

Abstract
Myeloid cell leukemia-1 (Mcl-1) is a structurally and functionally unique anti-apoptotic Bcl-2 protein. While elevated levels of Mcl-1 contribute to tumor cell survival and drug resistance, loss of Mcl-1 in cardiac myocytes leads to rapid mitochondrial dysfunction and heart failure development. Although Mcl-1 is an anti-apoptotic protein, previous studies indicate that its functions extend beyond regulating apoptosis. Mcl-1 is localized to both the mitochondrial outer membrane and matrix. Here, we have identified that Mcl-1 in the outer mitochondrial membrane mediates mitochondrial fission, which is independent of its anti-apoptotic function. We demonstrate that Mcl-1 interacts with Drp1 to promote mitochondrial fission in response to various challenges known to perturb mitochondria morphology. Induction of fission by Mcl-1 reduces nutrient deprivation-induced cell death and the protection is independent of its BH3 domain. Finally, cardiac-specific overexpression of Mcl-1OM, but not Mcl-1Matrix, contributes to a shift in the balance towards fission and leads to reduced exercise capacity, suggesting that a pre-existing fragmented mitochondrial network leads to decreased ability to adapt to an acute increase in workload and energy demand. Overall, these findings highlight the importance of Mcl-1 in maintaining mitochondrial health in cells.
Keywords: Mcl-1, Drp1, mitochondria, fission, heart
Graphical abstract
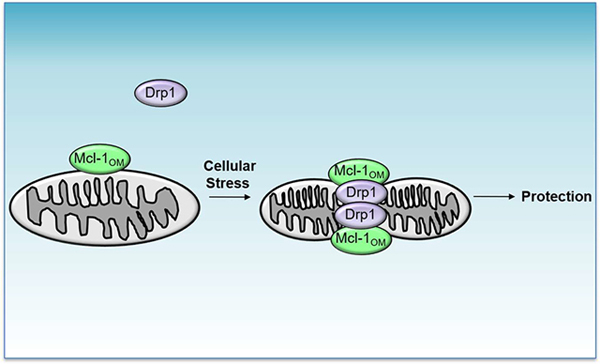
1. Introduction
The Bcl-2 family proteins are mostly known for their roles in regulating apoptosis. They function primarily at the mitochondria, where anti-apoptotic proteins preserve mitochondrial integrity while pro-apoptotic members cause permeabilization of the outer mitochondrial membrane and release of pro-apoptotic proteins. They are often deregulated in a variety of human cancers and elevated levels of the anti-apoptotic proteins contribute to tumor cell survival and drug resistance [1, 2]. In other tissues, such as the heart and brain, the anti-apoptotic proteins protect cells during stress [3, 4]. Preventing unnecessary cell death is particularly important in post-mitotic cells such as neurons and cardiac myocytes, which cannot be replaced when lost. Thus, concerns regarding potential harmful off-target effects with cancer therapies targeting the anti-apoptotic proteins exist.
Myeloid Cell Leukemia 1 (Mcl-1) is an anti-apoptotic Bcl-2 protein that has overlapping functions with Bcl-2 and Bcl-XL in preventing permeabilization of the outer mitochondrial membrane. However, Mcl-1 is also a structurally and functionally unique protein within this family. Bcl-2 proteins share up to four evolutionary conserved Bcl-2 homology (BH) domains, which are important for their function. While anti-apoptotic Bcl-2, Bcl-XL and Bcl-w share four evolutionary conserved Bcl-2 homology (BH) domains (BH1–4), Mcl-1 lacks the BH4 domain [5]. Mcl-1 also has a distinct N-terminal domain with regions enriched in proline, glutamic acid, serine, and threonine (PEST) residues, which ensures its rapid proteasomal degradation [5]. Thus, Mcl-1 has a much shorter half-life compared to other Bcl-2 family proteins [6]. Despite the redundant functions in the anti-apoptotic proteins, the phenotypes of mice deficient in each of the various anti-apoptotic proteins differ [7–10]. These findings clearly point to a functional specificity for these individual proteins.
There is strong evidence that the function of Mcl-1 extends beyond regulating apoptosis. For instance, Mcl-1 localizes to two distinct mitochondrial locations: the outer mitochondrial membrane (OMM) and the matrix [11]. In the matrix, Mcl-1 is involved in regulating bioenergetics while Mcl-1 in the OMM is responsible for inhibiting apoptosis [11]. Mcl-1 has also been reported to inhibit autophagy by sequestering Beclin1 [12] and to promote mitochondrial fission by interacting with the mitochondrial fission protein Drp1 [13, 14]. Moreover, we and others previously discovered that Mcl-1 is essential for maintaining cardiac homoeostasis and that cardiac-specific deletion of Mcl-1 in mice results in mitochondrial dysfunction and swelling with rapid development of heart failure [15, 16]. The absence of apoptotic myocytes in Mcl-1-deficient hearts indicates that the mitochondrial impairment originates from loss of other Mcl-1 key function(s).
Although the mitochondrial impairment caused by Mcl-1-deficiency appears to be independent of its anti-apoptotic function, why loss of Mcl-1 has such profound effects on mitochondrial structure and function is still unclear. There is currently a strong interest in developing small molecule inhibitors that target Mcl-1 in cancer cells. However, increased understanding of how Mcl-1 functions in various cells is still needed so that potential off-target effects can be minimized or avoided. In this study, we have explored alternative functions of Mcl-1 in cells and the heart. We report that Mcl-1 in the OMM induces mitochondrial fission via the large GTPase Drp1, which allows cells to adapt to stress. Importantly, this pro-survival function is independent of its traditional anti-apoptotic function.
2. Material and Methods
2.1. Animals
Mcl-1OM and Mcl-1Matrix transgenic mice were generated on a C57Bl/6 background at the UCSD Mouse Transgenic Core facility and overexpress Mcl-1OM or Mcl-1Matrix under the transcriptional control of the cardiomyocyte-specific α-myosin heavy chain (α-MHC) promoter. Age- and sex-matched 10–12 week-old mice were used for experimental procedures and data represents results obtained from both sexes. All experiments were approved by the Institutional Animal Care and Use Committee at the University of California, San Diego.
2.2. Cell Culture
Immortalized mouse embryonic fibroblasts (MEFs) were maintained in culture media composed of DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Gemini) and cultured at 37 °C in 5% CO2. For nutrient deprivation experiments, cells were cultured in glucose-and serum-free media (Thermo Fisher Scientific). For hypoxia experiments, cells were placed in cell culture media supplemented with 40 mM HEPES pH 7.4 (Thermo Fisher Scientific) and incubated in hypoxic pouches (GasPak EZ, BD Biosciences) equilibrated to 95% N2, 5% CO2.
2.3. Adenoviral Constructs and siRNA knockdown
Mouse Mcl-1, Mcl-1OM, and Mcl-1Matrix cDNA constructs were provided by Dr. Joseph Opferman from St. Jude Children’s Research Hospital [11]. The HA-Mcl-1-BH3-mutant (G198E D199A) replaces glycine 198 with glutamic acid and aspartic acid 199 with alanine and the Mcl-1-Bcl-2-chimera replaces Mcl-1’s BH1 (aa 234–253), BH2 (aa 285–300), and BH3 (aa 190–204) domains with those from Bcl-2 (aa 133–152, 184–199, and 190–204) and were synthesized by GenScript. Adenoviruses encoding the above proteins were generated using the pENTR directional TOPO cloning kit (Invitrogen) followed by recombination into the pAd/CMV/V5-DEST Gateway vector (Invitrogen).
Drp1 knockdown was performed by transfecting 50 nM Mission siRNA Universal Negative Control #1 (Sigma, SIC001) or Drp1 small interfering RNA (siRNA) (Sigma, SASI_Mm01_000125369) with Lipofectamine RNAiMax Transfection Reagent (Thermo Fisher Scientific) for 72 hours. Cells were infected with adenoviruses in DMEM+2% heat-inactivated serum for 3 hours and rescued with culture media.
2.4. Western Blot, Co-Immunoprecipitation and BN-PAGE Experiments
Western blots and immunoprecipitation experiments were performed as previously described [17]. Mitochondria were isolated from hearts by differential centrifugation as described [18]. Protein concentrations were determined by Bradford assay. Proteins were separated on Invitrogen NuPAGE Bis-Tris gels and transferred to nitrocellulose membranes. For immunoprecipitation experiments, samples were pre-cleared with protein A PLUS-agarose (Santa Cruz Biotechnology), and then incubated with 2.5 μg anti-Mcl-1 antibody (Rockland, 600–401-394) overnight. Immunocomplexes were captured with protein A PLUS-agarose beads.
The following antibodies were used to probe the membranes: Actin (1:1000; GeneTex, GTX11003), Bak (1:500, Millipore Upstate, 06–536), Bax (1:1000, Cell Signaling, 2772), Bcl-2 (1:1000, BD Biosciences, 610539), Bcl-XL (1:1000, Santa Cruz Biotechnology, sc-8392), Drp1 (1:1000, BD Biosciences, 611113), Fis1, (1:1000, Abcam, ab96764), Gapdh (1:1000; GeneTex, GTX627408), Mcl-1 (1:1000; Rockland, 600–401-394), Mff (1:1000, Proteintech, 17090–1-AP), Mfn1 (1:1000; Santa Cruz Biotechnology, sc-50330), Mfn2 (1:1000; MilliporeSigma, M6319), MitoProfile Total OXPHOS Rodent WB Antibody cocktail (1:1000; Abcam/MitoScience, MS604), MnSOD (1:1000; Millipore, 06–984), Opa1 (1:1000, BD Biosciences, 612607), phospho-Drp1 (Ser616) (1:1000, Cell Signaling, 4494s), Tim23 (1:1000; BD Biosciences, 611222), Tom20 (1:1000, Santa Cruz Biotechnology, sc-11415), and Tubulin (WB 1:1000; Sigma, T6074). Membranes were imaged using a ChemiDoc XRS+ System (Bio-Rad). Band densitometry quantification was performed using the Image Lab software (Bio-Rad).
Mitochondrial respiratory chain supercomplexes were assessed by BN-PAGE as previously described [19]. Briefly, isolated mitochondria were solubilized by digitonin (4g/g protein) for 20 minutes and then centrifuged at 20,000 g for 30 minutes. Supernatants were collected and resolved on Invitrogen NativePAGE 3–12% Bis-Tris gels and transferred to PVDF membranes. Membranes were probed with antibodies against Ndufb8 (1:1000, Abcam, ab192878), Uqcrfs1 (1:1000, Proteintech, 18443–1-AP), and CoxIV (1:1000, Proteintech, 11242–1-AP).
2.5. Immunofluorescence
Cells were fixed in 4% paraformaldehyde (Thermo Fischer Scientific), permeabilized with 0.2% Triton X-100 in PBS and blocked in 5% normal goat serum (Vector Laboratories Inc.). Cells were incubated with an antibody against Tom20 (Santa Cruz Biotechnology, sc-11415) overnight, washed and incubated with an Alexa Fluor 594 secondary antibody (Thermo Fisher Scientific, A11037). Cells were also stained with Hoechst 33342 to label nuclei (Thermo Fisher Scientific, H3570). Fluorescence images were captured using a Carl Zeiss Axio Observer Z1 or a Nikon Eclipse microscope. Mitochondrial morphology was assessed based on counts of at least 200 cells per dish repeated for 3 separate experiments. Cell viability was assessed by staining cells with YO-PRO-1 (1:1,000; Thermo Fisher Scientific, Y3603) plus Hoechst 33342 for 20 min. Cell death was determined by the number of YO-PRO-1-positive cells divided by the total number of Hoechst 33342-positive cells as previously described [20].
2.6. Echocardiography
Echocardiography was performed using a Vevo770 In Vivo Micro-Imaging System with an RMV707B 15–45 MHz imaging transducer (VisualSonics Inc.) as previously described [15]. While the measurements were being acquired, mice were maintained under light anesthesia (0.5%–1% isoflurane, 98%–99.5% O2) on a recirculating water warming pad. The VisualSonics software was used to analyze and quantify cardiac parameters.
2.7. Histology
Hearts were arrested in diastole with 200 mM KCl, fixed in 10% neutral buffered formalin and then dehydrated in 70% alcohol before processing in a tissue processor (Thermo Scientific STP 120) and embedding in paraffin. A microtome (Leica Biosystems) was used to cut the hearts into 6-μm sections. Hematoxylin and eosin (H&E) was used to stain deparaffinized and rehydrated sections. Images were collected using the 10x objective of a Nikon Eclipse microscope.
2.8. Transmission Electron Microscopy
Transmission electron microscopy was performed on heart sections from adult WT, Mcl-1OM and Mcl-1Matrix transgenic mice as previously described [21]. 2.5% glutaraldehyde in 0.1 M cacodylate buffer was used to fix hearts, followed by post-fixation with 1% osmium tetroxide and treatment with 0.5% tannic acid, 1% sodium sulfate. Hearts were then cleared in 2-hydroxypropyl methacrylate and embedded in LX112 (Ladd Research). Copper slot grids coated with parlodion were used to mount sections, and they were stained with uranyl acetate and lead citrate. A Philips CM100 electron microscope (FEI) was used to examine sections and acquire images. Mean mitochondrial area (μm2) was quantified using ImageJ from measurements of 300 mitochondria from 2 hearts using 14 different sections at 7900 X magnification.
2.9. Mitochondrial Respiration
A Seahorse XFp Analyzer (Seahorse Bioscience – Agilent Technologies) was used to measure mitochondrial respiration as described [21]. Briefly, hearts were homogenized in buffer containing 70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% fatty acid-free BSA using a Polytron at 11,000 g followed by further grinding with a Potter-Elvehjem Teflon tissue grinder. Respiring mitochondria were isolated by differential centrifugation and protein concentration of the final mitochondrial pellet was determined by Bradford assay. 1 μg mitochondria per well was added to a 96-well microplate in assay media (in mM: 70 sucrose, 220 mannitol, 10 KH2PO4, 5 MgCl2, 2 HEPES, 1 EGTA, 0.2% BSA, and 4 ADP). 10 mM pyruvate/1 mM malate and 10 mM glutamate/10 mM malate were used as substrates for Complex I-dependent oxygen consumption and 10 mM succinate was used as a substrate for Complex II. ATP synthase was inhibited by addition of 2 μM oligomycin A (Sigma) and FCCP was added to obtain maximal respiration rate. Wave for Desktop (Seahorse Bioscience – Agilent Technologies) was used to analyze the data.
2.10. Quantitative PCR
Gene expression and mitochondrial DNA copy number were assessed as previously described [18]. Briefly, RNA was isolated from hearts using the RNeasy Fibrous Tissue Mini kit (Qiagen) and genomic DNA was extracted from hearts of WT and αMHC-Mcl-1OM mice using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma). cDNA was synthesized from RNA using the QuantiTect Reverse Transcription Kit (Qiagen) and genomic DNA was PCR-amplified with TaqMan Universal Master Mix II. Pgc-1α, Tfam and Rn18s TaqMan primers were purchased from Life Technologies-Thermo Fisher Scientific. 18S rRNA and D-loop were used for mtDNA quantitation. A Bio-Rad CFX96 Real-Time PCR Detection System was used to perform qPCR. To calculate fold change in gene expression, relative amounts of mRNA were normalized to Rn18s and the 2(–ΔΔCt) method was employed.
2.11. Swimming Experiments
Mice were pre-trained for 5 minutes on day 1, 5 minutes on day 2, 10 minutes on day 3 and all experiments were conducted on day 4. For the swimming time course, the mice swam for 0, 15, 30, 45, or 60 minutes and the hearts were collected immediately after for Western blotting analysis. For the weight-loaded swimming endurance test, weights equivalent to 7.5% of the mouse’s body weight were attached to the tail stem [22]. The mice swam until exhaustion (as assessed by 5 seconds continuously submerged under water) and the time they spent swimming was recorded.
2.13. Statistical Analysis
All values are expressed as mean ± standard error of mean (SEM). Student’s t test was used to evaluate differences between 2 sets of data. ANOVA followed by Dunnett’s or Tukey’s multiple comparison test was used for data comparing more than 2 groups using GraphPad Software (GraphPad Software, Inc., USA). P values of less than 0.05 were considered statistically significant.
3. Results
3.1. Mcl-1OM Promotes Fragmentation and Perinuclear Clustering of Mitochondria
Mcl-1 is a mitochondrial protein that was recently identified to localize to both the outer mitochondrial membrane and the matrix [11]. Mcl-1 contains a transmembrane domain that anchors it in the outer or inner membranes where it either faces the cytosol or the matrix. Here, we have confirmed that Mcl-1 is also present at both locations in cardiac mitochondria and that Mcl-1OM is the more abundant form in the heart (Figure 1A). We treated mitochondria that had been isolated from mouse hearts with proteinase K to digest proteins in the outer and inner membranes, including Mcl-1OM, while proteins in the mitochondrial matrix are protected from proteinase K (Figure 1A).
Figure 1. Mcl-1 induces fragmentation and perinuclear clustering of mitochondria.

(A) Western blotting for Mcl-1 in mitochondria isolated from a mouse heart. The mitochondria were treated with 200 ng/μl proteinase K to digest proteins in the outer mitochondrial membrane. Relative levels of Mcl-1OM and Mcl-1Matrix in cardiac mitochondria (n=4). (B) Various Mcl-1 constructs used to dissect its function in cells. MTS is the mitochondrial targeting sequence from the ATP synthase. (C) Representative images of MEFs stained with anti-Tom20 (red) and Hoechst 33342 (blue) to label the mitochondria and nucleus, respectively. (D) Quantitation of cells exhibiting fragmentation and perinuclear clustering of mitochondria (n=3). (E) Representative images of MEFs stained with anti-Tom20 (red) and Hoechst 33342 (blue). (F) Quantitation of cells exhibiting fragmentation and perinuclear clustering of mitochondria (n=3). (G) Representative images of MEFs stained with anti-Tom20 (red) and Hoechst 33342 (blue) to label the mitochondria and nucleus, respectively. (H) Quantitation of cells exhibiting fragmentation and perinuclear clustering of mitochondria (n=3). Scale bars=10 μm. *p<0.05, ****p<0.0001. A minimum of 200 cells were counted for each n. Data are mean + SEM.
To dissect the specific functions of the Mcl-1 in regulating mitochondrial health and function, we utilized two different constructs of Mcl-1, which are targeted to either the outer mitochondrial membrane (Mcl-1OM) or matrix (Mcl-1Matrix), respectively (Figure 1B). First, we examined their effect on mitochondrial function, morphology and survival in mouse embryonic fibroblasts (MEFs). These cells exhibit a filamentous mitochondrial network at baseline, but overexpression of wild type Mcl-1 leads to increased fragmentation and perinuclear clustering of mitochondria (Figures 1C-D). We also examined the effect of Mcl-1 on morphology at its two distinct mitochondrial locations. Similar to wild type Mcl-1, Mcl-1OM induces fragmentation and perinuclear aggregation of mitochondria in MEFs. However, Mcl-1Matrix has little effect on mitochondrial morphology (Figures 1C-D). Changes in mitochondrial morphology have been linked to mitochondrial clearance [23], but we found that the mitochondrial fragmentation induced by Mcl-1 or Mcl-1OM has no effect on mitochondrial content. Western blotting for mitochondrial proteins Tim23 and MnSOD shows that their levels remain unchanged when Mcl-1 or Mcl-1OM is overexpressed (Supplemental Figures S1A-B). Also, the change in mitochondrial morphology induced by Mcl-1 or Mcl-1OM does not affect mitochondrial membrane potential. We found that overexpression of Mcl-1 does not affect mitochondrial uptake of tetramethyl rhodamine methyl ester (TMRM) (Supplemental Figure S1C), a dye that only accumulates in mitochondria with intact membrane potential. Taken together, these findings indicate that Mcl-1 in the outer membrane regulates mitochondrial morphology and network structure without altering mitochondrial content or membrane potential.
3.2. Mcl-1-mediated perinuclear clustering of mitochondria requires a functional BH3 domain
Next, we investigated whether Mcl-1’s ability to induce changes in mitochondrial morphology is unique to its BH domains or conserved among other anti-apoptotic Bcl-2 family members by generating a Mcl-1-Bcl-2-chimera, in which Mcl-1’s BH1, BH2, and BH3 domains are replaced with those from Bcl-2 (Figure 1B). We confirmed that the Mcl-1-Bcl-2-chimera is just as effective as Mcl-1 in inducing perinuclear clustering of mitochondria (Figures 1E-F) without disrupting mitochondrial membrane potential (Supplemental Figure S1C). Interestingly, we noted that the Mcl-1-Bcl-2-chimera is slightly more effective in promoting mitochondrial fragmentation compared to Mcl-1 (Figure 1F). This indicates that the mitochondrial fragmentation induced by Mcl-1 is either independent of its BH domains or that any functional BH domains are sufficient for this function.
Mcl-1 is a well-known anti-apoptotic protein that inhibits permeabilization of the outer mitochondrial membrane by preventing Bax/Bak activation [1]. To examine the connection between Mcl-1’s effect on mitochondrial morphology and its anti-apoptotic function, we also mutated glycine 198 to glutamic acid (G198E) and aspartic acid 199 to alanine (D199A) in the BH3 domain to disrupt Mcl-1’s anti-apoptotic function [24] (Figure 1B). Interestingly, we found that while the Mcl-1-BH3-mutant does not cause the same perinuclear clustering of mitochondria observed with Mcl-1 and the Mcl-1-Bcl-2-chimera, it is still effective at inducing mitochondrial fission (Figures 1G-H) without altering mitochondrial membrane potential (Supplemental Figure S1C). This suggests that while a functional BH3 domain in Mcl-1 is not necessary to facilitate fission, it is required for the redistribution of mitochondria to the perinuclear region. Unexpectedly, we also found that the mitochondrial perinuclear clustering induced by wild type Mcl-1 is only a temporary response and that by 48 hours the fragmented mitochondria are dispersed throughout the cytosol (Figures 1G-H). Autophagy and mitochondrial content remained constant within the same time frame as assessed by Western blotting for LC3 and Tim23 (Supplemental Figure S1D-E).
3.3. Mcl-1 interacts with the mitochondrial fission protein Drp1
Mitochondrial fission is mediated by the large GTPase Drp1, which translocates to mitochondria to induce membrane scission into two separate portions [25]. To investigate if Mcl-1 induces mitochondrial fission via Drp1, we performed co-immunoprecipitation experiments between Mcl-1 and Drp1. Co-immunoprecipitation experiments confirm that Mcl-1, Mcl-1-Bcl-2-chimera, and Mcl-1-BH3-mutant interact with endogenous Drp1 when overexpressed in cells (Figure 2A). Next, we investigated whether various challenges known to induce Drp1-mediated fission also lead to enhanced interaction between endogenous Drp1 and Mcl-1. We found that nutrient deprivation or exposure to hypoxia leads to increased mitochondrial fission in MEFs, which coincides with enhanced interaction between endogenous Mcl-1 and Drp1 (Figures 2B-E). We also examined whether specific damage to mitochondria would promote a similar effect. Indeed, we found that treatment with the mitochondrial uncoupler FCCP or the Complex I inhibitor rotenone also induces mitochondrial fission and increases interaction between endogenous Mcl-1 and Drp1 (Figures 2F-I).
Figure 2. Mcl-1 interacts with the mitochondrial fission protein Drp1.

(A) Immunoprecipitation of Mcl-1 and Western blotting for Mcl-1 and Drp1. (B) Representative images and quantitation of cells with mitochondrial fragmentation in response to nutrient deprivation (n=3). (C) Immunoprecipitation of Mcl-1 after nutrient deprivation and Western blotting for Drp1 and Mcl-1. (D) Representative images and quantitation of cells with mitochondrial fragmentation in response to hypoxia (n=3). Mitochondria are stained with anti-Tom20 and nuclei with Hoechst 33342. (E) Immunoprecipitation of Mcl-1 after exposure to hypoxia and Western blotting for Drp1 and Mcl-1. (F) Representative images and quantitation of cells with mitochondrial fragmentation in response to 25 μM FCCP (60 minutes). (G) Immunoprecipitation of Mcl-1 after treatment with 25 μM FCCP and Western blotting for Drp1 and Mcl-1. (H) Representative images and quantitation of cells with mitochondrial fragmentation in response to 1 μM rotenone (2 hours) (n=3). Mitochondria are stained with anti-Tom20 and nuclei with Hoechst 33342. (I) Immunoprecipitation of Mcl-1 after treatment with 1 μM rotenone and Western blotting for Drp1 and Mcl-1. Scale bars=10 μm. **p<0.01, ***p<0.001,****p<0.0001. A minimum of 200 cells were counted for each n. Data are mean + SEM.
To further confirm that Drp1 is involved in regulating Mcl-1-mediated mitochondrial fission and perinuclear clustering, we used siRNA to knockdown Drp1 in MEFs prior to Mcl-1 overexpression (Figure 3A). We found that knockdown of Drp1 leads to preservation of the mitochondrial network in cells overexpressing Mcl-1 (Figure 3B), confirming the link between Mcl-1 and Drp1. Mitochondrial fission can be either protective or detrimental in response to stress [26, 27]. Therefore, we assessed whether disrupting Mcl-1-mediated fission would affect cell viability following nutrient deprivation. We found that both Mcl-1 and the Mcl-1-BH3-mutant protect against cell death during nutrient deprivation (Figure 3C). However, the protection by Mcl-1 or the Mcl-1-BH3-mutant is abrogated in cells with Drp1 knockdown (Figure 3C). There were no significant changes in mitochondrial content in response to nutrient deprivation in cells overexpressing either β-gal or Mcl-1, indicating that mitophagy is not elevated (Supplemental Figure S1F). These findings suggest that Mcl-1-mediated mitochondrial fission via Drp1 is a pro-survival function that is distinct from its anti-apoptotic function.
Figure 3. Mitochondrial fission induced by Mcl-1 is dependent on Drp1.

(A) Western blot for Drp1 levels in MEFs after transfection with 50 nM control or Drp1 siRNA. (B) Representative images and quantitation of cells with fragmented mitochondria (n=3). (C) Cell death after 24 h of nutrient deprivation. Scale bars=20 μm. *p<0.05, ****p<0.0001. A minimum of 200 cells were counted for each n. Data are mean + SEM.
3.4. Mcl-1OM increases mitochondrial fission in vivo
To investigate if Mcl-1OM also promotes mitochondrial fission in vivo, we generated cardiac-specific Mcl-1OM transgenic mice using the α-MHC promoter (αMHC-Mcl-1OM) (Supplemental Figure S2A). A proteinase K protection assay confirmed that Mcl-1OM in transgenic hearts localizes to the outer mitochondrial membrane (Figure 4A). Hematoxylin and eosin (H&E) staining of heart sections show that the architecture of cardiac myocytes is similar in WT and αMHC-Mcl-1OM hearts (Figure 4B). The αMHC-Mcl-1OM transgenic mice also have similar cardiac structure and function to WT mice as measured by echocardiography. WT and αMHC-Mcl-1OM mice have similar ejection fraction (EF), fractional shortening (FS) (Figure 4C), and left ventricular internal end-diastolic and systolic dimensions (LVID;d and LVID;s) (Supplemental Figure S2B). WT and αMHC-Mcl-1OM mice also have similar heart size (Figure 4D and Supplemental Figure S2C). We also examined whether overexpressing Mcl-1 would lead to compensatory changes in other Bcl-2 proteins. However, we found no changes in anti-apoptotic Bcl-2 or Bcl-XL or in pro-apoptotic Bax or Bak protein levels in αMHC-Mcl-1OM mouse hearts compared to WT (Supplemental Figures S2E-F). Overall, our findings demonstrate that increased levels of Mcl-1OM have little effect on cardiac structure or function under baseline conditions.
Figure 4. Mcl-1OM increases mitochondrial fission.

in vivo (A) Western blot analysis of proteinase K treated cardiac mitochondria confirming that Mcl-1OM localizes to the outer mitochondrial membrane. Mitochondria from αMHC-Mcl-1OM hearts were treated with 200 ng/μl proteinase K. Tom20 was used as an outer mitochondrial membrane marker and MnSOD was used as a marker for the mitochondrial matrix. (B) WT and αMHC-Mcl-1OM heart sections stained with hematoxylin and eosin (H&E). Scale bars=500 μm. (C) Echocardiographic analysis revealed similar ejection fraction (EF) and fractional shortening (FS) in WT and αMHC-Mcl-1OM hearts (n=8). (D) Heart weight/body weight (HW/BW) ratio is similar in WT and αMHC-Mcl-1OM and WT mice (n=8). (E) Representative transmission electron micrographs of mitochondria in WT and αMHC-Mcl-1OM hearts. Quantitation of the mean mitochondrial area (μm2) (300 mitochondria were counted). (F) Representative Western blots of mitochondrial fission proteins in WT and αMHC-Mcl-1OM hearts. Quantitation of Drp1, Fis1, and Mff levels normalized to loading controls (n=5–6). (G) Representative Western blots of mitochondrial fusion proteins in WT and αMHC-Mcl-1OM hearts. Quantitation of Mfn1, Mfn2, and Opa1 protein levels normalized to loading control (n=5–6). (H) Western blot analysis and quantitation of Drp1 in mitochondrial fractions prepared from WT and αMHC-Mcl-1OM hearts (n=4). (I) Western blot analysis and quantitation of Drp1 and p-Drp1 (S616) in mitochondrial fractions prepared from WT and αMHC-Mcl-1OM hearts (n=6). (J) Immunoprecipitation of Mcl-1 from WT and αMHC-Mcl-1OM heart lysates and subsequent Western blotting with anti-Mcl-1 and anti-Drp1. IgG was used as a control. *p<0.05. Data are mean + SEM.
Next, we examined the effect of Mcl-1OM on mitochondrial morphology in cardiac myocytes. Transmission electron microscopy (TEM) showed that mitochondria in αMHC-Mcl-1OM hearts have decreased mean mitochondrial area (μm2) compared to WT (Figure 4E). Mitochondrial morphology is often dictated by levels of proteins involved in regulating fission and fusion. Interestingly, we found no significant changes in the overall levels of the mitochondrial fission proteins Drp1 and Fis1 (Figure 4F) or fusion proteins Mfn1, Mfn2, and Opa1 (Figure 4G) in hearts from αMHC-Mcl-1OM mice when compared to WT hearts. Of note, Mff, which is a mitochondrial receptor for Drp1, is significantly reduced in the transgenic mouse hearts (Figure 4F). Drp1 is normally localized in the cytosol, but phosphorylation of serine 616 by various kinases leads to its translocation to mitochondria and induction of fission [28]. Consistent with enhanced fission, Drp1 levels are significantly increased at the mitochondria in the hearts of αMHC-Mcl-1OM mice (Figure 4H). Unexpectedly, the levels of phosphorylated Drp1 at the mitochondria are not increased in the transgenic hearts (Figure 4I). Co-immunoprecipitation experiments also confirmed an enhanced interaction between Mcl-1 and Drp1 in αMHC-Mcl-1OM hearts (Figure 4I). Overall, these findings suggest that Mcl-1OM induces Drp1-mediated mitochondrial fission in hearts.
Given that mitochondrial morphology has been linked to mitochondrial bioenergetics and mitophagy [29, 30], we also examined the effect of elevated levels of Mcl-1OM on mitochondrial function and content in the heart. Using mitochondria isolated from WT and αMHC-Mcl-1OM hearts, we assessed mitochondrial respiration in the presence of substrates for Complex I (pyruvate/malate and palmitoyl carnitine/malate) or II (succinate/rotenone). The mitochondria had similar respiratory rates for all three substrates (Figure 5A). We also compared levels of various mitochondrial proteins and found that, with the exception of Tim23, the levels were similar in WT and αMHC-Mcl-1OM hearts (Figures 5B-C). We also observed no differences in Pgc-1α and Tfam transcript levels, markers of mitochondrial biogenesis, (Supplemental Figure S2D) or in mitochondrial DNA (mtDNA) copy number in these hearts (Figure 5D). Moreover, the electron transport chain complexes organize into various supercomplexes, which enhance the efficiency of electron transfer while limiting ROS production [31]. Although it has been previously observed that deletion of Mcl-1 in the liver leads to abnormal assembly of the supercomplexes [11], overexpression of Mcl-1OM has no effect on their formation in cardiac mitochondria (Figure 5E). Taken together, these findings confirm that elevated levels of Mcl-1OM in hearts have no major effect on baseline mitochondrial function or content.
Figure 5. αMHC-Mcl-1OM mice have no alterations in mitochondrial content or function.

(A) Mitochondrial respiration was assessed using a Seahorse XFp analyzer in response to three different substrates: pyruvate/malate, glutamate/malate, and succinate (n=6). OCR = oxygen consumption rate. (B) Representative Western blots and quantitation of mitochondrial proteins Tim23 and Tom20 in WT and αMHC-Mcl-1OM mouse hearts. (n=5–6). (C) Representative Western blot analysis and quantitation of mitochondrial OXPHOS proteins in WT and αMHC-Mcl-1OM mouse hearts (n=5–6). (D) Quantitation of mitochondrial DNA (mtDNA) content normalized to genomic DNA (nDNA) in hearts of WT and αMHC-Mcl-1OM mice (n=4). (E) Representative BN-PAGE immunoblots of individual ETC complexes and supercomplexes (SCs) in WT and αMHC-Mcl-1OM cardiac mitochondria (n=4). SCs were visualized by antibodies against subunits of complex I (CI, Ndufb8), complex III (CIII, Uqcrfs1), and complex IV (CIV, CoxIV). (F) Representative Western blots of mitochondrial Drp1 in hearts of WT and αMHC-Mcl-1OM mice after swimming. (G) Quantitation of mitochondrial Drp1 levels after swimming (n=3–5). (H) Swim time of WT and αMHC-Mcl-1OM mice in response to weight-loaded swimming endurance test (n=7). *p<0.05, ***p<0.001. Data are mean + SEM.
Next, we examined potential physiological consequences of enhanced cardiac mitochondrial fission in αMHC-Mcl-1OM mice. To distinguish between Mcl-1’s anti-apoptotic and pro-fission functions, we focused on exercise, which is associated with enhanced fission in heart and skeletal muscle without activation of apoptosis [27, 32]. In the heart, induction of fission has been reported to play a role in bioenergetic adaptation to exercise [27]. First, we performed a time course experiment to confirm induction of mitochondrial fission in hearts in response to swimming. Consistent with previous findings, we observed that Drp1 translocates to cardiac mitochondria after 60 minutes of swimming (Figure 5F-G). Interestingly, we failed to observe a significant increase in mitochondrial Drp1 levels in αMHC-Mcl-1OM hearts in response to swimming, likely due to the pre-existing increased fission at baseline. We also found much greater variability in Drp1 translocation after swimming in the αMHC-Mcl-1OM hearts compared to WT. With the existing shift in mitochondrial morphology, we investigated whether this affected the mice ability to adapt to exercise. We utilized a weight-loaded swimming test to examine exercise capacity in the mice [33]. The weight ensures continuous swimming of the mice and prevents floating. This test revealed that αMHC-Mcl-1OM mice have significantly reduced exercise capacity compared to WT mice (Figure 5H). Thus, a pre-existing shift in the balance towards fission in the heart might reduce the ability to adapt to intense exercise.
Finally, to compare the effects of Mcl-1OM and Mcl-1Matrix on cardiac mitochondrial morphology and exercise capacity, we also generated cardiac-specific Mcl-1Matrix transgenic (αMHC-Mcl-1Matrix) mice (Supplemental Figure S3A). Using isolated mitochondria from hearts of αMHC-Mcl-1Matrix mice, we confirmed that Mcl-1Matrix is protected against Proteinase K digestion (Figure 6A). Similar to αMHC-Mcl-1OM mice, we found that WT and αMHC-Mcl-1Matrix mice have similar heart size, cardiac structure and function as WT mice (Figures 6B-C and Supplemental Figure S3C). Additionally, hearts from WT and αMHC-Mcl-1Matrix mice have similar levels of anti-apoptotic Bcl-2 and Bcl-1XL and pro-apoptotic Bax and Bak as WT mice (Supplemental Figures S3D-E). Interestingly, TEM analysis to evaluate mitochondrial morphology at the ultrastructural level showed that many of the myocytes in αMHC-Mcl-1Matrix mice exhibited enlarged mitochondria (Figure 6D) but without altering mitochondrial content (Supplemental Figure 3F-G), respiration (Figure 6E), or respiratory chain supercomplex formation (Figure 6F). Although many mitochondria were enlarged in myocytes overexpressing Mcl-1Matrix, we observed no differences in fission and fusion proteins (Figure 6G-J). Levels of Drp1 at the mitochondria are also unaltered (Supplemental Figure S3H). Finally, we evaluated whether mice with elevated levels of Mcl-1Matrix and enlarged mitochondria in the heart affected their ability to adapt to intense exercise. Using the weight-loaded swimming test to examine exercise capacity in the mice, we observed no change in the exercise capacity in αMHC-Mcl-1Matrix mice compared to WT littermates (Figure 6K). This suggests that the reduced exercise capacity is caused by Mcl-1OM and possibly a shift towards mitochondrial fission.
Figure 6. Characterization of αMHC-Mcl-1Matrix mice.

(A) Proteinase K protection assay and Western blot analysis of mitochondria isolated from αMHC-Mcl-1Matrix mouse hearts. Isolated mitochondria were treated with 50 or 200 ng/μl proteinase K for 30 minutes to degrade proteins on the outer mitochondrial membrane. Tom20 was used as an outer mitochondrial membrane marker and MnSOD was used as a marker for the mitochondrial matrix. (B) Heart weight/body weight (HW/BW) ratios are similar in WT and αMHC-Mcl-1Matrix mice (n=7–8). (C) Echocardiographic analysis revealed similar ejection fraction (EF) and fractional shortening (FS) in WT and αMHC-Mcl-1Matrix TG hearts (n=7–8). (D) Representative electron micrographs of mitochondria in WT and αMHC-Mcl-1Matrix hearts. (E) Mitochondrial respiration was assessed using a Seahorse XFp analyzer in response to three different substrates: succinate, pyruvate/malate, and palmitoyl carnitine (n=5). OCR = oxygen consumption rate. (F) Representative BN-PAGE immunoblots of individual ETC complexes and supercomplexes (SCs) in WT and αMHC-Mcl-1Matrix cardiac mitochondria (n=4). SCs were visualized by antibodies against subunits of complex I (CI, Ndufb8), complex III (CIII, Uqcrfs1), and complex IV (CIV, CoxIV). (G) Western blots of mitochondrial fission proteins in WT and αMHC-Mcl-1Matrix hearts. (H) Quantitation of Drp1, Fis1, and Mff protein levels normalized to loading controls (n=4). (I) Western blots of mitochondrial fusion proteins in WT and αMHC-Mcl-1Matrix hearts. (J) Quantitation of Mfn1, Mfn2, and Opa1 protein levels normalized to loading controls (n=4). (K) Swim time of WT and αMHC-Mcl-1Matrix mice in response to the weight-loaded swimming endurance test (n=12–15). Data are mean + SEM.
4. Discussion
Here, we have identified a function for Mcl-1 in mitochondrial fission in cells and the heart which is independent of its anti-apoptotic function. First, we show that Mcl-1 interacts with Drp1 at the mitochondria to promote fission in vitro in response to various challenges. Furthermore, induction of fission by Mcl-1 is a protective response during stress. Finally, we confirmed that in the heart, Mcl-1OM, but not Mcl-1Matrix, promotes a shift in the balance towards fission. Enhanced Mcl-1OM levels in the heart has no effect on baseline cardiac function but leads to reduced exercise capacity, suggesting that a pre-existing fragmented mitochondrial network contributes to decreased ability to adapt to an acute increase in workload and energy demand. Overall, our findings provide important insights into Mcl-1’s function in regulating mitochondrial fission in cells and the heart.
Our findings show that overexpression of Mcl-1 leads to increased levels of mitochondrial Drp1 and enhanced fission both in vitro and in vivo. More importantly, challenges that induce mitochondrial fission in cells also promote increased interaction between endogenous Mcl-1 and Drp1. Combined with our previous finding that Mcl-1-deficient myocytes lack Drp1 at the mitochondria [15], this suggests that Mcl-1 might function as a mitochondrial receptor for Drp1. Drp1 is known to interact with Fis1, Mff, MiD51 and MiD49 at the mitochondria [34] and whether Mcl-1 is part of larger complex with the other Drp1 receptor proteins remains to be determined. Also, Drp1 resides in the cytosol and phosphorylation at Ser616 leads to its activation and translocation to mitochondria [28]. We did not investigate the effect of Mcl-1 on kinase activity and Drp1 phosphorylation status in vitro. Interestingly, although mitochondrial Drp1 levels increase in αMHC-Mcl-1OM mouse hearts, we did not observe a corresponding increase in phosphorylated Drp1 at the mitochondria. This suggests that the additional Drp1 at the mitochondria in the transgenic mouse hearts is not constitutively phosphorylated. Also, the balance between fission and fusion processes dictate overall mitochondrial morphology [26]. While we did not observe any changes in mitochondrial fission and fusion proteins in αMHC-Mcl-1OM mouse hearts, it is possible that Mcl-1OM also inhibits Mfn1/2 activity at the OMM. The intriguing possibility that Mcl-1OM simultaneously regulates both fission and fusion processes needs to be explored in future studies.
Mitochondrial fission can be either protective or detrimental depending on the context. Increased mitochondrial fission during stress is often associated with apoptosis [26], whereas deletion of Drp1 leads to loss of cardiac myocytes [35, 36]. Enlarged mitochondria have a lower threshold for opening of the mitochondrial permeability transition pore and necrotic cell death [37]. Drp1-mediated fission facilitated by Mcl-1 is a protective response during nutrient deprivation in cells that is independent of its anti-apoptotic activity. However, how Mcl-1-mediated fission protects cells against nutrient deprivation is still unclear. It is possible that the fragmented mitochondria are more protected against membrane permeabilization by Bax/Bak or mPTP opening. Mitochondrial fission is also closely linked to mitophagy [30, 38]. Although we found little evidence of mitophagy during nutrient deprivation, as mitochondrial content did not change, it is possible that Mcl-1-mediated fission can facilitate more efficient clearance of mitochondria during stress. However, mitophagy of depolarized mitochondria is mediated by the E3 ubiquitin ligase Parkin [39] and MEFs lack endogenous Parkin and are therefore relatively inefficient at clearing depolarized mitochondrial without overexpression of Parkin [40]. Finally, it has previously been reported that Mcl-1 interacts with Drp1 to promote fission. Morciano et al. found that Mcl-1 and Drp1 co-immunoprecipitated in HeLa cells, but this interaction was only observed when Drp1 was overexpressed [13]. A more recent study reported that induction of Drp1-mediated mitochondrial fission by Mcl-1 is important in promoting stem cells pluripotency [14]. Clearly, Mcl-1-mediated fission serves distinct functions depending on the context.
Accumulation of mitochondria in the perinuclear region after fission is commonly observed in stressed cells. The molecular basis for the mitochondrial clustering is not well understood but involves the movement of mitochondria along the cytoskeleton [41]. Here, we observed that overexpression of Mcl-1 alone in cells consistently led to perinuclear clustering of mitochondria. Thus, Mcl-1 and the BH3 domain might be involved in promoting the retrograde movement of mitochondria along microtubules. Whether Mcl-1 does this by regulating anchoring of mitochondria to microtubule motor proteins remains to be investigated. It is still unclear why mitochondria accumulate in this region during stress. It has been reported that perinuclear clustering of mitochondria establishes an oxidant-rich nuclear domain required for hypoxia-induced transcription [42]. However, the perinuclear clustering is not necessary for Mcl-1 to provide protection against nutrient-mediated cell death, as the BH3 mutant is just as effective in protecting cells as wild type Mcl-1. This suggests that the clustering of mitochondria might serve a different purpose in cells.
Mitochondrial fission and fusion are also involved in adapting to changes in bioenergetics and metabolism [43] and an imbalance in these processes can be detrimental to heart function. While we observed increased mitochondrial fission in the hearts of αMHC-Mcl-1OM mice, this has little effect on baseline mitochondrial or cardiac function. The lack of a cardiac phenotype in αMHC-Mcl-1OM mice is consistent with findings in cardiomyocyte-specific Drp1 transgenic mice which also have a normal cardiac phenotype at baseline [44]. The αMHC-Mcl-1Matrix mice with enlarged mitochondria also had normal mitochondrial respiration and cardiac function. This indicates that moderate shifts in fission/fusion are not inherently detrimental to the heart, unlike the more robust disruption induced by specific deletion of fission and fusion genes [35, 36, 38, 45]. While it has previously been reported that activation of Drp1-mediated fission in the heart is a beneficial response during exercise [27], our data suggests that a pre-existing fragmented mitochondrial network is not beneficial in response to high intensity exercise. The reason for this is unclear, but it is possible that fragmented mitochondria are less effective at increasing ATP production in response to the rapid increase in energy demand.
Alternatively, autophagy is activated during exercise in both skeletal muscle and heart and can provide energy through catabolism. Bcl-2 has been reported to inhibit induction of autophagy via its interaction with Beclin1 [46, 47] and it is possible that Mcl-1OM prevents activation of autophagy during exercise [15, 46]. Moreover, studies have reported that mitochondrial turnover is activated in skeletal muscle after exercise [48, 49]. Mitochondrial biogenesis and mitophagy are simultaneously activated in skeletal muscle after exercise to replace mitochondria that were damaged during exercise and to adapt to the increased energy demand. Here, we studied the effect of acute exercise using a weight-loaded swimming test and the short duration of the exercise (less than 10 minutes) makes it unlikely that alterations in mitochondrial biogenesis or mitophagy in hearts contribute to the reduced exercise tolerance in αMHC-Mcl-1OM transgenic mice. We also found no differences in mitochondrial content or biogenesis in WT and αMHC-Mcl-1OM transgenic mouse hearts at baseline, confirming that a pre-existing change in mitochondrial mass is not responsible for the phenotype.
In summary, our study demonstrates that Mcl-1 is involved in regulating mitochondrial fission and adaptation to stress. Our results implicate Mcl-1 in broader functional roles beyond regulating apoptosis, which may account for the rapid loss of myocytes in the absence of Mcl-1. Additionally, our findings add to the concern raised by previous Mcl-1 knockout studies [15, 16] that anticancer therapeutics directed toward Mcl-1 will lead to cardiotoxicity due to loss of multiple functions. In fact, the U.S. Food and Drug Administration (FDA) recently halted a phase I clinical trial involving an oral small molecule inhibitor of MCL-1 after cardiotoxicity signals were identified (https://www.ashclinicalnews.org/news/latest-and-greatest/fda-places-trials-mcl-inhibitor-clinical-hold/). This clearly demonstrates that additional studies to understand the biological functions of Mcl-1 in various tissues are urgently needed before small molecule inhibitors can safely be used in the clinic.
Supplementary Material
Highlights.
Mcl-1 interacts with Drp1 to promote mitochondrial fission during stress
Drp1-mediated fission is independent of Mcl-1’s traditional anti-apoptotic function
Overexpression of Mcl-1OM in heart leads to a shift in the balance towards fission
Pre-existing fragmented mitochondria is associated with reduced exercise capacity
Our findings highlight the importance of Mcl-1 in maintaining mitochondrial health
Acknowledgements
Dr. Joseph Opferman from St. Jude Children’s Research Hospital provided us with the mouse Mcl-1, Mcl-1OM, and Mcl-1Matrix constructs. This work was supported by National Institutes of Health grants R01HL138560 and R01HL132300 (to ÅBG), institutional cardiology training grant 5T32HL007444–32, AHA Predoctoral Fellowship 16PRE30260028, and National Institutes of Health Predoctoral Fellowship F31HL136228 (to AGM), National Institutes of Health NRSA Predoctoral Fellowship F31HL123309 (to AMO), National Institutes of Health NRSA Postdoctoral Fellowship F32HL139034 (to LJL), and TRDRP T30FT0846 (to WL).
Footnotes
Appendix A. Supplemental data
Disclosures
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kale J, Osterlund EJ and Andrews DW (2018) BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25:65–80. doi: 10.1038/cdd.2017.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell KJ and Tait SWG (2018) Targeting BCL-2 regulated apoptosis in cancer. Open Biol 8. doi: 10.1098/rsob.180002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonas EA, Porter GA and Alavian KN (2014) Bcl-xL in neuroprotection and plasticity. Front Physiol 5:355. doi: 10.3389/fphys.2014.00355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gustafsson AB and Gottlieb RA (2006) Bcl-2 Family Members and Apoptosis, Taken to Heart. Am J Physiol Cell Physiol. [DOI] [PubMed] [Google Scholar]
- 5.Senichkin VV, Streletskaia AY, Gorbunova AS, Zhivotovsky B and Kopeina GS(2020) Saga of Mcl-1: regulation from transcription to degradation. Cell Death Differ 27:405–419. doi: 10.1038/s41418-019-0486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maurer U, Charvet C, Wagman AS, Dejardin E and Green DR (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21:749–60. [DOI] [PubMed] [Google Scholar]
- 7.Rinkenberger JL, Horning S, Klocke B, Roth K and Korsmeyer SJ (2000) Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 14:23–7. [PMC free article] [PubMed] [Google Scholar]
- 8.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S and et al. (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267:1506–10. [DOI] [PubMed] [Google Scholar]
- 9.Veis DJ, Sorenson CM, Shutter JR and Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75:229–40. [DOI] [PubMed] [Google Scholar]
- 10.Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD and MacGregor GR (1998) Testicular degeneration in Bclw-deficient mice. Nat Genet 18:251–6. doi: 10.1038/ng0398-251 [DOI] [PubMed] [Google Scholar]
- 11.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR and Opferman JT (2012) Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 14:575–83. doi: ncb2488 [pii] 10.1038/ncb2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R and Pinkas-Kramarski R (2007) Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3:561–8. doi: 10.4161/auto.4713 [DOI] [PubMed] [Google Scholar]
- 13.Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M and Pinton P (2016) Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell 27:20–34. doi: 10.1091/mbc.E15-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen ML, Kline LA, Park KP, Ortolano NA, Romero-Morales AI, Anthony CC, Beckermann KE and Gama V (2018) A Non-apoptotic Function of MCL-1 in Promoting Pluripotency and Modulating Mitochondrial Dynamics in Stem Cells. Stem Cell Reports 10:684–692. doi: 10.1016/j.stemcr.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S and Gustafsson AB (2013) Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 27:1365–77. doi: 27/12/1365 [pii] 10.1101/gad.215871.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE and Opferman JT (2013) Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 27:1351–64. doi: 10.1101/gad.215855.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S and Gustafsson AB (2012) Microtubule-Associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J Biol Chem 287:19094–19104. doi: M111.322933 [pii] 10.1074/jbc.M111.322933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodall BP, Orogo AM, Najor RH, Cortez MQ, Moreno ER, Wang H, Divakaruni AS, Murphy AN and Gustafsson AB (2019) Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight 5. doi: 10.1172/jci.insight.127713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jha P, Wang X and Auwerx J (2016) Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr Protoc Mouse Biol 6:1–14. doi: 10.1002/9780470942390.mo150182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubli DA, Ycaza JE and Gustafsson AB (2007) Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN and Gustafsson AB (2013) Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288:915–26. doi: M112.411363 [pii] 10.1074/jbc.M112.411363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang WC, Hsu YJ, Wei L, Chen YJ and Huang CC (2016) Association of physical performance and biochemical profile of mice with intrinsic endurance swimming. Int J Med Sci 13:892–901. doi: 10.7150/ijms.16421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gustafsson AB and Dorn GW 2nd, (2019) Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol Rev 99:853–892. doi: 10.1152/physrev.00005.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G and Brady HJ (2006) Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem 281:5750–9. [DOI] [PubMed] [Google Scholar]
- 25.Smirnova E, Griparic L, Shurland DL and van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youle RJ and van der Bliek AM (2012) Mitochondrial fission, fusion, and stress.Science 337:1062–5. doi: 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, Hu DQ, Reddy S, Sandoval E, Stotland A, Gottlieb RA and Bernstein D (2018) Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ Res 122:282–295. doi: 10.1161/CIRCRESAHA.117.310725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitzig MT, Alleyn MD, Lockey RF and Kolliputi N (2018) A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 315:C80–C90. doi: 10.1152/ajpcell.00042.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mishra P and Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212:379–87. doi: 10.1083/jcb.201511036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE and Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J 27:433–46. doi: 7601963 [pii] 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT and Enriquez JA (2004) Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13:805–15. doi: S1097276504001248 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore TM, Zhou Z, Cohn W, Norheim F, Lin AJ, Kalajian N, Strumwasser AR, Cory K, Whitney K, Ho T, Ho T, Lee JL, Rucker DH, Shirihai O, van der Bliek AM, Whitelegge JP, Seldin MM, Lusis AJ, Lee S, Drevon CA, Mahata SK, Turcotte LP and Hevener AL (2019) The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol Metab 21:51–67. doi: 10.1016/j.molmet.2018.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei W, Li ZP, Zhu T, Fung HY, Wong TL, Wen X, Ma DL, Leung CH and Han QB (2017) Anti-Fatigue Effects of the Unique Polysaccharide Marker of Dendrobium officinale on BALB/c Mice. Molecules 22. doi: 10.3390/molecules22010155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loson OC, Song Z, Chen H and Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–67. doi: 10.1091/mbc.E12-10-0721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song M, Mihara K, Chen Y, Scorrano L and Dorn GW, 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21:273–85. doi: 10.1016/j.cmet.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M and Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–78. doi: 10.1161/CIRCRESAHA.116.303356 [DOI] [PubMed] [Google Scholar]
- 37.Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y,Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW 2nd, O’Rourke B and Kitsis RN (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 109:6566–71. doi: 1201608109 [pii] 10.1073/pnas.1201608109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ and Dorn GW 2nd, (2015) Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res 117:346–51. doi: 10.1161/CIRCRESAHA.117.306859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narendra D, Tanaka A, Suen DF and Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. doi: jcb.200809125 [pii] 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, Li H, Kitsis RN, Dorn Ii GW, Sadoshima J, Ellisman MH and Gustafsson AB (2017) A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8:14050. doi: 10.1038/ncomms14050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melkov A and Abdu U (2018) Regulation of long-distance transport of mitochondria along microtubules. Cell Mol Life Sci 75:163–176. doi: 10.1007/s00018-017-2590-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF and Gillespie MN (2012) Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5:ra47. doi: 10.1126/scisignal.2002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu T, Wang L and Yoon Y (2015) Morphological control of mitochondrial bioenergetics. Front Biosci (Landmark Ed) 20:229–46. doi: 10.2741/4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song M, Franco A, Fleischer JA, Zhang L and Dorn GW, 2nd (2017) Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab 26:872–883 e5. doi: 10.1016/j.cmet.2017.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Liu Y and Dorn GW 2nd, (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE and Levine B (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481:511–5. doi: nature10758 [pii] 10.1038/nature10758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD and Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–39. [DOI] [PubMed] [Google Scholar]
- 48.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M and Yan Z (2017) Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8:548. doi: 10.1038/s41467-017-00520-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka T, Nishimura A, Nishiyama K, Goto T, Numaga-Tomita T and Nishida M (2020) Mitochondrial dynamics in exercise physiology. Pflugers Arch 472:137–153. doi: 10.1007/s00424-019-02258-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.