

Abstract
Alzheimer's disease (AD) is a chronic neurodegenerative disease in the central nervous system that has complex pathogenesis in the elderly. The current review focuses on the epigenetic mechanisms of AD, according to the latest findings. One of the best-characterized chromatin modifications in epigenetic mechanisms is DNA methylation. Highly replicable data shows that AD occurrence is often accompanied by methylation level changes of the AD-related gene. Homocysteine (Hcy) is not only an intermediate product of one-carbon metabolism but also an important independent risk factor of AD; it can affect the cognitive function of the brain by changing the one-carbon metabolism and interfering with the DNA methylation process, resulting in cerebrovascular disease. In general, Hcy may be an environmental factor that affects AD via the DNA methylation pathway with a series of changes in AD-related substance. This review will concentrate on the relation between DNA methylation and Hcy and try to figure out their rule in the pathophysiology of AD.
1. Introduction
The increasing number of dementia patients in recent years is a serious problem, of which Alzheimer's disease (AD) is the most common type that accounts for an estimated 70% of dementia cases [1]. Data suggest that the prevalence of AD in people over 65 years old is approximately 10-30% and the estimated incidence is 1-3% [2]. Moreover, approximately 9.5 million people suffer from AD in China, accounting for an estimated 20% of the world in 2015 [3].
AD is a chronic neurodegenerative disease, which manifests as progressive memory loss and cognitive impairment [4, 5]. Senile plaques (SP) formed by extracellular amyloid-β (Aβ) peptide deposition and neurofibrillary tangles (NFTs) formed by excessive phosphorylation of intracellular tau protein constitute the hallmarks of AD [6, 7], which is also accompanied by the massive loss of neurons and synapses, as well as brain structural and functional abnormalities [8–10]. DNA methylation is an important part of epigenetics and is becoming a very attractive subject for researchers because it can shed light on unknown aspects of complex disease pathophysiology like AD. In addition, homocysteine (Hcy) is an environmental factor that seems related to AD through DNA methylation pathways.
2. Mechanisms
2.1. Alzheimer's Disease
Currently, the interaction of various factors such as genetics and environment affects the etiology and pathophysiological changes of AD [11, 12]. Multiple hypotheses are related to the pathogenesis of AD, such as the amyloid cascade hypothesis [13–15], tau protein hypothesis [16–18], cholinergic hypothesis [19], lipid metabolism disorder hypothesis [20], neuroinflammation hypothesis [21], and oxidative stress hypothesis [22], among which the amyloid cascade hypothesis and tau protein hypothesis provide the predominantly theoretical construct for AD.
The amyloid cascade hypothesis indicates that the β-amyloid precursor protein (APP) generates Aβ peptide under the cleavage of β-secretase and γ-secretase, which eventually forms SP [23]. Previous studies have found that excessive Aβ accumulated will cause synaptic damage, glial cell overactivation [24], and inflammatory reaction [25, 26], followed by SP formation (Figure 1) [27]. A series of landmark studies states that the role of targeting Aβ peptide in the treatment of AD will delay the progression of the disease [28, 29] by inhibiting monomer aggregation and preventing the formation of toxic species [30]. Zhai et al.'s [31] study revealed that the structural origin of β-sheet and plaque deposition, therefore, blocking and inhibiting the transmission of Aβ from the source, may effectively inhibit Aβ deposition, which may be an important method to prevent the formation of AD.
Figure 1.
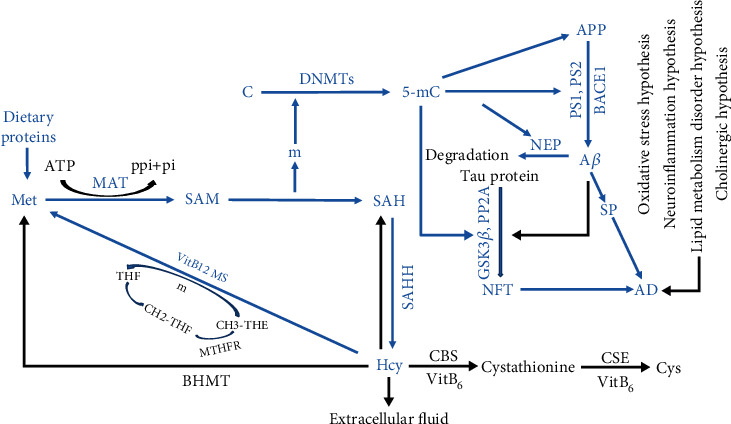
The mechanism of AD, DNA methylation and Hcy interaction. 5-mC: 5-methylcytosine; AD: Alzheimer's disease; Aβ: amyloid-β; APP: β-amyloid precursor protein; ATP: adenosine triphosphate; BACE1: β-secretase-1; BHMT: betaine-homocysteine methyltransferase; C: cytosine; CBS: cystathionine β synthase; CSE: cystathionine γ-lyase; Cys: cysteine; DMG: dimethylglycine; GSK3β: glycogen synthase kinase 3β; Hcy: homocysteine; m: methyl; MAT: methionine adenosyltransferase; Met: methionine; MTHFR: methylenetetrahydrofolate reductase; MS: methionine synthase; NFT: neurofibrillary tangle; NEP: neprilysin; PP2A: protein phosphatase 2A; PS1: presenilin-1; PS2: presenilin-2; SAH: S-adenosine homocysteine; SAHH: S-adenosine homocysteine hydrolase; SAM: S-adenosine methionine; SP: senile plaques; THF: tetrahydrofolate.
Phosphate-containing tau protein is part of the essential components of the cytoskeleton. In normal brain cells, a single tau molecule with 2 to 3 phosphate groups is related to the stability of microtubules and the axon transport of nerve cells [32]. Abnormal hyperphosphorylation of tau protein and intraneural aggregation are hallmark features of the early development of AD-related neurofibrillary pathology. The binding capacity of hyperphosphorylated tau protein and tubulin is reduced, paired helical filaments (PHFs) aggregated; while its conversion and clearance ability are reduced, formed NFTs are the early markers of AD [33–35]. The degree of tau protein phosphorylation is the consequence of the interaction of various protein kinases (PK) and protein phosphatases (PP) in the brain (Figure 1) [36–38]. The dysregulation of PP and PK activities reduces the binding capacity and stability of tau protein to microtubules, which result in neuronal dysfunction and neurodegeneration, and further to functional defects [39–41]. A notable exception study is that tau-specific site phosphorylation protects the brain in the early stages of AD by inhibiting Aβ toxicity [42]. The hyperphosphorylation of tau is considered independent of Aβ, but the final spread of tau throughout the neocortex is driven by Aβ [7]. AD is the interaction of multiple factors and mechanisms that exert multiple effects at different stages of disease progression, which is not possible to ascribe changes in one factor. It is debatable whether the existing hypothesis fully explains to the pathogenesis of AD. Therefore, it needs further exploration to identify.
2.2. Homocysteine
In 1933, Vincent du Vigneaud isolated a sulfur-containing nonprotein amino acid called Hcy from bladder stones. Methionine (Met) is metabolized to S-adenosine methionine (SAM) under the action of methionine adenosyltransferase (MAT). SAM is one of the major methyl donors that can be converted to S-adenosine homocysteine (SAH), a methyl removed in this process, which is involved in epigenetic modifications under the action of methyltransferase [43]. SAH removes adenosine by S-adenosine homocysteine hydrolase (SAHH) to form Hcy [44], substances well associated with the methionine cycle and energy metabolism (Figure 1) [45, 46]. A clinical trial suggests that oral Met load increased plasma Hcy from 12.8 ± 1.8 to 33.3 ± 3.4 micromol·L (-1) at 4 h [47]. Consequently, excessive Met can elevate the level of Hcy, which finally results in hyperhomocysteinemia (HHcy) [48, 49].
Subsequently predominant metabolism of Hcy occurs via three pathways (Figure 1): (1) Met cycle—under the action of methionine synthase, tetrahydrofolate metabolism provides a methyl group, and Hcy is remethylated to Met with assistance of vitamin B12 (VitB12) [50, 51]; (2) transsulfuration pathway—with vitamin B6 (VitB6) as a coenzyme, Hcy and serine are condensed into cystathionine under the catalysis of cystathionine β synthase (CBS), followed by cystathionine catalyzed by γ-cystathionine lyase to produce cysteine, which is oxidized to sulfate after a series of enzyme catalysis and excreted through the urine in the form of inorganic salts [52]; and (3) direct release into the extracellular fluid—excessive Hcy is thought to be released from the intracellular fluid to the extracellular fluid through the difference in internal and external concentrations and then exported to the systemic circulation to prevent its intracellular accumulation [53–55]. VitB6, VitB12, and folic acid are the main metabolic pathways of Hcy in the methylation cycle and transsulfuration pathway, the lack of which leads to the production of HHcy. Hence, the production and metabolism balance of Hcy is essential for maintaining the body's homeostasis. Genetic factors, nutritional factors, estrogen levels, and age all affect the Hcy plasma level [56–58]. Several recent fundamental discoveries highlight important pathological roles of HHcy in many diseases [59–62]. Studies suggest that HHcy induced hypertension by promoting TLR-4-driven chronic vascular inflammation and mitochondria-mediated cell death [63]. Moreover, HHcy aggravates atherosclerosis with elevated oxidative stress and reduced S-nitrosylation level of redox-sensitive protein residues in the vasculature [64], which also as a metabolic disorder parameter is independently associated with the severity of coronary heart disease [65]. Elevated plasma total Hcy level is associated with an increased risk of neurodegenerative disease [66].
2.3. DNA Methylation
Epigenetics is the study of genetic changes in gene expression that is not caused by the DNA sequence changes [67]. Among them, DNA methylation is one of the best-characterized epigenetic modification, which exerts an important role in maintaining cell function, genetic imprinting, and gene expression [68, 69]. DNA methylation occurs in cytosine-phosphate-guanine (CPG) fundamental sequence with catalyzing of DNA methyltransferase enzymes (DNMTs). Specific bases in the DNA sequence and cofactor proteins are jointly involved in maintaining and regulating the methylation pattern [70–72]. DNA methylation needs a series of DNMTs [73], such as maintenance methyltransferase DNMT1 and de novo methyltransferases DNMT3a and DNMT3b [74–77]. DNMT1 maintains the continuous methylation status of DNA, which is responsible for repeated methylation during cell division [78], and DNMT3a and DNMT3b methylate DNA strands that have not been methylated, which is responsible for de novo synthesis of DNA methylation [79].
In the genome, methylated CpG sites account for approximately 70% of human genes [80]. CpG sites are located in the first exon region, gene promoter region, or intron region and regulate the expression of downstream genes [81], where the covalent bonding of the methyl group with the 5th carbon atom of cytosine is considered to be the most stable epigenetic marker [82].
3. DNA Methylation in Alzheimer's Disease
Epigenetics studies have found an association between DNA methylation and AD [83], which is involved in the progression of the neurodegenerative disease [84]. The earliest accumulation of Aβ reduces the overall level of 5-hydroxymethylcytosine in vitro [85], resulting in DNA hypomethylation, and affects the pathological progress of AD [86]. Furthermore, DNA methylation is associated with Aβ and NFTs [87]. PS1 is a component of the γ-secretase that will cleave APP to produce various Aβ [88]. Increased expression of APP and induction of hypomethylation of APP and PS1 gene promoters will increase the production of Aβ in BV-2 cells [89]. Moreover, β-secretase-1 (BACE1) is also hypomethylated, which affects Aβ accumulation and accelerates AD pathology [87], as well as significantly reduces DNMT1 expression in cell experiments [90]. In short, the methylation or demethylation of key enzymes will increase Aβ synthesis and reduce Aβ degradation, eventually resulting in the development of AD.
Tau phosphorylation and dephosphorylation reactions are catalyzed by glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A), respectively; GSK3β and PP2A are two major kinds of enzymes that regulate hyperphosphorylated Tau. Sonawane and Chinnathambi's [91] study indicated the upregulation of GSK3β promoter demethylation expression and the downregulation of PP2A promoter methylation in the AD brain, both of which accelerated tau phosphorylation (Figure 1). In addition, the reduced expression of netrin-1-promoter hypermethylation may be related to memory loss [92].
DNA methylation is closely related to AD [93, 94]. Changes in DNA methylation are related to neural differentiation of the hippocampus [95], as well as across multiple brain regions. So far, DNA methylation exerts a central role in amyloid production, fibrogenesis, inflammation, and oxidative pathways. All the above studies suggest that DNA methylation is involved in the AD-related molecular mechanism [96].
4. Homocysteine in Alzheimer's Disease
With increasing age, the risk of AD increases under the interaction of genetic and environmental factors (obesity, smoking, and an unhealthy lifestyle) [97], of which Hcy is a risk factor of AD. Several studies are indicating that high Hcy concentrations cause cognitive dysfunction [98] and might be associated with dementia [99–101].
HHcy may promote dementia through a variety of mechanisms, including cerebral microangiopathy, endothelial dysfunction, oxidative stress, neuronal damage, and Aβ-mediated enhancement of vascular toxicity, neurotoxicity, and apoptosis [102]. The brain of AD patients is accompanied by cerebrovascular disease [103], and studies show a long-term high Hcy diet severely induces microbleeds, which may be the cause of memory deficits [104]. Although elevated Hcy does not induce lipid peroxidation in the whole brain of rats, similar physiological changes in levels are observed in both malondialdehyde (MDA) and superoxide anion (SOA), resulting in oxidative stress [105]. Not only does Hcy can increase the activity of MMP-9 and MMP-2 but also reduce the activity of arginase. Meanwhile, it is accompanied by nitrosative stress reaction that destroys the integrity of the blood-brain barrier (BBB), leading to cerebrovascular permeability and neurodegeneration [106, 107]. Lin et al.'s [108] study suggests that Hcy can affect nerve cell proliferation and Aβ deposit formation by inducing an increase in intracellular SAH [109–111]. Additionally, DNA damage-related genes are significantly upregulated and trigger oxidative and genotoxic stress [112]. Since high Hcy levels are a metabolic risk factor for neurodegenerative diseases, diet-induced Hcy levels not only increase aggravate Tau neuropathology in H-TAU mice but also affect synaptic integrity, neuroinflammation, and cognition function [113]. Moreover, the AD transgenic mouse model shows that Aβ content in cerebral blood vessels increased significantly, neurons died, and DNA damage of hippocampal neurons further reduce cognitive ability [114]. Excessive deposition of hyperphosphorylated Tau and neuropathy caused by synaptic inactivation lesions are also associated with the elevated Hcy level [115–117].
Hcy level changes AD development by inducing neuronal DNA damage, neuroinflammation, apoptosis, and autophagy abnormalities [117–119]. Genetic variation affects the relevant genes, which advances the age of onset and accelerates cognitive function decline [120, 121].
5. DNA Methylation and Homocysteine in Alzheimer's Disease
Dementia-like symptoms caused by HHcy are related to abnormal methylation and gene expression disorders [122]. One study found that HHcy can reduce the methylation level and increase cell damage by inhibiting the protein expression and enzyme activity of DNMT1, DNMT3A, and DNMT3B in the hippocampal neural stem cells of raw rat [123]. Another study also illustrates that HHcy enhances DNA damage by inducing methyl donor deficiency and disrupting DNA repair, resulting in neuronal cell death [124]. In addition, the upregulation of the 5lo enzyme pathway leads to hypomethylation of 5loDNA and promotes the formation of Aβ [125]. HHcy can decrease the activity of methylenetetrahydrofolate reductase (MTHFR) and tight connexin expression, while SAHH expression, BBB permeability, and oxidative stress are increased with DNA methyltransferase upregulation, resulting in neurodegeneration and synaptic toxicity [126]. Most importantly, the Met cycle and transsulfuration pathway are related to VitB family folic acid [127, 128].
Folic acid is involved in the regulation of one-carbon metabolism and methylation. In addition to this, the active form of folic acid is 5-methyltetrahydrofolate, which is a methyl donor for the remethylation of Hcy. HHcy is elicited by low folic acid, which damages hippocampal neurons and is an important factor of the high incidence of dementia in the elderly [129]. Furthermore, folic acid is not only positively related to the DNA methylation level of the cognitive impairment elderly but also related to the intensity of DNA methylation [130]. MTHFR is involved in folate metabolism, and the high level of Hcy caused by MTHFR deficiency will reduce the expression and methylation level of PP2A and leucine carboxylmethyltransferase 1 (LCMT1), resulting in tau dephosphorylation [131].
HHcy is a risk factor for AD and is also associated with VitB12 deficiency [132]. The accumulation of Hcy induced by VitB deficiency may impair the “methylation potential,” resulting in the upregulation of PS1, BACE, and increased Aβ [133, 134]. Several studies have implicated that the plasma Hcy level in the AD group increased while the folate and VitB12 levels decreased [135–138]. Moreover, abnormal Hcy metabolism causes plasma folic acid and VitB12 deficiency [139–142], which in turn affects the methylation level of AD-related genes via participating in AD development [143]. Mice lacking folic acid and VitB diets will have increased Hcy levels, Aβ levels, and tau phosphorylation, which is also accompanied by hypomethylation of the Alox5 promoter [144]. High Hcy-induced SAH increases [123, 124], and the SAM/SAH ratio decreases, both of which are related to the inhibition of methyltransferase [145]. Methylation analysis also further demonstrates the correlation between the SAM/Hcy cycle and DNA methylation, involved in PS1 and BACE1 methylation [141]. SAM is the predominant methyl donor; Scarpa et al. [145] analyzed the effect of SAM administration on the expression of 588 central nervous system genes in nerve cells and showed that among the seven genes treated by SAM, three genes had DNA methylation upregulated and four genes had DNA methylation downregulated [146]. SAM can regulate its products to take part in the methylation status of APP genes, which affects the formation of Aβ [147] by increasing APP and PS1 proteins expression; it can also induce hypomethylation of APP and PS1 gene promoters and increase Aβ production in BV-2 cells [148]. In short, Hcy can change the DNA methylation levels of key metabolic enzymes and cause brain damage [149].
6. Future Directions
Possible mechanisms for Hcy to induce AD are shown in Figure 1; high levels of Met intake produce excessive Hcy in the body which metabolizes through the Met cycle, during which the generated methyl adds to the five-bit carbon atom of the cytosine under the action of DNMTs, causing the methylation levels of the AD-related genes to change. The changes in methylation levels in turn affect the expression of the gene, resulting in the occurrence of AD. At the same time, excess accumulation of Hcy is regeneration to methionine under the action of methionine synthesis enzyme and VitB12, finally producing the Hcy. As a result, high levels of Hcy may induce AD along with changes in methylation levels of AD-related genes.
Unbalanced nutritional intake will not only increase Hcy levels but also affect DNA methylation and gene expression. At present, most researchers focus on the effect of Hcy on AD symptoms, rather than on molecular mechanisms. At the molecular level, studying the regulatory mechanism of Hcy and its metabolites on the expression of related genes in AD patients helps determine the appropriate nutritional requirements. Preventing the increase of the Hcy level caused by the imbalance of nutrition intake can either avoid or arrest the occurrence and aggravation of AD.
As AD progresses, treatment becomes difficult with little effect [150, 151]; the research and development of drugs also consume a lot of manpower and material resources [152]. So far, the main drugs used in AD treatment are donepezil, rivastigmine, galantamine, and memantine, which can only relieve symptoms but can not cure and reverse the development of AD [153–156]. In addition, some drugs must be used in combination to achieve the best therapeutic effect, which is also accompanied by increasing the risk of various adverse reactions [157]. Since 2003, the FDA has not approved a new drug for the treatment of AD [158]. Therefore, early diagnosis and treatment are essential. The current clinical early diagnosis depends on clinical observation, and cognitive testing is the first step to diagnose the complex disease characteristics in AD, which is time-consuming and has limitations. More definite diagnosis requires imaging (MRI or PET scan) or invasive lumbar puncture to measure CSF markers which is expensive. Thus, efficient diagnostic methods and early disease biomarkers are essential for the prevention and treatment of early AD [159].
Several researchers have reported that plasma Hcy levels are usually elevated in patients with AD [160]. HHcy is closely related to cortical atrophy and more severe cognitive decline [161, 162]. The high plasma Hcy concentrations are significantly associated with mild cognitive impairment (MCI) and AD, which is more strongly correlated with AD patients as compared to patients with MCI [163]. A meta-analysis included 34 studies with 9397 subjects and demonstrated a causal link between plasma total Hcy and the risk factor of AD [164]. More than 40% of patients with AD are associated with a high Hcy level in the plasma, which is associated with a more rapid neural atrophy than those with normal levels of Hcy [165]. Moreover, HHcy levels can predict a cognitive decline in healthy elderly patients [166, 167]. Therefore, HHcy also has the potential to predict AD, and preventing Hcy-induced neurotoxicity may become a novel strategy for AD prevention and treatment.
DNA methylation alteration in the hippocampus of AD patients occurs in specific regulatory regions that are critical to neurodifferentiation; this supports the idea that hippocampus neurogenesis may play a role in AD through epigenetic mechanisms [168]. The current findings suggest that the epigenetic modulation of DNA is vulnerable to the state of neurodegenerative diseases [169]. Moreover, brain DNA methylation is associated with AD pathology in multiple AD loci, and the results further prove that the destruction of DNA methylation is involved in the pathological process of AD [170]. Many researches have shown that DNA methylation is a useful marker for screening individuals at the risk of AD [171]. Therefore, AD-related gene methylation levels are a convenient and useful biomarker for AD diagnosing [172–174].
Proper nutrition not only changes Hcy levels but also prevents the development of AD and reduces cognitive impairment. Hcy levels may develop into AD biomarkers for diagnosis; moreover, factors that affect Hcy's production and metabolism not only increase Hcy levels but also affect DNA methylation levels of AD-related genes. Studying the mechanisms of DNA methylation in AD can help to explore the etiology and pathogenesis of AD, which can also be a very useful tool for researchers to identify AD biomarkers and even play an important role in early screening of patients in the future. Meanwhile, effective measures to reduce Hcy levels and DNA methylation will provide new ideas for the prevention and treatment of AD.
Acknowledgments
This work was supported by the Shijingshan's Tutor Studio of Pharmacology [GZS-2016(07)] and Funds for the Construction of National First Class Pharmacy Discipline [GESR (2017-85)].
Data Availability
No data were used to support this study.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Tingting Pi wrote the paper and Jing-Shan Shi reviewed drafts of the paper.
References
- 1.García-Blanco A., Baquero M., Vento M., Gil E., Bataller L., Cháfer-Pericás C. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. Journal of the Neurological Sciences. 2017;373(3):295–302. doi: 10.1016/j.jns.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Masters C. L., Bateman R., Blennow K., Rowe C. C., Sperling R. A., Cummings J. L. Alzheimer’s disease. Nature reviews. Disease Primers. 2015;1(1):50–56. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 3.Yee A., Avalon Genomics (HK) Limited, Shatin H. K., et al. Alzheimer's disease: insights for risk evaluation and prevention in the Chinese population and the need for a comprehensive programme in Hong Kong/China. Hong Kong Medical Journal. 2018;24(5):492–500. doi: 10.12809/hkmj187244. [DOI] [PubMed] [Google Scholar]
- 4.Lane C. A., Hardy J., Schott J. M. Alzheimer's disease. European Journal of Neurology. 2018;25(1):59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- 5.Minter M. R., Taylor J. M., Crack P. J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. Journal of Neurochemistry. 2016;136(3):457–474. doi: 10.1111/jnc.13411. [DOI] [PubMed] [Google Scholar]
- 6.Morgan A. R., Touchard S., Leckey C., et al. Inflammatory biomarkers in Alzheimer's disease plasma. Alzheimer's & Dementia. 2019;15(6):776–787. doi: 10.1016/j.jalz.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Kant R., Goldstein L. S. B., Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nature Reviews Neuroscience. 2020;21(1):21–35. doi: 10.1038/s41583-019-0240-3. [DOI] [PubMed] [Google Scholar]
- 8.Taga M., Minett T., Classey J., et al. Metaflammasome components in the human brain: a role in dementia with Alzheimer's pathology? Brain Pathology. 2017;27(3):266–275. doi: 10.1111/bpa.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattson M. P. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musi N., Valentine J. M., Sickora K. R., et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6, article e12840) doi: 10.1111/acel.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hachinski V. Dementia: paradigm shifting into high gear. Alzheimer's & Dementia. 2019;15(7):985–994. doi: 10.1016/j.jalz.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Madore C., Yin Z., Leibowitz J., Butovsky O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity. 2020;52(2):222–240. doi: 10.1016/j.immuni.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nature Neuroscience. 2015;18(6):794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 14.De Strooper B., Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164(4):603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 15.Hardy J. A., Higgins G. A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 16.Eftekharzadeh B., Daigle J. G., Kapinos L. E., et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer's disease. Neuron. 2018;99(5):925–940.e7. doi: 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbons G. S., Lee V. M. Y., Trojanowski J. Q. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurology. 2019;76(1):101–108. doi: 10.1001/jamaneurol.2018.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wegmann S., Bennett R. E., Delorme L., et al. Experimental evidence for the age dependence of tau protein spread in the brain. Science Advances. 2019;5(6):p. eaaw6404. doi: 10.1126/sciadv.aaw6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartus R., Dean R., Beer B., Lippa A. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 20.Sparks D. L., Scheff S. W., Hunsaker J. C., III, Liu H., Landers T., Gross D. R. Induction of Alzheimer-like β-Amyloid Immunoreactivity in the Brains of Rabbits with Dietary Cholesterol. Experimental Neurology. 1994;126(1):88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y., Su Y., Li B., et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Aβ42 by inhibiting Rho. Science. 2003;302(5648):1215–1217. doi: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]
- 22.Farr S. A., Ripley J. L., Sultana R., et al. Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radical Biology & Medicine. 2014;67:387–395. doi: 10.1016/j.freeradbiomed.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan C.-C., Yu J.-T., Tan M.-S., Jiang T., Zhu X.-C., Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiology of Aging. 2014;35(5):941–957. doi: 10.1016/j.neurobiolaging.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 24.Arranz A. M., De Strooper B. The role of astroglia in Alzheimer's disease: pathophysiology and clinical implications. Lancet Neurology. 2019;18(4):406–414. doi: 10.1016/S1474-4422(18)30490-3. [DOI] [PubMed] [Google Scholar]
- 25.Tuppo E. E., Arias H. R. The role of inflammation in Alzheimer's disease. The International Journal of Biochemistry & Cell Biology. 2005;37(2):289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Bettcher B. M., Johnson S. C., Fitch R., et al. Cerebrospinal fluid and plasma levels of inflammation differentially relate to CNS markers of Alzheimer's disease pathology and neuronal damage. Journal of Alzheimer's Disease. 2018;62(1):385–397. doi: 10.3233/JAD-170602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Awasthi M., Singh S., Pandey V. P., Dwivedi U. N. Alzheimer's disease: An overview of amyloid beta dependent pathogenesis and its therapeutic implications along with in silico approaches emphasizing the role of natural products. Journal of the Neurological Sciences. 2016;361:256–271. doi: 10.1016/j.jns.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Sevigny J., Chiao P., Bussière T., et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 29.Wes P. D., Sayed F. A., Bard F., Gan L. Targeting microglia for the treatment of Alzheimer’s disease. Glia. 2016;64(10):1710–1732. doi: 10.1002/glia.22988. [DOI] [PubMed] [Google Scholar]
- 30.Steffen J., Krohn M., Paarmann K., et al. Revisiting rodent models: Octodon degus as Alzheimer's disease model? Acta Neuropathologica Communications. 2016;4(1):p. 91. doi: 10.1186/s40478-016-0363-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhai L., Otani Y., Ohwada T. Uncovering the networks of topological neighborhoods in β-strand and amyloid β-sheet structures. Scientific Reports. 2019;9(1):p. 10737. doi: 10.1038/s41598-019-47151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong F. P., Ng K. Y., Koh R. Y., Chye S. M. Tau proteins and tauopathies in Alzheimer's disease. Cellular and Molecular Neurobiology. 2018;38(5):965–980. doi: 10.1007/s10571-017-0574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rüb U., Stratmann K., Heinsen H., Seidel K., Bouzrou M., Korf H. W. Alzheimer’s disease: characterization of the brain sites of the initial tau cytoskeletal pathology will improve the success of novel immunological anti-tau treatment approaches. Journal of Alzheimer's Disease. 2017;57(3):683–696. doi: 10.3233/JAD-161102. [DOI] [PubMed] [Google Scholar]
- 34.Horvath J., Burkhard P. R., Herrmann F. R., Bouras C., Kövari E. Neuropathology of parkinsonism in patients with pure Alzheimer's disease. Journal of Alzheimer's Disease. 2014;39(1):115–120. doi: 10.3233/JAD-131289. [DOI] [PubMed] [Google Scholar]
- 35.del Carmen Cárdenas-Aguayo M., Gómez-Virgilio L., DeRosa S., Meraz-Ríos M. A. The role of tau oligomers in the onset of Alzheimer's disease neuropathology. ACS Chemical Neuroscience. 2014;5(12):1178–1191. doi: 10.1021/cn500148z. [DOI] [PubMed] [Google Scholar]
- 36.Ma R. H., Zhang Y., Hong X. Y., Zhang J. F., Wang J. Z., Liu G. P. Role of microtubule-associated protein tau phosphorylation in Alzheimer's disease. Journal of Huazhong University of Science and Technology [Medical Sciences] 2017;37(3):307–312. doi: 10.1007/s11596-017-1732-x. [DOI] [PubMed] [Google Scholar]
- 37.Yang C., Li X., Gao W., et al. Cornel iridoid glycoside inhibits tau hyperphosphorylation via regulating cross-talk between GSK-3β and PP2A signaling. Frontiers in Pharmacology. 2018;9 doi: 10.3389/fphar.2018.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theendakara V., Bredesen D. E., Rao R. V. Downregulation of protein phosphatase 2A by apolipoprotein E: implications for Alzheimer's disease. Molecular and Cellular Neurosciences. 2017;83:83–91. doi: 10.1016/j.mcn.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Moszczynski A. J., Yang W., Hammond R., Ang L. C., Strong M. J. Threonine175, a novel pathological phosphorylation site on tau protein linked to multiple tauopathies. Acta Neuropathologica Communications. 2017;5(1):p. 6. doi: 10.1186/s40478-016-0406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bejanin A., Schonhaut D. R., La Joie R., et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140(12):3286–3300. doi: 10.1093/brain/awx243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He Z., Guo J. L., McBride J. D., et al. Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nature Medicine. 2018;24(1):29–38. doi: 10.1038/nm.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ittner A., Chua S. W., Bertz J., et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer's mice. Science. 2016;354(6314):904–908. doi: 10.1126/science.aah6205. [DOI] [PubMed] [Google Scholar]
- 43.Friso S., Choi S. W., Girelli D., et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mentch S. J., Locasale J. W. One-carbon metabolism and epigenetics: understanding the specificity. Annals of the New York Academy of Sciences. 2016;1363(1):91–98. doi: 10.1111/nyas.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakubowski H. Homocysteine editing, thioester chemistry, coenzyme A, and the origin of coded peptide synthesis †. Life. 2017;7(1):p. 6. doi: 10.3390/life7010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian S., Han J., Huang R., et al. Increased plasma homocysteine level is associated with executive dysfunction in type 2 diabetic patients with mild cognitive impairment. Journal of Alzheimer's Disease. 2017;58(4):1163–1173. doi: 10.3233/JAD-170162. [DOI] [PubMed] [Google Scholar]
- 47.Mandaviya P. R., Stolk L., Heil S. G. Homocysteine and DNA methylation: a review of animal and human literature. Molecular Genetics and Metabolism. 2014;113(4):243–252. doi: 10.1016/j.ymgme.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Coppola A., Astarita C., Liguori E., et al. Impairment of coronary circulation by acute hyperhomocysteinaemia and reversal by antioxidant vitamins. Journal of Internal Medicine. 2004;256(5):398–405. doi: 10.1111/j.1365-2796.2004.01389.x. [DOI] [PubMed] [Google Scholar]
- 49.Nappo F., de Rosa N., Marfella R., et al. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. JAMA. 1999;281(22):2113–2118. doi: 10.1001/jama.281.22.2113. [DOI] [PubMed] [Google Scholar]
- 50.Bellamy M. F., McDowell I. F. W., Ramsey M. W., et al. Hyperhomocysteinemia after an oral methionine load acutely impairs endothelial function in healthy adults. Circulation. 1998;98(18):1848–1852. doi: 10.1161/01.CIR.98.18.1848. [DOI] [PubMed] [Google Scholar]
- 51.Visram M., Radulovic M., Steiner S., et al. Homocysteine regulates fatty acid and lipid metabolism in yeast. The Journal of Biological Chemistry. 2018;293(15):5544–5555. doi: 10.1074/jbc.M117.809236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Škovierová H., Vidomanová E., Mahmood S., et al. The molecular and cellular effect of homocysteine metabolism imbalance on human Health. International Journal of Molecular Sciences. 2016;17(10):p. 1733. doi: 10.3390/ijms17101733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schalinske K. L., Smazal A. L. Homocysteine imbalance: a pathological metabolic marker. Advances in Nutrition. 2012;3(6):755–762. doi: 10.3945/an.112.002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannibal L., Blom H. J. Homocysteine and disease: causal associations or epiphenomenons? Molecular Aspects of Medicine. 2017;53:36–42. doi: 10.1016/j.mam.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 55.Genoud V., Quintana P. G., Gionco S., Baldessari A., Quintana I. Structural changes of fibrinogen molecule mediated by the N-homocysteinylation reaction. Journal of Thrombosis and Thrombolysis. 2018;45(1):66–76. doi: 10.1007/s11239-017-1574-1. [DOI] [PubMed] [Google Scholar]
- 56.Ueland P. M., Refsum H., Stabler S. P., Malinow M. R., Andersson A., Allen R. H. Total homocysteine in plasma or serum: methods and clinical applications. Clinical Chemistry. 1993;39(9):1764–1779. doi: 10.1093/clinchem/39.9.1764. [DOI] [PubMed] [Google Scholar]
- 57.Velazquez R., Ferreira E., Winslow W., et al. Maternal choline supplementation ameliorates Alzheimer's disease pathology by reducing brain homocysteine levels across multiple generations. Molecular Psychiatry. 2019;10 doi: 10.1038/s41380-018-0322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalani A., Kamat P. K., Givvimani S., et al. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. Journal of Molecular Neuroscience. 2014;52(2):202–215. doi: 10.1007/s12031-013-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tessari P., Cecchet D., Vettore M., Coracina A., Puricelli L., Kiwanuka E. Decreased homocysteine trans-sulfuration in hypertension with hyperhomocysteinemia: relationship with insulin resistance. The Journal of Clinical Endocrinology and Metabolism. 2018;103(1):56–63. doi: 10.1210/jc.2017-01076. [DOI] [PubMed] [Google Scholar]
- 60.Weber G. J., Pushpakumar S., Tyagi S. C., Sen U. Homocysteine and hydrogen sulfide in epigenetic, metabolic and microbiota related renovascular hypertension. Pharmacological Research. 2016;113(Part A):300–312. doi: 10.1016/j.phrs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miao L., Deng G. X., Yin R. X., et al. No causal effects of plasma homocysteine levels on the risk of coronary heart disease or acute myocardial infarction: a Mendelian randomization study, European Journal of Preventive Cardiology. 2019 doi: 10.1177/2047487319894679. [DOI] [PubMed] [Google Scholar]
- 62.McCully K. S. Homocysteine metabolism, atherosclerosis, and diseases of aging. Comprehensive Physiology. 2015;6(1):471–505. doi: 10.1002/cphy.c150021. [DOI] [PubMed] [Google Scholar]
- 63.Smith A. D., Refsum H. Homocysteine, B vitamins, and cognitive impairment. Annual Review of Nutrition. 2016;36(1):211–239. doi: 10.1146/annurev-nutr-071715-050947. [DOI] [PubMed] [Google Scholar]
- 64.Familtseva A., Chaturvedi P., Kalani A., et al. Toll-like receptor 4 mutation suppresses hyperhomocysteinemia-induced hypertension. American Journal of Physiology-Cell Physiology. 2016;311(4):C596–C606. doi: 10.1152/ajpcell.00088.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J., Zhang Y., Zhang Y., et al. GSNOR modulates hyperhomocysteinemia-induced T cell activation and atherosclerosis by switching Akt S-nitrosylation to phosphorylation. Redox Biology. 2018;17:386–399. doi: 10.1016/j.redox.2018.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Montecinos-Oliva C., Arrázola M. S., Jara C., Tapia-Rojas C., Inestrosa N. C. Hormetic-like effects of L-homocysteine on synaptic structure, function, and Aβ aggregation. Pharmaceuticals. 2020;13(2):p. 24. doi: 10.3390/ph13020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reik W., Kelsey G. Cellular memory erased in human embryos. Nature. 2014;511(7511):540–541. doi: 10.1038/nature13648. [DOI] [PubMed] [Google Scholar]
- 68.Wakeling L. A., Ions L. J., Escolme S. M., et al. SIRT1 affects DNA methylation of polycomb group protein target genes, a hotspot of the epigenetic shift observed in ageing. Human Genomics. 2015;9(1):p. 14. doi: 10.1186/s40246-015-0036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong J., Agha G., Baccarelli A. A. The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circulation Research. 2016;118(1):119–131. doi: 10.1161/CIRCRESAHA.115.305206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castillo-Aguilera O., Depreux P., Halby L., Arimondo P., Goossens L. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules. 2017;7(4):p. 3. doi: 10.3390/biom7010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeltsch A., Jurkowska R. Z. Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Research. 2016;44(18):8556–8575. doi: 10.1093/nar/gkw723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edwards J. R., Yarychkivska O., Boulard M., Bestor T. H. DNA methylation and DNA methyltransferases. Epigenetics & Chromatin. 2017;10(1):p. 23. doi: 10.1186/s13072-017-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bansal A., Pinney S. E. DNA methylation and its role in the pathogenesis of diabetes. Pediatric Diabetes. 2017;18(3):167–177. doi: 10.1111/pedi.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsai Y. T., Chang C. M., Wang J. Y., et al. Function of DNA methyltransferase 3a in lead (Pb(2+))-induced cyclooxygenase-2 gene. Environmental Toxicology. 2015;30(9):1024–1032. doi: 10.1002/tox.21976. [DOI] [PubMed] [Google Scholar]
- 75.Álvarez-Errico D., Vento-Tormo R., Ballestar E. Genetic and epigenetic determinants in autoinflammatory diseases. Frontiers in Immunology. 2017;8:p. 318. doi: 10.3389/fimmu.2017.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang R., Zheng Z., Chen Q., et al. The developmental regulator PKL is required to maintain correct DNA methylation patterns at RNA-directed DNA methylation loci. Genome Biology. 2017;18(1):p. 103. doi: 10.1186/s13059-017-1226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hedrich C. M., Mäbert K., Rauen T., Tsokos G. C. DNA methylation in systemic lupus erythematosus. Epigenomics. 2017;9(4):505–525. doi: 10.2217/epi-2016-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jeltsch A., Broche J., Bashtrykov P. Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes. 2018;9(11):p. 566. doi: 10.3390/genes9110566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chatterjee B., Lin M. H., Chen C. C., et al. DNA Demethylation by DNMT3A and DNMT3B in vitro and of Methylated Episomal DNA in Transiently Transfected Cells. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2018;1861(11):1048–1061. doi: 10.1016/j.bbagrm.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 80.Jones P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 81.Liu B., Song J., Luan J., et al. Promoter methylation status of tumor suppressor genes and inhibition of expression of DNA methyltransferase 1 in non-small cell lung cancer. Experimental Biology and Medicine. 2016;241(14):1531–1539. doi: 10.1177/1535370216645211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim M., Costello J. DNA methylation: an epigenetic mark of cellular memory. Experimental & Molecular Medicine. 2017;49(4):p. e322. doi: 10.1038/emm.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu J., He G., Zhu J., et al. Prenatal nutritional deficiency reprogrammed postnatal gene expression in mammal brains: implications for schizophrenia. International Journal of Neuropsychopharmacology. 2015;18(4) doi: 10.1093/ijnp/pyu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nicolia V., Lucarelli M., Fuso A. Environment, epigenetics and neurodegeneration: focus on nutrition in Alzheimer's disease. Experimental Gerontology. 2015;68:8–12. doi: 10.1016/j.exger.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 85.Shu L., Sun W., Li L., et al. Genome-wide alteration of 5-hydroxymenthylcytosine in a mouse model of Alzheimer's disease. BMC Genomics. 2016;17(1) doi: 10.1186/s12864-016-2731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Do Carmo S., Hanzel C. E., Jacobs M. L., et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer 's Model. Scientific Reports. 2016;6(1, article 34051) doi: 10.1038/srep34051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwata A., Nagata K., Hatsuta H., et al. Altered CpG methylation in sporadic Alzheimer's disease is associated with APP and MAPT dysregulation. Human Molecular Genetics. 2014;23(3):648–656. doi: 10.1093/hmg/ddt451. [DOI] [PubMed] [Google Scholar]
- 88.De Strooper B., Iwatsubo T., Wolfe M. S. Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2012;2, article a006304(1) doi: 10.1101/cshperspect.a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin H. C., Hsieh H. M., Chen Y. H., Hu M. L. S -Adenosylhomocysteine increases β-amyloid formation in BV-2 microglial cells by increased expressions of β-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology. 2009;30(4):622–627. doi: 10.1016/j.neuro.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 90.Li W., Jiang M., Zhao S., et al. Folic acid inhibits amyloid β-peptide production through modulating DNA methyltransferase activity in N2a-APP cells. International Journal of Molecular Sciences. 2015;16(10):25002–25013. doi: 10.3390/ijms161025002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sonawane S. K., Chinnathambi S. Prion-like propagation of post-translationally modified tau in Alzheimer's disease: a hypothesis. Journal of Molecular Neuroscience. 2018;65(4):480–490. doi: 10.1007/s12031-018-1111-5. [DOI] [PubMed] [Google Scholar]
- 92.Nuru M., Muradashvili N., Kalani A., Lominadze D., Tyagi N. High methionine, low folate and low vitamin B6/B12 (HM-LF-LV) diet causes neurodegeneration and subsequent short-term memory loss. Metabolic Brain Disease. 2018;33(6):1923–1934. doi: 10.1007/s11011-018-0298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Semick S. A., Bharadwaj R. A., Collado-Torres L., et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease. Acta Neuropathologica. 2019;137(4):557–569. doi: 10.1007/s00401-019-01966-5. [DOI] [PubMed] [Google Scholar]
- 94.Smith R. G., Hannon E., De Jager P. L., et al. Elevated DNA methylation across a 48-kb region spanning theHOXAgene cluster is associated with Alzheimer's disease neuropathology. Alzheimer's & Dementia. 2018;14(12):1580–1588. doi: 10.1016/j.jalz.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Altuna M., Urdánoz-Casado A., de Gordoa J. S.-R., et al. DNA methylation signature of human hippocampus in Alzheimer's disease is linked to neurogenesis. Clinical Epigenetics. 2019;11(1):p. 91. doi: 10.1186/s13148-019-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicolia V., Lucarelli M., Fuso A. Environment, epigenetics and neurodegeneration: focus on nutrition in Alzheimer's disease. Experimental Gerontology. 2015;68:8–12. doi: 10.1016/j.exger.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 97.Hu Q., Teng W., Li J., Hao F., Wang N. Homocysteine and Alzheimer's disease: evidence for a causal link from Mendelian randomization. Journal of Alzheimer's Disease. 2016;52(2):747–756. doi: 10.3233/JAD-150977. [DOI] [PubMed] [Google Scholar]
- 98.Nazef K., Khelil M., Chelouti H., et al. Hyperhomocysteinemia is a risk factor for Alzheimer's disease in an Algerian population. Archives of Medical Research. 2014;45(3):247–250. doi: 10.1016/j.arcmed.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 99.Sharma M., Tiwari M., Tiwari R. K. Hyperhomocysteinemia: impact on neurodegenerative diseases. Basic & Clinical Pharmacology & Toxicology. 2015;117(5):287–296. doi: 10.1111/bcpt.12424. [DOI] [PubMed] [Google Scholar]
- 100.Grossi E., Stoccoro A., Tannorella P., Migliore L., Coppedè F. Artificial neural networks link one-carbon metabolism to gene-promoter methylation in Alzheimer's disease. Journal of Alzheimer's Disease. 2016;53(4):1517–1522. doi: 10.3233/JAD-160210. [DOI] [PubMed] [Google Scholar]
- 101.Setién-Suero E., Suárez-Pinilla M., Suárez-Pinilla P., Crespo-Facorro B., Ayesa-Arriola R. Homocysteine and cognition: a systematic review of 111 studies. Neuroscience and Biobehavioral Reviews. 2016;69:280–298. doi: 10.1016/j.neubiorev.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 102.Love S., Miners J. S. Cerebrovascular disease in ageing and Alzheimer's disease. Acta Neuropathologica. 2016;131(5):645–658. doi: 10.1007/s00401-015-1522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pirchl M., Ullrich C., Humpel C. Differential effects of short- and long-term hyperhomocysteinaemia on cholinergic neurons, spatial memory and microbleedings in vivo in rats. The European Journal of Neuroscience. 2010;32(9):1516–1527. doi: 10.1111/j.1460-9568.2010.07434.x. [DOI] [PubMed] [Google Scholar]
- 104.Ataie A., Sabetkasaei M., Haghparast A., Moghaddam A. H., Ataee R., Moghaddam S. N. Curcumin exerts neuroprotective effects against homocysteine intracerebroventricular injection-induced cognitive impairment and oxidative stress in rat brain. Journal of Medicinal Food. 2010;13(4):821–826. doi: 10.1089/jmf.2009.1278. [DOI] [PubMed] [Google Scholar]
- 105.Nath N., Prasad H. K., Kumar M. Cerebroprotective effects of hydrogen sulfide in homocysteine-induced neurovascular permeability: involvement of oxidative stress, arginase, and matrix metalloproteinase-9. Journal of Cellular Physiology. 2018;234(3):3007–3019. doi: 10.1002/jcp.27120. [DOI] [PubMed] [Google Scholar]
- 106.Love S., Miners J. S. Cerebrovascular disease in ageing and Alzheimer's disease. Acta Neuropathologica. 2016;131(5):645–658. doi: 10.1007/s00401-015-1522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamat P. K., Kyles P., Kalani A., Tyagi N. Hydrogen sulfide ameliorates homocysteine-induced Alzheimer's disease-like pathology, blood-brain barrier disruption, and synaptic disorder. Molecular Neurobiology. 2016;53(4):2451–2467. doi: 10.1007/s12035-015-9212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin N., Qin S., Luo S., Cui S., Huang G., Zhang X. Homocysteine induces cytotoxicity and proliferation inhibition in neural stem cells via DNA methylation in vitro. The FEBS Journal. 2014;281(8):2088–2096. doi: 10.1111/febs.12764. [DOI] [PubMed] [Google Scholar]
- 109.Collins A. R. Measuring oxidative damage to DNA and its repair with the comet assay. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2014;1840(2):794–800. doi: 10.1016/j.bbagen.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 110.Li J. G., Barrero C., Gupta S., Kruger W. D., Merali S., Praticò D. Homocysteine modulates 5-lipoxygenase expression level via DNA methylation. Aging Cell. 2017;16(2):273–280. doi: 10.1111/acel.12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Di Meco A., Li J.-G., Praticò D. Perry G., Avila J., Tabaton M., Zhu X., editors. Dissecting the role of 5-lipoxygenase in the homocysteine-induced Alzheimer's disease pathology. Journal of Alzheimer's Disease. 2018;62(3):1337–1344. doi: 10.3233/JAD-170700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Currò M., Gugliandolo A., Gangemi C., Risitano R., Ientile R., Caccamo D. Toxic effects of mildly elevated homocysteine concentrations in neuronal-like cells. Neurochemical Research. 2014;39(8):1485–1495. doi: 10.1007/s11064-014-1338-7. [DOI] [PubMed] [Google Scholar]
- 113.Di Meco A., Li J.-G., Barrero C., Merali S., Praticò D. Elevated levels of brain homocysteine directly modulate the pathological phenotype of a mouse model of tauopathy. Molecular Psychiatry. 2019;24(11):1696–1706. doi: 10.1038/s41380-018-0062-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li J. G., Praticò D. High levels of homocysteine results in cerebral amyloid angiopathy in mice. Journal of Alzheimer's Disease. 2015;43(1):29–35. doi: 10.3233/JAD-141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kruman I. I., Kumaravel T. S., Lohani A., et al. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. The Journal of Neuroscience. 2002;22(5):1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shirafuji N., Hamano T., Yen S. H., et al. Homocysteine increases tau phosphorylation, truncation and oligomerization. International Journal of Molecular Sciences. 2018;19(3):p. 891. doi: 10.3390/ijms19030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Negahdar H., Hosseini S. R., Parsian H., et al. Homocysteine, trace elements and oxidant/antioxidant status in mild cognitively impaired elderly persons: a cross-sectional study. Romanian Journal of Internal Medicine. 2015;53(4):336–342. doi: 10.1515/rjim-2015-0043. [DOI] [PubMed] [Google Scholar]
- 118.Velazquez R., Ferreira E., Winslow W., et al. Maternal choline supplementation ameliorates Alzheimer's disease pathology by reducing brain homocysteine levels across multiple generations. Molecular Psychiatry. 2019 doi: 10.1038/s41380-018-0322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chaudhury S., Patel T., Barber I. S., et al. Polygenic risk score in postmortem diagnosed sporadic early-onset Alzheimer's disease. Neurobiology of Aging. 2018;62:244.e1–244.e8. doi: 10.1016/j.neurobiolaging.2017.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang J. C., Alinaghi S., Tafakhori A., et al. Genetic screening in two Iranian families with early-onset Alzheimer's disease identified a novel PSEN1 mutation. Neurobiology of Aging. 2018;62:244.e15–244.e17. doi: 10.1016/j.neurobiolaging.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hara J., Shankle W. R., Barrentine L. W., Curole M. V. Novel therapy of hyperhomocysteinemia in mild cognitive impairment, Alzheimer's disease, and other dementing disorders. The Journal of Nutrition, Health & Aging. 2016;20(8):825–834. doi: 10.1007/s12603-016-0688-z. [DOI] [PubMed] [Google Scholar]
- 122.Halil M., Cemal Kizilarslanoglu M., Emin Kuyumcu M., Yesil Y., Cruz Jentoft A. J. Cognitive aspects of frailty: mechanisms behind the link between frailty and cognitive impairment. The Journal of Nutrition, Health & Aging. 2015;19(3):276–283. doi: 10.1007/s12603-014-0535-z. [DOI] [PubMed] [Google Scholar]
- 123.Lin N., Qin S., Luo S., Cui S., Huang G., Zhang X. Homocysteine induces cytotoxicity and proliferation inhibition in neural stem cells via DNA methylation in vitro. The FEBS Journal. 2014;281(8):2088–2096. doi: 10.1111/febs.12764. [DOI] [PubMed] [Google Scholar]
- 124.Mattson M., Kruman I., Duan W. Folic acid and homocysteine in age-related disease. Ageing Research Reviews. 2002;1(1):95–111. doi: 10.1016/S0047-6374(01)00365-7. [DOI] [PubMed] [Google Scholar]
- 125.Li J. G., Barrero C., Gupta S., Kruger W. D., Merali S., Praticò D. Homocysteine modulates 5-lipoxygenase expression level via DNA methylation. Aging Cell. 2017;16(2):273–280. doi: 10.1111/acel.12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kalani A., Kamat P. K., Givvimani S., et al. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. Journal of Molecular Neuroscience. 2014;52(2):202–215. doi: 10.1007/s12031-013-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hannibal L., Blom H. J. Homocysteine and disease: causal associations or epiphenomenons? Molecular Aspects of Medicine. 2017;53:36–42. doi: 10.1016/j.mam.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 128.Tchantchou F. Homocysteine metabolism and various consequences of folate deficiency. Journal of Alzheimer's Disease. 2006;9(4):421–427. doi: 10.3233/JAD-2006-9408. [DOI] [PubMed] [Google Scholar]
- 129.Araújo J. R., Martel F., Borges N., Araújo J. M., Keating E. Folates and aging: role in mild cognitive impairment, dementia and depression. Ageing Research Reviews. 2015;22:9–19. doi: 10.1016/j.arr.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 130.Bednarska-Makaruk M., Graban A., Sobczyńska-Malefora A., et al. Homocysteine metabolism and the associations of global DNA methylation with selected gene polymorphisms and nutritional factors in patients with dementia. Experimental Gerontology. 2016;81:83–91. doi: 10.1016/j.exger.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 131.Sontag J.-M., Wasek B., Taleski G., et al. Altered protein phosphatase 2A methylation and tau phosphorylation in the young and aged brain of methylenetetrahydrofolate reductase (MTHFR) deficient mice. Frontiers in Aging Neuroscience. 2014;6:p. 214. doi: 10.3389/fnagi.2014.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nazef K., Khelil M., Chelouti H., et al. Hyperhomocysteinemia is a risk factor for Alzheimer's disease in an Algerian population. Archives of Medical Research. 2014;45(3):247–250. doi: 10.1016/j.arcmed.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 133.Fuso A., Cavallaro R. A., Zampelli A., et al. γ-Secretase is differentially modulated by alterations of homocysteine cycle in neuroblastoma and glioblastoma cells. Journal of Alzheimer's Disease. 2007;11(3):275–290. doi: 10.3233/JAD-2007-11303. [DOI] [PubMed] [Google Scholar]
- 134.Fuso A., Seminara L., Cavallaro R. A., D'Anselmi F., Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Molecular and Cellular Neurosciences. 2005;28(1):195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 135.Kamat P. K., Kyles P., Kalani A., Tyagi N. Hydrogen sulfide ameliorates homocysteine-induced Alzheimer's disease-like pathology, blood-brain barrier disruption, and synaptic disorder. Molecular Neurobiology. 2016;53(4):2451–2467. doi: 10.1007/s12035-015-9212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Janel N., Alexopoulos P., Badel A., et al. Combined assessment of DYRK1A, BDNF and homocysteine levels as diagnostic marker for Alzheimer's disease. Translational Psychiatry. 2017;7(6, article e1154) doi: 10.1038/tp.2017.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yadav B. K., Oh S. Y., Kim N. K., Shin B. S. Association of rs2075575 and rs9951307 Polymorphisms of AQP-4 Gene with Leukoaraiosis. Journal of Stroke and Cerebrovascular Diseases. 2014;23(5):1199–1206. doi: 10.1016/j.jstrokecerebrovasdis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 138.Tannorella P., Stoccoro A., Tognoni G., et al. Methylation analysis of multiple genes in blood DNA of Alzheimer's disease and healthy individuals. Neuroscience Letters. 2015;600:143–147. doi: 10.1016/j.neulet.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 139.Li J. G., Chu J., Barrero C., Merali S., Praticò D. Homocysteine exacerbates β-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Annals of Neurology. 2014;75(6):851–863. doi: 10.1002/ana.24145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rai V. Folate pathway gene methylenetetrahydrofolate reductase C677T polymorphism and Alzheimer disease risk in Asian population. Indian Journal of Clinical Biochemistry. 2016;31(3):245–252. doi: 10.1007/s12291-015-0512-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rai V. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism and Alzheimer disease risk: a meta-analysis. Molecular Neurobiology. 2017;54(2):1173–1186. doi: 10.1007/s12035-016-9722-8. [DOI] [PubMed] [Google Scholar]
- 142.Obeid R., Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Letters. 2006;580(13):2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- 143.Grossi E., Stoccoro A., Tannorella P., Migliore L., Coppedè F. Artificial neural networks link one-carbon metabolism to gene-promoter methylation in Alzheimer's disease. Journal of Alzheimer's Disease. 2016;53(4):1517–1522. doi: 10.3233/JAD-160210. [DOI] [PubMed] [Google Scholar]
- 144.Li J.-G., Barrero C., Merali S., Praticò D. Five lipoxygenase hypomethylation mediates the homocysteine effect on Alzheimer's phenotype. Scientific Reports. 2017;7(1, article 46002) doi: 10.1038/srep46002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Scarpa S., Cavallaro R. A., D'Anselmi F., Fuso A. Gene silencing through methylation: an epigenetic intervention on Alzheimer disease. Journal of Alzheimer's Disease. 2006;9(4):407–414. doi: 10.3233/JAD-2006-9406. [DOI] [PubMed] [Google Scholar]
- 146.Cavallaro R. A., Fuso A., D'Anselmi F., Seminara L., Scarpa S. The effect of S-adenosylmethionine on CNS gene expression studied by cDNA microarray analysis. Journal of Alzheimer's Disease. 2006;9(4):415–419. doi: 10.3233/JAD-2006-9407. [DOI] [PubMed] [Google Scholar]
- 147.Scarpa S., Fuso A., D'Anselmi F., Cavallaro R. A. Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Letters. 2003;541(1-3):145–148. doi: 10.1016/S0014-5793(03)00277-1. [DOI] [PubMed] [Google Scholar]
- 148.Lin H. C., Hsieh H. M., Chen Y. H., Hu M. L. S-Adenosylhomocysteine increases β-amyloid formation in BV-2 microglial cells by increased expressions of β-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology. 2009;30(4):622–627. doi: 10.1016/j.neuro.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 149.Kalani A., Kamat P. K., Tyagi S. C., Tyagi N. Synergy of homocysteine, microRNA, and epigenetics: a novel therapeutic approach for stroke. Molecular Neurobiology. 2013;48(1):157–168. doi: 10.1007/s12035-013-8421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Joe E., Ringman J. M. Cognitive symptoms of Alzheimer's disease: clinical management and prevention. BMJ. 2019;367, article l6217 doi: 10.1136/bmj.l6217. [DOI] [PubMed] [Google Scholar]
- 151.Panza F., Lozupone M., Bellomo A., Imbimbo B. P. Do anti-amyloid-β drugs affect neuropsychiatric status in Alzheimer's disease patients? Ageing Research Reviews. 2019;55:p. 100948. doi: 10.1016/j.arr.2019.100948. [DOI] [PubMed] [Google Scholar]
- 152.Knapp M., King D., Romeo R., et al. Cost-effectiveness of donepezil and memantine in moderate to severe Alzheimer's disease (the DOMINO-AD trial) International Journal of Geriatric Psychiatry. 2017;32(12):1205–1216. doi: 10.1002/gps.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Birks J. S., Harvey R. J. Donepezil for dementia due to Alzheimer's disease. Cochrane Database of Systematic Reviews. 2018;6(6, article CD001190) doi: 10.1002/14651858.CD001190.pub3. [DOI] [PubMed] [Google Scholar]
- 154.Kandiah N., Pai M. C., Senanarong V., et al. Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clinical Interventions in Aging. 2017;12:697–707. doi: 10.2147/CIA.S129145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wu Z., Zhao L., Chen X., Cheng X., Zhang Y. Galantamine attenuates amyloid-β deposition and astrocyte activation in APP/PS1 transgenic mice. Experimental Gerontology. 2015;72:244–250. doi: 10.1016/j.exger.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 156.Matsunaga S., Kishi T., Nomura I., et al. The efficacy and safety of memantine for the treatment of Alzheimer's disease. Expert Opinion on Drug Safety. 2018;17(10):1053–1061. doi: 10.1080/14740338.2018.1524870. [DOI] [PubMed] [Google Scholar]
- 157.Deardorff W. J., Grossberg G. T. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer's disease. Drug Design, Development and Therapy. 2016;Volume 10:3267–3279. doi: 10.2147/DDDT.S86463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Galvin J. E. Optimizing diagnosis and management in mild-to-moderate Alzheimer's disease. Neurodegenerative Disease Management. 2012;2(3):291–304. doi: 10.2217/nmt.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Arvanitakis Z., Shah R. C., Bennett D. A. Diagnosis and management of dementia: review. JAMA. 2019;322(16):1589–1599. doi: 10.1001/jama.2019.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Madsen S. K., Rajagopalan P., Joshi S. H., Toga A. W., Thompson P. M., Alzheimer's Disease Neuroimaging Initiative (ADNI) Higher homocysteine associated with thinner cortical gray matter in 803 participants from the Alzheimer's disease neuroimaging initiative. Neurobiology of Aging. 2015;36(1):S203–S210. doi: 10.1016/j.neurobiolaging.2014.01.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huang C. W., Chang W. N., Huang S. H., et al. Impact of homocysteine on cortical perfusion and cognitive decline in mild Alzheimer's dementia. European Journal of Neurology. 2013;20(8):1191–1197. doi: 10.1111/ene.12159. [DOI] [PubMed] [Google Scholar]
- 162.Tu M. C., Huang C. W., Chen N. C. Hyperhomocysteinemia in Alzheimer dementia patients and cognitive decline after 6 months follow-up period. Acta Neurologica Taiwanica. 2010;19(3):168–177. [PubMed] [Google Scholar]
- 163.Ma F., Wu T., Zhao J., et al. Plasma homocysteine and serum folate and vitamin B12 levels in mild cognitive impairment and Alzheimer's disease: a case-control study. Nutrients. 2017;9(7):p. 725. doi: 10.3390/nu9070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hu Q., Teng W., Li J., Hao F., Wang N. Homocysteine and Alzheimer's disease: evidence for a causal link from Mendelian randomization. Journal of Alzheimer's Disease. 2016;52(2):747–756. doi: 10.3233/JAD-150977. [DOI] [PubMed] [Google Scholar]
- 165.Kamat P. K., Kyles P., Kalani A., Tyagi N. Hydrogen sulfide ameliorates homocysteine-induced Alzheimer's disease-like pathology, blood-brain barrier disruption, and synaptic disorder. Molecular Neurobiology. 2016;53(4):2451–2467. doi: 10.1007/s12035-015-9212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McCaddon A., Hudson P., Davies G., Hughes A., Williams J. H. H., Wilkinson C. Homocysteine and cognitive decline in healthy elderly. Dementia and Geriatric Cognitive Disorders. 2001;12(5):309–313. doi: 10.1159/000051275. [DOI] [PubMed] [Google Scholar]
- 167.McCaddon A., Regland B. Homocysteine and cognition - No longer a hypothesis? Medical Hypotheses. 2006;66(3):682–683. doi: 10.1016/j.mehy.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 168.Altuna M., Urdánoz-Casado A., Sánchez-Ruiz de Gordoa J., et al. DNA methylation signature of human hippocampus in Alzheimer's disease is linked to neurogenesis. Clinical Epigenetics. 2019;11(1):p. 91. doi: 10.1186/s13148-019-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Blanch M., Mosquera J. L., Ansoleaga B., Ferrer I., Barrachina M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. The American Journal of Pathology. 2016;186(2):385–397. doi: 10.1016/j.ajpath.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 170.Yu L., Chibnik L. B., Srivastava G. P., et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 With pathological diagnosis of Alzheimer disease. JAMA Neurology. 2015;72(1):15–24. doi: 10.1001/jamaneurol.2014.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Di Francesco A., Arosio B., Falconi A., et al. Global changes in DNA methylation in Alzheimer's disease peripheral blood mononuclear cells. Brain, Behavior, and Immunity. 2015;45:139–144. doi: 10.1016/j.bbi.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 172.Sliwinska A., Sitarek P., Toma M., et al. Decreased expression level of BER genes in Alzheimer's disease patients is not derivative of their DNA methylation status. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2017;79, article Part B:311–316. doi: 10.1016/j.pnpbp.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 173.Kobayashi N., Shinagawa S., Nagata T., et al. Usefulness of DNA methylation levels in COASY and SPINT1 gene promoter regions as biomarkers in diagnosis of Alzheimer's disease and amnestic mild cognitive impairment. PLoS One. 2016;11(12, article e0168816) doi: 10.1371/journal.pone.0168816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kobayashi N., Shinagawa S., Nagata T., et al. Development of biomarkers based on DNA methylation in the NCAPH2/LMF2 promoter region for diagnosis of Alzheimer's disease and amnesic mild cognitive impairment. PLoS One. 2016;11(1, article e0146449) doi: 10.1371/journal.pone.0146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used to support this study.