

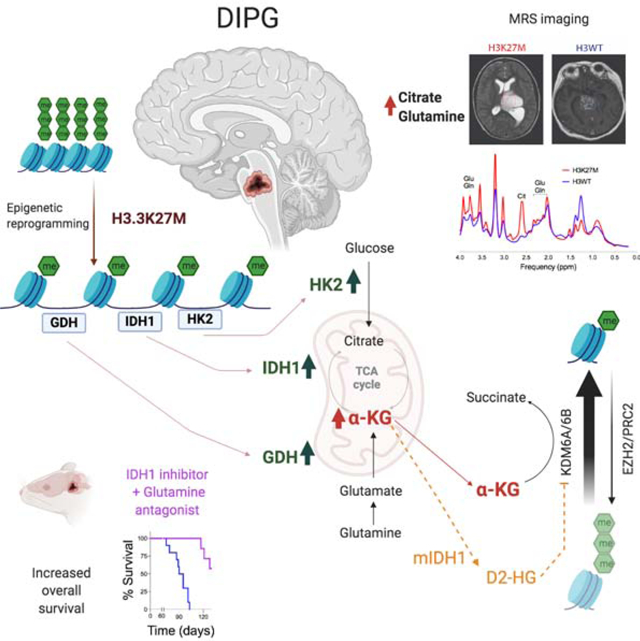
SUMMARY
H3K27M diffuse intrinsic pontine gliomas (DIPGs) are fatal and lack treatments. They mainly harbor H3.3K27M mutations resulting in H3K27me3 reduction. Integrated analysis in H3.3K27M-cells, tumors and in vivo imaging in patients showed enhanced glycolysis, glutaminolysis, and TCA cycle metabolism with high alpha-ketoglutarate (α-KG) production. Glucose and/or glutamine-derived α-KG maintained low H3K27me3 in H3.3K27M cells, and inhibition of key enzymes in glycolysis or glutaminolysis increased H3K27me3, altered chromatin accessibility, and prolonged survival in animal models. Previous studies have shown that mutant isocitrate-dehydrogenase (mIDH)1/2 glioma cells convert α-KG to D-2-hydroxyglutarate (D-2HG) to increase H3K27me3. Here we show H3K27M and IDH1 mutations are mutually exclusive and experimentally synthetic lethal. Overall, we demonstrate that H3.3K27M and mIDH1 hijack a conserved and critical metabolic pathway in opposing manners to maintain their preferred epigenetic state. Consequently, interruption of this metabolic/epigenetic pathway showed potent efficacy in pre-clinical models suggesting key therapeutic targets for much needed treatments.
Keywords: Histone mutation, DIPG, metabolism, epigenetics H3K27me3, glycolysis, glutaminolysis, α-KG, IDH mutation
Graphical Abstract
In brief
Chung et al. show that H3.3K27M mutation in DIPGs enhances glycolysis and TCA cycle metabolism to produce α-KG that is required to maintain a preferred epigenetic state of low H3K27me3. Inhibiting enzymes related to α-KG production increases H3K27me3 has anti-tumor activity in mouse models of DIPG.
INTRODUCTION
H3K27M-mutant gliomas, including diffuse intrinsic pontine gliomas (DIPGs), are lethal childhood brain tumors. DIPGs are inoperable due to their location within the pons/brainstem. Available treatment options, including chemo/radiotherapy, are ineffective, and over 90% of patients die within 1.5 years of diagnosis(Morales La Madrid et al., 2015). Over 80% of DIPGs harbor recurrent mutations in histone H3-H3F3A and HIST1H3B/C (~25%) where the lysine at position 27 is replaced by methionine (collectively H3K27M) (Fontebasso et al., 2013; Wu et al., 2012). H3K27M mutations result in a global H3K27me3 reduction via multiple mechanisms, including aberrant PRC2 interactions and hampered H3K27me3 spreading (Bender et al., 2013; Chan et al., 2013; Harutyunyan et al., 2019; Lewis et al., 2013; Stafford et al., 2018). Epigenetic approaches including increasing global H3K27me3 in H3K27M cells are a key therapeutic strategy leading to cell death of H3K27M cells in vitro and in vivo (Anastas et al., 2019; Grasso et al., 2015; Hashizume et al., 2014; Krug et al., 2019; Mohammad et al., 2017; Nagaraja et al., 2017; Piunti et al., 2017). While H3K27M mutations are thought to suppress neuroglial differentiation through deregulation of epigenetic modifications, it is not known what additional mechanisms, if any, drive pathogenesis.
Altered metabolism is a universal hallmark of cancer. Tumor cells take up and metabolize nutrients such as glucose and glutamine to support key anabolic processes and is actively driven by oncogenes and inactivated tumor suppressors (Venneti and Thompson, 2017). We hypothesized that H3K27M mutations rewire both glucose and glutamine metabolism. We also set out to determine if metabolic and epigenetic pathways are integrated in H3K27M tumors. Metabolic regulation of epigenetic modifications has been demonstrated in mIDH1/2 gliomas. Wild-type (WT) IDH1/2 converts isocitrate to α-KG that serves as a cofactor for α-KG-dependent dioxygenases, including DNA demethylases and Jumanji family of histone lysine demethylases (KDM) (Loenarz and Schofield, 2008). Gain-of-function mIDH1/2 metabolizes α-KG to D-2HG. D-2HG is structurally similar to α-KG and blocks its function, resulting in global hypermethylation of CpG islands (CpGi) and histone residues, including H3K27me3 (Duncan et al., 2012; Losman and Kaelin, 2013; Lu et al., 2012; Sasaki et al., 2012; Turcan et al., 2012). Based on this premise, we also hypothesized that global H3K27me3 levels are metabolically regulated in H3K27M cells and understanding these pathways could uncover therapeutic opportunities for treatment.
RESULTS
H3.3K27M exhibit enhanced glycolysis and TCA cycle metabolism compared to H3WT cells
We used an integrated approach to comprehensively determine metabolic alterations driven by H3.3K27M mutations using paired isogenic cell lines expressing H3.3K27M or H3.3WT, patient-derived tumor cell lines and tumor samples. We expressed either H3.3K27M or H3.3WT in immortalized mouse neuronal stem cells (NSC) (Johnson et al., 2010). H3.3K27M NSC exhibited global decreased H3K27me3 and an increase in the opposing mark H3K27ac without changes in global H3K36me3, H3K4me3, or H3K4me1 levels (Figures 1A, S1A). H3.3K27M NSC showed increased proliferation compared to H3.3WT cells (Figure S1B). H3K27me3 ChIP-seq confirmed global genomic H3K27me3 reduction in H3.3K27M versus H3.3WT NSC (Figures 1B). RNA-seq revealed many differentially regulated genes in H3.3K27M versus H3.3WT NSC, including downregulation of genes related to neuronal differentiation by GSEA (Figures 1C). Some of the most significantly up-regulated pathways in H3.3K27M NSC were related to carbohydrate metabolism and TCA-cycle regulation (Figure 1C). Similar results were observed in independent isogenic mouse cell lines expressing H3.3K27M or H3.3WT (Patel et al., 2019) (Figures S1C).
Figure 1. H3.3K27M show upregulation of glycolysis and TCA cycle metabolism compared to H3.3WT cells.
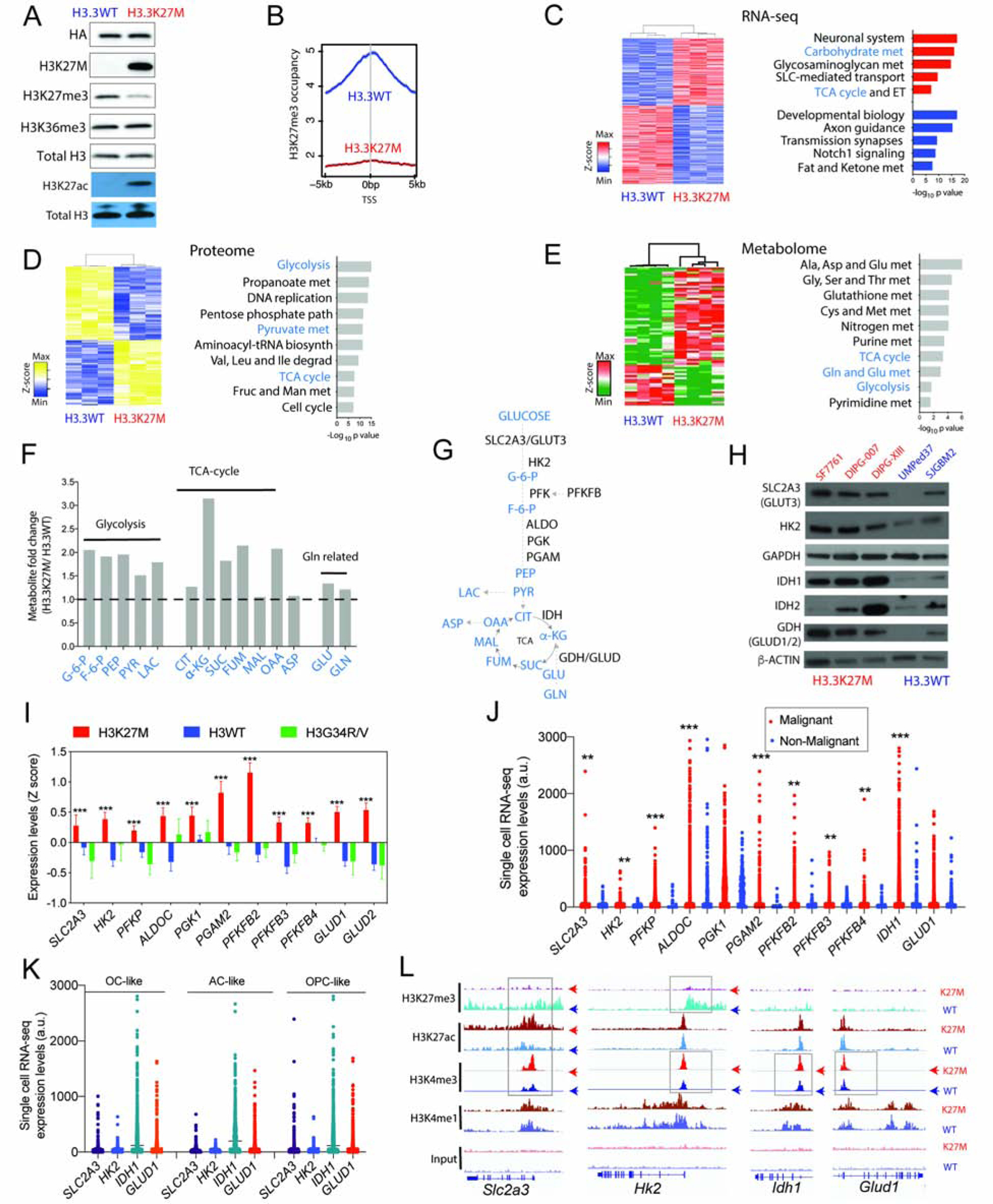
(A) Western blot (WB) of NSC stably transduced with H3.3K27M or H3.3WT for HA-tag, mutant-specific H3K27M, H3K27me3, H3K36me3, H3K27ac and total H3.
(B) H3K27me3 occupancy determined by ChIP-seq in genomic regions flanking transcriptional start site (+/− 5kb) in H3.3K27M and H3.3WT NSC.
(C) Heatmap and GSEA of differentially expressed genes determined by RNA-sequencing (RNA-seq) in H3.3WT and H3.3K27M NSC; up-(red) and down-(blue) regulated genes (n=3).
(D) Data from unbiased proteomics represented as heatmap and pathway analysis of differential protein levels in H3.3K27M versus H3.3WT NSC (n=3).
(E) Heatmap and enrichment analysis of differential metabolites in H3.3K27M versus H3.3WT NSC (n=4).
(F) Bar graph of key metabolites (H3.3K27M/H3.3WT NSC fold difference, Y-axis) related to glycolysis, TCA-cycle and glutaminolysis.
(G) Abbreviated schematic of glycolysis, TCA cycle and glutaminolysis (metabolites indicated in blue and ezymes in black).
(H) Representative WB of SLC2A3/GLUT3, HK2, IDH1, IDH2, GDH (GLUD1/2), GAPDH and β-ACTIN in H3.3K27M (SF7761, DIPG-007 and DIPG-XIII, red) and H3WT (UMPed37 and SJGBM2, blue) patient-derived cell lines.
(I) Bar graph of expression levels (Z-scores, Y-axis) of genes related to glycolysis and glutaminolysis in H3K27M (n=83), H3WT (n=101) and H3G34R/V (n=19) high-grade gliomas. Data plotted as mean ± SEM and analyzed by ANOVA, *** p<0.0001.
(J) Single cell RNA-seq expression scatter plot of genes related to glycolysis and glutaminolysis in H3K27M patient tumor samples.
(K) Single cell RNA-seq expression scatter plot of SLC2A3, HK2, IDH1 and GLUD1 in oligodendrocyte (OC)-like, astrocyte (AC)-like and oligodendrocyte precursor (OPC)-like cells in H3K27M patient tumor samples. Data in 1j–k derived from Filbin et al. 2018. (Filbin et al., 2018) and analyzed by non-parametric, 2-sided, unpaired, 2-tailed, Student’s t-test. ** p<0.001, *** p<0.0001.
(L) Representative Integrated Genomics Browser (IGV) tracks for H3K27me3, H3K27ac, H3K4me3, H3K4me1 and input in H3.3K27M and H3.3WT NSC at Slc2a3, Hk2, Idh1 and Glud1 gene loci. Top panel WT=H3.3WT, K27M= H3.3K27M NSC. Boxes indicate differential enrichment in H3.3K27M versus H3.3WT NSC.
(Ala, alanine; ALDOC, aldolase-C; Asp, aspartate; Cys, cystine; ET, electron transport; Fruc, fructose; F-6-P, fructose-6-phosphate; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; Glc, glucose; Gln, glutamine; G-6-P, glucose-6-phosphate; Glu, glutamate; Gly, glycine; Ile, isoleucine; Leu, leucine; Man, mannose; Met, methionine; PEP, phosphoenol pyruvate; PFKP, phosphofructokinase-platelet; PFKFB, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase; PGAM2, phospho-glyceromutase 2; PGK1, phosphoglycerate kinase1; Ser, serine; Val, valine)
WB are representative. Data are plotted as mean ± SD, n = biologic replicates
We used an unbiased approach by determining overall changes in the proteome and metabolome in H3.3K27M versus H3.3WT NSC. H3.3K27M showed differential regulation of 1069 (503 upregulated, 566 downregulated) proteins compared to H3.3WT NSC. Pathway analysis of the 503 upregulated proteins revealed glycolysis and TCA-cycle as top upregulated pathways (Figures 1D). Metabolite analysis revealed 78 metabolites that were differentially regulated (48 upregulated, 30 downregulated) between H3.3K27M and H3.3WT NSC (Figure 1E). Enrichment analysis of the 48 upregulated metabolites corroborated our proteomic findings to reveal upregulation of key glycolysis and TCA-cycle-associated metabolites including pyruvate, lactate and α-KG in H3.3K27M versus H3.3WT NSC (Figure 1E–G). We confirmed upregulation of key proteins related to glucose, glutamine and TCA-cycle metabolism in a panel of low-passage, patient-derived cell lines. The glucose transporter SLC2A3 (GLUT3), hexokinase 2 (HK2), IDH1, and glutamate dehydrogenase (GDH) were expressed at higher levels in H3.3K27M compared to H3WT human cell lines (Figures 1G–H). As controls, GAPDH, GOT1 and the cysteine/glutamate antiporter SLC7A11 were relatively unchanged in H3.3K27M versus H3WT cells (Figure 1H, S1D). Because glucose and glutamine are major carbon sources in glycolysis and the TCA cycle, we used isotope tracing with 13C-uniformly glucose and glutamine in vitro in both isogenic NSC and patient-derived cell lines. Isotope tracing revealed both increased glycolysis (Figures S1E–G) and glutaminolysis (Figures S1H–J) in H3.3K27M versus H3WT cells.
We evaluated the significance of our findings to patient tumors using multiple approaches. We queried data from a gene expression repository (PedcBioPortal) that included H3K27M (n=83), H3WT (n=101) and histone H3G34R/V hemispheric (n=19) pediatric high-grade gliomas (Mackay et al., 2017). Our analysis revealed increased expression of SLC2A3, several glycolytic enzymes including HK2, and GLUD1/2 (encoding GDH) in H3K27M versus either H3WT or H3G34R/V tumors (Figure 1I). We grouped high-grade pediatric gliomas, independent of H3K27M status, into high and low expression subgroups based on median expression value for SLC2A3/GLUT3, HK2, GLUD1 and PFKFB2. High expression tumors mainly localized to the pons/brainstem versus hemispheric regions, consistent with the primary location of H3K27M gliomas (Figure S2A). High expression of glycolysis-KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway genes, or HK2, or PFKFB2, was associated with poor overall prognosis independent of H3K27M status (Figure S2B–D). Expression levels of SLC2A3 and enzymes including HK2, IDH1 and GLUD1 were significantly higher in malignant compared to non-malignant cells in single-cell (sc) RNA-seq analyses from H3K27M tumors (Filbin et al., 2018) (Figure 1J). As observed in patient-derived cell lines, GAPDH, SCL7A11 and GOT1 levels were not significantly different between malignant and non-malignant cells (Figure S2E). Malignant cells in H3K27M tumors are heterogeneous and include highly proliferative oligodendrocyte precursor (OPC)-like cells along with oligodendrocyte (OC)-like and astrocyte (AC)-like cells (Filbin et al., 2018). All three subtypes of malignant cells showed high expression of IDH1 and GLUD1 compared to HK2 and SLC2A3 (Figure 1K).
To assess potential mechanisms of enhanced metabolism, we interrogated master regulators of glucose/glutamine metabolism including activation of mTor, N-Myc, C-Myc and Hif-1α. Western blotting did not reveal elevation of these factors, including Hif-1α in H3.3K27M versus H3.3WT NSC (Figure S2F–G). H3K27M tumor cells exhibit various epigenetic alterations at several gene promoters that can regulate gene expression. These include global reduction in H3K27me3, elevated H3K27ac levels and enrichment of the activating mark H3K4me3 (including bivalent H3K4me3/H3K27me3 at neurodevelopment-related gene promoters, Figure S2H) (Bender et al., 2013; Harutyunyan et al., 2019; Krug et al., 2019; Larson et al., 2019; Piunti et al., 2017). ChIP-seq analyses showed that promoters for Slc2a3, Hk2, Glud1 and Idh1 were enriched for the activation mark H3K4me3 and excluded H3K27me3 in H3.3K27M versus H3.3WT NSC (Figure 1L). Slc2a3 was also enriched for H3K27ac in H3.3K27M compared to H3.3WT NSC (Figure 1L). H3K4me1-enriched sites (enhancers) did not show changes near these gene loci (Figure 1L). Consistent with our control Western blot data (Figures 1H, S1C), Gapdh, Got1, and Slc7a11 did not show differences in enrichment between H3.3K27M and H3.3WT NSC for any of these marks (Figure S2I). The histone H3 mark enrichments at Slc2a3 in our NSC model system also aligned with earlier ChIP-seq studies in three human H3K27M DIPG cell lines (Piunti et al., 2017) (Figure S2J). Our data from multiple models including H3.3K27M versus H3.3WT isogenic cells, patient tumor cell lines, bulk and single cell RNA-seq analyses demonstrate that H3K27M cells show enhanced glycolysis and TCA-cycle metabolism.
H3K27M show higher glutamine and citrate levels in vivo compared to H3WT-midline gliomas
We used non-invasive, in vivo magnetic resonance spectroscopy (MRS) imaging to assess metabolite levels in patients with high-grade midline-gliomas in a retrospective, and blinded manner in 15 patients. As observed in midline-gliomas, 73% (11/15) were histopathologically confirmed to be H3K27M (n=11), while 27% (4/15) were H3WT midline gliomas (Figure 2A). Metabolites including glutamine (Gln), glutamate (Glu), myoinositol (mI), glycine (Gly), glucose (Glc), choline (Cho), citrate (Cit), Alanine (Ala) and Lactate (Lac) were assessed in all tumors. H3K27M midline gliomas contained significantly higher levels of citrate (p=0.0029) and glutamine (p=0.0227) compared to H3WT tumors (Figure 2B–C). Overall, in vivo MRS-imaging data paralleled our gene expression, protein and metabolite data derived from cell lines and tumor tissues.
Figure 2. In vivo Magnetic resonance spectroscopy (MRS) imaging in patients demonstrate higher glutamine and citrate levels in H3K27M compared to H3WT midline-gliomas.
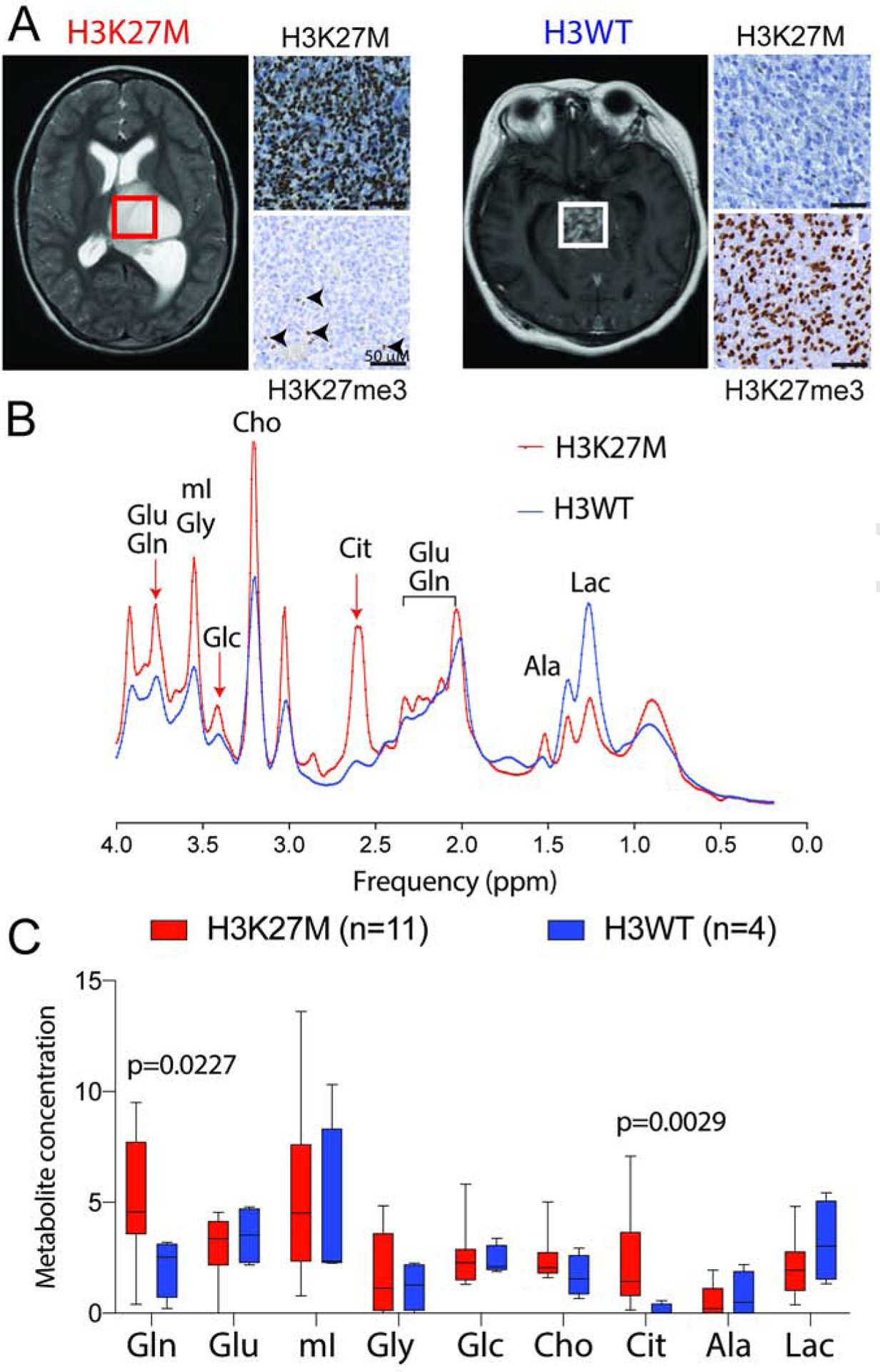
(A) Representative axial MRI images from H3K27M (thalamic/brainstem) and H3WT (brainstem) patients, with corresponding mutant-specific H3K27M and H3K27me3 (arrows, positive internal control) immunostaining on the right. Regions of interest (ROI) within the tumor, where MRS spectra were quantified, are indicated as boxes.
(B) Representative in vivo MRS spectra [TE (echo time) = 35 ms, TR (repetition time) = 2 s;] derived from H3K27M (red) and H3WT (blue) midline gliomas. Arrows indicate defined peaks for specific metabolites.
(C) MRS Quantification of indicated metabolite levels from H3K27M (n=11, red) and H3WT (n=4, blue) patients with midline-gliomas. Box plots show median and interquartile range, whiskers represent the highest and lowest observations, and were analyzed by 2-sided, non-parametric, unpaired, 2-tailed, t-test.
Gln=glutamine, Glu=glutamate, mI=myoinositol, Gly=glycine, Glc=glucose, Cho=choline, Cit=citrate, Ala=alanine, Lac=lactate.
Heterogeneous regulation of global H3K27me3 levels by glutamine and glucose metabolism in H3.3K27M cells
From our metabolomic studies, we noted that α-KG levels were higher in H3.3K27M compared to H3.3WT cells (Figure 1F). Histone KDMs, including the H3K27 demethylases KDM6A/6B use α-KG as a crucial co-factor, metabolizing it to succinate (Suc), while demethylating H3K27me3 (Figure 3A) (Loenarz and Schofield, 2008). A high α-KG/Suc ratio favors H3K27me3 demethylation (Carey et al., 2015), and H3.3K27M cells showed high α-KG/Suc ratios (Figure S3A). We therefore hypothesized that α-KG could regulate H3K27me3 levels in H3.3K27M cells. α-KG can be derived from both glutamine and glucose metabolism. Glucose carbons that enter the TCA cycle to generate citrate/isocitrate can give rise to α-KG via enzymatic activity of IDH1/2, while GDH metabolizes glutamine-derived glutamate to α-KG (Figure 3A).
Figure 3. Heterogeneous regulation of global H3K27me3 levels by glutamine and glucose metabolism in H3.3K27M cells.
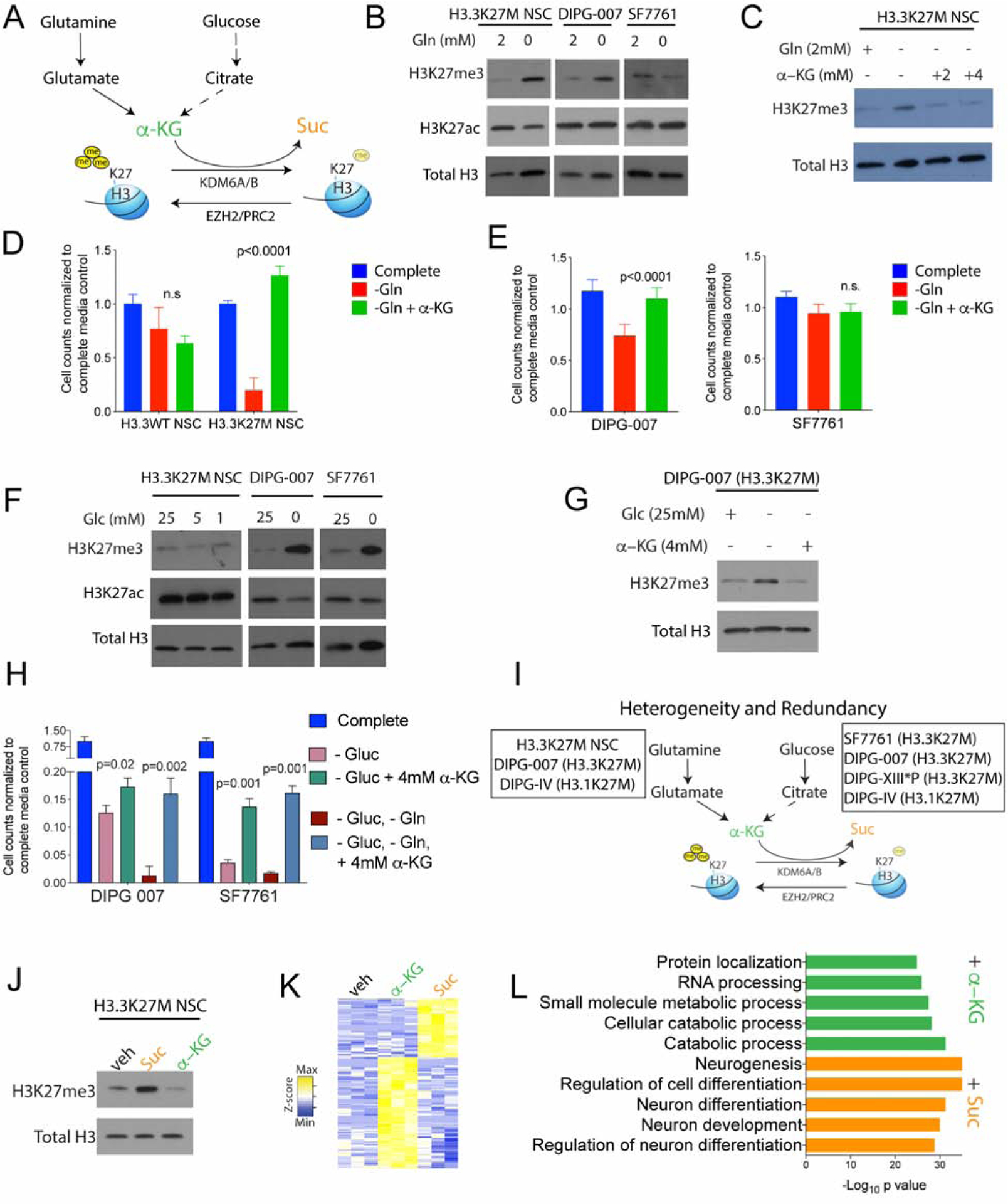
(A) Schematic depicting α-KG generation from glutamine and glucose metabolism: α-KG can promote H3K27me3 demethylation by serving as a critical co-factor for the H3K27 demethylases KDM6A/6B and is metabolized to succinate (Suc) during this reaction.
(B) H3.3K27M cells (H3.3K27M NSC, DIPG-007 and SF7761) were grown in complete or glutamine (Gln)-depleted media. Representative WB demonstrate changes in global H3K27me3 and H3K27ac in relation to total H3.
(C) Representative WB of H3.3K27M NSC showing alterations in global H3K27me3 levels in relation to total H3 in Gln-depleted media with/without 2 or 4mM cell-permeable α-KG.
(D–E) Cell counts (Y-axis) on Gln withdrawal with/without 4 mM cell-permeable α-KG in H3.3WT and H3.3K27M NSC (d), DIPG-007 and SF7761 (e) cells (n=4).
(F) H3.3K27M cells (H3.3K27M NSC, DIPG-007 and SF7761) were grown in complete or glucose (Glc)-depleted media. Representative WB demonstrates changes in global H3K27me3 and H3K27ac and total H3.
(G) Representative WB of H3.3K27M DIPG-007 cells showing changes in H3K27me3 levels in relation to total H3 in Glc-deprived media with/without 4 mM cell-permeable α-KG.
(H) Cell counts (Y-axis) in H3.3K27M DIPG-007 and SF7761 cells upon withdrawal of Glc, Gln or both with/without 4 mM cell-permeable α-KG (n=4).
(I) Schematic indicating heterogeneity and redundancy in the regulation of global H3K27me3 by glutamine (H3.3K27M NSC, DIPG-007 and DIPG-IV) and glucose (SF7761, DIPG-007, DIPG-XIII*P and DIPG-IV) metabolism in H3.3 and H3.1 K27M cell lines (see figure S3 for other cell lines).
(J) H3.3K27M NSC were treated with vehicle or 4 mM cell-permeable Suc or α-KG. Representative WB illustrating H3K27me3 in relation to total H3 levels.
(K–L) Heatmap of differentially regulated genes (K) and GSEA analysis of upregulated genes (L) in H3.3K27M NSC treated with α-KG or Suc (4 mM, n=3).
All experiments were conducted after 4 days in culture. WB are representative; n indicates biologic replicates; Data plotted as mean ± SD and analyzed by ANOVA.
To test if glutamine metabolism regulates H3K27me3 levels we used a panel of cells, including H3.3K27M NSC and patient-derived H3.3K27M and H3.1K27M cells. Glutamine deprivation from cell culture media resulted in increased H3K27me3 levels in H3.3K27M NSC, DIPG-007 and DIPG-IV cells but not SF7761 and DIPG-XIII*P cells (Figure 3B and S3A–N). This effect in H3K27M NSC was rescued by addition of cell-permeable α-KG (Figure 3C). Glutamine withdrawal did not significantly impact proliferation of H3.3WT NSC, but it inhibited that of H3.3K27M NSC, an effect reversed by downstream metabolites α-KG or glutamate (Figures 3D, S3B–C). In human cell lines, DIPG-007 cells were more sensitive to glutamine withdrawal than SF7761 cells and showed a partial decrease in proliferation that was reversed by α-KG (Figure 3E).
We next tested whether glucose metabolism regulated H3K27me3. Glucose withdrawal did not alter H3K27me3 levels in H3.3K27M and H3.3WT NSC, but it increased H3K27me3 levels in DIPG-007, SF7761, DIPG-IV and DIPG-XIII*P cells (Figure 3F, S3D–L). This effect was observed as early as 24 hours after glucose withdrawal and was rescued by α-KG in DIPG-007 cells (Figures 3G, S3E–F). Moreover, partial glucose withdrawal was sufficient to increase H3K27me3 levels in SF7661 but not in DIPG-007 cells (Figure S3G). This observation was borne out in cell viability studies, where maximal reduction in DIPG-007 cell numbers required both glucose and glutamine withdrawal but was achieved in SF7761 cells on glucose withdrawal alone (Figure 3H). Moreover, α-KG partially rescued viability on glucose withdrawal in both DIPG-007 and SF7761 cells but not in H3.3K27M NSC (Figures 3H, S3H). Glucose and glutamine withdrawal did not alter H3K27me3 levels in H3WT SJGBM2 cells (Figure S3I).
Overall, the impact of glutamine and glucose withdrawal on H3K27me3 levels showed both heterogeneity and redundancy amongst the tested cell lines (Figure 3I). Our data suggest that irrespective of the metabolic pathway used, α-KG is critical for maintaining low H3K27me3 in H3.3K27M cells. We verified this observation by adding cell-permeable α-KG to increase the α-KG/Suc ratio or cell-permeable Suc to lower the α-KG/Suc ratio in H3.3K27M NSC. Global H3K27me3 levels were further lowered on α-KG addition but were increased on Suc treatment (Figures 3J and S3M). As anticipated, addition of Suc to H3.3K27M NSC lowered proliferation while α-KG or the upstream metabolite glutamate increased proliferation that was abrogated by the H3K27 demethylase KDM6A/6B inhibitor GSK-J4 (Figure S3N). To determine the effects of α-KG and Suc on gene expression, we performed RNA-seq on H3.3K27M NSC treated with vehicle or with α-KG or Suc. Compared to vehicle treated controls, Suc induced pathways related to neuronal differentiation (consistent with increased H3K27me3), whereas α-KG induced gene expression pathways related to proliferation (Figures 3K–L).
GDH, HK2 and IDH1 inhibition increases H3K27me3 and suppresses proliferation of H3.3K27M cells
Having established that some H3K27M cells are reliant on glutamine to maintain low H3K27me3, we next sought to determine whether suppressing glutamine metabolism in these cells could impact global H3K27me3 levels and cell proliferation. We targeted GDH (Figure 4A) with two independent shRNAs, which increased H3K27me3 and slowed proliferation (Figures 4B–C). The glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) (Lemberg et al., 2018) increased H3K27me3 levels in glutamine-dependent H3.3K27M NSC and DIPG-007 but not in glucose-independent SF7761 cells (Figure 4D). Moreover, glutaminase (GLS) inhibitors CB-839 and BPTES increased H3K27me3 levels in DIPG-007 cells (Figures S4A–B). DON treatment in vivo, significantly suppressed growth compared to vehicle treated animals in H3.3K27M NSC xenografts (Figures 4E, S4C).
Figure 4. Inhibiting GDH and HK2 in vitro and in vivo increases global H3K27me3 levels and suppresses proliferation of H3.3K27M cells.
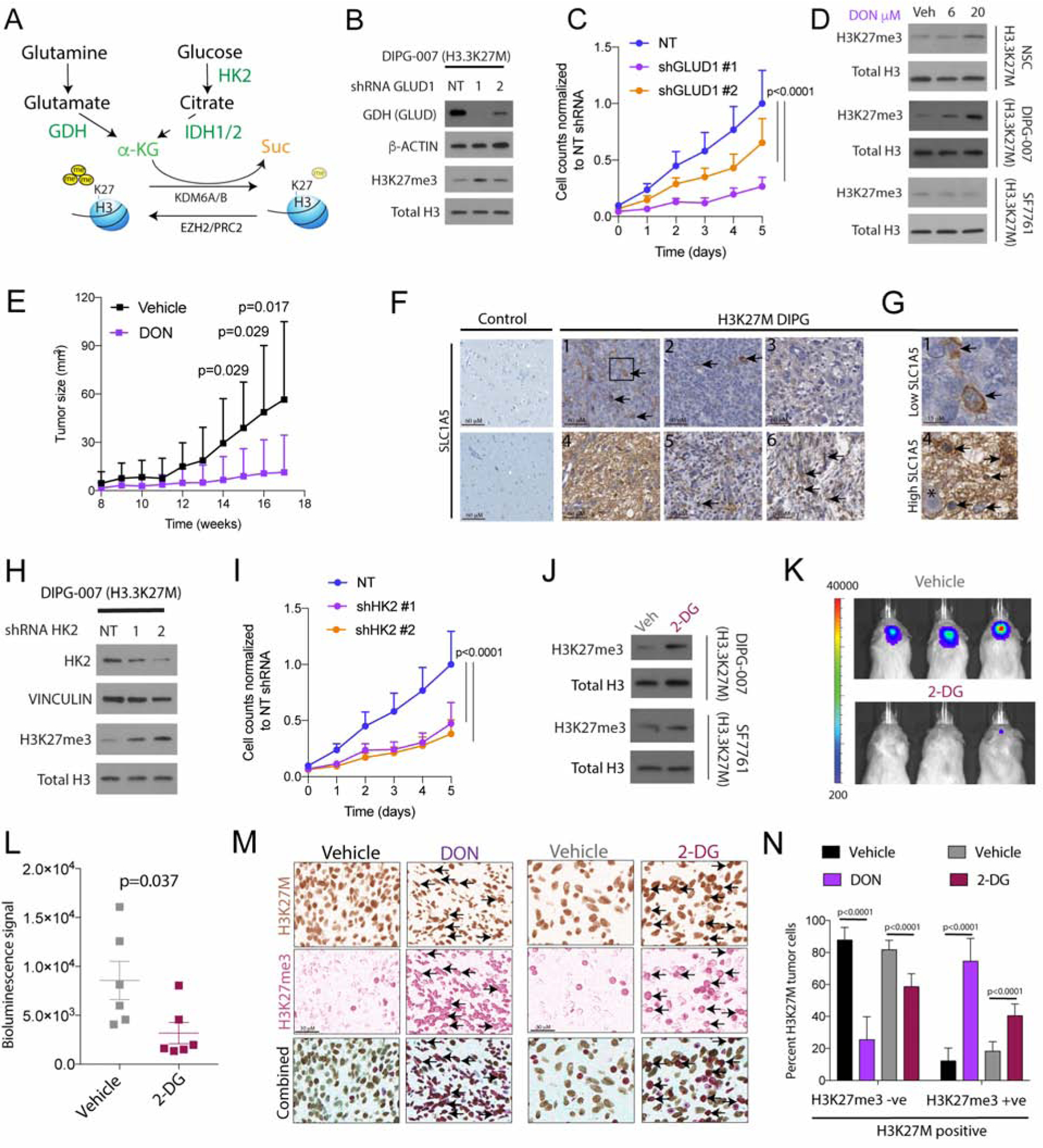
(A) Schematic of key enzymes related to α-KG generation in glutamine (GDH metabolizes glutamate to α-KG) and glucose (HK2, first and irreversible enzyme in glycolysis and IDH1 metabolizes isocitrate to α-KG) metabolic pathways.
(B–C) Representative WB of GDH, β-ACTIN, H3K27me3 and total H3 in DIPG-007 stably transduced with non-targeted (NT) or 2 independent GLUD1 (GDH) shRNAs (b). Proliferation of cells from 4b (normalized cell counts, Y-axis) plotted against time (days, X-axis) in (NT, light blue) or 2 independent GLUD1 shRNAs (orange and purple) (c, n=3).
(D) H3.3K27M cells (NSC, DIPG-007 and SF7761) cells were treated with vehicle (Veh), 6 or 20 μM of the glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) for 4 days. Representative WB of H3K27me3 and total H3 levels.
(E) Tumor volume (mm3, Y-axis) of H3.3K27M NSC xenografted into the flanks in mice treated with DON (purple, 1mg/kg, i.p., every other day for 6 weeks) or vehicle (black, n=10).
(F–G) Representative IHC images (f, scale bar=60μM) for the glutamine transporter SLC1A5 in control brains (n=6) or H3.3K27M DIPG tumor samples (n=6, #1–6). Insets from #1 and #4 shown on the right (g, scale bar, 15μM; Asterisk shows neuron surrounded by SLC1A5 positive tumor cells).
(H–I) Representative WB of HK2, VINCULIN, H3K27me3 and total H3 in DIPG-007 stably transduced with non-targeted (NT) or 2 independent HK2 shRNAs (h). Proliferation of cells from 4h (normalized cell counts, Y-axis) plotted against time (days, X-axis) in (NT, light blue) or 2 independent HK2 shRNAs (orange and purple) (i, n=3).
(J) H3.3K27M DIPG-007 and SF7761 cells were treated with vehicle (Veh) or 25 mM 2-DG for 2 days. Representative WB of H3K27me3 and total H3 levels.
(K–L) Representative bioluminescent images of mice with pontine H3.3K27M DIPG-007 orthotopic xenografts treated i.p. with vehicle or 2-DG (k, 500mg/kg, every other day for 3 weeks, n=6) and bioluminescent signal quantification (Y-axis, l).
(M) Representative single color or double color overlay IHC images from Vehicle/DON (from 4e) or vehicle/2-DG (from 4k) treated tumor samples stained with combined mutant-specific H3K27M (brown) and H3K27me3 (red) antibodies. Arrows indicate tumor cells that are positive for both H3K27M and H3K27me3.
(N) Quantification of H3K27M positive tumor cells that were negative or positive for H3K27me3 (from 4m) in vehicle (n=3, black)/DON (n=3, purple) and Vehicle (n=5, gray)/2-DG (n=5, maroon) treated animals (all biologic replicates).
Data plotted as mean ± SD and analyzed by ANOVA (4c, i and n) or 2-sided, unpaired, 2-tailed, Student’s t-test (4e and l); n indicates biologic replicates.
Cancer cells can take up glutamine via the transporter SLC1A5 and metabolize it to glutamate by GLS. Both SLC1A5 and GLS were elevated in glutamine-dependent DIPG-007 compared to non-dependent SF7761 cells (Figure S4D). GSK3α/β phosphorylation (pGSK3α/β) drives glutamine dependency in cancer cells (Momcilovic et al., 2018). Accordingly, glutamine sensitive DIPG-007, but not glutamine resistant SF7761, showed elevated pGSK3α/β (Figure S4D). To determine if this heterogeneity was mirrored in patient H3K27M DIPGs, we assessed SLC1A5 expression by immunohistochemistry (IHC) in tumor samples (n=6). Similar to cell lines, SLC1A5 expression was heterogeneous across H3.3K27M DIPGs (Figures 4F–G, S4E–F). Within tumors with low overall SLC1A5 levels, individual tumor cells still expressed high SLC1A5 levels (Figures 4G, arrows). Moreover, higher expression of SLC1A5 was associated with a poor prognosis in pediatric high-grade gliomas (Figure S4G). Patient-derived samples mirrored our cell culture results, suggesting that there is marked heterogeneity in glutamine metabolism in H3K27M DIPGs.
We next targeted HK2, which was elevated in H3.3K27M cells and tumor samples (Figures 1H–J). Two independent HK2 shRNAs increased H3K27me3 levels and suppressed proliferation in DIPG-007 cells (Figures 4H–I). Similarly, pharmacological inhibition with 2-deoxy-D-glucose (2-DG), an HK2 inhibitor, raised H3K27me3 levels in both DIPG-007 and SF7761 cells (Figure 4J). Other azole HK2 inhibitors (Agnihotri et al., 2019) produced a similar effect (Figure S4H–I). We took advantage of the blood-brain barrier (BBB) penetrability of 2-DG (Pardridge et al., 1982) and found it suppressed tumor growth in DIPG-007 orthotopic xenografts compared to vehicle treatment (Figures 4K–L). Furthermore, GLUD1 and HK2 knockdown significantly lowered α-KG/Suc ratios (Figure S4J).
We wanted to assess if inhibiting glutamine or glucose metabolism increased H3K27me3 levels in H3.3K27M tumors in vivo. To specifically examine changes in global H3K27me3 in H3K27M mutant cells, we combined IHC of mutant-specific H3K27M and H3K27me3 and compared results in vehicle versus DON or 2-DG treated animals. Both DON and 2-DG treated tumors exhibited significantly increased H3K27me3 levels in H3.3K27M mutant cells compared to vehicle treated animals in vivo (Figures 4M–N, S4K).
IDH1/2 enzymes generate α-KG from citrate-derived isocitrate (Figure 4A). H3K27M cells in vitro, and in vivo MRS in patient tumors, showed higher citrate levels in compared to H3WT (Figures 1F, 2A–C, S1F). IDH1 protein levels were higher in H3K27M compared to H3WT tumor samples (Figure 5A). IDH1 knockdown with three independent siRNAs increased H3K27me3 levels (Figure 5B). Similarly, shRNA-mediated IDH1 knockdown increased H3K27me3 levels, suppressed cell growth (Figures 5B–C) lowered α-KG/Suc ratios (Figure S5A). We also tested pharmacologically inhibiting IDH1 using a panel of recently described small molecules designed to covalently inhibit WT-IDH1 (WT-IDH1i) (Jakob et al., 2018). Of these, compound 13 was found to be the most effective WT-IDH1i (IC50~14 nM), while the inactive analogue, compound 18 (enone analogue), did not inhibit WT-IDH1 and served as control (Figure S5B). WT-IDH1i 13, but not the inactive compound 18, increased H3K27me3 levels and lowered proliferation of H3.3K27M but not of H3WT cells (Figures 5D–F and S5C–E). These data together suggest that inhibition of GDH, HK2 and IDH1 lower α-KG/Suc ratios, increases H3K27me3 and suppresses proliferation of H3.3K27M cells.
Figure 5. Inhibiting IDH1 increases H3K27me3 levels and is toxic, and GDH, HK2 and IDH1 knockdown results in altered chromatin accessibility at gene loci related to neuroglial differentiation.
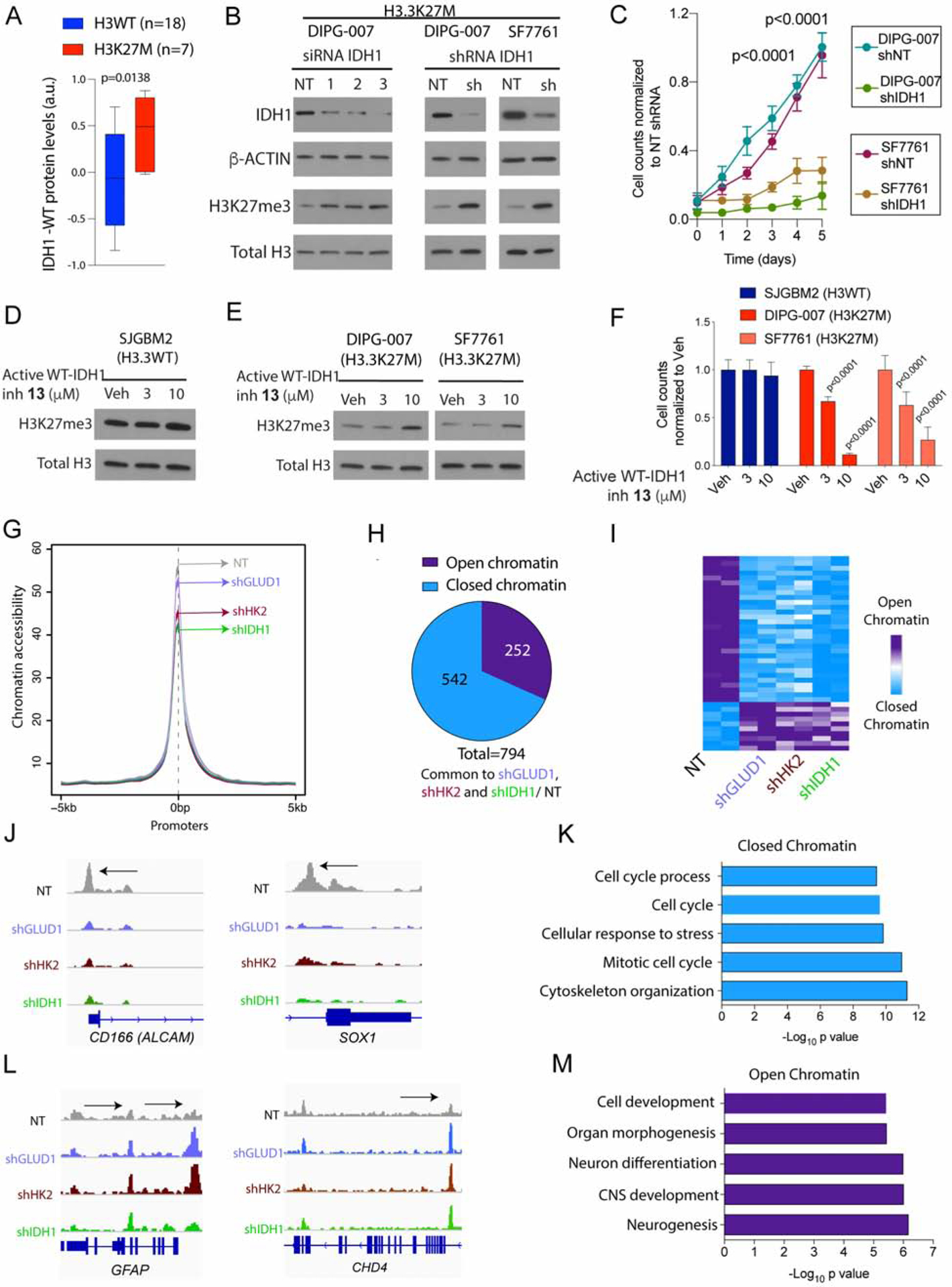
(A) IDH1-WT protein levels in H3WT (n=18) and H3K27M (n=7, red) pediatric high-grade gliomas (Pediatric Brain Tumor Atlas, PedcBioPortal). Box plots show median and interquartile range, whiskers represent the highest and lowest observations.
(B) H3.3K27M DIPG-007 cells were transfected with non-targeted (NT) or 3 independent IDH1 siRNAs and H3.3K27M SF7761 and DIPG-007 cells were stably transduced with NT or IDH1 shRNA. Representative WB of IDH1, β-ACTIN, H3K27me3, and total H3 levels.
(C) Cell proliferation (normalized cell counts, Y-axis) against time (days, X-axis) in NT or IDH1 shRNA in H3.3K27M SF7761 and DIPG-007 cells from (b) (n=3).
(D–F) SJGBM2 (H3WT, D) or DIPG-007 and SF7761 (H3.3K27M, E) cells were treated with vehicle (Veh) or indicated concentrations of the active WT-IDH1i 13. Representative WB of H3K27me3 and total H3 levels after treatment. In parallel, cell counts (normalized cell counts, Y-axis) was measured per condition after treatment (n=3, F).
(G) ATAC-seq was compared in H3.3K27M DIPG-007 cells stably transduced with non-targeted (NT, dark gray) or shGLUD1 (light blue), shHK2 (brown) and shIDH1 (green). Chromatin accessibility was compared at promoter regions (+/− 5kb from TSS, n=2).
(H–I) Pie chart (H) and heatmap (I) demonstrating significantly altered (adjusted p<0.05) ATAC-seq peaks commonly changed in shGLUD1/ NT, shHK2/ NT and shIDH1/ NT from indicating closed (significantly lowered compared to NT control, blue) or open (significantly higher compared to NT control, purple) chromatin.
(J–K) Representative ATAC-seq peaks at CD166/ALCAM and SOX1 loci in NT (dark gray), shGLUD1 (light blue), shHK2 (brown) and shIDH1 (green) DIPG-007 cells and GSEA analyses of common regions with closed chromatin (blue).
(L–M) Representative ATAC-seq peaks at GFAP and CHD4 loci in NT (dark gray), shGLUD1 (light blue), shHK2 (brown) and shIDH1 (green) DIPG-007 cells and GSEA analyses of common regions with open chromatin (purple).
Treatments were performed for 2 days. Data plotted as mean ± SD and analyzed by 2-sided, unpaired, 2-tailed, Student’s t-test (5a, c) or ANOVA (5f); n indicates biologic replicates.
GDH, HK2, and IDH1 knockdown results in altered chromatin accessibility at gene loci related to neuroglial differentiation
We determined if an increase in global H3K27me3 on enzyme inhibition was accompanied by alterations in genome-wide chromatin accessibility. We performed ATAC-seq in DIPG-007 cells with GDH, HK2 and IDH1 shRNA-knockdown or non-targeted (NT) controls. Integrated analysis revealed 794 significant and commonly altered regions in all three knockdown conditions (Figures S5G–H). Of these regions, 68% (542/794) were significantly downregulated corresponding to regions of lowered/closed chromatin accessibility, while 32% (252/794) were upregulated regions corresponding to regions with increased/open chromatin accessibility (Figures S5G–H, 5G–H). Regions with closed chromatin included stem-cell genes such as CD166 and SOX1 (Figures 5J–K, S5I). In contrast, regions with open chromatin included neuroglial differentiation-related genes including GFAP and the neurodevelopment chromatin regulator CHD4 (Figures 5L–M, S5I). PRC2 binding sites that show H3K27me3 retention in H3K27M tumors and H3.3K27M NSC including HOX loci and CDKN2A (Bender et al., 2013; Chan et al., 2013; Mohammad et al., 2017) did not show changes in chromatin-accessibility (Figures S5J–K). These data together suggest that commonly altered regions with GDH, HK2 and IDH1 knockdown mainly decreased chromatin accessibility, including at stem-cell factors and increased chromatin accessibility at far fewer loci including genes related to neuroglial differentiation.
Inhibition of IDH1 and glutamine metabolism is therapeutic in vivo
We sought to determine if metabolic inhibitors have therapeutic potential in preclinical DIPG animal models. Drug delivery to the brainstem, due to poor BBB-penetrability, is a major therapeutic hurdle. We took advantage of the high BBB-penetrability of the glutamine antagonist and DON analogue JHU-083 that achieves millimolar concentrations in the mouse brain (Hanaford et al., 2019). We also established that WT-IDH1i 13 penetrates the brainstem in mice (Figure S6A). We combined JHU-083 along with IDH1i 13 to address potential drug resistance pathways arising from inbuilt redundancies (Figure 3I). Mice were implanted with H3.3K27M DIPG-XIII*p or DIPG-007 cells in the pons. After tumor engraftment was confirmed by bioluminescence imaging, mice were treated with WT-IDH1i 13, or JHU-083, or a combination of both using two treatment regimens (for details see Figures S6B–C). Both WT-IDH1i 13 or JHU-083 compared to vehicle treated mice significantly increased overall survival in both animal models (Figure 6A–D). Moreover, maximal therapeutic effect was observed on combined treatment in both DIPG models (Figure 6A–D). Our results serve as proof-of-principle that combined inhibition of α-KG producing enzymes in glucose and glutamine metabolism pathways is therapeutic in DIPG animal models.
Figure 6. Inhibition of IDH1 and glutamine metabolism is therapeutic in vivo.
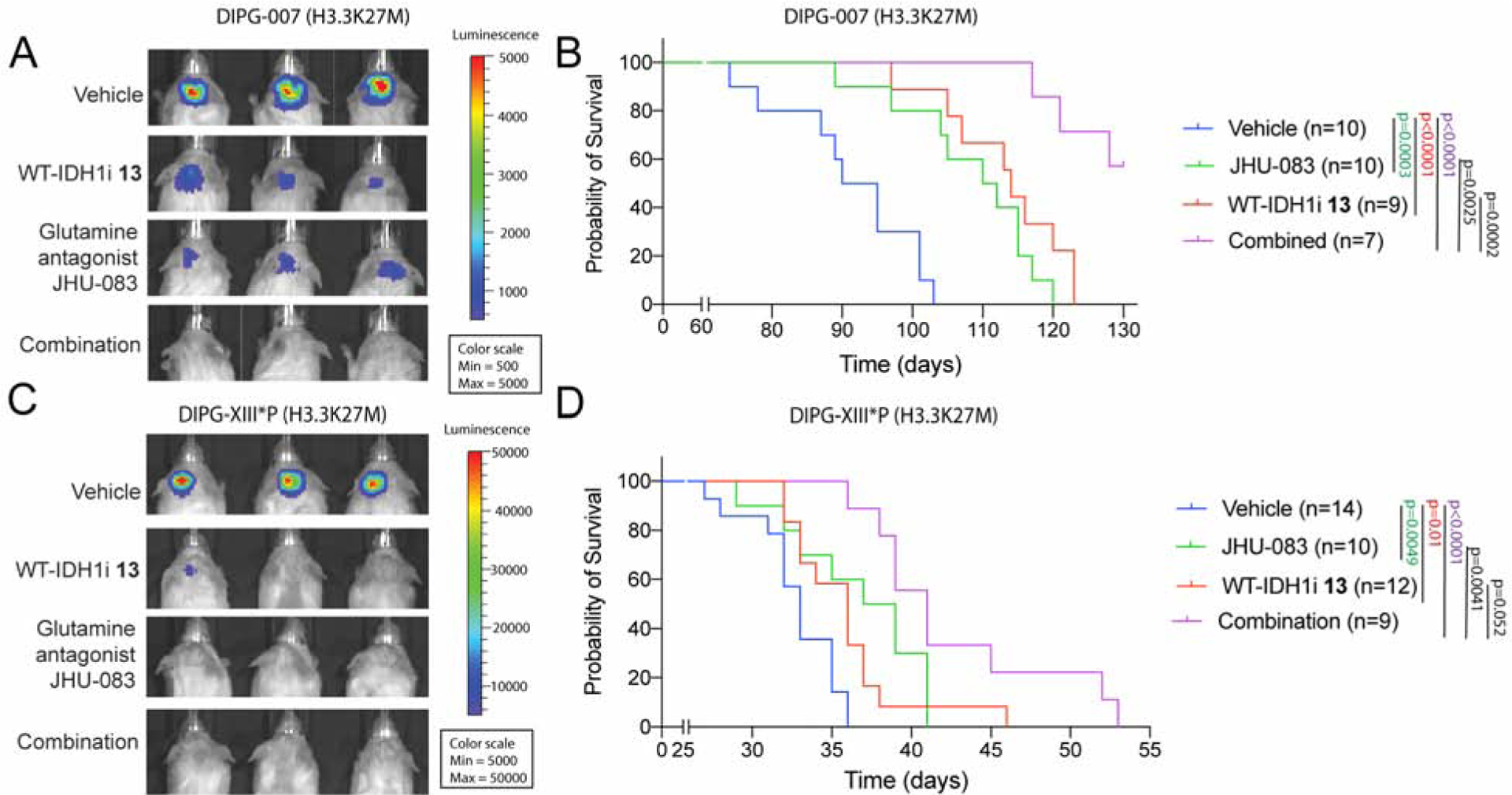
(A–B) Representative bioluminescence images (a) and Kaplan-Meier analysis (b) from animals implanted with H3.3K27M DIPG-007 cells in the pons and treated for four weeks (see Fig S6b) with vehicle (n=10), WT-IDH1i 13 (n=9) or the glutamine antagonist JHU-083 (n=10) or both (n=7).
(C–D) Representative bioluminescence images (c) and Kaplan-Meier analysis (d) from animals implanted with H3.3K27M DIPG-XIII*P cells in the pons and treated for three weeks (see Fig S6c) with vehicle (n=14), WT-IDH1i 13 (n=12) or the glutamine antagonist JHU-083 (n=10) or both (n=9).
See figure S6 for treatment details.
D-2HG increases H3K27me3 levels and is toxic to H3.3K27M cells and mutually exclusive H3.3K27M and IDH1 R132H mutations are synthetic lethal
mIDH1/2 gliomas metabolize α-KG to D-2HG, which competitively inhibits H3K27 demethylases to increase global H3K27me3 levels (Figure 7A) (Losman and Kaelin, 2013). Accordingly, H3K27me3 IHC demonstrated higher H3K27me3 levels in IDH1 R132H (n=8) compared to IDH WT (n=19) gliomas and confirmed global H3K27me3 reduction (n=12) in pediatric H3K27M compared to H3WT (n=24) gliomas (Figures 7B–C). Importantly, analysis of publicly available data sets (TCGA) in a glioma cohort (n=720) demonstrated that mIDH1/2 (n=463) and H3K27M (n=257) were mutually exclusive and occurred in divergent age groups (Figure 7D) (Schwartzentruber et al., 2012; Sturm et al., 2012).
Figure 7. D-2HG increases H3K27me3 levels and is toxic to H3.3K27M cells and mutually exclusive H3.3K27M and IDH1 R132H mutations are synthetic lethal.
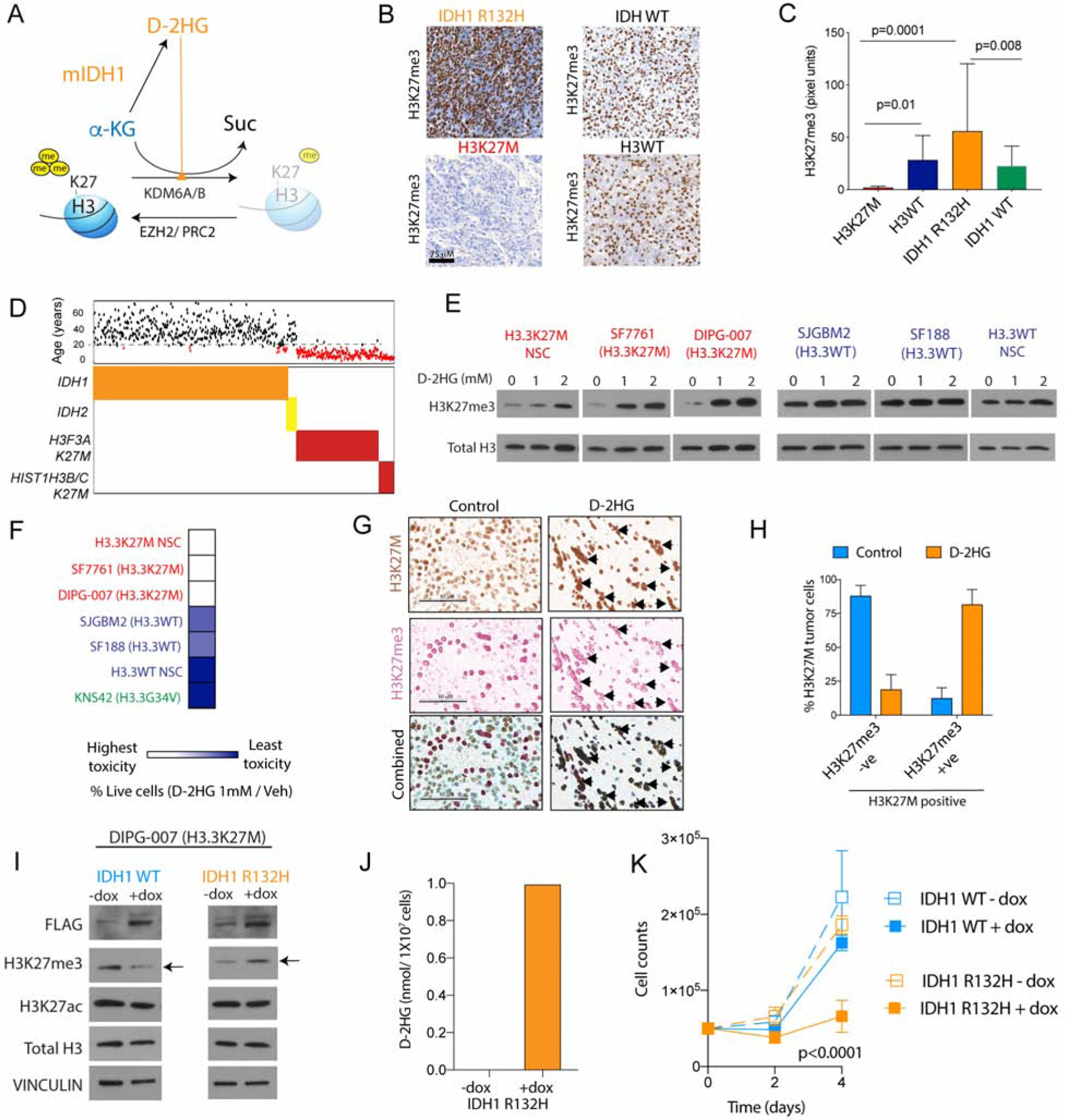
(A) Schematic depicting mIDH1 metabolizing α-KG to D-2HG. D-2HG competitively inhibits α-KG’s function as a co-factor for H3K27 demethylases KDM6A/6B to increase global H3K27me3 levels.
(B–C) Representative images (b) and blinded quantification (c, Y-axis, pixel units) of H3K27me3 stained H3K27M (n=12, red) and H3WT (n=24, dark blue), IDH1 R132H (n=8, orange) and IDH1/2 WT (n=19, light blue) patient tumor samples.
(D) Distribution of mIDH1(n=443) and mIDH2 (n=20); and K27M H3.3 (H3F3A, n=222) and H3.1 (HIST1H3B/C, n=35) gliomas. Corresponding age is graphed above (black >18y; red<18y).
(E) H3.3K27M (NSC, SF7761 and DIPG-007, red) or H3WT (NSC, SJGBM2 and SF188, blue) cells were treated with indicated concentrations of D-2HG. Representative WB of H3K27me3 and total H3 levels.
(F) Heat map of fold-change in cell numbers (% live cells) upon 1 mM D-2HG/Veh treatment in H3.3K27M (NSC, SF7761, DIPG-007; red), H3.3G34V (KNS42; green) and H3WT (SJGBM2 and SF188; blue) cells (n=3, see also Fig S6c).
(G) Representative single color or double color overlay IHC images from vehicle (n=3) or D-2HG (n=2, 25mg/kg, every other day for 2 weeks) treated H3.3K27M NSC xenograft tumor samples stained with combined mutant specific H3K27M (brown) and H3K27me3 (red) antibodies. Arrows indicate tumor cells that are positive for both H3K27M and H3K27me3, scale bar=60μM.
(H) Quantification of H3K27M positive tumor cells from H3.3K27M NSC xenografts that were either negative or positive for H3K27me3 (from g) in vehicle (light blue) or D-2HG (orange) treated animals.
(I–J) H3.3K27M DIPG-007 cells were stably transfected with doxycycline (dox) inducible FLAG-tagged WT IDH1 (light blue) or IDH1 R132H (orange). Representative WB of FLAG, H3K27me3, H3K27ac, total H3 and VINCULIN levels (I). D-2HG measured in DIPG-007 cells transduced with IDH1 R132H +/− dox (J, n=3).
(K) Cell proliferation (cell counts, Y-axis) plotted against time (days, X-axis, n=4).
Data expressed as mean ± SD and analyzed by ANOVA, n indicates biologic replicates.
While H3K27M (Figures 1–3) and mIDH1 gliomas rely on anabolic metabolic pathways to generate α-KG, we hypothesized that H3K27M and mIDH1 use α-KG in opposing manners to regulate global H3K27me3 levels. We tested this hypothesis using two approaches. First, treating H3.3K27M cells with cell-permeable D-2HG increased global H3K27me3 levels and decreased cell proliferation (Figures 7E–F, S7A–C). In contrast, treating H3WT or mutant H3.3G34V (KNS42) cells with D-2HG produced a marginal or no change in global H3K27me3 levels with a lesser effect on cell numbers (Figures 7E–F, S7A–C). Moreover, D-2HG administration resulted in higher global H3K27me3 levels in H3.3K27M tumor cells in vivo compared to vehicle treatment (Figures 7G–H, S7D). Second, we expressed mIDH1 R132H or WT IDH1 in H3.3K27M DIPG-007 cells and evaluated effects on global H3K27me3 and cell numbers. mIDH1, but not WT IDH1, expression resulted in D-2HG production, increased global H3K27me3 levels, and markedly reduced proliferation (Figures 7I–K). Conversely, forced expression of H3.3K27M in mIDH1 R132H TB-096 cells lowered global H3K27me3 levels and significantly lowered proliferation (Figures S7E–F). These data together demonstrate that D-2HG increases H3K27me3 and is toxic in H3.3K27M cells, and that mutually exclusive H3.3K27M and mIDH1 R132H are synthetic lethal.
DISCUSSION
The discovery of histone mutations in gliomas, including H3K27M in midline-gliomas such as DIPGs, has furthered our knowledge of these deadly childhood brain tumors. Using a comprehensive transcriptomic, proteomic and metabolomic approach, we discovered that H3.3K27M compared to H3WT cells showed enhanced glycolysis, glutaminolysis, and TCA-cycle metabolism accompanied by higher expression of key enzymes in these pathways including HK2, GLUD1 and IDH1 (Figure 8). We corroborated many of these findings in patient tumor samples and in vivo MRS imaging, which showed higher glutamine and citrate levels in H3K27M compared to H3WT gliomas. Metabolic reprogramming is a hallmark of cancer and our data furthers our knowledge of the pathogenesis of H3K27M mutant gliomas by demonstrating that H3.3K27M mutant tumors can enhance critical metabolic pathways to sustain their growth.
Figure 8. H3K27M mutations are dependent on a critical metabolic pathway also used by mIDH1 to regulate global H3K27me3 levels.
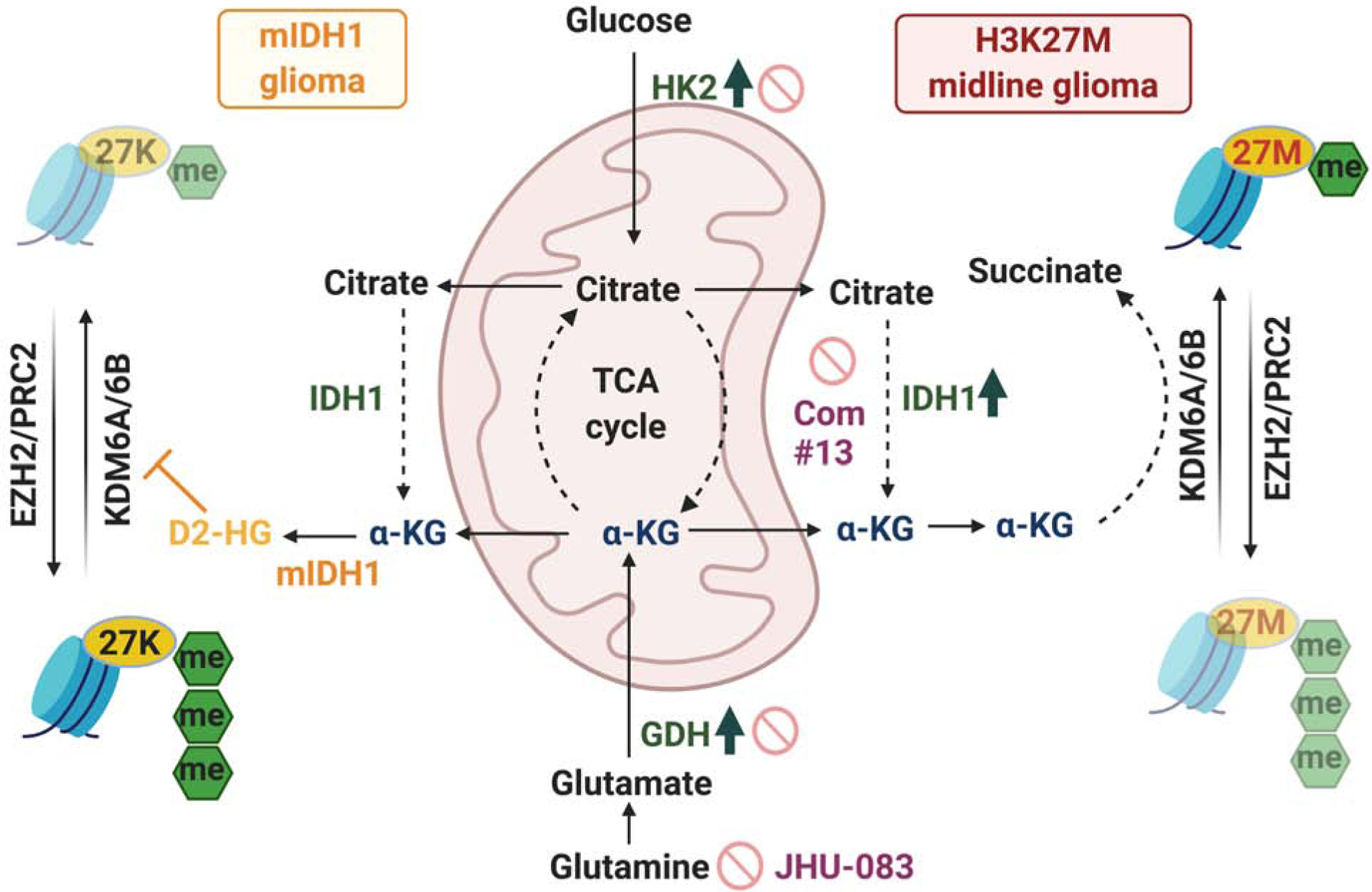
Schematic model of integrated metabolic and epigenetic pathways in H3K27M cells. HK2, IDH1 and GDH, enzymes that generate α-KG, were elevated in H3K27M tumors. H3K27M tumor cells use α-KG to maintain low global H3K27me3 levels. Inhibition of HK2, IDH1 and GDH increased global H3K27me3, altered chromatin accessibility and suppressed H3K27M cell proliferation in vitro and in vivo and are potential therapeutic targets (stop signs). Mutually exclusive mIDH1 use α-KG to generate D-2HG. D-2HG treatment or expression on mIDH1 in H3K27M cells was toxic and increased H3K27me3.
The TCA-cycle intermediate α-KG, which serves as a critical co-factor for H3K27M demethylases, was elevated in H3.3K27M cells. Importantly, α-KG was required to maintain low global H3K27me3 levels in H3.3K27M cells (Figure 8). Genetic or pharmacologic inhibition of enzymes related to α-KG generation including GDH, HK2 and IDH1 resulted in elevation of global H3K27me3, suppression of tumor cell proliferation and shifted chromatin accessibility to mainly closed states, consistent with the repressive nature of elevated H3K27me3. These data are in line with α-KG regulating cell-fate decisions, as previously reported in embryonic stem cells (Carey et al., 2015; TeSlaa et al., 2016). In melanoma and mouse embryonic stem cells, glutamine metabolism regulates global H3K27me3 levels (Carey et al., 2015; Pan et al., 2016; TeSlaa et al., 2016). Our studies suggest that in addition to glutamine, glucose metabolism can also regulate global H3K27me3 levels in H3.3K27M cells. H3.3K27M cells showed heterogeneity in their utilization of glucose or glutamine to regulate global H3K27me3 with dependence on one or both pathways. Furthermore, inhibition of IDH1 activity or antagonizing glutamine metabolism prolonged overall survival in DIPG animal models. Maximal beneficial effects were observed on combined inhibition of both pathways. This suggests that H3K27M glioma cells can use heterogenic microenvironmental metabolic cues such as glucose and glutamine availability to regulate chromatin. Inbuilt redundancies in this pathway may be of significance in understanding development of therapeutic resistance. Overall, we demonstrate that metabolic and epigenetic pathways are closely integrated in H3.3K27M cells, similar to that observed in mIDH1/2 gliomas.
mIDH1/2 metabolizes α-KG to D-2HG, which competitively inhibits the function of α-KG-dependent dioxygenases, including histone KDMs and DNA demethylases (Figure 8). D-2HG treatment in H3.3K27M cells increased H3K27me3 levels in vitro and in vivo and lowered cell numbers of H3.3K27M cells to a greater extent than H3WT cells. Importantly, mIDH1/2 and H3K27M mutations were mutually exclusive in patients samples and forced co-expression of mIDH1 in H3.3K27M cells led to D-2HG production, global H3K27me3 elevation and synthetic lethality. H3K27M and mIDH1 could be mutually exclusive because the acquisition of one creates a synthetic lethal context for the other based on the preferred epigenomic state. DNA-demethylases including TET1/2 and other histone KDMs also require α-KG to drive histone and DNA hypomethylation. While our data suggest that α-KG maintains low H3K27me3 in H3.3K27M cells, we cannot rule out the possibility that α-KG may also regulate methylation of other histone lysine residues and/or DNA. Another caveat is that inhibition of metabolic pathways can compromise bioenergetic and biosynthetic pathways in addition to altering epigenetics. Further studies are needed to comprehensively elucidate integrated metabolic and epigenetic pathways in H3K27M gliomas.
Metabolic regulation of global H3K27me3 in H3.3K27M cells led to heterogenous dependencies on GDH, HK2, and WT IDH1, which could be important leads for potential therapeutic development (Figure 8). Moreover, in vivo MRS imaging could serve as a non-invasive clinical tool to monitor patients and longitudinally assess efficacies of potential metabolic and other treatments. As proof-of-principle, pharmacologic inhibition of these ezymes resulted in elevated H3K27me3 and marked reduction in cell numbers. Suppression of either IDH1, or glutamine metabolism alone, or combined inhibition showed a significant increase in survival in preclinical DIPG animal models. Overall, our study fills a signficant gap in our knowledge of H3K27M-midline glioma biology by identifying integrated metabolic and epigenetic vultnerabilites that could be leveraged to develop future treatment strategies that are urgently needed for these fatal tumors.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Sriram Venneti (svenneti@med.umich.edu)
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
The datasets generated during this study are available at NCBI/ GEO (GSE135419)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Tumor samples and patients
All studies were performed after Institutional Review Board approval from respective institutions. H3K27M and H3WT tumor tissues were obtained from Children’s Hospital of Los Angeles (CHLA) and the University of Michigan in the form of formalin fixed-paraffin embedded samples. Patient identifiers were removed from all samples. Clinical features of these samples have been previously reported (Venneti et al., 2013; Venneti et al., 2014). MRS imaging in patients was conducted at Children’s Hospital Los Angeles and was compliant with the Health Insurance Portability and Accountability Act. The requirement to obtain informed consent was waived. H3K27M (n=11) and H3WT (n=4) patients with midline-gliomas were retrospectively identified and MRS data was analyzed in a blinded manner. H3K27M mutations were confirmed by sequencing or by immunohistochemistry with a mutant-specific H3K27M antibody and global reduction in H3K27me3. The median age for H3K27M patients was 4 years (range 0.4–9 years) and for H3WT patients was 12 years (range 4–16 years). For H3K27M patients, 8 were male and 3 were female. For H3WT patients, 3 were male and 1 was female. Gene expression and metadata from pediatric high-grade gliomas (n=203) were downloaded from the Pediatric cBioportal (https://pedcbioportal.org/). (Mackay et al., 2017) From available metadata, tumors were grouped according to histone status as H3K27M (n = 83), H3WT (n = 101) and H3G34R/V (n = 19) high-grade gliomas. Single cell RNA-seq data from patient tumor samples was obtained from publicly available data (https://singlecell.broadinstitute.org/single_cell) (Filbin et al., 2018). Expression levels of various metabolic enzymes were assessed in malignant, non-malignant, OC-like, AC-like and OPC-like cells as indicated by Filbin et. al.
Mice and housing conditions
Animal experiments were performed after approval from the University of Michigan Committee on Use and Care of Animals and were conducted as per NIH guidelines for animal welfare. All animal procedures were approved by Institutional Animal Care & Use Committee (IACUC) at the University of Michigan. Animals were housed and cared according to standard guidelines with free access to food and water. All experiments were performed on NOD-SCID-IL2R gamma chain-deficient (NSG) or Nude mice (8–10 weeks old) with male and female animals used equally. Animals including littermates of the same sex were randomly assigned to control or treatment conditions.
Cell lines and culture conditions
All cell lines were validated by STR profiling and tested negative for mycoplasma. Cells were cultured in a humidified incubator at 37°C and 5% CO2. HSJD-DIPG-007 (referred to as DIPG-007, H3.3K27M), SF7761 (H3.3K27M), DIPG-XIII*P (H3.3K27M), DIPG-IV (H3.1K27M), TB-096 (IDH1 R132H), SF188 (H3WT), SJGBM2 (H3WT), UMPed37 (H3WT), KNS42 (H3.3G34V) and immortalized mouse NSC (p16Ink4a − /p14Arf −) were obtained after MTA from the respective institutions as indicated in the resources table. DIPG-007, SF7761, SJGBM2, UMPed37 and mNSC (mouse) cells were cultured in Neurobasal A supplemented with N2, B27, L-glutamine (2 mM), Pen/strep (1X), heparin (2 μg/mL), human-EGF (20 ng/mL), human-bFGF (20 ng/mL), and BSA (45 ng/ml). DIPG-XIII cells were cultured in equal parts Neurobasal A and DMEM/F12 supplemented with B27 (without vitamin A), HEPES buffer (10 mM), MEM sodium pyruvate solution (1mM), MEM Non-Essential Amino Acids (1X), GlutaMAX-I Supplement (1X), Antibiotic-Antimycotic (1X), heparin (2 μg/mL), human-EGF (20 ng/mL), human-bFGF (20 ng/mL), PDGF-AA (10 ng/mL), and PDGF-BB (10 ng/mL). SF188 and KNS42 cells were cultured in DMEM supplemented with FBS (10%), L-glutamine (2 mM), and Pen/strep (1X). TB-096 cells were cultured in equally parts SF7761 media and SF188 media.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| HA (WB) | Biolegend | Cat# 901501 ; RRID: AB_2565006 |
| H3K27M (WB and immunohistochemistry) | EMD | Cat# ABE419 ; RRID: AB_2728728 |
| H3K27me3 (WB) | EMD | Cat# 07–449 ; RRID: AB_310624 |
| H3K27ac (WB) | abcam | Cat# ab177178 ; RRID: AB_2828007 |
| H3K4me1 (WB) | Cell signaling | Cat# 5326s ; RRID: AB_10695148 |
| H3K4me3 (WB) | Cell signaling | Cat# 9751s ; RRID: AB_2616028 |
| H3K36me3 (WB) | ACTIVE MOTIF | Cat# 61021 ; RRID: AB_2614986 |
| Total H3 (WB) | Cell signaling | Cat# 3638S ; RRID: AB_1642229 |
| H3K27me3 (ChIP-seq) | ACTIVE MOTIF | Cat# 39155 ; RRID: AB_2561020 |
| H3K27ac (ChIP-seq) | ACTIVE MOTIF | Cat# 39133 ; RRID: AB_2561016 |
| H3K4me1 (ChIP-seq) | ACTIVE MOTIF | Cat# 39297 ; RRID: AB_2615075 |
| H3K4me3 (ChIP-seq) | ACTIVE MOTIF | Cat# 39159 ; RRID: AB_2615077 |
| H3K27ac (immunohistochemistry) | Cell signaling | Cat# 8173 ; RRID: AB_10949503 |
| H3K27M (immunohistochemistry) | EMD | Cat# ABE419 ; RRID: AB_2728728 |
| H3K27me3 (immunohistochemistry) | EMD | Cat# 07–449 ; RRID: AB_310624 |
| SLC1A5 (immunohistochemistry) | Sigma | Cat# HPA035240 ; RRID: AB_10604092 |
| SLC2A3/GLUT3 (WB) | abcam | Cat# ab191071 ; RRID: AB_2736916 |
| HK2 (WB) | Santa Cruz bio | Cat# sc-374091;RRID:AB_10917915 |
| IDH1 (WB) | Cell signaling | Cat# 8137 ; RRID: AB_10950504 |
| IDH2 (WB) | Santa Cruz bio | Cat# sc-374476; RRID: AB_10986415 |
| GDH (GLUD1/2) (WB) | abcam | Cat# ab166618 ; RRID: |
| SLC7A11 (WB) | Cell signaling | Cat# 12691s ; RRID: AB_2687474 |
| β-ACTIN (WB) | Sigma | Cat# A5441 ; RRID: AB_476744 |
| GAPDH (WB) | Santa Cruz bio | Cat# sc-25778; RRID: AB_10167668 |
| p-s6rp (Ser240/244) (WB) | Cell signaling | Cat# 2215s ; RRID: AB_331682 |
| p-s6rp (Ser235/236) (WB) | Cell signaling | Cat# 4858s ; RRID: AB_916156 |
| s6rp (WB) | Cell signaling | Cat# 2317s ; RRID: AB_2238583 |
| N-myc (WB) | Santa Cruz bio | Cat# sc-53993; RRID: AB_831602 |
| C-myc (WB) | Santa Cruz bio | Cat# sc-40 ; RRID: AB_627268 |
| SLC1A5/ASCT2 (WB) | ThermoFisher | Cat# PA5–50527;RRID: AB_2635980 |
| GLS (WB) | abcam | Cat# ab156876 ; RRID: AB_2721038 |
| GSK3 a/b (WB) | Cell signaling | Cat# 5676s ; RRID: AB_10547140 |
| p-GSK3 a/b (Ser21/9) (WB) | Cell signaling | Cat# 9331s ; RRID: AB_329830 |
| VINCULIN (WB) | Sigma | Cat# V9264 ; RRID: AB_10603627 |
| FLAG (WB) | Cell signaling | Cat# 2368s ; RRID: AB_2217020 |
| GOT1 (WB) | Abcam | Cat# ab171939 |
| Hif-1α (WB) for mouse | Abcam | Cat# ab16066; RRID:AB_302234 |
| HIF-1α (WB) for human | Abcam | Cat# ab179483; RRID:AB_2732807 |
| Bacterial and Virus Strains | ||
| One-shot Stbl3 chemically competent E.coli | ThermoFisher | Cat# C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DM-succinate | Acros Organics | Cat# AC150731000 |
| DM-αKG | Sigma | Cat# 349631 |
| DMSO | Sigma | Cat# D2650 |
| Glucose | Gibco | Cat# A2494001 |
| L-Glutamine | Gibco | Cat# 25030–081 |
| Matrix-gel (Matrigel® Basement Membrane Matrix) | Corning | Cat# 356234 |
| D-Luciferin, Potassium Salt | GoldBio | Cat# LUCK-1G |
| GSK-J4 | Cayman Chem. | Cat# 12073 |
| GSK-J5 | Cayman Chem. | Cat# 12074 |
| DON (6-Diazo-5-oxo-L-norleucine) | Cayman Chem. | Cat# 17580 |
| 2-DG (2-Deoxy-D-glucose) | Cayman Chem. | Cat# 14325 |
| D-2HG ((2R)-Octyl-α-hydroxyglutarate) | Cayman Chem. | Cat# 16366 |
| CB-839 | Cayman Chem. | Cat# 22038 |
| BPTES | Cayman Chem. | Cat# 19284 |
| Lonidamine | Cayman Chem. | Cat# 14640 |
| Ketoconazole | Cayman Chem. | Cat# 15212 |
| Posaconazole | Cayman Chem. | Cat# 14737 |
| Desferrioxamine | Cayman Chem. | Cat# 14595 |
| JHU-083 | MCE | Cat# HY-122218 |
| IDH1 inhibitor 1 | Abbvie | |
| IDH1 inhibitor 2 | Abbvie | |
| IDH1 inhibitor 3 | Abbvie | |
| IDH1 inhibitor 13 | Abbvie | |
| Inactive enone analogue 18 | Abbvie | |
| Critical Commercial Assays | ||
| TransIT®-Lenti Transfection Reagent | Mirus | Cat# MIR 6650 |
| TMT10plex™ Isobaric Label Reagent Set | ThermoFisher | Cat# 90110 |
| D-2-Hydroxyglutarate Assay Kit | abcam | Cat# ab211070 |
| Succinate Assay Kit (Colorimetric) | abcam | Cat# ab204718 |
| Alpha Ketoglutarate (alpha KG) Assay Kit | abcam | Cat# ab83431 |
| Deposited Data | ||
| NCBI/ GEO (GSE135419), next generation sequencing files. | This paper | GEO: GSE135419 |
| Experimental Models: Cell Lines | ||
| HSJD-DIPG007 (H3.3K27M) | Dr. Rintaro Hashizume, Northwestern University; RRID: CVCL_VU70 | |
| SU-DIPG-XIII (H3.3K27M) | Dr. Michelle Monje, Stanford University; RRID: CVCL_IT41 | |
| SU-DIPG-IV (H3.1K27M) | Dr. Michelle Monje, Stanford University; RRID: CVCL_IT39 | |
| SF7761 (H3.3K27M) | Dr. Rintaro Hashizume, Northwestern University; RRID: CVCL_IT45 | |
| SJGBM2 (H3WT) | Dr. Carl Koschmann, University of Michigan; RRID: CVCL_M141 | |
| SF188 (H3WT) | Dr. Craig B. Thompson, Memorial Sloan Kettering Cancer Center; RRID: CVCL_6948 | |
| UMPed37 (H3WT) | Dr. Carl Koschmann, University of Michigan | |
| KNS42 (H3.3G34V) | Dr. Carl Koschmann, University of Michigan; RRID: CVCL_0378 | |
| TB-096 (IDH1 R132H) | Dr. Hai Yan, Duke University | |
| mNSC | Dr. Richard J. Gilbertson, St Jude’s Hospital | |
| Experimental Models: Organisms/Strains | ||
| Mouse: Hsd:Athymic Nude-Foxn1nu | Jackson Laboratories | Cat# 002019; RRID: IMSR_JAX:002019 |
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | Jackson Laboratories | Cat#:005557; RRID: IMSR_JAX:005557 |
| Oligonucleotides | ||
| siRNA IDH1 NT: ON-TARGETplus Non-targeting Control siRNAs | Dharmacon | Cat# D-001810–01-05 |
| siRNA IDH1–1: ON-TARGETplus IDH1 siRNA Targeted Region:3'UTR | Dharmacon | Cat# J-008294–09-0002 |
| siRNA IDH1–2: ON-TARGETplus IDH1 siRNA Targeted Region:ORF | Dharmacon | Cat# J-008294–10-0002 |
| siRNA IDH1–3: ON-TARGETplus IDH1 siRNA Targeted Region:ORF | Dharmacon | Cat# J-008294–11-0002 |
| Recombinant DNA | ||
| H3.3WT lentiviral plasmid | Dr. David Allis, The Rockefeller University | |
| H3.3K27M lentiviral plasmid | Dr. David Allis, The Rockefeller University | |
| shNT lentiviral plasmid | Sigma | Cat# SHC002V |
| shGLUD1–1 lentiviral plasmid | Sigma | Cat# TRCN0000343656 |
| shGLUD1–2 lentiviral plasmid | Sigma | Cat# TRCN0000028600 |
| shHK2–1 lentiviral plasmid | Sigma | Cat# TRCN0000037670 |
| shHK2–2 lentiviral plasmid | Sigma | Cat# TRCN0000195582 |
| shIDH1 lentiviral plasmid (V2LHS_217815) | Dharmacon | Cat# RHS4430–200189861 |
| pLenti PGK Blast V5-LUC | Addgene | Cat# 19166 ; RRID: Addgene_19166 |
| pSLIK-IDH1-FLAG | Addgene | Cat# 66802 ; RRID: Addgene_66802 |
| pSLIK-IDH1-R132H-FLAG | Addgene | Cat# 66803 ; RRID: Addgene_66803 |
| Software and Algorithms | ||
| PRISM 7.0/8.0 | GraphPad | |
| Image Studio software | LI-COR | |
| IVIS Living Image Software | Perkin Elmer | |
| IGV(Integrative Genomics Viewer) 2.5.0 | Broad Institute | |
| AperioImageScope/ AperioScan Scope | Aperio | |
| RSEM (RNA-Seq by Expectation-Maximization) | https://deweylab.github.io/RSEM/ | |
| CASAVA 1.8.2 pipeline | Illumina | |
| methylKit | https://bioconductor.org/packages/release/bioc/html/methylKit.html | |
| Proteome Discoverer v2.1 | Thermo Fisher | |
| SICER | http://www.lcmodel.com/ | |
| MACS | http://liulab.dfci.harvard.edu/MACS/ | |
| Agilent MassHunter Workstation Software Quantitative | Agilent | |
| GSEA | Broad Institute http://software.broadinstitute.org/gsea/index.jsp |
|
| MetaboAnalyst | https://www.metaboanalyst.ca/ | |
| Matlab’s image processing toolbox | Matlab | |
| Image Studio software | Licor | |
METHOD DETAILS
Cell counting
For manual counting, 500,000 cells were plated on in 24 well plates. After indicated treatment and time, cells were dissociated using trypsin or Accutase. Cells were counted using the Countess II FL Automated Cell Counter. For bioluminescent cell counting, 10,000 cells in 100ul were plated in 96 well plates. After indicated treatment and time, 10ul of 15 mg/ml D-luciferin was added into the wells and cells were incubated at room temperature on a rocker for 5min. The luminescent signal was measured using a Synergy HTX Multi-Modeplate reader.
Western blotting
Cells were lysed in RIPA buffer containing protease and phosphatase inhibitor. The same amount of proteins from whole cell lysate or histone extraction were electrophoresed on a 4–15% Mini-PROTEAN TGX (Tris-Glycine eXtended) Precast gel and transferred to a PVDF membrane using the Trans-Blot Turbo transfer system. Membranes were blocked with 5% skim milk TBST (TBS buffer containing 0.1% Tween 20) when using regular antibodies or with 5% BSA TBST when using phosphorylation-specific antibodies. Membranes were incubated with primary antibodies in 5% skim milk or BSA TBST at 4°C overnight and washed with TBST three times. Membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (HRP) for 2 hours at room temperature and washed with TBST three times and TBS one time. Immunoreactivity was detected by SuperSignal West Pico PLUS Chemiluminescent Substrate or TMA-6. The band signal was analyzed using Image Studio software (LI-COR) and normalized by each internal control gene.
Histone extraction
Cells were plated at sub-confluent densities. After indicated treatment, cells were washed with PBS. Cell pellet from centrifugation was suspended in 1.5 ml hypotonic lysis buffer (10mM Tris HCl pH8.0, 1mM KCl, and 1.5mM MgCl2, protease inhibitor and phosphatase inhibitor) and incubated for 30 min on rotator at 4°C. The pellet was collected by centrifugation at 10,000g, 4°C for 10 min. The pellet was suspended in 400 μl of 0.4 N H2SO4 and incubated on a rotator at 4°C overnight. After centrifuging, the supernatant was transferred to a new tube and 132 μl TCA was added dropwise. The mixed solution was incubated on ice for 30 min. The histone pellet was harvested by centrifuging at 16,000g, 4°C for 10 min and washed with ice-cold acetone. After centrifuging at 16,000g, 4°C for 5 min, acetone was removed. The histone pellet was washed using acetone again. Subsequently, the histone pellet was made dry with the caps open at room temperature for 20 minutes to evaporate remaining acetone. The dried histone pellet was suspended in an appropriate volume of ddH2O water and placed on ice.
ChIP-seq and ATAC-seq
H3.3WT and H3.3K27M mNSC cell lines were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield. An aliquot of chromatin (30 μg) was precleared with protein A agarose beads (Invitrogen). Soluble chromatin was spiked-in with soluble Drosophila chromatin equivalent to 5–10% of mouse chromatin as previously described. (Lu et al., 2016) The mixed soluble chromatin was incubated with 4 μg of antibody against H3K27me3, H3K27ac, H3K4me3 and H3K4me1 (see resources table). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65° C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation. Quantitative PCR (QPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad). The resulting signals were normalized for primer efficiency by carrying out QPCR for each primer pair using Input DNA.
Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. Steps were performed on an automated system (Apollo 342, Wafergen Biosystems/Takara). After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single end). Reads were aligned to the mouse genome (mm10) using the BWA algorithm (default settings). Duplicate reads were removed and only uniquely mapped reads (mapping quality >= 25) were used for further analysis. The number of test tags were normalized by the same number of spike-in Drosophila tags for each sample. Alignments were extended in silico at their 3’-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library, and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. For active histone marks (H3K4me3, H3K27Ac, H3K4me1), peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of p-value = 1e-7. For H3K27me3 enriched regions were identified using the SICER algorithm at a cutoff of FDR 1E-10 and a max gap parameter of 600 bp. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations. The data was visualized and analyzed using Integrative Genomics Viewer (IGV) genome browser.
For ATAC-Seq 100,000 cells were collected by centrifugation at 500g for 5min. Cell pellets were resuspended in lysis buffer, pelleted, and tagmented using the enzyme and buffer provided in the Nextera Library Prep Kit (Illumina). Tagmented DNA was then purified using the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified using Agencourt AMPure SPRI beads (Beckman Coulter). Resulting material was quantified using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems), and sequenced with PE42 sequencing on the NextSeq 500 sequencer (Illumina). Reads were aligned using the BWA algorithm (mem mode; default settings). Duplicate reads were removed, only reads mapping as matched pairs and only uniquely mapped reads (mapping quality >= 1) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peaks were identified using the MACS 2.1.0 algorithm at a cutoff of p-value 1e-7, without control file, and with the –nomodel option. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed.
RNA- sequencing
RNA-sequencing was performed as previously described. (Bayliss et al., 2016) RNA was isolated using Trizol (15596–026, Invitrogen) and treated with DNase (9003-98-9, Sigma). RNA sequencing libraries were prepared according to the Illumina TruSeq protocol and were sequenced on the HiSeq 2000. RNA-seq data generated were aligned to mouse reference genome using bowtie and analyzed using the RSEM software package with default parameters. Differentially expressed genes were defined using empirical Bayes hierarchical models (EBSeq) that factors for mapping ambiguity variance. Pathway analysis of differentially regulated genes was performed by GSEA (http://software.broadinstitute.org/gsea/index.jsp).
Tandem mass tags (TMT) proteomics
Cells were rinsed with PBS, 3 times and lysed using RIPA buffer containing 1mM phenylmethylsulfonyl fluoride (PMSF). Protein concentration was measured using BCA Protein Assay kit. Samples (80 μg/condition) were proteolyzed and labeled with TMT 10-plex essentially by following manufacturer’s protocol (ThermoFisher). Briefly, upon reduction and alkylation of cysteines, the proteins were precipitated by adding 6 volumes of ice cold acetone followed by overnight incubation at −20° C. The precipitate was spun down, and the pellet was allowed to air dry. The pellet was resuspended in 0.1M TEAB and overnight digestion with trypsin (1:50; enzyme: protein) at 37° C was performed with constant mixing using a thermomixer. The TMT 10-plex reagents were dissolved in 41 μl of anhydrous acetonitrile and labeling was performed by transferring the entire digest to TMT reagent vial and incubating at room temperature for 1 h. Reaction was quenched by adding 8 μl of 5% hydroxyl amine and further 15 min incubation. Labeled samples were mixed together, and dried using a vacufuge. An offline fractionation of the combined sample (~200 μg) into 10 fractions was performed using high pH reversed-phase peptide fractionation kit according to the manufacturer’s protocol (Pierce; Cat #84868). Fractions were dried and reconstituted in 12 μl of 0.1% formic acid/2% acetonitrile in preparation for LC-MS/MS analysis.
Liquid chromatography-mass spectrometry analysis (LC-multinotch MS3): In order to obtain superior quantitation accuracy, we employed multinotch-MS3 (Ref: McAlister GC) which minimizes the reporter ion ratio distortion resulting from fragmentation of co-isolated peptides during MS analysis. Orbitrap Fusion (Thermo Fisher Scientific) and RSLC Ultimate 3000 nano-UPLC (Dionex) was used to acquire the data. Two μl of the sample was resolved on a PepMap RSLC C18 column (Thermo Scientific) at the flow-rate of 300 nl/min using 0.1% formic acid/acetonitrile gradient system (2–22% acetonitrile in 110 min;22–40% acetonitrile in 25 min; 6 min wash at 90% followed by 25 min re-equilibration) and directly spray onto the mass spectrometer using EasySpray source (Thermo Fisher Scientific). Mass spectrometer was set to collect one MS1 scan (Orbitrap; 120K resolution; AGC target 2×105; max IT 50 ms) followed by data-dependent, “Top Speed” (3 seconds) MS2 scans (collision induced dissociation; ion trap; NCD 35; AGC 5×103; max IT 100 ms). For multinotch-MS3, top 10 precursors from each MS2 were fragmented by HCD followed by Orbitrap analysis (NCE 55; 60K resolution; AGC 5×104; max IT 120 ms, 100–500 m/z scan range).
Proteome Discoverer (v2.1; Thermo Fisher) was used for data analysis. MS2 spectra were searched against SwissProt mouse protein database (release 2016-04-13; 24861 sequences) using the following search parameters: MS1 and MS2 tolerance were set to 10 ppm and 0.6 Da, respectively; carbamidomethylation of cysteines (57.02146 Da) and TMT labeling of lysine and N-termini of peptides (229.16293 Da) were considered static modifications; oxidation of methionine (15.9949 Da) and deamidation of asparagine and glutamine (0.98401 Da) were considered variable. Identified proteins and peptides were filtered to retain only those that passed ≤1% FDR threshold. Quantitation was performed using high-quality MS3 spectra using the Reporter Ion Quantifier Node of Proteome Discoverer (Average signal-to-noise ratio of 10 and <30% isolation interference). Pathway analysis of differentially regulated proteins was performed by GSEA (http://software.broadinstitute.org/gsea/index.jsp).
Metabolite analysis and 13C-isotope tracing
For whole metabolite snapshot, 2 million cells were plated in T-25 flasks and cultured for 16 hrs in an incubator. A full media change was performed 2 hrs before collecting metabolites. Our liquid chromatography coupled-tandem mass spectrometry (LC-MS/MS) metabolomics analysis was performed as described previously. (Lee et al., 2019) In brief, Agilent 1290 UHPLC and 6490 Triple Quadrupole (QqQ) Mass Spectrometer (LC-MS) were used for label-free targeted metabolomics analysis. Agilent MassHunter Optimizer and Workstation Software LC-MS Data Acquisition for 6400 Series Triple Quadrupole B.08.00 was used for standard optimization and data acquisition. Agilent MassHunter Workstation Software Quantitative Analysis Version B.0700 for QqQ was used for initial raw data extraction and analysis. For each MRM transition, its retention time of left delta and right delta of 1 min was used. Additional parameters include mass extraction window of 0.05 Da right and left from the extract m/z, Agile2 integrator algorithm, peak filter of 100 counts, noise algorithm RMS, noise SD multiplier of 5 min, S/N 3, Accuracy Max 20% max %Dev, and Quadratic/Cubic Savitzky-Golay smoothing algorithm with smoothing function width of 14 and Gaussian width of 5.
For reversed-phase liquid chromatography (RPLC), a Waters Acquity UPLC BEH TSS C18 column (2.1 × 100mm, 1.7μm) was used in the positive ionization mode with mobile phase (A) consisting of 0.5 mM NH4F and 0.1% formic acid in water; mobile phase (B) consisting of 0.1% formic acid in acetonitrile. Gradient program: mobile phase (B) was held at 1% for 1.5 min, increased to 80% in 15 min, then to 99% in 17 min and held for 2 min before going to initial condition and held for 10 min. For hydrophilic interaction liquid chromatography (HILIC), a Waters Acquity UPLC BEH amide column (2.1 × 100mm, 1.7μm) was used in the negative ionization mode with mobile phase (A) consisting of 20 mM ammonium acetate (NH4OAc) in water at pH 9.6; mobile phase (B) consisting of acetonitrile (ACN). Gradient program: mobile phase (B) was held at 85% for 1 min, decreased to 65% in 12 min, then to 40% in 15 min and held for 5 min before going to the initial condition and held for 10 min.
Both columns were at 40 °C and 3 μl of each sample was injected into the LC-MS with a flow rate of 0.2 ml/min. Calibration was achieved through Agilent ESI-Low Concentration Tuning Mix. Optimization was performed on the 6490 QqQ in the RPLC-positive or HILIC-negative mode for each of 245 standard compounds (215 and 217 compounds for RPLC-positive and HILIC-negative, respectively) to obtain the best fragment ion and MS parameters such as fragmentation energy for each standard. Retention time (RT) for each standard was measured from a pure standard solution or a mix standard solution. The LC-MS/MS methods were created with dynamic MRM (dMRM) with RTs, RT windows, and transitions of all 245 standard compounds. Key parameters of electrospray ionization (ESI) in both the positive and the negative acquisition modes are: Gas temp 275 °C, Gas Flow 14 l/min, Nebulizer at 20 psi, SheathGasHeater 250 °C, SheathGasFlow 11 l/min, and Capillary 3000 V. For MS: Delta EMV 200V or 350V for the positive or negative acquisition mode respectively and Cycle Time 500ms and Cell Acc 4V for both modes. For 2-HG measurement, 5 million control or dox-treated cells were prepared and D-2HG levels measured according to established kit protocols (ab211070).
D-2HG, α-KG, and succinate measure measurement were performed by plating 5,000,000 cells (for D-2HG) and 800,000 cells (for α-KG and succinate). The samples preparation and assay procedure were performed as described in the kit protocol (D-2HG; ab211070, α-KG; ab83431, succinate; ab204718). The colorimetric signal was measured using a Synergy HTX Multi-Modeplate reader.
For 13C tracing, 2 million cells were plated on the 5 plates of T-25 and cultured using the media containing 13C-U-labeled glucose or glutamine for 16 hrs. Cells were collected by centrifugation and the medium was completely removed using an aspirator. 1ml of 80% methanol that was chilled on dry ice was added in the cell pellet. The cell pellet was suspended and placed at −80°C for 10 min. The metabolite was harvested by centrifuging at 700g, 4°C for 10 min and transferring to a chilled tube. Protein concentration was measured in parallel. The aliquot metabolite was normalized to protein concentration and transferred to a new tube. The metabolite pellet was obtained by speedvac at 4°C. Agilent 1290 UHPLC and 6530 Accurate-Mass Q-TOF LC/MS were used for stable 13C-labeled glucose or glutamine metabolomics tracing experiments. Agilent MassHunter Workstation Software LC/MS Data Acquisition for 6200 series TOF/6500 series Q-TOF (B.06.01) was used for calibration and data acquisition. For chromatography, a Phenomenex Luna NH2 column (5 μm,1.0 × 150mm, 1.7μm) was used with mobile phase (A) consisting of 5 mM ammonium acetate, pH 9.6 in water; mobile phase (B) consisting of acetonitrile with a flow rate of 0.075 ml/min. Gradient program: mobile phase B was started at 80%, decreased to 0% in 15 min and held for 4 min before going to the initial condition with an increased flowrate of 0.09 ml/min and held for 3 min. The column was held at 25 °C and 10 μL of the sample was injected into the LC-MS. Calibration of TOF MS was achieved through Agilent ESI-Low Concentration Tuning Mix. Key parameters were: mass range 100–1400 Da, Gas temp 285 °C, Fragmentor 148 V, Skimmer 80 V, Drying Gas 8 l/min, Nebulizer at 30 psi and Vcap 3500 V, Ref Nebulizer at 10 psi. Negative mode was used and the reference ions were at 119.0363 and 980.01637 Da.
Computational data post-processing, quality control, and statistical analysis
Pre-processed data with Agilent MassHunter Workstation Software Quantitative Analysis were post-processed for further quality control in the programming language R. First, we examined the distribution of sums of all metabolite abundance peak areas across individual samples in a given experiment as a measure for equal sample loading into the instrument. Next, we calculated coefficients of variation (CVs) in all biological replicate groups for each metabolite given a cut-off value of peak areas in each of the RPLC and HILIC methods. We then compared distributions of CVs for the whole dataset for a set of peak area cut-off values of 0, 1000, 5000, 10000, 15000, 20000, 25000 and 30000 in each method. A noise cut-off value of peak areas in each method was chosen by manual inspection of the CV distributions. The noise-filtered data of individual samples were then normalized by the total intensity of all metabolites. We retained only those metabolites with at least 2 technical replicate measurements for a given experimental variable. Then, each metabolite abundance level in each sample was divided by the mean of all abundance levels across all samples in a given experiment for comparisons, statistical analyses, and visualizations among metabolites. This normalization and scaling method has been used in our previous studies with biologically meaningful results. (Halbrook et al., 2019) Pathway analysis was done using the webtool, MetaboAnalyst (https://www.metaboanalyst.ca/)
For 13C-tracing analysis, we used Agilent MassHunter Workstation Software Profinder B.08.00 with Batch Targeted Feature Extraction and Batch Isotopologue Extraction and Qualitative Analysis B.07.00. Various parameter combinations, e.g. m/z and RT tolerance, were used to find the best peaks and signals by manual inspection. Key parameters were: mass tolerance = 15 ppm and RT tolerance = 1 or 0.5 min. Isotopologue ion thresholds, the anchor ion height threshold was set to 250 counts and the threshold of the sum of ion heights to 1000 counts. Co-elution correlation threshold was set to 0.3.
Magnetic resonance spectroscopy (MRS)
MRS was conducted as previously described. (Panigrahy et al., 2006) MRS studies were integrated with routine pre/post contrast MR imaging and were conducted on a 1.5T and 3T MR systems (Signa LX, GE Healthcare, Milwaukee, Wisconsin). Single-voxel point-resolved spectroscopy (PRESS) with a short echo time (TE) of 35 ms, a repetition time (TR) of 1.5 seconds, and 128 signal averages was used for all acquisitions. Sizes and shapes of the ROIs were adjusted to lesion size and typically varied between 5 to 10cm3. Total acquisition time, including scanner adjustments, was less than five minutes per spectrum. Spectra were processed with fully automated LCModel (Stephen Provencher Inc., Oakville, Ontario, Canada, LCModel Version 6.3–1L) software. (Provencher, 1993) T2-weighted fast spin-echo, FLAIR, and T1-weighted FLAIR images were acquired in all cases and the position of the region of interest (ROI) was documented on at least three MR images. The evaluation of MRS data included metabolites that are deemed to be measured reliably and have been reported in various previous studies (Bluml et al., 2011; Panigrahy et al., 2006). These were glutamate (Glu), glutamine (Gln), citrate (Cit), alanine (Ala) and glycine (Gly). Absolute concentrations (institutional units (i.u.)) were determined by using the unsuppressed water signal as reference signal and the default water content set by the LC Model software (65%). For each patient imaged, neuropathology, H3K27M and H3K27me3 immunostaining from corresponding biopsied tumor tissues were assessed by two neuropathologists in a blinded manner.
Immunohistochemistry and quantification
Immunohistochemistry was performed using serial sections from patient tumor sections or mouse brain sections. The sections were deparaffinized using xylene and rehydrated by decreasing concentrations of ethanol in the order of 100%, 95%, and water. Antigen retrieval was treated using a low-boiled 10mM citrate buffer, pH 6.0. Endogenous peroxidase activity was quenched by 3% hydrogen peroxide. Blocking was performed using 5% goat serum, 1% BSA. Sections were incubated for 5 h with one of the following rabbit polyclonal antibodies: anti-H3K27me3 (07–449, Millipore, 0.1 μg/mL), H3K27ac (D5E4 XP, Cell Signaling, 1:150), H3K27M (anti-H3 K27M, Millipore, #ABE419; 0.5 μg/mL) or rabbit polyclonal anti-SLC1A5 (#HPA035240; SIGMA, 0.5 μg/ml dilution). Tissue sections were then incubated for 60 min with biotinylated goat anti-rabbit IgG (PK6101, Vector Labs) at 1:200 dilution. The stained sections were visualized using 3,3’Diaminobenzidine (DAB) or Fast Red. The Slides were then rinsed, dehydrated, mounted and cover slipped.
Stained sections were scanned and quantified in a blinded manner. For automated scoring, each slide was scanned using an AperioScaNSCope Scanner (Aperio Vista) and viewed through AperioImageScope software program. For double staining experiments, the same stained section was visualized through the brown, red or combined channels with all parameters kept constant. An individual blinded to the experimental design captured JPEG images from each core (circular area of 315 cm2 corresponding to the entire core) at 10X magnification on the AperioImageScope viewing program. Cells that were positive for H3K27M were counted as H3K27me3 positive or H3K27me3 negative in three non-overlapping high power fields.
Quantification of immunostaining on each JPEG was conducted using an automated analysis program with Matlab’s image processing toolbox, based on previously described methodology. (Panwalkar et al., 2017) The algorithm used color segmentation with RGB color differentiation, K-Means Clustering, and background-foreground separation with Otsu’s thresholding. To arrive at a score, the numbers of extracted pixels were multiplied by their average intensity for each core (represented as pixel units). The final score for a given case and marker was calculated by averaging the score of two cores (for each case) for a given marker.
Knockdown of genes using shRNA virus and siRNA transfection
To knockdown genes using shRNA, 1.5 million HEK293T cells were plated on 100mm dishes 24 hrs before transfection. 2.5ug transfer DNA, 2.5ug Packaging DNA Premix, and 15ul TransIT-Lenti Reagent were mixed in 500ul Opti-MEM media using a pipette and incubated at room temperature for 10 min. The mixture was added dropwise to the HEK293T cell dish and swirled gently to distribute. The transfected cells were incubated at 37°C and 5% CO2 to produce virus for 72 hrs. Lentivirus particles were harvested by centrifugation at 300g for 5min and filtered using 0.45um PVDF filter. Lentivirus particles were plated onto dissociated target cells. After 18 hrs, lentiviral-particle-containing media was removed and target cell media was added. After 3 days, infected cells were treated with antibiotic for selection. To knockdown genes using siRNA, 3 million cells were plated in a T25-flask. 15ul of TransIT-X2 and 13ul of 10uM siRNA were mixed in 500ul Neurobasal A media and incubated for 30 min. The mixture was added dropwise to the cells and swirled gently to distribute. The transfected cells were incubated at 37°C and 5% CO2 for 72 hrs.
Xenograft and drug administration in vivo
For subcutaneous injection into flank, cells were isolated and counted. 100,000 cells in 50 μl of medium was mixed with 50 μl of Matrigel on ice. Cells were then injected subcutaneously into the flank of 8-week-old athymic nude mice. For drug treatment, mice were randomly divided into two groups at 8 weeks after implantation. The mice in the drug treatment group were injected intraperitoneally with DON (1mg/kg) every other day for 6 weeks or D-2HG (25mg/kg) every other day for 2 weeks. Tumor growth was measured weekly using calipers. Tumor volume (V) was calculated using the formula V= L × W × W / 2. V is volume (mm3), L is the longest diameter (mm), and W is the shortest diameter (mm).
For orthotopic xenografts into the pons, 400,000 cells/ 2μl of bioluminescent HSJD-DIPG-007 or SU-DIPG-XIII *P cells were injected into each mouse. NSG mice were anesthetized with 75mg/kg dexmedetomidine and 0.25mg/kg ketamine by intraperitoneal injection. 5mg/kg carprofen was used for analgesia. Mice were mounted on a stereotaxic device. A small sagittal incision was made using a scalpel and a small hole was created using a micro drill at 1.0 mm posterior and 0.8 mm lateral left from lambda. A sterile Hamilton syringe was used to inject cells. Half of the cells were injected at 5 mm depth from the inner base of the skull and the remaining cells were injected after 0.5 mm retraction in order to implant cells into the pontine tegmentum. After surgery, 1mg/kg atipamezole solution was intraperitoneally injected for anesthesia reversal. Tumor engraftment was confirmed by bioluminescence imaging. For 2-DG treatment, mice were randomly divided into two groups at 3-weeks after implantation and were injected intraperitoneally with vehicle or 2-DG (500mg/kg) every other day for 3 weeks. For inhibition of IDH1 and glutamine metabolism, orthotopic xenograft mice were administered with IDH1 inhibitor #13 (10mg/kg) by intraperitoneal injection or JHU-083 (20mg/kg) by oral gavage as described in Figure S6 b and c. Tumor size was measured using bioluminescent imaging (IVIS).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in consultation with the department of bioinformatics. The sample size (n) along with the statistical test performed and corresponding p-values are indicated in each figure and figure legend. No samples or animals were excluded from data analyses. Because H3K27M-midline gliomas are rare, sample sizes were determined based on number of tumor samples available. Similarly, sample size for MRS imaging was determined in a retrospective manner based on number of patients that were imaged. The exact n for each experiment is included in each figure legend. Data are represented as the means ± standard deviation. Box-and-Whisker plots indicate maximum, mean and minimum. Graphs were plotted and statistical analyses were performed using Prism software (version 7/8, GraphPad, La Jolla, CA). Unpaired, two-sided, two-tailed, Student’s t test, Chi square test, one-way or two-way analysis of variance (ANOVA) followed by post hoc Bonferroni multiple comparison analysis were used to analyze data as indicated. Survival analyses of data obtained from the PedcBioPortal was performed using median cut-off (high expression defined as tumor samples above median and low-expression defined as tumor samples below the median). Overall survival data including in animal models were plotted as Kaplan-Meier curves and the Log-rank test was used to examine the association of various factors with overall survival. Data were considered significant if p values were below 0.05 (95% confidence intervals).
Supplementary Material
HIGHLIGHTS.
H3.3K27M mutations enhance glucose, glutamine and TCA-cycle metabolism
TCA-cycle intermediate α-KG enables maintenance of H3K27 hypomethylation
Targeting enzymes related to α-KG synthesis including WT-IDH1 and/or GDH is therapeutic
H3.3K27M and mutant-IDH1 in gliomas are mutually exclusive and are synthetic lethal
ACKNOWLEDGMENTS
We would like to thank Dr. Ali Shilatifard for providing ChIP-seq data and Drs. Peter Lewis, Chao Lu and Lydia Finley for providing helpful comments. The authors would like to thank Drs. Venkatesha Basrur and AIexander Nesvizhskii for proteomics analyses and Dr. Masha G. Savelieff for expert editorial assistance.
We would like to thank the Chad Carr Foundation (led by Tammi Carr, Jason Carr and Lloyd Carr) and the University of Michigan Chad Carr Pediatric Brain Tumor Center (led by Drs. Valerie Opipari and Rajen Mody) for leading the fight against DIPG and supporting this work.
Funding: This work was supported by the Chad Tough Foundation (S.V.), NINDS R01NS110572 (S.V.), Mathew Larson Foundation (S.V.), St Baldrick’s Foundation (S.V.), Claire McKenna Foundation (S.V.), Alex Lemonade Stand Foundation (S.V.), Storm The Heavens Foundation (S.V.), and a joint Chad Tough Foundation and Michael Mosier Defeat DIPG Foundation fellowship award (C.C.). The Venneti lab is also supported by grants from the Sidney Kimmel, Doris Duke and Sontag Foundations and the Taubman Research Institute (S.V. is the Julie Reyes Taubman Scholar). The Blüml lab at Children’s Hospital Los Angeles has been supported by the Rudi Schulte Research Institute and the Ian’s Friends Foundation. C.K. acknowledges support from NIH/NINDS K08-NS099427-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
JBS and ZB declare the following competing financial interest(s): both are current employees of AbbVie. The small molecule WT-IDH1 inhibitors were provided by AbbVie after a material transfer agreement. AbbVie participated in the interpretation of inhibitor data, review, and approval of the publication. All other authors declare no competing interests.
REFERENCES
- Agnihotri S, Mansouri S, Burrell K, Li M, Mamatjan Y, Liu J, Nejad R, Kumar S, Jalali S, Singh SK, et al. (2019). Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin Cancer Res 25, 844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, Blanco MA, Giera S, Gillespie SM, Das J, et al. (2019). Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 36, 528–544 e510. [DOI] [PubMed] [Google Scholar]
- Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, Sabari B, Margol AS, Panwalkar P, Parolia A, et al. (2016). Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 8, 366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. [DOI] [PubMed] [Google Scholar]
- Bluml S, Panigrahy A, Laskov M, Dhall G, Krieger MD, Nelson MD, Finlay JL, and Gilles FH (2011). Elevated citrate in pediatric astrocytomas with malignant progression. Neuro Oncol 13, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, and Yan H (2012). A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, et al. (2018). Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Liu XY, Sturm D, and Jabado N (2013). Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol 23, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et al. (2009). The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A 106, 12932–12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, et al. (2019). Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab 29, 1390–1399 e1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaford AR, Alt J, Rais R, Wang SZ, Kaur H, Thorek DLJ, Eberhart CG, Slusher BS, Martin AM, and Raabe EH (2019). Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl Oncol 12, 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, et al. (2019). H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nature communications 10, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al. (2014). Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20, 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob CG, Upadhyay AK, Donner PL, Nicholl E, Addo SN, Qiu W, Ling C, Gopalakrishnan SM, Torrent M, Cepa SP, et al. (2018). Novel Modes of Inhibition of Wild-Type Isocitrate Dehydrogenase 1 (IDH1): Direct Covalent Modification of His315. J Med Chem 61, 6647–6657. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SE, White E, Eden C, et al. (2010). Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, Bertrand KC, Mikael LG, McConechy MK, Chen CCL, et al. (2019). Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 35, 782–797 e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, Fan Y, Shaw TI, Silveira AB, Qu C, et al. (2019). Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 35, 140–155 e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kremer DM, Sajjakulnukit P, Zhang L, and Lyssiotis CA (2019). A large-scale analysis of targeted metabolomics data from heterogeneous biological samples provides insights into metabolite dynamics. Metabolomics 15, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg KM, Vornov JJ, Rais R, and Slusher BS (2018). We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther 17, 1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, and Schofield CJ (2008). Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol 4, 152–156. [DOI] [PubMed] [Google Scholar]
- Losman J-A, and Kaelin WG (2013). What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27, 836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. [DOI] [PubMed] [Google Scholar]
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, et al. (2018). The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 33, 905–921 e905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales La Madrid A, Hashizume R, and Kieran MW (2015). Future Clinical Trials in DIPG: Bringing Epigenetics to the Clinic. Front Oncol 5, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al. (2017). TraNSCriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 31, 635–652 e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy A, Krieger MD, Gonzalez-Gomez I, Liu X, McComb JG, Finlay JL, Nelson MD Jr., Gilles FH, and Bluml S (2006). Quantitative short echo time 1H-MR spectroscopy of untreated pediatric brain tumors: preoperative diagnosis and characterization. AJNR Am J Neuroradiol 27, 560–572. [PMC free article] [PubMed] [Google Scholar]
- Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, Yip S, Hukin J, Sun Y, Schipper MJ, et al. (2017). Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Crane PD, Mietus LJ, and Oldendorf WH (1982). Kinetics of regional blood-brain barrier transport and brain phosphorylation of glucose and 2-deoxyglucose the barbiturate-anesthetized rat. J Neurochem 38, 560–568. [DOI] [PubMed] [Google Scholar]
- Patel SK, Hartley RM, Wei X, Furnish R, Escobar-Riquelme F, Bear H, Choi K, Fuller C, and Phoenix TN (2019). Generation of diffuse intrinsic pontine glioma mouse models by brainstem targeted in utero electroporation. Neuro Oncol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher SW (1993). Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30, 672–679. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. (2012). IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. (2012). Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 22, 425–437. [DOI] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, and Teitell MA (2016). alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab 24, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, and Judkins AR (2013). Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol 23, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Santi M, Felicella MM, Yarilin D, Phillips JJ, Sullivan LM, Martinez D, Perry A, Lewis PW, Thompson CB, and Judkins AR (2014). A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol 128, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, and Thompson CB (2017). Metabolic Reprogramming in Brain Tumors. Annu Rev Pathol 12, 515–545. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at NCBI/ GEO (GSE135419)