al. elucidate the effect of linker length on a minimized variant of an aptamer modified with LNA and 2′O-methyl RNA bases against TCR-CD3ε.
Introduction
Recently, monoclonal antibodies (mAbs) have been used successfully to design immunotherapeutic modalities.1,2 These modalities are aimed at treating various types of malignancies. However, an increasing amount of evidence suggests that mAb-based treatments against immunological markers lead to various autoimmune diseases and liver-specific toxicity in humans.3,4 These side effects arise from prolonged circulation times of mAbs because of their relatively high molecular weight, and this characteristic has been a challenge in all types of mAb-based therapeutic strategies.5 To address this problem, nucleic acid aptamers, considered synthetic mAbs, could be alternative molecules to design immunotherapies.6,7 In recent years, the potential of aptamers as immunostimulatory agents has been explored. For example, dimeric aptamers have been developed against 4-1BB, expressed on the surface of activated mouse T cells, and OX40, a stimulatory molecule belonging to the tumor necrosis factor (TNF) superfamily of receptors. Both dimeric aptamers showed superior stimulatory activity compared with corresponding mAbs against the same targets.8,9 In another study, the immunomodulatory ability of anti-CD28 aptamers has been shown to have antagonistic or agonistic ability via dimerization.10
Although aptamers show tremendous potential for designing various scaffolds for immunomodulation, the challenges lie in identification of versatile aptamers against key receptors expressed on the immune cell surface. One such critical receptor, which plays a significant role in immune cell activation, is the T cell receptor cluster of differentiation 3 (TCR-CD3) complex, expressed on human T cells. The TCR-CD3 complex consists of multiple domains.11 Identifying versatile aptamers against such complexes is difficult using the conventional systematic evolution of ligand by exponential enrichment (SELEX) process.12,13 Recently, however, we successfully addressed this challenge by introducing a variant of SELEX called ligand-guided selection (LIGS).14, 15, 16, 17 The LIGS method was designed to identify aptamers against complex receptor proteins in their native conformation.14, 15, 16, 17 Using LIGS, we reported identification of a panel of aptamers that can specifically recognize the TCR-CD3 complex expressed in human cultured T cells and T cells obtained from healthy individuals.16 So far, the only known ligands against CD3-TCR are mAbs, and to the best of our knowledge, no alternative synthetic ligands are available against TCR-CD3 other than the aptamer reported by our lab.
In the last decade, there has been increased interest in truncating aptamers to variants that show enhanced activity.18,19 In fact, we and others have shown that truncation and dimerization of aptamers results in enhanced binding properties of molecules against cellular targets.18,20,21 However, most dimeric aptamers are confined to enhancing affinity and stability to develop targeting agents to deliver RNA therapeutic agents. Given the vast potential of aptamers as synthetic immunomodulators, here we explored an aptamer called ZUCH-1, which was identified via LIGS against TCR-CD3, as a T cell activator. We followed post-SELEX modification strategies to systematically truncate the aptamer and modified it with nuclease-resistant RNA analogs to enhance structural and nuclease stability. Finally, dimeric analogs were designed with variable lengths to introduce an agent that could activate TCR-CD3 for design of aptamer-based immunotherapies. Interestingly, we found that linker length between the aptamers play an essential role in activating TCR-CD3, opening novel avenues for designing functional dimeric aptamers against cell surface receptors.
Results
Truncation, Locked Nucleic Acid (LNA), and 2′-O-methyl (2′OMe) RNA Incorporation
Recently, we introduced an aptamer, ZUCH-1 (Figure 1A; Table S1) against the human CD3ε receptor in its native functional state using LIGS, a variant of SELEX.16 With an apparent dissociation constant in the nanomolar range, ZUCH-1 is suitable for designing aptamer-based T cell activation agents. Thus, we sought to investigate the functionality of dimeric aptamers designed based on ZUCH-1 to activate TCR-CD3ε.We strategically designed shorter variants of ZUCH-1 to further enhance the aptamer’s affinity without compromising its specificity. Then the optimized monomeric aptamer was modified further with nuclease-resistant RNA bases, followed by design of a functional and stable dimeric aptamer against TCR-CD3ε.
Figure 1.

Truncation, Modification, and Dimerization of ZUCH-1 Aptamer Variants
(A) The original ZUCH-1 aptamer. (B) The truncated OSJ-T1 variant, with 14 bases removed from the 5′ and 3′ terminals. (C) The stem-modified OSJ-T2 variant, with adenosine added at the 45th position. (D) The truncated OSJ-T3 variant having the most optimized length with 39 nt. (E) The LNA- and 2′OMe RNA-modified variant. (F) The dimerized variants of OSJ-T3-LNA-OMe with variable linkers to assess biological function. nS, number of spacers.
The base length of the initially reported parent aptamer ZUCH-1 is 76 nt. However, as demonstrated by several studies, not all nucleotides in an aptamer play a significant role in aptamer folding, target recognition, or function. Building on this assumption, we designed shorter variants of ZUCH-1. We first removed 14 nt from ZUCH-1 at the 5′ and 3′ ends of the aptamer sequence, resulting in the OSJ-T1 variant, as shown in Figure 1B. The calculated affinity of OSJ-T1 was similar to that of the full-length aptamer with a KD of 2.3 nM (Table 1; affinity curve in Figure 2A, i). The insignificant change in the affinity of OSJ-T1 suggests that bases removed from the terminal ends of the parent aptamer were nonessential and that their removal did not disrupt the functional fold critical for aptamer binding or recognition. However, OSJ-T1 consists of 48 nt with one unpaired thymine base in the stem. Typically, unpaired bases in the stem region of a nucleic acid fold can destabilize the stem.22 To investigate whether stabilization of the stem improved affinity, we used OSJ-T1 as a template and modified the aptamer structure in a series of steps. First, we introduced a base modification by adding a complementary adenine to the 45th base position in the stem of the OSJ-T1 sequence to remove the kink, resulting in variant OSJ-T2 (Figure 1C). However, with a calculated KD of 2.7 nM (Table 1), stabilization of the stem did not lead to higher affinity, suggesting that stem stabilization did not directly result in enhancing the overall functional structure of the aptamer or the aptamer’s ability to recognize TCR-CD3ε (Figure 2A, ii). Second, we further truncated the stem sequence of OSJ-T2 by removing a total of 10 bases from both terminal ends, which resulted in variant OSJ-T3, consisting of 39 nt with an affinity of 2.1 nM (Table1; Figures 1D and 2A, iii).
Table 1.
Affinities of Truncated, Modified, and Dimerized ZUCH-1 Parent Aptamers
| Name | Apparent affnity Jurkat-ATCC (nM) |
|---|---|
| OSJ-T1 | 2.3 |
| OSJ-T2 | 2.7 |
| OSJ-T3 | 2.1 |
| OSJ-T3-LNA-OMe | 1.7 |
| OSJ-dimer-2S | 0.5 |
| OSJ-dimer-4S | 0.3 |
| OSJ-dimer-6S | 0.4 |
| OSJ-dimer-8S | 1.7 |
Figure 2.

Analysis of Affinity and Specificity of Truncated Monomers and Modified Monomeric Variants against TCR-CD3ε Expressed on Jurkat E6.1 Cells
(A) Affinity curves for the truncated variants and modified monomer with unnatural RNA analogs were plotted, and the KD value of each was calculated using GraphPad Prism with a nonlinear fit, one site total, binding. (B) The specificity of the truncated variants and the modified variant against TCR-CD3ε-positive Jurkat E6.1 cells (B, row 1) and TCR-CD3ε-negative engineered Jurkat E6.1 cells (B, row 2). Row 3 presents the overall conclusion of three independent specific binding experiments of the variant aptamers using one-way ANOVA with a t test performed with GraphPad Prism. ∗∗∗:0.0004 < p < 0.0008, ∗∗∗∗p ≤ 0.0001.
We next focused on increasing the robustness of the OSJ-T3 variant. We did this by improving nuclease resistance and structural stabilization by incorporating two different types of RNA analogs: LNAs and 2′-OMe RNA bases (Figure 1E).23 LNA bases are ribonucleotide (RNA) analogs containing a methylene link between the 2′-oxygen and 4’-carbon of the ribose ring.23 This constraint on the ribose moiety results in locked C3′ endo hybridization that increases affinity toward its complementary base, resulting in enhancement of duplex stability.23,24 The 2′-OMe group (2′-OMe RNA) is a naturally occurring modification found in RNA that enhances affinity for RNA-DNA targets because of the preference of 2′-OMe-modified ribose sugars to adopt a C3′ endo conformation.25,26 These two unnatural bases have been shown to provide a significant improvement in duplex stability as a consequence of the high affinity of the two bases, leading to enhanced structural stabilization, stability against nucleases, and increased melting temperature.27, 28, 29 Additionally, previous reports have demonstrated that the high affinity of LNA and 2′OMe RNA analogs lead to superior duplex stability in vitro and in vivo.27 Nonetheless, aptamer post-SELEX modification with LNA and 2′-OMe RNA to enhance the stability of a DNA aptamer has never been attempted. We predicted that modification with LNA and 2′-OMe RNA might lead to a highly stable aptamer under physiological conditions. Because RNA analogs lead to A-form RNA conformations, we systematically modified the variant OSJ-T3 with LNA and 2′-OMe RNA bases to avoid introducing structural destabilization to OSJ-T3. To investigate the specific effects of LNA and 2′-OMe RNA bases on the aptamer’s affinity, three variants of the OSJ-T3 aptamer were synthesized: (1) OSJ-T3-LNA, in which first and second bases from the 5′ end were modified with corresponding LNA bases (Figures S1 and 2A); (2) OSJ-T3-OMe, in which the 40th and 41st bases from the 5′ end were modified with 2′-OMe RNA bases (Figures S1 and 2B); and (3) OSJ-T3-LNA-OMe, with modifications to the first and second bases from the 3′ terminal with LNA and the 40th and 41st bases from the 5′-terminal base with 2′OMe RNA bases, respectively (Figure 1E). As expected, aptamer OSJ-T3-LNA-OMe with dual modification provided heightened stabilization of the stem, leading to formation of a functional secondary structure of the aptamer with a KD value of 1.7 nM (Figure 2A, iv; Table 1).
Specificity
The specificity of all truncated variants and modified monomers with RNA analogs were analyzed using the TCR-CD3-positive cell line Jurkat E6.1 and double knockout (DKO) negative cell line based on Jurkat E6.1, which lacks the TCR-CD3 complex (Figure 2B). The DKO cell line, generated via a CRISPR-Cas9 system, lacked the TRAC gene that encodes the alpha constant chain of the TCR and the CD3E gene that encodes the CD3ε polypeptide.30 The specificity of all truncated aptamers (OSJ-T1, OSJ-T2, and OSJ-T3 variants; Figure 2B, i–iii) was tested against the positive cell line Jurkat E6.1 and negative cell line CD3ε DKO (Figure 2B, i–iii). All aptamers were specific toward Jurkat E6.1 cells compared with CD3ε DKO cells.
Interestingly, we observed that modification with LNA or 2′-OMe RNA at the terminal ends of the aptamer led to reduced specificity despite careful optimization of the folding conditions. Thus, the resulting aptamers OSJ-T3-LNA and OSJ-T3-OMe did not show specific binding toward TCR-CD3-expressing Jurkat cells (Figure S2). One explanation for the observed loss of specificity of these two variants might be the undesirable hybridization of the modified bases to the natural bases on the hairpin, leading to loss of the aptamer’s functional secondary structure. Undesirable fold formation because of base modifications of the molecular beacon has been observed before.31,32 However, the dually modified aptamer variant OSJ-T3-LNA-OMe, consisting of LNA and 2′OMe RNA base modifications at both terminals (Figure 1E), did maintain its specificity (Figure 2B, iv), suggesting that modifications at both ends did not interfere with the aptamer’s functional fold formation.
Effect of Different Linker Lengths on Affinity and Specificity
In addition to modification with unnatural bases, previous studies have also shown that the dimerization of aptamers can lead to increased binding affinity.18,20 We therefore dimerized the variant OSJ-T3-LNA-OMe with variable spacer lengths consisting of phosphoramidite 18 with six glycol linkages (Figure 1E). We designed four dimeric arrangements with different spacer numbers, starting with two spacer modifiers consisting of 12 polyethylene glycol (PEG) units (2S) as the shortest and eight spacer modifiers consisting of 48 PEG units (8S) as the longest. Two more dimers with six spacers (6S) and four spacers (4S) were also designed. We tested the binding affinity of all four dimers against Jurkat E6.1 cells over a range of concentrations for each aptamer (Figure 3A, i–iv). The calculated apparent KD values (Table 1) were determined using GraphPad Prism software by plotting the specific median fluorescence intensity against the concentration of the aptamer variant. The binding affinities of the OSJ-D-2S, OSJ-D-4S, OSJ-D-6S, and OSJ-D-8S dimers showed KD values of 0.5 nM, 0.3 nM, 0.4 nM, and 1.7 nM, respectively (Table 1). The affinities exhibited by 2S, 4S, and 6S linkers averaged a more than 2-fold increase over the monomeric aptamer OSJ-T3-LNA-OMe, with a KD value of 1.7 nM, whereas the affinity of 8S was similar to that of the monomers (see affinity curves in Figure 3A).
Figure 3.

Analysis of Affinity and Specificity of Dimeric Variants against TCR-CD3ε Expressed on Jurkat E6.1 Cells
(A) Affinity curves for dimerized variants with 2 spacers consisting of 12 ethylene glycol units (i), four spacers with 24 ethylene glycol units (ii), six spacers with 36 ethylene glycol units (iii), and eight spacers with 48 ethylene glycol units (iv). Affinity curves were plotted, and the KD value of each was calculated using GraphPad Prism with a nonlinear fit, one site total, binding. (B) Analysis of the specificity of dimerized aptamer variants against TCR-CD3ε-positive wild-type (WT) Jurkat E6.1 cells (B, row 1) and CRISPR-Cas9-engineered Jurkat E6.1 cells, knocking out (KO) TCR-CD3ε (B, row2). Row 3 shows the overall conclusion from three independent specificity analyses against KO and WT using one-way ANOVA and t test performed with GraphPad Prism. ∗∗∗∗p ≤ 0.0001.
We concluded that the observed increased affinities resulted from the spacing in the 2S, 4S, and 6S dimeric aptamer arrangements. Each TCR possesses two epsilon domains, each capable of binding an aptamer domain.11 The dimeric aptamers with 2, 4, and 6 spacers may not be sufficient in length and/or flexibility for each aptamer in the dimeric scaffold to bind two different domains. However, the increase in local concentration could provide a tethered aptamer partner in close proximity that would result in an increase in affinity. Besides, dimerization reduces the entropic penalty compared to that of monomeric aptamers, leading to highly favorable binding and, consequently, an increase in avidity in dimeric aptamer designs.33,34
On the other hand, OSJ-D-8S showed an affinity similar to that of the monomeric variant of aptamer OSJ-T3-LNA-OMe. The spacer length in the OSJ-D-8S dimer is approximately similar to the distance observed in the antibody arms of immunoglobulin G (IgG) antibodies. IgG are a broad family of antibodies, of which the OKT-3-specific anti-CD3ε mAb was used in LIGS experiments to discover anti-TCR aptamers. Previous studies with microscopic imaging using OKT3 suggest that the distance between the two binding domains on the IgG antibody is approximately 150–180 Å.35, 36, 37 In the OSJ-D-8S dimer, the calculated distance between the two aptamers is approximately 168 Å, which falls between the optimal distances reported for an antibody.34,38,39 Similar dimensions for the OSJ-D-8S and OKT3 suggest that the two aptamer binding sites might interact with the two epsilon domains of the TCR-CD3 complex, leading to similar binding properties for OSJ-D-8S and an IgG-based OKT3 antibody. We next analyzed the specificity of dimeric aptamers using TCR-CD3 knockout cells. The specificity is retained toward TCR-CD3 in all four constructs, suggesting that dimerization enhanced aptamer affinity while retaining its specificity (Figure 3B). Furthermore, we tested the specificity of all four dimer aptamers using the TCR-CD3-positive Jurkat E6.1 cell line and DKO negative cell line based on Jurkat E6.1 cells (Figure 3B, i–iv). All dimers (OSJ-D-2S, OSJ-D-4S, OSJ-D-6S, and OSJ-D-8S) bind to Jurkat E6.1 cells (Figure 3B, i–iv, rows 1 and 3) with high specificity compared with binding toward CD3 DKO cells (Figure 3B, i–iv, rows 2 and 3). Figure 3B, rows 1 and 2, highlights the specificity of the dimers, where we see significant binding of the dimers toward Jurkat E6.1 cells and non-significant binding toward DKO cells because of a small level of non-specific binding. Figure 3B, row 3, presents an overall comparison of three independent specificity analyses of Jurkat E6.1 and knockout cells. These findings demonstrate that dimerization of the aptamer increased the binding affinity of the aptamers toward Jurkat cells while maintaining binding specificity.
Next, the nuclease resistance of all dimeric aptamers (OSJ-D-2S, OSJ-D-4S, OSJ-D-6S, and OSJ-D-8S) was investigated. LNA and 2′OMe-RNA modifications showed improved nuclease resistance. All dimeric aptamers were stable in 10% fetal bovine serum (FBS) (Figure 4) after 4 and 6 h of incubation, suggesting that these modifications led to enhanced nuclease resistance of dimeric aptamers (Figure 4).
Figure 4.

Analysis of Nuclease Resistance of LNA- and 2′OMe RNA-Modified Dimerized Aptamers
(A and B) Analysis of 4-h (A) and 6-h (B) incubation.
Cell Activation from the 8S Dimer
Because the designed dimeric aptamer variants target the TCR-CD3 complex, we next investigated whether a newly designed dimeric variant could activate TCR-CD3 on Jurkat E6.1 cells. It is widely known that antibodies against CD3 can induce T cell activation, and this concept has been used extensively for designing immunotherapeutic molecules.40 Antigenic activation of T cells leads to significant cellular changes essential for the immune response. One of the earliest cell surface antigens expressed by T cells following activation is CD69, a membrane-bound, type II C-lectin receptor.41,42 CD69 expression is an early hallmark of lymphocyte activation based on its rapid appearance on the surface of the plasma membrane after stimulation.43 CD69 is not expressed on resting T cells; therefore, we detected activation of T cells via expression of CD69.43 Because activation of Jurkat E6.1 cells require at least two signals to be fully activated, we used the anti-CD28 antibody as a costimulation agent.44,45 Activation was quantified using the CD69-labeled antibody (CD69 mouse anti-human Cy5.5). In the absence of anti-CD3 and anti-CD28, resting T cells do not express CD69, confirming that both antibodies are required for activation and, hence, expression of CD69 on the surface of T cells (Figure S3). We then investigated whether the TCR-CD3 complex could be activated by all four dimeric aptamer variants while using an anti-CD28 antibody as the secondary costimulator (Figure 5). Jurkat E6.1 cells were incubated in cell suspension buffer for 2, 4, and 6 h in a surface-modified, polystyrene96-well, flat-bottom plate with 0.15 nmol of each dimeric aptamer and 25 μg/mL unlabeled anti-human CD28 at 37°C with 5% CO2. Following cell incubation of the dimeric aptamers, the cells were incubated with fluorescently labeled (Cy5.5) anti-CD69 antibody, followed by flow cytometry analysis. T cell activation was calculated by comparing the number of cells expressing the CD69 receptor compared with the number of cells not undergoing treatment (Figure S4). Cells expressed CD69 after 4 h of incubation with the OSJ-D-8S dimeric aptamer, suggesting that OSJ-D-8S is an efficient activator of T cells. However, the dimeric variants OSJ-D-2S, OSJ-D-4S, and OSJ-D-6S did not activate the cells and showed no expression of CD69, although, the OSJ-D-6S dimer did show a low level of activation after 6 h. The positive control was included with the CD3ε antibody for 2, 4, and 6 h in a surface-modified, polystyrene 96-well, flat-bottom plate (Figure S4). Notably, the OSJ-D-8S aptamer showed activation of Jurkat E6.1 cells similar to that of the anti-human CD3ε antibody in 6 h (Figure 5, third row)
Figure 5.

Investigation of the Biological Function of Dimeric Aptamer Variants
Shown is activation of TCR-CD3 in Jurkat E6.1 cells after incubation with 0.15nmol dimer aptamers (D-2S, D-4S, D-6S, and D-8S, respectively) and 10 μL of 25 μg/mL unlabeled costimulatory anti-human CD28 antibody. Activation was studied for 2, 4, and 6 h at 37°C with 5% CO2, and the appearance of the early activation marker CD69 receptor was monitored using flow cytometry with 5 μL of 100 μg/mL anti-CD69 mAb labeled with Cy5.5. The y axis of the plots represents the side scatter (SSC) of the cells. The x axis represents the fluorescence signal of the anti-CD69 antibody. The contour plots correspond to each variant with variable spacers at three time points. The percentages beyond the cutoff line (bottom right quadrant) in the contour plots correspond to activated cells with positive expression of the CD69 receptor.
To compare activation with the corresponding anti-CD3 antibody, a second positive control experiment was performed using a 96-well plate pre-coated with 10 μg/mL CD3 for 2 h at 37°C and then washed twice, followed by adding cells with CD28 antibody (Figures S4A and 4B). In addition, we performed multiple negative control experiments: (1) a dimeric aptamer without anti-CD28 antibody and without costimulation; (2) a randomized dimeric control with anti-CD28 antibody in which aptamer-based stimulation was absent but the costimulatory effect of anti-CD28 was present; and (3) an anti-CD28 antibody with each experimental setup incubated for 2, 4, and 6 h. All showed no T cell activation, suggesting that the activation observed for OSJ-D-8S was specific (Figures S5A–S5D).
Discussion
Immunotherapeutic strategies, which involve directing T cells toward diseased cells, such as cancer, have revolutionized drug development approaches.46,47 Currently, multiple immunotherapy approaches are being developed beyond cancer treatment. However, current approaches to developing immunotherapeutic agents are confined to engineering T cells to express disease-targeting antigen in the form of chimeric antigen receptor T (CAR-T) cells or the use of engineered fragments of mAbs in the form of bispecific antibodies, or use of mAbs against immune checkpoint inhibitors.46, 47, 48 Although all three strategies are successful in treating disease, particularly cancer, growing evidence suggests that immunotherapy approaches have challenges. These challenges involve slow pharmacokinetics of mAbs, leading to immune-related adverse effects (iRAEs), as well as the high cost of therapy development.49 This calls for low-cost, easily modifiable synthetic analogs with favorable pharmacokinetics properties, using alternative molecules to develop immunotherapeutic agents. In this regard, nucleic acid aptamers are promising alternative molecules. Aptamers are synthetic, easily modifiable, and have tunable pharmacokinetics.50,51 There are multiple examples already regarding the feasibility of designing multimeric aptamers as immunomodulators. For example, antagonistic aptamers have been developed against CTLA-4 and PD1, and agnostic aptamers have been developed against 4-1BB.52 However, the challenge of designing aptamer-based immunotherapeutic agents heavily relies on discovery of aptamers capable of recognizing critical receptors on the immune cell surface. In particular, the TCR-CD3complex is one of the most sought-after receptors to develop synthetic immunotherapeutic agents. We successfully identified DNA aptamers against TCR-CD3ε using the LIGS method we developed in our lab. Although we discovered multiple aptamers against TCR-CD3ε, the aptamer ZUCH-1 showed the highest affinity. However, ZUCH-1 is 76 nt in length, preventing design of functional scaffolds. Additionally, because natural nucleic acid bases are susceptible to nuclease degradation, it is necessary to modify aptamers consisiting of native bases with unnatural nucleic acid analogs. It has been widely reported that addition of unnatural nucleic acid analogs, such as LNA or OMe-RNA bases, enhances nuclease stability. Particularly, LNA and OMe base modifications to natural DNA have been reported to show higher human serum stability compared with native DNA aptamers. In this study, we successfully demonstrated that truncation followed by modification of unnatural nucleic acid analogs could further improve the parent aptamer without compromising its specificity but enhancing its affinity. Although improving the biostability of aptamers is an essential step when designing functional aptamers, an important goal of the current study is to explore the applicability of anti-TCR-CD3ε aptamers as T cell activators. Of all molecules tested, neither monomeric aptamers nor dimeric aptamers with shorter linkers activated TCR-CD3. However, the construct with eight spacers and dimensions similar to those of an antibody could activate TCR-CD3, suggesting that design of functional dimeric aptamer scaffolds is predominantly governed by linker length, particularly against TCR-CD3ε.
Similarly, the monovalent Fab fragment of the anti-TCR antibody only poorly activated TCR-CD3, whereas the bivalent antibody could effectively activate TCR-CD3, suggesting that the dimeric aptamer analog against TCR-CD3ε might have activation characteristics similar to those of an antibody against the same receptor. A number of mechanisms of TCR-CD3 activation have been proposed. Still, the precise molecular interactions that explain how bivalent ligands, such as mAbs or dimeric aptamers, activate TCR-CD3 have not yet been defined, as indicated by the abundance of multiple controversial models.53,54 However, it has been shown that conformational change is required for optimal T cell activation, suggesting that a dimeric analog with eight spacers may induce a conformational switch to activate TCR-CD3.
In conclusion, here we report development of the first synthetic prototype of an anti-TCR-CD3 ligand that was modified with LNA and 2′-methyl-RNA nucleic acid analogs. All constructs described here specifically recognize TCR-CD3 with high affinity. The observed differential activation properties suggest that the dimensions of the bivalent design play a crucial role in activation of the TCR complex. Future work will investigate activation of TCR-CD3 using T cells obtained from healthy volunteers and mechanistic investigation of TCR-CD3 activation by dimeric aptamers. To the best of our knowledge, so far no studies have reported synthetic anti-TCR ligands capable of activating T cells.
Materials and Methods
Cell Culture and Reagents
Jurkat clone E6.1 (T lymphocyte) cells were purchased from the American Type Culture Collection (ATCC). Jurkat DKO cells generated via CRISPR-Cas9 targeting the CD3E and TRAC genes were purchased from Synthego (Redwood City, CA, USA). All cell lines were cultured in HyClone RPMI 1640 medium (with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES] and L-glutamine) supplemented with 100 units/mL penicillin and streptomycin 1% (Corning), 1% minimum essential medium (MEM) non-essential amino acids (Gibco), and 10% FBS (heat-inactivated, Gibco). All cell lines were routinely evaluated on a flow cytometer (Becton Dickinson, FACScan) for expression of the CD marker using an anti-hCD3ε (Phycoerythrin [PE]-conjugated mouse IgG1, R&D Systems) antibody to authenticate the cell line.
DNA Synthesis
All DNA reagents needed to perform DNA synthesis were purchased from Glen Research or ChemGenes. All aptamers were chemically synthesized by attaching 5′ 6-FAM (6-Carboxyfluorescein) using standard solid-phase phosphoramidite chemistry on an ABI394 DNA synthesizer (Biolytics) using a 1-μmol scale. The monomer OSJ-T3-LNA was synthesized using locked analog phosphoramidite bases (Glen Research) to modify the first and second bases (5′ end) of the aptamer with LNA, whereas OSJ-T3-OMe was synthesized by using the 2′-OMe-Ac-C-CE and 2′-OMe-U-CE phosphoramidites (Glen Research) to modify the 40th and 41st bases (Glen Research), respectively. OSJ-T3-LNA-OMe was synthesized using the locked analog phosphoramidite to modify the first and second bases as well as OMe-Ac-C-CE and 2′-OMe-U-CE phosphoramidites to modify the 40th and 41st bases, respectively.
Two monomeric OSJ-T3-LNA-OMe molecules were tethered using the spacer phosphoramidite 18 (Glen Research) to form dimers. Two-, four-, six-, and eight-spacer repeats were used to design the OSJ-D-2S, OSJ-D-4S, OSJ-D-6S, and OSJ-D-8S dimers, respectively. Synthesized DNA sequences were de-protected according to the base modification employed and purified using high-performance liquid chromatography (HPLC) (Waters) equipped with a C-18 reverse-phase column (Phenomenex) and UV detector using 0.1 M Triethylammonium Acetate (TEAA) as the mobile phase. The random single-stranded DNA (ssDNA) molecule and the dimeric random controls were purchased from Integrated DNA Technologies.
Preparation of Solutions
After purification of all aptamers, the DNA stock solution concentrations were determined by using an ultraviolet-visible (UV-vis) spectrophotometer (Thermo Scientific). Sub-stock solutions of 10 μM were prepared for all aptamer molecules by dilution of each of the respective stock solutions with RPMI 1640 medium (with 25 mM HEPES and L-glutamine). The 10-μM solutions were then diluted further in RPMI 1640 medium (with 25 mM HEPES and L-glutamine) to prepare various working solutions. The random control molecules were prepared as described previously.
Cell Suspension Buffer (CSB)/ Binding Buffer
All binding assays were performed in CSB composed of HyClone RPMI 1640 medium (with 25 mM HEPES and L-glutamine) containing 200 mg/L tRNA (Sigma-Aldrich), 2 g/L bovine serum albumin (BSA; Fisher Scientific), and 200 mg/L salmon sperm DNA solution (Invitrogen).
Aptamer Folding Conditions
Prior to aptamer binding with the cells, the truncated aptamers prepared in HyClone RPMI 1640 medium were denatured for 5 min at 95°C and allowed to fold into their secondary structure for 45 min at 37°C. Dually modified and dimeric aptamers prepared in HyClone RPMI 1640 medium (with 25 mM HEPES and L-glutamine) were denatured for 10 min at 95°C followed by 15 min of fold interruption on ice and then 30 min at 37°C.
Cell Binding Assays
Affinity Assay
Jurkat E6.1 cells were prepared by washing three times with 3 mL HyClone RPMI 1640 medium (with 25 mM HEPES and L-glutamine) prior to aptamer binding to the cells. All aptamers and random sequences were prepared at an initial concentration of 250 nM from 10 μM sub-stock solutions. Truncated and dually modified aptamers were denatured and allowed to fold into their secondary structures as described previously. The truncated monomers were initially prepared at a concentration of 250 nM and serially diluted to achieve the desired final concentrations of 62.5 nM, 25 nM, 12.5 nM, 5 nM, 1 nM, and 0.5 nM in 150 μL (75 μL of aptamer in RPMI 1640 medium and 75 μL of 1.5 × 105 Jurkat E6.1 cells in CSB). Dually modified and OSJ-D-2S, OSJ-D-4S, and OSJ-D-6S dimers were initially prepared at a concentration of 25 nM and serially diluted to achieve the desired final concentrations of 12.5 nM, 6.25 nM, 3.13 nM, 1.6 nM, 0.78 nM, and 0.02 nM in 150 μL (75 μL of aptamer in RPMI 1640 medium and 75 μL of 1.5 × 105 Jurkat E6.1 cells in CSB). The OSJ-D-8S dimer was initially prepared at a concentration of 50 nM and serially diluted to achieve the desired final concentrations of 25 nM, 12.5 nM, 6.25 nM, 3.13 nM, 1.6 nM, 0.39 nM, and 0.07 nM in 150 μL (75 μL of aptamer in RPMI 1640 medium and 75 μL of 1.5 × 105 Jurkat E6.1 cells in CSB). Following this, aptamers and cells were incubated at 37°C for 1 h. After incubation, cells were washed twice with 2 mL RPMI 1640 medium and reconstituted in 250 μL RPMI 1640 medium. Binding of each aptamer was analyzed using flow cytometry by counting 5,000 events for a given concentration. As a positive control, Jurkat E6.1 cell lines were incubated with 5 μL of 25 μg/mL anti-hCD3ε antibody (PE-conjugated mouse IgG1, R&D Systems) or 2 μL of 200 μg/mL isotype control (PE mouse IgG1, κ, BD Biosciences) for 30 min on ice, followed by a one-time wash with 2 mL of RPMI 1640 medium and reconstitution in 250 μL RPMI 1640 medium. Binding events were monitored in FL1 Green (515–545 nm) for the aptamer and in FL2 Yellow (565–605 nm, 564–606 nm) for the antibody by counting 5,000 events using flow cytometry.
The final amount of aptamer used to plot the truncated aptamer affinity ranged from 0.075–9.375 pmol and 0.003–3.75 pmol for the dually modified OSJ-T3-LNA-OMe. The dimeric aptamer final amount ranged from 0.0117–3.75 pmol.
All experiments were done in triplicate, and the specific binding for each concentration was calculated using the equation
These specific binding values were then used to generate an affinity curve using GraphPad Prism. The KD values were calculated based on a fit using GraphPad Prism software (one site-specific binding) using the equation Y = Bmax × X / (KD + X), where X is the x axis, Y is the y axis, and Bmax is the maximum number of binding sites.
Specificity Assay
Cells were prepared by washing three times with HyClone RPMI 1640 medium (with 25 mM HEPES and L-glutamine) prior to aptamer binding to the cells. Truncated and dually modified aptamers were denatured and allowed to fold into their secondary structures as described previously. Specific binding was investigated by incubating 75 μL of 0.0375 nmol of each aptamer or a random control in RPMI 1640 medium with 75 μL of 1.5 × 105 Jurkat E6.1 cells or TCR-CD3ε DKO Jurkat cells in CSB, resulting in a concentration of 250 nM. Following incubation, the cells were washed twice with 2 mL of RPMI 1640 medium and reconstituted in 250 μL RPMI 1640 medium. Binding events were monitored in FL1 Green (515–545 nm) for the aptamer using flow cytometry by counting 5,000 events for each concentration. As a positive control, Jurkat E6.1 cells were incubated with 5 μL of 25 μg/mL anti-hCD3ε (PE-conjugated mouse IgG1, R&D Systems) antibody or 2 μL of 200 μg/mL isotype control (PE mouse IgG1, κ, BD Biosciences) for 30 min on ice. Cells were then washed once with 2 mL of RPMI 1640 medium and reconstituted in 250 μL RPMI 1640 medium. Binding events were monitored in FL2 Yellow (565–605 nm, 564–606 nm) for the antibody by counting 5,000 events using flow cytometry.
All affinity experiments were done in triplicate, and the specific binding percentages were calculated using the following equation:
Stability of Aptamers
To analyze the stability and nuclease resistance of the dimers, 20 μL of 0.15 nmol dimer aptamers were incubated for 4 and 6 h in RPMI 1640 medium with 10% FBS at 37°C with 5% CO2. At the end of each incubation time point, 1.2 μL of each aptamer was mixed with 8.8 μL of 1% Tris-borate-EDTA (TBE) buffer and 2 μL of cyber green loading dye to make a dilution of 1:10. Samples along with a low-molecular-weight ladder were loaded on a 4% agarose gel and run for 30 min at 120 V using Fisherbrand electrophoresis power supplies (FB3000Q).
Cell Activation
Jurkat E6.1 cells were washed three times with 3 mL RPMI 1640 medium (with 25 mM HEPES and L-glutamine) prior to activation of TCR-CD3 in Jurkat E6.1 cells. Following the cell wash, 2 × 105 Jurkat E6.1 cells were incubated with 0.15 nmol of dimer aptamers or a dimer random control along with 10 μL of 25 μg/mL unlabeled anti-human CD28 (BioXCells) in 150 μL CSB. The cells, dimer aptamers, and anti-human CD28 antibody were incubated for 2, 4, and 6 h in a surface-modified, polystyrene 96-well, flat-bottom plate at 37°C with 5% CO2. This was followed by transferring the solution from the 96-well flat-bottom plate to Ria Tubes 12 × 75 (Falcon round-bottom polystyrene tubes) to stain for the CD69 receptor using 5 μL (100 μg/mL) of PreCP-Cy 5.5 mouse anti-human CD69 (BD Biosciences). Staining was done for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 Red (663–687 nM) at each time point using flow cytometry by counting 5,000 events.
Positive Controls
Activation of Cells with Anti-CD3 and Anti-CD28 Antibodies
Positive control experiments were performed by incubating cells with anti-human CD3ε and anti-human CD28 antibodies. The cells were washed three times with 3 mL RPMI 1640 medium (with 25 mM HEPES and L-glutamine), and then 2 × 105 Jurkat E6.1 cells were incubated for 2, 4 and 6 h with 10 μL of 10 μg/mL unlabeled anti-human CD3ε and 10 μL of 25 μg/mL unlabeled anti-human CD28 antibodies at 37°C with 5% CO2 in a surface-modified, polystyrene, 96-well, flat-bottom plate. Cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 Red (663–687 nM) for each time point using flow cytometry by counting 5,000 events.
Plate Binding Assay
A 96-well plate was pre-coated with 50 μL (10 μg/mL) anti-CD3ε antibody for 2 h at 37°C with 5% CO2. Following pre-coating, the 96-well plate was washed three times with sterile 1× PBS to remove the unbound antibodies. The cells were then prepared by washing twice with 2 mL RPMI 1640 medium. 2 × 105 Jurkat E6.1 cells were added to the plate along with 10 μL of 25 μg/mL anti-CD28 antibody at 37°C with 5% CO2 for 2, 4, and 6 h. Following this, cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 for each time point using flow cytometry by counting 5,000 events.
Negative Controls
Untreated Cells in the Absence of Anti-CD3ε Antibody or Anti-CD28 Antibody
Cells were prepared by washing three times with 3 mL RPMI 1640 medium. 2 × 105 Jurkat E6.1 cells were incubated for 2, 4, and 6 h in 150 μL CSB in a surface-modified, polystyrene 96-well, flat-bottom plate at 37°C with 5% CO2. After incubation for 2, 4, and 6 h, the cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice. Following staining, cells were washed once with 2 mL RPMI 1640 medium and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 at each time point using flow cytometry by counting 5,000 events.
Evaluating the Decrease in Activation of TCR-CD3 in Jurkat E6.1 Cells in the Presence of the Dimers (6S, 8S) and in the Absence of Anti-CD28 Antibody
Cells were prepared by washing three times with 3 mL RPMI 1640 medium. 2 × 105 Jurkat E6.1 cells were incubated with OSJ-D-6S or OSJ-D-8S dimer in 150 μL CSB in the absence of anti-CD28 antibody at 37°C with 5% CO2 in a surface-modified, polystyrene 96-well, flat-bottom plate. Cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 at each time point using flow cytometry by counting 5,000 events.
Evaluating the Lack of Activation of TCR-CD3 in Jurkat E6.1 Cells Using the Random Dimer
Cells were prepared by washing three times with 3 mL RPMI 1640 medium. 2 × 105 Jurkat E6.1 cells were incubated for 2, 4, and 6 h with 0.15 nmol random dimer in 150 μL CSB in the presence of 10 μL of 25 μg/mL unlabeled anti-CD28 at 37°C with 5% CO2 in a surface-modified, polystyrene, 96-well, flat-bottom plate. Following incubation, cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 at each time point using flow cytometry by counting 5,000 events.
Evaluating the Lack of Activation of TCR-CD3 in Jurkat E6.1 Cells Using Only Anti-CD28 Antibodies
Cells were prepared by washing three times with 3 mL RPMI 1640 medium. 2 × 105 Jurkat E6.1 cells were incubated for 2, 4, and 6 h in the presence of 10 μL of 25 μg/mL unlabeled anti-CD28 antibody only at 37°C with 5% CO2 in a surface-modified, polystyrene 96-well, flat-bottom plate. Following this, cells were stained with 5 μL of 100 μg/mL CD69 mouse anti-human Cy5.5 antibody for 45 min on ice, washed once with 2 mL RPMI 1640 medium, and reconstituted in 250 μL RPMI 1640 medium. Expression of the activation marker CD69 was monitored in FL4 at each time point using flow cytometry by counting 5,000 events.
Author Contributions
D.J. conducted initial affinity assays, purified molecules. N.W. repeated the specificity and affinity assays and helped write the manuscript. L.F. conducted all experiments, including the activation assays, performed experimental design, and wrote the manuscript. P.R.M. conceived and designed the experiments, edited the manuscript, and, as the principle investigator, supervised the experiments and directed the project.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors are grateful for funding for this work by NIGMS grant SC1 GM122648.
References
- 1.Weiner L.M., Murray J.C., Shuptrine C.W. Antibody-based immunotherapy of cancer. Cell. 2012;148:1081–1084. doi: 10.1016/j.cell.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 3.Sandigursky S., Mor A. Immune-Related Adverse Events in Cancer Patients Treated With Immune Checkpoint Inhibitors. Curr. Rheumatol. Rep. 2018;20:65. doi: 10.1007/s11926-018-0770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belli C., Zuin M., Mazzarella L., Trapani D., D’Amico P., Guerini-Rocco E., Achutti Duso B., Curigliano G. Liver toxicity in the era of immune checkpoint inhibitors: A practical approach. Crit. Rev. Oncol. Hematol. 2018;132:125–129. doi: 10.1016/j.critrevonc.2018.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Hansel T.T., Kropshofer H., Singer T., Mitchell J.A., George A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010;9:325–338. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 6.Dollins C.M., Nair S., Sullenger B.A. Aptamers in immunotherapy. Hum. Gene Ther. 2008;19:443–450. doi: 10.1089/hum.2008.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pastor F. Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals (Basel) 2016;9:64. doi: 10.3390/ph9040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNamara J.O., Kolonias D., Pastor F., Mittler R.S., Chen L., Giangrande P.H., Sullenger B., Gilboa E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dollins C.M., Nair S., Boczkowski D., Lee J., Layzer J.M., Gilboa E., Sullenger B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008;15:675–682. doi: 10.1016/j.chembiol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastor F., Soldevilla M.M., Villanueva H., Kolonias D., Inoges S., de Cerio A.L., Kandzia R., Klimyuk V., Gleba Y., Gilboa E., Bendandi M. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids. 2013;2:e98. doi: 10.1038/mtna.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhns M.S., Badgandi H.B. Piecing together the family portrait of TCR-CD3 complexes. Immunol. Rev. 2012;250:120–143. doi: 10.1111/imr.12000. [DOI] [PubMed] [Google Scholar]
- 12.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 13.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 14.Zumrut H.E., Ara M.N., Fraile M., Maio G., Mallikaratchy P. Ligand-Guided Selection of Target-Specific Aptamers: A Screening Technology for Identifying Specific Aptamers Against Cell-Surface Proteins. Nucleic Acid Ther. 2016;26:190–198. doi: 10.1089/nat.2016.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zumrut H.E., Ara M.N., Maio G.E., Van N.A., Batool S., Mallikaratchy P.R. Ligand-guided selection of aptamers against T-cell Receptor-cluster of differentiation 3 (TCR-CD3) expressed on Jurkat.E6 cells. Anal. Biochem. 2016;512:1–7. doi: 10.1016/j.ab.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zumrut H.E., Batool S., Argyropoulos K.V., Williams N., Azad R., Mallikaratchy P.R. Integrating Ligand-Receptor Interactions and In Vitro Evolution for Streamlined Discovery of Artificial Nucleic Acid Ligands. Mol. Ther. Nucleic Acids. 2019;17:150–163. doi: 10.1016/j.omtn.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zumrut H., Yang Z., Williams N., Arizala J., Batool S., Benner S.A., Mallikaratchy P. Ligand-Guided Selection with Artificially Expanded Genetic Information Systems against TCR-CD3ε. Biochemistry. 2020;59:552–562. doi: 10.1021/acs.biochem.9b00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallikaratchy P.R., Ruggiero A., Gardner J.R., Kuryavyi V., Maguire W.F., Heaney M.L., McDevitt M.R., Patel D.J., Scheinberg D.A. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2011;39:2458–2469. doi: 10.1093/nar/gkq996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moccia F., Riccardi C., Musumeci D., Leone S., Oliva R., Petraccone L., Montesarchio D. Insights into the G-rich VEGF-binding aptamer V7t1: when two G-quadruplexes are better than one! Nucleic Acids Res. 2019;47:8318–8331. doi: 10.1093/nar/gkz589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batool S., Argyropoulos K.V., Azad R., Okeoma P., Zumrut H., Bhandari S., Dekhang R., Mallikaratchy P.R. Dimerization of an aptamer generated from Ligand-guided selection (LIGS) yields a high affinity scaffold against B-cells. Biochim. Biophys. Acta, Gen. Subj. 2019;1863:232–240. doi: 10.1016/j.bbagen.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimoto M., Shermane Lim Y.W., Hirao I. Molecular affinity rulers: systematic evaluation of DNA aptamers for their applicabilities in ELISA. Nucleic Acids Res. 2019;47:8362–8374. doi: 10.1093/nar/gkz688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meroueh M., Chow C.S. Thermodynamics of RNA hairpins containing single internal mismatches. Nucleic Acids Res. 1999;27:1118–1125. doi: 10.1093/nar/27.4.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaur H., Babu B.R., Maiti S. Perspectives on chemistry and therapeutic applications of Locked Nucleic Acid (LNA) Chem. Rev. 2007;107:4672–4697. doi: 10.1021/cr050266u. [DOI] [PubMed] [Google Scholar]
- 24.Vester B., Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 25.Iribarren A.M., Sproat B.S., Neuner P., Sulston I., Ryder U., Lamond A.I. 2′-O-alkyl oligoribonucleotides as antisense probes. Proc. Natl. Acad. Sci. USA. 1990;87:7747–7751. doi: 10.1073/pnas.87.19.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beijer B., Sulston I., Sproat B.S., Rider P., Lamond A.I., Neuner P. Synthesis and applications of oligoribonucleotides with selected 2′-O-methylation using the 2′-O-[1-(2-fluorophenyl)-4-methoxypiperidin-4-yl] protecting group. Nucleic Acids Res. 1990;18:5143–5151. doi: 10.1093/nar/18.17.5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallikaratchy P., Gardner J., Nordstrøm L.U., Veomett N.J., McDevitt M.R., Heaney M.L., Scheinberg D.A. A self-assembling short oligonucleotide duplex suitable for pretargeting. Nucleic Acid Ther. 2013;23:289–299. doi: 10.1089/nat.2013.0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun B.W., Babu B.R., Sørensen M.D., Zakrzewska K., Wengel J., Sun J.S. Sequence and pH effects of LNA-containing triple helix-forming oligonucleotides: physical chemistry, biochemistry, and modeling studies. Biochemistry. 2004;43:4160–4169. doi: 10.1021/bi036064e. [DOI] [PubMed] [Google Scholar]
- 29.Yan Y., Yan J., Piao X., Zhang T., Guan Y. Effect of LNA- and OMeN-modified oligonucleotide probes on the stability and discrimination of mismatched base pairs of duplexes. J. Biosci. 2012;37:233–241. doi: 10.1007/s12038-012-9196-4. [DOI] [PubMed] [Google Scholar]
- 30.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang C.J., Wang L., Wu Y., Kim Y., Medley C.D., Lin H., Tan W. Synthesis and investigation of deoxyribonucleic acid/locked nucleic acid chimeric molecular beacons. Nucleic Acids Res. 2007;35:4030–4041. doi: 10.1093/nar/gkm358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim Y., Yang C.J., Tan W. Superior structure stability and selectivity of hairpin nucleic acid probes with an L-DNA stem. Nucleic Acids Res. 2007;35:7279–7287. doi: 10.1093/nar/gkm771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meares C.F. The chemistry of irreversible capture. Adv. Drug Deliv. Rev. 2008;60:1383–1388. doi: 10.1016/j.addr.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian L., Heyduk T. Bivalent ligands with long nanometer-scale flexible linkers. Biochemistry. 2009;48:264–275. doi: 10.1021/bi801630b. [DOI] [PubMed] [Google Scholar]
- 35.Saphire E.O., Stanfield R.L., Crispin M.D., Parren P.W., Rudd P.M., Dwek R.A., Burton D.R., Wilson I.A. Contrasting IgG structures reveal extreme asymmetry and flexibility. J. Mol. Biol. 2002;319:9–18. doi: 10.1016/S0022-2836(02)00244-9. [DOI] [PubMed] [Google Scholar]
- 36.Saphire E.O., Parren P.W., Pantophlet R., Zwick M.B., Morris G.M., Rudd P.M., Dwek R.A., Stanfield R.L., Burton D.R., Wilson I.A. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X., Zhang L., Tong H., Peng B., Rames M.J., Zhang S., Ren G. 3D Structural Fluctuation of IgG1 Antibody Revealed by Individual Particle Electron Tomography. Sci. Rep. 2015;5:9803. doi: 10.1038/srep09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue Y., O’Mara M.L., Surawski P.P., Trau M., Mark A.E. Effect of poly(ethylene glycol) (PEG) spacers on the conformational properties of small peptides: a molecular dynamics study. Langmuir. 2011;27:296–303. doi: 10.1021/la103800h. [DOI] [PubMed] [Google Scholar]
- 39.Clausen-Schaumann H., Rief M., Tolksdorf C., Gaub H.E. Mechanical stability of single DNA molecules. Biophys. J. 2000;78:1997–2007. doi: 10.1016/S0006-3495(00)76747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H., Rhodes M., Wiest D.L., Vignali D.A. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 41.Ziegler S.F., Ramsdell F., Alderson M.R. The activation antigen CD69. Stem Cells. 1994;12:456–465. doi: 10.1002/stem.5530120502. [DOI] [PubMed] [Google Scholar]
- 42.Cibrián D., Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simms P.E., Ellis T.M. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin. Diagn. Lab. Immunol. 1996;3:301–304. doi: 10.1128/cdli.3.3.301-304.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molinero L.L., Fuertes M.B., Rabinovich G.A., Fainboim L., Zwirner N.W. Activation-induced expression of MICA on T lymphocytes involves engagement of CD3 and CD28. J. Leukoc. Biol. 2002;71:791–797. [PubMed] [Google Scholar]
- 45.Bashour K.T., Gondarenko A., Chen H., Shen K., Liu X., Huse M., Hone J.C., Kam L.C. CD28 and CD3 have complementary roles in T-cell traction forces. Proc. Natl. Acad. Sci. USA. 2014;111:2241–2246. doi: 10.1073/pnas.1315606111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inthagard J., Edwards J., Roseweir A.K. Immunotherapy: enhancing the efficacy of this promising therapeutic in multiple cancers. Clin. Sci. (Lond.) 2019;133:181–193. doi: 10.1042/CS20181003. [DOI] [PubMed] [Google Scholar]
- 48.Brischwein K., Parr L., Pflanz S., Volkland J., Lumsden J., Klinger M., Locher M., Hammond S.A., Kiener P., Kufer P. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007;30:798–807. doi: 10.1097/CJI.0b013e318156750c. [DOI] [PubMed] [Google Scholar]
- 49.Lemiale V., Meert A.P., Vincent F., Darmon M., Bauer P.R., Van de Louw A., Azoulay E., Groupe de Recherche en Reanimation Respiratoire du patient d’Onco-Hématologie (Grrr-OH) Severe toxicity from checkpoint protein inhibitors: What intensive care physicians need to know? Ann. Intensive Care. 2019;9:25. doi: 10.1186/s13613-019-0487-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Batool S., Bhandari S., George S., Okeoma P., Van N., Zümrüt H.E., Mallikaratchy P. Engineered Aptamers to Probe Molecular Interactions on the Cell Surface. Biomedicines. 2017;5:54. doi: 10.3390/biomedicines5030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keefe A.D., Pai S., Ellington A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soldevilla M.M., Villanueva H., Pastor F. Aptamers: A Feasible Technology in Cancer Immunotherapy. J. Immunol. Res. 2016;2016:1083738. doi: 10.1155/2016/1083738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwashima M. Kinetic perspectives of T cell antigen receptor signaling. A two-tier model for T cell full activation. Immunol. Rev. 2003;191:196–210. doi: 10.1034/j.1600-065x.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 54.Minguet S., Swamy M., Alarcón B., Luescher I.F., Schamel W.W. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:43–54. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]