

Abstract
Earlier reports suggested that galantamine, a drug approved to treat mild-to-moderate Alzheimer’s disease (AD), and other centrally acting reversible acetylcholinesterase (AChE) inhibitors can serve as adjunct pretreatments against poisoning by organophosphorus compounds, including the nerve agent soman. The present study was designed to determine whether pretreatment with a clinically relevant oral dose of galantamine HBr mitigates the acute toxicity of 4.0×LD50 soman (15.08 µg/kg) in Macaca fascicularis posttreated intramuscularly with the conventional antidotes atropine (0.4 mg/kg), 2-pyridine aldoxime methyl chloride (30 mg/kg), and midazolam (0.32 mg/kg). The pharmacokinetic profile and maximal degree of blood AChE inhibition (∼25%–40%) revealed that the oral doses of 1.5 and 3.0 mg/kg galantamine HBr in these nonhuman primates (NHPs) translate to human-equivalent doses that are within the range used for AD treatment. Subsequent experiments demonstrated that 100% of NHPs pretreated with either dose of galantamine, challenged with soman, and posttreated with conventional antidotes survived 24 hours. By contrast, given the same posttreatments, 0% and 40% of the NHPs pretreated, respectively, with vehicle and pyridostigmine bromide (1.2 mg/kg, oral), a peripherally acting reversible AChE inhibitor approved as pretreatment for military personnel at risk of exposure to soman, survived 24 hours after the challenge. In addition, soman caused extensive neurodegeneration in the hippocampi of saline- or pyridostigmine-pretreated NHPs, but not in the hippocampi of galantamine-pretreated animals. To our knowledge, this is the first study to demonstrate the effectiveness of clinically relevant oral doses of galantamine to prevent the acute toxicity of supralethal doses of soman in NHPs.
SIGNIFICANCE STATEMENT
This is the first study to demonstrate that a clinically relevant oral dose of galantamine effectively prevents lethality and neuropathology induced by a supralethal dose of the nerve agent soman in Cynomolgus monkeys posttreated with conventional antidotes. These findings are of major significance for the continued development of galantamine as an adjunct pretreatment against nerve agent poisoning.
Introduction
Nerve agents, including soman, sarin, VX, and novichok, are highly toxic organophosphorus (OP) compounds that have been used with catastrophic consequences during the 1937–1945 Second Sino-Japanese War, the 1980s Iran-Iraq conflict, the 1995 Tokyo subway terrorist attack, the 2013 Syrian Civil War, and the recent 2017–2018 terrorist attacks against civilians in Syria, Malaysia, and the United Kingdom (reviewed in Romano and King, 2001, and Franca et al., 2019). The acute toxicity triggered by nerve agents and their chemically related OP insecticides can be fatal and results primarily from overstimulation of muscarinic and nicotinic receptors by acetylcholine (ACh) that accumulates in the peripheral and central nervous systems as OP compounds irreversibly inhibit acetylcholinesterase (AChE), the enzyme that catalyzes ACh hydrolysis. Thus, subjects poisoned with OP compounds develop a cholinergic crisis typically characterized by miosis, profuse secretions, diarrhea, bronchoconstriction, muscle fasciculation, tremors, and seizures (reviewed in Hurst et al., 2012). Death generally results from respiratory arrest due to central respiratory paralysis (Rickett et al., 1986).
The standard therapy used to treat OP intoxication includes atropine to block overactivation of muscarinic receptors, an oxime [generally 2-pyridine aldoxime methyl chloride (2-PAM), also known as pralidoxime] to reactivate AChE, and a benzodiazepine (midazolam or diazepam), as needed, to suppress seizures (reviewed in Newmark, 2019). However, this conventional antidotal therapy has some important limitations. Specifically, AChE inhibited by some OP compounds, particularly soman, becomes quickly refractory to reactivation by oximes (Marrs et al., 2006), and benzodiazepines progressively lose their anticonvulsant effectiveness if treatment is delayed after onset of OP-induced seizures (McDonough et al., 2010). Consequently, concerted efforts are underway for development of more effective posttreatments (Jett and Laney, 2019). Equally important, however, is the need for an effective pretreatment to protect military personnel and first responders at risk of exposure to high levels of OP compounds in a chemically contaminated environment. During the 1995 Tokyo sarin attacks, first responders were hospitalized with acute signs of toxicity due to their primary or secondary exposure to the nerve agent while attending the mass casualties (Nishiwaki et al., 2001). Yet, to date, personal protective equipment remains the only preventive countermeasure used by first responders attending victims of OP poisoning (Chai et al., 2017).
In 2006, galantamine, a centrally acting reversible AChE inhibitor approved for treatment of mild-to-moderate Alzheimer’s disease (AD), emerged as a potentially effective and safe pretreatment against OP poisoning. Used as a stand-alone pretreatment, intramuscularly delivered galantamine prevents the acute toxicity of a lethal dose (1.0×LD50) of nerve agents in rats, guinea pigs, and nonhuman primates (NHPs) (Albuquerque et al., 2006; Hilmas et al., 2009; Hamilton et al., 2017; Golime et al., 2018). Used as an adjuvant pretreatment and/or immediate posttreatment, intramuscularly delivered galantamine also increases the effectiveness of a high dose of atropine to mitigate the acute toxicity of supralethal doses of the OP insecticide paraoxon and the nerve agents soman, sarin, and VX in rodents (Albuquerque et al., 2006; Hilmas et al., 2009; Golime et al., 2018). The term supralethal dose refers to doses that are multiples of the median 1.0×LD50. The effectiveness of galantamine as a pretreatment against OP intoxication has been attributed, at least in part, to its ability to protect AChE in the peripheral and central nervous systems from OP-induced irreversible inhibition and to its neuroprotective properties (reviewed in Pereira et al., 2010).
To date, no study has focused on the more practical use of an oral galantamine pretreatment to prevent the acute toxicity of nerve agents. The present study was, therefore, designed to examine the effectiveness of an oral pretreatment with galantamine HBr in preventing the acute toxicity of a supralethal dose of soman in Cynomolgus monkeys (Macaca fascicularis) posttreated intramuscularly with human-equivalent doses of atropine, 2-PAM, and midazolam that are relevant for prehospital management of nerve agent poisoning. Results presented here reveal that oral pretreatment with clinically relevant doses of galantamine resulted in 100% survival of NHPs challenged with soman and posttreated with conventional antidotes. By contrast, given the same posttreatment, only 40% of the NHPs pretreated with pyridostigmine, a peripherally acting reversible AChE inhibitor specifically approved for use as pretreatment by military personnel at risk of exposure to soman (Aebersold, 2012), and none of those pretreated with saline survived the subsequent soman challenge. Pretreatment of NHPs with galantamine was also more effective than pretreatment with saline or pyridostigmine in preventing soman-induced neurodegeneration. These results lay the groundwork for advanced development of galantamine as an effective and safe adjunct pretreatment against nerve agent intoxication.
Materials and Methods
Subjects.
Twenty 4–6-year-old male Cynomolgus monkeys weighing 3.69–6.89 kg were obtained from a commercial vendor (AlphaGenesis, Yemassee, SC). The subjects were quarantined under the guidelines of the Centers for Disease Control for 3 months, during which time they were prescreened to exclude potential tuberculosis and simian retrovirus infections. Animals were individually housed in stainless steel cages with water available ad libitum and visual and auditory contact with each other. The cages, which were equipped with a squeeze-back mechanism, had 4.5 ft2 floor area and 32-in height. Behavioral enrichment was provided using foraging devices, challenge balls, and/or manipulative toys attached to the cage front. Animals received a commercial primate diet (Teklad 2050; Harlan Laboratories, Indianapolis, IN), with a daily supplement of fresh fruits and vegetables, in addition to supplemental food enrichment consisting of fruit and nut mix, popcorn, peanuts, and granola bars once or twice weekly. The colony was maintained at 20–22°C with a relative humidity of 50% (±15%) on a 12-hour light/dark cycle. Experiments were performed during the light phase. Subjects were exclusively used in this study and received no treatments other than those described below. All experimental procedures were approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee and were conducted in full accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Pharmaceutical-Grade and Nonpharmaceutical Chemicals.
A stock solution of 1.8–2.0 mg/ml soman (methylphosphonofluoridic acid 1,2,2-trimethylpropyl ester) in saline was obtained through a Chemical Agent Provisioning Agreement between the University of Maryland, Baltimore, and the US Army Materiel Command. The stock solution was stored at −80°C, handled, and disposed of according to standard operating procedures and following the regulations set forth by the US Army Materiel Command. On the day of the experiments, a vial containing an aliquot (0.2–0.5 ml) of the dilute stock solution of soman was thawed in ice and further diluted with cold sterile saline to appropriate concentrations. The diluted solution was kept on ice and used within 30 minutes after the stock solution was removed from storage. Any remaining diluted solution of soman and materials contaminated with the nerve agent were decontaminated with 10% sodium hydroxide before disposal.
Galantamine HBr and pyridostigmine bromide were purchased from Sanochemia Pharmazeutika (Vienna, Austria) and Sigma-Aldrich (St. Louis, MO), respectively. Pharmaceutical-grade solutions of saline (0.9% NaCl, injectable), atropine sulfate (15 mg/ml, injectable), and midazolam hydrochloride (5 mg/ml, injectable) were purchased from VetOne/MWI Veterinary Supply Co. (Boise, ID), Sparhawk Laboratories, Inc. (Shawnee Mission, KS), and Hospira, Inc. (Lake Forest, IL), respectively. Pharmaceutical-grade 2-PAM chloride for injection (Protopam, lyophilized 1 g) was purchased from Baxter Healthcare Corp. (Deerfield, IL). Injectable solutions of ketamine (Zetamine, 100 mg/ml ketamine HCl) and Euthasol were also purchased from VetOne/MWI Veterinary Supply Co.
Oral Treatments with Test and Control Articles.
Daily doses of 8–24 mg galantamine are currently approved for AD treatment. In the absence of knowledge of the pharmacokinetic profile of galantamine in Cynomolgus monkeys, allometric scaling (Reagan-Shaw et al., 2008) was used for a first approximation of the doses to be tested in these NHPs. Using the allometric factor 3.1 derived from the body surface area of humans and monkeys, human and nonhuman primate doses can be interconverted according to the relationship shown in eq. 1.
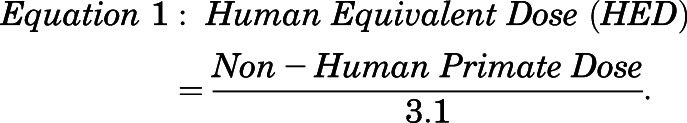 |
(1) |
Thus, the human doses of 8 (or 0.13 mg/kg) and 24 mg (or 0.4 mg/kg) galantamine translate to approximately 0.41 and 1.25 mg/kg galantamine base (or 0.52 and 1.6 mg/kg galantamine HBr), respectively, in monkeys. The oral doses of galantamine HBr selected for the pharmacokinetic experiments in NHPs were 0.5, 1.5, and 3.0 mg/kg, which include and exceed the allometrically converted doses. Subsequently, the results of the pharmacokinetic experiments performed in the present study guided the selection of the doses to be tested in the pharmacodynamic experiments designed to assess the effectiveness of galantamine HBr in preventing the acute toxicity of soman.
The oral dose of pyridostigmine bromide used here (1.2 mg/kg) was adapted from the study of Hayward et al. (1990). In that study, rhesus macaques were treated orally four times with 1.2 mg/kg pyridostigmine bromide every 8 hours, exposed to 5.0×LD50 soman ∼4 to 5 hours after the last dose of pyridostigmine, and then treated intramuscularly with 0.4 mg/kg atropine, 25.71 mg/kg 2-PAM, and 1 mg/kg midazolam or diazepam. At the time of the exposure to soman, pyridostigmine had generated ∼40% reversible inhibition of red blood cell AChE (Hayward et al., 1990). This degree of enzyme inhibition, which has been shown to significantly increase survival of primates that are subsequently exposed to soman (see Dunn and Sidell, 1989, and references therein), was likely to represent the steady-state degree of enzyme inhibition after the intermittent dose regimen. However, in the present study, animals were treated with a single oral dose of 1.2 mg/kg pyridostigmine and exposed 1 hour later to soman. It has been reported that a single oral treatment of nonhuman primates with 1.14 mg/kg pyridostigmine bromide produced ∼55% maximal inhibition of red blood cell AChE at approximately 1 hour after dosing (Joiner and Kluwe, 1988). Thus, in the present study, at the time the nonhuman primates were exposed to soman, the degree of reversible AChE inhibition produced by the pretreatment with pyridostigmine was likely to be within optimal levels. It is also noteworthy that, based on allometric conversion using eq. 1, the oral dose of pyridostigmine bromide (30 mg or 0.5 mg/kg) approved by the US Food and Drug Administration (FDA) as pretreatment for military personnel at risk of exposure to soman is equivalent to 1.5 mg/kg in nonhuman primates. In addition, based on its pharmacokinetic profile in both species, the FDA-approved oral dose of pyridostigmine is equivalent to 1.2 mg/kg in nonhuman primates.
Solutions of galantamine HBr (20 mg/ml) and pyridostigmine bromide (20 mg/ml) were prepared by quantitatively dissolving the salts in an appropriate volume of a pharmaceutical-grade solution of saline. The concentrations of both solutions were verified using the compendial methods described in the US Pharmacopeia. The volume of the solution needed to deliver a given dose of a test article (galantamine HBr or pyridostigmine bromide) or the control article (saline) was determined based on the animal’s body weight on the day of the experiment. A plain oatmeal cookie (Homekist) divided into four quarters that were preimpregnated with the adequate volume of a test solution was used to deliver the oral treatment to the animals in the morning after an overnight fast. Each animal was closely monitored to ascertain that it consumed the entire cookie. In case cookie crumbs were wasted by a monkey during treatment, they were collected on a mesh screen and weighed so that any wasted volume could be estimated and replaced with a new piece of cookie. Upon eating the required treat, animals received their normal diet.
Pharmacokinetic Experiment: Treatment and Blood Draws.
Animals were randomly assigned to the different treatment groups, which consisted of a single oral dose of 0.5, 1.5, or 3.0 mg/kg galantamine HBr (n = 5, 10, and 5, respectively). Twenty-four hours prior to a treatment, each animal was transferred to a stainless steel squeeze-back cage (61 cm width × 71 cm depth × 86 cm height) and transported to the treatment room, which was distant from the homeroom. After restraint, animals were sedated with 5–10 mg/kg ketamine (i.m.) and subsequently laid down on a procedure table. Their body temperature and respiratory rate were continuously monitored during sedation. One of the thighs was shaved, and 1 ml of blood was collected by venipuncture from the saphenous vein into heparinized tubes. After the blood draw, animals were transferred to the squeeze-back cage, where they were allowed to recover prior to being transferred to their home cage.
On the next day, animals were again transported to the treatment room, where they were treated, as described in the previous section, with an oral dose of galantamine HBr. Ten minutes after their oral treatment with galantamine, animals were sedated with ketamine (5–10 mg/kg, i.m.) and maintained under sedation, with a repeated ketamine dose, for the first hour of the pharmacokinetic experiment, enabling blood to be sampled by venipuncture at 20, 40, and 60 minutes after the treatment. Animals were allowed to recover from sedation and were then resedated with the same ketamine dose at each subsequent time point of blood sampling (2, 5, and 24 hours posttreatment). At the end of the pharmacokinetic study, Cynomolgus monkeys were housed for 1 month prior to the beginning of the pharmacodynamic experiment.
Measurements of Galantamine Concentrations in Plasma of Treated Cynomolgus Monkeys and Pharmacokinetic Analysis.
Blood samples drawn from all animals before and after a given treatment were separated into 0.5-ml aliquots that were transferred to each of two heparinized vials. One of the vials was centrifuged to separate plasma, which was then stored as 200-µl aliquots. Plasma from all animals treated with 0.5 and 3.0 mg/kg galantamine and from five animals randomly selected from the 10 animals treated with 1.5 mg/kg galantamine was used for determination of plasma concentrations of galantamine by liquid chromatography in tandem with mass spectrometry following the methods described by Steiner et al. (2012) and Suresh et al. (2014). In short, 200 µl of plasma samples were spiked and thoroughly mixed with 50 µl of a solution of the internal standard diphenhydramine HCl (4 µM; purity ≥99% by high-performance liquid chromatography; Sigma-Aldrich). After the addition of 0.8 ml of dichloromethane, the mixture was vortexed for 20 seconds and subsequently centrifuged at 16,000g for 2 minutes. After two more extractions of the pellets with dichloromethane, the organic phases were pooled and then evaporated to dryness under nitrogen at 50°C. The remaining residue was reconstituted with 200 µl of the liquid chromatography mobile phase consisting of mobile phase A (0.1% formic acid in ultraperformance liquid chromatography–grade water) and mobile phase B 90.1% formic acid in acetonitrile) at the ratio of 95:5. Then, 20 μl of the reconstituted material were used for a liquid chromatography in tandem with mass spectrometry analysis performed on Waters TQ-XS triple quadrupole mass spectrometer with ACQUITY UPLC platform (Thermo Scientific, San Jose, CA). The liquid chromatography separation was performed on an ACQUITY-Xselect CSH C18 130 Å column (4.6 × 100 mm, 1.7 μm) (Waters Corp., Milford, MA) connected to an ACQUITY CSH C18 130 Å VanGuard pre-column (2.1 × 5 mm; 1.7 µm) (Waters Corp.). Analytes were separated under isocratic conditions using mobile phases A and B at the fixed ratio of 95:5. The flow rate was set at 0.8 ml/min. Detection was performed in the positive-ion mode, and positive electrospray ionization data were acquired using multiple reaction monitoring of the precursor to product ion (M+H)+ transitions of 288-to-213 m/z and 256-to-167 m/z for galantamine and diphenhydramine, respectively. The assay was linear between 0.8 ng/ml and 1.6 µg/ml galantamine, and the limit of quantification was 5.4 ng/ml.
The area under the plasma concentration-time curve from time 0 to infinity (AUC) and half-life (t1/2) of galantamine were estimated through a noncompartmental analysis of the data using the linear-log trapezoidal fit in the software Kinetica 5.0 (Thermo Scientific Kinetica, Philadelphia, PA). Values of maximal plasma concentration of galantamine (Cmax) and time to reach Cmax (i.e., Tmax) were derived directly from the plasma concentrations measured at the various time points from each animal.
Analytical Method for Determination of AChE Activity.
AChE activity was measured in whole blood using a modification of the radiometric assay described by Johnson and Russell (1975). Briefly, 90 µl of whole blood samples pretreated with the butyrylcholinesterase inhibitor tetraisopropyl pyrophosphoramide (0.1 mM final concentration) were incubated at room temperature for 2 minutes with 10 μl of 0.1 M ACh chloride and [acetyl-3H]ACh iodide (20 μCi/ml, which produced approximately 100,000 cpm when totally hydrolyzed by eel AChE). At the end of the incubation time, the reaction was stopped by the addition of an aqueous solution (100 μl) consisting of chloroacetic acid (0.50 M), sodium chloride (1 M), and sodium hydroxide (0.25 M). After vortexing the mixture, samples were clarified by centrifugation, and the clarified samples (140 μl) were transferred to a scintillation vial containing 3.86 ml of the fluor cocktail, which consisted of 90% (v/v) toluene, 10% (v/v) 3-metyl-1-butanol, 0.03% (w/v) 1,4-bis(5-phenyl-2-oxazolyl), and 0.05% (w/v) 2,5-diphenyloxazole. The mix was vortexed for 60 seconds, and the amount of tritiated acetate in the organic phase was measured by liquid scintillation counting for 2 minutes (Tri-Carb 2900TR; Perkin Elmer, Waltham, MA ). Each sample was assayed in triplicate, and counts were corrected for background by subtraction of counts obtained using whole blood samples devoid of cholinesterase activity and treated in a similar fashion. Background-corrected counts obtained from the blood sample drawn from each animal prior to its treatment with galantamine were taken as 100% and used to normalize the background-corrected counts obtained from blood samples drawn at different times after the treatment of that animal. Percentage of inhibition of enzyme activity at each time point was then determined using the expression: % inhibition = 100% − normalized AChE activity .
The concentration-response relationship for galantamine to inhibit AChE in whole blood of Cynomolgus monkey was also analyzed in vitro using heparinized blood purchased from a commercial vendor (Alpha Genesis Inc., Yemassee, SC). Blood was incubated for 30 minutes with a known concentration of galantamine (ranging from 0.01 to 10 µM) at room temperature and subsequently treated as above for measurement of AChE activity. The assay was performed in triplicate, and the resulting concentration-response relationships were fitted using the Hill equation in the SigmaPlot software version 12 (Systat Software, Inc., San Jose, CA):
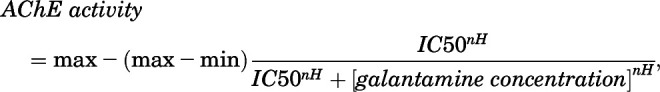 |
where min and max are the minimal and maximal enzyme activities, respectively, IC50 is the concentration of galantamine that inhibits AChE by 50%, and nH is the Hill coefficient.
Pharmacodynamic Experiment: Treatments, Soman Challenge, Functional Observational Battery, and Survival Data Analysis.
Animals were randomly assigned to one of four experimental oral pretreatments: saline (0.15 ml/kg; n = 4), 1.5 mg/kg galantamine (n = 5), 3.0 mg/kg galantamine (n = 6), or 1.2 mg/kg pyridostigmine (n = 5). Table 1 lists the animals assigned to each group and their respective body weights. On each experimental day, animals were transferred to a stainless steel squeeze-back cage and transported to the treatment room, where they were orally treated, as described above, with a test or control article. One hour after the oral treatment, each animal was constrained with the help of the squeeze mechanism, and soman (4.0×LD50 or 15.08 µg/kg; Adams, 1990) was administered intramuscularly in the quadriceps muscle. At 1 minute after the nerve agent challenge, animals were posttreated intramuscularly with atropine sulfate (0.4 mg/kg), 2-PAM chloride (30 mg/kg), and midazolam (0.32 mg/kg) in rapid succession. The interval between the pretreatments and the soman challenge was fixed at 1 hour because: 1) plasma concentrations of galantamine and whole blood AChE inhibition peak between 1 and 3 hours after treatment of Cynomolgus monkeys with galantamine HBr (see Results), 2) plasma concentrations of pyridostigmine and blood AChE inhibition reach peak at approximately 1 hour after an oral treatment with pyridostigmine bromide (Joiner and Kluwe, 1988), and 3) it was previously reported that a single oral dose of pyridostigmine bromide delivered between 1 and 8 hour prior to a subsequent exposure to soman effectively prevents lethality of NHPs that are repeatedly posttreated with different antidotal therapies (von Bredow et al., 1991).
TABLE 1.
Body weights and toxicity scores presented by soman-challenged Macaca fascicularis subjected to different pretreatments and posttreated with atropine, 2-PAM, and midazolam
Doses (i.m.): 15.08 µg/kg soman; 0.4 mg/kg atropine sulfate; 30 mg/kg 2-PAM chloride; and 0.32 mg/kg midazolam. Clinical signs that characterize each toxicity score are described in the text of the Materials and Methods section. In short, a score of 0 was assigned to subjects presenting normal behavior. Scores of 1 to 2, 3, and 4 to 5 were assigned to subjects presenting mild, moderate, and severe signs of toxicity, respectively, as described in the text. Subjects were humanely euthanized (euth) as soon as they developed life-threatening signs of toxicity (toxicity score 5).
| Subject ID | Body weight (kg) | Time of FOB observations after soman challenge | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 15 min | 30 min | 1 h | 2 h | 3 h | 4 h | 6 h | 8 h | 24 h | ||
| Oral pretreatment: saline | ||||||||||
| 12-092 | 4.38 | 4 | 4 | 4 | 4 | 4 | 5 (euth) | |||
| 24-098 | 4.63 | 4 | 4 | 4 | 5 (euth) | |||||
| 20-106 | 4.13 | 4 | 4 | 5 (euth) | ||||||
| 21-104 | 4.75 | 4 | 4 | 4 | 4 | 5 (euth) | ||||
| Oral pretreatment: 1.2 mg/kg pyridostigmine bromide | ||||||||||
| 23-099 | 6.15 | 4 | 4 | 4 | 4 | 3 | 3 | 3 | 3 | 2 |
| 15-102 | 5.71 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 5 (euth) | |
| 16-103 | 4.03 | 4 | 4 | 4 | 4 | 4 | 4 | 5 (euth) | ||
| 02-109 | 3.60 | 4 | 4 | 4 | 4 | 3 | 2 | 2 | 2 | 0 |
| 03-110 | 3.77 | 4 | 4 | 4 | 4 | 5 (euth) | ||||
| Oral pretreatment: 1.5 mg/kg galantamine HBr | ||||||||||
| 22-105 | 4.72 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 0 |
| 05-112 | 3.89 | 3 | 3 | 3 | 2 | 2 | 1 | 1 | 1 | 0 |
| 09-107 | 4.47 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 13-101 | 7.40 | 3 | 3 | 3 | 2 | 1 | 0 | 0 | 0 | 0 |
| 06-113 | 6.09 | 3 | 2 | 2 | 1 | 1 | 0 | 0 | 0 | 0 |
| Oral pretreatment: 3.0 mg/kg galantamine HBr | ||||||||||
| 08-095 | 5.34 | 0 | 1 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 07-097 | 4.44 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 17-100 | 4.45 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 11-093 | 4.52 | 4 | 4 | 4 | 3 | 4 | 4 | 4 | 3 | 3 |
| 10-108 | 5.10 | 3 | 3 | 4 | 4 | 4 | 3 | 3 | 3 | 2 |
| 01-096 | 4.34 | 3 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 |
Based on allometric conversion using eq. 1, the selected dose of atropine (0.4 mg/kg, i.m.) approximates that delivered to adults by three autoinjectors (6 mg or 0.1 mg/kg), as approved for prehospital management of OP poisoning. Likewise, the selected dose of 2-PAM (30 mg/kg) is equivalent to that delivered by one autoinjector (600 mg or 10 mg/kg) to OP-intoxicated humans. Previous studies of the effectiveness of adjuvant therapies in soman-exposed nonhuman primates have used similar doses of atropine and 2-PAM (Baze, 1993). The dose of midazolam (0.32 mg/kg) was selected because 1) it is halfway in the log scale between 0.1 and 1.0 mg/kg, which are doses that showed anticonvulsant activity in soman (5.0×LD50)–exposed Cynomolgus monkeys pretreated orally with pyridostigmine and posttreated with atropine (0.5 mg/kg)-plus-2-PAM (25.7 mg/kg) (Baze, 1993); and 2) it was shown to mitigate seizures induced by 2.0×LD50 soman in rhesus monkeys pretreated with pyridostigmine (0.024 mg/kg, i.m.) and posttreated with atropine (0.5 mg/kg)-plus-2-PAM (25.7 mg/kg) (McDonough, 2002).
After the injections, the squeeze mechanism was partially released, and animals were closely monitored. A functional observational battery (FOB) described earlier (Gauvin and Baird, 2008) was recorded at fixed time intervals before pretreatment, between pretreatment and the soman challenge, and after antidotal treatment. The FOB was recorded by two assistants, who worked independently, were unaware of the pretreatment given to the animals, and were involved neither in animal restraint nor in the injection procedure. The FOB consisted of scoring the animal’s posture, arousal, vocalization, stereotypical behavior, facial movements, visual and auditory responses, gross motor movements, ocular changes (nystagmus, pupil dilation), leg muscle fasciculations, and eye lid paralysis, in addition to the typical clinical signs of a cholinergic crisis, including salivation, lacrimation, bronchial secretions, increased urination, diarrhea, tremors, convulsions, and breathing distress.
The severity of the intoxication induced by 15.08 µg/kg soman in NHPs posttreated with atropine, 2-PAM, and midazolam was evaluated using a qualitative scoring system based on the clinical neurologic signs recorded in the FOB. The criteria used to assign toxicity scores were as follows. Animals presenting normal cage behavior were assigned a score of 0. A toxicity score of 1 was defined by a period of hypoactivity and/or low responsiveness to an external stimulus. A score of 2 was defined by hypoactivity in cage, low responsiveness to external stimuli, nystagmus, and clonic tremors or short-lasting (<10 minutes) mild whole-body tonic-clonic convulsions that did not recur within 24 hours. A score of 3 was defined by clonic tremors or mild tonic-clonic convulsions that also lasted <10 minutes but recurred two to three times within 1 to 2 hours after onset and were accompanied by muscle fasciculations, occasional nystagmus, and loss of posture. If signs of toxicity did not worsen, the severity of toxicity declined within 1 to 2 or 3 to 4 hours after reaching scores of 1 or 2 to 3, respectively. A score of 4 was defined by mixed periods of long lasting (10–20 minutes) whole-body motor convulsions and hypoactivity throughout the day, muscle fasciculations, and loss of posture, with some animals showing short-lasting periods of a comatose state after bouts of very strong convulsions. Animals could still recover from a toxicity score 4, as evidenced by periods of normal posture and normal responsiveness that were evident starting between 4 and 8 hours after the treatments. A score of 5, on the other hand, was defined by life-threatening signs of intoxication from which the animals were unlikely to recover. These signs included unremitting convulsions lasting >20 minutes, lack of response to external stimuli, severe muscle fasciculations, loss of posture, severe respiratory distress characterized by gasping, and low body temperature by palpation. Animals were euthanized humanely by terminal transcardial perfusion, as described below, at 24 hours after the soman challenge or at the time they presented life-threatening signs of toxicity.
Hippocampal Histopathology.
At 24 hours after the soman challenge or at the time life-threatening signs of toxicity became evident, animals were sedated with ketamine (10 mg/kg, i.m.), transferred to the procedure table, and given an intravenous injection of heparin to prevent blood coagulation. After the heparin injection, animals were euthanized with an intravenous overdose of Euthasol and transcardially perfused with PBS (pH = 7.4) followed by 4% paraformaldehyde in PBS. Their brains were then harvested, postfixed overnight at 4°C with freshly prepared 4% paraformaldehyde in PBS, cryoprotected with a solution of 30% sucrose in PBS, and frozen with crushed dry ice before storage at −80°C.
Using a Leica CM 3000 cryostat (Leica Biosystems, Buffalo Grove, IL), frozen brains were cut, according to the technique described by Pardo et al. (2012), in 30-µm thick sections [for Fluoro-Jade B (FJB) staining] or 50-µm sections (for NeuN immunohistochemistry) that were anatomically matched and contained the hippocampus. The sections were then transferred to wells containing PBS. This was followed by three washes in 70% ethanol, 50% ethanol, and water, each for 1 minute.
For FJB staining, two adjacent 30-µm sections interspaced by 120 µm were processed per animal. After being washed in PBS and water, slices were immersed in a potassium permanganate solution (0.06%) for 15 minutes, on a rotating platform, rinsed in water for 1 minute, and finally transferred to wells containing the FJB (0.0004%; Chemicon, Temecula, CA) staining solution prepared as described in Mamczarz et al. (2011). Sections were incubated with FJB for 30 minutes under gentle shaking in the dark. After being rinsed with water, slices were transferred to a microscope slide, dried on a slide warmer (50°C), immersed in xylene, and cover slipped with Cytoseal mounting media (Richard Allan Scientific, Kalamazoo, MI). Sections were then examined at 10× magnification under a Nikon Eclipse 80i upright microscope using a fluorescein isothiocyanate filter set (Excitation: 465 nm; Emission: 520 nm). Photomicrographs were taken using a Ds-FiZ microscope camera controlled by the NIS-Elements BR 3.0 SP4 software (Nikon Instruments Inc., Melville, NY). Images were captured using the same parameters of magnification, area, exposure, and gain and were used to qualitatively identify the presence of FJB-positive cells in the CA1 field of the hippocampus.
For immunohistochemistry, ethanol-permeabilized sections were anatomically matched across all treatment groups and processed in a single batch using an antibody that recognizes the neuron-specific marker NeuN. Briefly, free-floating sections were washed with PBS, blocked in 2% horse serum with 0.2% Triton-X, and subsequently incubated for 48 hours at 4°C with the anti-NeuN antibody (1:1000 dilution; catalog number MAB377; EDM Millipore, Temecula, CA). Then, sections were washed in PBS and incubated for 1 hour at room temperature with a biotinylated donkey anti-mouse IgG (1:1000 dilution; catalog number 715-065-150; Jackson Immuno Research Laboratory, West Grove, PA). After another wash in PBS, the sections were incubated at room temperature for 1 hour in peroxidase-labeled streptavidin (1:1000 dilution; Jackson). Subsequently, NeuN immunoreactivity was developed with 3′-diaminobenzidine tetrahydrochloride (Polysciences Inc., Warrington, PA). The free-floating sections were mounted on gelatin-coated slides, dried overnight at room temperature, and cover slipped on the next day.
Counts of NeuN-immunoreactive cells were obtained from the CA1 field of the left ventral hippocampus in every fifth section for a total of 9–12 sections per subject. Specifically, sections were examined under a Leica DM-6 B microscope (Leica), and images were captured at 10× magnification using the Leica Application Suite X (LAS-X) (Leica). Magnification, area, exposure, and gain parameters were kept constant for all sections. A square region of interest (ROI) of 1000 × 1000 pixels (0.7 mm2) was positioned on the CA1 field of the hippocampus as shown in Figure 7, and unbiased counts of NeuN-immunoreactive cells within the ROI were obtained using the cell count module of the ImageJ freeware (version 1.52; National Institutes of Health, Bethesda, MD). A single ROI was counted per slice. Neuronal density was estimated by dividing the mean number of NeuN-immunopositive cells counted in all slices from a given animal by the ROI area.
Fig. 7.

NeuN immunoreactivity in the CA1 field of the hippocampus of soman-challenged Cynomolgus that were pretreated with galantamine, pyridostigmine, or saline and posttreated with conventional antidotal therapy. (A–D) Representative photomicrographs of NeuN-immunopositive cells in the CA1 field of the hippocampus of soman-challenged NHPs pretreated orally with saline (A), 1.2 mg/kg pyridostigmine bromide (B), 1.5 mg/kg galantamine HBr (C), or 3.0 mg/kg galantamine HBr (D). The ROIs enclosed within the red squares in (A–D) are magnified in the respective (A’–D’). (E) Quantification of neuronal density. Graph and error bars represent means and S.D. of neuronal density in the CA1 field of the hippocampus of the soman-exposed NHPs subjected to the different treatments. Pairwise comparisons using the Tukey post hoc test: *P < 0.05; **P < 0.01. Scale bar in (A) (500 µm) applies to (A–D) and in (A’) (100 µm), applies to (A’–D’).
Statistical Data Analysis.
Pharmacokinetic parameters obtained from the different oral doses of galantamine as well as neuronal density measured in the hippocampi of NHPs subjected to different pretreatments were statistically analyzed using a one-way ANOVA followed by the Tukey post hoc test using the SigmaPlot software version 12 (Systat Software, Inc.). Pearson correlational analyses were also performed in Sigmaplot. Percentage of animals that survived the soman challenge was plotted against time, and the survival curves were compared among the treatment groups using a Kaplan-Meier analysis in the GraphPad Prism Software 8.3.1 (GraphPad Software, LLC, San Diego, CA).
Results
Plasma Levels of Galantamine After Oral Treatment of Cynomolgus Monkeys with Galantamine HBr.
As shown in Figure 1, after the oral administration of 0.5, 1.5, or 3.0 mg/kg galantamine HBr to Cynomolgus monkeys, plasma levels of galantamine increased up to a maximum between 1 and 3 hours posttreatment and declined subsequently. One of the five animals treated with 3.0 mg/kg galantamine had no quantifiable plasma concentrations of galantamine. Therefore, data from this animal were not included in the analysis.
Fig. 1.

Concentrations of galantamine measured in the plasma obtained at various times from non-human primates treated orally with galantamine. Filled symbols and error bars represent means and S.E.M., respectively, of results obtained from Cynomolgus monkeys treated with 0.5, 1.5, or 3.0 mg/kg galantamine HBr (n = 5, 5, and 4, respectively).
Table 2 shows the pharmacokinetic parameters derived for each dose studied here. Statistical analysis of the data using a one-way ANOVA revealed that neither t1/2 nor Tmax varied significantly with the dose of galantamine. However, both Cmax and AUC increased significantly as the dose of galantamine increased from 0.5 to 3.0 mg/kg [Cmax: F(2,13) = 33.19, P < 0.001; AUC: F(2,13) = 11.6, P = 0.002]. A Pearson correlation analysis revealed that increasing doses of galantamine correlated linearly with increasing Cmax (r2 = 0.70; P < 0.001; Fig. 2A) and AUC (r2 = 0.62; P < 0.001; Fig. 2B). A significant direct correlation was also traced between Cmax and AUC (r2 = 0.84; P < 0.001; Fig. 2B).
TABLE 2.
Pharmacokinetic profile of oral galantamine in Cynomolgus monkeys
Results are presented as means ± S.E.M. of 5, 5, and 4 Cynomolgus monkeys treated orally with 0.5, 1.5, or 3.0 mg/kg galantamine, respectively. One-way ANOVA revealed that Cmax and AUC increased significantly with increasing doses of galantamine (see text). Pairwise comparisons using the Tukey post hoc test: *P < 0.05; **P < 0.01; ***P < 0.001 (comparison with 0.5 mg/kg); ††P < 0.01 (comparison with 1.5 mg/kg).
| Pharmacokinetic results | Galantamine dose | ||
|---|---|---|---|
| 0.5 mg/kg | 1.5 mg/kg | 3.0 mg/kg | |
| Cmax (ng/ml) | 33.6 ± 4.8 | 106.6 ± 14.5** | 186.7 ± 20.3***,†† |
| AUC (ng/ml.h) | 159.7 ± 106.0 | 480.3 ± 24.0* | 701.8 ± 104.2** |
| Tmax (h) | 2.0 ± 0.5 | 1.7 ± 0.6 | 2.8 ± 0.9 |
| t1/2 (h) | 3.2 ± 2.3 | 3.2 ± 1.7 | 1.7 ± 0.3 |
Fig. 2.

Correlation between doses of galantamine and plasma Cmax or AUC and between Cmax and AUC. (A) Cmax of galantamine measured after treatment is linearly correlated with the dose of galantamine delivered to each animal (r2 = 0.70; P < 0.001). (B) AUC of plasma galantamine concentrations versus time is linearly correlated with the dose of galantamine (r2 = 0.62; P < 0.001). (C) Cmax and AUC are also linearly correlated. In all graphs, each data point represents results obtained from the individual subjects in the treatment groups presented in Fig. 1. The solid line represents the linear regression on the data points (r2 = 0.84; P < 0.001).
Fig. 3.

Degree of AChE inhibition measured in whole blood obtained at various times from non-human primates treated orally with galantamine. Filled symbols and error bars represent means and S.E.M., respectively, of results obtained from Cynomolgus monkeys treated with 0.5, 1.5, or 3.0 mg/kg galantamine HBr (n = 5, 10, and 4, respectively).
Blood AChE Activity After Oral Treatment of Cynomolgus Monkeys with Galantamine HBr.
Levels of blood AChE inhibition increased with time after the administration of galantamine HBr to the monkeys, peaked between 1 and 3 hours after the treatment, and declined subsequently. At 24 hours after the treatment, there was no significant inhibition of blood AChE (Fig. 3). The time-dependent rise and fall of blood AChE inhibition paralleled the rise and fall of the plasma galantamine concentration (see Figs. 1 and 3), as anticipated based on the reversibility of AChE inhibition by galantamine.
To determine whether the degree of AChE inhibition measured in the blood of galantamine-treated monkeys was driven by the plasma concentrations of galantamine in these animals, the concentration-response relationship for galantamine-induced inhibition of whole blood AChE activity was analyzed in vitro. As shown in Figure 4A, blood AChE activity decreased as the concentrations of galantamine increased from 0.01 to 10 µM. Fitting of the plot of galantamine concentrations versus blood AChE activity with the Hill equation revealed that, under the experimental conditions used in this study, the IC50 for galantamine to inhibit AChE activity in blood of Cynomolgus monkeys is 1.55 ± 0.15 µM (mean ± S.D., n = 3). The IC50 for galantamine to inhibit AChE activity in guinea pig blood assayed using the same method was reported to be 1.80 ± 0.38 μM (Albuquerque et al., 2006). Based on a different radiometric assay, the IC50 for galantamine to inhibit AChE in human red blood cells was found to be 0.35 μM (Thomsen et al., 1991). Direct comparisons of IC50s derived from different experiments should be made with caution because these values are largely influenced by numerous factors, including substrate concentration and tissue concentration, which vary depending on the assay (Mortensen et al., 1998).
Fig. 4.

Concentration-dependent inhibition of blood AChE by galantamine in vitro and correlation between the degree of AChE inhibition measured in blood of galantamine-treated animals and the degree of AChE inhibition estimated to be generated by plasma concentrations of galantamine in the treated animals. (A) Concentration-response relationship for galantamine-induced inhibition of AChE in whole blood of Cynomolgus monkeys. Filled symbols and error bars represent means and SEM, respectively, of results obtained from triplicate assays. (B) Maximal degree of AChE inhibition measured in blood of Cynomolgus monkeys treated with 0.5, 1.5, or 3.0 mg/kg galantamine correlated linearly with the degree of AChE inhibition estimated to be produced by the peak concentrations of galantamine measured in the plasma of the treated animals (r2 = 0.56, P = 0.001). Each symbol represents results obtained from one animal, and the solid line represents the linear regression on the data points.
The curve fitting was then used to estimate the degree of AChE inhibition that can be produced by the maximal concentrations of galantamine measured in the plasma of treated animals. The finding that the percentage of AChE inhibition estimated to be generated by maximal plasma concentrations of galantamine measured in treated animals significantly correlated with the percentage of AChE inhibition measured in the blood of those animals (r2 = 0.56, P = 0.001; Fig. 4B) confirmed that the degree of AChE inhibition measured in the blood of galantamine-treated animals depends on the blood concentrations of galantamine.
Effectiveness of Oral Galantamine Pretreatment to Counter Soman-Induced Acute Toxicity in Cynomolgus Monkeys Posttreated with Conventional Antidotes.
All four Cynomolgus monkeys pretreated orally with saline, injected intramuscularly with soman, and posttreated intramuscularly with atropine, 2-PAM, and midazolam presented clinical signs of intoxication within 10–15 minutes after the soman challenge (Table 1). Typical signs of intoxication included rapid onset of hyperexcitation, characterized by initial agitation that quickly evolved to frantic movement and jumps. In all animals, the severity of the intoxication evolved quickly. Specifically, between 15 and 20 minutes after the soman injection, intermittent clonic tremors developed and progressed to whole-body tonic-clonic motor convulsions. Approximately 20–30 minutes after the soman challenge, this severe excitatory phase gave way to a short-lasting comatose state that was followed by periods of hyper- and hyporeactivity. Approximately 1 hour after the soman challenge, motor convulsions recurred in all saline-pretreated animals. As time progressed, recurring convulsions increased in severity and duration and were accompanied by muscle fasciculations, salivation, lacrimation, increased urination, diarrhea, nystagmus, loss of posture, loss of body temperature, and severe respiratory distress characterized by gasping. Between 90 minutes and 4 hours after the soman challenge, all saline-pretreated animals presented a toxicity score of 5, which defined the endpoint for humane euthanasia (Fig. 5).
Fig. 5.

Effectiveness of pretreatment with galantamine, pyridostigmine, or saline to mitigate the acute toxicity of 4.0×LD50 soman in Cynomolgus monkeys posttreated with atropine, 2-PAM, and midazolam. (A) Percentage of animals presenting a given toxicity score after their challenge with 4.0×LD50 soman when they were pretreated with galantamine HBr (1.5 or 3.0 mg/kg, n = 5 and 6, respectively), pyridostigmine (1.2 mg/kg, n = 5), or saline (n = 4) and posttreated with atropine (0.4 mg/kg), 2-PAM (30 mg/kg), and midazolam (0.32 mg/kg). The higher the score is, the more severe the signs of acute toxicity are. (B) Kaplan-Meier plots of the survival of the soman-challenged animals subjected to the different pretreatments. The survival curves obtained from the four pretreatment groups were significantly different (χ2 = 26.84; df = 3, P < 0.0001).
Pyridostigmine-pretreated monkeys that were injected with soman and posttreated with conventional antidotes also developed typical clinical signs of intoxication within 10–15 minutes after the soman challenge (Table 1). However, in two animals, the pyridostigmine pretreatment prevented clinical signs of intoxication from becoming life-threatening. In these two animals, clinicals signs of intoxication progressed from the rapid onset of hyperexcitation to intermittent clonic tremors followed by bouts of whole-body motor convulsions lasting 15–20 minutes that, as in the saline-pretreated animals, gave way to a short-lasting comatose state. Approximately 1 hour after the soman challenge, motor convulsions recurred in these two pyridostigmine-pretreated animals, which also presented muscle fasciculations, salivation, lacrimation, increased urination, and diarrhea. Clinical signs of intoxication did not progress further in these two animals, which survived for 24 hours after the soman challenge. Between 3 and 8 hours after the soman challenge, the other three pyridostigmine-pretreated monkeys had to be euthanized as they presented the life-threatening signs of intoxication that defined the toxicity score of 5 (Fig. 5).
Pretreatment with 1.5 or 3.0 mg/kg galantamine delayed the onset and/or reduced the severity of clinical signs of toxicity in all soman-challenged monkeys that were posttreated with the conventional antidotes (Table 1). In the group of five animals that had been pretreated with 1.5 mg/kg galantamine HBr, challenged with soman, and posttreated with conventional antidotes, one, two, and two animals presented the clinical signs that defined a toxicity score of 1, 2, and 3, respectively (Fig. 5A). In the group of six animals that had been pretreated with 3 mg/kg galantamine HBr, one, two, one, and two animals presented the clinical signs that defined the toxicity scores of 1, 2, 3, and 4, respectively (Fig. 5A). No animal in these groups presented a toxicity score of 5. At approximately 4–6 hours after the soman challenge, all animals pretreated with 1.5 or 3.0 mg/kg galantamine were no longer presenting clinical signs of toxicity. All galantamine-pretreated animals survived 24 hours after the soman challenge (Fig. 5B).
A Kaplan-Meier analysis revealed that the survival curves obtained from the three pretreatment groups were significantly different (χ2 = 26.84; df = 3, P < 0.0001) (Fig. 5B). Pairwise comparisons revealed that the percentage of survival of soman-challenged monkeys that were pretreated with saline and posttreated with conventional antidotes was significantly lower than the percentage of survival of animals that had been pretreated with 1.5 mg/kg galantamine (uncorrected P = 0.0027; Bonferroni-corrected P = 0.0135) or 3.0 mg/kg galantamine (uncorrected P = 0.0011; Bonferroni-corrected P = 0.0055). Based on uncorrected P values, the percentage of survival of soman-challenged monkeys pretreated with saline was also lower than the percentage of survival of animals pretreated with pyridostigmine (P = 0.0108). In addition, the percentage of survival of animals pretreated with pyridostigmine was lower than the percentage of survival of animals pretreated with 1.5 mg/kg galantamine (P = 0.034) or 3.0 mg/kg galantamine (P = 0.049).
Effectiveness of Oral Galantamine Pretreatment to Counter Soman-Induced Neurodegeneration in the Hippocampus of Cynomolgus Monkeys Posttreated with Conventional Antidotes.
Neurodegeneration was evaluated in the hippocampi of soman-exposed NHPs, given that previous studies reported extensive neuronal damage in the hippocampi of rodents and NHPs that survived an acute exposure to convulsion-inducing doses of soman (Baze, 1993; Apland et al., 2010).
For a qualitative analysis of the presence or absence of extensive neuronal cell death, brain sections containing the hippocampus were stained with FJB, an anionic fluorescein derivative commonly used for visualization of neurons undergoing acute degeneration induced by physical or chemical insults (Conti et al., 1998; Schmued and Hopkins, 2000; Albuquerque et al., 2006; Apland et al., 2010). As shown in Figure 6, large numbers of FJB-positive cells were visualized in the CA1 field of the hippocampi of NHPs that were treated with saline or pyridostigmine before their exposure to soman and posttreatment with atropine, 2-PAM, and midazolam. In contrast, very few FJB-positive cells were detected in the hippocampi of animals that were pretreated with galantamine, challenged with soman, and posttreated with atropine, 2-PAM, and midazolam (Fig. 6).
Fig. 6.

Neurodegeneration in the hippocampi of soman-challenged Cynomolgus that were pretreated with galantamine, pyridostigmine, or saline and posttreated with conventional antidotal therapy. (A) Large numbers of Fluoro Jade-B (FJ-B)-positive (dead) cells were visualized in the CA1 field of the hippocampi of Cynomolgus monkeys that were pretreated orally with saline, challenged with 4.0×LD50 soman, and posttreated intramuscularly with 0.4 mg/kg atropine, 30 mg/kg 2-PAM, and 0.32 mg/kg midazolam. Similar results were obtained from Cynomolgus that were pretreated orally with 1.2 mg/kg pyridostigmine bromide and euthanized 24 hours after the soman challenge (B) or soon after presenting life-threatening signs of intoxication. Very few FJB-positive cells were visualized in the hippocampi of galantamine (1.5 or 3.0 mg/kg, oral)-pretreated animals that were euthanized 24 hours after their challenge with soman (C and D). Scale bar, 25 µm for larger panels and 2.5 µm for insets.
The density of NeuN-immunoreactive cells was analyzed for a more quantitative evaluation of the extent of neuronal damage in the hippocampi of soman-exposed NHPs subjected to different pretreatments. As shown in Figure 7, NeuN immunoreactivity in the CA1 field of the hippocampus was sparser among soman-exposed NHPs that had been pretreated with saline or pyridostigmine bromide than among those that had been pretreated with galantamine (Fig. 7, A–D and A’–D’). The one-way ANOVA revealed a significant effect of the pretreatment on neuronal density [F(3,19) = 6.239, P = 0.005]. Pairwise comparisons using the Tukey post hoc test indicated that the density of NeuN-immunoreactive neurons was significantly higher in the CA1 field of the hippocampus of NHPs pretreated with 3 mg/kg galantamine than in the same area of NHPs pretreated with saline or pyridostigmine bromide (Fig. 7E).
Discussion
The present study is the first to demonstrate that oral pretreatment with galantamine HBr effectively prevents the acute toxicity of 4.0×LD50 soman (15.08 µg/kg) in Cynomolgus monkeys posttreated with doses of atropine, 2-PAM, and midazolam relevant for prehospital management of nerve agent intoxication. The effectiveness of galantamine compared with that of pyridostigmine as an adjunct pretreatment to mitigate the acute toxicity of soman is discussed herein.
In 2003, the FDA approved the use of pyridostigmine, a reversible AChE inhibitor commonly prescribed to treat myasthenia gravis, as pretreatment for military personnel at risk of exposure to the nerve agent soman (Aebersold, 2012). The approval was granted after the demonstration that pretreatment with pyridostigmine significantly increased survival of guinea pigs and NHPs exposed to supralethal doses of the nerve agent soman and posttreated with atropine, oximes, and benzodiazepines (see Lorke and Petroianu, 2019, and references therein). Notably, in the absence of posttreatment with conventional antidotes, pyridostigmine is unable to mitigate the acute toxicity of any dose of soman, including doses ≤1.0×LD50 (von Bredow et al., 1991).
In the present study, 40% of Cynomolgus monkeys survived for 24 hours when they were pretreated with pyridostigmine bromide (1.2 mg/kg, p.o.), exposed to soman (4.0×LD50 or 15.08 µg/kg), and posttreated with atropine (0.4 mg/kg)-plus-2-PAM (30 mg/kg) in conjunction with 0.32 mg/kg midazolam. Given the same posttreatment, none of the Cynomolgus monkeys pretreated with saline survived for 24 hours after the soman challenge. It is possible that a higher dose of midazolam could have improved the survival outcomes in both groups. In fact, Hayward et al. (1990) reported that all rhesus monkeys pretreated orally with pyridostigmine bromide survived for 48 hours after they were challenged with 5.0×LD50 soman and posttreated with atropine (0.4 mg/kg), 2-PAM (25.71 mg/kg), and 1 mg/kg midazolam. Of major interest, however, in present study, using galantamine (1.5 or 3.0 mg/kg) as the pretreatment in association with posttreatment consisting of atropine, 2-PAM, and the low dose of midazolam (0.32 mg/kg) resulted in 100% survival of soman-challenged NHPs.
At the time their brains were harvested, soman-exposed NHPs orally pretreated with saline or pyridostigmine bromide presented extensive neurodegeneration in the CA1 field of the hippocampus, as revealed by FJB staining. By contrast, soman-exposed NHPs orally pretreated with 1.5 or 3.0 mg/kg galantamine HBr did not. Collombet et al. (2006) demonstrated that FJB-positive neurons in the hippocampus of rodents exposed to a convulsion-inducing dose of soman are devoid of NeuN immunoreactivity. They also suggested that acute loss of NeuN immunoreactivity could be used as a predictor of delayed neurodegeneration. Here, evidence is provided that the density of NeuN-immunoreactive neurons is significantly higher in the CA1 field of the hippocampus of NHPs orally pretreated with 3 mg/kg galantamine HBr than of those orally pretreated with saline or pyridostigmine bromide. This finding suggests that inclusion of galantamine in the treatment regimen affords significant protection against soman-induced neurodegeneration.
The effectiveness of galantamine to suppress the acute toxicity of soman can be attributed, at least in part, to its ability to promptly cross the blood brain barrier and protect central AChE from irreversible inhibition by soman. Although, as a reversible AChE inhibitor, pyridostigmine can protect a critical portion of AChE from irreversible inhibition by soman (Gordon et al., 1978; Dirnhuber et al., 1979; Heyl et al., 1980; Haigh et al., 2005; Eckert et al., 2006), this carbamate does not promptly cross the blood brain barrier. Thus, it does not protect AChE in the brain from irreversible inhibition by soman and, consequently, it does not prevent the toxic effects of this agent in the central nervous system (Anderson et al., 1992). However, the possibility cannot be ruled out that additional actions contribute to the effectiveness of galantamine as an adjuvant pretreatment to mitigate the acute toxicity of soman. For instance, galantamine inhibits AChE more selectively than butyrylcholinesterase (Thomsen et al., 1991) and, as such, may spare the natural OP scavenging property of the latter (Albuquerque et al., 2006). It is also possible that the nicotinic allosteric potentiating action of galantamine contributes to its effectiveness as a pretreatment against nerve agent poisoning. Specifically, acting as a nicotinic allosteric potentiating ligand, galantamine may prevent desensitization of nicotinic receptors by ACh that accumulates after the irreversible AChE inhibition by nerve agents and, thereby, increase nicotinic receptor signaling, which has anti-inflammatory and neuroprotective properties (see Albuquerque et al., 2009).
Although the results presented here demonstrate that the acute toxicity of soman in NHPs is effectively suppressed by pretreatment with galantamine HBr, earlier studies have raised concerns regarding the safety of centrally acting reversible AChE inhibitors, including galantamine (see Myhrer and Aas, 2016, and references therein). For instance, Sweeney et al. (1989) reported that mice presented reduced locomotor activity and impaired memory in the Morris water maze up to 2 and 4 hours, respectively, after an i.p. injection of 5 mg/kg galantamine HBr. These effects were likely due to the large degree of AChE inhibition (78%–83%) produced by the test dose of galantamine (Sweeney et al., 1989). Likewise, rats treated intraperitoneally with 3 mg/kg galantamine HBr presented reduced locomotor activity in open fields and memory impairment in a novel object recognition test (Myhrer et al., 2010). In rats, this dose of galantamine HBr also induce clear signs of cholinergic overactivation, including jaw tremors (Podurgiel et al., 2013). As presented here, however, the acute toxicity of soman in Cynomolgus monkeys was effectively mitigated with oral doses of galantamine HBr that reversibly inhibited AChE activity by approximately 25%–40%. This degree of reversible AChE inhibition, which is markedly lower than that induced by doses reported to cause detrimental neurobehavioral effects in rats and mice, is generally considered safe for humans (Dunn and Sidell, 1989). In fact, no clinical signs of cholinergic overactivation and no cognitive deficits have been observed in healthy human subjects administered an oral dose of galantamine (8 mg) that generates approximately 30% inhibition of AChE in red blood cells (Morasch et al., 2015). In addition to producing levels of reversible AChE inhibition considered to be safe for humans, oral doses of galantamine HBr that prevent soman-induced lethality in NHPs were found to translate, on the basis of pharmacokinetic equivalence (Reigner and Blesch, 2002), to human-equivalent doses that are in the range of FDA-approved doses of galantamine HBr to treat AD.
A single oral treatment of humans with 8 mg galantamine produces an AUC of 406 ± 77 ng*h/ml (Zhao et al., 2002; Huang and Fu, 2010). By comparison, a single oral treatment of Cynomolgus monkeys with 1.5 or 3.0 mg/kg galantamine HBr, which effectively protected the animals against the acute toxicity of 4.0×LD50 soman, produced AUCs of 480 ± 24 and 702 ± 104 ng*h/ml, respectively (Table 2). Using the equation 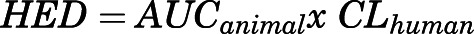 , where AUCanimal is the AUC produced by the target dose in an animal and CLhuman is the clearance of galantamine in humans, it was possible to estimate the human-equivalent dose of galantamine needed to prevent the acute toxicity of soman in humans. Considering that clearance of galantamine in humans is approximately 19.5 l/h (Lilienfeld, 2002), the oral doses of 1.5 and 3.0 mg/kg galantamine HBr in Cynomolgus monkeys translate to approximately 9.4 and 13.7 mg galantamine base in humans. These doses are clinically relevant because daily oral doses of galantamine base ranging from 8 to 24 mg are currently approved for treatment of AD, and the incidence of adverse events, which are primarily of gastrointestinal origin, is reportedly low among galantamine-treated AD patients (Lilienfeld, 2002; Carney et al., 2019). In addition, as mentioned above, Morasch et al. (2015) reported that healthy human subjects treated with 8 mg galantamine presented no neurobehavioral deficits and no clinical signs of untoward side effects. At doses clinically used to treat AD patients, the safety profile of galantamine is generally superior to that of other centrally acting reversible AChE inhibitors, including donepezil and rivastigmine (Carney et al., 2019).
, where AUCanimal is the AUC produced by the target dose in an animal and CLhuman is the clearance of galantamine in humans, it was possible to estimate the human-equivalent dose of galantamine needed to prevent the acute toxicity of soman in humans. Considering that clearance of galantamine in humans is approximately 19.5 l/h (Lilienfeld, 2002), the oral doses of 1.5 and 3.0 mg/kg galantamine HBr in Cynomolgus monkeys translate to approximately 9.4 and 13.7 mg galantamine base in humans. These doses are clinically relevant because daily oral doses of galantamine base ranging from 8 to 24 mg are currently approved for treatment of AD, and the incidence of adverse events, which are primarily of gastrointestinal origin, is reportedly low among galantamine-treated AD patients (Lilienfeld, 2002; Carney et al., 2019). In addition, as mentioned above, Morasch et al. (2015) reported that healthy human subjects treated with 8 mg galantamine presented no neurobehavioral deficits and no clinical signs of untoward side effects. At doses clinically used to treat AD patients, the safety profile of galantamine is generally superior to that of other centrally acting reversible AChE inhibitors, including donepezil and rivastigmine (Carney et al., 2019).
In a previous study, pretreatment with a single i.m. dose of 0.362 mg/kg galantamine, which was estimated to produce approximately 50% inhibition of blood AChE activity, protected Cynomolgus monkeys against the acute toxicity of 1.0×LD50 soman in the absence of any antidotal posttreatment (Hamilton et al., 2017). In that study, too, galantamine HBr was superior to pyridostigmine bromide as a pretreatment against soman intoxication. As important, however, was the finding that the effective antidotal dose of galantamine HBr had no detrimental effect on the cognitive function of the NHPs. Only doses that were ≥40% higher than the effective antidotal dose and produced >50% AChE inhibition induced a transient, yet statistically significant cognitive deficit in Cynomolgus monkeys (Hamilton et al., 2017).
In conclusion, the data presented here demonstrate that NHPs are effectively protected against the acute toxicity of a supralethal dose of soman (15.08 µg/kg or 4.0×LD50) when they are pretreated with a clinically relevant oral dose of galantamine HBr and posttreated with doses of conventional antidotes recommended for prehospital management of nerve agent poisoning. They also support the contention that this antidotal therapy is more effective than the standard posttreatments in association with vehicle or pyridostigmine bromide as pretreatment. Therefore, this study lays the groundwork for the continued advanced development of galantamine HBr as a safe and effective oral pretreatment against OP poisoning.
Acknowledgments
The authors are indebted to Mabel A. Zelle for her technical assistance.
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AD
Alzheimer’s disease
- AUC
area under the plasma concentration-time curve from time 0 to infinity
- FDA
US Food and Drug Administration
- FJB
Fluoro-Jade B
- FOB
functional observation battery
- NHP
nonhuman primate
- OP
organophosphorus
- 2-PAM
2-pyridine aldoxime methyl chloride
- ROI
region of interest
- t1/2
half-life
- Tmax
time to reach Cmax
Authorship Contributions
Participated in research design: Lane, Aracava, Basinger, Pereira, Albuquerque.
Conducted experiments: Lane, Carter, Pescrille, Aracava, Fawcett, Albuquerque.
Performed data analysis: Lane, Aracava, Fawcett, Pereira, Albuquerque.
Wrote or contributed to the writing of the manuscript: Aracava, Pereira.
Secured research funds: Basinger, Albuquerque.
Footnotes
This work was supported in part by the Countervail Corporation with funds from the CounterACT Program of National Institutes of Health National Institutes of Neurologic Disorders and Stroke via a Phase II SBIR Grant [R44-NS068049] and was presented during the poster session of the 10th Annual NIH CounterACT Network Research Symposium in 2016.
A preprint of this article was deposited in bioRxiv [https://doi.org/10.1101/2020.02.03.932798].
The use of galantamine as an antidote against organophosphorus poisoning is protected under the Patent Application PCT/US05/33789 filed by the University of Maryland Baltimore and the US Medical Research Institute of Chemical Defense on Sept 23, 2005 and licensed to Countervail Corp.
Edson X. Albuquerque died during the preparation of this article.
References
- Adams NL. (1990) The intramuscular lethality of soman (GD) in the cynomolgus monkey. USAMRICD-TR-90-06 US Army Medical Research Institute of Chemical Defense, Aberdeen Proving Ground, MD. ADA224887. NLM unique ID 9108742.
- Aebersold P. (2012) FDA experience with medical countermeasures under the animal rule. Adv Prev Med 2012:507571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EFR, Aracava Y, Fawcett WP, Oliveira M, Randall WR, Hamilton TA, Kan RK, Romano JA, Jr., Adler M. (2006) Effective countermeasure against poisoning by organophosphorus insecticides and nerve agents. Proc Natl Acad Sci USA 103:13220–13225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DR, Harris LW, Woodard CL, Lennox WJ. (1992) The effect of pyridostigmine pretreatment on oxime efficacy against intoxication by soman or VX in rats. Drug Chem Toxicol 15:285–294. [DOI] [PubMed] [Google Scholar]
- Apland JP, Figueiredo TH, Qashu F, Aroniadou-Anderjaska V, Souza AP, Braga MF. (2010) Higher susceptibility of the ventral versus the dorsal hippocampus and the posteroventral versus anterodorsal amygdala to soman-induced neuropathology. Neurotoxicology 31:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baze WB. (1993) Soman-induced morphological changes: an overview in the non-human primate. J Appl Toxicol 13:173–177. [DOI] [PubMed] [Google Scholar]
- Carney G, Bassett K, Wright JM, Maclure M, McGuire N, Dormuth CR. (2019) Comparison of cholinesterase inhibitor safety in real-world practice. Alzheimers Dement (N Y) 5:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai PR, Boyer EW, Al-Nahhas H, Erickson TB. (2017) Toxic chemical weapons of assassination and warfare: nerve agents VX and sarin. Toxicol Commun 1:21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombet JM, Masqueliez C, Four E, Burckhart M-F, Bernabé D, Baubichon D, Lallement G. (2006) Early reduction of NeuN antigenicity induced by soman poisoning in mice can be used to predict delayed neuronal degeneration in the hippocampus. Neurosci Lett 398:337–342. [DOI] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. (1998) Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci 18:5663–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnhuber P, French MC, Green DM, Leadbeater L, Stratton JA. (1979) The protection of primates against soman poisoning by pretreatment with pyridostigmine. J Pharm Pharmacol 31:295–299. [DOI] [PubMed] [Google Scholar]
- Dunn MA, Sidell FR. (1989) Progress in medical defense against nerve agents. JAMA 262:649–652. [PubMed] [Google Scholar]
- Eckert S, Eyer P, Mückter H, Worek F. (2006) Kinetic analysis of the protection afforded by reversible inhibitors against irreversible inhibition of acetylcholinesterase by highly toxic organophosphorus compounds. Biochem Pharmacol 72:344–357. [DOI] [PubMed] [Google Scholar]
- Franca TCC, Kitagawa DAS, Cavalcante SFA, da Silva JAV, Nepovimova E, Kuca K. (2019) Novichoks: the dangerous fourth generation of chemical weapons. Int J Mol Sci 20:E1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvin DV, Baird TJ. (2008) A functional observational battery in non-human primates for regulatory-required neurobehavioral assessments. J Pharmacol Toxicol Methods 58:88–93. [DOI] [PubMed] [Google Scholar]
- Golime R, Palit M, Acharya J, Dubey DK. (2018) Neuroprotective effects of galantamine on nerve agent-induced neuroglial and biochemical changes. Neurotox Res 33:738–748. [DOI] [PubMed] [Google Scholar]
- Gordon JJ, Leadbeater L, Maidment MP. (1978) The protection of animals against organophosphate poisoning by pretreatment with a carbamate. Toxicol Appl Pharmacol 43:207–216. [DOI] [PubMed] [Google Scholar]
- Haigh JR, Johnston SR, Peters BM, Doctor BP, Gordon RK, Adler M, Gall KJ, Deshpande SS. (2005) Inhibition of guinea pig hemi-diaphragm acetylcholinesterase activity by pyridostigmine bromide and protection against soman toxicity. Chem Biol Interact 157–158:381–382. [DOI] [PubMed] [Google Scholar]
- Hamilton LR, Schachter SC, Myers TM. (2017) Time course, behavioral safety, and protective efficacy of centrally active reversible acetylcholinesterase inhibitors in Cynomolgus macaques. Neurochem Res 42:1962–1971. [DOI] [PubMed] [Google Scholar]
- Hayward IJ, Wall HG, Jaax NK, Wade JV, Marlow DD, Nold JB. (1990) Decreased brain pathology in organophosphate-exposed rhesus monkeys following benzodiazepine therapy. J Neurol Sci 98:99–106. [DOI] [PubMed] [Google Scholar]
- Heyl WC, Harris LW, Stitcher DL. (1980) Effects of carbamates on whole blood cholinesterase activity: chemical protection against soman. Drug Chem Toxicol 3:319–332. [DOI] [PubMed] [Google Scholar]
- Hilmas CJ, Poole MJ, Finneran K, Clark MG, Williams PT. (2009) Galantamine is a novel post-exposure therapeutic against lethal VX challenge. Toxicol Appl Pharmacol 240:166–173. [DOI] [PubMed] [Google Scholar]
- Huang F, Fu Y. (2010) A review of clinical pharmacokinetics and pharmacodynamics of galantamine, a reversible acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease, in healthy subjects and patients. Curr Clin Pharmacol 5:115–124. [DOI] [PubMed] [Google Scholar]
- Hurst CG, Newmark J, Romano JA., Jr. (2012) Chemical terrorism: introduction, in Harrison’s Principles of Internal Medicine, 18th ed. (Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J. 1779–1788, McGraw-Hill, New York. [Google Scholar]
- Jett DA, Laney JW. (2019) Civilian research on chemical medical countermeasures. Toxicol Mech Methods DOI: 10.1080/15376516.2019.1669250 [published ahead of print]. [DOI] [PubMed] [Google Scholar]
- Johnson CD, Russell RL. (1975) A rapid, simple radiometric assay for cholinesterase, suitable for multiple determinations. Anal Biochem 64:229–238. [DOI] [PubMed] [Google Scholar]
- Joiner R, Kluwe W. (1988) Multiple animal studies for medical chemical defense program in soldier/patient decontamination and drug development. Task 85-18: Conduct of Pralidoxime chloride, atropine in citrate buffer and pyridostigmine bromide pharmacokinetic studies, and comparative evaluation of the efficacy of pyridostigmine plus atropine. pp 67-73. Available from the National Technical Information Service (https://ntrl.ntis.gov/NTRL/), accession number ADB127309.
- Lilienfeld S. (2002) Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev 8:159–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorke DE, Petroianu GA. (2019) Reversible cholinesterase inhibitors as pretreatment for exposure to organophosphates. A review. J Appl Toxicol 39:101–116. [DOI] [PubMed] [Google Scholar]
- Mamczarz J, Kulkarni GS, Pereira EFR, Albuquerque EX. (2011) Galantamine counteracts development of learning impairment in guinea pigs exposed to the organophosphorus poison soman: clinical significance. Neurotoxicology 32:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs TC, Rice P, Vale JA. (2006) The role of oximes in the treatment of nerve agent poisoning in civilian casualties. Toxicol Rev 25:297–323. [DOI] [PubMed] [Google Scholar]
- McDonough JH. (2002) Midazolam: an improved anticonvulsant treatment for nerve agent-induced seizures. Proceedings of the 2001 ECBC Scientific Conference on Chemical and Biological Defense Research; 6–8 March; Marriott’s Hunt Valley Inn, Hunt Valley, MD. ADA409494.
- McDonough JH, McMonagle JD, Shih TM. (2010) Time-dependent reduction in the anticonvulsant effectiveness of diazepam against soman-induced seizures in Guinea pigs. Drug Chem Toxicol 33:279–283. [DOI] [PubMed] [Google Scholar]
- Morasch KC, Aaron CL, Moon JE, Gordon RK. (2015) Physiological and neurobehavioral effects of cholinesterase inhibition in healthy adults. Physiol Behav 138:165–172. [DOI] [PubMed] [Google Scholar]
- Mortensen SR, Brimijoin S, Hooper MJ, Padilla S. (1998) Comparison of the in vitro sensitivity of rat acetylcholinesterase to chlorpyrifos-oxon: what do tissue IC50 values represent? Toxicol Appl Pharmacol 148:46–49. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Aas P. (2016) Pretreatment and prophylaxis against nerve agent poisoning: are undesirable behavioral side effects unavoidable? Neurosci Biobehav Rev 71:657–670. [DOI] [PubMed] [Google Scholar]
- Myhrer T, Enger S, Aas P. (2010) Behavioral side effects in rats treated with acetylcholinesterase inhibitors suggested used as prophylactics against nerve agents. Pharmacol Biochem Behav 95:338–343. [DOI] [PubMed] [Google Scholar]
- Newmark J. (2019) Therapy for acute nerve agent poisoning: an update. Neurol Clin Pract 9:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki Y, Maekawa K, Ogawa Y, Asukai N, Minami M, Omae K, Sarin Health Effects Study Group (2001) Effects of sarin on the nervous system in rescue team staff members and police officers 3 years after the Tokyo subway sarin attack. Environ Health Perspect 109:1169–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo ID, Garman RH, Weber K, Bobrowski WF, Hardisty JF, Morton D. (2012) Technical guide for nervous system sampling of the cynomolgus monkey for general toxicity studies. Toxicol Pathol 40:624–636. [DOI] [PubMed] [Google Scholar]
- Pereira EFR, Aracava Y, Alkondon M, Akkerman M, Merchenthaler I, Albuquerque EX. (2010) Molecular and cellular actions of galantamine: clinical implications for treatment of organophosphorus poisoning. J Mol Neurosci 40:196–203. [DOI] [PubMed] [Google Scholar]
- Podurgiel S, Collins-Praino LE, Yohn S, Randall PA, Roach A, Lobianco C, Salamone JD. (2013) Tremorolytic effects of safinamide in animal models of drug-induced parkinsonian tremor. Pharmacol Biochem Behav 105:105–111. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. (2008) Dose translation from animal to human studies revisited. FASEB J 22:659–661. [DOI] [PubMed] [Google Scholar]
- Reigner BG, Blesch KS. (2002) Estimating the starting dose for entry into humans: principles and practice. Eur J Clin Pharmacol 57:835–845. [DOI] [PubMed] [Google Scholar]
- Rickett DL, Glenn JF, Beers ET. (1986) Central respiratory effects versus neuromuscular actions of nerve agents. Neurotoxicology 7:225–236. [PubMed] [Google Scholar]
- Romano JA, Jr., King JM. (2001) Psychological casualties resulting from chemical and biological weapons. Mil Med 166 (Suppl):21–22. [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130. [DOI] [PubMed] [Google Scholar]
- Steiner WE, Pikalov IA, Williams PT, English WA, Hilmas CJ. (2012) An extraction assay analysis for galanthamine in guinea pig plasma and its Application to nerve agent countermeasures. J Anal Bioanal Tech 3:144. [Google Scholar]
- Suresh PS, Mullangi R, Sukumaran SK. (2014) Highly sensitive LC-MS/MS method for determination of galantamine in rat plasma: application to pharmacokinetic studies in rats. Biomed Chromatogr 28:1633–1640. [DOI] [PubMed] [Google Scholar]
- Sweeney JE, Puttfarcken PS, Coyle JT. (1989) Galanthamine, an acetylcholinesterase inhibitor: a time course of the effects on performance and neurochemical parameters in mice. Pharmacol Biochem Behav 34:129–137. [DOI] [PubMed] [Google Scholar]
- Thomsen T, Zendeh B, Fischer JP, Kewitz H. (1991) In vitro effects of various cholinesterase inhibitors on acetyl- and butyryl-cholinesterases of healthy volunteers. Biochemical Pharmacol 41:139–141. [DOI] [PubMed] [Google Scholar]
- von Bredow JD, Adams NL, Groff WA, Vick JA. (1991) Effectiveness of oral pyridostigmine pretreatment and cholinolytic-oxime therapy against soman intoxication in nonhuman primates. Fundam Appl Toxicol 17:761–770. [PubMed] [Google Scholar]
- Zhao Q, Brett M, Van Osselaer N, Huang F, Raoult A, Van Peer A, Verhaeghe T, Hust R. (2002) Galantamine pharmacokinetics, safety, and tolerability profiles are similar in healthy Caucasian and Japanese subjects. J Clin Pharmacol 42:1002–1010. [PubMed] [Google Scholar]