

Abstract

Microtubule affinity-regulating kinase 4 (MARK4), a member of the serine/threonine kinase family, is an emerging therapeutic target in anticancer drug discovery paradigm due to its involvement in regulation of microtubule dynamics, cell cycle regulation, and cancer progression. Therefore, to identify the novel chemical architecture for the design and development of novel MARK4 inhibitors with concomitant radical scavenging property, a series of small-molecule arylaldoxime/5-nitroimidazole conjugates were designed and synthesized via multistep chemical reactions following the pharmacophoric hybridization approach. Compound 4h was identified as a promising MARK4 inhibitor with high selectivity toward MARK4 inhibition as compared to the panel of screened 30 kinases pertaining to the serine/threonine family, which was validated by molecular docking and fluorescence binding studies. The comprehensive cell-based examination divulged the promising apoptotic, antiproliferative, and antioxidant potential for the chemotype 4h. The compound 4h was endowed with the Ka value of 3.6 × 103 M–1 for human serum albumin, which reflects its remarkable transportation and delivery properties to the target site via blood. The present study impedes that in the future, such compounds may stand as optimized pharmacological lead candidates in drug discovery for targeting cancer via MARK4 inhibition with a remarkable anticancer profile.
Introduction
Design and development of anticancer agents has been a challenging task for the scientific community and a wide range of active targets are being exploited to combat this devastating disease.1−3 Kinases are considered as one of the most intensively pursued targets in current pharmacological research, particularly for cancer, due to their critical roles in cellular signaling.4,5 Kinases catalyze the transfer of the γ-phosphate group of adenosine 5′-triphosphate (ATP) onto a substrate, mediate most signal transductions,6 and regulate various cellular activities, including proliferation, survival, apoptosis, metabolism, transcription, differentiation, and a wide array of other cellular processes.7,8 Microtubule affinity-regulatory kinase 4 (MARK4), a member of the Ser/Thr kinase family, has emerged as an important therapeutic target for anticancer drug development recently.9,10 MARK4 is coded by genes located on the chromosome number 19 in the human genome11 and it abets the breast cancer cell proliferation as well as migration through the inhibition of Hippo signaling.12 MARK4 enhances adipogenesis and triggers cellular apoptosis by activating JNK1 and by inhibiting the p38MAPK pathways.13 It gets overexpressed in cancerous cells including hepatocarcinoma and leukemia and plays a significant role in the promotion of breast and prostate cancers.14 Therefore, MARK4 has emerged as an important therapeutic target in the anticancer drug discovery paradigm.
MARK4 carries out the phosphorylation of the microtubule-associated proteins (MAPs) by using ATP at the binding domain Lys-Xaa-Gly-Ser (KXGS) motif.11 The process of phosphorylation maintains the cell polarity, microtubule stability, protein stability, intracellular signaling, cell cycle control, cell division specifically in the G1/S checkpoint, and many other complex cellular processes due to the detachment of MAPs from microtubule assembly.15,16 Phosphorylation of MARK4 at Thr214 residue activates the microtubule dynamics; however, the attachment of the phosphate group at Ser218 residue inhibits its function. Asp181 is the active site of MARK4, which in turn gets activated by phosphorylation of Thr214 residue.14 Mostly, the kinase-inhibiting agents are reversible, and ATP competitive inhibitors bind in the ATP-binding pocket of the kinase protein. Vemurafenib (1), S49076 (2), and nilotinib (3) (Figure 1) are some of the FDA-approved kinase inhibitors, which function by binding at the ATP binding domain of kinases.4,17 The imidazole motif is the important fragment of nilotinib and vemurafenib, which aid in promoting the function of these approved molecules with the binding pocket of their respective kinases.4,18 Over the years, electron-deficient nitroaromatic compounds have been investigated for use in cancer treatment and, likely, the nitroimidazole derivatives have been reported as potent mitogen-activated protein (MAP) kinase inhibitors.19,20 A perusal of literature reveals that oximes act as a core structural motif of myriad chemotypes endowed with anticancer activities including kinase inhibition.21−23 Designing hybrid molecules in which two or more pharmacophores are covalently linked is a rationally attractive approach toward the development of effective therapeutic molecules. The pharmacophoric hybridization approach results in the development of promising chemotherapeutic agents having different modes of action and reduced side effects.24
Figure 1.

Structural resemblance of the identified small-molecule MARK4 inhibitor 4h with some reported kinase inhibitors 1–3.
Further, the small molecules play a pivotal role in the inhibition of various kinases pertaining to the human kinome and are being utilized extensively in the drug development.8,25 Considering the important pharmacological properties, viz., ease in cellular diffusion, membrane permeability, transport, bioavailability, binding affinity, and potency associated with small ring heterocyclic motifs,26−28 the molecular hybridization approach was employed to graft a 5-nitroimidazole scaffold with a pharmacologically important oxime core to construct novel hybrid miniature chemotypes as a new class of potential MARK4 inhibitors. Also, as the MARK4 expression plays a crucial role in the induction of oxidative stress in adipocytes and, therefore, the inhibition of MARK4 may result in the reduction of reactive oxygen species (ROS) generation, which therefore relieves the oxidative stress in cells.29−31 Therefore, considering the above-mentioned facts and our earlier reports on microtubule affinity-regulating kinase 4,32−34 we herein report the design and synthesis of novel arylaldoxime/5-nitroimidazole conjugates as potent MARK4 inhibitors and antioxidant agents with remarkable anticancer activity.
Results
Design
Human kinases possess the β sheet protein folding pattern dominating on N-terminal side and α helix pattern in the C-terminal lobe, which are connected by a hinge region sequence. The ATP carries out the phosphorylation by binding in the cleft formed between the N- and C-terminal lobes of the kinase protein.4,35 The X-ray crystallography established that the MARK4 protein comprises of a catalytic domain, short linker sequence, ubiquitin-associated (UBA) domain bound to the N-terminal lobe of the catalytic domain, spacer domain, and globular kinase-associated domain (KA). The catalytic domain is the site of interest for targeting the phosphorylation process.36 The smaller ATP competitors have the major access to interact with the active residues of the kinase protein and hence show maximum inhibition potential.8,37
Oxime-derived compounds have been established to inhibit the kinase function by interacting at the ATP binding site and therefore prevent the phosphorylation process, which in turn disrupts the microtubule assembly.38,39 The nitroimidazole ring is the fragment of diverse promising kinase inhibitors20,40,41 and interacts with the various cleft residues via π electron cloud and the oxygen atoms, hence showing appreciable binding affinity with MARK4 as per in silico screening (Table S1). The docking simulation technique was employed for binding affinity assessment of our designed compounds with MARK4 and then the attempt was made for their synthesis. In silico screening of our initial compound 4a of Scheme 1 showed that one of the oxygen atoms of the nitro group interacts via hydrogen bonding with Asp196 and the other oxygen atom interacts by van der Waals interactions with Lys85 besides making a hydrogen bond with Asp196 residue of the catalytic domain of MARK4 protein. The five-membered heterocyclic aromatic imidazole ring makes the π–sigma interactions with Val70. The phenyl substituent interacts via π–alkyl interactions with Ala195 and Ala83 and via the π–sigma interaction with Val70. The nitrogen atom of oxime core shows polar interactions with Asp196, whereas its oxygen-bonded hydrogen atom forms the hydrogen bond with the Asp196 residue (Figure S1). Convinced by in silico analysis of the designed compounds, the remarkable affinity of small molecules toward kinase inhibition, and the potency of the chosen scaffolds, we intended to synthesize novel arylaldoxime hybrids of 5-nitroimidazole heterocycle following the molecular hybridization approach to formulate and identify the small MARK4 inhibitors with the optimization of enhanced bioeffects.
Scheme 1. Synthetic Route Adopted for the Preparation of Arylaldoxime/5-Nitroimidazole Hybrids 4a–4h.

Reagents and conditions: (a) NH4OH·HCl, 3 N NaOH, EtOH, reflux, 14–20 h; (b) NCS, DMF, 60 °C, 8–12 h; (c) 2-methyl-4(5)-nitroimidazole, TBAB, K2CO3, ACN, reflux, 3–4 h.
Synthesis
A series of arylaldoxime/5-nitroimidazole hybrid molecules (Scheme 1) were synthesized starting from the synthesis of different arylaldoximes (2a–2h), which were further converted into their corresponding carboximidoyl chlorides (3a–3h) via N-chlorosuccinimide (NCS)-mediated chlorination. Initially, the carboximidoyl chlorides (3a–3h) were reacted with 2-methyl-4(5)-nitroimidazole in the presence of K2CO3 using DMF as the solvent under reflux, but the reaction time was exceeding 72 h with the formation of side products as well and the overall yield was very low (20–25%). To overcome these hurdles, tetra-n-butylammonium bromide (TBAB) was employed as a phase transfer catalyst. Mechanistically, the solid–liquid phase transfer catalyst TBAB replaces the potassium ion from imidazolium-potassium salt to form the quaternary ammonium salt of 2-methyl-4(5)-nitroimidazole, which is much more dissociated in the organic phase (ACN) than its potassium salt and hence more reactive. The quaternary salt reacts with the carboximidoyl chlorides (electrophile in the organic phase) to form the target molecules (4a–4h).
The TBAB increases the nucleophilicity and transport of the imidazolium ion into the organic phase and therefore leads to the increase in yield and reduced reaction time.42 Therefore, the attempt of employing the TBAB as a phase transfer catalyst (solid–liquid phase-transfer system) increased the credible yield of desired products, reduced the reaction completion time, predominantly prevented the occurrence of other side reactions, and resulted in the preferable nucleophilic substitution at the desired position of the nitroimidazole ring.43,44 Structures of the synthesized target compounds were elucidated by 1H NMR, 13C NMR, ESI-MS spectral data, and X-ray, and their purity was confirmed by elemental analysis. The compounds were of ≥95% purity grade.
Single-Crystal Data
Single-crystal structures of compounds 4c and 4e were isolated and were suitable to measure by the X-ray method. The compound 4c crystallizes in a monoclinic crystal system and compound 4e in an orthorhombic crystal system (Table 1). The compound 4e, which crystallize in the orthorhombic crystal system, contains two molecules in the asymmetric unit, which correspond with two atropoisomers. These atropoisomers are present in all the structures. Friedel pairs were determined and their number was 25,280. ORTEP diagrams for compounds 4c and 4e are shown in Figure 2. The molecular structures present similar bond lengths and angles with that of the other compounds having similar functional groups (Table S4). Intermolecular hydrogen bonds and van der Waals forces predominate in their crystal packings (Table S5), and they can be the key of their interaction with the biomolecules and their bioactivity.
Table 1. Crystal Data and Structure Refinement for Compounds 4c and 4e.
| compound | 4c | 4e |
|---|---|---|
| formula | C12 H12 N4 O3 | C13 H14 N4 O3 |
| formula weight | 260.26 | 274.28 |
| T (K) | 100(2) | 100(2) |
| wavelength (Å) | 0.71073 | 0.71073 |
| crystal system | monoclinic | orthorhombic |
| space group | P21/n | P21212 |
| a (Å) | 6.2087(5) | 14.154(5) |
| b (Å) | 15.2664(10) | 14.763(5) |
| c (Å) | 13.4466(9) | 13.021(4) |
| α (°) | 90 | 90 |
| β (°) | 100.463(3) | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 1253.34(15) | 2720.8(17) |
| Z | 4 | 8 |
| F000 | 544 | 1152 |
| Dcalc (g cm–3) | 1.379 | 1.339 |
| μ (mm–1) | 0.103 | 0.098 |
| θ (°) | 2.67 to 28.32 | 2.53 to 26.38 |
| Rint | 0.0913 | 0.0646 |
| crystal size (mm3) | 0.08 × 0.10 × 0.20 | 0.07 × 0.07 × 0.24 |
| goodness-of-fit on F2 | 1.050 | 1.091 |
| R1[I > 2σ(I)]a | 0.0449 | 0.0468 |
| wR2 (all data)b | 0.1151 | 0.1113 |
| largest difference peak and hole (e Å–3) | 0.347 and −0.299 | 0.364 and −0.398 |
R1 = Σ||Fo| – |Fc||/Σ|Fo|.
wR2 = {Σ|[w(||Fo|2 – |Fc|2|)2]|/Σ[w(Fo2)2]}1/2.
Figure 2.

ORTEPs diagrams for the arylaldoxime-nitroimidazole hybrids 4c and 4e. All the nonhydrogen atoms are presented by their 50% probability ellipsoids. Drawing was done with the SHELXL package.
Molecular Docking
Binding affinities and the mode of binding of all the synthesized molecules with MARK4 were evaluated by the help of molecular docking. Autodock Vina combined with PyRx software was used to check out the interactions offered by the active site residues of MARK4 with each synthesized molecule (ligand).45,46 Our first objective was to validate the binding of the initial designed compound 4a in the active site of MARK4. Compound 4a showed an efficient binding with the active site of MARK4 (Figure S1). After the validation of binding affinity of the designed initial compound 4a, we have synthesized further derivatives incorporating various aryl substituents. The binding energy and list of MARK4 residues interacting to the ligands are given in Table S1, Figure 3, and Figures S1–S3. All synthesized hybrid molecules showed an appreciable binding energy ranging between −6.8 and −7.6 kcal/mol (Table S1). Docking results showed that each MARK4–ligand (4a–4h) complex is stabilized by a significant number of noncovalent interactions (hydrogen bond and van der Waals) offered by the residues lying in the active site region of MARK4 (Figure 3 and Figures S1–S3). These results supported our motivation to synthesize such a class of MARK4 inhibitors that bind efficiently to the active site residues and consequently diminish the enzymatic activity. The consistency of docking protocol was evaluated by redocking the known inhibitor of MARK4 (pyrazolopyrimidine inhibitor 5RC) into the active site of the MARK4.
Figure 3.

Compound 4h binds to the active site of MARK4. (A) 3D presentation of MARK4 docked ligand complex of compound 4h (purple) to the active site residues of MARK4. (B) Focused view of MARK4 binding pocket with compound 4h shows the hydrogen bond donor–acceptor residues of protein. (C) 2D representation of residues involved in different interactions like van der Waals interactions, hydrogen bonding, charge, or polar interactions (each type of interaction is represented by respective color; see inset).
MARK4 Inhibition and Structure–Activity Relationship (SAR)
After the in silico validation of binding affinities of synthesized molecules, we performed MARK4 enzyme inhibition assay using ATPase hydrolysis. To peruse this study, we have expressed and purified the recombinant MARK4 protein, as described earlier.29 All the synthesized molecules were evaluated against MARK4 at a single dose of 20 μM and it was found that only one compound (4h) showed significant inhibition. On the basis of initial screening, compound 4h was being considered for further MARK4 inhibition assay purposes with its increasing concentrations (0–20 μM) (Figure 4). Enzyme inhibition studies revealed that compound 4h inhibited the activity of MARK4 with IC50 values of 1.74 μM. The results of docking and enzyme assay showed that compound 4h binds to the active site of MARK4 and significantly inhibits its activity.
Figure 4.

Compound 4h inhibited the kinase activity of MARK4. (A) Hydrolysis of Pi from ATP. The position of Pi and ATP spots are indicated. Lane 1, negative control (no protein); lane 2, 100 nM MARK4 (positive control); lanes labeled as 0.5, 1, 2, 5, 10, and 20, concentration of the compound. (B) ATPase inhibition (% hydrolysis of Pi) with increasing concentrations of compound 4h is shown as a function of concentration calculated by comparing with the positive control.
Since the variation was done on the either side of the oxime core for the determination of the impact of the varied electronic arrangement on the activity of the target molecules. All the evaluated compounds of Scheme 1 share two similar structural features: (i) central oxime core and (ii) hydrophobic fragment (ring A, Figure 5). In Scheme 1, the −NOH group was coupled with 2-methyl-4(5)-nitroimidazole scaffold (ring B) (Figure 5). The initial compound (4a) bearing an unsubstituted phenyl ring was observed to be less potent (Table 2) when evaluated for MARK4 inhibitory activity. However, the introduction of electron-releasing groups (ERGs), viz., OCH3 (4b), CH3 (4c), C2H5 (4d), OC2H5 (4e), and Cl (4f), at the C-4 position in ring A as R1 substituents still leads to the minimal MARK4 inhibition. The replacement of these ERGs with the strongly electron-withdrawing NO2 group (4g) at the same position resulted in the slight enhancement in the potency. Surprisingly, multifold MARK4 inhibition was observed when the CF3 group was incorporated as the R2 substituent in ring A, keeping the C-4 position unsubstituted (4h, IC50 = 1.47 μM, Table 2).
Figure 5.

General structure of the designed chemotypes depicting the strategic variation of nitroaromatic scaffold (ring B) with a central oxime core bearing the hydrophobic fragment (ring A).
Table 2. MARK4 Inhibition and Human Cancer Cell Viability Profile of Compounds 4a–4h.
| compound | MARK4 inhibition IC50 (μM) | MCF-7 IC50 (μM) | HepG2 IC50 (μM) |
|---|---|---|---|
| 4a | >20 | 37.32 | 38.53 |
| 4b | >20 | 34.44 | 32.66 |
| 4c | >20 | 32.54 | 33.22 |
| 4d | >20 | 33.27 | 35.42 |
| 4e | 18.45 | 23.32 | 21.54 |
| 4f | >20 | 28.82 | 24.77 |
| 4g | 16.22 | 22.54 | 25.32 |
| 4h | 1.47 | 3.74 | 5.99 |
The inhibitory activity shown by the electron-withdrawing groups was more effective as compared to the electron-releasing groups in the entire series. Therefore, in terms of structure–activity relationship (SAR), it can be concluded that the MARK4 inhibition of studied hybrids was substituent-dependent. The compounds having the −CF3 group were the most potent among all the chemotypes, which supported the earlier reports that the compounds bearing the trifluoromethyl group occupy prime importance in medicinal chemistry due to their significant drug-like properties, viz., remarkable lipophilicity, absorption, and metabolic stability.47,48 Moreover, the presence of −CF3 group may have drifted the activity of the compound 4h due to its inevitable physiological significance mostly in terms of conferring the excellent lipophilicity and absorption to the molecules across the cellular plasma membrane.
Kinase Selectivity of Compound 4h
Kinase inhibitors generally showed off-target activities with same/different kinase families. Therefore, to check out the selectivity of compound 4h, we evaluate it against a panel of 30 kinases of the same family (CAMK family). The results of selectivity showed that compound 4h inhibited MARK4 more strongly as compared to other kinases of the same family (Figure 6). The inhibition results indicated that at the tested concentration, the compound 4h moderately inhibits MARK1, CAMKIV, MAPKAPK2, and DAPK1 (Table S2). It may be due to the similar/conserved binding pockets of these kinases. Importantly, kinase selectivity results suggested that compound 4h possesses high selectivity toward MARK4 inhibition.
Figure 6.

Compound 4h has specificity toward MARK4. (A) Kinase inhibition results with a panel of 30 kinases for compound 4h. (B) Pie chart presentation for selectivity data. The selected panel of 30 kinases was incubated with 10 μM dose of each compound and analyzed for percent activity values. The percent kinase activity was calculated and presented as a percentage of inhibition (%). In a total of 30 kinases, 23 kinases showed <20% inhibition, 6 kinases showed 20–40% inhibition, and only MARK4 exhibited >80% inhibition.
Binding Affinity toward MARK4 and HSA
To estimate the actual binding affinity of compound 4h toward MARK4 and human serum albumin (HSA), fluorescence binding assay was performed. In each case, the protein sample was excited at 280 nm and the emission spectra were recorded in the range of 300–400 nm with increasing concentrations of the selected compound. A decrease in the fluorescence intensity of MARK4/HSA with increasing concentrations of compound 4h was analyzed using a modified Stern–Volmer equation to estimate the binding constant Ka (Figure 7). For MARK4, the value of Ka for the compound 4h was found to be 8.7 × 106 M–1. However, the binding constant for HSA was observed to be 3.6 × 103 M–1 for this compound. The results clearly indicated that compound 4h binds strongly with MARK4. However, modest binding affinity was observed for HSA, indicating a desired transport affinity as drugs/drug-like molecules. HSA is the main carrier component of the bloodstream responsible for the transport of different molecules and the molecules possessing HSA binding affinities will be easy to transport.32 Thus, the moderate HSA binding affinity of the compound 4h supported its transportable properties.
Figure 7.

Compound 4h binding affinities with MARK4 and HSA. (A) Fluorescence emission spectra of MARK4 (10 μM) and HSA (25 μM) with increasing concentrations of 4h, showing quenching of MARK4/HSA fluorescence with increasing concentration of 4h; excitation wavelength is 280 nm and emission range is 300–400 nm. (B) Modified Stern–Volmer plot conquered from the quenching of MARK4/HSA fluorescence with increasing concentration of 4h, used to calculate the binding affinity (Ka).
Inhibition of Cancer Cell Proliferation
To evaluate the antiproliferation potential of compound 4h on human cancer cell lines (MCF-7 and HepG2), MTT-based cell viability assay was carried out and consequential IC50 values were calculated. We observed that compound 4h was toxic to MCF-7 and HepG2 cancerous cells. The molecule inhibited the growth of these cells in a concentration-dependent manner (Figure 8A). In the case of MCF-7 cells, the IC50 value for compound 4h was 3.74 μM, while for HepG2 cells, the IC50 value of 4h was 5.99 μM. However, the toxicity of the compound was evaluated on noncancerous cells (HEK293), and up to 100 μM, the compound did not affect the viability of noncancerous cells (Figure 8B). These results advocate that compound 4h is selectively inhibiting the growth of cancer cells (Table 2).
Figure 8.
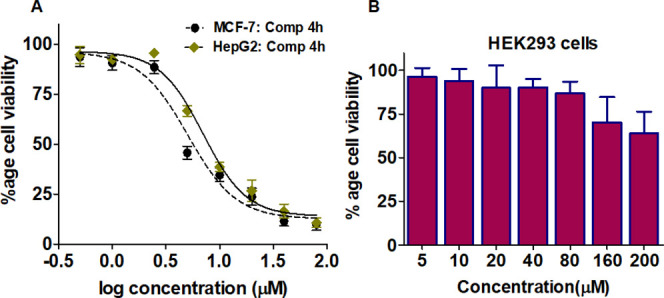
Cell viability studies. (A) Effect of compound 4h on the viability of MCF-7 and HepG2 vs log concentration of compound 4h. (B) Graphical representation for cell viabilities of HEK293 cells. Each of the selected cells was treated with increasing concentrations of compound 4h for 48 h. Cell viabilities were presented as the percentage of the number of viable cells to that of the control. Each data point shown is the mean ± SD from n = 3. (For anticancer activities, paclitaxel has been taken as the positive control.)
Apoptosis Studies
After getting remarkable results from binding, enzyme assay, and cell viability studies on these compounds, we have conducted an experiment to evaluate the potential of apoptosis induction to understand the mechanism of the selected compound before its therapeutic implications. The MCF-7 and HepG2 cells were incubated with IC50 dose of compound 4h. Annexin-V staining and PI staining were used to study the apoptosis induction, and we found that compound 4h-treated cells underwent apoptosis as compared to untreated controls (Figure 9). Compound 4h induces apoptosis in 11% and 26% of MCF-7 cells, whereas in the case of HepG2, it induces apoptosis in 28.1% and 20.8% of cells as compared to untreated control cells at the studied concentration. These results suggested that the selected compound caused the death of cancerous cells through induction of apoptosis. Apoptosis studies were consistent with earlier reports, which suggested that the inhibitors/inhibition of MARK4 induces the cell death in MCF-7 and other cancerous cells.29,33,49 All these observations indicated the predominant therapeutic potential of compound 4h.
Figure 9.

Compound 4h treatment induces apoptosis in human cancer cell lines. The MCF-7 and HepG2 cells were treated with IC50 concentrations of compound 4h for 10 h and processed for apoptosis analysis using Annexin-V/PI apoptosis kit. (A) Histogram showing the anti-FITC-Annexin-V- and PI-stained cells after the treatment of compound 4h. The name of the cell line is indicated on the right side of histogram. (B) Bar graphs represent the percentage of apoptotic cells stained with Annexin-V for duplicate measurements ± SD. Statistical analysis was performed using t-test for unpaired samples, and the p-value is calculated by comparing the treated cells with vehicle control (V.C.).
Determination of Reactive Oxygen Species (ROS) Level
Metabolic pathways and respiration lead to the production of a variety of free radicals.50 The redox state of cancerous cells plays an important role in cancer cells growth and proliferation.51 To check out the redox potential of the compound 4h, the level of ROS was measured after the treatment of MCF-7 and HepG2 cells with this compound. It was observed that after the treatment of the compound 4h, the DCF fluorescence decreases, which suggests a decrease in the level of ROS production (Figure 10). It was also reported previously that MARK4 promoted the oxidative stress and inhibition of MARK4 decreases the ROS level.30 Thus, our results of ROS estimation are also in agreement with the earlier reports that inhibition of MARK4 may lead to the decrease in ROS generation and thus controls cancer growth and proliferation.
Figure 10.

Compound 4h decreases the ROS production in human cancer cell lines. (A) Bar graphs represent the relative intensity of DCF fluorescence for triplicate measurements. Cells were treated with IC50 dose of compound 4h for 5–6 h and processed for ROS measurements using DCFDA staining by spectrofluorimetry. Statistical analysis was performed using t-test for unpaired samples; the p-value is calculated by comparing the treated cells with vehicle control (V.C.).
Discussion
In the quest for potent and selective MARK4 inhibitors, a series (Scheme 1) of arylaldoxime/5-nitroimidazole hybrids (4a–4h) were designed and synthesized following multistep chemical reactions. All these compounds have a free N–OH group as a backbone of the molecules coupled with varied terminal aryl groups. In Scheme 1, the nitroimidazole ring was grafted with various arylaldoximes for the determination of the effect of the electron-deficient nitroaromatic fragment on the overall activity of the target compounds. On the basis of molecular docking, fluorescence binding studies, and enzymatic assays, a small hybrid chemotype 4h (IC50 = 1.47 μM) was identified as the promising MARK4 inhibitor. The compound 4h was found to be more potent probably due to the additional interactions of the electron-withdrawing nitroimidazole ring with the Asp196, Lys85, and Val70 residues besides the other core interactions as per molecular docking studies. This compound showed the prominent inhibition of MARK4 protein among the panel of 30 kinases belonging to the serine/threonine family, which indicates the remarkable selectivity toward MARK4 inhibition. The HSA binding constant estimated as 3.6 × 103 M–1 for the compound 4h indicates that such molecules may be easily transported and delivered via blood to the target site for the desired pharmacological effect.
Further, the compound 4h showed a considerable antiproliferative potential against human cancer cell line MCF-7 (IC50 = 3.74 μM) and HepG2 cell line (IC50 = 5.99 μM). The compound 4h was found to induce apoptosis in 11% MCF-7 and 28.1% HepG2 cancerous cells, indicating that the designed compounds may be used as the significant lead candidates for the further development of MARK4 inhibitors with appreciable apoptotic potential. The strong antioxidant potential of the 4h compound in MCF 7 and HepG2 cells is most probably due to the combined effect of the presence of a free −NOH group, structural features of molecules, and their MARK4 inhibition potential. In the present study, we consider the combined antioxidant effect of oximes and MARK4 down-regulation responsible for the antioxidant activity as both oximes and MARK4 inhibition have been independently reported for the reduction of ROS in the cancer cells.29,31,52 MARK4 overexpression has been reported for the induction of oxidative stress in adipocytes, and inhibition of MARK4 helps relieve oxidative stress.30 Interestingly, our results also suggested that inhibition of MARK4 by selected compounds reduces the ROS levels. Thus, present results also support the observations of previous studies in relation to the association of ROS, MARK4 expression, and cancer progression.29,30,34 In addition, all the compounds were observed to have appreciable water solubility in the range of 10 ± 0.01 to 103 ± 2.1 mg/mL, indicating their efficient bioavailability (Table S3).
Most kinase inhibitors exhibit off-target interactions that occur through a pathway cross-talk or lack of selectivity, causing unanticipated side effects.53−55 Our synthesized compounds exerted selective MARK4 inhibitory potential having a minimal effect over other 30 kinases of the serine/threonine family, which indicates minor off-target interactions. Further, ROS generation causes DNA damage and mutation and facilitates tumor progression. It is therefore plausible that compounds having an antioxidant potential could minimize the genotoxic effects caused by ROS and reduce the incidence of carcinogenesis.56,57 Compound 4h showed the appreciable antioxidant potential apart from selective inhibition of MARK4 protein kinase. In addition, the efficient binding affinity with HSA and fair aqueous solubility renders such compounds remarkable therapeutic potential. Often, the kinase inhibitors/drugs are difficult to synthesize, possess low yield, and involve harsh chemical reactions, making them expensive.58 The ease of synthesis, small-molecule nature, least off-target interactions, and significant antioxidant potential associated with our synthesized molecules adds them as a new class of compounds as potent inhibitors of MARK4. In nutshell, the molecular hybridization of the 5-nitroimidazole scaffold with the substituted oxime core led to the identification and emergence of novel potent MARK4 inhibitors, having appreciable apoptotic activity, antiproliferative effect, bioavailability, and significant antioxidant potential.
Conclusions
We have synthesized water-soluble novel arylaldoxime/5-nitroimidazole small-molecule hybrids as potent MARK4 inhibitors and antiproliferative agents. The molecule 4h has been identified as the predominant apoptotic and significant antioxidant agent besides having a promising selective MARK4 inhibition profile. Compound 4h can be easily transported via blood to the target site and holds the efficient binding toward MARK4 as compared to the 30 screened CAMK enzymes. The observed potency of the compound 4h indicates that the presence of the trifluoromethyl group owns the remarkable significance for the designing of potent MARK4 inhibitors. The pharmacological effectiveness assessed signifies the valuable importance of oxime derived small molecules in medicinal chemistry for targeting the MARK4 enzyme with concomitant cellular free radical scavenging property and efficient bioavailability. The results divulged that the pharmacophoric hybridization approach is an effective tool for the design and development of promising MARK4 inhibitors. The synthesized arylaldoxime/5-nitroimidazole hybrid chemotypes may act as lead molecules for the designing of new drug candidates having an improved therapeutic profile for the future clinical development in anticancer drug paradigm in terms of MARK4 inhibition.
Experimental Section
Materials and Methods
All the required chemicals were purchased from Merck and Aldrich Chemical Company (USA). The reagents were of analytical grade and were used as supplied. Percolated aluminum sheets (Silica gel 60 F254, Merck Germany) were employed for thin-layer chromatography (TLC). The synthesized compounds were visualized on TLC using ultraviolet (UV) light (λ = 254 nm). The melting points of all the compounds were observed on a Veego instrument with model specifications REC-22038 A2 and are uncorrected. 1H NMR and 13C NMR were recorded on Bruker Spectrospin DPX 300 MHz and Bruker Spectrospin DPX 75 MHz spectrometers, respectively, using DMSO-d6 as a solvent and trimethylsilane (TMS) as the internal standard. Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; Ar, aromatic; imid, imidazole. The mass spectra of all the compounds were recorded by ESI-MS (AB-Sciex 2000, Applied Biosystem).
Synthesis of Compounds (2a–2h and 3a–3h)
The synthesis of intermediates (2a–2h) and carboximidoyl chlorides (3a–3h) is given in the Supporting Information.
Synthesis Protocol Employed for the Preparation of Arylaldoxime/5-Nitroimidazole Hybrids (4a–4h)
2-Methyl-4(5)-nitroimidazole (0.9 mmol) was dissolved in ACN (5 mL). Anhydrous K2CO3 (2.7 mmol) and phase-transfer catalyst TBAB (0.03 mmol) were added to the mixture followed by the dropwise addition of ACN solutions of synthesized carboximidoyl chlorides (4a–4h, 0.9 mmol). The reaction mixture was refluxed for 3–4 h and the progress of reaction was monitored by employing thin-layer chromatography (TLC) visualized under 254 nm UV irradiation. After completion, the reaction mixture was cooled down to room temperature and then partitioned between chloroform and water. The aqueous layer was extracted three times with chloroform. The combined organic phase was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo under reduced pressure. The product was recrystallized from chloroform and absolute ethanol to afford the target compounds (4a–4h).
N-[(2-Methyl-5-nitro-1H-imidazol-1-yl)(phenyl)methylidene]hydroxylamine (4a)
Yield: 93%; white solid; m.p.: 253 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.63 (s, 1H, N-OH), 8.56 (s, 1H, imid-H), 7.55–7.45(m, 5H, Ar-H), 2.18 (s, 3H, imid-CH3). 13C NMR (75 MHz, DMSO-d6): 147.07, 145.38, 143.28, 141.35, 130.22, 128.37, 123.93, 121.93, 13.53. ESI-MS (m/z): [M+ + 1] 106.22. Anal. calcd. for C11H10N4O3: C, 53.66; H, 4.09; N, 22.75. Found: C, 53.46; H, 3.99, N 22.55.
N-[(4-Methoxyphenyl)(2-methyl-5-nitro-1H-imidazol-1-yl)methylidene]hydroxylamine (4b)
Yield: 93%; white solid; m.p.: 257–258 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.35 (s, 1H, N-OH), 8.51 (s, 1H, imid-H), 7.41 (d, 2H, J = 9 Hz, Ar-H), 7.04 (d, 2H, J = 9 Hz, Ar-H), 3.80 (s, 3H, −OCH3), 2.18 (s, 3H, imid-CH3). 13C NMR (75 MHz, DMSO-d6): δ 161.74, 147.14, 145.11, 141.86, 127.72, 123.43, 121.91, 115.10, 55.87, 13.30. ESI-MS (m/z): [M+ + 1] 277.2. Anal. calcd. for C12H12N4O4: C, 52.17; H, 4.38; N, 20.28. Found: C, 52.13; H, 4.31; N, 20.45.
N-[(2-Methyl-5-nitro-1H-imidazol-1-yl)(4-methylphenyl)methylidene]hydroxylamine (4c)
Yield: 92%; white solid; m.p.: 250 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.48 (s, 1H, N-OH), 8.51 (s, 1H, imid-H), 7.36–7.27 (m, 4H, Ar-H), 2.35 (s, 3H, imid-CH3), 2.27 (s, 3H, −CH3). 13C NMR (75 MHz, DMSO-d6): 147.17, 145.28, 142.08, 141.25, 130.22, 128.37, 125.98, 121.93, 21.35, 13.30. ESI-MS (m/z): [M+ + 1] 261.10. Anal. calcd. for C12H12N4O3: C, 55.38; H, 4.65; N, 21.53. Found: C, 55.25; H, 4.60; N, 21.67.
N-[(4-Ethoxyphenyl)(2-methyl-5-nitro-1H-imidazol-1-yl)methylidene]hydroxylamine (4d)
Yield: 88%; yellow solid; m.p.: 261 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.35 (s, 1H, N-OH), 8.53 (s, 1H, imid-H), 7.39 (d, 2H, J = 7.8 Hz, Ar-H), 7.02 (d, 2H, J = 7.8 Hz, Ar-H), 4.10–4.04 (q, 2H, −CH2), 2.18 (s, 3H, imid-CH3), 1.36–1.31 (t, 3H, J = 13.5 Hz, −CH3). 13C NMR (75 MHz, DMSO-d6): 161.02, 147.13, 145.31, 141.86, 127.70, 123.26, 121.91, 115.50, 63.88, 14.94, 13.31. ESI-MS (m/z): [M+ + 1] 291.02. Anal. calcd. for C13H14N4O4: C, 53.79; H, 4.86; N, 19.30. Found: C, 53.84; H, 4.80; N, 19.45.
N-[(4-Ethylphenyl)(2-methyl-5-nitro-1H-imidazol-1-yl)methylidene]hydroxylamine (4e)
Yield: 85%; yellow solid; m.p.: 258–259 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.34 (s, 1H, N-OH), 8.52 (s, 1H, imid-H), 7.38 (d, 2H, J = 8.4 Hz, Ar-H), 7.03 (d, 2H, J = 8.4 Hz, Ar-H), 2.17 (s, 3H, imid-CH3), 1.76–1.70 (q, 2H, −CH2), 0.99–0.94 (t, 3H, J = 7.5 Hz, −CH3). 13C NMR (75 MHz, DMSO-d6): 161.19, 147.14, 145.30, 141.86, 127.70, 123.26, 121.91, 115.50, 63.88, 14.94, 13.31. ESI-MS (m/z): [M+ + 1] 275.01. Anal. calcd. for C13H14N4O3: C, 56.93; H, 5.14; N, 20.43. Found: C, 56.87; H, 5.23; N, 20.38.
N-[(4-Chlorophenyl)(2-methyl-5-nitro-1H-imidazol-1-yl)methylidene]hydroxylamine (4f)
Yield: 90%; white solid; m.p.: 255 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.11 (s, 1H, N-OH), 8.50 (s, 1H, imid-H), 7.41(d, 2H, J = 8.7 Hz, Ar-H), 7.04 (d, 2H, J = 8.7 Hz, Ar-H), 2.18 (s, 3H, imid-CH3). 13C NMR (75 MHz, DMSO-d6): 162.74, 147.14, 145.11, 141.86, 127.72, 123.43, 121.91, 115.10, 55.87, 13.11. ESI-MS (m/z): [M+ + 1] 281.44. Anal. calcd. for C11H9ClN4O3: C, 46.87; H, 3.23; N, 19.96. Found: C, 47.07; H, 3.21; N, 19.85.
N-[(2-Methyl-5-nitro-1H-imidazol-1-yl)(4-nitrophenyl)methylidene]hydroxylamine (4g)
Yield: 87%; yellow solid; m.p.: 260 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.25 (s, 1H, N-OH), 8.60 (s, 1H, imid-H), 8.31 (d, 2H, J = 8.7 Hz, Ar-H), 7.76 (d, 2H, J = 8.7 Hz, Ar-H), 2.20 (s, 3H, imid-CH3). 13C NMR (75 MHz, DMSO-d6): 147.34, 145.48, 140.97, 137.09, 131.05, 127.36, 110.76, 110.28, 13.33. ESI-MS (m/z): [M+ + 1] 292.07. Anal. calcd. for C11H9N5O5: C, 45.37; H, 3.11; N, 10.05. Found: C 45.42; H, 3.06; N, 10.16.
N-{(2-Methyl-5-nitro-1H-imidazol-1-yl)[2-(trifluoromethyl)phenyl]methylidene}hydroxyl Amine (4h)
Yield: 94%; white solid; m.p.: 270 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.04 (s, 1H, N-OH), 8.39 (s, 1H, imid-H), 7.95–7.92 (m, 1H, Ar-H), 7.83–7.76 (m, 2H, Ar-H), 7.62 (d, 1H, J = 7.8 Hz, Ar-H), 2.18 (s, 3H, imid-CH3). 13C NMR (75 MHz, DMSO-d6): 145.67, 139.07, 133.58, 111.43, 131.76, 129.73, 127.91, 127.49, 125.79, 122.16, 121.90, 14.43. ESI-MS (m/z): [M+ + 1] 315.21. Anal. calcd. for C12H9F3N4O3: C, 45.87; H, 2.89; N, 17.83. Found: C, 45.92; H, 2.72; N, 17.97.
X-ray Crystal Structure Determination
Three-dimensional X-ray data were collected on a Bruker Kappa Apex CCD diffractometer at low temperature for compounds 4c and 4e by the ϕ−ω scan method. Reflections were measured from a hemisphere of data collected from frames, each of them covering 0.3° in ω. A total of 31,157 reflections measured for 4c and 130,390 for 4e were corrected for Lorentz and polarization effects and for absorption by multiscan methods based on symmetry-equivalent and repeated reflections. Of the total, 2360 independent reflections for 4c and 4869 for 4e exceeded the significance level (|F|/σ|F|) > 4.0 > 4.0. After data collection, in each case, the multiscan absorption correction (SADABS)59 was applied, and the structure was solved by direct methods and refined by full-matrix least squares on F2 data using SHELX suite of programs.60 Hydrogen atoms were located in a difference Fourier map and left to refine freely, except for C(11) and C(12) in 4c and C(11), C(12), C(13), C(24), C(16A), C(16B), C(17A), C(17B), C(25A), C(25B), C(26A), and C(26B) in 4e, which were included in the calculation position and refined in the riding mode. Refinements were done with allowance for thermal anisotropy of all nonhydrogen atoms. A final difference Fourier map showed no residual density outside: 0.347 and −0.299 e Å–3 for 4c; 0.364 and −0.398 e Å–3 for 4e. w = 1/[σ2(Fo2) + (0.044300P)2 + 0.660000P] for 4c and 1/[σ2(Fo2) + (0.037200P)2 + 1.466400P] for 4e, where P = (|Fo|2 + 2|Fc|2)/3, were used in the latter stages of refinement. An important disorder on one molecule of the two present in the asymmetric unit appears in the crystal of 4e. This disorder has been refined and two atomic sites have been observed and refined with the anisotropic atomic displacement parameters for the ethyl-phenyl group. More specifically, this disorder was refined using 142 restraints (ISOR, SADI, SIMU, and DELU restraints were used). The site occupancy factor was 0.51705 for C(16A), C(17A), C(18A), C(24A), and C(25A). Further details of the crystal structure determination are given in Table 1. CCDC 1965977 and 1965978 numbers contain the supplementary crystallographic data for the structures reported in this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/ or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: (+44) 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk.
Molecular Docking
Molecular docking of the synthesized arylaldoxime hybrids was carried out using Autodock Vina and AutoDock 4 package.45,61 The atomic coordinates of MARK4 were taken from RCSB Protein Data Bank (PDB ID 5ES1).36 The 2D and 3D structures of all the synthesized hybrid molecules were drawn in ChemBio3D Ultra 12.0. For viewing of the binding affinity, bound conformations, and possible protein–ligand interactions, the coordinate files of MARK4 and respective synthesized arylaldoxime hybrids were subjected to molecular docking. For visualization and structure analysis of the docked complexes of MARK4 and also to generate 2D docking for the analysis of hydrogen bonds and hydrophobic interactions, PyMOL viewer (Schrödinger, LLC) and “Receptor-Ligand Interactions” modules of BIOVIA/Discovery Studio 2017R2 were used.62
Enzyme Inhibition Assay
To evaluate the enzymatic activity of MARK4, ATPase assay was performed in the presence of synthesized arylaldoxime hybrids, as described earlier.29,33 In brief, we have measured 32Pi liberated from MARK4 arbitrated hydrolysis of [γ-32P] ATP. First, MARK4 was incubated along with ice-cold ATP (1 mM) having radioactive-labeled [γ-32P] ATP (specific activity, 222 TBq mmol–1) for 2 h at 37 °C and, subsequently, TLC was performed. Initially, the experiments were conducted for screening of active compounds after the initial screening activity of MARK4 was monitored with ascending concentrations of the selected compound. The inference of MARK4 inhibition in terms of percentage hydrolysis of ATP was established using ImageJ software (https://imagej.nih.gov/ij/index.html).
Single-Dose Kinase Inhibition Profiling
In vitro biochemical profiling of compound 4h was evaluated with the 26 members of CAMK family (CAMK-1 and CAMK-2) of kinases using kinase screening kit by following the manufacturer’s protocols (Promega, Madison, USA), as described previously.32,63 Briefly, 10 μM of compound was added to the corresponding well of 384-well assay plate. Serially, 2 μL of kinase solution was added to each well of the plate. The reaction mixture was mixed gently using a plate shaker for 2 min. The plate was centrifuged and incubated at room temperature (25 °C) for 10 min. Following the incubation period, 2 μL of working stocks of ATP/substrate was added carefully to the respective well. Shake the assay plate, centrifuge, and incubate at room temperature (25 °C) for 60 min. Consequently, 5 μL of ADP-Glo reagent was added to all reaction wells of the assay plate. The plate was shaken for 2 min and incubated further at room temperature for 40 min. As a final point, 10 μL of Kinase Detection Reagent was added to each reaction well and the plate was incubated at room temperature for 30 min. After the completion of reaction, luminescence was measured using an integration time of 0.5 s per well. Using net luminescence of the no-compound control (negative control) reactions to represent 100% kinase activity, the percent kinase activity was calculated in the compound-containing reactions and plotted in terms of percent kinase activity inhibition. Additionally, as our laboratory is working on different human kinases named calcium–calmodulin-dependent protein kinase IV (CAMKIV), Fas-activated serine/threonine kinase (FASTK), PDK3, and integrin-linked kinase (ILK), so we have also studied the selectivity/inhibition potential of compound 4h by malachite green assay, as per our previously published protocol.64
Fluorescence Measurements
Binding affinities of the synthesized arylaldoxime hybrid 4h with MARK4 were determined using the fluorescence intensity change in the emission spectrum of MARK4, as per our published protocol.64 The titration of protein was done in triplicates and, for analysis, their average was taken. A significant decrease in the fluorescence intensity of protein with increasing concentration of the selected arylaldoxime hybrid was used to calculate the binding constant (Ka) and the number of binding sites (n) present on the protein molecule by using the modified Stern–Volmer equation.65
| 1 |
where Fo is the fluorescence intensity of native protein, F is the fluorescence intensity of protein in the presence of ligand, Ka is the binding constant, n is the number of binding sites, and L is the concentration of ligand. The binding constant (Ka) and number of binding sites (n) were obtained from the intercept and slope, respectively.
Cell Viability Assay
To estimate the cancerous cell growth inhibitory potential of synthesized compounds, MTT assay was carried out.29,64 Briefly, cells were plated at a density of 8000–9000 cells/well in a 96-well cell culture plate. After 10 h, cells were incubated with different concentrations (0.1–80 μM) of compound 4h for 48 h at 37 °C in a humidified CO2 incubator. After 48 h, the mixture of media and compound was removed, and 20 μL of MTT solution (from 5 mg/mL stock solution in PBS, pH 7.4) and 100 μL of DMEM were added to each well of the culture plate. The plates were incubated for 4–5 h at 37 °C in the CO2 incubator. The supernatant of each well was removed and the formazan crystals were dissolved by adding 100 μL of DMSO to each well. The absorbance (A) of the reaction product was measured at 570/590 nm on a multiplate ELISA reader (Bio-Rad). The percentage cell viability was calculated and it was used to estimate the IC50 (50% inhibitory concentration) value for compound 4h. For cell proliferation and anticancer activities, paclitaxel was used as a positive control.
Apoptosis Assay
Induction of apoptosis was analyzed using Annexin-V staining, as described previously.3,64 Briefly, 2.5 × 105 cells/well of a six-well culture plate were plated. Once the cells were grown to 80% confluency, they were incubated with IC50 concentration of 4h for 10 h at 37 °C. The control cells were treated with media only. After 10 h treatment, cells were harvested by trypsinization and collected by centrifuging them at 2000–3000 rpm for 4 min. Cells were washed three times with PBS and incubated in Annexin binding buffer with FITC-Annexin-V and PI using FITC-Annexin-V kit following the manufacturer’s guidelines (BD Biosciences, USA). Around 10,000 events were collected for each sample using flow cytometry (BD LSR II Flow Cytometry Analyzer) and analyzed by FlowJo.
Estimation of ROS Production
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) staining was used for measuring the redox state of cells, as described previously.29,64 Briefly, the MCF-7 and HepG2 cells were plated at a density of 2.5 × 105 cells/well of the six-well plate. On the next day, cells were respectively treated with IC50 dose of compound 4h in a 10-well culture plate for 5–6 h (H2O2 is used as a positive control). Cells were washed with 500–1000 μL of prewarmed (at 37 °C) Krebs Ringer buffer (20 mM HEPES, 2 mM MgSO4, 10 mM dextrose, 127 mM NaCl, 1 mM CaCl2, and 5.5 mM KCl). Subsequently, cells were incubated with DCFDA (10 μM) for 30 min in the dark at 37 °C in a humidified CO2 incubator. After 30 min incubation, the cells were harvested by trypsinization and collected by centrifugation. ROS levels were assessed by measuring the fluorescence at a Jasco spectrofluorimeter (FP-6200) using a 5 mm quartz cuvette. The excitation and emission filters were set at 485/500–550 nm, respectively.
Solubility Assay
Twenty milligrams of each the target compound was taken in a polypropylene microcentrifuge tube (2 mL strength) and 100 μL of citrate-buffered solution (pH 3.3) was added and then incubated on an Eppendorf Thermomixer (1000 rpm) at 25 °C. The compounds were centrifuged at 20,800g for the time period of 2 min after incubation and then subjected to filtration through a 0.22 μm polyvinylidene difluoride (PVDF) membrane. Aliquots of the obtained filtrates were diluted by solvents DMSO/acetonitrile (ACN)/trifluoroacetic acid in the ratio of 80:20:0.1 and then kept at 4 °C. An Agilent 1100 HPLC equipped with a Waters Symmetry IS C18 column, (3.5 μm, 2.1 × 20 mm) accompanied with a wavelength detection of 254 nm was employed for further analysis. Water (0.1% trifluoroacetic acid) and ACN (0.1% trifluoroacetic acid) systems were used as mobile phases during analysis. The column was eluted with 10% ACN (0.1% trifluoroacetic acid) for 0.5 min at first, then with 10% to 55% ACN (0.1% trifluoroacetic acid) over 9.5 min, and finally at the gradient of 70% ACN (0.1% trifluoroacetic acid) for 1 min at the flow rate of 1 mL/min. Peaks obtained were calibrated with the known standards and then the solubility was assessed.
Statistical Analysis
Data were expressed as mean ± standard error from three independent experiments. The statistical analysis of each data was performed using the two-tailed Student t-test for unpaired samples and values of p < 0.05 were considered as significant.
Acknowledgments
M.N.P. is thankful to the Council of Scientific and Industrial Research, New Delhi, India, for the award of senior research fellowship (no. 09/466 (0220) 2K19 EMR-I). This work is supported by the financial assistance from the Science and Engineering Research Board, Government of India (project no. EMR/2015/002372). P.K. thanks the Department of Biotechnology, Government of India, for the fellowship (BT/PR12828/AAQ/1/622/2015). N.S.K. acknowledges the Indian Council of Medical Research for the award of senior research fellowship (no. 45/9/2019-PHA/BMS). We sincerely acknowledge the Harvard University plasmid-providing facility for providing the MARK4 gene. We thank Almanac Life Sciences India Private Limited and Piquant Scientific Private Limited for helping us in kinase profiling studies.
Glossary
Abbreviations
- MARK4
microtubule affinity-regulating kinase 4
- MAPs
microtubule-associated proteins
- HSA
human serum albumin
- ATP
adenosine triphosphate
- UBA domain
ubiquitin-associated domain
- KA
kinase-associated domain
- TBAB
tetra-n-butylammonium bromide
- DMF
dimethylformamide
- DCM
dichloromethane
- ACN
acetonitrile
- DCFH-DA
2′,7′-dichlorodihydrofluorescein diacetate
- DMSO
dimethyl sulfoxide
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MARK1
microtubule affinity-regulating kinase 1
- MAPKAPK2
mitogen-activated protein kinase-activated protein kinase 2
- DAPK1
death-associated protein kinase 1
- HEK293
human embryonic kidney 293
- CAMKIV
calcium–calmodulin-dependent protein kinase IV
- FASTK
Fas-activated serine/threonine kinase
- PDK3
pyruvate dehydrogenase kinase 3
- ILK
integrin-linked kinase
- TEA
triethylamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01703.
Procedure for the synthesis of intermediates (2a–2h) and carboximidoyl chlorides (3a–3h), molecular docking studies of compounds 4a–4g, serine/threonine kinase panel inhibition data of compound 4h, solubility data of compounds 4a–4h, single-crystal data, bond lengths and angles, hydrogen bonds, and 1H NMR and 13C NMR spectra of title compounds 4a–4h (PDF)
Author Contributions
Authors M. N. Peerzada and P. Khan have contributed equally to the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Solier S.; Müller S.; Rodriguez R. Whole-Genome Mapping of Small-Molecule Targets for Cancer Medicine. Curr. Opin. Chem. Biol. 2020, 56, 42–50. 10.1016/j.cbpa.2019.12.005. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Prencipe F.; Oliva P.; Baraldi S.; Baraldi P. G.; Schiaffino Ortega S.; Chayah M.; Kimatrai Salvador M.; Lopez-Cara L. C.; Brancale A.; et al. Design, Synthesis, and Biological Evaluation of 6-Substituted Thieno[3,2- d ]Pyrimidine Analogues as Dual Epidermal Growth Factor Receptor Kinase and Microtubule Inhibitors. J. Med. Chem. 2019, 62, 1274–1290. 10.1021/acs.jmedchem.8b01391. [DOI] [PubMed] [Google Scholar]
- Peerzada M. N.; Khan P.; Ahmad K.; Hassan M. I.; Azam A. Synthesis, Characterization and Biological Evaluation of Tertiary Sulfonamide Derivatives of Pyridyl-Indole Based Heteroaryl Chalcone as Potential Carbonic Anhydrase IX Inhibitors and Anticancer Agents. Eur. J. Med. Chem. 2018, 155, 13–23. 10.1016/j.ejmech.2018.05.034. [DOI] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Zhu J.-Y.; Cuellar R. A.; Berndt N.; Lee H. E.; Olesen S. H.; Martin M. P.; Jensen J. T.; Georg G. I.; Schönbrunn E. Structural Basis of Wee Kinases Functionality and Inactivation by Diverse Small Molecule Inhibitors. J. Med. Chem. 2017, 60, 7863–7875. 10.1021/acs.jmedchem.7b00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. N.; Lewis R. J. Structural Basis for Control by Phosphorylation. Chem. Rev. 2001, 101, 2209–2242. 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Adams J. A. Kinetic and Catalytic Mechanisms of Protein Kinases. Chem. Rev. 2001, 101, 2271–2290. 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. III; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; et al. A Small Molecule-Kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Tassan J.-P.; Le Goff X. An Overview of the KIN1/PAR-1/MARK Kinase Family. Biol. cell 2004, 96, 193–199. 10.1016/j.biolcel.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Naz F.; Khan F. I.; Mohammad T.; Khan P.; Manzoor S.; Hasan G. M.; Lobb K. A.; Luqman S.; Islam A.; Ahmad F.; et al. Investigation of Molecular Mechanism of Recognition between Citral and MARK4: A Newer Therapeutic Approach to Attenuate Cancer Cell Progression. Int. J. Biol. Macromol. 2018, 107, 2580–2589. 10.1016/j.ijbiomac.2017.10.143. [DOI] [PubMed] [Google Scholar]
- Trinczek B.; Brajenovic M.; Ebneth A.; Drewes G. MARK4 Is a Novel Microtubule-Associated Proteins/Microtubule Affinity-Regulating Kinase That Binds to the Cellular Microtubule Network and to Centrosomes. J. Biol. Chem. 2004, 279, 5915–5923. 10.1074/jbc.M304528200. [DOI] [PubMed] [Google Scholar]
- Heidary Arash E.; Shiban A.; Song S.; Attisano L. MARK4 Inhibits Hippo Signaling to Promote Proliferation and Migration of Breast Cancer Cells. EMBO Rep. 2017, 18, 420–436. 10.15252/embr.201642455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M.; Tian L.; Gan L.; Liu Z.; Sun C. Mark4 Promotes Adipogenesis and Triggers Apoptosis in 3T3-L1 Adipocytes by Activating JNK1 and Inhibiting P38MAPK Pathways. Biol. Cell 2014, 106, 294–307. 10.1111/boc.201400004. [DOI] [PubMed] [Google Scholar]
- Naz F.; Anjum F.; Islam A.; Ahmad F.; Hassan M. I. Microtubule Affinity-Regulating Kinase 4: Structure, Function, and Regulation. Cell Biochem. Biophys. 2013, 67, 485–499. 10.1007/s12013-013-9550-7. [DOI] [PubMed] [Google Scholar]
- Drewes G.; Ebneth A.; Preuss U.; Mandelkow E. M.; Mandelkow E. MARK, a Novel Family of Protein Kinases That Phosphorylate Microtubule-Associated Proteins and Trigger Microtubule Disruption. Cell 1997, 89, 297–308. 10.1016/S0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Matenia D.; Mandelkow E.-M. The Tau of MARK: A Polarized View of the Cytoskeleton. Trends Biochem. Sci. 2009, 34, 332–342. 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Rodon J.; Postel-Vinay S.; Hollebecque A.; Nuciforo P.; Azaro A.; Cattan V.; Marfai L.; Sudey I.; Brendel K.; Delmas A.; et al. First-in-Human Phase I Study of Oral S49076, a Unique MET/AXL/FGFR Inhibitor, in Advanced Solid Tumours. Eur. J. Cancer 2017, 81, 142–150. 10.1016/j.ejca.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Patrawala S.; Puzanov I. Vemurafenib (RG67204, PLX4032): A Potent, Selective BRAF Kinase Inhibitor. Future Oncol. 2012, 8, 509–523. 10.2217/fon.12.31. [DOI] [PubMed] [Google Scholar]
- Kizaka-Kondoh S.; Konse-Nagasawa H. Significance of Nitroimidazole Compounds and Hypoxia-Inducible Factor-1 for Imaging Tumor Hypoxia. Cancer Sci. 2009, 100, 1366–1373. 10.1111/j.1349-7006.2009.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X.-X.; Duan Y.-T.; Chen L.-W.; Tang D.-J.; Yang M.-R.; Wang P.-F.; Xu C.; Zhu H.-L. Design, Synthesis and Biological Evaluation of Pyrazolyl-Nitroimidazole Derivatives as Potential EGFR/HER-2 Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 677–683. 10.1016/j.bmcl.2015.11.040. [DOI] [PubMed] [Google Scholar]
- Qin H.-L.; Leng J.; Zhang C.-P.; Jantan I.; Amjad M. W.; Sher M.; Naeem-Ul-Hassan M.; Hussain M. A.; Bukhari S. N. A. Synthesis of α,β-Unsaturated Carbonyl-Based Compounds, Oxime and Oxime Ether Analogs as Potential Anticancer Agents for Overcoming Cancer Multidrug Resistance by Modulation of Efflux Pumps in Tumor Cells. J. Med. Chem. 2016, 59, 3549–3561. 10.1021/acs.jmedchem.6b00276. [DOI] [PubMed] [Google Scholar]
- Cao J.; Gao H.; Bemis G.; Salituro F.; Ledeboer M.; Harrington E.; Wilke S.; Taslimi P.; Pazhanisamy S.; Xie X.; et al. Structure-Based Design and Parallel Synthesis of N-Benzyl Isatin Oximes as JNK3 MAP Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2891–2895. 10.1016/j.bmcl.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Bharate S. B.; Sawant S. D.; Singh P. P.; Vishwakarma R. A. Kinase Inhibitors of Marine Origin. Chem. Rev. 2013, 113, 6761–6815. 10.1021/cr300410v. [DOI] [PubMed] [Google Scholar]
- Viegas-Junior C.; Danuello A.; da Silva Bolzani V.; Barreiro E. J.; Fraga C. A. M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Pollard J. R.; Mortimore M. Discovery and Development of Aurora Kinase Inhibitors as Anticancer Agents. J. Med. Chem. 2009, 52, 2629–2651. 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- Comess K. M.; McLoughlin S. M.; Oyer J. A.; Richardson P. L.; Stöckmann H.; Vasudevan A.; Warder S. E. Emerging Approaches for the Identification of Protein Targets of Small Molecules - A Practitioners’ Perspective. J. Med. Chem. 2018, 61, 8504–8535. 10.1021/acs.jmedchem.7b01921. [DOI] [PubMed] [Google Scholar]
- Lawson A. D. G.; MacCoss M.; Heer J. P. Importance of Rigidity in Designing Small Molecule Drugs To Tackle Protein-Protein Interactions (PPIs) through Stabilization of Desired Conformers. J. Med. Chem. 2018, 61, 4283–4289. 10.1021/acs.jmedchem.7b01120. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- Khan P.; Rahman S.; Queen A.; Manzoor S.; Naz F.; Hasan G. M.; Luqman S.; Kim J.; Islam A.; Ahmad F.; et al. Elucidation of Dietary Polyphenolics as Potential Inhibitor of Microtubule Affinity Regulating Kinase 4: In Silico and In Vitro Studies. Sci. Rep. 2017, 7, 9470. 10.1038/s41598-017-09941-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Gan L.; Chen Y.; Luo D.; Zhang Z.; Cao W.; Zhou Z.; Lin X.; Sun C. Mark4 Promotes Oxidative Stress and Inflammation via Binding to PPARγ and Activating NF-ΚB Pathway in Mice Adipocytes. Sci. Rep. 2016, 6, 21382. 10.1038/srep21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özen T.; Taş M. Screening and Evaluation of Antioxidant Activity of Some Amido-Carbonyl Oxime Derivatives and Their Radical Scavenging Activities. J. Enzyme Inhib. Med. Chem 2009, 24, 1141–1147. 10.1080/14756360802669981. [DOI] [PubMed] [Google Scholar]
- Aneja B.; Khan N. S.; Khan P.; Queen A.; Hussain A.; Rehman M. T.; Alajmi M. F.; El-Seedi H. R.; Ali S.; Hassan M. I.; et al. Design and Development of Isatin-Triazole Hydrazones as Potential Inhibitors of Microtubule Affinity-Regulating Kinase 4 for the Therapeutic Management of Cell Proliferation and Metastasis. Eur. J. Med. Chem. 2019, 163, 840–852. 10.1016/j.ejmech.2018.12.026. [DOI] [PubMed] [Google Scholar]
- Parveen I.; Khan P.; Ali S.; Hassan M. I.; Ahmed N. Synthesis, Molecular Docking and Inhibition Studies of Novel 3-N-Aryl Substituted-2-Heteroarylchromones Targeting Microtubule Affinity Regulating Kinase 4 Inhibitors. Eur. J. Med. Chem. 2018, 159, 166–177. 10.1016/j.ejmech.2018.09.030. [DOI] [PubMed] [Google Scholar]
- Khan P.; Queen A.; Mohammad T.; Smita; Khan N. S.; Hafeez Z. B.; Hassan M. I.; Ali S. Identification of α-Mangostin as a Potential Inhibitor of Microtubule Affinity Regulating Kinase 4. J. Nat. Prod. 2019, 82, 2252–2261. 10.1021/acs.jnatprod.9b00372. [DOI] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discovery 2018, 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Sack J. S.; Gao M.; Kiefer S. E.; Myers J. E. Jr.; Newitt J. A.; Wu S.; Yan C. Crystal Structure of Microtubule Affinity-Regulating Kinase 4 Catalytic Domain in Complex with a Pyrazolopyrimidine Inhibitor. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 129–134. 10.1107/S2053230X15024747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer C.; Abadleh M.; Bischof J.; Hauser D.; Schattel V.; Hirner H.; Knippschild U.; Laufer S. 3,4-Diaryl-Isoxazoles and -Imidazoles as Potent Dual Inhibitors of P38alpha Mitogen Activated Protein Kinase and Casein Kinase 1delta. J. Med. Chem. 2009, 52, 7618–7630. 10.1021/jm9005127. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Skaltsounis A.-L.; Magiatis P.; Polychronopoulos P.; Knockaert M.; Leost M.; Ryan X. P.; Vonica C. A.; Brivanlou A.; Dajani R.; et al. GSK-3-Selective Inhibitors Derived from Tyrian Purple Indirubins. Chem. Biol. 2003, 10, 1255–1266. 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Leclerc S.; Garnier M.; Hoessel R.; Marko D.; Bibb J. A.; Snyder G. L.; Greengard P.; Biernat J.; Wu Y. Z.; Mandelkow E. M.; et al. Indirubins Inhibit Glycogen Synthase Kinase-3 Beta and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease. A Property Common to Most Cyclin-Dependent Kinase Inhibitors?. J. Biol. Chem. 2001, 276, 251–260. 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- Takle A. K.; Brown M. J. B.; Davies S.; Dean D. K.; Francis G.; Gaiba A.; Hird A. W.; King F. D.; Lovell P. J.; Naylor A.; et al. The Identification of Potent and Selective Imidazole-Based Inhibitors of B-Raf Kinase. Bioorg. Med. Chem. Lett. 2006, 16, 378–381. 10.1016/j.bmcl.2005.09.072. [DOI] [PubMed] [Google Scholar]
- Sun J.; Liu H.-Y.; Xu R.-F.; Zhu H.-L. Identification of Nitroimidazole-Oxime Derivatives Targeting the Polo-Box Domain of Polo-like Kinase 1. Bioorg. Med. Chem. 2017, 25, 6581–6588. 10.1016/j.bmc.2017.10.035. [DOI] [PubMed] [Google Scholar]
- Vander Zwan M. C.; Hartner F. W. Solid-Liquid Phase-Transfer Catalysis by a Quaternary Ammonium Salt. A Comparison with Crown Ethers and Polyalkylamines. J. Org. Chem. 1978, 43, 2655–2657. 10.1021/jo00407a020. [DOI] [Google Scholar]
- Liu Z.-Z.; Chen H.-C.; Cao S.-L.; Li R.-T. Solid — Liquid Phase — Transfer Catalytic Method for N-Alkylation of Nitroimidazole. Synth. Commun. 1993, 23, 2611–2615. 10.1080/00397919308012596. [DOI] [Google Scholar]
- Kim P.; Zhang L.; Manjunatha U. H.; Singh R.; Patel S.; Jiricek J.; Keller T. H.; Boshoff H. I.; Barry C. E.; Dowd C. S. Structure-Activity Relationships of Antitubercular Nitroimidazoles. 1. Structural Features Associated with Aerobic and Anaerobic Activities of 4- and 5-Nitroimidazoles. J. Med. Chem. 2009, 52, 1317–1328. 10.1021/jm801246z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Yale H. L. The Trifluoromethyl Group in Medical Chemistry. J. Med. Pharm. Chem. 1959, 1, 121–133. 10.1021/jm50003a001. [DOI] [PubMed] [Google Scholar]
- Chu L.; Qing F.-L. Oxidative Trifluoromethylation and Trifluoromethylthiolation Reactions Using (Trifluoromethyl)Trimethylsilane as a Nucleophilic CF3 Source. Acc. Chem. Res. 2014, 47, 1513–1522. 10.1021/ar4003202. [DOI] [PubMed] [Google Scholar]
- Li L.; Guan K.-L. Microtubule-Associated Protein/Microtubule Affinity-Regulating Kinase 4 (MARK4) Is a Negative Regulator of the Mammalian Target of Rapamycin Complex 1 (MTORC1). J. Biol. Chem. 2013, 288, 703–708. 10.1074/jbc.C112.396903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Circu M. L.; Aw T. Y. Reactive Oxygen Species, Cellular Redox Systems, and Apoptosis. Free Radical Biol. Med. 2010, 48, 749–762. 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacker P. T. Reactive Oxygen Species in Cancer Cells: Live by the Sword, Die by the Sword. Cancer Cell 2006, 10, 175–176. 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Bergström M. A.; Luthman K.; Karlberg A.-T. Metabolic Epoxidation of an Alpha,Beta-Unsaturated Oxime Generates Sensitizers of Extreme Potency. Are Nitroso Intermediates Responsible?. Chem. Res. Toxicol. 2007, 20, 927–936. 10.1021/tx7000114. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Honma M.; Suzuki H. Off-Target Serine/Threonine Kinase 10 Inhibition by Erlotinib Enhances Lymphocytic Activity Leading to Severe Skin Disorders. Mol. Pharmacol. 2011, 80, 466–475. 10.1124/mol.110.070862. [DOI] [PubMed] [Google Scholar]
- Hantschel O. Unexpected Off-Targets and Paradoxical Pathway Activation by Kinase Inhibitors. ACS Chem. Biol. 2015, 10, 234–245. 10.1021/cb500886n. [DOI] [PubMed] [Google Scholar]
- Wynn M. L.; Ventura A. C.; Sepulchre J. A.; García H. J.; Merajver S. D. Kinase Inhibitors Can Produce Off-Target Effects and Activate Linked Pathways by Retroactivity. BMC Syst. Biol. 2011, 5, 156. 10.1186/1752-0509-5-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isnaini I.; Permatasari N.; Mintaroem K.; Prihartini B.; Widodo M. A. Oxidants-Antioxidants Profile in the Breast Cancer Cell Line MCF-7. Asian Pac. J. Cancer Prev. 2018, 19, 3175–3178. 10.31557/APJCP.2018.19.11.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedűs C.; Kovács K.; Polgár Z.; Regdon Z.; Szabó É.; Robaszkiewicz A.; Forman H. J.; Martner A.; Virág L. Redox Control of Cancer Cell Destruction. Redox Biol. 2018, 16, 59–74. 10.1016/j.redox.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rageot D.; Beaufils F.; Borsari C.; Dall’Asen A.; Neuburger M.; Hebeisen P.; Wymann M. P. Scalable, Economical, and Practical Synthesis of 4-(Difluoromethyl)Pyridin-2-Amine, a Key Intermediate for Lipid Kinase Inhibitors. Org. Process Res. Dev. 2019, 23, 2416–2424. 10.1021/acs.oprd.9b00312. [DOI] [Google Scholar]
- Sheldrick G. M.SADABS, Version 2.10. University of Göttingen: Germany, 2003.
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C, Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biovia G. M.SADABS, Version 2.10. University of Gottingen, Germany: 2003.
- Voura M.; Khan P.; Thysiadis S.; Katsamakas S.; Queen A.; Hasan G. M.; Ali S.; Sarli V.; Hassan M. I. Probing the Inhibition of Microtubule Affinity Regulating Kinase 4 by N-Substituted Acridones. Sci. Rep. 2019, 9, 1676. 10.1038/s41598-018-38217-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N. S.; Khan P.; Ansari M. F.; Srivastava S.; Hasan G. M.; Husain M.; Hassan M. I. Thienopyrimidine-Chalcone Hybrid Molecules Inhibit Fas-Activated Serine/Threonine Kinase: An Approach To Ameliorate Antiproliferation in Human Breast Cancer Cells. Mol. Pharmaceutics 2018, 15, 4173–4189. 10.1021/acs.molpharmaceut.8b00566. [DOI] [PubMed] [Google Scholar]
- Boaz H.; Rollefson G. K. The Quenching of Fluorescence. Deviations from the Stern-Volmer Law. J. Am. Chem. Soc. 1950, 72, 3435–3443. 10.1021/ja01164a032. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.