

Abstract
Aims
This study aimed to investigate the potential interaction between Schisandra sphenanthera, imatinib and bosutinib combining in vitro and in silico methods.
Methods
In vitro metabolism of imatinib and bosutinib using recombinant enzymes and human liver microsomes were investigated in the presence and absence of Schisandra lignans. Physiologically‐based pharmacokinetic (PBPK) models for the lignans accounting for reversible and mechanism‐based inhibitions and induction of CYP3A enzymes were built in the Simcyp Simulator (version 17) and evaluated for their capability to predict interactions with midazolam and tacrolimus. Their potential effect on systemic exposures of imatinib and bosutinib were predicted using PBPK in silico simulations.
Results
Schisantherin A and schisandrol B, but not schisandrin A, potently inhibited CYP3A4‐mediated metabolism of imatinib and bosutinib. All three compounds showed a strong reversible inhibition on CYP2C8 enzyme with k i of less than 0.5 μmol L−1. The verified PBPK models were able to describe the increase in systemic exposure of midazolam and tacrolimus due to co‐administration of S. sphenanthera, consistent with the reported changes in the corresponding clinical interaction study (AUC ratio of 2.0 vs 2.1 and 2.4 vs 2.1, respectively). The PBPK simulation predicted that at recommended dosing regimens of S. sphenanthera, co‐administration would result in an increase in bosutinib exposure (AUC ratio 3.0) but not in imatinib exposure.
Conclusion
PBPK models for Schisandra lignans were successfully developed. Interaction between imatinib and Schisandra lignans was unlikely to be of clinical importance. Conversely, S. sphenanthera at a clinically‐relevant dose results in a predicted three‐fold increase in bosutinib systemic exposure.
Keywords: drug metabolism, herb–drug interactions, modelling and simulation, physiologically‐based pharmacokinetic (PBPK)
What is already known about this subject
Physiologically‐based pharmacokinetic (PBPK) modelling approaches combined with in vitro to in vivo extrapolation techniques have been extensively used for predicting clinically meaningful pharmacokinetic drug interactions.
Schisandrin A, schisantherin A and schisandrol B, the major bioactive lignans in Schisandra sphenanthera are potent CYP3A modulators with evidence for clinically‐significant drug interactions.
What this study adds
PBPK models for the perpetrator compounds in S. sphenanthera were developed with a capability to predict the interactions of Schisandra constituents with midazolam and tacrolimus.
Schisandra lignans at clinically‐relevant concentrations were predicted to have little to no effect on imatinib pharmacokinetics but were expected to lead to a three‐fold increase in the systemic exposure of bosutinib.
1. INTRODUCTION
Schisandra sphenanthera has long been an integral part of traditional Chinese medicine, while other closely‐related species, Schisandra chinensis can be found in many Western countries. 1 These natural products (typically administered as dried ethanol extracts) are used mainly for their putative hepatoprotective effect 2 , 3 , 4 and have been explored for anti‐cancer properties. 5 , 6 Interestingly, S. sphenanthera alleviated tacrolimus‐induced diarrhoea in liver transplant recipients, although the underlying mechanism is not clear yet. 4 Several biologically‐active dibenzocyclooctadiene lignans, namely schisantherin A, schisandrin A and schisandrol B (Table 1) have been identified in S. sphenanthera extract with evidence for cytochrome P450 (CYP)3A modulations. 13 , 15
TABLE 1.
Input parameters for PBPK models of the perpetrator compounds in S. sphenanthera
| Parameter | Schisantherin A | Schisandrin A | Schisandrol B |
|---|---|---|---|
| Physicochemical and blood‐binding properties | |||
| Chemical structure a |
|
|
|
| Molecular weight b | 536.6 | 416.5 | 416.5 |
| Log P o:w b | 1.7 c | 4.8 | 3.4 |
| Ionisation pattern b | Neutral | Neutral | Neutral |
| B/P d | 0.63 | 0.69 | 0.55 |
| fu p d | 0.08 | 0.03 | 0.08 |
| Absorption phase | |||
| Model | First‐order | CAT | First‐order |
| f a d | 1 | 1 | 1 |
| k a (h−1) d | 1.8 | 3.1 | 6.0 e |
| t lag (h) | 0.15 | ||
| P eff (10−4 cm s−1) d | 4.0 | 7.1 | 4.5 |
| fu G f | 1 | 1 | 1 |
| Q Gut (L h−1)d | 14.1 | 16.9 | 14.7 |
| Distribution phase | |||
| Prediction method | Poulin and Theil 7 | Poulin and Theil | Rodgers and Rowland 8 |
| V ss (L kg−1) | 0.4 | 11.6 | 4.7 |
| Elimination phase | |||
| CL int,CYP3A4 (μL min−1 mg protein−1) | 2.5 g | 170 h | |
| V max,CYP3A4 (pmol min−1 pmol CYP−1) | 12.3 i | ||
| K m,CYP3A4 (μmol L−1) | 5.6 i | ||
| V max,CYP3A5 (pmol min−1 pmol CYP−1) | 1.3 i | ||
| K m,CYP3A5 (μmol L−1) | 7.8 i | ||
| CL int,others (μL min−1 mg protein−1) | 21.5 g | 52 h | |
| Drug interactions | |||
| Reversible inhibition | |||
| k i,CYP3A for midazolam (μmol L−1) | 0.05 j | 1.51 c | 0.93 k |
| k i,CYP3A for tacrolimus (μmol L−1) | 0.25 c | 1.51 c | 0.93 k |
| k i,u,CYP3A for imatinib (μmol L−1) l | 0.07 | 40.10 | 4.37 |
| fu inc d | 0.50 | 0.66 | 0.91 |
| k i,u,CYP2C8 for imatinib (μmol L−1) l | 0.22 | 0.10 | 0.48 |
| fu inc d | 0.37 | 0.41 | 0.78 |
| k i,u,CYP3A for bosutinib (μmol L−1) l | 0.03 | 2.72 | 0.51 |
| fu inc d | 0.93 | 0.25 | 0.64 |
| k i,ABCB1 (μmol L−1) | 12.8 m | 12.2 n | > 100 |
| Mechanism‐based inhibition c | |||
| k inact,CYP3A (h−1) | 14.28 | 1.44 | |
| K I (μmol L−1) | 7.30 | 1.37 | |
| Induction o | |||
| Ind max,CYP3A | 11.50 | 11.50 | |
| IndC 50 (μmol L−1) | 0.08 | 0.08 | |
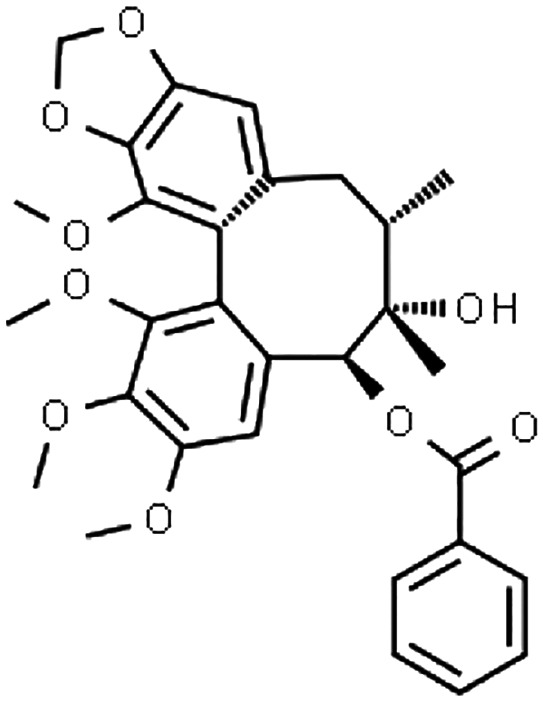
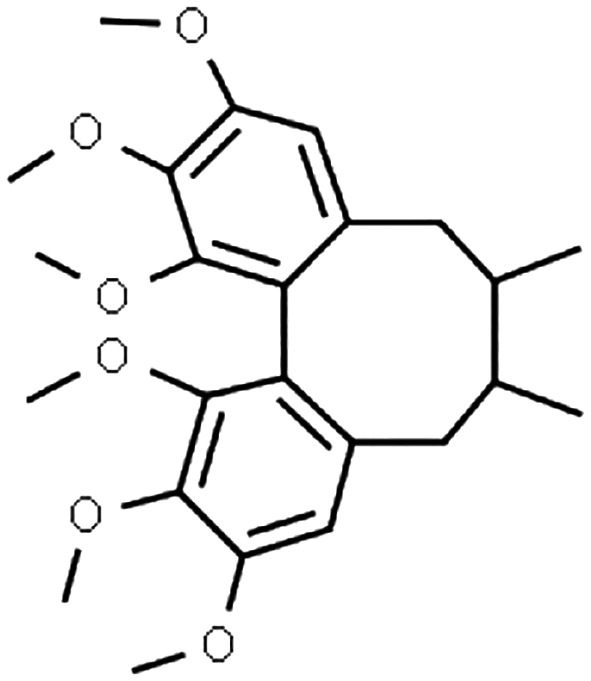
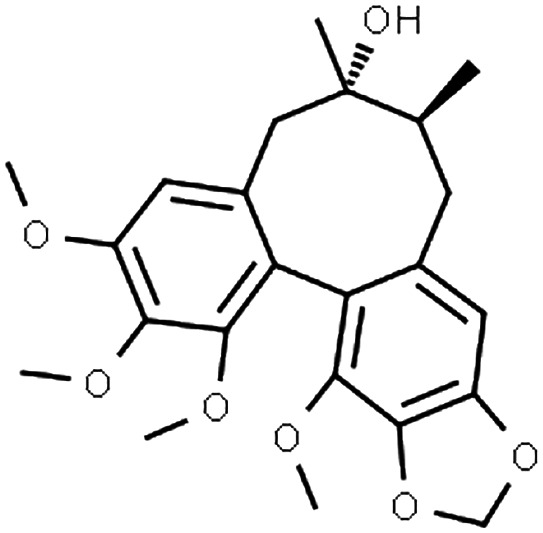
ABCB1, multidrug resistance protein 1 or p‐glycoprotein; B/P, blood to plasma ratio; CAT, compartmental absorption and transit model; CL int, hepatic intrinsic clearance; CYP, cytochrome P450; f a, fraction absorbed from the intestinal tract; fu G, unbound fraction in the enterocytes; fu inc, unbound fraction in incubation medium; fu p, unbound fraction in plasma; IndC 50, the concentration of inducer at half of Ind max; Ind max, maximum fold of induction; k a, first‐order absorption rate constant; k i, inhibitory constant; K I, the concentration that provides half of k inact; k inact, maximum inactivation rate of CYP enzyme; K m, substrate concentration giving half of V max; log P o:w, the partition coefficient in oil and water; P eff, the effective intestinal permeability; Q Gut, the gut blood flow rate; t lag, lag time; V max, maximum rate of reaction; V ss, volume of distribution at steady‐state based on total tissue volumes.
Retrieved from ChemSpider (www.chemspider.com).
Accessed from PubChem (pubchem.ncbi.nlm.gov) and ChEMBL (ebi.ac.uk/chembl).
Experimental data from Zhang et al. 9
Predicted in the Simcyp Simulator.
Optimised through a sensitivity analysis.
Assumed equal to 1. 10
Total CL int value was extracted from Zhang et al. 9 and f m , CYP3A4 was estimated based on the extent of inhibition by ketoconazole. 11
Total CL int was determined from substrate depletion in human liver microsomes and f m , CYP3A4 was based on percent of inhibition by ketoconazole. 11
In vitro metabolism data in recombinant CYP3A4 and CYP3A5. 12
Reversible inhibition towards CYP3A enzymes using erythromycin as a probe substrate. 13
K i was derived from IC50 towards metabolism of midazolam in recombinant CYP3A4, assuming a competitive inhibition. 12
In vitro inhibition data obtained from this study.
Inhibition of transport of rhodamine 123 in Caco‐2 cell line. 14
Inhibition of digoxin transport in Caco‐2 cells. 14
Parameters related to CYP3A induction were estimated from the increase of CYP3A4 mRNA in hepatocytes as a result of exposure to S. sphenanthera extract (see Section 2 for more details). 15
Clinically‐important herb–drug interactions with Schisandra lignans as the perpetrators have been reported for midazolam, 16 sirolimus 17 and tacrolimus. 4 , 18 , 19 Administration of S. sphenanthera extract (equivalent to 14, 19 and 1 mg day−1 of schisandrin A, schisantherin A and schisandrol B, respectively) in Chinese transplant recipients expressing CYP3A5 enzyme reduced by 40% the tacrolimus dose required to achieve the predefined target of trough blood concentrations 19 and thus was more cost‐effective. 20 This has led to the investigation of S. sphenanthera as a promising pharmacokinetic enhancer (booster) to increase the bioavailability of CYP3A substrates. 19 Conversely, there is also a potential for unwanted herb–drug interactions with S. sphenanthera, e.g. a higher risk for exposure‐related adverse events of the victim drugs. However, it is not possible to cover all the various permutations of combinations between natural products and conventional medicines by conducting dedicated clinical interaction studies. 21 Clearly, a reliable and systematic approach to predict these herb–drug interactions is needed.
As for drug–drug interactions, physiologically‐based pharmacokinetic (PBPK) modelling and simulation has been advocated for quantitative prediction of drug interactions with natural products. 22 Nevertheless, PBPK prediction of herb–drug interactions is more challenging than that with conventional perpetrator drugs. 23 Prior knowledge of perpetrator compounds in a complex mixture (extract) and their typical maintenance dosing regimens are necessary for an accurate in vitro to in vivo extrapolation of herb–drug interactions or lack thereof. There is also a possibility for interactions among compounds in the extract which may alter their systemic exposure. Hence, well‐characterised natural products are essential for implementing this quantitative framework. PBPK modelling has been successfully implemented in describing a number of potential herb–drug interactions, including grapefruit juice with midazolam and simvastatin, 24 milk thistle with midazolam, warfarin 23 and raloxifene, 25 and St John's wort with a range of CYP substrates. 26
Imatinib and bosutinib are both approved as the first‐line treatments for Philadelphia chromosome‐positive (Ph+) chronic myeloid leukaemia (CML). 27 , 28 , 29 People with CML and receiving treatment with tyrosine kinase inhibitors are known to concomitantly use complementary medicines, 30 including S. sphenanthera, with a purported aim to help manage adverse effects. The common side effects for patients being treated with these kinase inhibitors are gastrointestinal disturbances, e.g. diarrhoea, 28 , 31 while elevation of alanine transaminase is more frequent for bosutinib. 28 S. sphenanthera may be beneficial for ameliorating these adverse events, 3 , 4 in addition to its potential anti‐cancer activity. 5 , 6 Imatinib and bosutinib have distinct pharmacokinetic characteristics. Imatinib is a substrate for both CYP3A4 and CYP2C8 enzymes and has a low hepatic extraction ratio (EH), 32 while bosutinib is a high EH drug almost exclusively metabolised by CYP3A4. 33 Therefore, both medicines are prone to CYP3A modulation by S. sphenanthera. The development and evaluation of PBPK models for the Schisandra lignans will provide a greater understanding and allow the prediction of interactions with both drugs. The aim of this study was to build, evaluate and implement PBPK models of key perpetrator compounds found in S. sphenanthera extract for investigating the potential herb–drug interactions with imatinib and bosutinib.
2. METHODS
2.1. Inhibition of in vitro metabolism of imatinib and bosutinib by Schisandra lignans
2.1.1. Materials
Imatinib (mesylate salt), bosutinib, schisandrin A, schisantherin A, schisandrol B, β‐nicotinamide adenine dinucleotide 2′‐phosphate reduced (NADPH) tetrasodium salt, azamulin, dipotassium hydrogen phosphate trihydrate, potassium dihydrogen phosphate, dimethyl sulfoxide (DMSO) and pooled human liver microsomes (M0317) were purchased from Sigma‐Aldrich (Castle Hill, NSW, Australia). N‐desmethyl imatinib was obtained from Toronto Research Chemicals (North York, ON, Canada). Recombinant CYP3A4 enzyme were procured from Corning (Woburn, MA; distributed by In Vitro Technologies, Noble Park, VIC, Australia).
2.1.2. In vitro inhibition study on metabolism of imatinib and bosutinib
The rate of formation of N‐desmethyl imatinib (NDMI) from imatinib was evaluated in recombinant CYP3A4 (rCYP3A4) and human liver microsomes (HLM) at a concentration of 12.5 pmol CYP mL−1 and 0.25 mg microsomal protein mL−1, respectively. Azamulin (5 μmol L−1) was added to the latter to specifically inactivate CYP3A4 enzyme (mechanism‐based inhibition, MBI). 34 This system was assumed to represent a CYP2C8 pathway since CYP enzymes other than CYP3A4 and CYP2C8 had a very minor contribution (3%) to imatinib metabolism. 35 Incubation medium consisted of potassium phosphate buffer (0.1 mol L−1, pH 7.4), either HLM with azamulin or rCYP3A4, DMSO (2%), NADPH (2 mmol L−1) and milli Q water in a final volume of 200 μL. DMSO was used at a concentration that has a minimum impact on imatinib N‐demethylation (data not shown).
An extensive in vitro study showed that recombinant CYP2C8 enzyme (supersomes) was less sensitive than human liver microsomes (HLM) to identify potential mechanism‐based inhibitors and was characterised by a high lot‐to‐lot variability. Conversely, HLM are more reliable sources of CYP2C8 enzyme to evaluate mechanism‐based inhibition of the enzyme. 36 This is also likely to be the case for reversible inhibition of CYP2C8 enzyme. Therefore, CYP3A4‐inactivated HLM was chosen to evaluate potential reversible inhibitions of CYP2C8‐mediated metabolism of imatinib by Schisandra lignans. Meanwhile, recombinant CYP3A4 enzyme was used to evaluate in vitro inhibitions of CYP3A4‐mediated N‐demethylation of imatinib. This was due to the lack of inhibitors capable of completely inhibiting CYP2C8 enzyme in HLM (gemfibrozil glucuronide inhibited the enzyme by up to 85%). 37 An overlap between CYP3A4 and CYP3A5 substrates has been recognised. However, the contribution of CYP3A5 for imatinib metabolism has been ruled out from thorough in vitro work 35 and clinical studies, including in a large number of patients genotyped for the CYP3A5 gene. 38 , 39
Although bosutinib has a high in vivo extraction ratio (EH), its intrinsic clearance in HLM was low (<20% reduction after a 60‐min incubation). Thus, the CYP3A4‐mediated formation of its main metabolite, N‐desmethyl bosutinib, also known as M5, 33 was followed instead. Incubation medium for this reaction was as described for NDMI formation, except that HLM (0.5 mg protein mL−1) in a final volume of 100 μL was used.
The formation rates of NDMI and M5 were determined over the concentration range of 5–100 μmol L−1 for the respective parent compounds in the absence and presence of schisantherin A (0.1–25 μmol L−1), schisandrin A and schisandrol B (0.5–25 μmol L−1) without pre‐incubation to evaluate their potential for reversible inhibition. All incubations were performed in triplicate in 1.5 mL tubes in a shaking water bath at 37°C for 60 min. The temperature of incubation mixtures was equilibrated by placing the tubes in the water bath for 5 min. The reaction was initiated by the addition of NADPH and terminated by placing the tubes on a cooling block (Selleckchem, Houston, TX, USA). Sample preparation and analytical methods for NDMI and M5 are described in the supplementary materials.
The inhibitory constants (k i) for a selective inhibitor of CYP enzymes are expected to be similar across different substrates of the enzyme only for purely competitive inhibitions. 40 This is, however, not generally the case due to the possibility of non‐competitive and mixed inhibition. 41 Thus, specific k i values for Schisandra lignans on CYP‐mediated metabolism of imatinib and bosutinib were determined in the current study. The k i values were estimated using a nonlinear regression implemented in GraphPad Prism version 7.02 (GraphPad Software, La Jolla, CA, USA). The competitive, noncompetitive and mixed‐inhibition models (Equations 1–3, respectively) were considered. The best‐fit equations for each of the ki estimations were selected based on visual inspections of model predictions versus experimental data and Akaike information criterion (AIC).
| (1) |
| (2) |
| (3) |
where v and V max are the rate of metabolite formation and maximum value of v, respectively; K m denotes the Michaelis–Menten constant or substrate concentration ([S]) at half of V max; [I] represents the concentration of inhibitor; and α is a number equal to or larger than 1 that indicates the ‘mix’ of competitive and noncompetitive mechanisms. It is noteworthy that the reversible enzyme inactivation in the Simcyp Simulator is modelled solely by competitive inhibition. Theoretically, the discrimination between competitive and noncompetitive inhibitions is not critical when concentrations of substrates are much lower than their K m values, which is often the case in the clinical setting. 41 , 42 Nevertheless, assigning the best‐fit model is necessary for an accurate estimation of k i.
Nonspecific binding to microsomal membrane and CYP proteins may lead to an overestimation of k i values. 43 Thus, the k i values for Schisandra lignans were corrected for nonspecific binding during incubation (fu inc) based on their partition coefficients (log P) and protein concentrations ([P]) in incubation mixtures (Equation 4). 44
| (4) |
Stability of Schisandra lignans in incubation medium was determined by measuring the remaining concentrations after 60‐min incubation (supplementary materials). Evaluations with a more intensive sampling time were performed for those showing significant depletion compared to control (one‐way analysis of variance with Tukey post‐hoc test, GraphPad Prism version 7.02). Potential depletion of the inhibitors was modelled by first‐order kinetics as reported previously. 45 Corrections for k i values accounting for the concentrations of inhibitor at the end 45 , 46 and mid‐time of incubation 46 have been proposed, the latter of which was adopted for this study.
2.2. PBPK models for Schisandra lignans
All population‐based PBPK modelling and simulations were conducted using the Simcyp Simulator (version 17 release 1, Certara UK Limited, Simcyp Division, Sheffield, UK) using the ‘healthy Chinese’ and ‘general North European Caucasian’ population library data, which represent typical healthy adult people from the respective ancestry. The description of Simcyp Simulator workflow, basic algorithm and ordinary differential equations have been detailed previously. 47 , 48
The drug‐related input parameters for Schisandra lignans are summarised in Table 1. The first‐order absorption model parameterised by the fraction of oral dose that enters the gut wall (f a), and the first‐order absorption rate constant (k a) was used for schisantherin A and schisandrol B, while a more sophisticated model, that is the compartmental absorption and transit (CAT) model was employed for schisandrin A. A whole‐body PBPK model was used to describe the distribution of the three lignans. Drug partitioning to each of the tissue compartments was predicted in silico using the algorithms by Poulin and Theil (for schisandrin A and schisantherin A) 7 or Rodgers and Rowland (schisandrol B). 8 Partition coefficients (log P) of neutral compounds are generally the most influential parameter dictating the extent of distribution to different tissues. 8 , 49 A global sensitivity analysis suggested that uncertainties in tissue composition (that is concentrations of neutral lipids, neutral phospholipids, albumin and intracellular and extracellular water), but not tissue volumes may affect the in silico predictions of V ss of neutral compounds. Inter‐correlation between tissue composition and log P also had a significant impact on the estimation of K p (tissue‐to‐plasma partition coefficients). 49 For neutral compounds with a relatively small log P, e.g. schisantherin A, both prediction methods by Rodgers and Rowland 8 and Poulin and Theil 7 performed similarly. Therefore, the latter was selected for schisantherin A due to its simplicity. Interestingly, the Rodgers and Rowland algorithm provided a better estimate of K p of schisandrol B to different tissues, reflected by a better predictive performance of its pharmacokinetic profile compared to that of Poulin and Theil despite similar overall extent of distributions (V ss). However, the Rodgers and Rowland method tends to overestimate V ss and K p for highly lipophilic neutral compound, e.g. schisandrin A and thus the algorithm by Poulin and Theil is preferable for this lignan.
The intrinsic clearance (CL int) of schisandrin A was obtained from re‐analysis of its depletion in HLM, while contribution of CYP3A enzymes (f m,CYP3A = 0.76) was estimated from the percent of inhibition by ketoconazole. 11 CL int of schisantherin A 9 and schisandrol B 12 were also specified based on their metabolic pathways. 11 The mechanism‐based inhibition and induction of CYP3A4 following the chronic exposure to S. sphenanthera extract were modelled by an enzyme turnover model, depicted by an increase in degradation and synthesis of the active enzyme, respectively. 48 , 50 The maximum fold of induction (Ind max) of schisandrin A was based on the fold‐increase of CYP3A4 mRNA in hepatocytes after exposure to S. sphenanthera extract (contains 2.7% of schisandrin A) 15 and was normalised to the difference of fold‐induction by rifampicin in this study compared to its standard value. 50 The associated IndC 50 was around 3 μg mL−1 of S. sphenanthera extract, which was equivalent to 0.2 μmol L−1 of schisandrin A. This was further corrected for the nonspecific binding during incubation (estimated fu inc of 0.4, Equation 4) assuming that 0.32 mg protein was present per million of hepatocytes (according to the hepatocellularity and content of microsomal protein per gram of liver 51 ).
Since schisantherin A is also a major constituent of S. sphenanthera extract, 52 its induction parameters towards CYP3A enzymes were assumed to be the same as those for schisandrin A. Induction of CYP3A4 enzyme is not analogous to a reversible inhibition due to a possibility for different binding sites on the enzyme (that is competitive, noncompetitive and mixed‐inhibition) in the latter. This results in a high variability of inhibitory constants (k i) across different substrates, even for a selective CYP inhibitor, as has been observed with ketoconazole. 40 Conversely, induction of CYP3A4 enzyme involves binding and stimulation of pregnane X receptor (PXR), a nuclear receptor that regulates the expression of CYP3A genes. 53 This receptor binds to a variety of compounds (ligands) in a relatively nonspecific manner 54 and thus, the variability of in vitro induction parameters of CYP3A4 enzyme between structurally similar compounds is expected to be lower than that of reversible enzyme inhibition.
PBPK simulations for Schisandra lignans were performed (10 virtual trials each) with trial designs (number of people, ethnicity, age range, proportion of male to female and dosing regimens) matched to the corresponding clinical studies (Table 2). Clinically‐observed concentrations were superimposed to simulated profiles to allow visual inspection of the predictive performance. Prediction differences of pharmacokinetic parameters for the three lignans, expressed as the ratio of PBPK model prediction to clinically‐reported parameter values were also evaluated. PBPK simulations were also carried out to evaluate the capability of PBPK models of Schisandra lignans in predicting the extent of interactions with midazolam and tacrolimus with trial designs as detailed in Table 3. PBPK models for perpetrator and victim compounds were linked through competitive inhibition, MBI and induction of hepatic and intestinal CYP3A enzymes as described previously. 48 The PBPK model of tacrolimus based on clinical pharmacokinetic data in people from European ancestry 57 was utilised (Table S1). The default PBPK model of midazolam within the Simcyp Simulator was initially used for PBPK prediction of the interaction with S. sphenanthera. 16 However, the apparent clearance (CL/F) of midazolam in the associated clinical study was around two‐ to three‐fold lower than clinically‐reported values in Chinese people. 58 The reason for a far lower clearance in this cohort is not yet clear. Therefore, input parameters related to elimination of midazolam in the model were adjusted with f m for CYP3A4, and CYP3A5‐mediated pathways were fixed at the same values as those defined in the default model (Table S1).
TABLE 2.
Comparison of PBPK model prediction and clinically‐observed values for pharmacokinetic parameters of the three Schisandra lignans
| Dosing regimens a | Population | Pharmacokinetic parameter | PBPK model prediction b | Clinically‐observed value | Prediction fold‐difference | Reference |
|---|---|---|---|---|---|---|
| Schisandrin A | ||||||
| 14.50 mg, single dose | Healthy males from Chinese ancestry (n = 8, aged 23–24 years) | C max (ng mL−1) | 56.5 | 63.2 ± 29.9 | 0.89 | 55 |
| t max (h) | 0.7 | 1.3 ± 0.3 | 0.54 | |||
| AUC0–24h (ng h−1 mL −1) | 286.9 | 295.0 ± 157.7 | 0.97 | |||
| Schisantherin A | ||||||
| 14.65 mg, single dose | Healthy males from Chinese ancestry (n = 8, aged 23–24 years) | C max (ng mL−1) | 348.2 | 400.3 ± 105.3 | 0.87 | 55 |
| t max (h) | 0.8 | 1.2 ± 0.5 | 0.67 | |||
| AUC0–24h (ng h−1 mL −1) | 2324.7 | 2589.4 ± 885.4 | 0.90 | |||
| Schisandrol B | ||||||
| 0.36 mg, single dose | Healthy males from Korean ancestry (n = 10, aged 24–31 years) c | C max (ng mL−1) | 7.9 | 9.0 ± 0.8 | 0.88 | 56 |
| t max (h) | 0.3 | 0.3 ± 0.1 | 1.00 | |||
| AUC0–24h (ng h−1 mL −1) | 23.2 | 23.5 ± 4.8 | 0.99 | |||
| CL/F (L h−1) | 16.8 | 19.8 ± 2.6 | 0.85 | |||
AUC0–24h, area under the plasma concentration‐time curve during 24 h after dose; CL/F, apparent clearance; C max, peak plasma concentration; t max, time to reach C max.
Given as Schisandra sphenanthera extract.
Reported as geometric mean values of PBPK model prediction.
Population data for people from Chinese ancestry within the Simcyp Simulator were used for the PBPK simulation.
TABLE 3.
PBPK model predictions and clinically‐reported pharmacokinetic parameters of midazolam and tacrolimus in the presence and absence of S. sphenanthera extract in healthy people
| C max (ng mL−1) a | AUC0–24h (ng h−1 mL −1) | CL/F (L h−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Victim drug alone | Along with S. sphenantera | Ratio | Victim drug alone | Along with S. sphenantera | Ratio | Victim drug alone | Along with S. sphenantera | Ratio> | |
| Interaction with a single dose of midazolam (15 mg) based on the study by Xin et al. 16 , b | |||||||||
| PBPK prediction | 138.2 | 217.1 | 1.57 | 551.2 | 1,117.5 | 2.03 | 27.2 | 13.4 | 0.49 |
| Clinically‐reported values | 171.4 ± 69.8 | 282.3 ± 152.2 | 1.65 | 577.7 ± 226.8 | 1,185.6 ± 319.8 | 2.05 | 30.9 ± 16.7 | 13.8 ± 5.8 | 0.45 |
| Prediction fold‐difference c | 0.81 | 0.77 | 0.95 | 0.95 | 0.94 | 0.99 | 0.88 | 0.97 | 1.09 |
| Interaction with a single dose of tacrolimus (2 mg) based on the study by Xin et al. 18 , d | |||||||||
| PBPK prediction | 21.5 | 60.5 | 2.81 | 125.2 | 305.1 | 2.44 | 16.0 | 6.6 | 0.41 |
| Clinically‐reported values | 22.2 ± 10.6 | 66.4 ± 12.3 | 2.99 | 129.5 ± 65.1 | 274.8 ± 64.9 | 2.12 | 17.7 ± 12.6 | 6.5 ± 1.5 | 0.37 |
| Prediction fold‐difference | 0.97 | 0.91 | 0.94 | 0.97 | 1.11 | 1.15 | 0.90 | 1.02 | 1.11 |
AUC0–24h, area under the plasma concentration‐time curve during 24 h after dose; CL/F, apparent clearance; C max, peak concentration.
C max was reported as peak plasma and blood concentrations for midazolam and tacrolimus, respectively.
A cross‐over study in healthy Chinese males (n = 12) with age ranged from 21 to 23 years. A single oral dose of midazolam (15 mg) was given with and without S. sphenanthera extract containing schisandrin A (7.25 mg), schisantherin A (7.33 mg) and schisandrol B (0.12 mg) twice daily for 7 days prior to midazolam administration.
Prediction‐fold differences were expressed as the ratio of PBPK prediction to clinically‐observed values.
A cross‐over study in healthy Chinese males (n = 12) with age ranged from 22 to 25 years. A single oral dose of tacrolimus (2 mg) was given in the presence and absence of S. sphenanthera extract containing schisandrin A (7.25 mg), schisantherin A (7.33 mg) and schisandrol B (0.12 mg) twice daily for 13 days prior to tacrolimus administration.
2.3. PBPK prediction of interactions with imatinib and bosutinib
Input parameters for the PBPK model of imatinib and bosutinib 59 are summarised in Table S1. Development and verification of the PBPK model of imatinib in adults from European ancestry at both single‐dose administration and steady‐state have been detailed elsewhere. 60 The PBPK model was also verified against clinical pharmacokinetic data in people from Chinese ancestry (unpublished). Verification of the PBPK model of bosutinib in healthy people receiving the drug alone and with rifampicin, a strong CYP3A inducer is summarised in Figure S1 and Table S3.
PBPK simulations were carried out with n = 100 (40% female, aged 35–65 years) in European and Chinese populations. Imatinib was given at hypothetical single‐ and multiple‐dose of 400 mg, while bosutinib was administered at a single oral dose of 400 mg. The typical maintenance dosing regimens were assigned for each Schisandra lignan based on their contents in commercial product 61 and recommended dose of S. sphenanthera extract. Schisantherin A (9.4 mg), schisandrin A (6.8 mg) and schisandrol B (0.5 mg) were administered every 8 h for 14 days (steady‐state concentrations were assumed to be achieved within this timeframe). The simulated ratios of area under the plasma concentration‐time curve (AUC) of imatinib and bosutinib and ratio of trough concentrations (C min,ss) of imatinib with and without the lignans were used to predict the magnitude of interactions.
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 62 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18. 63 , 64
3. RESULTS
3.1. Inhibition of in vitro metabolism of imatinib and bosutinib by Schisandra lignans
Inhibitions of metabolite formation from imatinib and bosutinib by Schisandra lignans mainly followed a competitive inhibition model as depicted in Figure 1. For model selection, the proposed models (competitive, noncompetitive and mixed‐inhibitions) were fitted to the experimental data, and their goodness‐of‐fit based on AIC is summarised in Table S2. The Dixon plot was used to estimate k i of schisandrol B towards CYP3A4 metabolism pathway of imatinib owing to a better fit to experimental data compared to the models detailed in Equations 1–3 (Figure 1C). Schisantherin A exhibited the strongest inhibitory activity towards CYP3A4‐mediated metabolisms of imatinib and bosutinib (k i,u of 0.07 and 0.03 μmol L−1, respectively). Accordingly, the ratios of unbound therapeutic plasma concentration of schisantherin A (0.05–0.06 μmol L−1, Figure 2) to its k i,u values far exceeds 0.02, the cut‐off for screening of potential reversible drug interactions. 65 Unlike bosutinib, the CYP3A4‐mediated metabolism of imatinib was not reversibly inhibited by schisandrin A to a significant extent (k i,u of 2.7 vs 40.1 μmol L−1, respectively). Overall, k i,u values for bosutinib were substantially lower than those for imatinib. All the lignans exerted potent inhibitory activities towards the CYP2C8 enzyme, with k i,u of less than 0.5 μmol L−1 (Figure 1D–F). In vitro inhibition of the CYP2C8 metabolism pathway of imatinib were not enhanced by pre‐incubation with the lignans, implying a non‐time‐dependent mechanism. Schisandrin A is a substrate for CYP3A enzymes 11 and both schisantherin A 11 and schisandrol B 12 are also metabolised by these isoenzymes, albeit to a lesser extent than schisandrin A. As expected, concentration of schisandrin A, but neither that of schisantherin A nor schisandrol B was reduced during the incubations with HLM and rCYP3A4 (Figure 3). The associated k i,u values were corrected for this depletion over the course of incubation (Figure 1).
FIGURE 1.
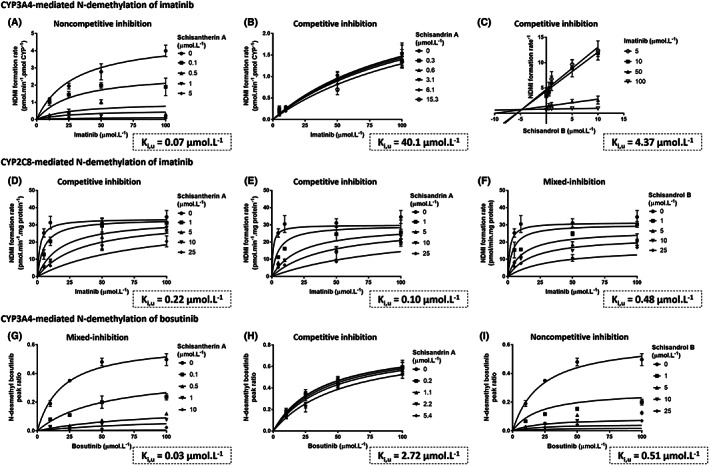
Determination of inhibitory constants (k i,u) of Schisandra lignans on CYP3A4‐ (A–C) and CYP2C8‐mediated N‐demethylation of imatinib (D–F) and CYP3A4‐mediated formation of N‐desmethyl bosutinib (G–I). Formation rates of N‐desmethyl bosutinib were expressed as peak ratios to the internal standard due to the absence of an authentic standard. Inhibition models that fitted best to each set of experimental data and their associated k i,u values are summarised. Experimental data are depicted as the mean values and standard deviations (n = 3)
FIGURE 2.
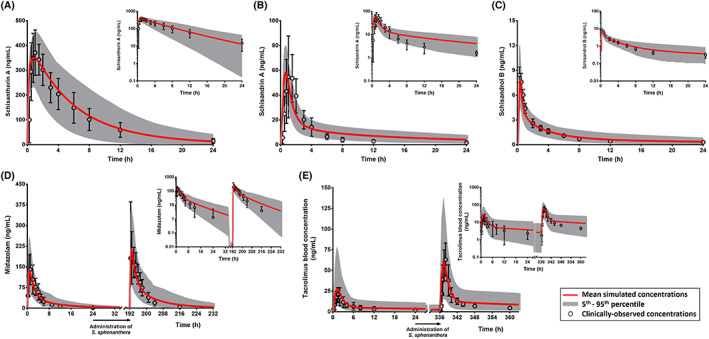
Comparison of PBPK simulations and clinically‐observed concentrations of schisantherin A (A), schisandrin A (B) and schisandrol B (C). PBPK model prediction of interactions between Schisandra lignans and midazolam (D) and tacrolimus (E). Clinically‐observed data (mean concentrations with associated standard deviations) are overlaid on simulated pharmacokinetic profiles shown in linear scales with the corresponding semi‐logarithmic plots as insets. All clinical pharmacokinetic data were from studies in healthy Chinese people, except for schisandrol B which were from people of Korean ancestry
FIGURE 3.
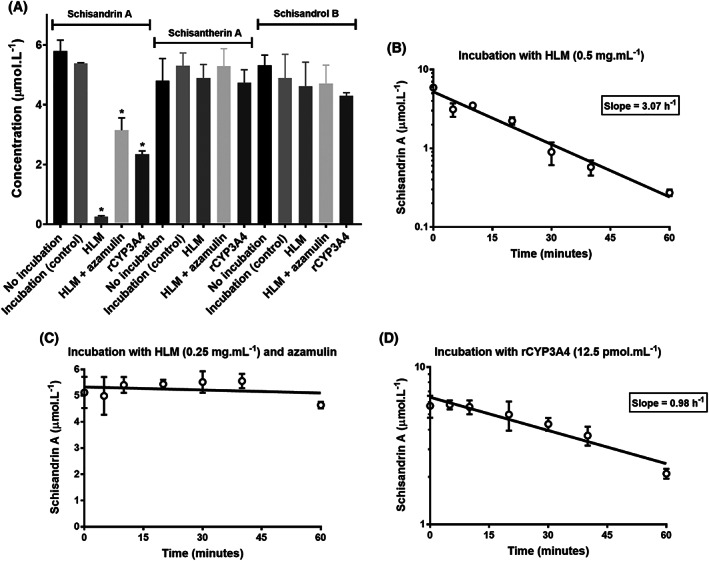
Evaluation of stability of Schisandra lignans during incubations with human liver microsomes (HLM, in the presence and absence of azamulin) and recombinant CYP3A4 (rCYP3A4) compared to control (A). Concentrations of schisandrin A declined mono‐exponentially over 60‐min incubations with HLM (B) and rCYP3A4 (D). Conversely, schisandrin A was relatively stable in HLM in the presence of azamulin (C). * means significantly different to control (P < .01; one‐way analysis of variance)
3.2. PBPK models for Schisandra lignans
PBPK predictions of pharmacokinetics of schisandrin A, schisantherin A and schisandrol B following a single‐dose administration of S. sphenanthera extract were similar to the clinically‐observed values (Figure 2, Table 2). However, substantial deviations from the 1.25‐fold criterion were observed for prediction of t max of schisandrin A and schisantherin A. There was good agreement between PBPK model predictions and clinical pharmacokinetic data of midazolam and tacrolimus with and without concomitant administration of S. sphenanthera extract. The clinically‐observed concentrations of both victim drugs all lay within the 5th to 95th percentiles of PBPK prediction (Figure 2). In line with that, the prediction differences for pharmacokinetic parameters of midazolam and tacrolimus and more importantly, ratios of C max and AUC0–24h in the presence and absence of interactions with Schisandra lignans were within 1.25‐fold (range: 0.94–1.15), except for C max of midazolam (Table 3).
3.3. PBPK prediction of interactions with imatinib and bosutinib
Schisandra lignans at their recommended dose were predicted to increase systemic exposure (AUC) of a single dose of imatinib in people from Chinese or European ancestry by 40% and 33%, respectively (Figure 4A, D). Conversely, interactions with imatinib at steady‐state were predicted to be of no clinical importance in either population (less than 5% increase in AUC0–24h,ss and C min,ss of imatinib). Systemic exposure of bosutinib on day 1 (AUC0–∞) represents the values at steady‐state (AUC0–24h,ss) since it lacks auto‐inhibitory activity towards CYP3A4 and thus the extent of interactions with Schisandra lignans were predicted to be similar between single‐ and multiple‐dose administrations of bosutinib. PBPK simulations enable prediction and interpretation of clinical consequences from different mechanism of interactions in isolation. PBPK model prediction of AUC ratios of bosutinib in European people assuming reversible and MBI (with induction) alone were 2.2 and 1.6, respectively (Figure 4F) which suggested a larger contribution of the former to overall interaction between Schisandra lignans and bosutinib.
FIGURE 4.
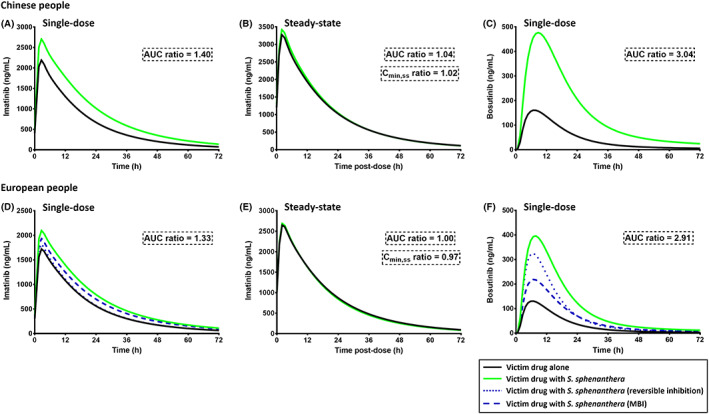
PBPK model prediction of the interactions between Schisandra sphenanthera extract and imatinib (400 mg) at single‐ (A, D) and multiple‐dosing regimens (B, E) and single 400 mg of bosutinib (C, F). PBPK simulations were performed in both European and Chinese populations and depicted as mean predicted concentrations in the presence and absence of co‐administration with S. sphenanthera. Schisantherin A (9.4 mg), schisandrin A (6.8 mg) and schisandrol B (0.5 mg) were given every 8 h as recommended in the product information. MBI, mechanism‐based inhibition
4. DISCUSSION
PBPK models of the three lignans in S. sphenanthera responsible for interactions with CYP3A substrates were successfully developed in this study. The PBPK models have an excellent performance in predicting the interactions with midazolam 16 and tacrolimus 18 with Schisandra lignans as observed in the corresponding clinical studies. The verified models were then implemented to predict (a priori) the extent of interactions with imatinib and bosutinib. It is noteworthy that S. sphenanthera is less commonly used in Western countries compared to Schisandra chinensis. 1 However, there is a paucity of clinical pharmacokinetic data for the latter. Despite a substantial difference in major constituents for both Schisandra species, 52 the trend and extent of interactions with midazolam, a probe drug for CYP3A enzymes at an equivalent dose of the respective extracts, are likely to be similar. 15
Although the PBPK models for schisandrin A and schisantherin A have been described before, 9 , 66 the evaluation of their capabilities in describing the clinically‐observed interactions with victim drugs is lacking. And, more importantly, PBPK predictions from these models were strictly limited to interactions with single isolated compounds and not as a combination of Schisandra lignans, the latter of which is more relevant in the clinical setting. Inhibition of tacrolimus metabolism by the combination of schisandrin A and schisantherin A was considerably overestimated using the previously reported models. 9 This limitation was inherently circumvented by the incorporation of CYP3A induction 15 to the models as described in this study (Table 1). Upregulation of pregnane X receptor (PXR), which underlies the induction of CYP3A enzymes by constituents in the S. sphenanthera extract, has been highlighted in sandwich‐cultured human hepatocytes and animal models, 2 , 15 , 67 although the predominant net outcome following a chronic exposure to the extract in clinical pharmacokinetic studies was CYP3A inhibition. 16 , 17 , 18 , 19 In this complex interaction scenario (simultaneous reversible and mechanism‐based inhibitions and induction of CYP3A enzymes by multiple modulators), a PBPK modelling approach is best positioned to provide a more mechanistic prediction of the extent of interaction. 48 A comprehensive characterisation of metabolic pathways for each of the perpetrator compounds in S. sphenanthera extract 11 , 12 also enables auto‐inhibition and potential interactions among Schisandra lignans to be accounted for in the PBPK models. However, a lack of clinical pharmacokinetic data following multiple‐dosing regimens of S. sphenanthera extract limited further verification of the models at steady state.
It is noteworthy that while clinical pharmacokinetic data for schisandrin A and schisantherin A were extracted from a study by Wei et al. 55 clinical data of schisandrol B were taken from another study. 56 This was necessary because concentration of schisandrol B in S. sphenanthera extract used in the former was questionably far lower than that normally found in commercially available extracts (0.01 55 vs 0.05–0.07% 56 , 61 ). This resulted in unrealistically high and low values of AUC and the corresponding apparent clearance (CL/F) of schisandrol B, respectively 55 and thus, clinical pharmacokinetic data from the study by Kim et al. 56 was used instead to verify the PBPK model of schisandrol B. Unfortunately, clinical data of both schisandrin A and schisantherin A were not reported in this study.
The interactions between Schisandra lignans and both midazolam and tacrolimus in Chinese people were predicted with excellent accuracy (Figure 2, Table 3). The simulation results implied that the PBPK model for tacrolimus, which was originally developed in European people, 57 could seamlessly be extrapolated to a Chinese population relying on the differences in body size, amount of CYP3A enzymes and frequency of CYP3A5 expression in the populations. 68 Conversely, the elimination components for the default PBPK model of midazolam in the Simcyp Simulator had to be adjusted due to a far lower CL/F of midazolam in the associated clinical interaction study, compared to previously reported values. 58 Despite this modification, the overall trend of interaction between Schisandra lignans and midazolam was well captured by the PBPK simulation, shown by similar ratios of AUC and C max compared to the clinically‐reported parameter values (Table 3). S. sphenanthera was predicted to affect midazolam given intravenously to a lesser extent than that by oral administration (AUC ratios of 1.4 vs 2.0, respectively; simulations not shown), suggesting a more extensive inhibition of intestinal CYP3A compared to the corresponding enzymes in hepatocytes.
PBPK simulations highlighted that the recommended oral dosing regimen of schisantherin A, schisandrin A and schisandrol B increased the systemic exposure of imatinib by 30 to 40% after a single dose of the drug. Conversely, it was predicted to have no effect on imatinib concentrations at steady state (Figure 4). This disparity was attributed to the mechanism‐based inactivation of CYP3A4 69 by a reactive metabolite of imatinib. 70 This leads to a lower sensitivity towards CYP3A inhibition at steady‐state, as demonstrated by a lack of clinically‐significant interaction between imatinib and ritonavir. 71 Although steady‐state CL/F of imatinib is relatively susceptible to CYP3A induction, e.g. by carbamazepine and phenytoin, 72 the predominant outcome from a chronic exposure to Schisandra lignans is a CYP3A inhibition and not induction. 15 , 19 Inhibition of CYP2C8 enzyme by Schisandra lignans was evaluated given the importance of this enzyme on imatinib metabolism. 73 This, however, was predicted to have a minimum impact on steady‐state concentrations of imatinib, possibly due to a low EH of imatinib.
To our knowledge, this is the first study to highlight a reversible CYP2C8 inhibition by Schisandra lignans which may have clinical implications for other CYP2C8 substrates. The extent of interactions between imatinib and S. sphenanthera extract in Chinese and European people were predicted to be similar. It is worth mentioning that the PBPK models of Schisandra lignans have not been verified in European people due to the lack of clinical pharmacokinetic data in this population.
Bosutinib is primarily metabolised by CYP3A4 enzyme with a contribution of this pathway (f m,CYP3A4) of approximately 0.9, estimated from a clinical interaction study with ketoconazole 74 based on the ratio of systemic exposure of bosutinib in the presence and absence of the CYP3A inhibitor. 75 This was in concordance with in vitro metabolic phenotyping which hinted at a significant role of CYP3A4 enzyme (but not CYP3A5) in bosutinib metabolism with minor contribution from flavin‐containing monooxygenase (FMO) and uridine 5′‐diphospho‐glucuronosyltransferase (UGT) enzymes. 33 , 76 Renal clearance accounts for up to 4% of total clearance of bosutinib. 77 N‐desmethyl bosutinib (M5) and oxydechlorinated bosutinib (M2) are the main circulating metabolites in plasma with systemic exposures (AUC) of approximately 25 and 20% of the parent drug, respectively. 76 , 77 Bosutinib did not inhibit its own CYP3A4‐mediated metabolism, 78 thus Schisandra lignans was expected to interact with bosutinib at single‐ (Figure 4) and multiple‐dosing regimens (results not shown) to a similar extent. Unlike imatinib, bosutinib has a high hepatic extraction ratio and its metabolism relies heavily on CYP3A4 enzyme. 33 Therefore, it was unsurprising that bosutinib was predicted to be more affected by Schisandra lignans compared to imatinib (Figure 4). PBPK simulation indicated a more important role of reversible compared to mechanism‐based inactivation of CYP3A enzymes to the overall interaction between Schisandra lignans and bosutinib. However, given a long turnover half‐life for CYP3A4 (approximately 1.5 days 79 ), the effect of MBI towards this enzyme may persist beyond cessation of S. sphenanthera consumption.
There appears to be a relationship between systemic exposure of bosutinib, represented by an AUC metric, and the occurrence of non‐haematologic adverse events. The target AUC0–24h for bosutinib is likely to be around 2.5–3 μg h−1 mL −1, above which the probability of developing diarrhoea and rash reaches a maximum without significant improvement in clinical responses. 80 Based on this apparent threshold for target AUC, the PBPK simulations suggested that daily dose of bosutinib should be reduced from 400 to 150–200 mg in the presence of interaction with Schisandra lignans at clinically‐relevant concentrations.
One of the limitations of the current work is that the MBI 81 and induction of CYP3A enzymes by schisandrol B 82 were not included in the PBPK model. The available data for a key related parameter, that is the ratio of K inact to K I of schisandrol B 81 is unrealistically higher than that of schisandrin A and schisantherin A 9 (336 vs 1 and 2 L μmol−1 h−1, respectively). Nevertheless, this exclusion was likely to have a minimum impact on the accuracy of PBPK predictions due to a substantially lower plasma concentration of schisandrol B compared to the other two lignans. Another limitation of the current study is the reliance on the assumption that Schisandra lignans are solely responsible for the interactions with CYP substrates. However, this is reasonable as other constituents of S. sphenanthera extract are either unlikely to interact with CYP3A4 and CYP2C8 enzymes (e.g. lipids or triacylglycerols and essential oils) or are present at very low concentrations, e.g. β‐sitosterol (0.5%). 83 Although β‐sitosterol has been reported as a CYP3A inhibitor (IC50 = 421 μmol L−1), 84 the steady‐state concentrations that may be attained from a standard clinical dose of S. sphenanthera extract is lower than its IC50 by five orders of magnitude. 85 A further limitation to this study is a lack of consideration for pharmacodynamic‐based interactions with both imatinib and bosutinib. A clinical study in Chinese liver transplant recipients suggested that S. sphenanthera extract may alleviate diarrhoea associated with treatment with tacrolimus. 4 Given diarrhoea is also common in patients receiving imatinib or bosutinib, 28 , 31 , 80 Schisandra lignans may help ameliorate this adverse event. Constituents of S. sphenanthera extract also exhibited anti‐cancer activity, although with evidence strictly limited to in vitro data in different cancer cell lines. 5 , 6 Potential clinical benefits for patients with CML remain to be investigated.
5. CONCLUSIONS
PBPK models for the constituents of S. sphenanthera extract responsible for interactions were successfully developed and verified using existing clinical pharmacokinetic and herb–drug interaction data with midazolam and tacrolimus. S. sphenanthera extract at a clinically‐relevant dosing regimen was predicted to have little to no effect on imatinib concentrations, but to increase bosutinib systemic exposure by up to three‐fold. Further clinical studies to confirm the extent of interaction with bosutinib and implications for the clinical outcomes are warranted.
COMPETING INTERESTS
There are no competing interests to declare. No funding was received for this work. J.A. is receiving a postgraduate scholarship from the Indonesia Endowment Fund for Education (LPDP), Ministry of Finance of the Republic of Indonesia. Certara UK Limited (Simcyp Division) is gratefully acknowledged for providing access to the Simcyp Simulator.
CONTRIBUTORS
J.A., A.V.B. and A.J.M. designed the research and contributed to the interpretation. J.A. performed the simulations and in vitro experiments, analysed the data and drafted the manuscript. A.V.B. and A.J.M. critically revised the manuscript.
Supporting information
Figure S1. Clinically‐observed concentrations of bosutinib alone (A) [8] and co‐administered with rifampicin (B) [9] overlaid with mean and 5th to 95th percentiles of the PBPK model predicted concentrations. The predictions are depicted in linear scale with the corresponding semi‐logarithmic plots as insets.
Table S1. Input parameters for PBPK models of the victim drugs used in the simulations
Table S2. Goodness‐of‐fit of the models used to describe in vitro inhibitions by Schisandra lignans
Table S3. Comparison of PBPK model predictions and clinically‐observed values for pharmacokinetic parameters of bosutinib in the presence and absence of rifampicin in healthy people
Adiwidjaja J, Boddy AV, McLachlan AJ. Potential for pharmacokinetic interactions between Schisandra sphenanthera and bosutinib, but not imatinib: in vitro metabolism study combined with a physiologically‐based pharmacokinetic modelling approach. Br J Clin Pharmacol. 2020;86:2080–2094. 10.1111/bcp.14303
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- 1. Szopa A, Barnas M, Ekiert H. Phytochemical studies and biological activity of three Chinese Schisandra species (Schisandra sphenanthera, Schisandra henryi and Schisandra rubriflora): current findings and future applications. Phytochem Rev. 2019;18:109‐128. [Google Scholar]
- 2. Zeng H, Li D, Qin X, et al. Hepatoprotective effects of Schisandra sphenanthera extract against lithocholic acid‐induced cholestasis in male mice are associated with activation of the pregnane x receptor pathway and promotion of liver regeneration. Drug Metab Dispos. 2016;44(3):337‐342. [DOI] [PubMed] [Google Scholar]
- 3. Chiu HF, Chen TY, Tzeng YT, Wang CK. Improvement of liver function in humans using a mixture of Schisandra fruit extract and sesamin. Phytother Res. 2013;27(3):368‐373. [DOI] [PubMed] [Google Scholar]
- 4. Jiang W, Wang X, Xu X, Kong L. Effect of Schisandra sphenanthera extract on the concentration of tacrolimus in the blood of liver transplant patients. Int J Clin Pharmacol Ther. 2010;48(3):224‐229. [DOI] [PubMed] [Google Scholar]
- 5. Liu HW, Yu XZ, Padula D, et al. Lignans from Schisandra sphenathera Rehd. et Wils. and semisynthetic schisantherin A analogues: absolute configuration, and their estrogenic and anti‐proliferative activity. Eur J Med Chem. 2013;59:265‐273. [DOI] [PubMed] [Google Scholar]
- 6. Casarin E, Dall'Acqua S, Smejkal K, Slapetova T, Innocenti G, Carrara M. Molecular mechanisms of antiproliferative effects induced by Schisandra‐derived dibenzocyclooctadiene lignans (+)‐deoxyschisandrin and (−)‐gomisin N in human tumour cell lines. Fitoterapia. 2014;98:241‐247. [DOI] [PubMed] [Google Scholar]
- 7. Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci. 2002;91(5):1358‐1370. [DOI] [PubMed] [Google Scholar]
- 8. Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918‐933. [DOI] [PubMed] [Google Scholar]
- 9. Zhang H, Bu F, Li L, et al. Prediction of drug–drug interaction between tacrolimus and principal ingredients of Wuzhi capsule in Chinese healthy volunteers using physiologically‐based pharmacokinetic modelling. Basic Clin Pharmacol Toxicol. 2018;122(3):331‐340. [DOI] [PubMed] [Google Scholar]
- 10. Yang J, Jamei M, Rowland‐Yeo K, Tucker GT, Rostami‐Hodjegan A. Prediction of intestinal first‐pass drug metabolism. Curr Drug Metab. 2007;8(7):676‐684. [DOI] [PubMed] [Google Scholar]
- 11. Wu R, Xiao Z, Zhang X, Liu F, Zhou W, Zhang Y. The cytochrome P450‐mediated metabolism alternation of four effective lignans from Schisandra chinensis in carbon tetrachloride‐intoxicated rats and patients with advanced hepatocellular carcinoma. Front Pharmacol. 2018;9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu JJ, Ge GB, He YQ, et al. Gomisin A is a novel isoform‐specific probe for the selective sensing of human cytochrome P450 3A4 in liver microsomes and living cells. AAPS J. 2016;18(1):134‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iwata H, Tezuka Y, Kadota S, Hiratsuka A, Watabe T. Identification and characterization of potent CYP3A4 inhibitors in Schisandra fruit extract. Drug Metab Dispos. 2004;32(12):1351‐1358. [DOI] [PubMed] [Google Scholar]
- 14. Yoo HH, Lee M, Lee MW, Lim SY, Shin J, Kim DH. Effects of Schisandra lignans on P‐glycoprotein‐mediated drug efflux in human intestinal Caco‐2. Planta Med. 2007;73(5):444‐450. [DOI] [PubMed] [Google Scholar]
- 15. Jackson JP, Freeman KM, Friley WW, et al. Prediction of clinically relevant herb–drug clearance interactions using sandwich‐cultured human hepatocytes: Schisandra spp. case study. Drug Metab Dispos. 2017;45(9):1019‐1026. [DOI] [PubMed] [Google Scholar]
- 16. Xin HW, Wu XC, Li Q, Yu AR, Xiong L. Effects of Schisandra sphenanthera extract on the pharmacokinetics of midazolam in healthy volunteers. Br J Clin Pharmacol. 2009;67(5):541‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li R, Guo W, Fu Z, Ding G, Wang Z, Fu H. A study about drug combination therapy of Schisandra sphenanthera extract and rapamycin in healthy subjects. Can J Physiol Pharmacol. 2012;90(7):941‐945. [DOI] [PubMed] [Google Scholar]
- 18. Xin HW, Wu XC, Li Q, et al. Effects of Schisandra sphenanthera extract on the pharmacokinetics of tacrolimus in healthy volunteers. Br J Clin Pharmacol. 2007;64(4):469‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Chen S, Qin X, et al. Wuzhi tablet (Schisandra sphenanthera extract) is a promising tacrolimus‐sparing agent for renal transplant recipients who are CYP3A5 expressers: a two‐phase prospective study. Drug Metab Dispos. 2017;45(11):1114‐1119. [DOI] [PubMed] [Google Scholar]
- 20. Sun Z, Ren M, Wu Q, Du X. Co‐administration of Wuzhi capsules and tacrolimus in patients with idiopathic membranous nephropathy: clinical efficacy and pharmacoeconomics. Int Urol Nephrol. 2014;46(10):1977‐1982. [DOI] [PubMed] [Google Scholar]
- 21. Rowland‐Yeo K, Jamei M, Rostami‐Hodjegan A. Predicting drug–drug interactions: application of physiologically based pharmacokinetic models under a systems biology approach. Expert Rev Clin Pharmacol. 2013;6:143‐157. [DOI] [PubMed] [Google Scholar]
- 22. Brantley SJ, Argikar AA, Lin YS, Nagar S, Paine MF. Herb–drug interactions: challenges and opportunities for improved predictions. Drug Metab Dispos. 2014;42(3):301‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brantley SJ, Gufford BT, Dua R, et al. Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb–drug interaction. CPT Pharmacometrics Syst Pharmacol. 2014;3:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fenneteau F, Poulin P, Nekka F. Physiologically based predictions of the impact of inhibition of intestinal and hepatic metabolism on human pharmacokinetics of CYP3A substrates. J Pharm Sci. 2010;99(1):486‐514. [DOI] [PubMed] [Google Scholar]
- 25. Gufford BT, Barr JT, Gonzalez‐Perez V, et al. Quantitative prediction and clinical evaluation of an unexplored herb–drug interaction mechanism in healthy volunteers. CPT Pharmacometrics Syst Pharmacol. 2015;4(12):701‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adiwidjaja J, Boddy AV, McLachlan AJ. Physiologically based pharmacokinetic modelling of hyperforin to predict drug interactions with St John's wort. Clin Pharmacokinet. 2019;58(7):911‐926. [DOI] [PubMed] [Google Scholar]
- 27. Hochhaus A, Larson RA, Guilhot F, et al. Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cortes JE, Apperley JF, DeAngelo DJ, et al. Management of adverse events associated with bosutinib treatment of chronic‐phase chronic myeloid leukemia: expert panel review. J Hematol Oncol. 2018;11(1):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cortes JE, Gambacorti‐Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol. 2018;36(3):231‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fleischer T, Chang TT, Chiang JH, Chang CM, Hsieh CY, Yen HR. Adjunctive Chinese herbal medicine therapy improves survival of patients with chronic myeloid leukemia: a nationwide population‐based cohort study. Cancer Med. 2016;5(4):640‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guilhot F, Hughes TP, Cortes J, et al. Plasma exposure of imatinib and its correlation with clinical response in the tyrosine kinase inhibitor optimization and selectivity trial. Haematologica. 2012;97(5):731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barratt DT, Somogyi AA. Role of pharmacogenetics in personalised imatinib dosing. Trans Cancer Res. 2017;6:S1541‐S1557. [Google Scholar]
- 33. Abbas R, Hsyu PH. Clinical pharmacokinetics and pharmacodynamics of bosutinib. Clin Pharmacokinet. 2016;55(10):1191‐1204. [DOI] [PubMed] [Google Scholar]
- 34. Parmentier Y, Pothier C, Delmas A, et al. Direct and quantitative evaluation of the human CYP3A4 contribution (fm) to drug clearance using the in vitro SILENSOMES model. Xenobiotica. 2017;47(7):562‐575. [DOI] [PubMed] [Google Scholar]
- 35. Filppula AM, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response. Drug Metab Dispos. 2013;41(1):50‐59. [DOI] [PubMed] [Google Scholar]
- 36. Kahma H, Filppula AM, Launiainen T, et al. Critical differences between enzyme sources in sensitivity to detect time‐dependent inactivation of CYP2C8. Drug Metab Dispos. 2019;47(4):436‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parmentier Y, Pothier C, Hewitt N, et al. Direct and quantitative evaluation of the major human CYP contribution (fmCYP) to drug clearance using the in vitro Silensomes model. Xenobiotica. 2019;49(1):22‐35. [DOI] [PubMed] [Google Scholar]
- 38. Yamakawa Y, Hamada A, Nakashima R, et al. Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit. 2011;33(2):244‐250. [DOI] [PubMed] [Google Scholar]
- 39. Gardner ER, Burger H, van Schaik RH, et al. Association of enzyme and transporter genotypes with the pharmacokinetics of imatinib. Clin Pharmacol Ther. 2006;80(2):192‐201. [DOI] [PubMed] [Google Scholar]
- 40. Greenblatt DJ, Venkatakrishnan K, Harmatz JS, Parent SJ, von Moltke LL. Sources of variability in ketoconazole inhibition of human cytochrome P450 3A in vitro. Xenobiotica. 2010;40(10):713‐720. [DOI] [PubMed] [Google Scholar]
- 41. Greenblatt DJ, Zhao Y, Venkatakrishnan K, et al. Mechanism of cytochrome P450‐3A inhibition by ketoconazole. J Pharm Pharmacol. 2011;63(2):214‐221. [DOI] [PubMed] [Google Scholar]
- 42. Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57(4):473‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burns K, Nair PC, Rowland A, Mackenzie PI, Knights KM, Miners JO. The nonspecific binding of tyrosine kinase inhibitors to human liver microsomes. Drug Metab Dispos. 2015;43(12):1934‐1937. [DOI] [PubMed] [Google Scholar]
- 44. Turner DB, Rostami‐Hodjegan A, Tucker GT, Rowland‐Yeo K. Prediction of non‐specific hepatic microsomal binding from readily available physicochemical properties. 2006; www.certara.com/wp-content/uploads/Resources/Posters/DavidISSX2006.pdf.
- 45. Berry LM, Zhao Z, Lin MH. Dynamic modeling of cytochrome P450 inhibition in vitro: impact of inhibitor depletion on IC50 shift. Drug Metab Dispos. 2013;41(7):1433‐1441. [DOI] [PubMed] [Google Scholar]
- 46. Gufford BT, Chen G, Lazarus P, Graf TN, Oberlies NH, Paine MF. Identification of diet‐derived constituents as potent inhibitors of intestinal glucuronidation. Drug Metab Dispos. 2014;42(10):1675‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jamei M, Marciniak S, Edwards D, et al. The Simcyp population based simulator: architecture, implementation, and quality assurance. In Silico Pharmacol. 2013;1(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rowland‐Yeo K, Jamei M, Yang J, Tucker GT, Rostami‐Hodjegan A. Physiologically based mechanistic modelling to predict complex drug–drug interactions involving simultaneous competitive and time‐dependent enzyme inhibition by parent compound and its metabolite in both liver and gut: the effect of diltiazem on the time‐course of exposure to triazolam. Eur J Pharm Sci. 2010;39(5):298‐309. [DOI] [PubMed] [Google Scholar]
- 49. Yau E, Olivares‐Morales A, Gertz M, et al. Global sensitivity analysis of the Rodgers and Rowland model for prediction of tissue:plasma partitioning coefficients: assessment of the key physiological and physicochemical factors that determine small‐molecule tissue distribution. AAPS J. 2020;22(2):41. [DOI] [PubMed] [Google Scholar]
- 50. Almond LM, Mukadam S, Gardner I, et al. Prediction of drug–drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44(6):821‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barter ZE, Bayliss MK, Beaune PH, et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab. 2007;8(1):33‐45. [DOI] [PubMed] [Google Scholar]
- 52. Lu Y, Chen DF. Analysis of Schisandra chinensis and Schisandra sphenanthera . J Chromatogr A. 2009;1216(11):1980‐1990. [DOI] [PubMed] [Google Scholar]
- 53. Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102(5):1016‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. The structural basis of pregnane x receptor binding promiscuity. Biochemistry. 2009;48(48):11572‐11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wei H, Miao H, Yun Y, et al. Validation of an LC‐MS/MS method for quantitative analysis of the 5 bioactive components of Wuzhi capsule in human plasma samples. Ther Drug Monit. 2014;36(6):781‐788. [DOI] [PubMed] [Google Scholar]
- 56. Kim SJ, Shin H, Cheon SM, et al. A sensitive UHPLC‐MS/MS method for the simultaneous quantification of three lignans in human plasma and its application to a pharmacokinetic study. J Sep Sci. 2017;40(17):3430‐3439. [DOI] [PubMed] [Google Scholar]
- 57. Emoto C, Johnson TN, Hahn D, et al. A theoretical physiologically‐based pharmacokinetic approach to ascertain covariates explaining the large interpatient variability in tacrolimus disposition. CPT Pharmacometrics Syst Pharmacol. 2019;8(5):273‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang HY, Chen X, Jiang J, Shi J, Hu P. Evaluating a physiologically based pharmacokinetic model for predicting the pharmacokinetics of midazolam in Chinese after oral administration. Acta Pharmacol Sin. 2016;37(2):276‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ono C, Hsyu PH, Abbas R, Loi CM, Yamazaki S. Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug–drug and drug–disease interactions. Drug Metab Dispos. 2017;45(4):390‐398. [DOI] [PubMed] [Google Scholar]
- 60. Adiwidjaja J, Boddy AV, McLachlan AJ. Implementation of a physiologically based pharmacokinetic modelling approach to guide optimal dosing regimens for imatinib and potential drug interactions in paediatrics. Front Pharmacol. 2020;10:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qin XL, Yu T, Li LJ, et al. Effect of long‐term co‐administration of Wuzhi tablet (Schisandra sphenanthera extract) and prednisone on the pharmacokinetics of tacrolimus. Phytomedicine. 2013;20(3‐4):375‐379. [DOI] [PubMed] [Google Scholar]
- 62. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alexander SPH, Cidlowski JA, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol. 2017;174(Suppl 1):S208‐S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Food and Drug Administration, Center for Drug Evaluation and Research . In vitro metabolism‐ and transporter‐mediated drug‐drug interaction studies: Guidance for industry. 2017.
- 66. Fan J, Chen L, Lu X, Li M, Zhu L. The pharmacokinetic prediction of cyclosporin A after coadministration with Wuzhi capsule. AAPS PharmSciTech. 2019;20:247. [DOI] [PubMed] [Google Scholar]
- 67. Fan S, Liu C, Jiang Y, et al. Lignans from Schisandra sphenanthera protect against lithocholic acid‐induced cholestasis by pregnane x receptor activation in mice. J Ethnopharmacol. 2019;245:112103. [DOI] [PubMed] [Google Scholar]
- 68. Barter ZE, Tucker GT, Rowland‐Yeo K. Differences in cytochrome P450‐mediated pharmacokinetics between Chinese and Caucasian populations predicted by mechanistic physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2013;52(12):1085‐1100. [DOI] [PubMed] [Google Scholar]
- 69. Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Potent mechanism‐based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787‐2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paludetto MN, Puisset F, Chatelut E, Arellano C. Identifying the reactive metabolites of tyrosine kinase inhibitors in a comprehensive approach: implications for drug–drug interactions and hepatotoxicity. Med Res Rev. 2019;39(6):2105‐2152. [DOI] [PubMed] [Google Scholar]
- 71. van Erp NP, Gelderblom H, Karlsson MO, et al. Influence of CYP3A4 inhibition on the steady‐state pharmacokinetics of imatinib. Clin Cancer Res. 2007;13(24):7394‐7400. [DOI] [PubMed] [Google Scholar]
- 72. Pursche S, Schleyer E, von Bonin M, et al. Influence of enzyme‐inducing antiepileptic drugs on trough level of imatinib in glioblastoma patients. Curr Clin Pharmacol. 2008;3(3):198‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nebot N, Crettol S, d'Esposito F, Tattam B, Hibbs DE, Murray M. Participation of CYP2C8 and CYP3A4 in the N‐demethylation of imatinib in human hepatic microsomes. Br J Pharmacol. 2010;161(5):1059‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abbas R, Hug BA, Leister C, Burns J, Sonnichsen D. Effect of ketoconazole on the pharmacokinetics of oral bosutinib in healthy subjects. J Clin Pharmacol. 2011;51(12):1721‐1727. [DOI] [PubMed] [Google Scholar]
- 75. Hua L, Chiang CW, Cong W, et al. The cancer drug fraction of metabolism database. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):511‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bosulif (bosutinib), Pharmacology review . US Food and Drug Administration, Center for Drug Evaluation and Research. 2012; https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203341Orig1s000PharmR.pdf.
- 77. Abbas‐Borhan R, Chaudhary I, Hug BA, et al. Mass balance, metabolic disposition, metabolite characterization, and pharmacokinetics of oral 14C‐labeled bosutinib in healthy subjects. 9th International ISSX Meeting 2010; https://issx.confex.com/issx/intl9/webprogram/Paper20292.html
- 78. Filppula AM, Neuvonen PJ, Backman JT. In vitro assessment of time‐dependent inhibitory effects on CYP2C8 and CYP3A activity by fourteen protein kinase inhibitors. Drug Metab Dispos. 2014;42(7):1202‐1209. [DOI] [PubMed] [Google Scholar]
- 79. Yang J, Liao M, Shou M, et al. Cytochrome P450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9(5):384‐394. [DOI] [PubMed] [Google Scholar]
- 80. Hsyu PH, Mould DR, Upton RN, Amantea M. Pharmacokinetic‐pharmacodynamic relationship of bosutinib in patients with chronic phase chronic myeloid leukemia. Cancer Chemother Pharmacol. 2013;71(1):209‐218. [DOI] [PubMed] [Google Scholar]
- 81. Zhai J, Zhang F, Gao S, et al. Time‐ and NADPH‐dependent inhibition on CYP3A by gomisin A and the pharmacokinetic interactions between gomisin A and cyclophosphamide in rats. Molecules. 2017;22(8):1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zeng H, Jiang Y, Chen P, et al. Schisandrol B protects against cholestatic liver injury through pregnane x receptors. Br J Pharmacol. 2017;174(8):672‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Huyke C, Engel K, Simon‐Haarhaus B, Quirin KW, Schempp CM. Composition and biological activity of different extracts from Schisandra sphenanthera and Schisandra chinensis . Planta Med. 2007;73(10):1116‐1126. [DOI] [PubMed] [Google Scholar]
- 84. Vijayakumar TM, Kumar RM, Agrawal A, Dubey GP, Ilango K. Comparative inhibitory potential of selected dietary bioactive polyphenols, phytosterols on CYP3A4 and CYP2D6 with fluorometric high‐throughput screening. J Food Sci Technol. 2015;52(7):4537‐4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Duchateau G, Cochrane B, Windebank S, et al. Absolute oral bioavailability and metabolic turnover of beta‐sitosterol in healthy subjects. Drug Metab Dispos. 2012;40(10):2026‐2030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinically‐observed concentrations of bosutinib alone (A) [8] and co‐administered with rifampicin (B) [9] overlaid with mean and 5th to 95th percentiles of the PBPK model predicted concentrations. The predictions are depicted in linear scale with the corresponding semi‐logarithmic plots as insets.
Table S1. Input parameters for PBPK models of the victim drugs used in the simulations
Table S2. Goodness‐of‐fit of the models used to describe in vitro inhibitions by Schisandra lignans
Table S3. Comparison of PBPK model predictions and clinically‐observed values for pharmacokinetic parameters of bosutinib in the presence and absence of rifampicin in healthy people
Data Availability Statement
Data available on request from the authors.