

Abstract
Boron neutron capture therapy (BNCT) is a radiation method used for cancer therapy. Cellular uptake of boron-10 (10B) atoms induces cancer cell death by the generation of alpha particles and recoiling lithium-7 (7Li) nuclei when the cells are irradiated with low-energy thermal neutrons. Current BNCT technology shows effective therapeutic benefits in refractory cancers such as brain tumors and head and neck cancers. However, improvements to cancer targeting and the cellular uptake efficacy of the boron compounds and the expansion of the diseases treatable by BNCT are highly desirable. In this research, we aimed to develop an antibody-based drug delivery method for BNCT through the use of the Z33 peptide, which shows specific recognition of and interaction with the Fc domain of human IgG, for on-demand receptor targeting. In addition, we determined with an in vitro assay that macropinocytosis induction during antibody-based drug delivery is crucial for the biological activity of BNCT.
Introduction
Boron neutron capture therapy (BNCT) is nuclear capture-based radiotherapy. In BNCT, 10B (nonradioactive) compounds, including arylboronic acids [(l)-4-dihydroxyborylphenylalanine, BPA] and polyhedral borane anion (disodium mercaptoundecahydro-closo-dodecaborate, BSH), which are currently being tested in clinical trials, are used because they generate α particles (4He) and recoiling 7Li nuclei in the range of 5–9 μm, which corresponds to the diameter of a cell, when irradiated by thermal neutrons (energy less than 0.5 eV) in the cells that internalize them, leading to cancer cell-killing activity.1−9 BNCT has the ability to effectively treat malignant brain cancer and head and neck cancer;7−9 however, its use as a cancer treatment is limited because of its particularly low cancer cell targeting efficacy, its poor cellular uptake efficiency, and the poor retention of boron compounds inside cancer cells.7,8 To attain effective BNCT biological activity, ∼20 μg/g tumor weight of 10B must be selectively taken up by cancer cells (∼109 atoms/cell), and enough neutrons must be absorbed to sustain a lethal 10B(n, α)7Li capture reaction.8 Additional important still requirements for BNCT include low intrinsic toxicity and little uptake by normal tissues at tumor/normal tissue and tumor/blood boron concentration ratios of >3:1 in tailor-made therapy.8,10 In recent years, BNCT technology has been proceeded including, for example, block copolymer boron conjugate for the EPR effect for enhanced tumor accumulation.11 However, further technical improvements are greatly needed for future BNCT-based cancer therapy.
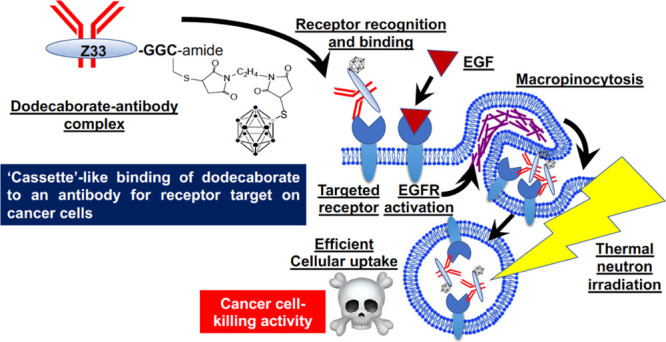
Antibody-based cancer receptor targets and intracellular delivery of therapeutic molecules are strong criteria for effective cancer treatment. Preparation of an antibody-therapeutic molecule conjugate without reducing the efficacy of antibody recognition is very important for generating a sophisticated cancer targeting system. The Z33 peptide, which is derived from the B-domain of protein A, has been shown to efficiently bind to an Fc of human IgG1 (Kd: 43 nM).12 The peptide has been applied for antibody/protein purification, the assembly of treatment compounds, and drug delivery systems.13−15 In this study, we used the functional Z33 peptide as the receptor target in the BNCT of cancer cells. The Z33 peptide was adopted for BNCT in an on-demand “cassette” antibody-based cancer cell targeting system bearing therapeutic boron compounds to expand the cancer species to which it can be applied (Figure 1). In addition, enhanced cellular uptake of the antibody-boron conjugates by effective macropinocytosis induction was shown to be important to increase the biological activity in BNCT after accumulation of boron compounds on plasma membrane using a receptor-target antibody (Figure 1).
Figure 1.
Experimental concept of “cassette” binding of dodecaborate to an antibody receptor target on a cancer cell. The Z33 peptide is conjugated to dodecaborate (Z33-DB) and binds to the Fc of the objective antibody. The antibody can recognize and bind to target receptors on the cancer membrane. Next, EGFR activation by EGF induces macropinocytosis, leading to efficient cellular uptake of the Z33-DB compound. After Z33-DB cellular uptake, thermal neutron radiation induces effective cancer cell-killing activity through BNCT.
Results and Discussion
Figure 1 shows the experimental concept of “cassette”-like binding of dodecaborate to an antibody receptor target on a cancer cell. Dodecaborate is conjugated to the Z33 peptide (Z33-DB), which binds to an Fc of the objective antibody. After Z33-DB is produced, the objective antibody is mixed with Z33-DB to easily form the “cassette” complex, which recognizes and binds to target receptors on the cancer membrane. On the other hand, cancer cells expressing high levels of macropinocytosis-inducing receptors, such as epidermal growth factor receptor (EGFR) and the CXCR4 chemokine receptor, can induce macropinocytotic cellular uptake pathways by receptor activation.16−18 Macropinocytotic pathways are accompanied by clathrin-independent and actin-dependent plasma membrane ruffling and engulfment of large volumes (>1 μm) of extracellular fluids.19,20 For example, high expression of oncogenic K-Ras in pancreatic adenocarcinoma cells induces macropinocytosis to increase the cellular uptake of nutrition from outside cells, leading to malignant progression, and treatment of these cells with the macropinocytosis inhibitor 5-(N-ethyl-N-isopropyl)amiloride, which is not an anticancer reagent, reduced their proliferation in vivo.21 Next, EGFR receptor activation by EGF induces macropinocytosis, leading to efficient cellular uptake of Z33-DB. As described below in detail, only receptor targeting by an antibody specifically localizes to the therapy-relevant molecules on plasma membranes, and the active induction of macropinocytosis and efficient cellular uptake is needed to induce therapeutic biological activities. After cellular uptake, thermal neutron irradiation induces effective cancer cell-killing activity through BNCT (Figure 1).
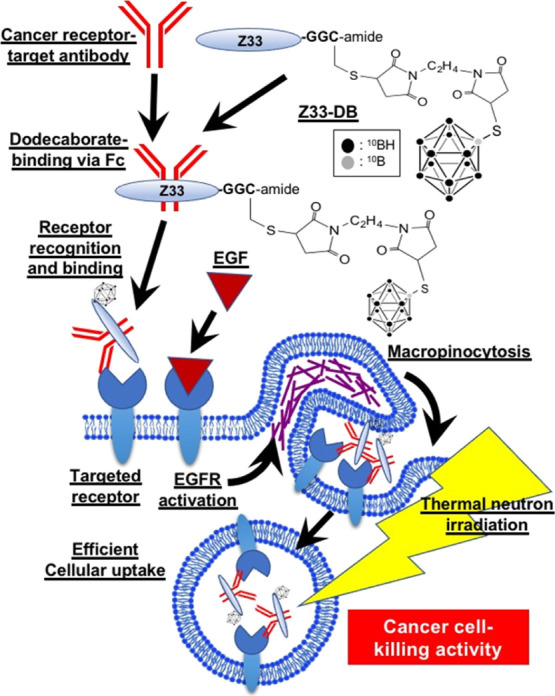
Figure 2 shows the preparation methods for the conjugate of the Z33 peptide and dodecaborate. The Z33 peptide was synthesized by Fmoc-solid-phase synthesis methods, and the sulfhydryl group of cysteine residue at the C-terminus was subjected to a Michael addition reaction with a BMOE linker (Z33-BMOE). Dodecaborate (BSH) was then subjected to another Michael addition reaction with Z33-BMOE, leading to the Z33-DB conjugate (Figure 2). The Z33-DB compound was used for the “cassette” binding of the objective antibody and the receptor target in BNCT.
Figure 2.

Synthesis of Z33 peptide-conjugated dodecaborate (Z33-DB). The Z33 peptide was prepared by Fmoc-solid-phase peptide synyhesis methods. The sulfhydryl group of Cys in the Z33 peptide sequence was conjugated with BMOE (Z33-BMOE), and the linker-conjugated Z33 reacted with the BSH leading to the Z33-dodecaborate conjugate (Z33-DB).
Before assessment of the Z33-DB-based receptor targets, we examined receptor recognition of the fluorescently labeled Z33 peptide (Z33-Alexa660) (Figure S1). In the experiments, A431 cells (derived from human epidermoid carcinoma), which express high levels of human EGFR on the plasma membrane, and CHO-K1 cells (derived from Chinese hamster ovary), which do not express human EGFR, were used for determining the recognition and accumulation of the Z33-Alexa660 that is in complex with the anti-EGFR antibody, cetuximab. Figure S1 shows the confocal laser microscopic observation of the A431 cells and CHO-K1 cells treated with Z33-Alexa660 (200 nM) for 30 min at 4 °C to prevent endocytosis. According to our results, the complex formed with cetuximab (100 nM) greatly enhanced the accumulation of Z33-Alexa660 on the plasma membranes of the A431 cells (Figure S1). In the case of the CHO-K1 cells, the Z33-Alexa660 and cetuximab complex did not accumulate on the plasma membrane, suggesting that the Z33-Alexa660/cetuximab complex specifically targeted the objective receptor (Figure S1). We also synthesized a fluorescently labeled random amino acid sequence of the Z33 peptide (rZ33-Alexa660) and tested whether it accumulated on the membrane. The randomized Z33 peptide sequence did not accumulate on the A431 cells even with the addition of cetuximab (Figure S1).
Next, we evaluated the effects of EGF cotreatment in inducing the cellular uptake of the Z33-Alexa660/FITC-labeled cetuximab (FITC-cetuximab) complex through macropinocytosis. EGFR activation by treatment with the receptor ligand EGF induced signal transduction that initiates macropinocytosis via the activation of Rac (Rho family of small GTPase), which leads to actin organization and lamellipodia formation.16,17 First, we assessed macropinocytosis induction through treatment with EGF, and we found that the cellular uptake of the macropinocytosis marker FITC-labeled dextran (70 kDa)22,23 was significantly increased by treatment with EGF, as analyzed using confocal laser microscopy and flow cytometry under experimental conditions (Figures S2 and S3). Enhanced macropinocytotic cellular uptake was confirmed at >12 h after the EGF co-treatment (Figure S2). Therefore, our experimental condition of cellular Z33-Alexa660/FITC-cetuximab complex uptake was fixed as 24 h treatment. Figures 3a, S4, and S5 show confocal laser microscope captured images of the A431 cells treated with the Z33-Alexa660 (200 nM)/FITC-cetuximab (100 nM) complex for 24 h at 37 °C, and cotreatment with EGF (100 nM) substantially increased the cellular uptake of the Z33-Alexa660/FITC-cetuximab complex compared to the uptake by cells not cotreated with EGF. In addition, the random amino acid sequence of the Z33 peptide did not increase the cellular uptake, even after the EGF was used to stimulate macropinocytosis induction (Figure 3a). In addition, we confirmed the induction of macropinocytosis by EGF treatment even in the presence of cetuximab under experimental conditions using a macropinocytosis marker (Figure S3). Figure 3b also shows the results from the flow cytometry analysis of Z33-Alexa660 binding to the A431 cell membrane and the cellular uptake based on the l-ethylenediaminetetraacetic acid disodium salt solution (EDTA) methods, as described in the Experimental Section. Specifically, the complex consisting of Z33-Alexa660 and cetuximab with and without EGF was shown to have significantly enhanced membrane binding ability. These results suggest that EGF treatment did not affect the binding efficacy of the Z33 peptide/cetuximab complex to the plasma membranes and that treatment with EGF increased the cellular uptake of the Z33 peptide/cetuximab complex bound to the plasma membrane. Trypsinization is very difficult to completely detach antibody from targeted receptors and plasma membrane. However, we conducted flow cytometer analysis in the methods of trypsinization, as described in the Experimental Section, and EGF treatment increased cellular fluorescent intensity of Z33-Alexa660/cetuximab (Figure S6). In addition, EGF treatment also increased cellular fluorescent intensity of Z33-Alexa660 without cetuximab. However, increased cellular uptake level of Z33-Alexa660 by EGF treatment was lower than that of the Z33-Alexa660/cetuximab complex without EGF treatment. In this in vitro assay, co-treatment of the Z33-Alexa660/cetuximab complex was focused on because in flow circumstances in body, Z33 peptides without the antibody might not be accumulated on targeted tumor cells and might be eliminated. Therefore, in this research, we focus on further experiments of cellular uptake of boron compounds and thermal neutron irradiation using the Z33 peptides/cetuximab complex and EGF in vitro assay.
Figure 3.
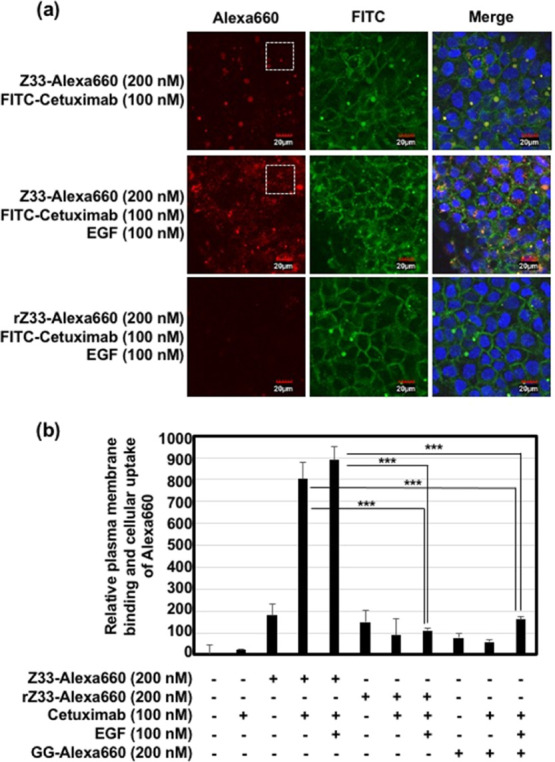
(a) Confocal laser microscopic images of A431 (human EGFR high expression) cells treated with Alexa660-labeled Z33 or rZ33 (each 200 nM) and FITC-labeled cetuximab (100 nM) with or without EGF (100 nM) in cell culture medium containing 10% FBS for 24 h at 37 °C. Red: Alexa660, green: FITC, blue: Hoechst 33342. Scale bar: 20 μm. Enlarged pictures of (a) (areas within the white dotted square) are shown in Figure S4. (b) Relative plasma membrane binding and cellular uptake of Alexa660 after treatment with Alexa660-labeled Z33, rZ33, or GG (each 200 nM) with or without cetuximab (100 nM) and/or EGF (100 nM) in cell culture medium containing 10% FBS for 24 h at 37 °C prior to cell detachment by EDTA treatment and flow cytometer analysis. The data are expressed as the mean (±SD) of three experiments. ***P < 0.001.
In addition, in our experiments, we adopted and used cetuxumab anti-EGFR antibody, which is an antagonist and blocks the activation of EGFR. Therefore, binding of cetuximab to the targeted EGFR blocks the receptor activation and cellular uptake of the cetuximab-bound EGFR by clathrin-mediated endocytosis. However, EGF activates the EGFR without binding of cetuximab on the plasma membrane leading to induction of macropinocytosis, and then, the cetuximab-bound EGFR might be taken up by cells by macropinocytosis, which can induce membrane ruffling, nonspecific engulfment, and cellular uptake.
We next assessed the cellular receptor recognition and cellular uptake of Z33-DB. Figures 4a and S7 show confocal laser microscopic captured images of the A431 cells treated with the Z33-DB (200 nM)/cetuximab (100 nM) complex for 24 h at 37 °C, and cotreatment with EGF (100 nM) to induce macropinocytosis greatly enhanced the cellular uptake of the DB stained with the BSH antibody, a finding similar to the results shown in Figure 3. In cells not cotreated with EGF, plasma membrane accumulation of only Z33-DB was confirmed (Figures 4a and S7). In addition, ELISA experiments showed internalized average amount of boron <0.001 μg (BSH), 0.0063 μg (Z33-DB), 0.0089 μg (complex of Z33-DB and cetuximab without EGF), and 0.0199 μg (complex of Z33-DB and cetuximab with EGF) in 1.0 × 107 cells of A431 (Figure 4b). These results suggest that macropinocytosis induction significantly enhances the cellular uptake of dodecaborate after receptor recognition of the antibody. In addition, cell viability was not affected after treatment with the Z33-DB/cetuximab complex and EGF, as determined by a WST-8 assay and colony assay (Figure S8). We also checked the binding concentration of Z33-DB to the cetuximab using ultrafiltration and high-performance liquid chromatographic (HPLC) separation, as described in the Experimental Section, and we confirmed binding of Z33-DB (72 nM) to cetuximab (100 nM) in our experimental condition for forming the Z33-DB/cetuximab complex.
Figure 4.
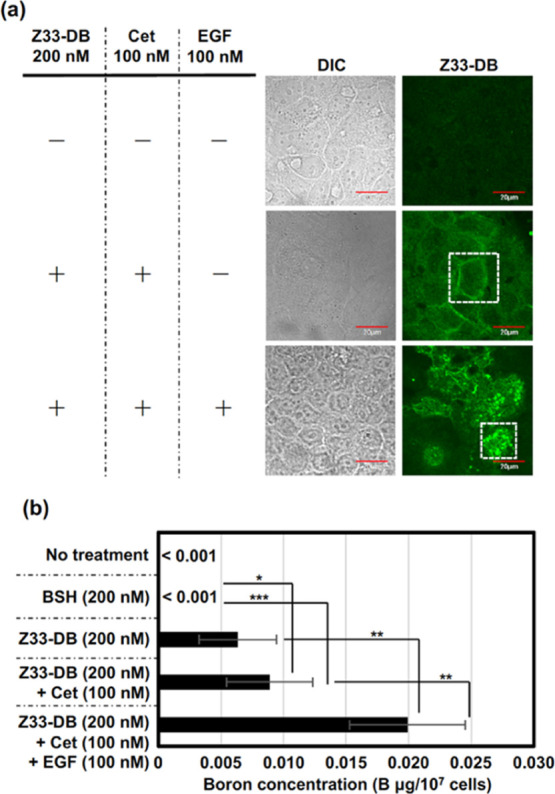
(a) Confocal laser microscopic images of the A431 cells treated with Z33-DB (each 200 nM) with or without cetuximab (cet) (100 nM) and/or EGF (100 nM) in cell culture medium containing 10% FBS for 24 h at 37 °C. Before being observed, the cells were fixed with 4% PFA and 0.1% Triton-X 100 and then stained with an anti-BSH antibody [first antibody: A9H3 anti-BSH-Mab, second antibody: Alexa-Fluor 488 goat anti-mouse IgG(H + L)]. Green: Z33-DB. Scale bar: 20 μm. Enlarged pictures (areas within the white dotted square) are shown in Figure S7. (b) ELISA assay for detecting boron concentration in 1.0 × 107 cells of A431 treated with Z33-DB (each 200 nM) with or without cetuximab (cet) (100 nM) and/or EGF (100 nM) in cell culture medium containing 10% FBS for 24 h at 37 °C. The data are expressed as the mean (±SD) of three experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, we conducted an in vitro BNCT assay. In this assay, the effects of macropinocytotic uptake of dodecaborate in this enhanced cancer cellular uptake system on BNCT activity were assessed. The A431 cells were treated with the Z33-DB (200 nM)/cetuximab (100 nM) complex for 24 h at 37 °C prior to thermal neutron irradiation (90 min at 25 °C), and a colony assay (14 days) was used to assess the cancer cell-killing activity, as described in the Experimental Section. Figure 5 shows the results of the in vitro BNCT assay: the macropinocytosis induction by EGF treatment enhanced the cancer cell-killing activity of the Z33-DB/cetuximab treatment. Interestingly, Z33-DB/cetuximab treatment without EGF stimulation did not increase the cancer cell-killing activity, as indicated by the BNCT assay (Figure 5), suggesting that macropinocytosis induction and efficient cellular uptake of boron compounds are very important for attaining effective cancer cell-killing activity, even after receptor recognition and accumulation of the compound on the membrane. Under the experimental condition created without EGF treatment, efficient accumulation of dodecaborate through the use of cetuximab was attained; however, the binding of dodecaborate to the membrane did not induce cancer cell-killing activity after thermal neutron irradiation because of possible detachment and/or digestion by proteases outside the cells, suggesting the importance of cellular uptake for achieving sophisticated BNCT biological activity. As described above, cancer receptors [e.g., CXCR4 and proteoglycan (syndecan-4)], which induce macropinocytosis and are highly expressed on cancer cells, are thus considered to be applicable on the basis of the BNCT methodology we developed. Although, as a next step, the experimental technique using noncovalent binding of the objective antibody to Z33-DB should be assessed in tumor-xenograft animals in vivo and covalent binding systems may also be needed not to induce the exchange of antibodies in the body;24 the experimental model we developed will contribute to further development of practical BNCT technologies.
Figure 5.

(a) Surviving fraction of the A431 cells treated with or without BSH (200 nM), Z33-DB (200 nM), cetuximab (100 nM), and EGF (100 nM) for 24 h at 37 °C, prior to thermal neutron irradiation (90 min), and a colony assay, as described in the Experimental Section. The data are expressed as the mean (±SD) of three experiments. (b) Surviving fraction of A431 cells in the same experimental conditions (thermal neutron fluence 3.7 × 107). The data are expressed as the mean (±SD) of three experiments. *P < 0.05, **P < 0.01.
Conclusions
In this research, we developed a “cassette”-like antibody binding technique for the receptor-targeted delivery of boron compounds based on BNCT. Active induction of macropinocytosis was crucial for the efficient cellular uptake of the boron compounds in targeted cancer cells and their biological activity after thermal neutron irradiation. Prof. Barth et al. pointed out that development of BNCT technology for delivery of boron compounds to targeted tumor cells on the bases of e.g., porphyrins, polyamines, nucleosides, liposomes nanoparticles, however, the best way to further improve the clinical efficacy of BNCT would be to optimize the dosing paradigms and delivery of boron compounds.25 Of course, cell-targeted delivery is one of the very important techniques to specifically deliver the objective boron compounds into tumor cells in BNCT to prevent damaging on normal cells and inducing side effects. As active targeting to objective tumor cells, antibodies against tumor receptors have been applied and conjugated to each carrier molecule with boron compounds. As shown in our research results, anti-EGFR antibody can recognize and accumulate on the plasma membrane via EGFR; however, only accumulation of the antibodies on the plasma membranes can be observed without effective cellular uptake of the antibody-conjugated carrier molecules with boron compounds. In our experiments, even in vitro assessments, only accumulation of boron compounds on the plasma membrane shows low cancer-killing activity after thermal neutron irradiation. Enhanced cellular uptake of the antibody-boron conjugates by effective macropinocytosis induction might be needed to induce the sophisticated biological activity in BNCT. The “cassette-like” antibody binding system to boron compounds is considered to be very useful for development of the tailor-made tumor target system with very easy handling. The system is considered to be possibility to attain future tumor targeting on demand. Our main focus in this research is the analysis and evaluation of the delivery mechanism of the Z33-antibody complex system, and the experiments on the boron carrier can be interpreted as an example of a demonstration experiment for the proof of concept with being applicable for improving previous approaches using various boron compound-carrier techniques. Our findings contribute not only to the development of the intracellular delivery of boron compounds in BNCT but also to an understanding of the importance of the effective cellular uptake of therapeutic molecules in the strategy of cancer receptor targeting.
Experimental Section
Peptide Synthesis
All peptides were chemically synthesized via the 9-fluorenylmethyloxycarbonyl (Fmoc) solid-phase synthesis method on a Rink amide resin with coupling reagents of 1-hydroxybenzotriazole (HOBt)/2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (Peptide Institute, Osaka, Japan)/N-methylmorpholine (NMM) (Nacalai Tesque, Kyoto, Japan) in dimethylformamide (DMF) (Nacalai Tesque) as previously described.26,27 The Rink amide resin and the Fmoc-amino acid derivatives were purchased from Shimadzu Biotech (Kyoto, Japan) and the Peptide Institute (Osaka, Japan), respectively. Deprotection of the protected peptide and cleavage from the resin were conducted via treatment with a trifluoroacetic acid/ethanedithiol mixture (95:5) for 3 h at 25 °C, followed by reverse-phase HPLC purification. The purification of each peptide was estimated to be >97% on the basis of the analytical HPLC peaks. The structures of the synthesized peptides were confirmed using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOFMS) (Microflex, Bruker, Billerica, MA, USA).
Z33-GGC (NH2-Phe-Asn-Met-Gln-Gln-Gln-Arg-Arg-Phe-Tyr-Glu-Ala-Leu-His-Asp-Pro-Asn-Leu-Asn-Glu-Glu-Gln-Arg-Asn-Ala-Lys-Ile-Lys-Ser-Ile-Arg-Asp-Asp-Gly-Gly-Cys-amide): MALDI-TOFMS: 4321.4 [calcd. for (M + H)+: 4321.7]. Retention time in HPLC, 11.0 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 2.3%.
rZ33-GGC (NH2-Asp-Gln-Ala-Leu-Asn-Glu-Met-His-Gln-Arg-Asp-Leu-Asn-Phe-Glu-Asp-Pro-Ile-Asn-Lys-Ser-Glu-Lys-Tyr-Arg-Gln-Arg-Phe-Asn-Ala-Ile-Gln-Arg-Gly-Gly-Cys-amide): MALDI-TOFMS: 4321.5 [calcd. for (M + H)+: 4321.7]. Retention time in HPLC, 11.0 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.2%.
For the preparation of fluorescently labeled peptides, synthesized and purified peptides were subjected to reactions with Alexa Flour 660 C2 maleimide (Thermo Fisher Scientific Inc., Rockford, IL, USA) in the presence of NMM (0.5% v/v) in methanol/DMF (1:1) for 3 h at 25 °C and then purified by HPLC. The purity of each peptide was estimated to be >97% on the basis of the analytical HPLC results. The structures of the synthesized peptides were confirmed using MALDI-TOFMS.
Z33-GGC-Alexa660 [(NH2-Phe-Asn-Met-Gln-Gln-Gln-Arg-Arg-Phe-Tyr-Glu-Ala-Leu-His-Asp-Pro-Asn-Leu-Asn-Glu-Glu-Gln-Arg-Asn-Ala-Lys-Ile-Lys-Ser-Ile-Arg-Asp-Asp-Gly-Gly-Cys (Alexa 660)-amide)]: MALDI-TOFMS: 5224.8 [calcd. for (M + H)+: 5224.0 (Figure S9)]. Retention time in HPLC, 10.9 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.05%
rZ33-GGC-Alexa660 (NH2-Asp-Gln-Ala-Leu-Asn-Glu-Met-His-Gln-Arg-Asp-Leu-Asn-Phe-Glu-Asp-Pro-Ile-Asn-Lys-Ser-Glu-Lys-Tyr-Arg-Gln-Arg-Phe-Asn-Ala-Ile-Gln-Arg-Gly-Gly-Cys (Alexa 660)-amide): MALDI-TOFMS: 5225.0 [calcd. for (M + H)+: 5224.0 (Figure S9)]. Retention time in HPLC, 10.8 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.01%
GGC-Alexa660 (NH2-Gly-Gly-Cys (Alexa 660)-amide): MALDI-TOFMS: 1136.6 [calcd. for (M + H)+: 1137.6 (Figure S9)]. Retention time in HPLC, 13.0 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.03%.
Preparation of the Dodecaborate-Z33 Peptide Conjugate
For the preparation of the disodium mercaptoundecahydrododecaborate (BSH)-Z33 peptide conjugate, synthesized and purified Z33-GGC peptide (1 equiv) was first subjected to reaction with bismaleimide ethane (BMOE) (3 equiv) in the presence of NMM (0.5% v/v) in methanol/DMF (1:1) for 6 h at 25 °C and then was purified by HPLC purification. The BMOE-linked Z33 peptide was then subjected to react with 10BSH [provided by Stella Pharma Corporation (Osaka, Japan)] in methanol/DMF (1:1) for 1 h at 25 °C (Figure S10) and then was purified by HPLC purification. The purity of each peptide was estimated to be >97% on the basis of the analytical HPLC results. The structures of the synthesized peptides were confirmed using MALDI-TOFMS.
Z33-BMOE (NH2-Phe-Asn-Met-Gln-Gln-Gln-Arg-Arg-Phe-Tyr-Glu-Ala-Leu-His-Asp-Pro-Asn-Leu-Asn-Glu-Glu-Gln-Arg-Asn-Ala-Lys-Ile-Lys-Ser-Ile-Arg-Asp-Asp-Gly-Gly-Cys (BMOE)-amide): MALDI-TOFMS: 4543.1 [calcd. for (M + H)+: 4542.8]. Retention time in HPLC, 11.1 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.3%.
Z33-BMOE-DB [(NH2-Phe-Asn-Met-Gln-Gln-Gln-Arg-Arg-Phe-Tyr-Glu-Ala-Leu-His-Asp-Pro-Asn-Leu-Asn-Glu-Glu-Gln-Arg-Asn-Ala-Lys-Ile-Lys-Ser-Ile-Arg-Asp-Asp-Gly-Gly-Cys (BMOE-DB)-amide)]: MALDI-TOFMS: 4706.5 [calcd. for (M + H)+: 4705.9 (Figure S9)]. Retention time in HPLC, 12.0 min (column: Cosmosil 5C18-AR-II (4.6 × 150 mm); gradient: 5–95% B in A (A = H2O containing 0.1% CF3COOH; B = CH3CN containing 0.1% CF3COOH) over 30 min; flow: 1 mL/min; detection 220 nm). The yield from the starting resin was 0.03%.
Preparation of Fluorescently Labeled Cetuximab
Cetuximab (Erbitux, Merck Biopharma Japan, Tokyo, Japan) was purified by washing with Dulbecco’s phosphate-buffered saline (PBS) (Nacalai Tesque) and filtration using Amicon Ultra centrifugal filters (100 K device, Merck Millipore, Billerica, MA, USA). The concentrations of cetuximab were detected using a Pierce BCA protein assay kit (Thermo Fisher Scientific Inc.). Purified cetuximab in PBS was fluorescently labeled using fluorescein isothiocyanate isomer I (FITC) (Sigma-Aldrich, St. Louis, MO, USA) (1 equiv) in the presence of NMM (1 equiv) in PBS for 1 h at 25 °C, prior to filtration using Amicon Ultra centrifugal filters (100 K device, Merck Millipore). The antibody concentration of the FITC-labeled cetuximab was measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific Inc.).
Cell Cultures
Human epidermoid carcinoma-derived A431 cells and Chinese hamster ovary (CHO)-K1 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). Each cell line was cultured in MEM (Gibco, Life Technologies Corporation, Grand Island, NY, USA) for A431 cells and F-12 nutrient mixture (Ham’s F-12) for CHO-K1 cells (Gibco, Life Technologies Corporation) containing 10% heat-inactivated FBS (Gibco, Life Technologies Corporation). Each cell was grown on 100-mm dishes incubated at 37 °C in 5% CO2.
Confocal Laser Scanning Microscopic Images of Cellular Uptake
A431 cells (4.7 × 104 cells/well, 200 μL) were plated onto an 8-well plate (μ-slide 8-well, ibidi GmbH, Gräfelfing, Germany) and incubated in MEM containing 10% FBS for 24 h at 37 °C under 5% CO2. After completely adhering, the cells were washed with MEM containing 10% FBS (170 μL/well). Before they were observed under a confocal microscope, the cells were stained with Hoechst 33342 dye (Thermo Fisher Scientific Inc.; 5 μg/mL) for 15 min at 37 °C. The cells were then washed with fresh cell culture medium and analyzed using an FV1200 confocal laser scanning microscope (Olympus, Tokyo, Japan) equipped with a 40× objective without cell fixation.
Flow Cytometry Analysis of the Membrane to Detect the Binding and Cellular Uptake of the Fluorescently Labeled Z33 Peptide and Dextran
A431 cells (1.4 × 105 cells/well, 1 mL) were plated into a 24-well microplate (Iwaki, Tokyo, Japan) and incubated in MEM containing 10% FBS for 24 h at 37 °C in 5% CO2. After completely adhering to the wells, the cells were washed with MEM containing 10% FBS and treated with each sample (510 μL/well) for 24 h at 37 °C in 5% CO2 before being washed with PBS (triple washing, 200 μL). The cells were then treated with 2 mM EDTA or 0.1 g/L-Trypsin/0.1 mmol/L/l-EDTA (200 μL/well) at 37 °C for 10 min. The cells were then added to PBS (200 μL/well) and subjected to fluorescence analysis with a Guava easyCyte (Merck Millipore) flow cytometer using 488 nm laser excitation and a 525 nm emission filter. In the case of trypsinization, after detachment of the cells, the cells were washed with PBS (400 μL) and centrifuged (800g at 4 °C for 10 min). This washing cycle was repeated, the cells were suspended in PBS (400 μL), and subjected to the fluorescent analysis. The quantification of live cells (10,000 cells/sample) was based on their cellular fluorescence intensity as measured through forward-scattering and side-scattering analyses.
Immunostaining and Confocal Laser Scanning Microscopic Images (Anti-BSH Antibody Staining)
A431 cells (4.7 × 104 cells/well, 200 μL) were plated onto an 8-well plate (μ-slide 8-well, ibidi GmbH) and incubated in MEM containing 10% FBS for 24 h at 37 °C in 5% CO2. After completely adhering, the cells were washed with MEM containing 10% FBS and treated with each Z33-BSH sample in MEM containing 10% FBS (170 μL/well) for 24 h at 37 °C. The cells were then fixed with 4% paraformaldehyde at 25 °C for 30 min and treated with 0.1% Triton X-100 in PBS at 25 °C for 15 min (200 μL/well). After washing with PBS, the cells were stained with anti-BSH antibody (A9H3 anti-BSH-Mab)28 in PBS (100 μL/well) for 1 h at 25 °C. Then, the cells were stained with Alexa-Flour 488 goat anti-mouse IgG (H + L) (100 μL/well) for 1 h at 25 °C. After washing with PBS, the cells were analyzed using an FV1200 confocal laser scanning microscope (Olympus) equipped with a 40× objective.
ELISA
A431 cells (1.0 × 106 cells/well) were plated onto a 60 mm cell culture dish for 24 h in 10% FBS-containing MEM. The medium was replaced with an equivalent medium containing each Z33-BSH sample in 10% FBS-containing MEM (900 μL). After incubating for 24 h at 37 °C under 5% CO2, the cells were harvested with 0.05% trypsin/0.02% EDTA in PBS and suspended in total 2 mL of MEM. After cell counting, the cells were collected by centrifugation (370g) for 5 min. The pelleted cells were resuspended in 0.05% Tween-20 and allowed to stand for 10 min. The boron concentration of the obtained cell lysate was determined by competitive ELISA using anti-BSH antibody A9H3 (N = 3).28
Cell Viability (WST-8 Assay)
Cell viability was analyzed using a WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] assay. A431 cells (1.4 × 104 cells/100 μL) were incubated in 96-well microplates in MEM containing 10% FBS for 24 h at 37 °C in 5% CO2. The cells were then treated with each sample (50 μL) at 37 °C in 5% CO2. After the sample treatment, the WST-8 reagent (10 μL) was added to each well, and the samples were incubated for 40 min at 37 °C. The absorbance at 450 nm (A450) and 620 nm (A620) was measured, and the viable cell number was calculated by subtracting the A620 value from the A450 value.
Binding Assay of Z33-DB and Cetuximab
Z33-DB (final 200 nM) and cetuximab (final 100 nM) were complexed in PBS buffer for 30 min at 25 °C, prior to ultrafiltration using Amicon Ultra-0.5 centrifugal filters (100,000 molecular weight cut-off, Merck) (10,000g at 4 °C for 10 min) to remove nonbound Z33-DB to cetuximab. Bound Z33-DB concentration to cetuximab was detected using HPLC (Hitachi Chromaster and Chromassist data station).
Thermal Neutron Irradiation and Assessment of the Cell-Killing Effect (Surviving Fraction)
A431 cells (2.0 × 105 cells/well, 2 mL) were plated into a 6-well microscope plate (Iwaki) and incubated in MEM containing 10% FBS for 24 h at 37 °C in 5% CO2. After completely adhering, the cells were washed with MEM containing 10% FBS and treated with each sample (720 μL/well) for 24 h at 37 °C in 5% CO2. The cells were then washed with PBS and detached by treatment with 0.5% trypsin for 7 min at 37 °C. The number of detached cells was counted (6000 cells), and then, MEM containing 10% FBS was added to a total volume of approximately 6 mL. The cells were added to a column-shaped tube (1 mL/tube) and irradiated with thermal neutrons for 90 min at the Institute for Integrated Radiation and Nuclear Science, Kyoto University, Kyoto, Japan. The thermal neutron fluence was determined by averaging two gold foils that were symmetrically attached to the surface of the column-shaped tube along the direction of incidence of the thermal neutrons. The cells were then plated onto a 6-well microscope plate (Iwaki) (1000 cells/well) and incubated in MEM containing 10% FBS for 14 days at 37 °C to examine colony formation. The colonies were fixed with ethanol and stained with 0.1% crystal violet for quantitative visualization by the naked eye.
Statistical Analyses
All statistical analyses were performed using GraphPad Prism software (ver. 5.00; GraphPad, San Diego, CA, USA). One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used. Differences were considered significant when the calculated p-value was <0.05.
Acknowledgments
This study was supported in part by JSPS KAKENHI (JP16H02612 and JP20K20463 for I.N.). This study was also supported by the Leading University as a Base for Human Resource Development in Nanoscience and Nanotechnology, Osaka Prefecture University (OPU). Experiments based on thermal neutron irradiation were conducted with the assistance of Professor Minoru Suzuki (Kyoto University). Professor Ikuo Fujii (OPU) assisted in the experiments of MALDI-TOFMS. Kayo Hirano (OPU) assisted in the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01377.
Confocal laser microscopic images of the cells treated with Alexa 660-labeled Z33; relative cellular uptake of FITC-dextran; with cetuximab and Z33-DB; enlarged pictures (Z33-Alexa660) of Figure 3a; enlarged pictures (FITC-cetuximab) of Figure 3a; trypsin treatment and flow cytometer analysis; enlarged pictures (stained with anti-BSH antibody) of Figure 4a; cell viability; MALDI-TOFMS data; and HPLC data (PDF)
Author Contributions
I.N., Y.H., and M.K. designed the study. I.N., A.A., Y.S., S.H., M.I., T.T.-N., and Y.H. performed the experiments. Statistical analysis was conducted by T.T.-N. The manuscript was written by I.N., Y.H., and M.K. All authors discussed and analysed the obtained results.
The authors declare no competing financial interest.
Supplementary Material
References
- Soloway A. H.; Hatanaka H.; Davis M. A. Penetration of brain and brain tumor. VII. Tumor-binding sulfhydryl boron compounds. J. Med. Chem. 1967, 10, 714–717. 10.1021/jm00316a042. [DOI] [PubMed] [Google Scholar]
- Hatanaka H. A revised boron-neutron capture therapy for malignant brain tumors. II. Interim clinical result with the patients excluding previous treatments. J. Neurol. 1975, 209, 81–94. 10.1007/bf00314601. [DOI] [PubMed] [Google Scholar]
- Mishima Y.; Honda C.; Ichihashi M.; Obara H.; Hiratsuka J.; Fukuda H.; Karashima H.; Kobayashi T.; Kanda K.; Yoshino K. Treatment of malignant melanoma by single thermal neutron capture therapy with melanoma-seeking 10B-compound. Lancet 1989, 334, 388–389. 10.1016/s0140-6736(89)90567-9. [DOI] [PubMed] [Google Scholar]
- Wongthai P.; Hagiwara K.; Miyoshi Y.; Wiriyasermkul P.; Wei L.; Ohgaki R.; Kato I.; Hamase K.; Nagamori S.; Kanai Y. Boronophenylalanine, a boron delivery agent for boron neutron capture therapy, is transported by ATB0,+, LAT1 and LAT2. Cancer Sci. 2015, 106, 279–286. 10.1111/cas.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luderer M. J.; de la Puente P.; Azab A. K. Advancements in tumor targeting strategies for boron neutron capture therapy. Pharm. Res. 2015, 32, 2824–2836. 10.1007/s11095-015-1718-y. [DOI] [PubMed] [Google Scholar]
- Nedunchezhian K.; Aswath N.; Thiruppathy M.; Thirugnanamurthy S. Boron neutron capture therapy - a literature review. J. Clin. Diagn. Res. 2016, 10, ZE01–ZE04. 10.7860/JCDR/2016/19890.9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhood B.; Samadian H.; Ghorbani M.; Zakariaee S. S.; Knaup C. Physical, dosimetric and clinical aspects and delivery systems in neutron capture therapy. Rep. Practical Oncol. Radiother. 2018, 23, 462–473. 10.1016/j.rpor.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth R. F.; Mi P.; Yang W. Boron delivery agents for neutron capture therapy of cancer. Canc. Commun. 2018, 38, 35. 10.1186/s40880-018-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M. Boron neutron capture therapy (BNCT): a unique role in radiotherapy with a view to entering the accelerator-based BNCT era. Int. J. Clin. Oncol. 2020, 25, 43–50. 10.1007/s10147-019-01480-4. [DOI] [PubMed] [Google Scholar]
- Barth R. F.; Coderre J. A.; Vicente M. G.; Blue T. E. Boron neutron capture therapy of cancer: current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. 10.1158/1078-0432.ccr-05-0035. [DOI] [PubMed] [Google Scholar]
- Mi P.; Yanagie H.; Dewi N.; Yen H.-C.; Liu X.; Suzuki M.; Sakurai Y.; Ono K.; Takahashi H.; Cabral H.; Kataoka K.; Nishiyama N. Block copolymer-boron cluster conjugate for effective boron neutron capture therapy of solid tumors. J. Controlled Release 2017, 254, 1–9. 10.1016/j.jconrel.2017.03.036. [DOI] [PubMed] [Google Scholar]
- Braisted A. C.; Wells J. A. Minimizing a binding domain from protein A. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5688–5692. 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pille J.; Cardinale D.; Carette N.; Di Primo C.; Besong-Ndika J.; Walter J.; Lecoq H.; van Eldijk M. B.; Smits F. C. M.; Schoffelen S.; van Hest J. C. M.; Mäkinen K.; Michon T. General strategy for ordered noncovalent protein assembly on well-defined nanoscaffolds. Biomacromolecules 2013, 14, 4351–4359. 10.1021/bm401291u. [DOI] [PubMed] [Google Scholar]
- van Eldijk M. B.; Smits F. C. M.; Thies J. C.; Mecinović J.; van Hest J. C. M. Thermodynamic investigation of Z33-antibody interaction leads to selective purification of human antibodies. J. Biotechnol. 2014, 179, 32–41. 10.1016/j.jbiotec.2014.03.023. [DOI] [PubMed] [Google Scholar]
- Muguruma K.; Yakushiji F.; Kawamata R.; Akiyama D.; Arima R.; Shirasaka T.; Kikkawa Y.; Taguchi A.; Takayama K.; Fukuhara T.; Watabe T.; Ito Y.; Hayashi Y. Novel hybrid compound of a plinabulin prodrug with an IgG binding peptide for generating a tumor selective noncovalent-type antibody-drug conjugate. Bioconjugate Chem. 2016, 27, 1606–1613. 10.1021/acs.bioconjchem.6b00149. [DOI] [PubMed] [Google Scholar]
- Araki N.; Hamasaki M.; Egami Y.; Hatae T. Effect of 3-methyladenine on the fusion process of macropinosomes in EGF-stimulated A431 cells. Cell Struct. Funct. 2006, 31, 145–157. 10.1247/csf.06029. [DOI] [PubMed] [Google Scholar]
- Dise R. S.; Frey M. R.; Whitehead R. H.; Polk D. B. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G276–G285. 10.1152/ajpgi.00340.2007. [DOI] [PubMed] [Google Scholar]
- Tanaka G.; Nakase I.; Fukuda Y.; Masuda R.; Oishi S.; Shimura K.; Kawaguchi Y.; Takatani-Nakase T.; Langel Ü.; Gräslund A.; Okawa K.; Matsuoka M.; Fujii N.; Hatanaka Y.; Futaki S. CXCR4 stimulates macropinocytosis: implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. 10.1016/j.chembiol.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Swanson J. A.; Watts C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- Swanson J. A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C.; Davidson S. M.; Soydaner-Azeloglu R. G.; Parker S. J.; Kamphorst J. J.; Hackett S.; Grabocka E.; Nofal M.; Drebin J. A.; Thompson C. B.; Rabinowitz J. D.; Metallo C. M.; Vander Heiden M. G.; Bar-Sagi D. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone S.; Cocucci E.; Podini P.; Kirchhausen T.; Clementi E.; Meldolesi J. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. J. Cell Sci. 2006, 119, 4758–4769. 10.1242/jcs.03238. [DOI] [PubMed] [Google Scholar]
- Nakase I.; Hirose H.; Tanaka G.; Tadokoro A.; Kobayashi S.; Takeuchi T.; Futaki S. Cell-surface accumulation of flock house virus-derived peptide leads to efficient internalization via macropinocytosis. Mol. Ther. 2009, 17, 1868–1876. 10.1038/mt.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Ito Y. Recent chemical approaches for site-specific conjugation of native antibodies: technologies toward next-generation antibody-drug conjugates. Chembiochem 2019, 20, 2729–2737. 10.1002/cbic.201900178. [DOI] [PubMed] [Google Scholar]
- Barth R. F.; Mi P.; Yang W. Boron delivery agents for neutron capture therapy of cancer. Canc. Commun. 2018, 38, 35. 10.1186/s40880-018-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futaki S.; Ohashi W.; Suzuki T.; Niwa M.; Tanaka S.; Ueda K.; Harashima H.; Sugiura Y. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjugate Chem. 2001, 12, 1005–1011. 10.1021/bc015508l. [DOI] [PubMed] [Google Scholar]
- Nakase I.; Tadokoro A.; Kawabata N.; Takeuchi T.; Katoh H.; Hiramoto K.; Negishi M.; Nomizu M.; Sugiura Y.; Futaki S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2007, 46, 492–501. 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- Kirihata M.; Uehara K.; Asano T.. Hapten compound and antibody. WO 2007097065 A1, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.