

Abstract
BACKGROUND
Arachidyl amido cholanoic acid (Aramchol) is a potent downregulator of hepatic stearoyl-CoA desaturase 1 (SCD1) protein expression that reduces liver triglycerides and fibrosis in animal models of steatohepatitis. In a phase IIb clinical trial in patients with nonalcoholic steatohepatitis (NASH), 52 wk of treatment with Aramchol reduced blood levels of glycated hemoglobin A1c, an indicator of glycemic control.
AIM
To assess lipid and glucose metabolism in mouse hepatocytes and in a NASH mouse model [induced with a 0.1% methionine and choline deficient diet (0.1MCD)] after treatment with Aramchol.
METHODS
Isolated primary mouse hepatocytes were incubated with 20 μmol/L Aramchol or vehicle for 48 h. Subsequently, analyses were performed including Western blot, proteomics by mass spectrometry, and fluxomic analysis with 13C-uniformly labeled glucose. For the in vivo part of the study, male C57BL/6J mice were randomly fed a control or 0.1MCD for 4 wk and received 1 or 5 mg/kg/d Aramchol or vehicle by intragastric gavage for the last 2 wk. Liver metabolomics were assessed using ultra-high-performance liquid chromatography-time of flight-MS for the determination of glucose metabolism-related metabolites.
RESULTS
Combination of proteomics and Western blot analyses showed increased AMPK activity while the activity of nutrient sensor mTORC1 was decreased by Aramchol in hepatocytes. This translated into changes in the content of their downstream targets including proteins involved in fatty acid (FA) synthesis and oxidation [P-ACCα/β(S79), SCD1, CPT1A/B, HADHA, and HADHB], oxidative phosphorylation (NDUFA9, NDUFB11, NDUFS1, NDUFV1, ETFDH, and UQCRC2), tricarboxylic acid (TCA) cycle (MDH2, SUCLA2, and SUCLG2), and ribosome (P-p70S6K[T389] and P-S6[S235/S236]). Flux experiments with 13C-uniformely labeled glucose showed that TCA cycle cataplerosis was reduced by Aramchol in hepatocytes, as indicated by the increase in the number of rounds that malate remained in the TCA cycle. Finally, liver metabolomic analysis showed that glucose homeostasis was improved by Aramchol in 0.1MCD fed mice in a dose-dependent manner, showing normalization of glucose, G6P, F6P, UDP-glucose, and Rbl5P/Xyl5P.
CONCLUSION
Aramchol exerts its effect on glucose and lipid metabolism in NASH through activation of AMPK and inhibition of mTORC1, which in turn activate FA β-oxidation and oxidative phosphorylation.
Keywords: Nonalcoholic fatty liver disease, Steatohepatitis, Methionine and choline deficient diet, Tricarboxylic acid cycle, Hemoglobin A1c, Stearoyl-CoA desaturase 1
Core Tip: Arachidyl amido cholanoic acid (Aramchol), a phase III investigational drug for the treatment of nonalcoholic steatohepatitis (NASH), has been demonstrated to be able to reduce liver steatosis, inflammation, and fibrosis in various animal models of steatohepatitis. In a phase IIb clinical trial, it reduced blood levels of glycated hemoglobin A1c, an indicator of glycemic control. In this study, we showed that Aramchol-treated hepatocytes had activated AMPK and inhibited mTORC1, which in turn activated fatty acid β-oxidation and oxidative phosphorylation, inhibiting de novo lipogenesis, gluconeogenesis, and cataplerosis. These results explain the mechanism by which Aramchol exerts its effect on glucose and lipid metabolism in NASH.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a growing health problem with a global prevalence of 25% and is the most common liver disorder in Western countries, with an economic burden estimated at more than $100 billion per year in the United States alone[1,2]. Currently, nonalcoholic steatohepatitis (NASH) is the second leading cause for liver transplant waitlist registration or liver transplantation overall, and the leading cause in females. NASH will probably become the leading indication for liver transplant in males as well, given the high rate of increase[3]. NAFLD describes a spectrum of disorders including simple deposition of lipid droplets in the cytoplasm of hepatocytes (steatosis), nonalcoholic steatohepatitis (NASH) with varying degrees of fibrosis, cirrhosis, and hepatocellular carcinoma (HCC)[4]. Hepatic steatosis can result from diverse dysregulations including impaired lipid uptake, mitochondrial fatty acid (FA) β-oxidation alterations, defective very-low-density lipoprotein (VLDL) secretion and assembly, and/or increased de novo lipogenesis (DNL). NASH, the advanced form of NAFLD, is characterized by the presence of inflammation, hepatocyte ballooning, and fibrosis, in addition to the steatosis[5,6]. The combination of multiple hits such as steatosis, oxidative stress, mitochondrial dysfunction, apoptosis, inflammation, hepatic stellate cell activation, and collagen production results in the progression of NASH[5,7], which increases the risk for cirrhosis and HCC[8].
Despite being the most common cause of chronic liver disease in Western countries, and the great efforts made by the pharmaceutical industry, there are currently no treatments for NAFLD approved by the Food and Drug Administration agency or the European Medicine Agency. Medical recommendations are aimed at weight loss and diabetes control, which have been shown to slow the progression of the disease[9]. At present, many drugs targeting NASH are in clinical development[10,11], including arachidyl-amido cholanoic acid (Aramchol).
Aramchol has been demonstrated to be able to reduce NASH and fibrosis in two NASH preclinical mice models [induced with a methionine and choline deficient diet (MCD) and thioacetamide][12,13]. Previous work performed in MCD-fed mice showed that improvement of hepatic lipid accumulation, inflammation, and fibrosis by Aramchol was mediated by stearoyl-CoA desaturase 1 (SCD1) downregulation, the rate-limiting step in monounsaturated fatty acids (MUFA) synthesis[14,15], decreasing hepatic FA and triglycerides (TG), improving FA β-oxidation, and increasing glutathione (GSH), the main cellular antioxidant, and the GSH/oxidized GSH ratio[12]. In a phase IIb clinical trial, Aramchol resulted in a reduction in liver fat, NASH resolution, and fibrosis improvement. Aramchol was proven to be safe and well tolerated[16,17]. Aramchol is currently being studied in NASH patients with fibrosis stage 2-3 for a Phase III clinical study (ARMOR).
The present work showed that mouse hepatocytes responded to Aramchol treatment by promoting AMPK pathway activation and mTORC1 pathway inhibition, which in turn activated FA β-oxidation and oxidative phosphorylation, inhibiting DNL, gluconeogenesis, and cataplerosis. In the MCD mouse model of NASH, this dual effect of Aramchol on AMPK and mTORC1 activity translated into an improvement of liver glucose metabolism, which in humans is associated with a reduction of the blood levels of glycated hemoglobin A1c (HbA1c), reflecting the mean blood sugar concentration of the previous weeks to months[18,19].
MATERIALS AND METHODS
ARREST study
The ARREST study (ClinicalTrials.gov NCT02279524) enrolled 247 NASH patients who were overweight/obese and had prediabetes/diabetes with a mean baseline value of HbA1c of about 6.6%. More than 50% of the patients were hypertensive and had dyslipidemia. Baseline histology demonstrated a population with advanced disease, with 60% having stages 2 and 3 fibrosis and 70% having a NASH activity score ≥ 5. Eligible subjects were enrolled into three treatment arms: Aramchol 400 and 600 mg tablets/day, and placebo tablets in a ratio of 2:2:1. The subjects were evaluated at study sites for 11 scheduled visits during one year (52 wk). After completion of the study treatment period, the subjects were followed for an additional period of 13 wk without study medication (until visit 11, week 65). Blood HbA1c levels were determined at baseline, 8, 24, 40, and 52 wk.
Animal experiments
Eight-week-old C57BL/6J male mice were purchased from Charles River, St Germain sur l'Arbresle, France. After allowing them to acclimate for 1 wk, mice were randomly distributed into two groups. One of the groups consisting of 10 mice was fed a normal rodent maintenance diet (Teklad Global 14% Protein; Envigo RMS Spain, Sant Feliu de Codines, Spain) containing 0.3% methionine and 1030 mg choline/kg diet. The other group consisting of 30 animals was maintained on a diet with 0.1% methionine and free of choline (0.1MCD diet) during 4 additional weeks. Two weeks after the start of the 0.1MCD diet, mice were divided into groups of 10 and treated by intragastric gavage with a formulation of 1 or 5 mg/kg/d of Aramchol, similar to the human 600 mg/d dose, or vehicle (0.2% sodium lauryl sulfate and 1.7% carboxymethyl cellulose). Animals kept on the normal diet were also administered the vehicle preparation. Animal procedures were designed to minimize animal discomfort or pain. Animal experiments were approved by the County of Bizkaia and CIC bioGUNE's Biosafety Bioethics Committee.
Primary mouse hepatocytes isolation and culture
Mouse hepatocytes from 2- to 3-mo-old male C57BL/6J mice were isolated by collagenase perfusion. Briefly, animals were anesthetized with isofluorane (1.5% isofluorane in O2), the abdomen was opened, a catheter was inserted into the inferior vena cava, and the portal vein was immediately cut. The liver was perfused with 20 mL of buffer I and after that, with 20 mL of buffer II. Finally, the liver was digested with collagenase buffer III. Once digested, the liver was carefully extracted and the gallbladder was removed. The digested liver was broken up in a petri dish and filtered. Isolated hepatocytes were seeded over collagen-coated culture dishes at a density of 7600 cells/mm2 in 10% FBS and 1% PSG supplemented MEM and placed at 37 °C in a humidified atmosphere of 5% CO2-95% air.
Perfusion buffers composition: Stock solution 1 was prepared with 3.97g D-Glucose, 7.14 g NaCl, 0.37 g KCl, 2.1 g NaHCO3, 27.2 g KH2PO4, and 38.2 g MGSO4(7H2O) in a final volume of one liter H2O. For perfusion buffer I, 400 mL of EGTA 250 mmol/L (pH = 8) was added to 330 mL 1 × stock solution. Perfusion buffer II was prepared with 1 × stock solution, and perfusion buffer III was prepared with 0.09 g of CaCl2 in 330 mL of 1 × stock solution and 50 mg of Worthington Collagenase (LS004196) per 50mL of buffer (digestion buffer).
In vitro Aramchol treatment
Aramchol was provided by Galmed Pharmaceuticals. It was originally produced by WIL Research Europe B.V. as test substance (number 204147/Y). The batch number used was CS11-153Am-1402. All analytes were within the limit of quantifications. In addition, heavy metals and ions were also below the upper limits (irons < 10 ppm; arsenites < 1 ppm; heavy metals < 10 ppm; sulfate < 1% and chloride < 0.1%). Aramchol was prepared in DMSO, at a concentration of 100 mM, and frozen at -20 °C. A 20 mmol/L running stock preparation of Aramchol was prepared by diluting the 100 mM stock for in vitro treatments. The Aramchol preparations were stored at -20 °C until each use.
Primary mouse hepatocytes were isolated as described above and allowed to attach for 3 h. After this, culture medium was removed and replaced by serum-free MEM with or without DMSO (vehicle) and Aramchol (20 µmol/L), and cultured for an additional 48 h.
Western blot analysis
Frozen liver tissue samples or cultured cells were homogenized in lysis buffer (10 mmol/L Tris/HCl pH 7.6, 5 mmol/L ethylene diamine tetraacetic acid, 50 mmol/L NaCl, 1% Triton X-100, complete protease inhibitor cocktail, and 50 mmol/L NaF) and a centrifugation step was performed (10000g, 20 min, 4 °C). Protein concentration was determined in supernatants by using the Bradford-based Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, United States). After protein determination, 10-30 μg protein was loaded on sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE). Electrophoresis was performed and proteins were transferred onto nitrocellulose membranes. Western blotting was performed following standard protocols (Amersham BioSciences, Piscataway, NJ) using primary antibodies for ACCα/β, phosphorylated ACCα/β (S79) (from Cell Signaling, Danvers, MA, United States), β-actin, AMPKα1 (from MERCK), phosphorylated AMPKα (T172), CPT1A/B, S6, Phospho-S6 (S235/236), SCD1, p70S6K and phosphorylated p70S6K (T389) (from Cell Signaling, Danvers, MA, United States).
Proteomic analysis
Primary mouse hepatocyte samples were incubated in 7 mol/L urea, 2 mol/L thiourea, 4% CHAPS, and 5 mmol/L DTT containing buffer at room temperature under agitation for 30 min and digested following the filter-aided FASP protocol. Trypsin was added at a trypsin:protein ratio of 1:50, and the mixture was incubated overnight at 37 °C, dried out in a RVC2 25 Speedvac concentrator (Christ), and resuspended in 0.1% FA. Peptides were desalted and resuspended in 0.1% FA using C18 stage tips (Millipore). Samples were analyzed in a hybrid trapped ion mobility spectrometry – quadrupole time of flight mass spectrometer (timsTOF Pro, Bruker Daltonics) powered with Parallel Accumulation–Serial Fragmentation scan mode and coupled online to a nanoElute liquid chromatograph (Bruker). Protein identification and quantification were carried out using PEAKS software (Bioinformatics solutions). Intensity data corresponding to proteins identified with at least two different peptides at FDR < 5% was loaded onto Perseus platform[20] and further processed (log2 transformation, imputation). A t-test was applied to determine the statistical significance of the differences detected and volcano plot was generated.
Fluxomic analysis
Following hepatic perfusion and once the mouse hepatocytes were attached, 3 h after the seeding, culture medium was replaced with FBS free MEM supplemented with 20 µmol/L Aramchol or DMSO (vehicle). Primary hepatocytes were incubated during 48 h and after this time, the medium was replaced with FBS free DMEM without glucose, supplemented with 11 m mol/L fully 13C-labeled glucose (Cambridge Isotope Laboratories, MA, United States). Plates were washed with PBS three times and snap frozen with liquid nitrogen after 120 and 240 min. Samples were stored at -80 °C until metabolite extraction. Extracted ion traces of labeled and unlabeled species were obtained for glucose, citric acid, and malic acid in a 20 mDa window. These traces were subsequently smoothed and integrated with QuanLynx software (Waters, Manchester, United Kingdom).
Metabolomic analysis
A semiquantitative ultra-high performance liquid chromatography (UHPLC)-time of flight-MS based platform was used for the determination of the liver metabolic profiles[21,22]. Briefly, methanol was added to the liver tissue (15 mg) for protein precipitation. The methanol was spiked with metabolites not detected in unspiked cell extracts that were used as internal standards. After protein precipitation, samples were homogenized (6500 rpm for 23 s × 1 round) using a Precellys 24 homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France), and then centrifuged at 18000 × g for 10 min at 4 ºC. An aliquot of 500 µL was collected and mixed with chloroform. After 10 min of agitation, a second centrifugation of the samples was performed (18000 × g for 15 min at 4 ºC). Supernatants were dried under vacuum and then, reconstituted in water. After a third centrifugation (18000 × g for 15 min at 4 ºC), the extracts were transferred to plates for UHPLC-MS analysis. Metabolic features were identified prior to the analysis. LC-MS data pre-processing was performed using the TargetLynx application manager for MassLynx 4.1 software (Waters Corp., Milford, United States). Peak detection, noise reduction, and data normalization steps followed the procedures previously described[23].
Statistical analysis
Data are indicated as the mean ± SEM. Student’s t-test was used for evaluating the differences between groups (significance defined as P < 0.05). Software package R v.3.1.1 (R Development Core Team, 2011; https://cran.r-project.org/) was used for all calculations.
RESULTS
Aramchol improves glycemic control in NASH patients
ARREST is a multicenter, phase IIb, randomized, double blind, placebo-controlled study designed to evaluate the efficacy and safety of two Aramchol doses (400 and 600 mg tablets/d) and placebo tablets in subjects with NASH, confirmed by liver biopsy, who were overweight or obese, and had prediabetes or type II diabetes. Blood HbA1c content, which reflects glycemic control, was determined at baseline, 8, 24, 40, and 52 wk. At week 52, both Aramchol doses resulted in a decrease in HbA1c, as compared to baseline levels, while patients in the placebo arm showed an increase. The differences from placebo were statistically significant (P = 0.0061 and P = 0.0008 for Aramchol 400 mg vs placebo and 600 mg vs placebo, respectively, Figure 1).
Figure 1.

Arachidyl amido cholanoic acid improves glycemic control in nonalcoholic steatohepatitis patients. ARREST is a multicenter, placebo-controlled study designed to evaluate the efficacy and safety of two arachidyl amido cholanoic acid (Aramchol) doses (400 and 600 mg tablets/d) and placebo tablets in subjects with nonalcoholic steatohepatitis, confirmed by liver biopsy, who were overweight or obese, and had prediabetes or type II diabetes. Blood hemoglobin A1c (HbA1c) content was determined at baseline and weeks 8, 24, 40, and 52. At week 52, both Aramchol doses resulted in a decrease in HbA1c, while NASH patients on the placebo arm showed an increase. The differences from placebo were statistically significant (aP = 0.0061 and bP = 0.0008 for Aramchol 400 mg vs placebo and 600 mg vs placebo, respectively).
Aramchol differentially regulates AMPK and mTORC1 in hepatocytes
In order to understand glucose metabolism improvement in Aramchol treated patients in the ARREST study, we analyzed its role in the anabolic and catabolic pathways mediated by AMPK and mTOR.
Primary hepatocytes of 4- to 6-mo-old C57BL/6J mice were isolated and allowed to attach during 3 h. After this, culture medium was removed and replaced by serum-free MEM with DMSO (vehicle) or Aramchol (20 µmol/L), and cultured for an additional 48 h. As expected, Aramchol addition markedly reduced SCD1 protein content in cultured hepatocytes (Figure 2A and B).
Figure 2.

Arachidyl amido cholanoic acid differentially regulates AMPK and mTORC1 in cultured hepatocytes. Mouse primary hepatocytes were isolated and allowed to attach for 3 h, after which culture medium was replaced by serum-free MEM with DMSO (vehicle) or Arachidyl amido cholanoic acid (Aramchol) (20 µM), and the cells were cultured for an additional 48 h. A: Effect of Aramchol treatment on SCD1, CPT1A/B, AMPK, P-AMPKα (Thr172), ACCα/β, P-ACCα/β (Ser79), p70S6K, P-p70S6K (T389), S6, and P-S6 (Ser235/236) protein levels in cultured mouse hepatocytes by Western blot analysis. β-actin was used as a loading control; B: Densitometric analysis of the Western blot protein levels. Effect of Aramchol treatment on the ratios of phosphorylated/total protein levels of AMPK, ACC and p70S6K is shown. In the case of SCD1 and CPT1A/B, the protein level is normalized against β-actin (loading control). Results are expressed as fold of total specific protein levels. At least triplicates were used per experimental condition. Data is shown as the mean ± SEM. aP < 0.05.
Due to AMPK's role as a central regulator of both glucose and lipid metabolism, we evaluated the effect of Aramchol on AMPK and AMPK's α-subunit phosphorylated at T172 (P-AMPK), its active form. The ratio P-AMPK/AMPK was increased (P < 0.05) in culture medium incubated with Aramchol (Figure 2A and B). In relation to this, the β-oxidation rate-limiting enzyme CPT1A/B protein content was increased in hepatocytes treated with Aramchol. The ratio between the long chain fatty acid synthetic enzyme ACC and its inactive form P-ACC (P-ACC/ACC ratio) was increased by 2-fold in Aramchol treated hepatocytes (Figure 2A and B).
p70S6K is a downstream target of mTORC1, and the phosphorylation of p70S6K at T389 has been used as a hallmark of activation by mTORC1 of its target substrate, the S6 ribosomal protein[24]. P-p70S6K/p70S6K ratio levels were diminished (P < 0.05) in hepatocytes after Aramchol addition (Figure 2A and B). In relation to p70S6K protein levels and its phosphorylated form, we analyzed the levels of S6 ribosomal protein. Total S6 protein content was diminished in hepatocytes cultured with Aramchol. Moreover, P-S6/S6 ratio was reduced by about 80% in Aramchol treated primary hepatocytes (Figure 2A and B).
Proteomic studies
The effect of Aramchol on the proteome of mouse hepatocytes was also analyzed. A total of 3220 proteins were identified by proteomics analysis, of which the contents of 219 changed (6.80%, P < 0.05) between the two experimental groups (vehicle vs Aramchol 20 µmol/L). Hierarchical clustering classified samples into two differentiated groups, vehicle and Aramchol-treated hepatocytes (Figure 3A). Figure 3B shows a volcano plot of the identified proteins. The differentially expressed proteins according with the up- or down-regulation were classified with colors depending on their functions. Moreover, selected differentially expressed proteins (Table 1) were analyzed with STRING software and classified in key biological functions showing a reduction of translation (RBM27, EIF3A, and RPL22L1) and fibrosis (vimentin, filaminA/B, and KRT18); and activation of lipid droplet clearance (PLIN3 and APOB), fatty acid oxidation (CPT1A/B, HADHA, and HADHB), oxidative phosphorylation (NDUFA9, NDUFB11, NDUFS1, NDUFV1, ETFDH, and UQCRC2), antioxidant response (GSTK1 and CAT), and the tricarboxylic acid (TCA) cycle (MDH2, SUCLA2, and SUCLG2) (Figure 3C). A complete list of the differentially expressed proteins classified in the above-mentioned biological functions can be found in Supplemental Table 1.
Figure 3.
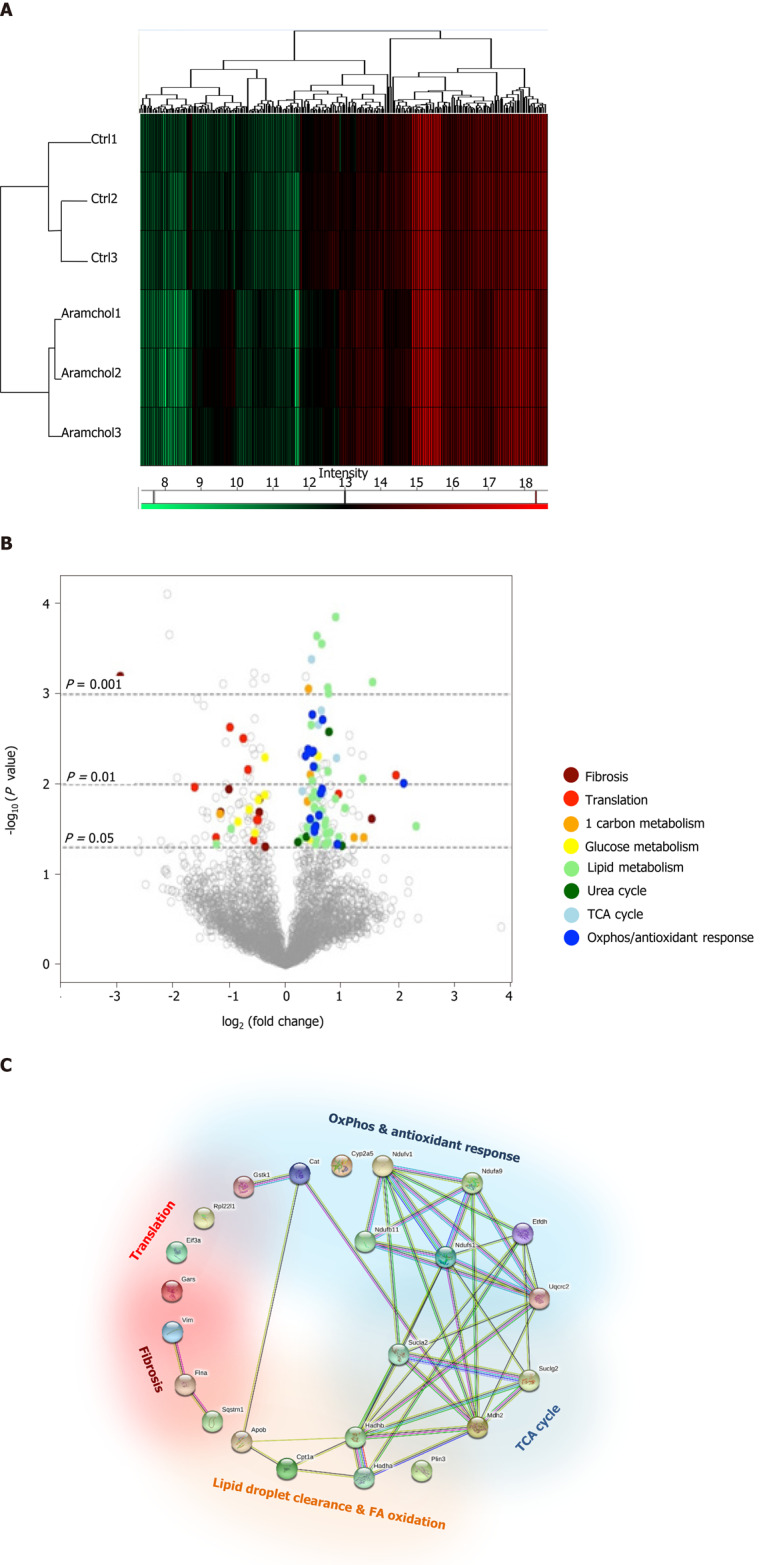
Effect of Arachidyl amido cholanoic acid on the proteome of cultured mouse hepatocytes. A total of 3220 proteins were identified by proteomics analysis, of which the contents of 219 changed significantly (6.80%, P < 0.05) between the two experimental groups [vehicle vs arachidyl amido cholanoic acid (Aramchol) 20 µmol/L]. A: Hierarchical clustering showing classification of the samples into two differentiated groups: Control and Aramchol treated; B: The differentially expressed proteins according to the up- or down-regulation were classified with colors in a volcano plot depending on their biological functions; C: Classification of the selected differentially expressed proteins in key biological functions. STRING software was used for the analysis.
Table 1.
Selected proteins whose content is significantly modified in hepatocytes by Arachidyl amido cholanoic acid
| Accession | Protein name | P value | Fold change achol/ctrl |
| Fibrosis | |||
| P20152 | Vimentin GN = Vim PE = 1 SV = 3 | 6.29E-04 | 0.13 |
| Q8BTM8 | Filamin-A GN = Flna PE = 1 SV = 5 | 2.02E-02 | 0.45 |
| Translation | |||
| Q9D7S7 | 60S ribosomal protein L22-like 1 GN = Rpl22l1 PE = 1 SV = 1 | 3.86E-02 | 0.42 |
| P23116 | Eukaryotic translation initiation factor 3 subunit A GN = Eif3a PE = 1 SV = 5 | 3.10E-03 | 0.59 |
| Q9CZD3 | Glycine-tRNA ligase GN = Gars PE = 1 SV = 1 | 6.87E-03 | 0.63 |
| Lipid droplet clearance & FA oxidation | |||
| Q9DBG5 | Perilipin-3 GN = Plin3 PE = 1 SV = 1 | 3.09E-02 | 0.51 |
| E9Q414 | Apolipoprotein B-100 GN = Apob PE = 1 SV = 1 | 3.50E-02 | 1.65 |
| Q64337 | Sequestosome-1 GN = Sqstm1 PE = 1 SV = 1 | 2.90E-02 | 5.01 |
| P97742 | Carnitine O-palmitoyltransferase 1 liver isoform GN = Cpt1a PE = 1 SV = 4 | 3.02E-02 | 1.32 |
| Q99JY0 | Trifunctional enzyme subunit beta mitochondrial GN = Hadhb PE = 1 SV = 1 | 1.40E-02 | 1.38 |
| Q8BMS1 | Trifunctional enzyme subunit alpha mitochondrial GN = Hadha PE = 1 SV = 1 | 1.78E-02 | 1.42 |
| TCA cycle | |||
| P08249 | Malate dehydrogenase mitochondrial GN = Mdh2 PE = 1 SV = 3 | 6.58E-03 | 1.46 |
| Q9Z2I8 | Succinate-CoA ligase (GDP-forming) subunit beta mitochondrial GN = Suclg2 PE = 1 SV = 3 | 1.52E-03 | 1.55 |
| Q9Z2I9 | Succinate-CoA ligase (ADP-forming) subunit beta mitochondrial GN = Sucla2 PE = 1 SV = 2 | 5.11E-03 | 1.87 |
| OxPhos & antioxidant response | |||
| Q91VD9 | NADH-ubiquinone oxidoreductase 75 kDa subunit mitochondrial GN = Ndufs1 PE = 1 SV = 2 | 4.28E-03 | 1.41 |
| Q9DB77 | Cytochrome b-c1 complex subunit 2 mitochondrial GN = Uqcrc2 PE = 1 SV = 1 | 6.36E-03 | 1.41 |
| Q921G7 | Electron transfer flavoprotein-ubiquinone oxidoreductase mitochondrial GN = Etfdh PE = 1 SV = 1 | 3.06E-02 | 1.43 |
| Q9DC69 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 9 mitochondrial GN = Ndufa9 PE = 1 SV = 2 | 1.26E-02 | 1.54 |
| Q91YT0 | NADH dehydrogenase (ubiquinone) flavoprotein 1 mitochondrial GN = Ndufv1 PE = 1 SV = 1 | 1.92E-03 | 1.58 |
| O09111 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 11 mitochondrial GN = Ndufb11 PE = 1 SV = 2 | 4.61E-02 | 1.89 |
| P24270 | Catalase GN = Cat PE = 1 SV = 4 | 4.84E-03 | 1.28 |
| Q9DCM2 | Glutathione S-transferase kappa 1 GN = Gstk1 PE = 1 SV = 3 | 3.31E-02 | 1.43 |
| P20852 | Cytochrome P450 2A5 GN = Cyp2a5 PE = 2 SV = 1 | 9.73E-03 | 4.29 |
See text for experimental details.
Aramchol regulates TCA cycle activity
Acetyl-CoA oxidation to CO2 by the TCA cycle is the central process in energy metabolism. Nevertheless, the TCA cycle additionally operates in biosynthetic pathways in which intermediates abandon the cycle to be converted mainly to glucose, FA, or non-essential amino acids. In order to measure the effect of Aramchol on the TCA cycle, murine hepatocytes were incubated with Aramchol for 48 h, after which uniformly labeled 13C6-glucose was added. The 13C-label dispersion in the TCA cycle was determined by measuring the ratios of the different isotopomers of malate at steady-state by mass spectrometry (Figure 4A). After Aramchol treatment, we found an increase in the number of rounds that malate remained in the cycle, indicated by upregulation of the +4 labeled malate species compared to control (Figure 4B).
Figure 4.

Arachidyl amido cholanoic acid regulates tricarboxylic acid cycle activity. Glucose flux in the tricarboxylic acid (TCA) cycle was investigated in order to determine glucose utilization in the liver after arachidyl amido cholanoic acid (Aramchol) treatment (20 µmol/L). Murine hepatocytes were cultured with vehicle or Aramchol (20 µmol/L) for 48 h, after which uniformly labeled 13C6-glucose was added. A: Diagram of the label dispersion of fully 13C-labeled glucose for the initial cycle of the TCA cycle. Carbon atoms are either white, grey-shaded, or black depending on the origin of labelled and non-labelled carbons. Grey-shaded carbons originate directly from pyruvate (Pyr). Pyr is converted in oxaloacetate by pyruvate carboxylase after introduction of a carboxyl group coming from a carboxylate ion. Black coloured carbons originate from acetyl-CoA and are introduced via citrate synthase. White carbons are not labeled. The mass increases of the TCA cycle metabolites due to the incorporation of 13C are indicated and follow the same colour coding. The numbers indicated in blue are the total mass increase due to incorporation of labels via both pathways. Note that the formation of isocitrate (iCit) from citrate causes the formation of two distinct isotopomers because the hydroxyl group can shift equally likely to both sides. The formation of malate (Mal) from fumarate again doubles the amount of isotopomers for the same reason. In subsequent cycles, the isotopomer patterns take forms that are more complex. The transformation from iCit to succinate causes the loss of two carbon dioxide molecules; B: The 13C-label dispersion in the TCA cycle was determined by measuring the ratios of the different isotopomers of Mal at steady-state by mass spectrometry. After Aramchol treatment, an upregulation of the +4 labeled Mal species was found compared to control. aP < 0.002; bP < 0.0002 Aramchol treated vs control. TCA: Tricarboxylic acid; Pyr: Pyruvate; OA: Oxaloacetate; PC: Pyruvate carboxylase; CO3-: Carboxylate ion; Ac-CoA: Acetyl-CoA; CS: Citrate synthase; Cit: Citrate; iCit: Isocitrate; Mal: Malate; Fum: Fumarate; Suc: Succinate; CO2: Carbon dioxide.
Aramchol treatment improves glucose homeostasis in mice fed a 0.1MCD diet
C57BL6/J mice were fed a 0.1MCD diet for 2 wk. After this, mice were divided into groups of 10 and treated by intragastric gavage with 1 or 5 mg/kg/d of Aramchol or vehicle alone. Animals kept on a normal diet were also provided with the vehicle preparation.
Western blot analysis revealed an increased P-AMPK/AMPK ratio in the Aramchol treated animals (5 mg/kg/d) compared with the 0.1MCD non-treated mice (Figure 5A). Together with this, a reduction in P-p70S6K/p70S6K ratio was observed in the Aramchol-treated animals (Figure 5A).
Figure 5.

Arachidyl amido cholanoic acid treatment improves glucose homeostasis in mice fed a 0.1MCD diet. C57BL6/J mice were fed a 0.1MCD diet for 2 wk. After this, mice were divided into groups of 10 and treated by intragastric gavage with 1 or 5 mg/kg/d of Arachidyl amido cholanoic acid (Aramchol) or vehicle alone. Animals kept on a normal diet were also provided with the vehicle preparation. A: Effect of 5 mg/kg/d Aramchol treatment on AMPK, P-AMPKα (Thr172), p70S6K, and P-p70S6K (T389) protein levels in the livers by Western blot analysis. β-actin was used as loading control. Densitometric analysis of phosphorylated/total AMPK and p70S6K ratio was represented. Results are expressed as fold of total specific protein levels. Experiment was performed in cuadruplicates per experimental condition. Data is shown as the mean ± SEM. aP < 0.05 vs MCDD; B: Metabolomic analysis of livers from the 0.1MCD fed mice showed a reduction of glucose, glucose 6-phosphate, fructose 6-phosphate, UDP-glucose, ribulose 5-phosphate, fructose 1, 6-bisphosphate, and pyruvate, as compared to mice fed a normal diet. The treatment with Aramchol tended to normalize the concentration of these metabolites in a dose dependent manner. aP < 0.05; bP < 0.01; cP < 0.001.
Metabolomic analysis of livers from the 0.1MCD fed mice showed a reduction of glucose, glucose 6-phosphate (G6P), fructose 6-phosphate (F6P), UDP-glucose, and ribulose 5-phosphate/xylulose 5-phosphate (Rbl5P/Xyl5P), while the contents of FBP and pyruvate remained normal, as compared to mice fed a normal diet. Treatment with Aramchol, following the protocol described above, tended to normalize the concentration of glucose, G6P, F6P, UDP-glucose, and Rbl5P/Xyl5P in a dose dependent manner, while FBP and pyruvate remained basically unchanged (Figure 5B).
DISCUSSION
NAFLD, and particularly its advanced form NASH, have become major global health concerns. Despite the many drugs currently under development, no pharmacological treatment has been approved for NAFLD or NASH. Aramchol is a novel investigational drug, synthesized as a fatty acid bile acid conjugate, currently under clinical phase III study for the treatment of NASH. In the phase IIb ARREST clinical study, all patients were prediabetic or had type II diabetes at baseline with a mean HbA1c value (reflecting glycemic control) of around 6.5%. At the completion of the study (52 wk), Aramchol showed a statistically significant decrease in HbA1c which was dose-dependent.
Studies in preclinical animal models of steatohepatitis have identified SCD1, the first step committing FA into TG synthesis, as the main target of Aramchol[12], explaining its anti-steatotic activity in patients with NAFLD[16]. Similar studies for other metabolic pathways, mainly those involved in glucose metabolism, are lacking. The present study tried to elucidate the effect of Aramchol on AMPK and mTOR pathways, key regulators of catabolism and anabolism.
AMPK activates catabolic pathways that generate ATP, such as FA β-oxidation, while inhibiting cell growth and biosynthesis and other processes that consume ATP[25,26]. The ratio P-AMPK/AMPK, which indicates activation of the pathway, was significantly increased by Aramchol treatment (Figure 2A and B). In relation to this, CPT1A/B, a catalyzer of the rate-limiting step of FA β-oxidation by delivering long-chain FA from the cytoplasm into mitochondria[27-29], was increased. In addition, the protein contents of ACC, which is considered the key regulatory enzyme in the conversion of citrate to long-chain FA[30,31], and its inactive phosphorylated form (P-ACC), were also examined (Figure 2A and B). The ratio between them was increased in Aramchol-treated hepatocytes corresponding with the observed activation of catabolism.
mTORC1 plays a central role in regulating the synthesis of lipids, proteins, and nucleotides, and also suppresses catabolic pathways. The analysis of downstream targets of mTORC1 (Figure 2A and B) indicated an inhibition of the pathway and therefore, a decrease of anabolism, and an increase of catabolic processes.
The effect of Aramchol on the proteome of mouse hepatocytes reveals two clearly separate groups (Figure 3A) and the analysis (Table 1) of selected differentially expressed proteins resulted in key biological functions showing (Figure 3B) a reduction of translation and fibrosis and activation of the TCA cycle, lipid droplet clearance, fatty acid oxidation, oxidative phosphorylation, and antioxidant response. These results agree with the hypothesis that Aramchol promotes a catabolic response and inhibits anabolic processes. The antifibrotic effect of Aramchol confirms previous results[12,13].
In order to determine glucose utilization in the liver after Aramchol treatment, glucose flux in the TCA cycle was investigated. In energy metabolism, acetyl-CoA oxidizes to CO2 by the TCA cycle as the central process. Moreover, TCA cycle intermediates abandon the cycle to be converted primarily to glucose, FA, or non-essential amino acids, which constitutes the biosynthetic function of the TCA. The replacement of TCA cycle anions removed from the cycle to allow its continued function is called anaplerosis. On the contrary, intermediates coming from amino acid catabolism and not fully oxidized in the TCA cycle must be removed by a process called cataplerosis. In the analysis of the TCA cycle activity, the obtained results show an increase in the number of rounds that malate remained in the TCA cycle (Figure 4A and B), indicating a reduction in cataplerosis in response to Aramchol treatment, which is linked to reduced gluconeogenesis and DNL.
In summary, the in vitro results obtained in the present study show that Aramchol regulates hepatocyte metabolism from anabolism to catabolism by activating AMPK and inhibiting mTORC1 pathways, which in turn activate FA β-oxidation and oxidative phosphorylation, inhibiting DNL and gluconeogenesis and reducing cataplerosis.
We also studied the effect of Aramchol in vivo in the 0.1MCD murine model of NASH (Figure 5). Compared to the canonic MCD mouse model of NASH, where the diet is devoid of methionine and choline, in the 0.1MCD model the diet is devoid of choline but contains 0.1% methionine. Mice subjected to the 0.1MCD diet develop steatosis, inflammation, and fibrosis comparable to those induced by a diet completely devoid of methionine and choline without the association of weight loss[12]. Increased FA uptake and reduced VLDL export represent the main mechanism by which the MCD diet induces intrahepatic TG accumulation in this model[32,33]. We have previously observed that Aramchol treatment in 0.1MCD fed mice markedly reduced the protein content of SCD1, which was associated with a reduction of hepatic TG accumulation[12]. Here we observed that, similarly to the results observed in isolated hepatocytes, Aramchol regulated hepatocyte metabolism from anabolism to catabolism by activating AMPK and inhibiting mTORC1 pathways (Figure 5A), which may explain the anti-steatotic effect of this molecule. MCD fed mice present a blockade in the activation of Akt, as indicated by reduced phosphorylation[34]. Accordingly, 0.1MCD fed mice showed a reduction in the liver contents of glucose and G6P, the first product of glucose metabolism (Figure 5B). The synthesis of G6P is one of the most critical steps in glucose metabolism, as it is at the convergence point of glycolysis, glycogen synthesis, and the pentose phosphate pathway. Consistent with this, the reduction of G6P in 0.1MCD fed mice was associated with a decrease in the content of F6P, which results from the isomerization of G6P, the first step in glycolysis; UDP-glucose, the glucose donor in the biosynthesis of glycogen; and Rbl5P/Xyl5P, two intermediates of the PPP that are in equilibrium. Of note, Aramchol administration to 0.1MCD fed mice tended to normalize the contents of glucose, G6P, F6P, UDP glucose and Rbl5P/Xyl5P, this effect being dose-dependent. The levels of FBP and pyruvate, which were not altered in 0.1MCD fed mice, did not change in response to Aramchol.
CONCLUSION
Aramchol improves not only the alterations in lipid metabolism that characterize NASH, but also hepatic glucose metabolism, offering an explanation for the reduction of blood HbA1c observed in NASH patients treated with Aramchol in the clinical ARREST study.
ARTICLE HIGHLIGHTS
Research background
Although nonalcoholic liver disease (NAFLD) represents the most common cause of chronic liver disease in Western countries, currently there is no approved treatment for NAFLD by the Food and Drug Administration agency or the European Medicine Agency. Arachidyl amido cholanoic acid (Aramchol) has been shown to reduce liver triglycerides and fibrosis in animal models of steatohepatitis. In a phase IIb clinical trial, Aramchol reduced blood levels of glycated hemoglobin A1c (HbA1c), an indicator of glycemic control.
Research motivation
Providing evidence of Aramchol's effect on glucose metabolism will explain the results observed in patients enrolled in the phase IIb clinical trial and, moreover, it will highlight the influence of Aramchol on glucose homeostasis for NAFLD treatment.
Research objectives
To elucidate the mechanisms by which Aramchol treatment improves lipid and glucose metabolism, using primary mouse hepatocytes and a 0.1% methionine and choline deficient diet (0.1MCD) nonalcoholic steatohepatitis (NASH) mouse model.
Research methods
Primary mouse hepatocytes were treated with 20 μmol/L Aramchol or vehicle for 48 h. Western blot, proteomics, and glucose fluxomic analyses were performed. The in vivo study involved 0.1MCD-fed mice for 4 wk which received 1 or 5 mg/kg/d Aramchol or vehicle by intragastric gavage for the last 2 wk. Liver metabolomics were assessed for the determination of glucose metabolism-related metabolites.
Research results
We discovered that in vitro primary hepatocytes treated with Aramchol increased AMPK activity and decreased the activity of nutrient sensor mTORC1. These effects led to changes in fatty acid (FA) synthesis and oxidation, oxidative phosphorylation, tricarboxylic acid and ribosome, together with reduced cataplerosis. In vivo liver metabolomic analysis indicated an improvement of glucose homeostasis, with normalization of glucose, G6P, F6P, UDP-glucose, and Rbl5P/Xyl5P.
Research conclusions
Aramchol improves glucose and lipid metabolism in NASH by activating FA β-oxidation and oxidative phosphorylation. These effects occur through AMPK activation and mTORC1 inhibition.
Research perspectives
In the future, it would be important to elucidate the exact molecular mechanisms by which Aramchol inhibits SCD1 and affects AMPK and mTORC1 pathways. Moreover, after completion of phase III clinical trial, data from a large patient population is expected to show the importance of glucose homeostasis improvement by Aramchol in NAFLD patients.
Footnotes
Institutional review board statement: The study was reviewed and approved by the Food and Drug Agency at United States (Form Approved: OMS No. 0910-0014, IND No. 079200), and Ministry of Health at Israel (Institutional Committee application number: 0488-14). More information is available at ClinicalTrials.gov NCT02279524.
Institutional animal care and use committee statement: All work performed with animals was approved by an Órgano Habilitado (Comité de Bioética y Bienestar Animal, CBBA/IACUC, at CIC bioGUNE) and the competent authority (Diputación de Bizkaia) following European and Spanish directives, under the following protocol: P-CBG-CBBA-0515. CIC bioGUNE’s Animal Facility is accredited by AAALAC Intl., The Institutional Animal Care and Use Committee at CIC bioGUNE, Derio, Bizkaia, Spain).
Conflict-of-interest statement: Dr. Mato is a Galmed Pharmaceuticals and OWL Metabolomics consultant and/or speaker. Dr. Hayardeny and Dr. Tzivirkun are Galmed Pharmaceuticals employees. Dr. Iruarrizaga-Lejarreta and Dr. Alonso are OWL Metabolomics employees. All other authors have nothing to disclose.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Manuscript source: Unsolicited manuscript
Peer-review started: May 19, 2020
First decision: June 4, 2020
Article in press: August 13, 2020
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: Spain
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Li CP S-Editor: Zhang H L-Editor: Wang TQ P-Editor: Wang LL
Contributor Information
David Fernández-Ramos, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain; CIBERehd - Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas, Madrid 28029, Spain.
Fernando Lopitz-Otsoa, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain.
Laura Delacruz-Villar, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain.
Jon Bilbao, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain.
Martina Pagano, Department of Precision Medicine, University of Campania "Luigi Vanvitelli", Naples 80138, Italy.
Laura Mosca, Department of Precision Medicine, University of Campania "Luigi Vanvitelli", Naples 80138, Italy.
Maider Bizkarguenaga, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain.
Marina Serrano-Macia, Liver Disease Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Spain.
Mikel Azkargorta, Proteomics Platform, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Spain.
Marta Iruarrizaga-Lejarreta, OWL Metabolomics, Derio 48160, Spain.
Jesús Sot, Instituto Biofisika (UPV/EHU, CSIC), Leioa 48940, Spain; Departamento de Bioquímica y Biología Molecular, Universidad del País Vasco, Leioa 48940, Spain.
Darya Tsvirkun, Pre-clinical and Chemistry, Manufacturing and Controls, Galmed Pharmaceuticals, Tel Aviv 6578317, Israel.
Sebastiaan Martijn van Liempd, Metabolomics Platform, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Spain.
Felix M Goni, Instituto Biofisika (UPV/EHU, CSIC), Leioa 48940, Spain; Departamento de Bioquímica y Biología Molecular, Universidad del País Vasco, Leioa 48940, Spain.
Cristina Alonso, OWL Metabolomics, Derio 48160, Spain.
María Luz Martínez-Chantar, CIBERehd - Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas, Madrid 28029, Spain; Liver Disease Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Spain.
Felix Elortza, Proteomics Platform, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Spain.
Liat Hayardeny, Pre-clinical and Chemistry, Manufacturing and Controls, Galmed Pharmaceuticals, Tel Aviv 6578317, Israel.
Shelly C Lu, Division of Digestive and Liver Diseases, Cedars-Sinai Medical Center, Los Angeles, CA 90048, United States.
José M Mato, Precision Medicine and Metabolism Laboratory, Centro de Investigación Cooperativa en Biociencias (CIC bioGUNE), Derio 48160, Bizkaia, Spain; CIBERehd - Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas, Madrid 28029, Spain. director@cicbiogune.es.
Data sharing statement
No additional data are available.
References
- 1.Younossi ZM, Henry L, Bush H, Mishra A. Clinical and Economic Burden of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Clin Liver Dis. 2018;22:1–10. doi: 10.1016/j.cld.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, Setiawan VW, Tran T, Ayoub WS, Lu SC, Klein AS, Sundaram V, Nissen NN. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am J Gastroenterol. 2018;113:1649–1659. doi: 10.1038/s41395-018-0088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, Federico A, Persico M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med Cell Longev. 2018;2018:9547613. doi: 10.1155/2018/9547613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76:99–128. doi: 10.1007/s00018-018-2947-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed M. Non-alcoholic fatty liver disease in 2015. World J Hepatol. 2015;7:1450–1459. doi: 10.4254/wjh.v7.i11.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassidy S, Syed BA. Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov. 2016;15:745–746. doi: 10.1038/nrd.2016.188. [DOI] [PubMed] [Google Scholar]
- 12.Iruarrizaga-Lejarreta M, Varela-Rey M, Fernández-Ramos D, Martínez-Arranz I, Delgado TC, Simon J, Juan VG, delaCruz-Villar L, Azkargorta M, Lavin JL, Mayo R, Van Liempd SM, Aurrekoetxea I, Buqué X, Cave DD, Peña A, Rodríguez-Cuesta J, Aransay AM, Elortza F, Falcón-Pérez JM, Aspichueta P, Hayardeny L, Noureddin M, Sanyal AJ, Alonso C, Anguita J, Martínez-Chantar ML, Lu SC, Mato JM. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun. 2017;1:911–927. doi: 10.1002/hep4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golan-Gerstl R, Valitsky M, Oren R, Brazovski EE, Hayardeny L, Reif S. The anti Fibrotic effect of Aramchol on liver Fibrosis in TAA animal model. J Hepatol. 2017;66:S655–6. [Google Scholar]
- 14.Leikin-Frenkel A, Gonen A, Shaish A, Goldiner I, Leikin-Gobbi D, Konikoff FM, Harats D, Gilat T. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non-atherogenic. Arch Med Res. 2010;41:397–404. doi: 10.1016/j.arcmed.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 2014;53:124–144. doi: 10.1016/j.plipres.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, Oren R FLORA Group. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2085–91.e1. doi: 10.1016/j.cgh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 17.V. Ratziu, L.d.G., R. Safadi, F. Poordad, F. Fuster, J. Flores-Figueroa, S.A. Harrison, M. Arrese, S. Fargion, D. Ben Bashat, C. Lackner, T. Gorfine, S. Kadosh, R. Oren RL and AJS on behalf of the A investigator study group. One-Year Results of the Global Phase 2b Randomized Placebo-Controlled Arrest Trial of Aramchol, a Stearoyl CoA Desaturase Inhibitor, inPatients with Nash. Late-Breaking Abstracts. Hepatology. 2018;68:1447A–1448A. [Google Scholar]
- 18.Schnell O, Crocker JB, Weng J. Impact of HbA1c Testing at Point of Care on Diabetes Management. J Diabetes Sci Technol. 2017;11:611–617. doi: 10.1177/1932296816678263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia W. Standardising HbA1c-based diabetes diagnosis: opportunities and challenges. Expert Rev Mol Diagn. 2016;16:343–355. doi: 10.1586/14737159.2016.1133299. [DOI] [PubMed] [Google Scholar]
- 20.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 21.Alonso C, Fernández-Ramos D, Varela-Rey M, Martínez-Arranz I, Navasa N, Van Liempd SM, Lavín Trueba JL, Mayo R, Ilisso CP, de Juan VG, Iruarrizaga-Lejarreta M, delaCruz-Villar L, Mincholé I, Robinson A, Crespo J, Martín-Duce A, Romero-Gómez M, Sann H, Platon J, Van Eyk J, Aspichueta P, Noureddin M, Falcón-Pérez JM, Anguita J, Aransay AM, Martínez-Chantar ML, Lu SC, Mato JM. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology. 2017;152:1449–1461.e7. doi: 10.1053/j.gastro.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbier-Torres L, Delgado TC, García-Rodríguez JL, Zubiete-Franco I, Fernández-Ramos D, Buqué X, Cano A, Gutiérrez-de Juan V, Fernández-Domínguez I, Lopitz-Otsoa F, Fernández-Tussy P, Boix L, Bruix J, Villa E, Castro A, Lu SC, Aspichueta P, Xirodimas D, Varela-Rey M, Mato JM, Beraza N, Martínez-Chantar ML. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6:2509–2523. doi: 10.18632/oncotarget.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Arranz I, Mayo R, Pérez-Cormenzana M, Mincholé I, Salazar L, Alonso C, Mato JM. Enhancing metabolomics research through data mining. J Proteomics. 2015;127:275–288. doi: 10.1016/j.jprot.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 24.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin SC, Hardie DG. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018;27:299–313. doi: 10.1016/j.cmet.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor RS, Guo L, Ghassemi S, Snyder NW, Worth AJ, Weng L, Kam Y, Philipson B, Trefely S, Nunez-Cruz S, Blair IA, June CH, Milone MC. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci Rep. 2018;8:6289. doi: 10.1038/s41598-018-24676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moody L, Xu GB, Chen H, Pan YX. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim Biophys Acta Gene Regul Mech. 2019;1862:141–152. doi: 10.1016/j.bbagrm.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Briant LJB, Dodd MS, Chibalina MV, Rorsman NJG, Johnson PRV, Carmeliet P, Rorsman P, Knudsen JG. CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets. Cell Rep. 2018;23:3300–3311. doi: 10.1016/j.celrep.2018.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gnoni A, Giudetti AM. Dietary long-chain unsaturated fatty acids acutely and differently reduce the activities of lipogenic enzymes and of citrate carrier in rat liver. J Physiol Biochem. 2016;72:485–494. doi: 10.1007/s13105-016-0495-3. [DOI] [PubMed] [Google Scholar]
- 31.Paglialunga S, Dehn CA. Clinical assessment of hepatic de novo lipogenesis in non-alcoholic fatty liver disease. Lipids Health Dis. 2016;15:159. doi: 10.1186/s12944-016-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montandon SA, Somm E, Loizides-Mangold U, de Vito C, Dibner C, Jornayvaz FR. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci Rep. 2019;9:16810. doi: 10.1038/s41598-019-53346-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park HS, Jeon BH, Woo SH, Leem J, Jang JE, Cho MS, Park IS, Lee KU, Koh EH. Time-dependent changes in lipid metabolism in mice with methionine choline deficiency-induced fatty liver disease. Mol Cells. 2011;32:571–577. doi: 10.1007/s10059-011-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, Okanoue T, Yoshikawa T. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Invest. 2010;90:1169–1178. doi: 10.1038/labinvest.2010.75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.