

to the editor: The Aicardi–Goutières syndrome is a genetic interferonopathy that is associated with severe disability and death.1 Most children with this syndrome are unable to walk or talk and have multisystemic complications, including skin inflammation (Fig. 1A). Janus kinase (JAK) inhibitors may be effective in blocking interferon activation in patients with the Aicardi–Goutières syndrome.2–4
Figure 1. Changes in Symptoms, Interferon Gene Expression, and Developmental Skills in Children with the Aicardi–Goutières Syndrome.
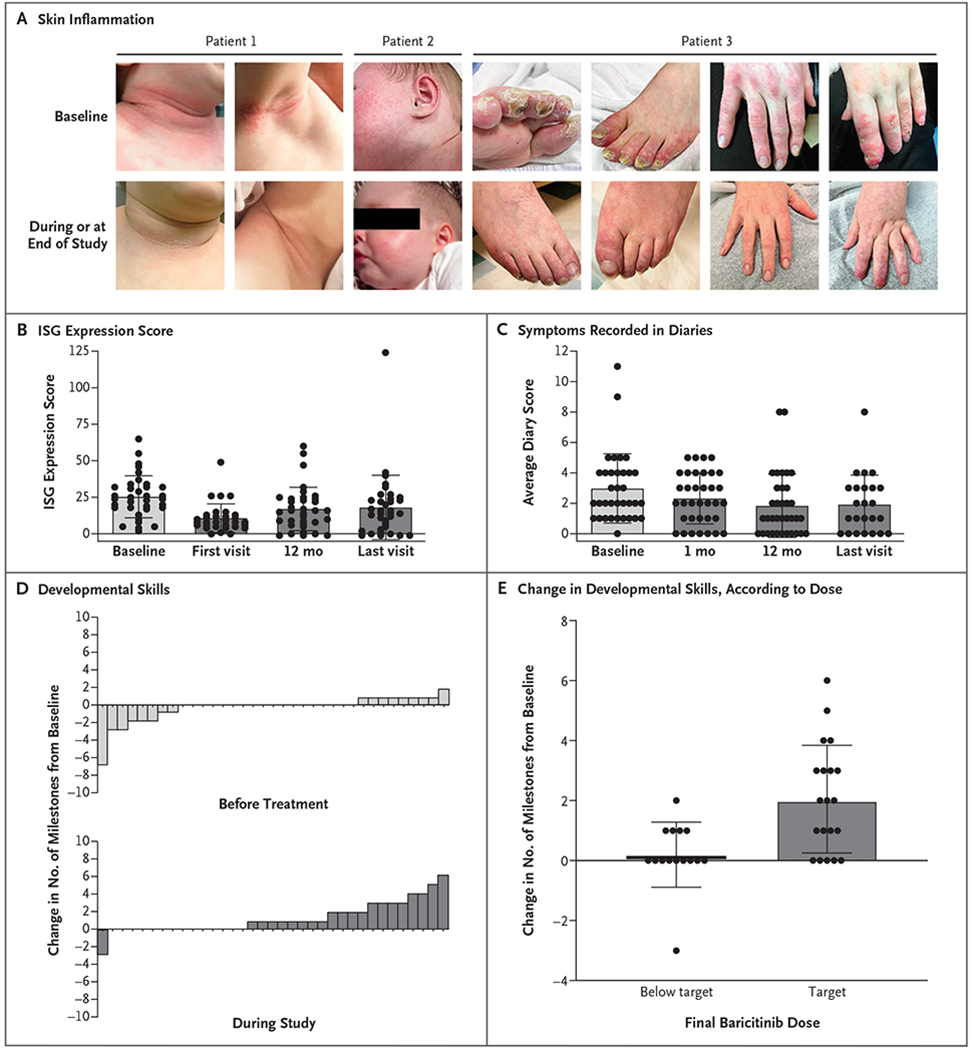
As shown in Panel A, a decrease in symptoms during the study was indicated by reduced skin inflammation in the neck, face, toes, and hands of the patients. In Patient 1, the findings shown are from 3 days after the initiation of treatment; in Patient 2, from 1 month after the initiation of treatment; and in Patient 3, from the 18-month visit (the clinical cutoff date for data analysis). As shown in Panel B, the interferon-signaling gene (ISG) expression scores decreased during treatment. This score is calculated from the expression of six interferon-signaling genes normalized to housekeeping genes. The six-gene interferon signature is the sum of the median z scores of these genes. The interferon signature is positive (i.e., type 1 interferon signaling is high) if it is at least 1.96 (>98th percentile) on a one-tailed test (see Section 3.1.6 in the Supplementary Appendix). As shown in Panel C, symptom scores recorded in diaries by the patients’ parents decreased during treatment. Recorded symptoms were neurologic disability, crying, sleep disturbances, irritability, seizures, fever, and skin inflammation of the trunk, arms, and legs (see the Supplemental Methods section and Table S5 in the Supplementary Appendix). As shown in Panel D, developmental milestones in the individual patients were met during treatment. Each bar represents an individual patient. As shown in Panel E, the gain of new milestones correlated with the dose of baricitinib. Target doses varied according to weight-based norms (see the Supplementary Methods section in the Supplementary Appendix). In Panels B, C, and E, each circle represents an individual patient. Horizontal lines indicate means, and I bars standard deviations.
We conducted an open-label study of a single-center, expanded-access program (ClinicalTrials.gov numbers, NCT01724580 and NCT03047369; see the protocol, available with the full text of this letter at NEJM.org) involving 35 patients with genetically confirmed Aicardi–Goutières syndrome. The patients received baricitinib, an oral JAK1 and JAK2 inhibitor. Eli Lilly provided the medication for the study and provided support for safety monitoring.
The median age at the onset of symptoms was 6.0 months, and the median age at the initiation of baricitinib was 2.9 years (range, 0.2 to 21.8). The patients participated in the study for a minimum of 12.0 months (range, 11.8 to 43.8). The dose of baricitinib, which ranged from 0.1 to 0.6 mg per kilogram of body weight per day, administered in 2 to 4 dosing increments per day, was determined according to the patient’s renal function, age, and symptoms.
JAK inhibitors are associated with infection, anemia, lymphopenia, and thrombosis. Two patients died during the study: one who had been receiving glucocorticoids for a decade, including a 7-year period before the study, had Aicardi–Goutières syndrome–related multisystem organ failure with an opportunistic infection, and the other had Aicardi–Goutières syndrome–related pulmonary hypertension with thrombotic microangiopathy.5 Clinical variables were measured with laboratory tests and graded according to Common Terminology Criteria for Adverse Events, version 5.0, in which grades range from 1 to 5, with higher grades indicating more severe adverse events. Among these variables, only thrombocytosis increased in severity during the study (mean change in grade from baseline, 0.30; 95% confidence interval [CI], 0.04 to 0.60).
A biomarker for interferon signaling, the interferon-signaling gene-expression score, indicated a response to treatment (Fig. 1B). This score is calculated from the expression of six interferon-signaling genes normalized to housekeeping genes. The six-gene interferon signature is the sum of the median z scores of these genes. The interferon signature is positive (i.e., type 1 interferon signaling is high) if it is at least 1.96 (>98th percentile) on a one-tailed test (see the Supplemental Methods section in the Supplementary Appendix, available at NEJM.org).
The parents of the patients used diaries to record the children’s Aicardi–Goutières syndrome–related symptoms, including neurologic disability, crying, sleep disturbances, irritability, seizures, fever, and skin inflammation of the trunk, arms, and legs (see the Supplemental Methods section and Table S5 in the Supplementary Appendix). The diary score included a total of 24 possible points, assigned every day, with daily scores averaged across the time period before each visit (1 month after the initiation of baricitinib and then every 3 months). Scores obtained during the study ranged from 0 to 10.6, with higher scores indicating more severe symptoms. The skin subscore included a total of 6 possible points, with higher scores indicating more severe symptoms. The overall diary scores improved within 1 month after the initiation of therapy (mean change in overall score, −0.7 diary points; 95% CI, −1.2 to −0.2) and were sustained (mean change in overall score, −1.4; 95% CI, −2.2 to −0.6) (Fig. 1A and 1C). The percentage of patients with a lower (improved) skin subscore of only 0 or 1 increased from 66% (23 of 35 patients) to 83% (29 of 35 patients) from the baseline visit to the final visit (difference, 17 percentage points; 95% CI, 2 to 32).
We evaluated the patients’ developmental histories from disease onset (median age since disease onset, 16.5 months; interquartile range, 7.4 to 41.5) to the clinical data cutoff date (Fig. 1D). We extracted information on key milestones, including head control, sitting, rolling, assisted and independent ambulation, grasp, smiling, babbling, and the use of single words and word combinations. Before treatment, 26 of the 35 patients had stable or declining neurologic function, and 9 of the 35 patients gained one or two skills after disease onset. During the study, 20 patients met new milestones, and 12 patients gained two to seven new skills. This improvement was noted by 3 months (mean change in the number of milestones, 0.5; 95% CI, 0.2 to 0.9) and persisted (mean change, 1.3; 95% CI, 0.6 to 1.9). One patient lost skills during an acute illness while participating in the study. Children in a higher daily dose category of baricitinib met more milestones (mean increase, 1.3 milestones; 95% CI, 0.6 to 1.9) than those in a lower daily dose category according to weight-based calculations (Fig. 1E).
The primary risks associated with baricitinib among patients with the Aicardi–Goutières syndrome were thrombocytosis, leukopenia, and infection (see Section 4.1.2 in the Supplementary Appendix). Patients who are receiving baricitinib should be monitored closely, especially those with underlying thrombotic risk factors or those who are receiving systemic glucocorticoids or immunosuppressive regimens. Because this study drew from an international population, we anticipate that our findings will be generalizable. The measurement of neurologic improvement is complex, but our data suggest improvement in neurologic function, even in patients with severe and long-standing disease.
Supplementary Material
THIS WEEK’S LETTERS.
986 Janus Kinase Inhibition in the Aicardi–Goutières Syndrome
989 Neuropathological Features of Covid-19
992 Remdesivir for the Treatment of Covid-19 — Preliminary Report
e66 Invasive or Conservative Strategy for Stable Coronary Disease
Acknowledgments
The content of this letter is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supported by Eli Lilly and by grants from the National Institute of Neurological Disorders and Stroke (NINDS U01NS106845, to Drs. Vanderver, Adang, Gavazzi, Collins, Keller, Takanohashi, and Shults; and K23NS114113, to Dr. Adang); the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD U01HD082806, to Drs. Vanderver and Takanohashi); the National Center for Advancing Translational Sciences (KL2TR001879, to Dr. Adang); the National Heart, Lung, and Blood Institute (K08-HL140129, to Dr. Frank); the Commonwealth Universal Research Enhancement Program of the State of Pennsylvania (SAP 4100077047, to Drs. Vanderver and Adang, Ms. McDonald, and Dr. Shults); the J.A. Kamens Chair in Translational Neurotherapeutics from the Children’s Hospital of Philadelphia (to Dr. Vanderver); the Parker B. Francis Foundation (to Dr. Frank); and the Department of Pediatrics at the Children’s Hospital of Philadelphia (to Dr. Jaffe).
Footnotes
Disclosure forms provided by the authors are available with the full text of this letter at NEJM.org.
Contributor Information
Adeline Vanderver, Children’s Hospital of Philadelphia, Philadelphia, PA
Laura Adang, Children’s Hospital of Philadelphia, Philadelphia, PA
Francesco Gavazzi, Children’s Hospital of Philadelphia, Philadelphia, PA
Katherine McDonald, Children’s Hospital of Philadelphia, Philadelphia, PA
Guy Helman, Royal Children’s Hospital Melbourne, Melbourne, VIC, Australia
David B. Frank, Children’s Hospital of Philadelphia, Philadelphia, PA
Nicole Jaffe, Children’s Hospital of Philadelphia, Philadelphia, PA
Sabrina W. Yum, Children’s Hospital of Philadelphia, Philadelphia, PA
Abigail Collins, Children’s Hospital Colorado, Aurora, CO
Stephanie R. Keller, Children’s Healthcare of Atlanta, Atlanta, GA
Pierre Lebon, Faculté de Médecine Université Paris Descartes, Paris, France
Jean-François Meritet, Hôpital Cochin, Paris, France
Jullie Rhee, Children’s National Health System, Washington, DC
Asako Takanohashi, Children’s Hospital of Philadelphia, Philadelphia, PA
Thais Armangue, Sant Joan de Déu Children’s Hospital, Barcelona, Spain
Nicole Ulrick, Children’s Hospital of Philadelphia, Philadelphia, PA
Omar Sherbini, Children’s Hospital of Philadelphia, Philadelphia, PA.
Jamie Koh, Children’s Hospital of Philadelphia, Philadelphia, PA
Kyle Peer, Children’s Hospital of Philadelphia, Philadelphia, PA
Constance Besnier, Children’s Hospital of Philadelphia, Philadelphia, PA
Carly Scher, Children’s Hospital of Philadelphia, Philadelphia, PA
Katherine Boyle, Children’s Hospital of Philadelphia, Philadelphia, PA
Holly Dubbs, Children’s Hospital of Philadelphia, Philadelphia, PA
Julia Kramer-Golinkoff, Children’s Hospital of Philadelphia, Philadelphia, PA
Amy Pizzino, Children’s Hospital of Philadelphia, Philadelphia, PA
Sarah Woidill, Children’s Hospital of Philadelphia, Philadelphia, PA
Justine Shults, University of Pennsylvania, Philadelphia, PA
References
- 1.Rice G, Patrick T, Parmar R, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet 2007;81:713–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Briand C, Frémond M-L, Bessis D, et al. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann Rheum Dis 2019;78:431–3. [DOI] [PubMed] [Google Scholar]
- 3.Kothur K, Bandodkar S, Chu S, et al. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation. Neurology 2018;90:289–91. [DOI] [PubMed] [Google Scholar]
- 4.Montealegre Sanchez GA, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128:3041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adang LA, Frank DB, Gilani A, et al. Aicardi Goutières syndrome is associated with pulmonary hypertension. Mol Genet Metab 2018;125:351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.