

Summary
Glucose homeostasis is maintained through organ crosstalk that regulates secretion of insulin to keep blood glucose levels within a physiological range. In type 2 diabetes, this coordinated response is altered, leading to a deregulation of beta cell function and inadequate insulin secretion. Reprogramming of white adipose tissue has a central role in this deregulation, but the critical regulatory components remain unclear. Here, we demonstrate that expression of the transcriptional coregulator GPS2 in white adipose tissue is correlated with insulin secretion rate in humans. The causality of this relationship is confirmed using adipocyte-specific GPS2 knockout mice, in which inappropriate secretion of insulin promotes glucose intolerance. This phenotype is driven by adipose-tissue-secreted factors, which cause increased pancreatic islet inflammation and impaired beta cell function. Thus, our study suggests that, in mice and in humans, GPS2 controls the reprogramming of white adipocytes to influence pancreatic islet function and insulin secretion.
Keywords: type 2 diabetes, adipose tissue, organ crosstalk, pancreas, beta cells, insulin, transcriptional coregulator, G protein pathway suppressor 2, GPS2
Graphical Abstract
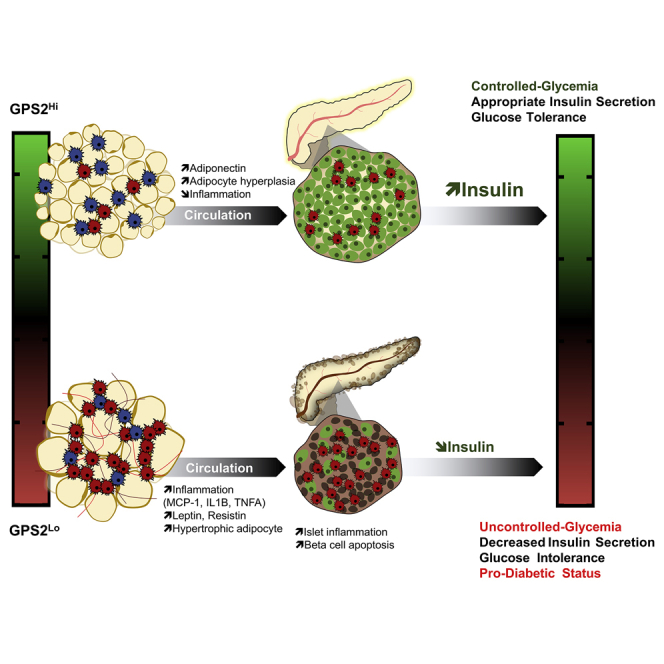
Highlights
-
•
GPS2 expression in adipose tissue is associated with insulin secretion rate in humans
-
•
Loss of GPS2 in adipocytes impacts on insulin secretion upon diet-induced obesity
-
•
Beta cell dysfunction in Gps2 KO mice is governed by islet inflammation
-
•
This beta cell maladaptation in Gps2 KO mice is mediated by adipose tissue secretome
Appropriate insulin secretion is governed through organ crosstalk. Drareni et al. show that GPS2 expression in adipose tissue is correlated with insulin secretion rate in humans. The causality of this relationship is confirmed using adipocyte-specific GPS2 knockout mice, in which inappropriate secretion of insulin promotes glucose intolerance in obese mice.
Introduction
Metabolic homeostasis is regulated by the coordinated action of multiple organ systems. The central principle of glucose homeostasis is that pancreatic beta cells secrete insulin in response to glucose, and insulin acts upon target tissues, such as the liver, muscle, and adipose tissue, to reduce blood glucose levels. However, in addition to glucose, beta cells respond to a vast array of metabolic and endocrine signals, including lipids, hormones, and cytokines (Scharfmann et al., 2019). Therefore, beta cells integrate a plethora of systemic signals that reflect the metabolic status and secrete appropriate amounts of insulin to maintain metabolic homeostasis. In type 2 diabetes, this metabolic homeostasis is disturbed, leading to the maladaptation of beta cells characterized by inappropriate secretion of insulin and glucose intolerance. In this pathophysiological context, white adipose tissue (WAT) is an essential endocrine organ that contributes to maintain glucose homeostasis by secreting factors, such as adiponectin, that optimize insulin secretion and action (Romacho et al., 2014). However, in obesity, WAT plays a key function in disturbing glucose homeostasis by secreting factors that affect the function of cells and tissues throughout the body, including beta cells (Kita et al., 2019; Marcelin et al., 2019). During the development of obesity, WAT undergoes morphological and cellular changes, leading to adipocyte hypertrophy, WAT inflammation, and hypoxia, which represent hallmarks of maladaptive WAT expansion in obesity. This maladaptive WAT expansion has been associated with numerous deleterious cardio-metabolic complications, including type 2 diabetes in humans (Muir et al., 2016; Romacho et al., 2014). Others and we recently proposed that the transcriptional coregulator GPS2, subunit of the chromatin-modifying HDAC3 corepressor complex (Treuter et al., 2017), influences adipocyte functions in the context of obesity and type 2 diabetes (Cardamone et al., 2012, 2014, 2018; Cederquist et al., 2016; Drareni et al., 2018; Fan et al., 2016; Toubal et al., 2013). By studying human WAT, we previously reported that reduced expression of GPS2 in adipocytes coincides with the increased expression of inflammatory (interleukin-6 [IL-6] and MCP1) and hypoxic (HIF1A) genes and hypertrophic adipocytes (Drareni et al., 2018; Toubal et al., 2013). Our functional analysis further revealed that adipocyte-specific loss of GPS2 triggers transcriptional alterations of inflammatory and remodeling gene signatures in WAT, leading to a pro-diabetic status characterized by insulin resistance and glucose intolerance.
Here, we discover a previously unrecognized adipocyte-specific role of GPS2 in the regulation of insulin secretion from beta cells. We report that GPS2 mRNA levels in WAT correlate with the insulin secretion rate in humans. Mechanistic studies revealed that the loss of GPS2 in adipocyte-specific knockout (AKO) mice provokes an inadequate adaption of pancreatic beta cells, leading to decreased insulin secretion in obese mice. Ex vivo assays revealed that the WAT secretome of obese GPS2 AKO mice interferes with beta cell function, leading to a reduction of insulin secretion upon glucose stimulation. Collectively, our study reveals that the loss of GPS2 in adipocytes impacts beta cell adaptation and insulin secretion during the progression of obesity and type 2 diabetes.
Results
Potential Impact of GPS2 Function in WAT in Insulin Secretion Rate in Humans
To evaluate the impact of GPS2 action in WAT onto insulin secretion rates (ISRs) in humans, we measured the mRNA levels of GPS2 in subcutaneous WAT (scWAT) of 23 clinical study participants (8 with normal glucose tolerance and 15 with type 2 diabetes; aged 48 ± 12 years; men 68%), which were subjected to graded glucose infusion in order to measure ISR (Table S1). Each subject received a stepped intravenous (i.v.) glucose infusion at 2, 4, 6, 8, and 10 mg/kg/min, each step lasting for 40 min. ISR was derived by deconvolution of peripheral C-peptide levels. Participants were stratified according to GPS2 mRNA expression in scWAT, quantified by qRT-PCR, into two categories, low (Lexp) or high expression (Hexp), defined by values below or above the median, respectively (Table S1). Basal glucose levels and ISR were higher in Lexp than in Hexp participants: 8.0 ± 0.5 versus 5.6 ± 0.3 mmol/L, mean ± SEM, p = 0.0008, and 3.04 ± 0.19 versus 2.12 ± 0.17 pmol/kg/min, p = 0.002, respectively (Figure S1). Following glucose infusion, glucose levels increased 178% ± 17% and 174% ± 23% (p = 0.88) in Lexp and Hexp participants, with average values of 14.9 ± 0.6 and 11.4 ± 0.78 mmol/L (p = 0.002), respectively. Concomitant ISR levels increased 236% ± 40% and 487% ± 75% (p = 0.006) in Lexp and Hexp participants. Average ISR following glucose infusion was 5.40 ± 0.67 versus 8.49 ± 0.75 pmol/kg/min, p = 0.01, respectively (Figure S1). Analysis of the glucose/ISR response curve demonstrated significantly higher ISR in Hexp than in Lexp participants at all glucose levels (p = 0.005, adjusted for sex, age, BMI, and glycemic status; Figure 1A). It was ∼52%, ∼60%, and ∼45% higher at 7, 10, and 15 mmol/L glucose, respectively. Average ISR (time 10–200 min) was well correlated with GPS2 mRNA expression in scWAT (Figure 1B).
Figure 1.

GPS2 Expression Level in WAT Influences Insulin Secretion Rate in Humans
(A) Insulin secretion rate (ISR) by capillary blood glucose during a graded glucose infusion. Participants were stratified by GPS2 mRNA expression as low or high expression as per values below (n = 12) or above the median (n = 11), respectively. Data are expressed as mean ± SEM. Statistics were computed in a mixed regression model with random effects and adjusted for sex, age, insulin sensitivity index (M), and glycemic status. p < 0.05 is significant. Symbols represent mean ± SEM at baseline and at each glucose infusion step for low or high expression groups of participants. For each participant, the average values of the four samples obtained at 10-min intervals at each glucose infusion step were used. Curves represent a log-fit. p < 0.05 is significant.
(B) Leverage plot for the correlation between average ISR (time 10–200 min) during a graded glucose infusion and GPS2 mRNA expression. Symbols represent data from individual participants. Correlation was adjusted for sex, age, baseline C-peptide, and insulin sensitivity index (M) in a multiple regression analyses R2 = 0.34, p = 0.004 for ISR and GPS2 mRNA expression correlation and R2 = 0.56, p = 0.0003 for the whole model.
(C) Correlation of acute insulin response (AIR) during a glucose-dependent arginine stimulation and adipocyte GPS2 mRNA expression in multivariate analyses. Correlation was adjusted for sex, age, glycemic status (T2DM or ND), and pre-arginine injection glucose levels. R2 = 0.31, p = 0.03 for AIR and GPS2 mRNA R2 = 0.80, p < 0.0001 for the whole model. Acute insulin response is defined as the mean of the 3 higher values from time 2 to 5 min.
Acute insulin, glucagon, and C-peptide were also quantified during glucose-dependent arginine stimulation. We observed that the acute secretion of insulin and C-peptide in participants with high GPS2 expression in WAT was more pronounced than in participants with low expression. In mirror, glucagon secretion was less pronounced in the high-expression group than in the low-expression group (Figure S1). Importantly, multivariate correlative analyses between GPS2 expressions in WAT with acute insulin secretion demonstrated a significant positive correlation between WAT GPS2 mRNA levels and acute insulin secretion (R2 = 0.31; p = 0.03, adjusted for sex, age, glycemic status, and pre-arginine glucose levels; Figure 1C). However, we did not observe correlation between WAT GPS2 mRNA levels and acute glucagon secretion (R2 = 0.04; p = 0.88, adjusted for sex, age, glycemic status, and pre-arginine glucose levels; Figure S1). Collectively, these results suggest a potential impact of GPS2 function in WAT on the insulin secretion response to glucose, but not to the secretion of glucagon.
Similar associations were observed in mice. Stratification of 18 high-fat-fed mice according to Gps2 expression in WAT revealed that Gps2 low expressers are more glucose intolerant than Gps2 high expressers (Figures 2A, 2B, S2A, and S2B). The glucose intolerance observed in Gps2 low expressers correlated with a decreased glucose-stimulated insulin secretion (Figures 2C and 2D), although the insulin resistance status (based on HOMA-IR) was similar between the groups (Figure S2C). Importantly, we could not observe these correlations by stratifying mice based on Gps2 expression in brown adipose tissue (BAT) (Figures S2D–S2F) or in liver (data not shown). Altogether, the data suggest that GPS2 action in WAT may influence insulin secretion from beta cells in humans and in mice.
Figure 2.

Adipocyte-Specific GPS2 Deficiency in Mice Leads to an Impaired Insulin Secretion in Response to Glucose
(A) qRT-PCR analysis of GPS2 in eWAT from WT C57BL6/J under HFD feeding for 12 weeks (n = 18), classified into 2 groups: high GPS2 expression (n = 9) and low GPS2 expression (n = 9).
(B) Oral glucose tolerance test (OGTT) in WT C57BL6 after 12 weeks of HFD classified into 2 groups: high GPS2 expression (n = 9) and low GPS2 expression (n = 9).
(C) Measurements of insulin secretion during the OGTT in WT C57BL6/J after 12 weeks of HFD classified into 2 groups: high GPS2 expression and low GPS2 expression (high GPS2 n = 9 and low GPS2 n = 9).
(D) Glucose-stimulated insulin secretion calculated using the ratio of insulin secretion during OGTT at 15 min (T15) to basal (T0) in WT C57BL6/J after 12 weeks of HFD classified into 2 groups: high GPS2 expression and low GPS2 expression (high GPS2 n = 9 and low GPS2 n = 9).
(E) Fasting insulin concentration in WT and GPS2 AKO mice in 4 and 12 weeks of HFD (n = 5 to 6).
(F) Measurements of insulin secretion during the OGTT in WT and GPS2 AKO mice in 4 and 12 weeks of HFD (4 weeks HFD n = 6; 12 weeks HFD n = 5 to 6).
(G) Glucose-stimulated insulin secretion calculated using the ratio of insulin secretion during OGTT at 15 min (T15) to basal (T0) in WT and GPS2 AKO mice in 4 and 12 weeks of HFD (4 weeks HFD n = 6; 12 weeks HFD n = 5 to 6).
All data are represented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Specific Depletion of GPS2 in Adipocytes Impacts Insulin Secretion upon Diet-Induced Obesity
To assess the impact of adipocyte GPS2 deficiency on the regulation of insulin secretion upon diet-induced obesity, GPS2 AKO mice and their wild-type (WT) littermates were subjected to either normal chow diet (CD) or high-fat diet (HFD) for 4 and 12 weeks and then glucose homeostasis and insulin secretion were monitored. Depletion of GPS2 was efficient in WAT and BAT depots in GPS2 AKO mice although GPS2 expression was not altered in liver and pancreatic islets, as expected (Figure S2G). As previously described (Drareni et al., 2018; Fan et al., 2016), GPS2 AKO mice were more glucose intolerant than WT controls after 4 weeks and 12 weeks of HFD, although no difference was observed in CD (Figure S3A). Fasting insulin concentration was lower (trend) in the blood of GPS2 AKO mice compared to WT mice only under obese conditions (Figure 2E). In agreement with this observation, insulin secretion, measured during the oral glucose tolerance test (OGTT), was altered in the GPS2 AKO mice compared to WT mice after 4 weeks and 12 weeks of HFD, although no difference was observed under CD conditions (Figures 2F, S3B, and S3C). Indeed, insulin secretion calculated using the ratio between insulin concentrations at 15 min after glucose ingestion to baseline was lower in the GPS2 AKO mice compared to WT mice upon HFD (Figure 2G).
Impaired insulin secretion by the pancreas in response to glucose can be the result of decreased beta cell mass and/or impaired function of beta cells to produce insulin in response to glucose (Dalmas, 2019b; Donath et al., 2013). In order to identify the mechanisms underlying the inadequate insulin secretion caused by GPS2 deficiency in mature adipocytes, we evaluated the size and number of pancreatic islets in our mice (Figures 3A–3C). We did not observe significant changes in the number of islets in both genotypes under CD and HFD (Figures 3B and S3D). Unexpectedly, the islet size (surface) was significantly reduced in GPS2 AKO mice compared to WT only upon HFD (Figures 3A–3C). The inadequate adaptation of pancreatic islets to HFD could have been provoked by an increased local inflammation and beta cell apoptosis (Dalmas, 2019a). In support of this possibility, we found that pancreatic islets from GPS2 AKO mice exhibited increased macrophage infiltration and beta cell apoptosis in conjunction with decreased beta cell proliferation compared to WT mice under HFD conditions, as assessed by Mac2+ staining, TUNEL assay, and Ki67+ staining, respectively (Figures 3D–3H). Inflammatory and apoptotic signaling molecules were also increased at the mRNA level in pancreatic islets from HFD-fed GPS2 AKO mice compared to WT controls (Figures 3I and 3J). Of note, proliferative markers were also deregulated in islets from HFD-fed GPS2 AKO mice, suggesting maladaptation to energy surplus (Figure 3K). We also observed that expression of beta cell differentiation markers, such as Ins2, Mafb, Glut2, Nkx6-1, and Pdx-1, was reduced in islets of HFD-fed GPS2 AKO mice compared to WT mice (Figure 3L). Consistent with potential increased local inflammation, beta cell apoptosis, and dedifferentiation, pancreatic islets isolated from HFD-fed GPS2 AKO mice secreted less insulin in response to glucose stimulation than WT controls (Figure 3M).
Figure 3.

GPS2 Deficiency in Adipocytes Provokes Pancreatic Islet Inflammation and Beta Cell Apoptosis
(A) Representative images of insulin immunofluorescence staining in pancreas from WT and GPS2 AKO mice in 4 and 12 weeks of HFD (4 weeks HFD n = 6; 12 weeks HFD n = 6).
(B and C) Measurements of islets number (B) and surface (C) in pancreas from WT and GPS2 AKO mice in 4 and 12 weeks of HFD (4 weeks HFD n = 6; 12 weeks HFD n = 6).
(D) Representative images of Mac2 immunofluorescence staining in pancreas from WT and GPS2 AKO mice in 4 and 12 weeks of HFD (4 weeks HFD n = 5; 12 weeks HFD n = 5).
(E) Quantification of Mac2-positive cells in pancreatic islets from WT and GPS2 AKO mice on HFD (4 weeks HFD n = 5; 12 weeks HFD n = 5).
(F) Representative images of TUNEL+ immunofluorescence staining in pancreas from WT and GPS2 AKO mice on HFD (n = 3).
(G) Quantification of TUNEL-positive cells in pancreatic islets from WT and GPS2 AKO mice in 12 weeks of HFD (n = 3).
(H) Representative images of Ki67+insulin+ immunofluorescence staining and quantification in pancreas from WT and GPS2 AKO mice fed with an HFD.
(I–L) Measurement of inflammatory, apoptotic, proliferative, and differentiation genes by qRT-PCR in pancreatic islets from WT and GPS2 AKO mice after 12 weeks of HFD (n = 3–5).
(M) Glucose-stimulated insulin secretion (GSIS) of islets from WT and GPS2 AKO mice after 4 weeks of HFD treated with low glucose (low Glc, 2.8 mM) and high glucose (high Glc, 16.7 mM; n = 5). Results are expressed as % of insulin content.
All data are represented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
WAT Secretory Products from GPS2 AKO Mice Provoke Islet Dysfunction
Above data suggested that beta cell mass and function are altered in obese GPS2 AKO mice. We therefore considered that secreted factors from GPS2-deficient WAT depots may alter islet function. To further investigate this adipocyte-islet crosstalk, isolated pancreatic islets from healthy WT mice were incubated with medium from WAT or BAT explants or serum from HFD-fed WT and GPS2 AKO mice for 24 h, followed by measurement of glucose-stimulated insulin secretion (Figures 4A and 4B). We observed that islets incubated with WAT explant medium or serum from GPS2 AKO mice were less potent in secreting insulin than islets treated with WAT explant medium or serum from WT mice. Despite efficient GPS2 depletion in the BAT of AKO mice, we did not observe any inhibitory effect of BAT explant medium from AKO mice, as compared to medium from WT mice, on insulin secretion (Figure S4F). Importantly, 1-h stimulation with WAT explant from GPS2 KO mice did not alter insulin secretion in comparison to WAT explant from WT mice (data not shown). This result suggests that WAT secretory products did not interfere with glucose-induced depolarization of the plasma membrane required for secretion of insulin. Islet dysfunction, mediated by medium from GPS2 AKO mice, was characterized by an increased expression of inflammatory (Emr1 (F4/80), Il1-β, and TNF-α) and apoptotic (Fas and Cas3) genes (Figure 4C). Interestingly, the inhibitory signal from WAT explant medium or serum from GPS2 AKO mice was lost after heat inactivating the medium or the serum (Figure 4D), suggesting that secreted proteins and peptides (such as adipokines, cytokines, and hormones) rather than metabolites may link WAT dysfunction to impaired insulin secretion.
Figure 4.

The WAT Secretome of GPS2 AKO Mice Promotes Beta Cell Failure
(A and B) GSIS of islets from WT C57BL6/J mice cultured with scWAT, eWAT, mesWAT explant media, or serum and treated with low glucose (low Glc, 2.8 mM) and high glucose (high Glc, 16.7 mM). Results are expressed in % of insulin content (n = 3).
(C) Expression of inflammatory and apoptotic genes in isolated islets from WT C57BL6/J mice cultured with eWAT explant media for 12 h (n = 3).
(D) GSIS of islets from WT C57BL6/J mice cultured with scWAT, eWAT, and serum heat inactivated culture medium for 12 h and then treated for 2 h with low glucose (low Glc, 2.8 mM) and high glucose (high Glc, 16.7 mM). Results are expressed in % of insulin content (n = 3).
(E) Transcriptome analyses by RNA sequencing of scWAT, epiWAT, and mesWAT from WT and GPS2 AKO mice after 12 weeks of HFD (n = 3). Heatmap and Gene Ontology terms of the differentially expressed genes in WT and GPS2 AKO fat pads are shown.
(F and G) WAT gene expression and secretome analysis of scWAT and eWAT from WT and GPS2 AKO mice after 12 weeks of HFD (n = 4 to 5).
All data are represented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
In order to characterize the GPS2-dependent gene signature in WAT depots that may contribute to beta cell dysfunction, we performed transcriptome analysis by RNA sequencing and qRT-PCR in 3 different WAT depots (subcutaneous WAT [scWAT], epididymal WAT [eWAT], and mesenteric WAT [mesWAT]). This analysis identified around 500 genes that were differentially expressed in all the 3 fat pads of GPS2 AKO mice, compared to WT mice, after 12 weeks of HFD (Figure 4E). Gene ontology analysis revealed that the most upregulated genes in WATs of the GPS2 AKO mice are involved in immune responses and insulin signaling (Figure 4E), in agreement with our previous study (Drareni et al., 2018). This pathologic WAT gene signature from GPS2 AKO mice was confirmed at the protein level in serum and WAT explant media by measuring adipokines (adiponectin, leptin, and resistin) and inflammatory cytokines (IL-6, TNFA, MCP-1/CCL2, and IL-1B; Figures 4F and 4G). Importantly, the BAT of GPS2 AKO mice did not present the same phenotype as the WAT, which might explain the lack of inhibitory capacity of BAT medium on insulin secretion (Figure S4E). Further, mesWAT seemed to act differently from eWAT and scWAT, because the inhibitory effect on islet function was not altered after heat inactivation, and the inflammatory profile of mesWAT from GPS2 AKO mice was less pronounced than eWAT and scWAT (Figures S4A and S4B). This difference is consistent with the proposed unique contribution of mesenteric fat, in comparison to other WAT depots, in obesity complications (Dowal et al., 2017).
Discussion
The pathogenesis of type 2 diabetes is a multifactorial disease involving interactions between all metabolic tissues. Here, we report that the dysfunction of white adipose tissue, driven by the depletion of the transcriptional coregulator GPS2 in adipocytes, induces a pancreatic dysfunction reflected by the failure of beta cells to adapt to diet-induced obesity. We have previously shown that GPS2 AKO mice develop adipocyte hypertrophy and glucose intolerance (Drareni et al., 2018). However, despite systemic glucose intolerance, AKO mice did not develop systemic insulin resistance, suggesting that the pancreatic response to glucose was impaired. Our current study elucidates the underlying mechanism by demonstrating that adipocyte dysfunction and reprogramming in AKO mice leads to a reduction of pancreatic islet size and an impaired insulin secretion in response to glucose upon HFD.
Beta cell mass is characterized by its plasticity and its ability to adapt to different situations, such as pregnancy and weight gain (Golson et al., 2010; Sorenson and Brelje, 1997). In response to HFD, beta cells produce more insulin, both by secreting more insulin per cell and by increasing cell mass. Surprisingly, both mechanisms seem to be impaired in the GPS2 AKO mice islets in response to HFD. Treating pancreatic islets from C57BL6 WT mice with adipose tissue-conditioned media from GPS2 AKO mice for 12 h was sufficient to inhibit insulin secretion, although no effect was seen after 1 h. In addition to the defects in insulin secretion, islets from GPS2 AKO mice also showed an increase of pro-apoptotic markers. Both alterations could be the result of the exposure of the islets to pro-inflammatory cytokines (IL-1B and TNFA) and adipokines (leptin and resistin). It has been shown that short-time (12–48 h) treatment of pancreatic beta cells with pro-inflammatory cytokines or adipokines results in significant inhibition of insulin secretion (Cantley, 2014; Kiely et al., 2007; Ying et al., 2019; Zhang and Kim, 1995). Furthermore, chronic exposure of beta cells to the same pro-inflammatory cytokines and adipokines provokes increased inflammation and apoptosis, resulting in decreased islet size and function (Dalmas et al., 2017; Demine et al., 2020; Ying et al., 2019).
Our results suggest that the pancreatic phenotype of GPS2 AKO mice is driven by mediators, likely proteins and peptides, released by the dysfunctional and hypertrophic adipose tissue from the AKO mice. This hypothesis is reinforced by the normal insulin secretion observed in WT C57BL6 islets treated with a pre-heated adipose-tissue-conditioned medium from GPS2 AKO mice. We believe that the phenotype of our GPS2 AKO mice is not due to one single molecule but to the combination of several altered molecules, including inflammatory cytokines and adipokines. In fact, a previous study from our laboratories demonstrated that myeloid deficiency of GPS2 provokes severe WAT inflammation without hypertrophy (Fan et al., 2016), which is not characterized by alteration of insulin secretion but by an increase of insulin production (adaptation to insulin resistance). These data suggest that inflammatory cytokines, secreted from macrophages and/or adipocytes, are not the main mediators of the GPS2 AKO phenotype. Increased WAT inflammation in combination with deregulation of adipokine secretion, such as adiponectin, leptin, and resistin, as observed in WAT of GPS2 AKO mice is more likely to be the main reason for a maladaptation of insulin secretion in GPS2 AKO upon HFD feeding.
Whereas WAT is well known to be characterized by a low-grade inflammation, increased macrophage accumulation, and pro-inflammatory cytokine and adipokine release during obesity, BAT in contrast seems to be more resistant to inflammation, as it displays less immune cell infiltration and pro-inflammatory cytokine secretion than WAT (Dowal et al., 2017; Fitzgibbons et al., 2011; Roberts-Toler et al., 2015). Specifically relevant for our study is the notion that the infiltration of immune cells in the BAT begins only after 16 months of HFD in mice (Dowal et al., 2017; Roberts-Toler et al., 2015), although GPS2 AKO mice were exposed to HFD for either 4 or 12 weeks. Consistent with that, treating islets from WT mice with GPS2-AKO-BAT-conditioned media did not alter their insulin secretion after glucose stimulation. Furthermore, GPS2 expression levels in the BAT of WT mice did not show any correlation with their metabolic status (i.e., glucose tolerance and insulin sensitivity and secretion). Therefore, our results support a mechanism in which the islet phenotype seen in GPS2 AKO mice is governed by dysfunction and inflammation of the WAT, but not the BAT.
Although it seems clear that WAT dysfunction in GPS2 AKO mice and in humans with lower GPS2 expression triggers pancreatic beta cell failure, there might be several mechanisms by which the loss of GPS2 in adipocytes leads to that phenotype. However, the coincidence of pancreatic dysfunction with adipocyte hypertrophy in the GPS2 AKO mice suggests that these phenotypes are interconnected and the result of the same adipocyte reprogramming upon disruption of specific regulatory transcriptional networks. This is supported by other mouse models of adipocyte hypertrophy, showing that the mice were glucose intolerant, but not insulin resistant, thus pointing at pancreatic dysfunction (Rohm et al., 2013). In our previous study (Drareni et al., 2018), we discovered that GPS2 represses the activity of the transcription factor HIF1. Interestingly, adipocyte-specific overexpression of HIF1A is characterized by hypertrophic adipocytes, WAT insulin resistance, and glucose intolerance (Halberg et al., 2009). Despite the increased insulin-resistant status of mice overexpressing HIF1A in adipocytes, no change in glucose-induced insulin release was observed in comparison to control mice, suggesting an inadequate adaptive response in pancreatic islets to produce insulin (Halberg et al., 2009). By contrast, genetic or pharmacological inhibition of HIF1A in adipocytes in obese mouse models is characterized by decreased adipose cell size, improvement of glucose tolerance and insulin sensitivity, and a reduction of glucose-induced insulin release (Jiang et al., 2011; Sun et al., 2013).The interplay between WAT phenotype, including hypertrophy, and islet remodeling upon obesity is further supported by very recent studies in humans (Kusminski et al., 2020; Gao et al., 2020). Together, these studies emphasize a potentially significant role of the GPS2 regulatory network in adipocytes for linking the WAT phenotype to the regulation of adaptive islet responses upon energy surplus.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal insulin antibody (Alexa 647 Fluor conjugated) | Cell signaling | Cat# C27C9; RRID: AB_2687822 |
| Mac2 Monoclonal antibody | Cedarlane | Cat# CL8942AP; RRID: AB_10060357 |
| In situ Cell death detection kit, TMR red | Sigma-Aldrich | Cat# 12156792910; RRID: N/A |
| KI67 Monoclonal antibody | e-bioscience | Cat# 13-5698-82; RRID: AB_2572794 |
| Biological Samples | ||
| Mouse adipose tissue explants | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Insulin (HUMALOG 100 unites/ml) | lilly | N/A |
| Insulin, Human Recombinant | Sigma | Cat#I-9278 |
| Rosiglitazone | Sigma | Cat#R-2408 |
| Iso-butyl-methylxanthine | Sigma | Cat#I5879-SG |
| Dexamethasone | Sigma | Cat#D1756 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma | Cat#C2920 |
| Critical Commercial Assays | ||
| MULTIPLEX Adipocyte kit | Millipore, | Cat# MADCYMAG-72K |
| Ultrasensitive mouse/rat insulin kit | Meso scale discovery | Cat# K152B2C-3 |
| M-MLV Reverse Transcriptase kit | invitrogen | Cat# 28025-021 |
| RNeasy RNA Mini Kit | qiagen | Cat# 74136 |
| Sybergreen | Eurogentec | Cat# RT-SY2X-03+WOUFL |
| Taquman | Promega | Cat# A6102 |
| Deposited Data | ||
| Data files for RNA sequencing | NCBI Gene expression: https://www.ncbi.nlm.nih.gov/geo/ | GEO: GSE111647 |
| Cell lines | ||
| Primary pre-adipocytes | This paper | N/A |
| Isolated mouse islets | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Adipoq-Cre | Jackson Laboratory | 010803 |
| Mouse: Gps2flox/flox mice | Fan et al., 2016 | Generated at Ozgene Pty, Ltd. (Bentley DC, Australia) |
| Mouse: c57bl6 | The Jackson laboratory | 000664 |
| Oligonucleotides | ||
| See Table S2 for the list of Oligos | N/A | N/A |
| Software and Algorithms | ||
| FIJI | FIJI Software | https://fiji.sc/ |
| Graphpad 7 | GraphPad Software | https://www.graphpad.com/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts: Nicolas Venteclef (nicolas.venteclef@inserm.fr)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the RNA-Seq gene expression data and raw fastq files in this paper is GEO: GSE111647 (https://www.ncbi.nlm.nih.gov/geo/).
Experimental Model and Subject Details
Clinical study
Human population
Biopsies from subcutaneous adipose tissue (SAT) were obtained from participants admitted to the Lariboisière hospital, University center of diabetes and its complications, Paris, France. Clinical and anthropometric data are summarized in Table S1. The study was conducted in accordance with the Helsinki Declaration and was registered in a public trial registry (Clinicaltrials.gov; NCT02368704). The Ethics Committee of CPP Ile-de-France approved the clinical investigations for all individuals, and written informed consent was obtained from all individuals. The principal investigator of this clinical trial is Prof. Gautier Jean-François: jean-francois.gautier@aphp.fr.
Measurement of insulin secretion
Insulin secretion was evaluated during the administration of graded infusions of glucose. Participants were instructed to eat a diet in which carbohydrates comprised at least 50% of total calories for at least five days preceding the test. Studies were started at 8:00-9:00 AM with subjects in the recumbent position after a 12h overnight fast. Intravenous catheters were placed in the right and in the left forearms for glucose administration and blood sampling. Following a 30 min baseline sampling period, a graded intravenous infusion of 30% glucose was then started at a rate of 2 mg/kg body weight.min-1, followed by infusions at 4, 6, 8 and 10 mg/kg body weight.min-1. Each infusion rate was maintained for a period of 40 min. Blood samples were drawn every 10 min throughout the experiment for measurement of glucose and C-peptide concentrations.
Measurement of acute insulin and glucagon secretions
At the end of the glucose ramping, the 20% glucose infusion rate was adjusted to obtain a blood glucose level of approximately 400 mg/dL (22 mmol/L). Glucose infusion was then maintained and an i.v. bolus of 5 g of arginine chlorhydrate was administered in 45 s, with venous blood sample collection before and 2, 3, 4, 5, 10, and 15 minutes after the bolus for measurements of insulin, glucagon et C-peptide.
Data analysis
Relationships between glucose levels and pre-hepatic insulin secretion rates (ISR) during graded intravenous glucose infusions were explored. ISR and glucose levels used in the analysis represented the average of the values for each individual at each glucose infusion rate step. ISR was then plotted against the corresponding glucose level to define a dose-response relationship. ISR were derived by deconvolution of circulating C-peptide concentrations (Eaton et al., 1980) using version 3.4a of the ISEC software. Individual kinetics parameters of C-peptide clearance are computed by ISEC from standard kinetic parameters, taking into account the age, sex, body surface area, and glucose tolerance status of the subject. The M index obtained during a hyperinsulinemic euglycemic clamp (data not shown) was used as an index of insulin sensitivity.
Animals
Gps2flox/flox mice were generated at Ozgene Pty, Ltd. (Bentley DC, Australia) using a targeting construct, which contained loxP sites flanking exons 2 and 5, followed by a FRT site and a neomycin cassette inserted between exons 5 and 6. The targeting vector was electroporated into C57BL/6 Bruce4 embryonic stem (ES) cells. The correctly recombined ES colony was then injected into C57BL/6 blastocysts. Male chimeras were mated with female C57BL/6 mice to get mice with a targeted Gps2 allele. The mice were crossbred with C57BL/6 flp-recombinase mice to remove the neomycin cassette to create heterozygous Gps2flox/+ mice. The mice were then crossbred with C57BL/6 mice for nine generations before being bred with heterozygous Gps2flox/+ mice to get the Gps2flox/flox mice (Fan et al., 2016). To generate adipocyte-specific Gps2 KO mice, Gps2flox/flox mice were crossed with adiponectin-Cre mice (B6;FVB-Tg (Adipoq-Cre) 1Evdr/J; Jackson Laboratory stock no. 010803). KO mice were bred for at least nine generations before the experiments were started. Adipoq-Cre-Gps2flox/flox littermates were used as control. All mice used in the studies were male, between 7– 8 weeks old at the time of the experiment starting point, and randomized before any experiment was started. All animal experiments were approved by the French ethical board (Paris-Sorbonne University, Charles Darwin N°5, 01026.02) and conducted in accordance with the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, developed by the Council for International Organizations of Medical Sciences (CIOMS). All mice strains were bred and maintained at the “Centre exploration fonctionnel (CEF)” at Paris Sorbonne University (UMS_28).
Method Details
In vivo treatments, metabolic measurements
Diet-induced obesity and insulin resistance
7–8-weeks-old WT and GPS2 AKO mice were fed with a 60%-fat diet (HFD, Research Diets, D12492) for 4 and 12 weeks.
Oral glucose tolerance test (OGTT)
Mice were fasted overnight before receiving an oral gavage of glucose (2 g/kg). Blood glucose levels were measured directly from tail vein blood at 0, 15, 30, 45, 60 and 90 min using a glucometer (Accu-Chek Performa, Roche). Blood samples were taken from the tail vein at 0, 15, 30 and 60 min.
Analysis of different metabolic circulating factors in explant medium
Adiponectin, leptin, IL6, MCP-1, PA-1 and resistin concentrations were determined by using MULTIPLEX Adipocyte kit according to the manufacturer’s instructions (Millipore, MADCYMAG-72K).
Histology, immunofluorescence and microscopy
Pancreatic tissue samples were fixed in 10% formaldehyde solution overnight and embedded in paraffin. For beta cell area analysis, epitope-specific antibody was used for IF detection of insulin (Cell Signaling Technology,1:200). For macrophages infiltration detection, specific Mac2 antibody was used (Cell Signaling Technology 1:200). For IF analysis, cleared and rehydrated sections were quenched with 3% H2O2 for 15 min at room temperature then washed twice for 5 min each with TBS + 0.1% (vol/vol) Tween-20. Sections were then blocked with TBS + 3% (vol/vol) BSA for 30 min at room temperature. Sections were then incubated for one hour at room temperature or overnight at 4°C with diluted primary antibodies, washed 3 times for 5 min with TBS + 0.1% (vol/vol) Tween-20, followed by incubation with appropriate fluorophore-conjugated secondary antibodies (Invitrogen). Sections were then washed twice for 5 min with TBS then mounted with hard-set DAPI-containing mounting media (Vectashield, Vector Labs). Images were acquired on an Axiovert 200M microscope or through whole slide scanning with ZEISS AxioScan.
Mac2+ cells and TUNEL+cells were quantified based on the average number of Mac2+/TUNEL+ cells per islets in all islets identified in histological sections collected from WT and AKO mice. Ki67+ cells was quantified based on the average number of Ki67+ Insulin+ cells per islets (normalized by the islets area) in all islets identified in histological sections collected from WT and AKO mice.
Isolation of pancreatic islets from mice
To isolate islets, pancreas was perfused through the common bile duct with a HBSS collagenase solution (1.4 g/L; collagenase type 4 Worthington) and digested in the same solution in a 37°C water bath for 15-18 min as previously described (Dalmas et al., 2017). After shaking for 15 s, pancreas were washed three times with HBSS supplemented with 0.5% bovine serum albumin (BSA) and filtrated through 500 mm and 70 mm cell strainers (Corning). Islets were retained on the 70 mm cell strainer while the cell mixture passing through the 70 mm cell strainer represented the exocrine stoma. Islets from the same condition were double handpicked and pooled into a Petri dish with RPMI-1640 (GIBCO) containing 11.1 mM glucose, 100 units/ml penicillin, 100 mg/ml streptomycin, 2 mM Glutamax, 50 mg/ml gentamycin, 10 mg/ml Fungison and 10% FCS (Invitrogen). Islets were used directly for RNA isolation or cell culture in humid environment containing 5% CO2.
Glucose-stimulated insulin secretion assay
Islets were seeded in 60 mm culture dishes for 24 h in RPMI-1640 containing mouse islet medium. 25 islets were transferred to 35 mm culture dishes and then pre-incubated for 1 h in modified Krebs-Ringer bicarbonate buffer (KRB; 115 mM NaCl, 4.7 mM KCl, 2.6 mM CaCl2 2H2O, 1.2 mM KH2PO4, 1.2 mM MgSO4 7H2O, 10 mM HEPES, 0.5% BSA [pH 7.4]) with no glucose, KRB was then replaced by KRB 2.8 mM glucose for 2 h (basal insulin release) or by 2 h in KRB (16.7 mM glucose) (stimulated insulin release). Insulin content was harvested by extraction with 0.18 N HCl in 70% EtOH. Insulin concentrations were determined using a mouse insulin ultrasensitive mouse/rat insulin kit (Meso Scale Discovery, Rockville, MD, USA).
Explants medium collection and islets treatment
Pieces eWAT, scWAT and MesWat pads and liver were isolated from mice and sliced into equally sized pieces of 50-100ug. Explants were washed twice in PBS and cultured in ECBM (endothelial cell basal medium) supplemented with 1% svf and 1% PS for the fat pads and Williams Medium E for liver supplemented with 1% svf and 1% PS during 24H. Islets from C57BL6/J mice were isolated (as previously described) and treated with explant medium diluted 1:8 during 12H.
RT–qPCR analysis
RNA was extracted from tissues or purified cells using the RNeasy RNA Mini Kit (QIAGEN). Complementary DNAs were synthesized using M-MLV Reverse Transcriptase kit (Promega) for the adipose tissue and using super script kit (Promega) for the islets. RT–qPCR was performed using the QuantStudio 3 Real-Time PCR Systems (ThermoFisher Scientific). 18S was used for normalization to quantify relative mRNA expression levels. Relative changes in mRNA expression were calculated using the comparative cycle method (2−ΔΔCt).
RNA-sequencing analyses
Library preparation and Illumina sequencing were performed at the Ecole normale superieure genomic core facility (Paris, France). Messenger (polyA+) RNAs were purified from 400 μg of total RNA using oligo(dT). Libraries were prepared using the strand specific RNA-Seq library preparation TruSeq Stranded mRNA kit (Illumina). Libraries were multiplexed by 12 on 2 runs. A 75 bp single read sequencing was performed on a NextSeq 500 (Illumina). A mean of 25,44 ± 6,01 million passing Illumina quality filter reads was obtained for each of the 12 samples. The analyses were performed using the Eoulsan pipeline (Jourdren et al., 2012), including read filtering, mapping, alignment filtering, read quantification, normalization and differential analysis: Before mapping, poly N read tails were trimmed, reads ≤ 40 bases were removed, and reads with quality mean ≤ 30 were discarded. Reads were then aligned against the Mus musculus genome from Ensembl version91 using STAR (version 2.6.1b) (Dobin et al., 2013). Alignments from reads matching more than once on the reference genome were removed using Java version of samtools (Li et al., 2009). To compute gene expression, Mus musculus GTF genome annotation version 91 from Ensembl database was used. All overlapping regions between alignments and referenced exons were counted using HTSeq-count 0.5.3 (Anders et al., 2015). The sample counts were normalized using DESeq2 1.8.1 (Love et al., 2014). Statistical treatments and differential analyses were also performed using DESeq2 1.8.1. Mouse individuals, sampled tissues and mouse genotypes were taken into account in the DESeq2 as well as the interaction between individuals, genotype and tissues.
Quantification and Statistical Analysis
Human data are expressed as mean ± SD or as mean ± SEM as indicated in the text, tables and figures. Analyses of variance (ANOVA) or covariance (ANCOVA) were used for group comparisons. Repeated-measures analyses of variance (MANOVA) were employed to compare glucose, insulin and C-peptide values across a period of time during the graded intravenous infusions of glucose. Mixed regression model with random effects was employed to compare glucose/ISR response curves. Mouse data are expressed as mean ± SEM. Experiments were performed at least 3 times. Student’s t test and ANOVA were employed for comparisons between groups. Statistics were performed with JMP (SAS Institute Inc, Cary, NC) or with GraphPad software (Prism). p ≤ 0.05 is significant.
Acknowledgments
N.V. was supported by grants from the French National Agency of Research (PROVIDE and PUMAS), the French and European Foundation for Diabetes (SFD and EFSD), and the European Union H2020 framework (ERC-EpiFAT 725790). Human study was performed at the Clinical Investigation Centre (Groupe Hospitalier Saint-Louis/Lariboisière, Paris) and was supported by Assistance Publique des Hôpitaux de Paris (APHP) (PHRC AOR09087; J.-F.G., principal investigator [PI]). E.D. was supported by grants from ATIP-AVENIR and the French and European Foundation for Diabetes (SFD and EFSD). E.T. was supported by grants from the Swedish Research Council, the Swedish Diabetes Foundation, the Novo Nordisk Foundation, and the Center for Innovative Medicine (CIMED) at the Karolinska Institutet. RNA sequencing and transcriptome analysis were supported by the France Génomique national infrastructure, funded as part of the “Investissements d’Avenir” program managed by the French National Agency of Research(ANR-10-INBS-09).
Author Contributions
Karima Drareni and N.V. designed research studies, analyzed data, and wrote the manuscript. Karima Drareni, F.A., and R.B. conducted the in vivo experiments and acquired and analyzed data with the help of C.C., S.B., A.G., and E.D. R.R., G.V., J.-L.N., and J.-F.G. conducted human analysis and wrote the manuscript. Karine Dias and S.L. conducted transcriptomic analyses. E.T., E.D., and F.A. helped in the experimental design, data interpretation, and manuscript writing.
Declaration of Interests
The authors declare no competing interests.
Published: September 15, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108141.
Contributor Information
Karima Drareni, Email: karimadrareni@gmail.com.
Nicolas Venteclef, Email: nicolas.venteclef@inserm.fr.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley J. The control of insulin secretion by adipokines: current evidence for adipocyte–beta cell endocrine signalling in metabolic homeostasis. Mamm. Genome. 2014;25:442–454. doi: 10.1007/s00335-014-9538-7. [DOI] [PubMed] [Google Scholar]
- Cardamone M.D., Krones A., Tanasa B., Taylor H., Ricci L., Ohgi K.A., Glass C.K., Rosenfeld M.G., Perissi V. A protective strategy against hyperinflammatory responses requiring the nontranscriptional actions of GPS2. Mol. Cell. 2012;46:91–104. doi: 10.1016/j.molcel.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardamone M.D., Tanasa B., Chan M., Cederquist C.T., Andricovich J., Rosenfeld M.G., Perissi V. GPS2/KDM4A pioneering activity regulates promoter-specific recruitment of PPARγ. Cell Rep. 2014;8:163–176. doi: 10.1016/j.celrep.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardamone M.D., Tanasa B., Cederquist C.T., Huang J., Mahdaviani K., Li W., Rosenfeld M.G., Liesa M., Perissi V. Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol. Cell. 2018;69:757–772.e7. doi: 10.1016/j.molcel.2018.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederquist C.T., Lentucci C., Martinez-Calejman C., Hayashi V., Orofino J., Guertin D., Fried S.K., Lee M.J., Cardamone M.D., Perissi V. Systemic insulin sensitivity is regulated by GPS2 inhibition of AKT ubiquitination and activation in adipose tissue. Mol. Metab. 2016;6:125–137. doi: 10.1016/j.molmet.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmas E. Innate immune priming of insulin secretion. Curr. Opin. Immunol. 2019;56:44–49. doi: 10.1016/j.coi.2018.10.005. [DOI] [PubMed] [Google Scholar]
- Dalmas E. Role of innate immune cells in metabolism: from physiology to type 2 diabetes. Semin. Immunopathol. 2019;41:531–545. doi: 10.1007/s00281-019-00736-5. [DOI] [PubMed] [Google Scholar]
- Dalmas E., Lehmann F.M., Dror E., Wueest S., Thienel C., Borsigova M., Stawiski M., Traunecker E., Lucchini F.C., Dapito D.H. Interleukin-33-activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity. 2017;47:928–942.e7. doi: 10.1016/j.immuni.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Demine S., Schiavo A.A., Marín-Cañas S., Marchetti P., Cnop M., Eizirik D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020;11:7. doi: 10.1186/s13287-019-1523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath M.Y., Dalmas É., Sauter N.S., Böni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 2013;17:860–872. doi: 10.1016/j.cmet.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Dowal L., Parameswaran P., Phat S., Akella S., Majumdar I.D., Ranjan J., Shah C., Mogre S., Guntur K., Thapa K. Intrinsic properties of brown and white adipocytes have differential effects on macrophage inflammatory responses. Mediators Inflamm. 2017;2017:9067049. doi: 10.1155/2017/9067049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drareni K., Ballaire R., Barilla S., Mathew M.J., Toubal A., Fan R., Liang N., Chollet C., Huang Z., Kondili M. GPS2 deficiency triggers maladaptive white adipose tissue expansion in obesity via HIF1A activation. Cell Rep. 2018;24:2957–2971.e6. doi: 10.1016/j.celrep.2018.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton R.P., Allen R.C., Schade D.S., Erickson K.M., Standefer J. Prehepatic insulin production in man: kinetic analysis using peripheral connecting peptide behavior. J. Clin. Endocrinol. Metab. 1980;51:520–528. doi: 10.1210/jcem-51-3-520. [DOI] [PubMed] [Google Scholar]
- Fan R., Toubal A., Goñi S., Drareni K., Huang Z., Alzaid F., Ballaire R., Ancel P., Liang N., Damdimopoulos A. Loss of the co-repressor GPS2 sensitizes macrophage activation upon metabolic stress induced by obesity and type 2 diabetes. Nat. Med. 2016;22:780–791. doi: 10.1038/nm.4114. [DOI] [PubMed] [Google Scholar]
- Fitzgibbons T.P., Kogan S., Aouadi M., Hendricks G.M., Straubhaar J., Czech M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1425–H1437. doi: 10.1152/ajpheart.00376.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Luo Z., Jin Z., Ji Y., Ying W. Adipose tissue macrophages orchestrate β cell adaptation in obesity through secreting miRNA-containing extracellular vesicles. bioRxiv. 2020 doi: 10.1101/2020.06.12.148809. [DOI] [Google Scholar]
- Golson M.L., Misfeldt A.A., Kopsombut U.G., Petersen C.P., Gannon M. High fat diet regulation of β-cell proliferation and β-cell mass. Open Endocrinol. J. 2010;4 doi: 10.2174/1874216501004010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N., Khan T., Trujillo M.E., Wernstedt-Asterholm I., Attie A.D., Sherwani S., Wang Z.V., Landskroner-Eiger S., Dineen S., Magalang U.J. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C., Qu A., Matsubara T., Chanturiya T., Jou W., Gavrilova O., Shah Y.M., Gonzalez F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes. 2011;60:2484–2495. doi: 10.2337/db11-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdren L., Bernard M., Dillies M.A., Le Crom S. Eoulsan: a cloud computing-based framework facilitating high throughput sequencing analyses. Bioinformatics. 2012;28:1542–1543. doi: 10.1093/bioinformatics/bts165. [DOI] [PubMed] [Google Scholar]
- Kiely A., McClenaghan N.H., Flatt P.R., Newsholme P. Pro-inflammatory cytokines increase glucose, alanine and triacylglycerol utilization but inhibit insulin secretion in a clonal pancreatic beta-cell line. J. Endocrinol. 2007;195:113–123. doi: 10.1677/JOE-07-0306. [DOI] [PubMed] [Google Scholar]
- Kita S., Maeda N., Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Invest. 2019;129:4041–4049. doi: 10.1172/JCI129193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski C.M., Ghaben A.L., Morley T.S., Samms R.J., Adams A.C., An Y., Johnson J.A., Joffin N., Onodera T., Crewe C. A novel model of diabetic complications: adipocyte mitochondrial dysfunction triggers massive β-cell hyperplasia. Diabetes. 2020;69:313–330. doi: 10.2337/db19-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G., Silveira A.L.M., Martins L.B., Ferreira A.V., Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J. Clin. Invest. 2019;129:4032–4040. doi: 10.1172/JCI129192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir L.A., Neeley C.K., Meyer K.A., Baker N.A., Brosius A.M., Washabaugh A.R., Varban O.A., Finks J.F., Zamarron B.F., Flesher C.G. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity (Silver Spring) 2016;24:597–605. doi: 10.1002/oby.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Toler C., O’Neill B.T., Cypess A.M. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity (Silver Spring) 2015;23:1765–1770. doi: 10.1002/oby.21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohm M., Sommerfeld A., Strzoda D., Jones A., Sijmonsma T.P., Rudofsky G., Wolfrum C., Sticht C., Gretz N., Zeyda M. Transcriptional cofactor TBLR1 controls lipid mobilization in white adipose tissue. Cell Metab. 2013;17:575–585. doi: 10.1016/j.cmet.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Romacho T., Elsen M., Röhrborn D., Eckel J. Adipose tissue and its role in organ crosstalk. Acta Physiol. (Oxf.) 2014;210:733–753. doi: 10.1111/apha.12246. [DOI] [PubMed] [Google Scholar]
- Scharfmann R., Staels W., Albagli O. The supply chain of human pancreatic β cell lines. J. Clin. Invest. 2019;129:3511–3520. doi: 10.1172/JCI129484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson R.L., Brelje T.C. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- Sun K., Halberg N., Khan M., Magalang U.J., Scherer P.E. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 2013;33:904–917. doi: 10.1128/MCB.00951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubal A., Clément K., Fan R., Ancel P., Pelloux V., Rouault C., Veyrie N., Hartemann A., Treuter E., Venteclef N. SMRT-GPS2 corepressor pathway dysregulation coincides with obesity-linked adipocyte inflammation. J. Clin. Invest. 2013;123:362–379. doi: 10.1172/JCI64052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuter E., Fan R., Huang Z., Jakobsson T., Venteclef N. Transcriptional repression in macrophages-basic mechanisms and alterations in metabolic inflammatory diseases. FEBS Lett. 2017;591:2959–2977. doi: 10.1002/1873-3468.12850. [DOI] [PubMed] [Google Scholar]
- Ying W., Lee Y.S., Dong Y., Seidman J.S., Yang M., Isaac R., Seo J.B., Yang B.-H., Wollam J., Riopel M. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 2019;29:457–474.e5. doi: 10.1016/j.cmet.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Kim K.H. TNF-alpha inhibits glucose-induced insulin secretion in a pancreatic beta-cell line (INS-1) FEBS Lett. 1995;377:237–239. doi: 10.1016/0014-5793(95)01272-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-Seq gene expression data and raw fastq files in this paper is GEO: GSE111647 (https://www.ncbi.nlm.nih.gov/geo/).