

Abstract
While zebrafish has for some time been regarded as a powerful model organism with which to study early events in hematopoiesis, recent evidence suggests that it also ideal for unraveling the molecular requirements for T cell development in the thymus. Like mammals, zebrafish possess an adaptive immune system, comprising B lymphocytes as well as both the γδ and αβ lineages of T cells, which develop in the thymus. Moreover, the molecular processes underlying T cell development in zebrafish appear to be remarkably conserved. Thus, findings in the zebrafish model will be of high relevance to the equivalent processes in mammals. Finally, molecular processes can be interrogated in zebrafish far more rapidly than is possible in mammals because the zebrafish possesses many unique advantages. These unique attributes, and the methods by which they can be exploited to investigate the role of novel genes in T cell development, are described here.
Keywords: Zebrafish, T cell development, Morpholino, Functional rescue, in situ hybridization
1. Introduction
Zebrafish is becoming a powerful vertebrate model for in vivo genetic studies of human development [1–3]. Zebrafish and mammals share similar blood cells and use common molecular pathways to regulate the production of blood cells including thymocytes [4–6]. In zebrafish, the first definitive HSCs derive from the ventral wall of the dorsal aorta at about 28–30 hours post fertilization (hpf), then migrate to the caudal hematopoietic tissue, and finally home to the thymus and the kidney, where T cell development and adult hematopoiesis occur, respectively [7]. T cell progenitors marked by ikaros expression appear in the thymus by 3 days post fertilization (dpf) [7]. As in mammals, zebrafish T cell development gives rise to two T lineages, γδ and αβ and is accompanied by the rearrangement of four T-cell receptor (TCR) loci (TCRα, TCRβ, TCRγ and TCRδ) [8]. As in mammals, TCR genes in zebrafish are assembled by a V(D)J recombination process that is dependent upon the recombination activating genes, rag1 and rag2, [9]. The T cell specific nonreceptor tyrosine kinase lck is also found in the lymphoid precursors and maturing T lymphocytes in the bilateral thymic lobes of zebrafish [3,10]. A recent review summarizes additional genes that play an important role in zebrafish and serve as markers of T cell development (c-myb, ikaros, rag1, rag2, lck, TCRα, TCRβ, TCRγ, TCRδ, IL7R, jak3, ccr9a, ccr9b, zap70, gata3, runx1, foxn1, etc.) [11].
Although the mouse remains the gold standard for immunological research, the zebrafish model provides a number of distinct advantages that complement the use of mice. Specifically, zebrafish females lay a large number of externally fertilized eggs that develop ex utero and this allows easy kinetic analysis of the effect of mutations that are lethal at early embryonic stages and would thus be very difficult to analyze during mouse development. Most zebrafish genes have mammalian orthologs, and the roles they play in hematopoiesis in general, and T cell development in particular, are highly conserved. Accordingly, insights gained into processes underlying T cell development in zebrafish will be applicable to mammalian biology [12]. Another significant advantage of using the zebrafish model is that embryos are transparent for the first 3 weeks of life. The optical clarity of zebrafish embryos allows visualization of development using fluorescent lineage tracers. These tracers enable real time imaging to study migration, colonization and cell-fate determination of T progenitors without the need for sophisticated intravital two-photon microscopy [13,14]. Transgenic fluorescent lineage tracers available for studying T cell development are shown in Table 1. For gene expression analysis, both the expression level and cell type distribution of transcripts can be easily assessed using whole mount in situ hybridization with anti-sense probes. Finally, gene function can be rapidly assessed using gain- and loss-of-function approaches. Indeed, the requirement of a gene can be readily examined by loss-of-function using antisense oligonucleotides called “morpholinos”, which interfere with either splicing or translation of mRNA [15]. Morpholinos can be obtained by providing Gene Tools with the target sequence, following which they will design sequence-specific oligos to block either mRNA translation or splicing (https://oligodesign.gene-tools.com/request/). Conversely, gain-of-function, overexpression experiments can be done either by simply injecting the egg with mRNA encoding the protein of interest or by using heat-inducible plasmids that allow manipulation of both the timing or level of induction. These advantages, which make zebrafish such a powerful model for the dissection of T cell development, will be described here. Expected experimental outcomes will be illustrated using our recent analysis of the role of the ribosomal protein Rpl22 in T cell development.
Table 1.
Transgenic lines for studying thymocyte development in zebrafish
2. Materials
2.1. Zebrafish facility, husbandry and culture of embryos
AHAB zebrafish housing systems (Aquatic Habitats, Aquatic Eco-Systems, Inc.): The fish room is maintained on a 14-hour day/10-hour night cyle. We maintain and breed Zebrafish at 28.5 °C under the standard aquaculture conditions published in The Zebrafish Book (http://zfin.org/zf_info/zfbook/zfbk.html). Embryos were staged as described previously [16].
Egg water stock (60×): pH 7.0, 300 mM NaCl, 10.2 mM KCl, 19.8 mM CaCl2, 19.8 mM MgSO4. Prepare 60x Stock by adding 17.5 g of NaCl, 0.76 g of KCl, 2.8 g of CaCl2, and 4.9 g of MgSO4 to 1 Liter Millipore Q water. Adjust pH to 7.0, and sterilize by autoclaving. Make a 1× working solution by diluting 60x stock into distilled water. Add methylene blue to a final concentration of 0.01%. The solution can be stored for several weeks at room temperature (RT). [See Note 1]
Phenylthiourea (PTU, Sigma, P7629): Prepare the 60×stock solution by dissolving 1.8 g powder in 1L Millipore Q water and heating to 60°C for several hours under agitation. Store at 4°C. Make up the working solution by dilution to 1× with egg water. The use of PTU can prevent the embryos from developing pigment, thereby prolonging the developmental window during which they embryos are transparent.
Pronase (Roche, Cat# 11459643001): Dissolve 1g powder in 100ml egg water to make 10mg/ml stock solution. Aliquot into 15ml tubes and store at −20 °C.
Petri dishes (100 × 20 mm).
Transfer pipettes (Samco Scientific).
Incubator for zebrafish embryos (VWR scientific).
2.2. Zebrafish lines
Wild-type and transgenic zebrafish lines are available from Zebrafish International Resource Center (ZIRC, Eugene, OR). For examples described in this chapter, we employed the following fish lines: AB wild-type fish; and 2) Tg(lck:EGFP)cz1[17].
2.3. Mopholino design and preparation
Morpholinos (MO)(Gene Tools, Philomath, OR): We use morpholinos to knock down gene expression. The 25-base morpholinos are complementary with their target RNAs and can either inhibit mRNA translation or mRNA splicing. Translation-blocking morpholinos often decrease protein expression to levels undetectable by Western blot and begin to act immediately in fertilized embryos. Splice-blocking morpholinos act after the switch from maternal to zygotic transcription at 3.5dpf and allow selective deletion of particular protein domains.
Danieau buffer: pH 7.6, 5.0 mM HEPES, 58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, dissolved in nuclease free water.
The morpholino stock is dissolved in either Danieau buffer or nuclease free water at a concentration of 1–3 mM in 5 μl aliquots. Stocks should be stored frozen at –80°C or in airtight microtubes at RT to prevent evaporation [See Note 2]. These are sample sequences for MOs targeting the zebrafish rpl22 start codon (5’-CCGACAGTTTTGGCAGAAAGCCAGT-3’, designated ‘Rpl22 MO’) and as well as a 5-base mismatch control (5’-CCCACACTTTTCGCACAAACCCAGT-3’, designated ‘Rpl22 MM’).
2.4. Microinjection
Needle puller (Flaming/brown micropipette puller, Model P-97, Sutter Instrument Co.).
Borosilicate filamented glass capillaries (Cat: 1402328, World Precision instruments, Inc.).
Microinjector (PM1000 cell microinjector, Microdata Instrument, Inc.). This microinjector requires a separate compressed air pump (Senco, Model: PC1010).
Micromanipulator holder (GJ-8 magnetic stand, Narishige Scientific Instruments).
Micromanipulator arm: M-152 (Narishige Scientific Instruments).
Dissecting stereomicroscope (Nikon SMZ100).
Plastic Mold for making injection plates: See Figure 1A for a description of the plastic mold to make injection chambers. This can also be found in the Zebrafish Book (http://zfin.org/zf_info/zfbook/chapt5/5.1.html).
Stage micrometers (Klarmann rulings, Cat #: KR-842).
Straight probes for micromanipulating zebrafish embryos (Fine Science Tools, Cat#: 10140–01).
Petri dishes (100 × 20 mm).
Fig.1. Microinjection and dechorionation for zebrafish embryos.
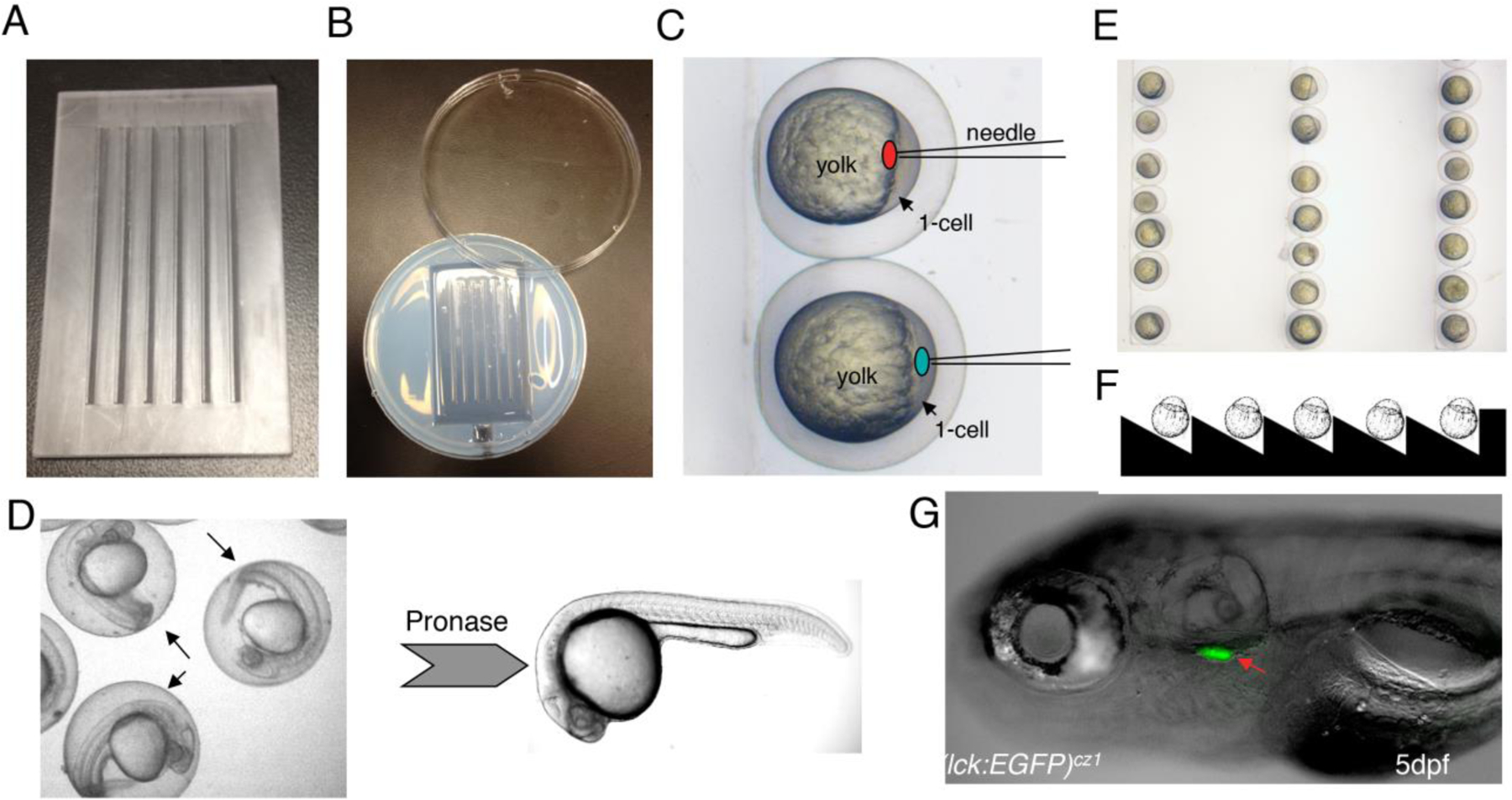
(A) Injection mold. (B) Injection chamber/plate. (C) Schematic of morpholino or plasmid injection. For morpholino knockdown, we inject the liquid (red spot) to the interface between cell and yolk. For plasmid overexpression, we directly inject the solution to the cytoplasm (green spot) at 1-cell stage. (D) Dechorionation process. The arrows indicate the chorions around the 24hpf embryos. (E) Alignment of the 1- cell stage embryos in the trenches of the injection plate. (F) Schematic of the vertical section of the injection plate. (G) Lateral view of a 5dpf Tg(lck:EGFP)cz1 embryo. Arrow indicates the EGFP+ T cells in the thymus.
2.5. Transient rescue experiments
Plasmid DNA preparation kit for highly purified DNA.
Waterbath.
Construct: For heat shock mediated inducible expression, clone cDNA encoding the gene of interest into the heat-inducible pSGH2 vector [18,6]. The pSGH2 vector contains two I-SceI restriction sites.
I-SceI meganuclease: (New England Biolabs, R0694S) Aliquot 2 µl each upon arrival and store at −80°C) along with I-SceI buffer (New England Biolabs), supplied with the NEBuffer I-SceI pack. [See Note 3].
2.6. Western Blot [19].
Deyolking buffer: 55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3.
Wash buffer: 10 mM Tris-HCl pH8.5, 110 mM NaCl, 3.5 mM KCl, 2.7 mM CaCl2.
200 μl Precision tips.
Protease inhibitor cocktail (Roche, Cat #: 11836170001).
PMSF (Sigma, P7626).
SDS lysis buffer: 100mM Tris pH 6.8, 4% SDS, 200mM DTT and 20% glycerol, supplemented with complete mini protein inhibitor cocktail (Roche).
MOPS running buffer (Novex, Life technologies, NP0001)
Transfer buffer: 20 mM Tris, 150 mM glycine, 20% methanol, and 0.038% SDS.
20 x TBST solution (Fisher Scientific, Cat#: 03–500-537): 500mM Tris pH 7.4, 60mM KCl, 2.8M NaCl, and 1.0% Tween-20 in high purity H20. Make 1 X TBST by diluting 20x TBST solution with distilled H20.
Blocking buffer: 5% non-fat milk powder in TBST.
Antibodies as needed. The following antibodies were used in the experiments described in this chapter: Anti-human Rpl22 serum (detects N-terminus of human Rpl22, Transduction Labs, San Jose, CA [20]), dilution: 1:1000; Anti-Actin (Sigma, AC-40), dilution: 1:2000. Secondary anti-rabbit or mouse IgG HRP-conjugated Antibodies (Cell Signaling), dilution: 1:1000.
0.45 μm PVDF transfer membrane (Millipore, Cat#: IPFL00010).
ECL substrate and chemiluminescent reagents (Millipore, P36599A).
Protein gel running and transfer equipment (e.g. Novex XCell SureLock® Mini-Cell and XCell II™ Blot Module,Life technologies).
Autoradiography films.
2.7. Whole-mount TUNEL assays
In Situ Cell Death Detection Kit, TMR red (Roche, Cat#: 12156792910).
Acetone.
Permeabilization solution (0.1% v/v Triton X-100 and 0.1% w/v sodium citrate in PBS): 100μl TritonX-100 and 1ml 10% sodium citrate in 98 ml of pH 7.4 PBS, freshly prepared.
2.8. Whole-Mount In Situ Hybridization
Fixative: 4% paraformaldehyde in PBS. Dissolve 40g of powder in 1L of PBS by heating to 60°C, then adjust the pH to 7.4. Fixative can be aliquotted into 50-ml tubes and stored at − 20 °C (up to ~6 months).
Nuclease free water.
PBS (10× Stock solution, pH 7.4).
Tween-20.
PBST: 1 x PBS plus 0.1% Tween-20.
Methanol.
Proteinase K stock: 10 mg/ml of proteinase K (Roche Diagnostics, Cat #: 03115836001) in PBST.
RNA polymerase (T7, Sp6, or T3) and transcription buffer (Ambion).
DIG RNA labeling mix (Roche Diagnostics, Cat#: 11277073910).
RNAse inhibitor (Ambion, Cat#: AM2682).
NucAway™ spin columns (Ambion, Cat#: AM10070).
UltraPure formamide (Life technologies, Cat#: 15515–026).
20×SSC stock solution (Cellgro, Cat #: 46–020-CM).
Prehybridization buffer (Hyb−): 50% formamide, 5× SSC, 0.1% Tween-20. Store at −20°C.
Hybridization buffer (Hyb+): Hyb (−) plus 5 mg/ml RNA from Bakers Yeast (Sigma, R-6750) and 50 μg/mL heparin (Sigma, H3149). Store at −20°C.
2× SSCT : 2× SSC, 0.1% Tween-20.
2× SSCT/50% formamide: 2× SSCT, 50% formamide.
0.2× SSCT: 20× SSC diluted to 0.2× with nuclease free water, 0.1% Tween-20.
Blocking reagent for nucleic acid hybridization (Roche, Cat#: 11096176001).
Heat inactivated FBS: Incubate FBS at 60°C for 1 hour, filter sterilize, then store at −20°C.
MAB buffer: 10mM Tris pH 9.5, 100mM maleic acid (Sigma, M0375), 150 mM NaCl.
MABT: MAB plus 0.1% Tween-20.
10% BMB Block: 10% blocking reagent in MAB solution.
Complete Block: MABT plus 10% heat inactivated FBS, 2% BMB Block.
Anti-digoxigenin–alkaline phosphatase antibody (Roche Diagnostics, Cat # 11093274910).
Staining buffer: 100 mM Tris–HCl pH 9.5, 50 mM MgCl2, 100 mM NaCl, 0.1% Tween-20, 1mM levamisol (Sigma, Cat: L9756, add fresh from a 0.5M stock).
Vector BCIP/NBT Staining solution: We use BCIP/NBT AP substrate kit IV (Vector Laboratories, SK-5400). Add 2 drops of kit solution per 5 mL of staining solution, mix; 2 drops solution 2, mix; and 2 drops solution 3, mix well.
15mm Netwell™ Insert with 74 µm Mesh Size Polyester Membrane (Corning, Cat #3477).
Orbital shaker.
Tecan NanoQuant machine for RNA/DNA quantitation (Tecan, Infinite M200).
Hybridization oven (Boekel Scientific, Model 136400).
2.9. Mounting embryos for observation
Fluorescence stereomicroscope (Nikon SMZ1500) equipped with green fluorescent protein (GFP) and DsRed filters, DS-Fi1 digital camera, and Nikon Ar imaging software.
Microscope depression slides (Science Enthusiast, cat#: 26430).
Tricaine (Sigma, A5040): Prepare a 25× solution (4 mg/ml) by dissolving 1.2 g in 300 ml of water. Store at 4°C.
Methylcellulose (Sigma, M0387): Prepare a 3% solution in egg water. The living or stained embryos are mounted in 3% methylcellulose and photographed.
2.10. Fluorescence-activated cell sorting (FACS)
1× trypsin/EDTA.
Trizol reagent.
35 mm culture dish.
40 μm nylon mesh filter.
FACSAria II (Becton Dickinson Immunocytometry Systems, San Jose, CA).
3. Methods
3.1. Identifying zebrafish ortholog.
Many mammalian genes have zebrafish orthologs. We use both homology and synteny-based approaches to identify the zebrafish orthologs of mammalian genes. Conserved synteny (i.e., contiguous genes or ESTs with conserved map order on the chromosomes of different species) is a prominent feature of vertebrate genomes. Analysis of zebrafish genomic mapping data has revealed conserved rearrangements and homology segments between zebrafish and human genomes [21]. Thus, the homology based syntenic analysis can be used to effectively predict and find orthologous genes between zebrafish and human.
Go to http://www.ensembl.org/Homo_sapiens/Info/index.html and enter the name of the human gene (e.g., RPL22) in the SEARCH section.
Click on ContigView, then the gene name, to get the amino acid sequence as well as genomic position.
Go to http://www.ncbi.nlm.nih.gov/, select tBLASTn and paste the amino acid sequence in the dialog box. Select ‘Nucleotide selection (nr/nt)’ in the ‘Choose search set- Database’ window, and ‘zebrafish’ in the ‘Organism’ window, and click on BLAST, followed by clicking on ‘Formatting’ in a new window to get the search summary.
Record the zebrafish gene name (e.g., zgc:123327, rpl22), then go to http://www.genome.ucsc.edu/ to identify the genomic position of this gene.
Repeat these steps for other genes located near the gene of interest until it is clear that there is a good syntenic relationship.
Next align the amino acid sequences of the human, mouse and zebrafish orthologs of your gene of interest. The Clustal W2 algorithm is an effective tool for this task (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
3.2. Preparation of Microinjection Plates/ Needles
Prepare a 1.5% agarose solution with 1x egg water.
Pour enough molten agarose into a 100cc petri dish to cover the bottom with a thin layer, then allow to solidify at room temperature. When the agarose is solid, pour another layer of molten agarose on top and place the plastic injection mold (Figure 1A) into the agarose solution. Make sure no bubbles are trapped under the mold. Cover and allow it to cool.
Overlay the injection plate with ddH2O and store at 4°C (See Figure 1B).
Pull microinjection needles using the P-97 micropipetter puller. Load the glass capillary tubes into the heating chamber with the middle part of the capillary surrounded by a platinum heating film. The platinum heating film electrically heats up the glass capillary, enabling a horizontal linear force to pull the heated glass apart to produce two separate needles. Store needles into a Petri dish on a row of plasticine to prevent the needles from breaking.
Under a stereomicroscope, use a blade to cut the tip off of the needle. A good microinjection needle should be long and sharp but not flexible. This will minimize damage to the embryo and help to pierce the chorions.
3.3. Preparation of zebrafish embryos
The procedures to follow to set up matings are described in detail in the zebrafish book (http://zfin.org/zf_info/zfbook/zfbk.html). Briefly, set up well-fed fish (6–18months old) the night before in pairs (2 males and 2 females) in the mating chamber with the divider in place. The next morning, remove the divider to allow the fish to spawn.
Collect embryos approximately 20 minutes after female and male zebrafish begin to mate. Visually inspect the embryos to ensure that the clutch is uniformly at the early single cell stage. For gene knockdown analysis, as well as for most other manipulations of zebrafish embryos, it is best to inject into the cytoplasm of 1-cell stage embryos, which produces the most consistent results.
3.4. Dechorionation
The chorion is the membrane around the developing zebrafish embryo (See Figure 1D, arrows). Embryos should be removed from their protective chorions to facilitate further observation, fixation, or other manipulations. Removal of the chorion, or dechorionation, is accomplished by pronase digestion of embryos older than 18hpf. We generally dechorionate at 24hpf.
Dilute 10mg/ml pronase stock with egg water to 2mg/ml working solution.
Transfer the embryos to a Petri dish, removing as much egg water as possible. Add 2ml of the diluted pronase solution to the dish and swirl the embryos. Incubate at room temperature for 10 min.
Use a Pasteur pipette to gently transfer the embryos to a fresh Petri dish full of egg water. Most of chorions will have been removed at this time (See Figure 1D).
Rinse the embryos thoroughly (at least three times) with egg water to remove the pronase.
3.5. FACS sorting of lineage marked T cell progenitors
Transgenic zebrafish embryos produced as above from appropriate lines [Table I; e.g., Tg(lck:EGFP)cz1] are dechorionated by pronase treatment. For FACS sorting of fluorescently labeled progenitors, we use about 100 transgenic embryos (5dpf , See Figure 1E) that have been grown in the 28.5°C incubator.
To anesthetize the embryos, transfer them to a petri dish containing tricane in egg water at 0.2 mg/ml and incubate until they are no longer swimming.
Transfer anesthetized embryos to deyolking buffer (keep on ice) and pass them through a 200 μl pipette tip several times to remove their yolk.
Dissect and macerate deyolked whole embryos in ice-cold 0.9 ×PBS plus 5% FBS using a scalpel blade to facilitate digestion.
Transfer embryos to a 35 mm culture dish containing 1× trypsin/EDTA solution and incubate for 30 to 60 min at 32°C. During incubation, gently agitate by pipetting every 10 min.
Terminate the digestion by adding CaCl2 to a final concentration of 1 mM and fetal calf serum to 10%.
Pellet the cells for 5 min at 400×g, then wash twice with 0.9 ×PBS at 400×g.
Filter the suspension through 40 μm nylon mesh to eliminate debris (Keep cold).
Pellet and resuspend the cells in 0.9 ×PBS/5% FBS. Stain with 1μg/ml propidium iodide (PI) 30 minutes on ice to identify dead cells and debris.
Perform flow cytometry on the resulting single cell suspensions at room temperature. We use a FACSAria II (Becton Dickinson). Use non-transgenic AB strain zebrafish cells to set the gate control, then sort the progenitors of interest (e.g., lck-GFP+ T cell progenitors) and GFP− cells directly in Trizol, if you are preparing RNA.
Isolate total RNA from the purified cells by Trizol extraction (see manual from Sigma). Quantitative or conventional PCR can then be used to assess mRNA expression of the gene of interest.
3.6. Morpholino microinjection.
Before injection, heat the morpholino (MO; antisense oligonucleotide) in a thermocycler for 10 min at 65 °C to dissolve any precipitates that can clog the microinjection needle.
Assemble the needle to the microinjection arm.
Turn on air the compressor pump and PM1000 microinjector.
Press the ‘Timer’ and ‘BALN’ buttons to activate balance pressure. Press the ‘VENT’ button.
Use the ‘Fill’ button to load the needle with the MO targeting your gene of interest.
Use the ‘Inject’ knob on the injector panel to adjust the injection air pressure and set the injection time. For the PM1000 cell microinjector, we use 60 ms injection time and 10-psi injection pressure as the starting point. Use the ‘Balance’ knob to adjust the balance pressure between 0–0.4psi to prevent both liquid from leaking out of the needle and medium from flowing back into the needle.
Place a drop of mineral oil on the microscope stage micrometer. Submerge the tip of the injection needle into the mineral oil and press the foot pedal to finish the injection. The released droplet will form a perfect sphere in the oil. Measure the diameter of the droplet and calculate the volume according to 4/3πR3. Adjust the injection pressure and injection time to produce the desired injection volume and dose of morpholino (see Table 2).
Transfer embryos into the trenches of the injection plate using a Pasteur pipette (Figure 1E).
Use the fine probe to align the embryos, with the animal pole facing you (See Figures 1E, F).
Manipulate the injection arm to advance the needle, gently piercing the chorion of the embryos. Inject the morpholino into the interface between cytoplasm and yolk of 1-cell stage embryos (See Figure 1C, lower panel and Note 4).
Move the injection plate and repeat until all of the embryos on the plate have been injected, then transfer the embryos to a clean dish containing egg water, and place in 28.5°C incubator.
Table 2.
Morpholino (MO) Dose per Microinjection
| Diameter of drop (μm) | Radius of drop (μm) | Volume (pL) 4/3πR3 | MO Dose (ng) of 0.5mM = 4.15ng/nl | MO Dose (ng) of 2mM = 16.6ng/nl |
|---|---|---|---|---|
| 1 | 0.5 | 0.52 | 0.00 | 0.01 |
| 2 | 1 | 4.19 | 0.02 | 0.07 |
| 3 | 1.5 | 14.13 | 0.06 | 0.23 |
| 4 | 2 | 33.49 | 0.14 | 0.56 |
| 5 | 2.5 | 65.42 | 0.27 | 1.09 |
| 6 | 3 | 113.04 | 0.47 | 1.88 |
| 7 | 3.5 | 179.50 | 0.74 | 2.98 |
| 8 | 4 | 267.95 | 1.11 | 4.45 |
| 9 | 4.5 | 381.51 | 1.58 | 6.33 |
| 10 | 5 | 523.33 | 2.17 | 8.69 |
| 11 | 5.5 | 696.56 | 2.89 | 11.56 |
| 12 | 6 | 904.32 | 3.75 | 15.01 |
| 13 | 6.5 | 1149.76 | 4.77 | 19.09 |
| 14 | 7 | 1436.03 | 5.96 | 23.84 |
| 15 | 7.5 | 1766.25 | 7.33 | 29.32 |
3.7. Western Blot Analysis
To examine protein expression, dechorionate embryos using pronase as above, then wash three times in egg water to eliminate residual pronase.
Transfer both control and morpholino injected embryos to 1.5 ml microcentrifuge tubes. We found that approximately 5 embryo equivalents were required to detect Rpl22, but this will vary with protein abundance and antibody quality.
Centrifuge at 300×g for 5min and remove all residual egg water. Add 200μl deyolking buffer and pipet up and down with a 200μl tip until the yolk is disrupted. 200μl deyolking buffer is enough for up to 100 embryos and the volume should be adjusted in proportion as the number of embryos is varied.
Agitate the tubes on the orbital shaker for 5 min at 1100 rpm to dissolve the yolk.
Centrifuge at 300×g for 30 seconds and discard the supernatant. Wash two more times, then add 0.5 ml wash buffer to the pellets, agitate for 2 min at 1100 rpm, and pellet as above.
Solubilize the pelleted embryos in 2 μl 2× SDS-sample buffer per embryo and heat for 5 min at 95°C. No homogenization is necessary, as the cells should dissolve rapidly in the buffer.
Remove insoluble material by centrifugation at 4°C for 10 min at 12,000×g. After collection, the supernatant can be immediately run on an SDS-PAGE gel or stored at −80°C in aliquots .
Approximately 5 embryo equivalents are loaded per lane, resolved by SDS-PAGE, transferred to PVDF membrane, and immunoblotted. The sample immunoblot employed mouse anti-actin monoclonal antibody and rabbit anti-Rpl22 serum, followed by incubation with appropriate secondary HRP-conjugated-Abs. Bound antibody was visualized by enhanced chemiluminescence (Figure 2B). [See Note 5].
Fig.2. Using zebrafish model to investigate the role of Rpl22 in T cell development.
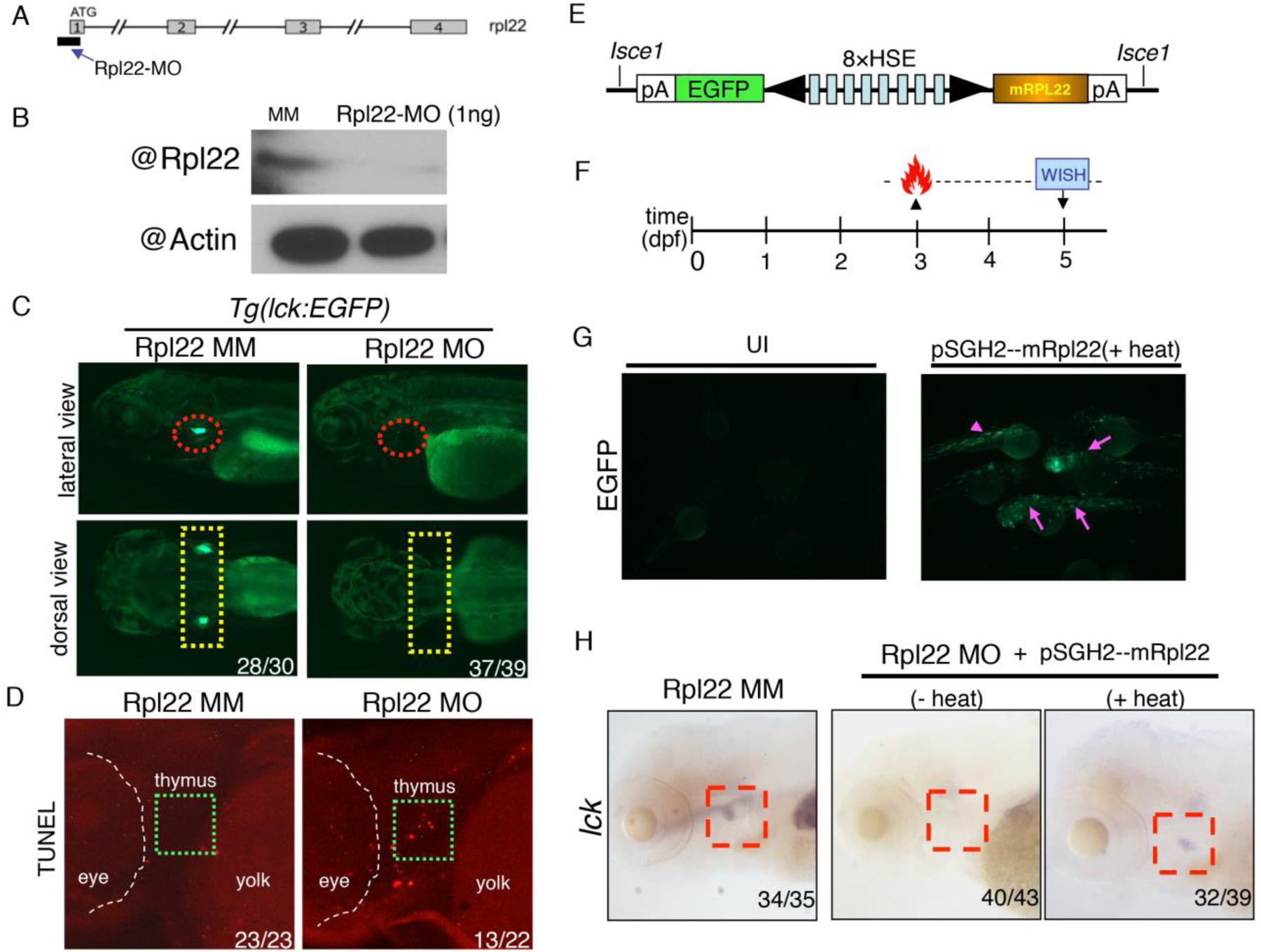
(A, B) Targeted knockdown of Rpl22 in zebrafish embryos. (A) For Rpl22, the MO is designed to specifically block the initiation of translation at the start codon in exon 1. The short black line and blue arrow indicate the positions of sequences targeted. (B) The effect of 1ng Rpl22-MO knockdown on expression of endogenous Rpl22 is assessed by immunoblotting on 5dpf embryos. (C) Rpl22-MO injected (1 ng), but not MM (5-base pair mismatch) control injected, Tg(lck:EGFP) fish exhibit a loss of EGFP-marked T cells at 5 dpf (lateral view, red circles; dorsal view, yellow rectangles). Numbers refer to fraction of morphants with the depicted phenotypes. (D) Injection of 1 ng Rpl22-MO causes apoptosis in the thymus at 3.5 dpf, while injection of the same quantity of 5-base pair mismatch control morpholino (Rpl22-MM) caused no change in the thymus (green dashed rectangles). The white dashed line delimits the eye. Numbers refer to fraction of morphants with the depicted phenotypes. (E-H) The use of a heat-shock inducible, bidirectional expression construct to restore T cell development in rpl22 morphants. (E) Upper panels depict schematics of the heat-inducible expression plasmid pSGH2-mRpl22. (F) The heat shock-inducible expression plasmid associated with I-sceI mediated transient overexpression is injected into 1-cell stage embryos. At 3dpf, the embryos are heat shocked at 37°C for 1hr and then effects on T cell development are analyzed at 5 dpf using WISH for lck, a marker of thymic progenitors. (G) After heat-shock, GFP+ embryos are selected for subsequent WISH analysis (pink arrowheads). (H) Heat shock induction of mouse Rpl22 expression rescues T cell development in rpl22 morphants (5dpf lck WISH staining, red rectangles). Numbers refer to fraction of morphants with the depicted phenotypes.
3.8. Fluorescent microscopy
T cell development in Tg(lck:EGFP)cz1 embryos is clearly evident by 5dpf. To examine the effect of gene knockdown on T cell development in this model, the embryos must be immobilized. To accomplish this, place 5dpf morphant embryos (i.e., morpholino-injected) in a petri dish containing tricane in egg water at 0.2 mg/ml and incubate until the embryos are no longer swimming. This usually requires about 5 min.
3% methylcellulose works well for mounting 5dpf embryos for regular microscopic observation and photography. To mount embryos, add a drop of 3% methylcellulose to the microscope depression slide.
Transfer the embryo using a pasteur pipette and position it in the methylcellulose.
Use the steel probe to precisely orient the embryo for lateral or dorsal view.
Photograph using a fluorescence stereomicroscope (See Figure 2C).
3.9. Whole-mount TUNEL assays
To determine if thymocytes are undergoing apoptosis, we employ TUNEL staining.
Fix Embryos in 4% PFA overnight at 4°C.
Dehydrate in graded methanol/PBS (50%, 70%, 95%, 100%), incubating for 5 min at room temperature at each step and then storing at –20°C overnight.
Incubate for 10 min in acetone at –20°C.
Wash embryos twice in PBS for at least 5 min per wash.
Permeabilize the embryos for 15 min at RT in 0.1% Triton X-100 plus 0.1% sodium citrate/PBS (freshly prepared). Longer incubation times may be necessary when working on older embryos.
Wash twice in PBS, 10min per wash.
Add 50μl enzyme solution (containing terminal deoxynucleotidyl transferase) to 450μl label solution (containing nucleotide mixture in reaction buffer) to obtain a total of 500μl TUNEL reaction mixture, then mix well (Roche TMR Kit instructions).
Incubate embryos for 1 hour at 37°C in the dark in TUNEL reaction mixture in a 1.5ml, round-bottom microcentrifuge tube. 100–150μl should be enough for up to 50 embryos.
In the dark, wash for 2hr (4x30’ in PBST), then photograph (Figure 2D).
3.10. Whole-Mount In Situ Hybridization.
Whole mount in situ hybridization (WISH) employs an antisense ribonucleotide probe and can be used for a number of purposes including determining the tissue distribution and gross expression level of an mRNA species and/or the presence of a particular cell population marked by a lineage-restricted mRNA.
To prepare DNA for making riboprobes, linearize the plasmid encoding your probe and purify by two rounds of phenol/chrloroform extraction. Check 1μl of digested DNA on a 1% agarose gel to ensure the DNA template is linear. Quantify the DNA concentration of the template (e.g. Tecan NanoQuant).
To transcribe the riboprobes, add the following to the transcription reaction: 1 μg of linearized DNA template, 2 μl of 10× transcription buffer, 2 μl of DIG-labeling RNA mix, 1 μl of RNase inhibitor (40 U/μL), 2 μl of RNA polymerase (T7, T3 or Sp6) and enough nuclease free water to bring the reaction to 20 μl. Incubate the reaction at 37°C for 2 h, then purify the riboprobe using a Ambion NucAway spin column. Assess the quality and yield of the probe yield by running 1 μl of the probe on an agarose gel and quantifying the concentration. Probes can be stored at −80°C for up to 2 years.
Dechorionate and fix 24h embryos using pronase. After dechorionation, grow the embryos in egg water containing 0.003% PTU to prevent pigmentation, which starts to develop at around 24 hpf. Fix embryos at 5dpf (or another developmental stage appropriate for your experiment) with 4% fix solution and rock overnight at 4°C (See Note 6). Thymocyte seeding can be observed by 3.5–4dpf, but the optimal time to assess whether knockdown of a gene blocks development is 5dpf. Wash embryos in PBST three times, 5 min each. Wash with 100% methanol, incubate for 5 min, then transfer the embryos to fresh methanol and store at −20°C.
Rehydrate embryos for 5–10 min each in 75% Methanol/PBST, 50% Methanol/PBST, and 25% Methanol/PBST. Wash embryos in PBST once quickly, then twice more for 5 min each.
Digest embryos with 10μg/ml Proteinase K in PBST for 30 min at room temperature with gentle rocking. These conditions work well for zebrafish embryos from 3–5 dpf. Proteinase K is made from a 10mg/ml stock stored at −20°C.
Remove the proteinase K with three quick PBST washes.
Re-fix embryos for 1hr at room temperature. This step is necessary to eliminate residual proteinase K.
Remove the fixative with three quick PBST washes.
Hybridization: Preheat in situ hybridization buffer Hyb(−) and Hyb(+) solutions to 65 °C. The temperature should be consistent throughout the hybridization procedure. Add Hyb(−) to the 1.5ml tubes containing embryos and shake gently for 15 min at 65 °C. Replace the Hyb(−) with Hyb(+) and shake gently for 1 hour at 65 °C. Add probes (typical range is 0.3–1.0 ng/μl) and rock gently overnight at 65 °C.
Wash embryos in 2× SSCT/50% formamide twice for 30 min at 65°C.
Wash embryos in 2× SSCT twice for 15 min at 65°C.
Wash embryos in 0.2× SSCT twice for 30 min at 65°C.
Wash embryos in MABT 3 times at room temperature, 10 min each.
Transfer the embryos from MABT to complete blocking solution in 12-well plates containing 15mm Netwell™ inserts with polyester membrane and incubate with gentle shaking for at least 2 h at room temperature. [See Note 7].
Remove complete blocking solution and replace it with anti-digoxygenin-AP antibody at 1:5,000 dilution in complete blocking solution and rock overnight at 4°C.
Wash embryos in complete blocking solution, 1hr at room temperature.
Wash embryos in MABT solution, 30 min at room temperature.
Wash embryos in staining buffer at room temperature, 3 × 5min, with gentle rocking.
Wash in 0.1M Tris pH9.5 for 5 min at RT with gentle rocking.
Add Vector BCIP/NBP staining solution. Wrap the plate in aluminum foil and incubate in the dark at room temperature for 20–60min monitoring the development of staining every 20min. Alternatively, the embryos can be stained overnight at 4°C.
Stop the staining reaction by washing in PBST with gentle rocking for 3× for 5 min at room temperature.
Replace PBST with 50% glycerol/PBS and wash for 15min at room temperature. Glycerol is the mild clearing reagent that makes the stained embryos more transparent for photography. Transfer embryos to a six-well plate containing 100% glycerol, then store at 4°C.
Photograph. Effects of gene knockdown on development of the T lineages can be assessed using the lck probe (See Figure 2H).
3.11. Heat inducible gene expression to rescue developmental arrest caused by gene knockdown
In zebrafish, it is critical to perform rescue experiments to ensure that phenotypes resulting from MO-mediated gene knockdown are truly due to the absence of the intended target, rather than due to off-target effects. This can be conveniently accomplished by injecting MO treated embryos with MO-resistant forms of the mRNA encoding the intended target; however, because injected mRNA lasts only a few days, mRNA overexpression is only effective for phenotypes at early embryonic stages. For later developmental stages more than 3dpf, heat-shock inducible DNA constructs can be used to temporally over-express exogenous genes to study T cell development [18,6]. The I-SceI meganuclease was originally utilized in Medaka to induce stronger promoter activity in the F0 founder. Coinjection of I-SceI significantly improves the specificity of transient expression and decreases the mosaicism in zebrafish [22]. We have employed this approach to ensure that ectopic expression of Rpl22 can restore the block in T cell development observed in rpl22 morphants at 5dpf. In doing so, we heat induced expression of mouse Rpl22 at 3dpf and we identified phenotypic rescue of thymocyte development by performing WISH with an lck probe at 5dpf (Figure 2E–H).
To enable heat-inducible expression of a gene of interest, clone the cDNA of the gene in question into pSGH2 [18]. pSGH2 enable simultaneous expression of the gene of interest as well as GFP to mark embryos that have productively incorporated the vector (See Figure 2E).
Upon cloning, prepare purified pSGH2 expressing the gene of interest, using a high purity plasmid preparation kit.
Collect the 1-cell stage embryos and align them on the microinjection plate. [See Note 8].
Mix pSGH2 plasmid with 0.5×I-SceI buffer and 0.5 units/μl I-SceI meganuclease (New England Biolabs) and microinject (100pg) directly into the cytoplasm of one-cell stage embryos.
At 24hpf, check the quality of the injected embryos and remove the dead embryos. Dechorionate the embryos by pronase treatment and then culture at 28.5°C to 3dpf.
At 3dpf, transfer the injected embryos into the 1.5ml tubes filled with 1ml egg water, heat shock at 37°C waterbath for 1hr, then resume culturing the embryos at 28.5°C [See Note 9].
pSGH2 vector has the heatshock inducible bidirectional promoter consisting of multimerized heat shock elements (HSE). Thus, this heat-shock inducible expression construct allows temporal control of ectopic expression of the gene of interest, coupled with EGFP marking of expressing embryos (See Figure 2E–G). After heat-shock, GFP+ embryos are selected for analysis (Figure 2G). In Figure 2H, WISH with an lck probe was performed to identify developing T cells. This revealed that the developmental arrest caused by Rpl22 knockdown can be rescued by a morpholino resistant form of Rpl22 (pSGH2-Rpl22) in a heat-inducible manner.
Acknowledgments
We are grateful to the support and help from Dr. Jennifer Rhodes, Allison Ulrich and Alison N. Bilbee for the zebrafish work. We gratefully acknowledge the assistance of the following core facilities of the Fox Chase Cancer Center: Flow Cytometry, DNA Sequencing, Imaging and Laboratory Animal/Zebrafish. This work was supported by NIH grants AI081814, AI073920, NIH core grant P01CA06927, Center grant P30-DK-50306. Y.Z. is a W.J. Avery Postdoctoral Fellow of Fox Chase Cancer Center.
4. Notes
Methylene blue is used to prevent fungal growth in egg water; however, it can also induce autofluorescence, especially for embryos after 3dpf. Therefore, for observation under fluorescent microscopy, we recommend that embryos should be cultured in egg water without methylene blue.
For morpholino storage, GeneTools recommends to store aliquots at RT. We use airtight microtubes to prevent evaporation. If morpholino aliquots are stored for a long time, GeneTools recommends freeze-drying the oligo. MOs should be heated up at 65°C for 10 min before microinjection to dissolve possible precipitates.
Due to the low stability of I-sceI meganuclease, aliquots of enzyme should be prepared (e.g. 2µl) upon arrival and stored at −80°C. The microinjection mix should be prepared immediately before injection and kept on ice.
We inject the morpholino at 1-cell stage to get more consistent results for T cell phenotypes at 5dpf.
We have an excellent antibody for testing the efficiency of Rpl22 translation-blocking MO. Fig. 2B shows a sample immunoblot of Rpl22 expression in controls and embryos treated with 1ng of a morpholino that blocks translation (Figure 2A,B). However, sometimes antibodies are not available for detection of zebrafish proteins. Thus, an alternative way of evaluating the efficacy of start-site morpholinos that do not alter mRNA size, is to produce a chimeric mRNA comprising the target sequence surrounding the target genes’ ATG fused in frame with GFP-coding sequence. Co-injection of this fusion mRNA with MO will reveal whether the morpholino blocks GFP synthesis, which serves as a surrogate marker for expression of the endogenous target protein. Splice-blocking morpholinos can also be used to knockdown target gene, and the efficiency can be checked by assessing the predicted size shift by RT-PCR.
For embryos older than 18 hpf, the chorion should be removed before fixation to avoid a curved tail after fixation.
We use inserts with a polyester membrane to conveniently transfer the embryos between wells during WISH washing and staining steps.
The heat-inducible plasmid should be directly injected into the cytoplasm of the cell. We strictly use only 1-cell stage embryos to ensure that the transient overexpression has consistent effects on phenotypic rescue.
Temperature or incubation time of heat-shock treatment may be varied for different ectopically expressed genes. Increasing the temp/incubation time may increase the extent of overexpression; however, excessive heating may also kill embryos. Different conditions should be tested to identify the best compromise between overexpression and survival.
References
- 1.Amatruda JF, Zon LI (1999) Dissecting hematopoiesis and disease using the zebrafish. Dev Biol 216 (1):1–15. doi: 10.1006/dbio.1999.9462 [DOI] [PubMed] [Google Scholar]
- 2.Lieschke GJ, Currie PD (2007) Animal models of human disease: zebrafish swim into view. Nat Rev Genet 8 (5):353–367. doi: 10.1038/nrg2091 [DOI] [PubMed] [Google Scholar]
- 3.Trede NS, Langenau DM, Traver D, Look AT, Zon LI (2004) The use of zebrafish to understand immunity. Immunity 20 (4):367–379 [DOI] [PubMed] [Google Scholar]
- 4.Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI (2005) Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes Dev 19 (19):2331–2342. 10.1101/gad.1337005 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhodes J, Hagen A, Hsu K, Deng M, Liu TX, Look AT, Kanki JP (2005) Interplay of pu.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev Cell 8 (1):97–108. 10.1016/j.devcel.2004.11.014 [doi] [DOI] [PubMed] [Google Scholar]
- 6.Bajoghli B, Aghaallaei N, Hess I, Rode I, Netuschil N, Tay BH, Venkatesh B, Yu JK, Kaltenbach SL, Holland ND, Diekhoff D, Happe C, Schorpp M, Boehm T (2009) Evolution of genetic networks underlying the emergence of thymopoiesis in vertebrates. Cell 138 (1):186–197. doi: 10.1016/j.cell.2009.04.017 [DOI] [PubMed] [Google Scholar]
- 7.Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin HF, Handin RI, Herbomel P (2006) Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity 25 (6):963–975. doi: 10.1016/j.immuni.2006.10.015 [DOI] [PubMed] [Google Scholar]
- 8.Schorpp M, Bialecki M, Diekhoff D, Walderich B, Odenthal J, Maischein HM, Zapata AG, Boehm T (2006) Conserved functions of Ikaros in vertebrate lymphocyte development: genetic evidence for distinct larval and adult phases of T cell development and two lineages of B cells in zebrafish. J Immunol 177 (4):2463–2476 [DOI] [PubMed] [Google Scholar]
- 9.Willett CE, Cherry JJ, Steiner LA (1997) Characterization and expression of the recombination activating genes (rag1 and rag2) of zebrafish. Immunogenetics 45 (6):394–404 [DOI] [PubMed] [Google Scholar]
- 10.Langenau DM, Zon LI (2005) The zebrafish: a new model of T-cell and thymic development. Nat Rev Immunol 5 (4):307–317. doi: 10.1038/nri1590 [DOI] [PubMed] [Google Scholar]
- 11.Ma D, Wei Y, Liu F (2013) Regulatory mechanisms of thymus and T cell development. Dev Comp Immunol 39 (1–2):91–102. doi: 10.1016/j.dci.2011.12.013 [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Duc AC, Rao S, Sun XL, Bilbee AN, Rhodes M, Li Q, Kappes DJ, Rhodes J, Wiest DL (2013) Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev Cell 24 (4):411–425. doi: 10.1016/j.devcel.2013.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kissa K, Murayama E, Zapata A, Cortes A, Perret E, Machu C, Herbomel P (2008) Live imaging of emerging hematopoietic stem cells and early thymus colonization. Blood 111 (3):1147–1156. doi: 10.1182/blood-2007-07-099499 [DOI] [PubMed] [Google Scholar]
- 14.Bertrand JY, Kim AD, Teng S, Traver D (2008) CD41+ cmyb+ precursors colonize the zebrafish pronephros by a novel migration route to initiate adult hematopoiesis. Development 135 (10):1853–1862. doi: 10.1242/dev.015297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nasevicius A, Ekker SC (2000) Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet 26 (2):216–220. doi: 10.1038/79951 [DOI] [PubMed] [Google Scholar]
- 16.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203 (3):253–310. doi: 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- 17.Langenau DM, Ferrando AA, Traver D, Kutok JL, Hezel JP, Kanki JP, Zon LI, Look AT, Trede NS (2004) In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc Natl Acad Sci U S A 101 (19):7369–7374. doi: 10.1073/pnas.0402248101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bajoghli B, Aghaallaei N, Heimbucher T, Czerny T (2004) An artificial promoter construct for heat-inducible misexpression during fish embryogenesis. Dev Biol 271 (2):416–430. doi: 10.1016/j.ydbio.2004.04.006 [DOI] [PubMed] [Google Scholar]
- 19.Link V, Shevchenko A, Heisenberg CP (2006) Proteomics of early zebrafish embryos. BMC Dev Biol 6:1. doi: 10.1186/1471-213X-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson SJ, Lauritsen JP, Hartman MG, Foushee AM, Lefebvre JM, Shinton SA, Gerhardt B, Hardy RR, Oravecz T, Wiest DL (2007) Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity 26 (6):759–772. doi: 10.1016/j.immuni.2007.04.012 [DOI] [PubMed] [Google Scholar]
- 21.Rhodes J, Hagen A, Hsu K, Deng M, Liu TX, Look AT, Kanki JP (2005) Interplay of pu.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev Cell 8 (1):97–108. doi: 10.1016/j.devcel.2004.11.014 [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Zhu H, Zon LI, Evans T (2006) BMP signaling restricts hemato-vascular development from lateral mesoderm during somitogenesis. Development 133 (11):2177–2187. doi: 10.1242/dev.02386 [DOI] [PubMed] [Google Scholar]
- 23.Hess I, Boehm T (2012) Intravital imaging of thymopoiesis reveals dynamic lympho-epithelial interactions. Immunity 36 (2):298–309. doi: 10.1016/j.immuni.2011.12.016 [DOI] [PubMed] [Google Scholar]
- 24.Langenau DM, Feng H, Berghmans S, Kanki JP, Kutok JL, Look AT (2005) Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 102 (17):6068–6073. doi: 10.1073/pnas.0408708102 [DOI] [PMC free article] [PubMed] [Google Scholar]