

For ca. 20 years, the HCV protease NS3 has been implicated in NS5A hyperphosphorylation. We now show that it is the NS3-mediated cis cleavage at the NS3-4A junction that permits NS5A phosphorylation at serines 2201, 2208, 2211, and 2214, leading to hyperphosphorylation, which is a necessary condition for genotype 2 HCV replication. We further show that NS5A may already be phosphorylated at these serine residues right after NS3-4A cleavage and before NS5A is released from the NS4A-5A polyprotein. Our data suggest that the dual-functional NS3, a protease and an ATP-binding RNA helicase, could have a direct or indirect role in NS5A hyperphosphorylation.
KEYWORDS: NS5A, hepatitis C virus, proteases, protein phosphorylation
ABSTRACT
Replication of the genotype 2 hepatitis C virus (HCV) requires hyperphosphorylation of the nonstructural protein NS5A. It has been known that NS5A hyperphosphorylation results from the phosphorylation of a cluster of highly conserved serine residues (S2201, S2208, S2211, and S2214) in a sequential manner. It has also been known that NS5A hyperphosphorylation requires an NS3 protease encoded on one single NS3-5A polyprotein. It was unknown whether NS3 protease participates in this sequential phosphorylation process. Using an inventory of antibodies specific to S2201, S2208, S2211, and S2214 phosphorylation, we found that protease-dead S1169A mutation abrogated NS5A hyperphosphorylation and phosphorylation at all serine residues measured, consistent with the role of NS3 in NS5A sequential phosphorylation. These effects were not rescued by a wild-type NS3 protease provided in trans by another molecule. Mutations (T1661R, T1661Y, or T1661D) that prohibited proper cleavage at the NS3-4A junction also abolished NS5A hyperphosphorylation and phosphorylation at all serine residues, whereas mutations at the other cleavage sites, NS4A-4B (C1715S) or NS4B-5A (C1976F), did not. In fact, any combinatory mutations that prohibited NS3-4A cleavage (T1661Y/C1715S or T1661Y/C1976F) abrogated NS5A hyperphosphorylation and phosphorylation at all serine residues. In the C1715S/C1976F double mutant, which resulted in an NS4A-NS4B-NS5A fusion polyprotein, a hyperphosphorylated band was observed and was phosphorylated at all serine residues. We conclude that NS3-mediated autocleavage at the NS3-4A junction is critical to NS5A hyperphosphorylation at S2201, S2208, S2211, and S2214 and that NS5A hyperphosphorylation could occur in an NS4A-NS4B-NS5A polyprotein.
IMPORTANCE For ca. 20 years, the HCV protease NS3 has been implicated in NS5A hyperphosphorylation. We now show that it is the NS3-mediated cis cleavage at the NS3-4A junction that permits NS5A phosphorylation at serines 2201, 2208, 2211, and 2214, leading to hyperphosphorylation, which is a necessary condition for genotype 2 HCV replication. We further show that NS5A may already be phosphorylated at these serine residues right after NS3-4A cleavage and before NS5A is released from the NS4A-5A polyprotein. Our data suggest that the dual-functional NS3, a protease and an ATP-binding RNA helicase, could have a direct or indirect role in NS5A hyperphosphorylation.
INTRODUCTION
The hepatitis C virus (HCV) is a single-stranded positive-sense RNA virus that affects more than 70 million people worldwide (1). Its genome encodes a polyprotein comprising 3 structural (core, E1, and E2) and 7 nonstructural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins that have to be released as individual functional proteins via cleavages by host and viral proteases (2). The structural proteins plus p7 are released by host signal peptidase and signal peptide peptidase (3). The release of NS2 is catalyzed by the NS2 cysteine protease (4), whereas the release of the rest of the nonstructural proteins is catalyzed by the NS3 serine protease (5, 6). Cleavage at the NS3-4A junction occurs first and occurs only in cis, i.e., an intramolecular event (7). In contrast, cleavages at the rest of the three junctions occur later and can occur in trans, i.e., intermolecular events (5, 6), in the following order: NS5A-5B, NS4B-5A, and NS4A-4B (8). However, one study found no particular trans-cleavage order (9).
All nonstructural proteins carry out unique functions. NS2 and NS3 are proteases, as described above (4–6). NS4A is a cofactor of NS3 (6, 10). NS4B induces formation of membranous structures where the HCV genome replicates (11). NS5B is an RNA-dependent RNA polymerase required for viral genome replication (12). NS5A binds a plethora of host and viral proteins and executes versatile functions, including host-virus interaction, viral genome replication, and viral particle assembly (13). When analyzed with SDS-PAGE and immunoblotting, NS5A appears as two bands at 56 and 58 kDa, representing hypo- and hyperphosphorylated states, respectively (13). Both states are critical to the viral life cycle (14–18). Recent studies have shown that NS5A transits between hypo- and hyperphosphorylated states via the phosphorylation of a cluster of highly conserved serine residues, i.e., S2201, S2205, S2208, S2211, and S2214, in a sequential manner (19–21). NS5A transition between the two states is thought to regulate its intracellular distribution in large and small structures required for a complete viral life cycle (20, 22). Of the serine residues, S2205 was shown to undergo phosphorylation and dephosphorylation cycles that fire the casein kinase I α-mediated sequential S2208/S2211/S2214 phosphorylation cascade, leading to the hyperphosphorylated state (20, 23). However, a phosphorylation-ablating alanine mutation at S2205 did not completely abolish sequential phosphorylation (19). Thus, even though S2205 phosphorylation plays a key role in NS5A sequential phosphorylation, it is not absolutely required. In contrast, an alanine mutation at S2201 abolishes, whereas a phosphorylation-mimicking aspartate mutation rescues, NS5A sequential phosphorylation (19, 24) in the absence of S2205 phosphorylation (20). Therefore, S2201 phosphorylation could bypass S2205 phosphorylation to regulate NS5A sequential phosphorylation.
Replication of the HCV genome is regulated by the phosphorylation status of NS5A (15–18, 21, 24–26). In the case of genotypes 1, 3, 4, and 5, reduced NS5A hyperphosphorylation is critical to HCV genome replication (15, 26). In the case of genotype 2, NS5A hyperphosphorylation is indispensable for HCV genome replication (16–18, 21, 24, 25). Many protein kinases are reported to be responsible for NS5A phosphorylation (16, 23, 27–30). The lipid kinase phosphatidylinositol 4-kinase III α has been reported to reduce NS5A hyperphosphorylation, thereby enhancing replication of HCV genotypes 1, 3, 4, and 5 (15, 26). Of particular interest is the involvement of the NS3 protease in NS5A hyperphosphorylation (31, 32). An alanine mutation at the catalytic serine residue (S1169) in NS3 abolishes NS5A hyperphosphorylation, which cannot be rescued with an active NS3 provided in trans (31). Given these observations, it remained unknown how the NS3 protease participates in NS5A hyperphosphorylation and on which particular serine residues. Following the NS5A sequential phosphorylation cascade mentioned above, the NS3 protease could hypothetically participate in S2201 phosphorylation that bypasses S2205 phosphorylation to fire sequential 2208/S2211/S2214 phosphorylation and lead to NS5A hyperphosphorylation. To test this, we made an antibody specific to S2201 phosphorylation and confirmed that NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214 require an active NS3 protease carried in one single NS3-NS4A-NS4B-NS5A polyprotein. The above-described phosphorylation events depend on a successful cleavage at the NS3-4A junction. Finally, for the first time, we found that NS5A can be hyperphosphorylated in an NS4A-NS4B-NS5A polyprotein whose cleavages were blocked via mutations.
RESULTS
Production of an S2201 phosphorylation-specific antibody.
In order to measure phosphorylation at serine 2201 in the NS5A of genotype 2 HCV (Fig. 1A), we generated an S2201 phosphorylation-specific antibody. Figure 1B shows a dot blot analysis of its specificity. The antibody detected only the S2201-phosphorylated peptide in a dose-dependent manner; it did not detect other peptides regardless of phosphorylation. On immunoblots (Fig. 1C), S2201 phosphorylation was detected in the hyperphosphorylated NS5A band in NS3-5A-transfected HEK293T cells. In HCV (J6/JFH1, genotype 2a)-infected Huh7.5.1 cells (Fig. 1D), S2201 phosphorylation became detectable after 48 h of infection.
FIG 1.
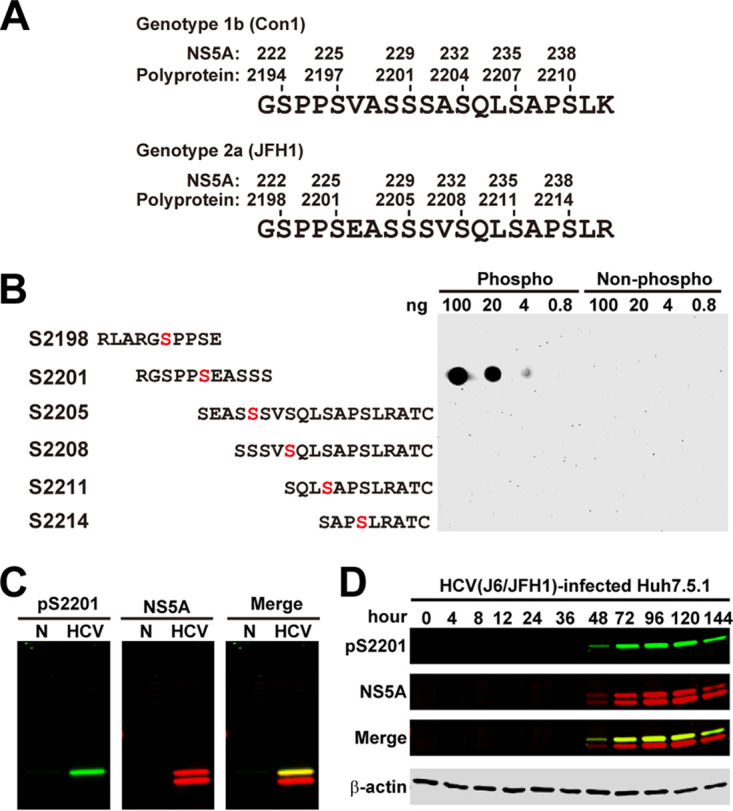
Characterization of the NS5A S2201 phosphorylation-specific antibody. (A) Numbering systems for amino acid positions starting at NS5A or the polyprotein of HCV genotype 1 (Con1, UniProt no. Q9WMX2) versus genotype 2 (JFH1, UniProt no. Q99IB8). (B) Dot blot analysis. Synthetic phosphorylated (Phospho) versus nonphosphorylated (Non-phospho) peptides were serially diluted and dotted on a membrane before detection with the S2201 phosphorylation-specific antibody. Serine residues of interests are colored red. (C and D) Immunoblotting for S2201 phosphorylation in NS5A (pS2201) in NS3-5A-transfected HEK293T cells (HCV, NS3-5A transfected; N, nontransfected) and in genotype 2a (J6/JFH1)-infected Huh7.5.1 cells.
NS5A hyperphosphorylation at serines 2201, 2208, 2211, and 2214 requires NS3-mediated cis cleavage at the NS3-4A junction.
We first confirmed that NS5A hyperphosphorylation requires an active NS3 protease encoded on the same NS3-5A polyprotein in the HEK293T cells transfected with either a wild-type or a protease-dead mutant NS3-5A expression construct whose catalytic serine residue was mutated to alanine (Fig. 2A, S1169A) (31, 32). We have shown in many prior publications that NS5A phosphorylation occurs similarly in the HEK293T cells and the Huh7.5.1 cells (19–21, 27). In the wild-type NS3-5A-transfected HEK293T cells, we observed a single NS3 band with a correct molecular weight of about 67 kDa (Fig. 2B, lane WT) and two NS5A bands at about 56 and 58 kDa, corresponding to hypo- and hyperphosphorylation (Fig. 2C, lane WT). In contrast, in the protease-dead S1169A mutant-transfected cells, we observed a single uncleaved NS3-5A polyprotein at about 156 kDa (Fig. 2B, lane S1169A), presumably lacking hyperphosphorylation (Fig. 2C, lane S1169A). To test whether NS5A hyperphosphorylation in the S1169A mutant can be rescued by NS3 provided in trans, the S1169A mutant was cotransfected with an S2211A mutant NS3-5A that has functional NS3 and a greatly reduced hyperphosphorylation level (21). When the S2211A mutant was transfected alone, an NS3 band (Fig. 2B lane S2211A) was observed, indicating intact protease activity. NS5A was also properly processed; however, it was barely hyperphosphorylated (Fig. 2C, lane S2211A). In the S1169A-plus-S2211A double-transfected cells (Fig. 2B, lane S1169A+S2211A), there was not an NS3-5A polyprotein band as observed when S1169A was transfected alone (lane S1169A). Thus, the active NS3 provided by S2211A can cleave the S1169A polyprotein, i.e., functioning in trans. Under these conditions, NS5A was properly cleaved but not hyperphosphorylated (Fig. 2C, lane S1169A+S2211A). Hence, NS5A hyperphosphorylation in the protease-dead S1169A mutant cannot be rescued by an active protease provided by the S2211A mutant.
FIG 2.

NS5A hyperphosphorylation requires NS3-mediated autocleavage at the NS3-4A junction. (A) Schematics of five expression constructs: WT, wild-type NS3-5A; S1169A, NS3 protease-dead NS3-5A mutant; S2211A, NS3-5A mutant that eliminates most NS5A hyperphosphorylation; NS3-4A. (B and C) Representative immunoblots for NS3 and NS5A in the HEK293T cells transfected with the expression constructs. β-Actin served as a loading control. (D) Summary of the immunoblotting results in panel C. Summarized are changes (percent) in non-p56 over total NS5A (p56 plus p58). Values are means ± standard errors of the means (SEM) (n = 3). *, P < 0.05 (determined by t test against the values for the wild type).
In the S1169A-plus-S2211A double-transfected cells, there were two NS3 bands: an NS3 band and an NS3-4A band (about 73 kDa) (Fig. 2B, lane S1169A+S2211A). The NS3 band came from the S2211A mutant because it was not observed in the S1169A mutant-transfected cells (Fig. 2B, lane S1169A). The higher NS3-4A band came from the S1169A construct because it was not observed in the S2211A-transfected cells (Fig. 2B, lane S2211A). This is consistent with the NS3-mediated cis cleavage at the NS3-4A junction (6) because the NS3 protease provided in trans can cleave the NS4A-4B and NS4B-5A junctions but not the NS3-4A junction. The fact that the NS3-4A band was not observed in the NS3-4A-transfected cells supports this view (Fig. 2B, lane NS3-4A). Like the NS3 provided by S2211A, the NS3 provided by NS3-4A also failed to cleave the NS3-4A junction in the S1169A mutant (Fig. 2B, lane S1169A+NS3-4A). Under these conditions, only the hypophosphorylated NS5A was observed (Fig. 2C, lane S1169A+NS3-4A). Thus, NS5A hyperphosphorylation requires an active NS3 protease in cis. Cleavage at the NS3-4A junction occurs only in cis. When NS3-4A cleavage does not occur, NS5A is not hyperphosphorylated. Figure 2D summarizes the results.
Not only did NS5A hyperphosphorylation not occur in the S1169A mutant NS3-5A, but NS5A phosphorylation at S2201, S2208, S2211, and S2214 did not occur either (Fig. 3A to D, lanes S1169A). In contrast, phosphorylation at these residues was observed in the wild-type NS3-5A-transfected cells and corresponded to the hyperphosphorylated NS5A bands (Fig. 3A to D, lanes WT). An active NS3 protease provided in trans did not rescue phosphorylation at these residues (Fig. 3A to D, lanes S1169A+S2211A and lanes S1169A+NS3-4A). Therefore, NS5A phosphorylation at S2201, S2208, S2211, and S2214 requires an active NS3 protease in cis. Figure 3E summarizes the results. The weak phosphorylation at S2201 (Fig. 3A) and S2208 (Fig. 3B) observed in the S2211A mutant is in line with the sequential phosphorylation cascade previously reported (19, 20).
FIG 3.

NS5A phosphorylation at serines 2201, 2208, 2211, and 2214 requires NS3-mediated autocleavage at the NS3-4A junction. (A to D) Representative immunoblotting for NS5A and NS5A phosphorylated at S2201 (pS2201), S2208 (pS2208), S2211 (pS2211), and S2214 (pS2214) in HEK293T cells transfected with the expression constructs. (E) NS5A phosphorylation at S2201, S2208, S2211, and S2214 normalized with total NS5A and relative to values for the wild type. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type).
Mutations that block cleavage at the NS3-4A junction block NS5A hyperphosphorylation at S2201, S2208, S2211, and S2214.
To check whether NS5A hyperphosphorylation is dependent on the cleavage at the NS3-4A junction T1661 (33–35), we measured NS5A hyperphosphorylation in several NS3-5A mutants that either block or permit cleavage at the junction. As seen in Fig. 4A, cysteine or serine mutation at T1661 permitted NS3-4A cleavage and resulted in a single NS3 band (lanes C and S), as in the wild type (lane WT). In contrast, arginine, tyrosine, or aspartate mutation at T1661 blocked NS3-4A cleavage and resulted in an NS3-4A band plus an NS3-like band (Fig. 4A, lanes R, Y, and D). The lower NS3-like band with a molecular weight similar to that of the wild-type NS3 is likely due to promiscuous cleavage in the vicinity of the cleavage junction, as has been shown previously (34). Figure 4B summarizes cleavage efficiency at the NS3-4A junction. Among all constructs, as long as the NS3-4A junction was properly cleaved (Fig. 4A, lanes WT, C, and S), NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214 were observed (Fig. 4C to F, lanes WT, C, and S). When the NS3-4A junction was not properly cleaved (Fig. 4A, lanes R, Y, and D), NS5A hyperphosphorylation and phosphorylation at the above sites were not observed (Fig. 4C to F, lanes R, Y, and D). Figures 4G and H summarize the results. Thus, NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214 require a proper cleavage at the NS3-4A junction.
FIG 4.

Mutations that block NS3-4A cleavage block NS5A hyperphosphorylation. (A) Representative immunoblotting for NS3 in HEK293T cells transfected with NS3-5A constructs with various mutations at the NS3-4A cleavage junction T1661: C, cysteine; D, aspartate; R, arginine; S, serine; Y, tyrosine. β-Actin served as a loading control. (B) Changes (percent) in NS3 over total NS3 (NS3 plus NS3-NS4A). Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type). (C to F) Representative immunoblotting for NS5A and NS5A phosphorylated at S2201 (pS2201), S2208 (pS2208), S2211 (pS2211), and S2214 (pS2214). (G) Changes (percent) in p58 over total NS5A (p56 plus p58) from 3 independent experiments. (H) NS5A phosphorylation at S2201, S2208, S2211, and S2214 normalized with total NS5A and relative to values for the wild type. *, P < 0.05 (determined by t test against the values for the wild type).
NS5A hyperphosphorylation does not require a cleaved NS4A.
The above data (Fig. 4) can also imply a role of a cleaved NS4A in NS5A hyperphosphorylation. That is, when NS4A was not cleaved from the NS3-4A bands (Fig. 4A, lanes R, Y, and D), NS5A was not hyperphosphorylated (Fig. 4C, lanes R, Y, and D). When NS4A was cleaved (Fig. 4A, lanes WT, C, and S), NS5A was hyperphosphorylated (Fig. 4C, lanes WT, C, and S). To test this possibility, we inserted a P2A autoproteolytic sequence between NS3 and NS4A. The P2A sequence should release NS3 from the NS3-5A polyprotein and allow the release of NS4A via trans cleavage by NS3. Figure 5 summarizes the results. As seen, the insertion of the P2A sequence resulted in a rather unstable polyprotein because none of our NS5A antibodies were able to detect the proteins (Fig. 5A to D, lanes P2A). This was perhaps due to random cleavage by the NS3 protease because when the P2A was inserted into the NS3 protease-dead NS3-P2A-5A mutant (P2A/S1169A), two polyprotein bands (NS3-P2A-5A and NS4A-5A) were detected with the NS5A antibody (Fig. 5A to D, lanes P2A/S1169A). The appearance of the NS4A-5A band in the P2A/S1169A mutant (Fig. 5A to D, lanes P2A/S1169A) indicates partial autoproteolysis mediated by the P2A peptide. Ideally, the P2A sequence would be inserted into mutants that block NS3-4A cleavage (i.e., T1661R, T1661Y, or T1661D) for the test. Due to the random cleavage activity of NS3, the experiments were not performed. Nevertheless, very weak hyperphosphorylation at S2201, S2208, S2211, and S2214 was detected in the NS3-P2A-5A polyprotein, indicating that NS5A hyperphosphorylation does not require a cleaved NS4A.
FIG 5.

NS5A hyperphosphorylation does not require a cleaved NS4A. (A to D) Representative immunoblotting for NS5A phosphorylation at S2201, S2208, S2211, and S2214 in HEK293T cells transfected with the wild-type (WT), NS3 protease-dead (S1169A), NS3-4A autoproteolytic (P2A, inserted between NS3 and NS4A), or P2A/S1169A double mutant NS3-5A construct. (E) Bar diagram summary. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type).
NS5A hyperphosphorylation at S2201, S2208, S2211, and S2214 requires cleavage at the NS3-4A, not the NS4A-4B or NS4B-5A, junction.
We next examined whether cleavage at the NS4A-4B (C1715) or NS4B-5A (C1976) junction is required for NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214 (Fig. 6A). For comparison, T1661R and T1661Y mutations, which block proper NS3-4A cleavage and NS5A hyperphosphorylation (Fig. 4A), were included in the analysis. Again, T1661R and T1661Y produced an NS3-4A band plus an NS3-like band (Fig. 6B, lanes T1661R and T1661Y). Although NS5A was properly processed in these mutants, it was not hyperphosphorylated at S2201, S2208, S2211 and S2214 (Fig. 6C to F, lanes T1661R and T1661Y). In the C1715S mutant, which blocks NS4A-4B junction cleavage, NS5A was successfully cleaved and was hyperphosphorylated at the above-mentioned serine residues (Fig. 6C to F, lane C1715S). In the C1976F mutant, which blocks NS4B-5A cleavage, NS5A fused with NS4B and appeared as two bands at about 83 and 85 kDa (Fig. 6C to F, lanes C1976F), i.e., with hypo- and hyperphosphorylation. Under these conditions, NS5A phosphorylation at S2201, S2208, S2211, and S2214 was observed in the NS4B-5A fusion protein at the upper band (Fig. 6C to F, lanes C1976F). Figures 6G and H summarize the results. Thus, successful cleavage at the NS3-4A junction and not the NS4A-4B or NS4B-5A junction is required for NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214. Moreover, NS5A hyperphosphorylation at these serine residues could occur in an NS4B-5A fusion protein.
FIG 6.

Mutation at the NS3-4A, but not the NS4A-4B or NS4B-5A, junction blocks NS5A hyperphosphorylation. (A) Schematics of NS3-5A expression constructs with mutations at the NS3-4A, NS4A-4B, or NS4B-5A cleavage junction. The sites of mutation are numbered and indicated with one-letter amino acid symbols: C, cysteine; D, aspartate; F, phenylalanine; R, arginine; S, serine; T, threonine; Y, tyrosine. (B to F) Representative immunoblotting for NS3, NS5A, and NS5A phosphorylated at S2201 (pS2201), S2208 (pS2208), S2211 (pS2211), and S2214 (pS2214) in HEK293T cells transfected with the constructs. (G) Changes (percent) in p58 over total NS5A (p56 plus p58). Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type). (H) NS5A phosphorylation at S2201, S2208, S2211, and S2214 normalized with total NS5A and relative to values for the wild type. *, P < 0.05 (determined by t test against the values for the wild type).
Combinatory mutations reveal an extra hyperphosphorylated NS4A-NS4B-NS5A polyprotein.
We then asked whether NS5A could be hyperphosphorylated at an even higher fusion level, i.e., NS4A-5A. To this end, we measured NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214 in various mutants that fuse nonstructural proteins at different levels (Fig. 7A). In the T1661Y/C1715S double mutant, which blocks cleavage at the NS3-4A and NS4A-4B junctions, an NS3-4B fusion protein was observed at about 100 kDa (Fig. 7B, lane T1661Y/C1715S). An NS3-like band was also observed again, most likely due to promiscuous cleavage at the vicinity of the NS3-4A junction (34). In this mutant, NS5A was correctly processed but was not hyperphosphorylated or phosphorylated at the serine residues (Fig. 7C to F, lanes T1661Y/C1715S). This is consistent with the requirement of a proper NS3-4A cleavage for NS5A hyperphosphorylation. In the T1661Y/C1976F double mutant, which blocks cleavage at the NS3-4A and NS4B-5A junctions, an NS3-4A fusion protein band plus an NS3-like band was observed (Fig. 7B, lane T1661Y/C1976F), indicating improper NS3-4A cleavage. In this mutant, NS5A fused with NS4B, appeared at 97 kDa and was not hyperphosphorylated (Fig. 7C to F, lanes T1661Y/C1976F). This is in sharp contrast to the results obtained with the C1976F single mutant, which permits proper NS3-4A junction cleavage and hyperphosphorylation of the NS4B-5A fusion protein (Fig. 6C to F, lanes C1976F). Therefore, proper NS3-4A cleavage is a prerequisite to the hyperphosphorylation of an NS4B-5A fusion protein. In the C1715S/C1976F double mutant, which blocks cleavages at the NS4A-4B and NS4B-5A junctions, NS3 was properly cleaved (Fig. 7B, lane C1715S/C1976F), whereas NS5A fused with NS4A and NS4B at about 89 kDa and was hyperphosphorylated at about 91 kDa (Fig. 7C to F, lanes C1715S/C1976F). Thus, NS5A could be hyperphosphorylated in an NS4A-5A fusion protein when NS3-4A is properly cleaved. Triple T1661Y/C1715S/C1976F mutations resulted in an unstable NS3-5A fusion protein barely detected on immunoblots (Fig. 7B to F). In summary, successful cleavage at the NS3-4A junction is required for NS5A hyperphosphorylation and phosphorylation at S2201, S2208, S2211, and S2214. NS5A could be hyperphosphorylated in an NS4A-5A fusion protein.
FIG 7.

Combinatory mutations that block NS3-4A cleavage block NS5A hyperphosphorylation. (A) Schematics of NS3-5A expression constructs with any two or three mutations at the cleavage junctions. The sites of mutations are numbered and indicated with one-letter amino acid symbols. (B to F) Representative immunoblotting for NS3, NS5A, and NS5A phosphorylated at S2201 (pS2201), S2208 (pS2208), S2211 (pS2211), and S2214 (pS2214) in HEK293T cells transfected with the constructs.
NS5A hyperphosphorylation at serines 2201, 2208, 2211, and 2214 requires NS3-mediated cis cleavage at the NS3-4A junction in native HCV host liver cells.
Since all the above observations (Fig. 2 to 7) were made in the transfection-friendly human embryonic kidney cells (HEK293T), we repeated key experiments in the native HCV host liver cells (T7-Huh7) that express T7 polymerase. Figure 8A depicts our pTM constructs that carry the T7 promoter to drive gene expression. Mutation that blocked NS3-4A cleavage (Fig. 8B, lane T1661R) blocked NS5A hyperphosphorylation at S2201, S2208, S2211, and S2214 (Fig. 8C to F, lanes T1661R). In contrast, mutations that permitted NS3-4A cleavage permitted NS5A hyperphosphorylation (Fig. 8B to F, lanes WT, T1661C, C1715S, and C1976F). Figures 8G and H summarize the results. Thus, NS3-4A cleavage is required for NS5A hyperphosphorylation at S2201, S2208, S2211, and S221 in the T7-Huh7 native HCV host liver cells.
FIG 8.

NS5A phosphorylation at serine 2201, 2208, 2211, and 2214 requires NS3-mediated autocleavage at the NS3-4A junction in the native HCV host liver cells. (A) Schematics of four pTM constructs that permit expression of nonstructural proteins in the T7-Huh7 native HCV host liver cells: T1661C (NS3-4A cleavable), T1661R (NS3-4A noncleavable), C1715S (NS4A-4B noncleavable), and C1976F (NS4B-5A noncleavable). (B to F) Immunoblotting for NS3, NS5A, and NS5A phosphorylation at S2201, S2208, S2211, and S2214. (G and H) Summary diagrams. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type [WT]).
NS5A hyperphosphorylation at serines 2204, 2207, and 2210 requires proper cleavage at the NS3-4A junction for genotype 1b HCV.
As all the above-described observations were made with genotype 2a, we examined whether NS5A hyperphosphorylation requires proper cleavage at the NS3-4A junction in genotype 1b. We first compared phosphorylation levels of NS5A in genotype 1b versus 2a. The total NS5A protein level was significantly lower in genotype 1b than in genotype 2a (Fig. 9A to E), reflecting a low expression level of NS5A protein or a poor detection ability of the NS5A antibody (9E10). The genotype 1b NS5A appeared at the lower portion of the immunoblots because it is 19 amino acids shorter than the genotype 2a NS5A. The phosphorylation levels of genotype 1b NS5A at various serine residues were also lower than those of genotype 2a NS5A (Fig. 9A to D). The low S2197 (numbering shift to genotype 1b) phosphorylation level (Fig. 9A) is likely a result of a poor detection ability of the antibody that was raised against the genotype 2a NS5A sequence (RGSPPSEASSS), where a negatively charged glutamate was replaced with a hydrophobic valine in the genotype 1b NS5A sequence (RGSPPSVASSS). Theoretically, the detection abilities of the rest of the three phosphorylation-specific antibodies should be similar for genotypes 1b and 2a because the sequences are identical in these regions except for a positively charged lysine in 1b that was replaced with a positively charged arginine in 2a (Fig. 1A). However, the phosphorylation levels in S2204, S2207, and S2210 were still much lower in genotype 1b than in genotype 2a (Fig. 9B to D), likely due to a low NS5A expression level. Nevertheless, the relative phosphorylation levels against total NS5A were similar for genotypes 1b and 2a (Fig. 9F).
FIG 9.
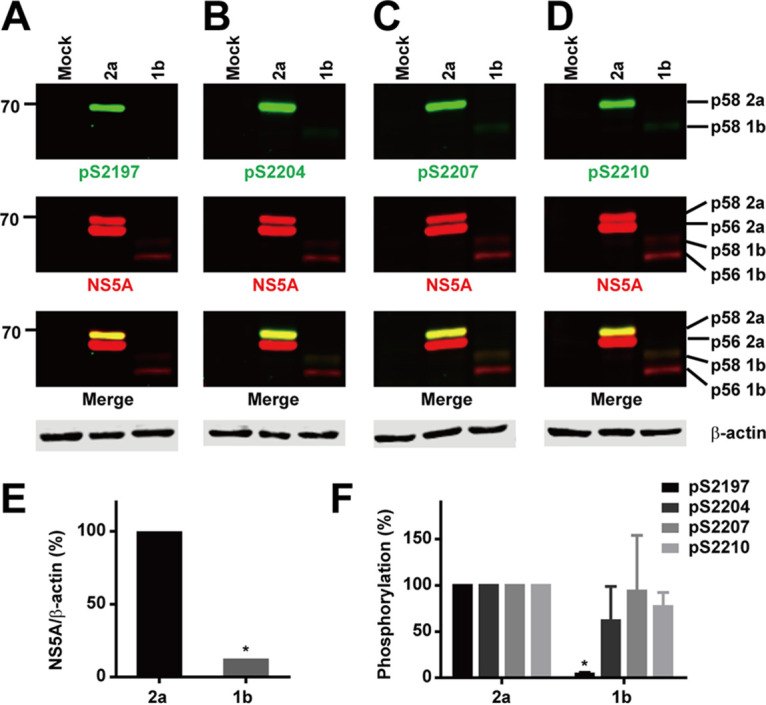
Similar NS5A phosphorylation levels at serines 2204, 2207, and 2210 in genotype 1b. (A to D) Immunoblotting for NS5A and NS5A phosphorylation at S2197, S2204, S2207, and S2210 in HEK293T cells transfected with wild-type NS3-5A constructs from genotype 2a (JFH1) or 1b (Con1). (E and F) Summary diagrams. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for genotype 2a).
We then examined whether NS5A hyperphosphorylation requires proper cleavage at the NS3-4A junction in genotype 1b. Due to its low NS5A expression level, we enhanced the signals, and hence noises appeared in some images. As seen in Fig. 10A, phosphorylation at S2197 was barely detected, whereas phosphorylation at S2204, S2207, and S2210 was apparent (Fig. 10B to D). Here, NS5A hyperphosphorylation was observed as long as proper NS3-4A cleavage was permitted (Fig. 10B to D, lanes WT, T1657C, C1711S, and C1972F). A protease-dead mutation (Fig. 10B to D, lanes S1165A) or a noncleavable mutation at the NS3-4A junction (lanes T1657R) blocked NS5A hyperphosphorylation. Thus, proper cleavage at the NS3-4A junction is required for NS5A hyperphosphorylation for both genotypes 1b and 2a.
FIG 10.

Genotype 1b NS5A phosphorylation at serines 2204, 2207, and 2210 requires NS3-mediated autocleavage at the NS3-4A junction. (A to D) Immunoblotting for NS5A and NS5A phosphorylation at S2197, S2204, S2207, and S2210 in HEK293T cells transfected with genotype 1b NS3-5A constructs: WT (wild type), S1165A (NS3 protease dead), T1657C (cleavable NS3-4A), T1657R (noncleavable NS3-4A), C1711S (noncleavable NS4A-4B), or C1972F (noncleavable NS4B-5A). (E) Summary diagram. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type).
Reporter HCV activity requires proper processing of the nonstructural proteins.
We next examined how cleavage at the junctions might affect reporter HCV (RLuc-J6/JFH-1) activity. As summarized in Fig. 11, the reporter activity increased with time in the Huh7.5.1 cells transfected with the wild-type HCV. Similar increases were observed in the cells transfected with the T1661C mutant HCV, which permits proper NS3 cleavage (Fig. 4B) and NS5A hyperphosphorylation (Fig. 4G and H). In contrast, the T1661R mutant, which blocks proper NS3 processing (Fig. 4B) and NS5A hyperphosphorylation (Fig. 4G and H), failed to produce reporter activity. Mutants that block cleavage at the NS4A-4B or NS4B-5A also failed to produce reporter activity. Thus, a complete HCV life cycle requires proper processing of all nonstructural proteins.
FIG 11.
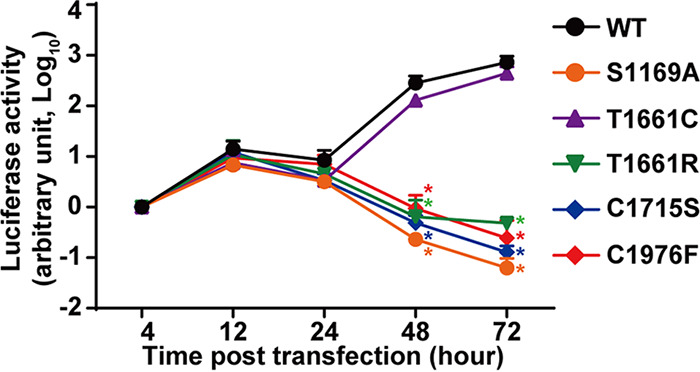
The HCV life cycle requires proper processing of the NS3-5A polyprotein. Luciferase activity of the reporter wild-type HCV and HCV with the indicated mutations is shown. Cell lysates from Huh 7.5.1 cells transfected with the reporter HCV in vitro transcripts were collected at the indicated time points for measurements. The luciferase activities were normalized against those at the 4-h time point. Values are means ± SEM (n = 3). *, P < 0.05 (determined by t test against the values for the wild type [WT] at the same time points).
DISCUSSION
Hyperphosphorylation of the NS5A protein is a necessary condition for a productive HCV life cycle, especially for the genotype 2 virus (16–18, 21, 24, 25). We and others have uncovered that NS5A transits from a hypophosphorylated to a hyperphosphorylated state via sequential phosphorylation of a series of highly conserved serine residues (19–21, 23, 36). Specifically, we found that S2201, phosphorylated by an unknown protein kinase, fires casein kinase I α-mediated sequential S2208/S2211/S2214 phosphorylation, thereby leading to NS5A hyperphosphorylation (19, 20). of special note, Lohmann’s group recently uncovered T2218 phosphorylation in a small fraction of hyperphosphorylated NS5A species (37). T2218 phosphorylation requires phosphorylation at T2220 and T2223 at the COOH terminus, suggesting an alternative phosphorylation cascade in the COOH- to NH2-terminal direction. Protein phosphorylation status is often balanced by protein kinases and phosphatases. In the case of HCV NS5A protein phosphorylation, other proteins, including the lipid kinase phosphatidylinositol 4-kinase III α and the NS3 serine protease, are also involved (15, 26, 31, 32). In the present study, we first showed that the NS3 protease on the very same NS3-5A polyprotein molecule is required for NS5A hyperphosphorylation (Fig. 2) (31, 32). In addition, we found that proper NS3-mediated cis cleavage at the NS3-4A junction is required for NS5A hyperphosphorylation (Fig. 2). As long as mutations permit proper in cis NS3-4A junction cleavage, NS5A hyperphosphorylation occurs (Fig. 4). Mutations that block NS4A-4B or NS4B-5A cleavage do not affect NS5A hyperphosphorylation (Fig. 6). With an inventory of phosphorylation-specific antibodies (Fig. 1) (16, 19, 21), we further showed that NS3-mediated cis cleavage at the NS3-4A junction is required for NS5A phosphorylation at S2201, S2208, S2211, and S2214 (Fig. 3). This is in line with the role of in cis NS3-4A junction cleavage in S2201 phosphorylation, which subsequently fires sequential S2208/S2211/S2214 phosphorylation. Consistent with sequential S2201/S2208/S2211/S2214 phosphorylation, an upstream alanine mutation at S2201 eliminates downstream phosphorylation at S2208, S2211, and S2214 (19). A downstream alanine mutation at S2211 does not eliminate upstream S2201 and S2208 phosphorylation (Fig. 3). Overall, we offered a new S2201 phosphorylation-specific antibody to the field (Fig. 1) and uncovered the requirement of NS3-mediated in cis NS3-4A cleavage in NS5A sequential phosphorylation and hyperphosphorylation for both genotypes 2a (Fig. 2 to 8) and 1b (Fig. 9 and 10).
For the first time, we found that NS5A can be hyperphosphorylated and phosphorylated at S2201, S2208, S2211, and S2214 in a mutant NS4A-5A polyprotein fused via mutations that block cleavages at the NS4A-4B and NS4B-5A junctions (Fig. 7). These results suggest that NS5A sequential phosphorylation and hyperphosphorylation could probably occur before or while NS5A is released from the NS4A-5A polyprotein.
It is interesting to note that NS5A hyperphosphorylation and phosphorylation at the above-mentioned serine residues also occur in a mutant NS4B-5A polyprotein fused via a mutation that blocks cleavage at the NS4B-5A junction (Fig. 6). It has been reported that the cis cleavage at the NS3-4A junction occurs prior to all trans cleavages at the NS4A-4B, NS4B-5A, and NS5A-5B junctions (7). While Tanji et al. reported trans cleavages occurring in the order NS5A-5B, NS4B-5A, and NS4A-4B (8), Lin et al. found no particular order in the trans cleavages (9). Specifically, the latter study found that NS4A was a required cofactor of NS3 for NS4B-5A cleavage; i.e., NS4A-4B cleavage should have already occurred in order to release NS4A prior to NS4B-5A cleavage. Thus, the order of NS3-mediated trans cleavages is inconclusive. Regardless of the cleavage order, our data suggest that NS5A could be hyperphosphorylated and phosphorylated at S2201, S2208, S2211, and S2214 before trans cleavages. The virological significance of our findings requires further investigation.
We cannot comment on the effects or timing of NS5B cleavage on NS5A hyperphosphorylation because we did not include NS5B in the constructs. NS5B has been implicated in NS5A hyperphosphorylation (15, 38). For example, polymerase-disabled NS5B has been shown to increase NS5A hyperphosphorylation by enhancing the translation of nonstructural proteins (38). Through interactions with NS5A and the lipid kinase phosphatidylinositol-4 kinase IIIα, NS5B activates the lipid kinase, which through intricate mechanisms influences NS5A phosphorylation (15). Our decision not to include NS5B in the study was based upon the fact that NS5A hyperphosphorylation requires NS3-5A carried on one single polyprotein (31, 32). In those studies, NS5B was found to be dispensable for NS5A hyperphosphorylation. Thus, we could not infer any direct or indirect influence of NS5B on NS5A hyperphosphorylation from our experimental design.
There are several possible mechanisms by which cis cleavage at the NS3-4A junction could affect NS5A hyperphosphorylation. One possibility is the recruitment of protein kinases during the cis-cleavage process. NS3 interacts with nonstructural proteins, including NS5A, for the HCV genome replication complex (2). When protein kinases temporarily bind to the transitional NS3 structure during NS3-4A junction cleavage, they gain access to phosphorylate NS5A. The fact that NS3-4A undergoes drastic architectural changes to regulate its proteolytic activity, membrane association, and virological functions (39) supports this view. The detection of NS5A hyperphosphorylation in the P2A/S1169A NS3-P2A-5A mutant polyprotein (Fig. 5, lanes P2A/S1169A) also supports the above view. In the P2A/S1169A mutant, the protease-dead NS3 bends over and attempts to cleave the NS3-4A junction. This creates a transitional structure that could recruit protein kinases for NS5A phosphorylation. After phosphorylation, the kinases dissociate from the transitional NS3 structure and the cleavage is completed. Another possibility is that the cleavage of the NS3-4A junction induces a global conformational change in the NS4A-5A polyprotein (40), exposing the serine residues, preferentially S2201, for phosphorylation followed by sequential S2208/S2211/S2214 phosphorylation. Identification of the kinase responsible for S2201 phosphorylation is of great translational potential.
In conclusion, we showed that NS3-mediated cis cleavage at the NS3-4A junction permitted NS5A hyperphosphorylation at serines 2201, 2208, 2211, and 2214, thereby leading to hyperphosphorylation, which is a necessary condition for genotype 2 HCV replication. NS5A may already be phosphorylated at these serine residues right after NS3-4A cleavage or even before NS5A is released from the NS4A-5A polyprotein. These data suggest that the dual-functional NS3, i.e., a protease and an ATP-binding RNA helicase, could have a direct or indirect role in NS5A hyperphosphorylation.
MATERIALS AND METHODS
Cell culture.
Human hepatocarcinoma 7.5.1 (Huh7.5.1), T7 polymerase-harboring human hepatocellular carcinoma (T7-Huh7), and human embryonic kidney 293 (HEK293T) cells were cultured in Dulbecco’s modified minimal essential medium (DMEM) (Invitrogen, catalog 12100-046) with 10% fetal bovine serum (Biological Industries, catalog 040011B) without antibiotics in a 5% CO2 incubator (Astec Inc.) at 37°C.
Phosphorylation-specific antibody.
An antibody specific to NS5A S2201 phosphorylation was custom-made by GeneTex Corporation using a synthetic phosphorylated peptide (RGSPPpSEASSS) injected into two rabbits. Rabbit sera went through two affinity purification steps. In the first step, the phosphorylated peptide was used to capture antibodies in the eluate fraction that recognize phosphorylated epitopes and nonphosphorylated epitopes in the immunizing phosphorylated peptide. In the second step, a nonphosphorylated peptide (RGSPPSEASSS) was used to deplete antibodies that recognize nonphosphorylated epitopes from the flowthrough fraction. Antibody specific to NS5A phosphorylation at S2208 (19), S2211 (16), or S2214 (21) was made in a similar way and was characterized previously in our laboratory. The specificity of these antibodies was examined with dot blot analysis using phosphorylated and nonphosphorylated peptides spotted onto a nitrocellulose membrane prior to detection.
Immunoblotting.
Immunoblotting was carried out as described previously (16). Primary antibodies included those for NS5A (9E10 [41]), NS5A (7B5, BioFront Technologies), β-actin (A5316, Sigma-Aldrich), and NS3 (E3, BioFront Technologies). Secondary antibodies included anti-mouse IgG IRDye680 (P/N 926-68070) and anti-rabbit IgG IRDye800 (P/N 926-32213) from Li-Cor (NE, USA). Proteins of interest were visualized and quantified with the Li-Cor Odyssey scanner and software.
HCV infection.
Infectious HCV (J6/JFH-1, genotype 2a) was produced as described previously (27). It was used to infect Huh7.5.1 cells at a multiplicity of infection of 0.01. Proteins were harvested at various time points postinfection for immunoblotting.
Plasmids and transfection.
The wild-type NS3-5A (pcDNA3.1-NS3-5A), NS3-4A (pcDNA3.1-NS3-4A), and pTM NS3-5A expression constructs for the genotype 2a JFH1 isolate (16, 21) or 1b BM4-5 variant (42) were made in the First Core Facility, National Taiwan University College of Medicine, Taipei, Taiwan. The BM4-5 variant lacks serine 2201 (S2205 in genotype 2a), which was added during our construction processes. Site-directed mutations were made from these constructs. In some cases, an autoproteolytic P2A sequence (ATNFSLLKQAGDVEENPGP) was inserted between NS3 and NS4A in the pcDNA3.1-NS3-5A construct. The entire NS3-5A region was sequenced to ensure that only the intended site was mutated. Plasmid transfection was done with the Lipofectamine 3000 reagent (Invitrogen, L3000015) following the manufacturer’s instruction.
Reporter HCV assay.
Procedures for the reporter HCV assay were described in detail previously (16). The reporter HCV construct, to which site-directed mutations were made, was a gift from Charles Rice (43). The reporter HCV construct was linearized for in vitro transcript production and purification prior to transfection. On the day of the experiment, the Huh 7.5.1 cells were seeded at a density that would reach about 80% confluence on the next day for transfection. The viral transcript was mixed with DMRIE-C (Invitrogen, 10459-014) at a ratio of 1 μg to 3 μl for transfection. Cell lysates were collected at various time points posttransfection for luciferase assay using a luciferase assay kit (Promega, E1500) following the manufacturer’s instructions.
ACKNOWLEDGMENTS
We thank the First Core Facility, National Taiwan University College of Medicine, for expertise in molecular biology.
This work was supported by grant NHRI-EX108-10620BI, National Health Research Institutes.
REFERENCES
- 1.Gotte M, Feld JJ. 2016. Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat Rev Gastroenterol Hepatol 13:338–351. doi: 10.1038/nrgastro.2016.60. [DOI] [PubMed] [Google Scholar]
- 2.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. 2006. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature 442:831–835. doi: 10.1038/nature04975. [DOI] [PubMed] [Google Scholar]
- 5.Tomei L, Failla C, Santolini E, De Francesco R, La Monica N. 1993. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J Virol 67:4017–4026. doi: 10.1128/JVI.67.7.4017-4026.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. 1994. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J Virol 68:5045–5055. doi: 10.1128/JVI.68.8.5045-5055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lohmann V, Koch JO, Bartenschlager R. 1996. Processing pathways of the hepatitis C virus proteins. J Hepatol 24:11–19. [PubMed] [Google Scholar]
- 8.Tanji Y, Hijikata M, Hirowatari Y, Shimotohno K. 1994. Hepatitis C virus polyprotein processing: kinetics and mutagenic analysis of serine proteinase-dependent cleavage. J Virol 68:8418–8422. doi: 10.1128/JVI.68.12.8418-8422.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin C, Pragai BM, Grakoui A, Xu J, Rice CM. 1994. Hepatitis C virus NS3 serine proteinase: trans-cleavage requirements and processing kinetics. J Virol 68:8147–8157. doi: 10.1128/JVI.68.12.8147-8157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolk B, Sansonno D, Krausslich HG, Dammacco F, Rice CM, Blum HE, Moradpour D. 2000. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J Virol 74:2293–2304. doi: 10.1128/jvi.74.5.2293-2304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/jvi.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behrens SE, Tomei L, De Francesco R. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J 15:12–22. doi: 10.1002/j.1460-2075.1996.tb00329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross-Thriepland D, Harris M. 2015. Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol 96:727–738. doi: 10.1099/jgv.0.000009. [DOI] [PubMed] [Google Scholar]
- 14.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong WM, Hsu SC, Kao WT, Lo CW, Lee KY, Shao JS, Chen YH, Chang J, Chen SS, Yu MJ. 2016. Phosphoproteomics identified an NS5A phosphorylation site involved in hepatitis C virus replication. J Biol Chem 291:3918–3931. doi: 10.1074/jbc.M115.675413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, Gao M. 2013. Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J Virol 87:2320–2329. doi: 10.1128/JVI.02861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masaki T, Matsunaga S, Takahashi H, Nakashima K, Kimura Y, Ito M, Matsuda M, Murayama A, Kato T, Hirano H, Endo Y, Lemon SM, Wakita T, Sawasaki T, Suzuki T. 2014. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-alpha in infectious virus production. J Virol 88:7541–7555. doi: 10.1128/JVI.03170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu SC, Tsai CN, Lee KY, Pan TC, Lo CW, Yu MJ. 2018. Sequential S232/S235/S238 phosphorylation of the hepatitis C virus nonstructural protein 5A. J Virol 92:e01295-18. doi: 10.1128/JVI.01295-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai CN, Pan TC, Chiang CH, Yu CC, Su SH, Yu MJ. 2019. Serine 229 balances the hepatitis C virus nonstructural protein NS5A between hypo- and hyperphosphorylated states. J Virol 93:e01028-19. doi: 10.1128/JVI.01028-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu SC, Lo CW, Pan TC, Lee KY, Yu MJ. 2017. Serine 235 is the primary NS5A hyperphosphorylation site responsible for hepatitis C virus replication. J Virol 91:e00194-17. doi: 10.1128/JVI.00194-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolk B, Buchele B, Moradpour D, Rice CM. 2008. A dynamic view of hepatitis C virus replication complexes. J Virol 82:10519–10531. doi: 10.1128/JVI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quintavalle M, Sambucini S, Summa V, Orsatti L, Talamo F, De Francesco R, Neddermann P. 2007. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J Biol Chem 282:5536–5544. doi: 10.1074/jbc.M610486200. [DOI] [PubMed] [Google Scholar]
- 24.Goonawardane N, Gebhardt A, Bartlett C, Pichlmair A, Harris M. 2017. Phosphorylation of serine 225 in hepatitis C virus NS5A regulates protein-protein interactions. J Virol 91:e00805-17. doi: 10.1128/JVI.00805-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross-Thriepland D, Mankouri J, Harris M. 2015. Serine phosphorylation of the hepatitis C virus NS5A protein controls the establishment of replication complexes. J Virol 89:3123–3135. doi: 10.1128/JVI.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harak C, Meyrath M, Romero-Brey I, Schenk C, Gondeau C, Schult P, Esser-Nobis K, Saeed M, Neddermann P, Schnitzler P, Gotthardt D, Perez-Del-Pulgar S, Neumann-Haefelin C, Thimme R, Meuleman P, Vondran FW, De Francesco R, Rice CM, Bartenschlager R, Lohmann V. 2016. Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat Microbiol 2:16247. doi: 10.1038/nmicrobiol.2016.247. [DOI] [PubMed] [Google Scholar]
- 27.Lee KY, Chen YH, Hsu SC, Yu MJ. 2016. Phosphorylation of serine 235 of the hepatitis C virus nonstructural protein NS5A by multiple kinases. PLoS One 11:e0166763. doi: 10.1371/journal.pone.0166763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan TC, Lo CW, Chong WM, Tsai CN, Lee KY, Chen PY, Liao JC, Yu MJ. 2019. Differential proteomics reveals discrete functions of proteins interacting with hypo- versus hyper-phosphorylated NS5A of the hepatitis C virus. J Proteome Res 18:2813–2825. doi: 10.1021/acs.jproteome.9b00130. [DOI] [PubMed] [Google Scholar]
- 29.Chen YC, Su WC, Huang JY, Chao TC, Jeng KS, Machida K, Lai MM. 2010. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J Virol 84:7983–7993. doi: 10.1128/JVI.00068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neddermann P, Clementi A, De Francesco R. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J Virol 73:9984–9991. doi: 10.1128/JVI.73.12.9984-9991.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch JO, Bartenschlager R. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J Virol 73:7138–7146. doi: 10.1128/JVI.73.9.7138-7146.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Lahser FC, Yi M, Wright-Minogue J, Xia E, Weber PC, Lemon SM, Malcolm BA. 2004. Conserved C-terminal threonine of hepatitis C virus NS3 regulates autoproteolysis and prevents product inhibition. J Virol 78:700–709. doi: 10.1128/jvi.78.2.700-709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou X, Yang W, Zhao Y, Agarwal A, Huang M. 2009. Internal cleavages of hepatitis C virus NS3 induced by P1 mutations at the NS3/4A cleavage site. Virology 383:271–278. doi: 10.1016/j.virol.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 35.Bartenschlager R, Ahlborn-Laake L, Yasargil K, Mous J, Jacobsen H. 1995. Substrate determinants for cleavage in cis and in trans by the hepatitis C virus NS3 proteinase. J Virol 69:198–205. doi: 10.1128/JVI.69.1.198-205.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross-Thriepland D, Harris M. 2014. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J Virol 88:1421–1432. doi: 10.1128/JVI.03017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schenk C, Meyrath M, Warnken U, Schnolzer M, Mier W, Harak C, Lohmann V. 2018. Characterization of a threonine-rich cluster in hepatitis C virus nonstructural protein 5A and its contribution to hyperphosphorylation. J Virol 92:e00737-18. doi: 10.1128/JVI.00737-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick CJ, Brown D, Griffin S, Challinor L, Rowlands DJ, Harris M. 2006. A link between translation of the hepatitis C virus polyprotein and polymerase function; possible consequences for hyperphosphorylation of NS5A. J Gen Virol 87:93–102. doi: 10.1099/vir.0.81180-0. [DOI] [PubMed] [Google Scholar]
- 39.Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc Natl Acad Sci U S A 105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7:1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 41.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 42.Guo JT, Bichko VV, Seeger C. 2001. Effect of alpha interferon on the hepatitis C virus replicon. J Virol 75:8516–8523. doi: 10.1128/jvi.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]