

Summary
Stem cells or their closely related committed progenitor cells are the likely founder cells of most neoplasms. In the continually renewing and hierarchically organized epithelia of the oesophagus, stomach and intestine, homeostatic stem cells are located at the beginning of the cell flux, in the basal layer of the oesophagus, the isthmic region of gastric oxyntic glands and at the bottom of gastric pyloric‐antral glands and colonic crypts. The introduction of mutant oncogenes such as Kras G12D or loss of Tp53 or Apc to specific cell types expressing the likes of Lgr5 and Mist1 can be readily accomplished in genetically engineered mouse models to initiate tumorigenesis. Other origins of cancer are discussed including ‘reserve’ stem cells that may be activated by damage or through disruption of morphogen gradients along the crypt axis. In the liver and pancreas, with little cell turnover and no obvious stem cell markers, the importance of regenerative hyperplasia associated with chronic inflammation to tumour initiation is vividly apparent, though inflammatory conditions in the renewing populations are also permissive for tumour induction. In the liver, hepatocytes, biliary epithelial cells and hepatic progenitor cells are embryologically related, and all can give rise to hepatocellular carcinoma and cholangiocarcinoma. In the exocrine pancreas, both acinar and ductal cells can give rise to pancreatic ductal adenocarcinoma (PDAC), although the preceding preneoplastic states are quite different: acinar‐ductal metaplasia gives rise to pancreatic intraepithelial neoplasia culminating in PDAC, while ducts give rise to PDAC via. mucinous cell metaplasia that may have a polyclonal origin.
Keywords: clonality, Cre recombinase, field cancerization, Lgr5, metaplasia, stem cells
1. INTRODUCTION
While a great deal is known about established tumours in terms of the likes of their intratumour genetic heterogeneity, invasion and metastatic properties, along with likely clinical course, relatively little is known about tumour origins—from which cell(s) do individual tumours arise from? The classic 2‐stage model of epidermal carcinogenesis developed by Berenblum 1 , 2 in which incomplete carcinogens such as the genotoxin 9,10‐dimethyl‐1,2‐benzanthracene (DMBA) were administered to mice demonstrated that tumour yields were not necessarily reduced by delaying for up to 43 weeks the time of the subsequent application of the necessary hyperplasiogenic promoter, in this case the phorbol ester, 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA), found in croton oil. Thus, it was considered that tumour initiation was largely irreversible. Since cell migration from the basal layer to the surface of the mouse epidermis only takes up to 9 days, 3 , 4 an entirely reasonable conclusion is that the transformation events must have occurred in long‐lived cells in the basal layer that do not join the upward‐moving cell escalator, presumably the epidermal stem cells. Stem cells with their in‐built powers of continued self‐renewal are clearly prime targets for permanent transformation, but as we shall see, numerous studies also highlight the critical importance of cell proliferation in general to the carcinogenic process. 5 Indeed, chronic inflammatory conditions in humans such as hepatitis, oesophagitis, gastritis and ulcerative colitis are all associated with regenerative hyperplasia, posing an increased, if somewhat unpredictable risk of tumour development.
In terms of cell organization, it is believed that some tumours conform to a stochastic model whereby all tumour cells are endowed with the potential to propagate the tumour and recapitulate the tumour's heterogeneity. On the other hand, many believe that tumours are sustained by a minority cell population of so‐called cancer stem cells, also known as tumour initiating cells. 6 , 7 Cancer stem cells are thought to be largely responsible for drug treatment resistance and for tumour relapse. 8 Whether cancer stem cells are direct descendants of somatic stem cells is a moot point, though some murine studies are consistent with this notion. For example, when the Apc gene is selectively deleted in Lgr5 (leucine‐rich‐repeat‐containing G protein‐coupled receptor 5) expressing intestinal stem cells (ISCs) in mice, the resultant adenomas are similarly sustained by Lgr5‐positive cells 9 ; remarkably, the adenoma Lgr5‐positive cells were intermingled with adenoma Paneth cells, essentially recapitulating the normal crypt stem cell niche. 10
As one might expect, some tumours with a phenotype resembling that of the embryonic/adult stem cells of their tissue of origin (eg liver) are associated with a poorer prognosis 11 ; on the other hand, a stem cell signature in a series of colonic tumours, while supporting a colonic stem cell origin, may not have any prognostic significance. 12 Concluding these introductory remarks, it is worth clarifying what we mean by ‘cancer stem cells’: the term should be restricted to the malignant cells within a tumour that perform stem‐like functions; 13 this review concerns the founder cells of cancers, most likely, but certainly not exclusively, the somatic stem cells (Figure 1). While the bulk of studies support the monoclonal origin (derivation from a single transformed cell) of most tumours, some studies suggest an origin from more than one cell (polyclonal). 14
Figure 1.
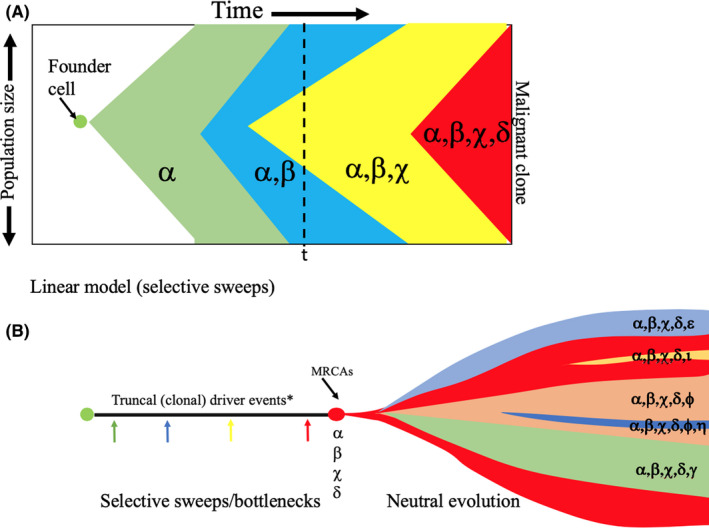
A possible model of tumour evolution combining selective sweeps followed by effectively neutral evolution in the cancer. A, The founder cell is likely to be a somatic stem cell. This cell multiplies to form a preinvasive lesion in which there could be a stepwise progression through the continued selection of ever‐fitter clones (The Selective Sweep Model). Each ‘driver’ mutation induces a clone within the tumour that, if positively selected, sweeps to fixation, replacing much of the population. Rounds of so‐called public mutation and selective sweeps can occur until the accrual of a sufficient mutational burden, including chromosome copy number alterations (aneuploidy) for malignancy to occur. Intratumoral genetic heterogeneity (IGH) will largely be due to the remnants of evolutionary older clones before the new clone has swept to fixation (time ‘t’). In this model, major clonal populations will reflect the driver mutations that are selected. B, At some point in time, before or after malignant potential is attained, a switch to ‘punctuated or branched evolution’ can occur under neutral evolutionary growth conditions, whereby subclones develop over time through the acquisition of new (private) genetic changes, thus creating substantial IGH. In this so‐called Big Bang Model, clones arise continuously during tumour expansion and are not significantly constrained by clonal selection, selective sweeps are rare, both public and private mutations co‐exist during tumour growth, and subclones can remain rare but may fuel drug resistance and relapse if only clonally dominant truncal somatic changes are targeted by therapy. Most driver mutations are likely to be ancestral events, and selection is minimal once cancer has been initiated; subclonal ‘passenger’ mutations might become drivers as a consequence of treatment. In the Big Bang Model, the frequency of a clone is a consequence of its time of appearance rather than its relative fitness. 151 , 152 , 153 , 154 , 155 MRCAs: most recent clonal ancestors. *A driver event (α, β......) can be any genetic change brought about by the likes of mutation, chromosomal loss/addition, epigenetic event
2. METHODS FOR INVESTIGATING CLONALITY AND THE CELL OF ORIGIN OF TUMOURS
2.1. Exploring clonality by X‐inactivation
One of the tenets of tumour biology is that most tumours have their origin in a single cell—they have a monoclonal origin. However, there is now a body of literature that suggests tumorigenesis in some tissues requires the co‐operation of several different cells or clones. Methods to study clonality have been critically reviewed by Garcia et al 14 and include looking at patterns of viral integration in EBV‐associated nasopharyngeal carcinomas and hepatitis B or C–associated hepatocellular carcinoma (HCC), affirming a monoclonal origin of these tumours. More commonly, clonality in epithelial tumours was assessed by exploiting the polymorphisms in X chromosome‐linked genes in females. The method relies on the fact that early in embryogenesis (day 16 in the human female), most of the genes on either the maternal or paternal X chromosome in a single cell are inactivated by methylation of CpG islands (lyonization). These changes are stable and heritable, and thus, females heterozygous for X‐linked polymorphisms are mosaic at the mRNA and protein level. Notwithstanding several potential problems with this approach, for example abnormal methylation in malignancy, X‐inactivation occurs at an early stage of embryogenesis such that the pool of embryonic stem cells for a given tissue is relatively small. Thus, during further development, the progeny of a single X‐inactivated cell become clustered together in what is called a ‘patch’. The patch may also be formed from the clustering together of the progeny of several embryonic cells with the same inactivation pattern. A tumour arising within a monophenotypic patch will appear clonal, but in reality could be either monoclonal or polyclonal (Figure 2); only polyclonal tumours arising at patch boundaries will be detectable. This confounding factor was explored by Novelli et al 15 by studying Sardinian women heterozygous for a heat‐sensitive mutation in glucose‐6‐phosphate dehydrogenase (G6PD) activity; patches as large as 450 crypts were observed in the colon. Thus, studies based on X‐inactivation cannot reliably answer questions about tumour clonality without knowledge of their location to patch boundaries, and a large patch size will be heavily biased towards showing tumours are monoclonal. However, examining the colon of an XO/XY sex chimeric patient who also suffered from familial adenomatous polyposis (FAP) with a Y chromosome‐specific probe, where the patch size was very small (1.24 crypts) with thus a very high probability of adjacent discordant crypts, it was suggested that a surprisingly high proportion of microadenomas (76%) were polyclonal. 16 Explanations proffered for this observation included that an early adenoma could have promoted growth in a neighbouring crypt since in the FAP patient all cells would already have had a mutation in one APC gene, or that the polyclonality was the result of the clustering (‘collision’) of two neighbouring newborn adenomas. However, further studies using APC mutations as clonal markers in colonic polyps from both FAP and sporadic cases have also found instances of polyclonality, particularly in FAP. 17 Clearly, polyclonality could have a profound bearing on the way we view tumour initiation, though studies such as those described do not necessarily shed light on the cell(s) of origin.
Figure 2.
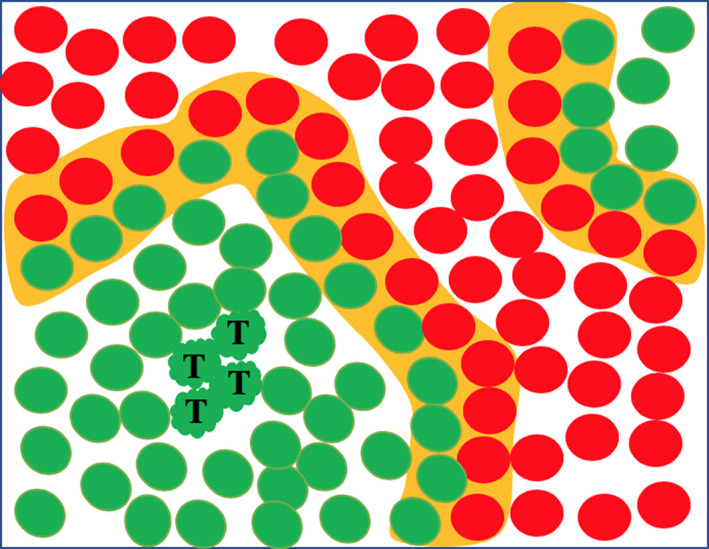
Schematic of an epithelial sheet of cells in a female that is heterozygous for an X chromosome‐linked trait. A polyclonal tumour (T) arising in a large monophenotypic patch will not be detected by this technique, and polyclonality will only be detected when a tumour arises in a patch boundary (orange shading) where divergent cells are close to one another
As noted out by Garcia et al, 14 clonality is also commonly assessed by looking at mutations in oncogenes and tumour suppressor genes. However, they point out that such assessments are usually constrained to the time of analysis, and, of course, we know clonal composition can change in the course of clonal evolution (Figure 1). Thus, a mutation in a marker gene may be altered in a subclone, hence indicating polyclonality in a monoclonally derived population, or alternatively regression of a polyclonal proliferation to a monoclonal population can occur with the outgrowth of a new dominant tumour clone. Many early studies of clonality failed to appreciate these dynamic changes.
2.2. Exploring clonality through mitochondrial DNA (mtDNA) mutation analysis in humans
Heritable genetic lineage tracing is well established in experimental animals 18 , 19 , 20 but such techniques are not available for human studies. However, the mitochondrial genome is highly prone to non‐pathogenic (passenger) mutations that can be used as clonal markers in human tissue. 21 , 22 Such mutations take many years to be established in a cell and are thus only likely to occur in stem cells (Figure 3), but once established the cell lineage from such a cell can be traced. Partially cytochrome c oxidase (CCO)‐deficient crypts (blue‐stained) can be seen in histological transverse sections of human small and large intestinal crypts 23 , 24 indicative of multiple stem cells within each crypt (Figure 4A), but with age, totally mutated crypts become more common, often occurring in patches (Figure 4B). The conversion of a partially mutated crypt to a fully mutated crypt is thought to be through a process of ‘neutral drift’ through the Lgr5‐positive stem cell pool, whereby neutral competition between equipotent symmetrically dividing stem cells eventually leads to the dominance of one stem cell (monoclonal conversion). 25 , 26 Patches of mutated crypts, on the other hand, evolve through crypt fission, initially shown by Greaves et al 27 who observed that bifurcating and neighbouring mutant colonic crypts had a common mitochondrial mutation. As we shall see (see Intestines), monoclonal conversion and crypt/gland fission could be powerful driving forces for the establishment and spread of nuclear genomic driver mutations within the gastrointestinal tract, in effect a mechanism for field cancerization. As illustrated in Figure 4, not only can small intestinal crypts be monoclonally derived, but since they contain the complete repertoire of intestinal cell lineages, they clearly must have their origin in a multipotential stem cell. Likewise, the branched gastric glands (units) of the proximal human stomach have been shown to have a monoclonal origin deriving from a multipotential stem cell 28 ; here, also neighbouring glands can have a common mtDNA mutation, again indicating gland fission was responsible.
Figure 3.
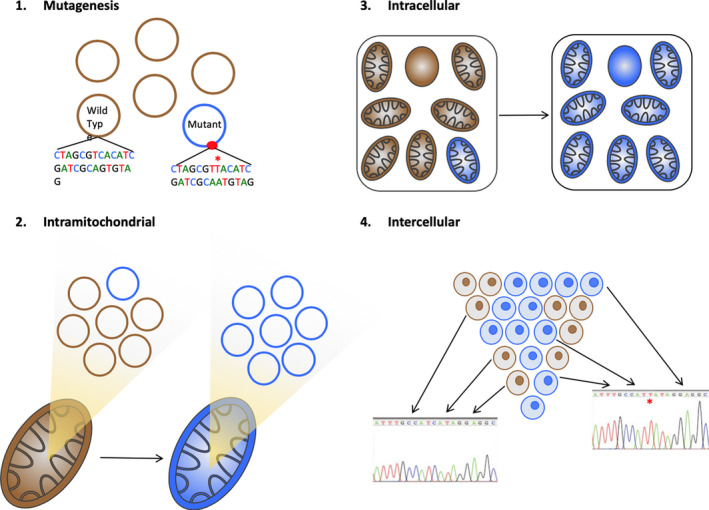
Tracing lineages through heritable genetic changes in mtDNA. (1) mtDNA mutations can occur spontaneously in a single circular genome and (2) through random genome turnover can be present in all copies of the genome within a single mitochondrion and then (3) within the many mitochondria in a single cell, evolving from a heteroplasmic state to a near‐homoplasmic state, a process known as ‘genetic drift’. Spontaneously arising mutations in the cytochrome c oxidase (CCO1) gene that resulted in a demonstrable deficiency of CCO activity (complex IV in the respiratory electron transport chain) were highlighted by Taylor et al, 24 showing that combined histochemistry for CCO activity (staining brown, Figure 4A) and normal nuclear‐encoded succinate dehydrogenase (SDH, complex II) activity (staining blue) was an excellent technique for detecting mtDNA defects. (4) Laser‐capture microdissection of the blue (CCO‐deficient) and brown cells and subsequent sequencing of the mitochondrial genome can identify the mutation. If all CCO‐negative cells in a blue patch have an identical mutation, then it is reasoned the patch has arisen from a single cell, since the minimal odds of exactly the same mutation arising separately in two cells are 2.48 × 109:1 (Reprinted with permission from Ref.22 Copyright 2016, John Wiley and Sons)
Figure 4.
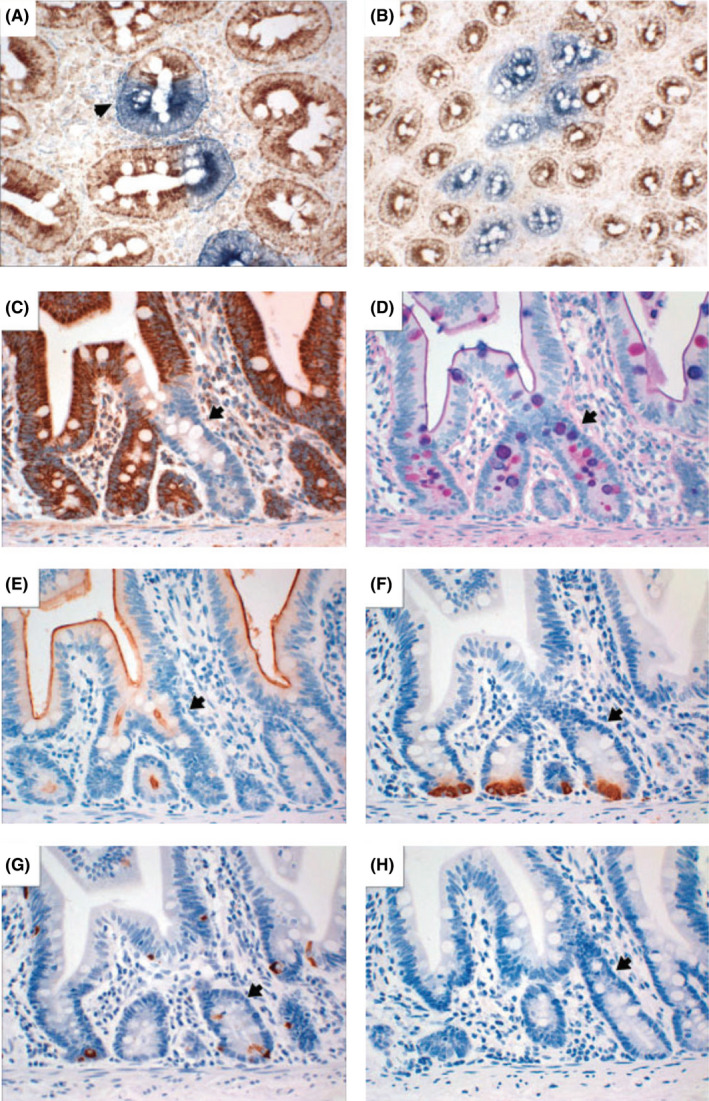
Human small bowel crypts contain multiple stem cells and are clonal. A, A mixed crypt in transverse section (arrow). B, A patch of CCO‐deficient crypts derived from a founder crypt through crypt fission (A,B, CCO (brown) and SDH (blue) histochemistry). Longitudinal serial sections through a crypt showing (C) a CCO‐negative crypt (arrow) stained by immunohistochemistry that contains (D) PAS/Alcian blue/diastase‐stained goblet cells, (E) CD10‐positive enterocytes, (F) lysozyme‐positive Paneth cells and (G) chromogranin A‐positive neuroendocrine cells. (H) An isotype‐matched negative control. All intestinal lineages are present in the CCO‐deficient crypt (Reprinted with permission from Ref.23 Copyright 2009, John Wiley and Sons)
This technique also lends itself to exploration of tumour progression. In the stomach, it is widely believed that there is a stepwise progression through atrophic gastritis, intestinal metaplasia (IM) and low‐/high‐grade dysplasia, culminating in intestinal‐type gastric cancer. By mtDNA mutation analysis, intestinalized metaplastic gastric glands are clonally derived and able to spread by fission. 28 , 29 Moreover, where a large patch of 15 clonally related metaplastic glands were found in conjunction with an area of dysplastic epithelium, an identical mutation in APC was shared by all dysplastic crypts and by just one of the metaplastic crypts. 29 Thus, one possible mechanism for the development of gastric cancer was proposed: metaplastic glands are clonal and spread by fission forming a patch of glands each containing the same mtDNA mutation (therefore derived from a founder gland), a mutation in a tumour suppressor gene occurs in a single metaplastic gland that quickly becomes dysplastic, which itself then undergoes a rapid rate of fission to form a clonal field of dysplasia. Further mutations on top of the founder mutation could lead to subclonal diversity and clonal competition, and the more diversity, the greater the likelihood of cancer development.
2.3. Genetic approaches to explore the origins of tumours in genetically engineered mouse models
The discovery of site‐specific recombination systems has revolutionized lineage tracing in vertebrates, 18 , 20 one of the most common systems being the Cre‐lox system, allowing DNA modifications to be targeted to a specific cell type. LoxP sites are short (34 bp) DNA stretches that under the catalysing influence of Cre recombinase undergo DNA cleavage and reciprocal strand exchange with another loxP site. One of three approaches is commonly used for identifying the particular cells targeted by carcinogens (Figure 5). For lineage labelling, the cells of interest (and subsequently their progeny) are heritably labelled with a label such as the enzyme β‐galactosidase encoded by the lacZ gene or a fluorescent dye such as GFP or tdTomato and the effect of exposure to a known carcinogen can be monitored; if the tumour cells are subsequently similarly labelled, then the carcinogen target cells have been identified (see Figure 10). More commonly, the strategy is designed to activate a suspected oncogene or inactivate a tumour suppressor gene in a particular cell type, if tumours resembling their human counterpart are produced, then a potential carcinogen target cell has been identified. Problems with site‐specific recombination are often due to the specificity of the drivers used for Cre expression, for example promiscuity of Cre expression leading to unwanted (off‐target) labelling, and these are discussed elsewhere. 18 , 20
Figure 5.
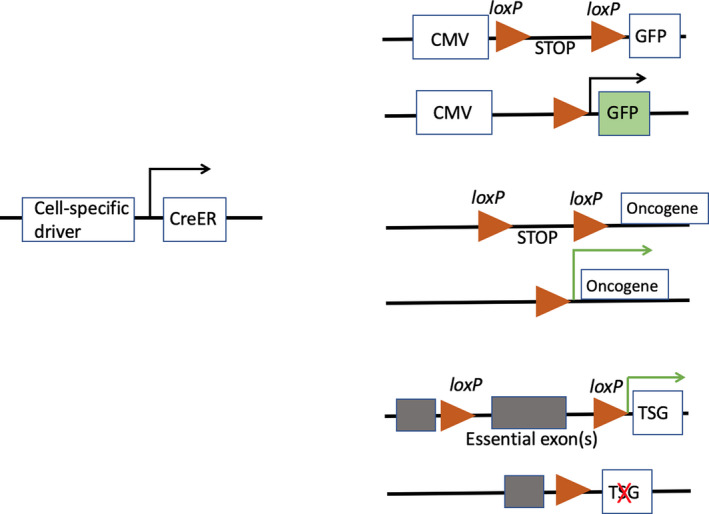
A, For lineage labelling, a stable transcriptional driver such as the cytomegalovirus (CMV) or ROSA26 promoter is engineered upstream of the reporter gene, and a loxP flanked stop signal (loxP‐STOP‐loxP [LSL]) is placed between these two sequences. In the presence of Cre, recombination of the loxP sites removes the LSL sequence to allow expression of the reporter gene. Crucially, Cre expression can be induced by a cell‐specific transcriptional driver (eg for albumin if labelling hepatocytes, see Figure 10), and temporal control is achieved by the use of a CreER fusion gene whereby Cre is fused with a mutant ligand‐binding domain of the oestrogen receptor that can only be activated by deliberate exposure to a synthetic ligand such as tamoxifen (CreERT2). Thus, in animals, translocation of CreER to the nucleus only occurs after tamoxifen injection where it induces recombination of the DNA at loxP sites. B,C, A similar strategy can be employed to excise the stop cassette preceding a mutant oncogene or essential exons in a tumour suppressor gene (TSG). This will lead to activation of the oncogenic program in the selected cell lineage
Figure 10.

Tamoxifen (TM)‐injected Alb‐Cre‐ERT2;R26RlacZ/+ and CK19‐Cre‐ERT2;R26RlacZ/+ mice were all exposed to thioacetamide (TAA) in the drinking water for 30 wk. Liver tumour nodules developed in all mice, but only the CK19‐positive cells in the iCCA from the Alb‐Cre‐ERT2;R26RlacZ/+mice expressed β‐galactosidase, indicating that the iCCA arose from transdifferentiated hepatocytes. Data from Ref.60
2.4. Does a tumour's phenotype (behaviour) indicate its cell of origin
In continually renewing systems (Figure 6) such as those of the gastrointestinal tract and haematopoietic system, experimental evidence supports the belief that mutations in stem cells are the most likely to successfully initiate vigorous malignant growth. The pioneering studies of Fialkow in 1967 30 showed that CML is probably initiated in a multipotential haematopoietic stem cell (HSC) because the Philadelphia (Ph) chromosome resulting in the BCR:ABL1 fusion gene was found in all erythrocytes and granulocytes, while monoclonality was inferred by finding only one enzyme type in CML cells in females heterozygous for G6PD. Numerous more recent studies also point to the HSC or an early progenitor as the cell of origin of most haematological malignancies. In AML, recurrent DNMT3a (DNA methyltransferase 1) mutations (but not the NPM1 [nucleophosmin 1] mutations found in AML blasts) have been found in HSCs from patients in remission. 31 Since these mutated HSCs were able to regenerate the entire haematopoietic hierarchy in xenografts in a manner superior to non‐mutated HSCs, it was suggested that the former cells were preleukaemic HSCs clonally expanded from a normal HSC, and from which AML evolves. Myelodysplastic syndrome (MDS), a preleukaemic haematological disorder that frequently progresses to AML and less frequently to lymphoblastic leukaemia, is also thought to be a clonal disease, initiated in a normal stem cell, progressing to a preMDS stem cell state with a heavy myeloid bias but retained lymphoid potential. 32 , 33 , 34
Figure 6.
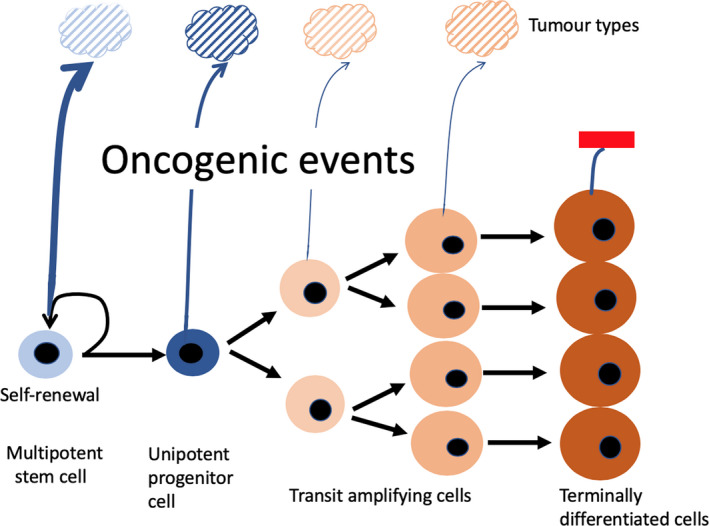
Does tumour phenotype inform on tumour histogenesis? Current dogma suggests that most tumours arising in continually renewing systems with a hierarchical organization (as illustrated) are initiated in stem cells or closely related progenitor cells, since ordinarily these are the only cells with a sufficient lifespan to accrue the mutational burden required for a malignant habitus. Moreover, stem cells possess the ability for seemingly continual turnover and are anchored to the niche. Inter‐tumoural diversity is unlikely to be because tumours can be initiated stochastically from any stage of lineage differentiation, and is most likely a reflection of either further differentiation and/or blocked differentiation/maturation arrest imposed by the oncogenic regime. Thus, tumours are unlikely to be similar to their cell of origin, but may possess some of their features, for example stemness or multipotentiality. The cell of origin may be further disguised by antecedent metaplasia, for example intestinal metaplasia in the oesophagus and stomach, mucinous cell metaplasia in the pancreas or by proceeding epithelial‐mesenchymal transition (EMT)
However, in the haematopoietic system we cannot rule out the possibility of more mature committed progenitors being a cell of origin of cancer. For example, using murine MOZ‐TIF2 and MLL‐AF9 AML models, leukaemic transformation of granulocyte‐monocyte precursors can be achieved. 35 , 36 , 37 On the other hand, these same models clearly show that the more mature lineages are less permissive for transformation; for example, when head‐to‐head comparisons are made of the tumours resulting from retroviral transduction with the MLL‐AF9 (MA9) fusion gene, HSC‐derived AMLs were the most aggressive, while committed progenitor cell‐derived AMLs were the least aggressive, at least in the xenograft scenario. 38 , 39 In a similar vein, introducing the BRAFV600E mutation, found in almost all cases of hairy cell leukaemia (HCL) and in the accompanying phenotypically normal HSCs, to murine HSCs, results in a haematopoietic malignancy similar to HCL, while similar targeting to B‐cell lineages did not produce a haematopoietic abnormality despite similar levels of MAPK signalling. 40 , 41
It is also worth noting that as with carcinomas, the phenotypes of haematological malignancies do not necessarily resemble their cell of origin. For example, ETV6‐RUNX1+ childhood acute lymphoblastic leukaemia is characterized by a CD19+ B‐cell lineage precursor phenotype, but the initiating events occur prenatally in a cell downstream in the lineage hierarchy: the tumour phenotype reflecting the stage of maturation arrest inflicted by the fusion gene. 42 The idea that the degree of stem cell maturation arrest could determine tumour phenotype was first proposed by Sell and Pierce in 1994. 43
The cell of origin of epithelial tumours (carcinomas) commonly influences their phenotype, but not necessarily so in the liver where three resident hepatic cell populations could contribute to the two major types of primary liver cancer (PLC)—HCC and intrahepatic cholangiocarcinoma (iCCA). While a number of phenotypically and anatomically seemingly distinct hepatocyte populations with regenerative capacity 44 , 45 could give rise to HCC with an appearance reminiscent of normal hepatocytes, 46 , 47 the liver also has a facultative stem cell compartment emanating from the smallest branches of the intrahepatic biliary tree. 46 These biliary epithelial cells (BECs), also known as hepatic progenitor cells (HPCs), are bipotential but are only activated in situations of regenerative stress when hepatocytes are reproductively senescent. 48 , 49 HCC is a paradigm for inflammation‐driven cancer, accompanied by hepatocyte senescence; thus, activation of HPCs is a common feature. Bipotential HPCs have been mooted to be the cells of origin of liver tumours with both hepatocytic and cholangiocytic differentiation, so‐called combined hepatocellular‐cholangiocarcinoma (cHCC‐iCCA). 50 , 51 Additionally, tumours with marked expression of stem/progenitor cell features (AFP, CK/7/19, Sox9, EpCAM) may be similarly derived and appear to carry a poorer prognosis. 11 , 52 , 53 , 54 In rodent models of tumorigenesis, HPCs can certainly be the founder cells of HCC. 55 , 56 While it is presumed that BECs are the cells of origin of most iCCAs and, indeed, this can be established by lineage tracing, 57 the ability of hepatocytes to be readily reprogrammed to BECs 58 somewhat confounds attempts to predict the cell of origin of both HCCs and iCCAs by simple appearance alone. 59 , 60 , 61 , 62 , 63
As shown for the haematopoietic system, the position of the cell of origin of intestinal cancer within the normal hierarchy can have a significant bearing on tumour phenotype. In the mouse small intestine, targeted deletion of the Apc (adenomatous polyposis coli) gene in the Lgr5‐positive crypt base columnar stem cells is associated with the development of large adenomas having nuclear‐associated β‐catenin. 64 On the other hand, targeted loss of the same gene in the short‐lived transit amplifying crypt cells only results in small adenomas with stalled growth. In the same system, lineage tracing revealed that not only did each of these Lgr5‐positive cells give rise to a clonal population, but additionally, lineage retracing from Lgr5‐positive cells from within adenomas demonstrated clonal outgrowth, supporting the belief that tumours can be sustained by stem cells. 9
In the epidermis, another continually renewing system, there is also persuasive evidence that the position of the initiated cell within the hierarchy has a profound bearing on subsequent tumour growth. Basal cell carcinoma (BCC) is the most common skin cancer in man and hedgehog (Hh) signalling is critical to its formation. In mice, when either ‘loss of function’ mutations in the Patched (Ptch) receptor or ‘gain of function’ mutations in Smoothened (Smo) were introduced into the stem cells/committed progenitors of the interfollicular epidermal basal layer via. K14‐CreER or the more differentiated cells expressing the keratinocyte protein involucrin via. Inv‐CreER, 65 BCCs were readily induced in the K14‐positive target cells, but were very much reduced in size and number in the involucrin‐positive population. Concluding this section, it is quite clear that cancer can be readily induced in stem cells but as cells progress towards maturation, conditions for initiation/progression soon become more or less unattainable.
3. CANCER ORIGINS IN SPECIFIC TISSUES/ORGANS
3.1. Oesophagus
The oesophagus in man is normally lined by a stratified squamous epithelium that gives rise to two major histological tumour subtypes, oesophageal squamous carcinoma (OSCC) and oesophageal adenocarcinoma (OAC), the latter preceded by an IM (Barrett's oesophagus)/dysplasia sequence. In the oesophagus, 90% of tumours are SCCs that occur mainly in the upper and middle regions, while OAC occurs in the lower regions of the oesophagus close to the gastric oesophageal junction (GOJ), often associated with acid reflux and obesity. Much of our knowledge of these two diseases comes from genetically engineered mouse models (GEMMs), so it is worth noting that while the human oesophagus is lined by a stratified non‐keratinizing epithelium, having submucosal glands and a basal layer thrown into papillary folds, the mouse oesophagus has no such glands, being keratinized and extending well into the forestomach (Figure 7).
Figure 7.
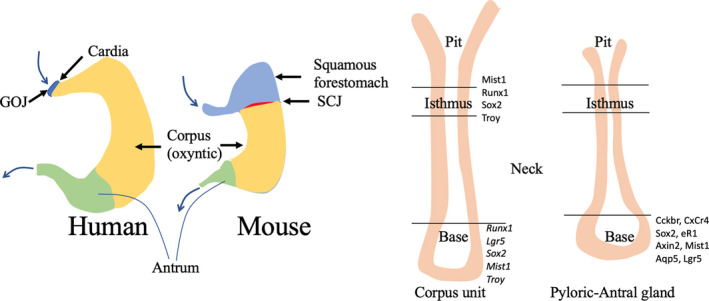
A, Anatomical differences between a human and mouse stomach. In particular, note the positioning of the gastro‐oesophageal junction (GOJ) in man and the ‘equivalent’ squamous cell junction (SCJ) in the mouse. B, The postulated stem cell zones in the corpus (oxyntic) and pyloric/antral gastric glands. 96 Cells at the base of corpus glands with stem cell markers are likely to be reserve stem cells, only displaying complete multipotentiality after damage to the more luminally located isthmic stem cells
The basal layer in humans is characterized by the uniform expression of the β1‐integrin, suggesting the entire basal layer is in an undifferentiated progenitor state, and indeed, all basal cells exhibit similar clonogenicities in vitro irrespective of their location along the basement membrane. 66 On the other hand, label‐retaining cells, a supposed marker of slow‐cycling stem cells, have been found to be more numerous in the papillary basal layer when patients were injected with 5‐iodo‐2’‐deoxyuridine 29 days beforehand. 67 Studies of the origin of SCC in the mouse oesophagus point to CK5‐positive basal cells being the cell of origin. CK5‐CreER mice overexpressing Sox2 exhibit a massive basal cell expansion but no SCC, but when these basal cells are isolated and infected with activated Stat3 lentivirus and transplanted to NSG mice, then SCC arises; suprabasal cells expressing Sox2 and Stat3 do not give rise to tumours. 68 Notch mutations are common in OSCC where Notch signalling promotes differentiation, thus acting as a tumour suppressor, 69 , 70 and Notch inhibition in a GEMM supports this belief. If mice express a dominant‐negative mutant of mastermind‐like 1 (Maml1), then Notch signalling is inhibited as NICD is unable to bind to Maml1; this inhibition results in a change in the dynamics of basal cell divisions in that fewer daughter cells undergo differentiation leading to massive mutant progenitor cell expansion—in effect a field cancerization effect. 71 The source of oesophageal high‐grade dysplasia (HGD) in mice was explored in a model of chemical carcinogenesis in the Confetti reporter mouse where basal cells were stochastically labelled with one of four fluorescent proteins; observing clones of different colours in the HGD suggested to the authors that HGD was polyclonal. 72 However, the widespread exposure to a mutagenic chemical (DEN) makes the initiation of several cells highly likely; hence, extrapolation to man is not justified.
Oesophageal adenocarcinoma can also be modelled in GEMMs. A major line of enquiry has been the origin of IM known as Barrett's oesophagus, since this is considered to be the precursor of dysplasia and OAC. 73 One such study noted a transitional zone at the squamous:columnar junction (SCJ) composed of two cell types, a basal layer expressing p63, CK5 and CK7, and a luminal cell layer only expressing CK7. 74 , 75 Re‐directing the bile over this area to mimic severe acid reflux resulted in an expansion of this area with cells expressing CDX2 (essential for normal intestinal‐type differentiation) and having the presence of mucin‐secreting goblet cells. However, the relevance of such studies is questionable since the SCJ in mice lies between the squamous forestomach and the glandular hindstomach, while in man the equivalent area is the GOJ, though a distinctive cell type with features of both columnar and squamous cells has been observed at the junction of the squamous epithelium with Barrett's epithelium. 76
The study of mtDNA and nuclear genomic mutations has been informative in studying the histogenesis of human Barrett's oesophagus. It was originally thought that Barrett's oesophagus evolved according to the selective sweep model (Figure 1A). Here, a founder genetic change such as a p16 point mutation in a progenitor cell gives that cell a selective advantage such that it sweeps to fixation extinguishing all other clones. Successive rounds of mutation and selective sweeps result in progression along the metaplasia‐dysplasia pathway. However, analysing genetic changes on a crypt‐by‐crypt basis across an entire Barrett's segment failed to find a common founder mutation, suggesting that clonal heterogeneity was due to the mutation of multiple progenitor cells that give rise to clones that evolve independently. 77 Moreover, the finding of an identical p16 point mutation in a oesophageal gland squamous duct and the adjoining metaplastic epithelium suggested that the gland ducts could be at least one of the locations for a stem cell niche from which Barrett's epithelium arises. Using mtDNA mutations as clonal markers, the metaplastic crypts in the oesophagus bear many similarities with normal intestinal crypts; they can occur in patches with all cells having a common mtDNA mutation, suggestive of crypt fission being a mode of spread and, importantly from a cell of origin perspective, areas of squamous epithelium and metaplasia were found to be clonally related suggesting a common cell of origin. 78 On the other hand, observations that the proliferative and functional organization of Barrett's glands can resemble both intestinal and gastric lineages (incomplete metaplasia) 79 lead to the reasonable conclusion that gastric gland stem cells, presumably migrated from the cardia, could also serve as a cell‐of origin. 80 Given incomplete metaplasia precedes complete metaplasia, the presence of gastric‐type epithelium could follow a 2‐step process, (a) migration of cardia stem cells into the oesophagus followed (b) by acquisition of intestinal‐type properties by the stem cells to generate metaplasia. A mouse model of Barrett‐like metaplasia and dysplasia caused by IL‐1β overexpression would support such a scenario 81 ; here, tracing the cell lineage from Lgr5+ cells abutting the SCJ (mouse homologue of cardia) observed reporter labelling in Barrett‐like epithelium at 4 months after tamoxifen injection.
In summary, considerable debate continues on the cell of origin of Barrett's oesophagus, 82 , 83 though it is widely accepted that it is the requisite precursor lesion for the development of OAC. At least four candidate founder cells present themselves: basal cells of the squamous epithelium, stem cells of submucosal ducts, stem cells of the proximal stomach and transitional cells at the SCJ.
3.2. Stomach
The relationship between OAC and intestinal gastric cancer (IGC) has become blurred as their histological and anatomical features are clearly intertwined. 84 IGC is associated with Helicobacter pylori (H pylori) infection, but with reductions in infection, there has been a shift in prevalence from the distal stomach towards the GOJ. 85 Thus, proximal IGC and OAC could represent subtle variations of a single tumour type from a common cell of origin.
Gastric cancer is classically believed to develop through a sequence of atrophic gastritis, metaplasia, low‐/high‐grade dysplasia and finally to cancer. Gastritis results in loss of parietal cells and chief cells and the development of a metaplastic mucous cell lineage that resembles antral glands, possibly originating from the transdifferentiation of chief cells, though mucous neck cells could also be involved. 86 These metaplastic mucous cells express spasmolytic polypeptide (now designated trefoil factor family 2 [TFF2]), hence SPEM (spasmolytic polypeptide‐expressing metaplasia); IM is believed to develop from pre‐existing SPEM glands. 87 As SPEM and the more differentiated lesion, IM, are strongly associated with IGC, the question arises whether they, and thus chief cells or mucous neck cells, can be precursors of IGC, in the experimental setting the answer is yes. Chief cells express the bHLH transcription factor Mist1 (muscle, intestine and stomach expression 1), and expression of activated Kras (G12D) in Mist1‐CreER2 mice leads to both types of metaplasia in the corpus of the stomach, eventually progressing to IGC. 88 A relationship of IM with IGC progression has also been seen in humans, where an identical APC point mutation was found in a metaplastic crypt and a field of dysplastic glands. 29 IGC can be generated from Mist1‐CreERT2 mice both in the corpus and in the antrum (Figure 7). In the isthmic region of the corpus, isthmic Mist1+Lgr5−ve cells are responsible for lineage tracing and induction of mutant Kras G12D results in widespread IM/dysplasia; however, using Mist1‐CreERT2;LSL‐Kras G12D;Apc flox/flox mice, it was clear that IGC could only be induced from these cells by Apc loss in the context of Kras (G12D)‐induced IM. 89 Moreover, these isthmic Mist1+ cells were also the origins of diffuse‐type gastric cancer upon loss of the E‐cadherin encoding gene (cdh1). Mist1 expression is also seen in cells located in the progenitor region of antral gastric glands, just above the basal Lgr5+ cells, and lineage tracing reveals their multipotentiality, contributing to all antral gland lineages. 90 Interestingly, large antral tumours were induced in all Mist1‐Cre‐ERT;Apc flox/flox mice within 60 days of tamoxifen injection, but no tumours were found in the corpus or the duodenum where there are also Mist1+ cells, suggesting antral Mist1+ cells are uniquely susceptible to Apc/Wnt‐β‐catenin tumorigenesis.
Lgr5 expression identifies multipotential stem cells at the base of antral glands, and targeted loss of Apc in Lgr5‐EGFP‐ires‐CreERT2;Apcfl/fl mice leads to highly proliferative, nuclear β‐catenin+ adenomas within several weeks. 91 Expression of the water channel protein, aquaporin 5 (Aqp5), appears to co‐localize with the major sub‐population of these Lgr5+ cells, and, again, lineage tracing from these cells indicates they are multipotential and persistent. 92 Using Aqp5‐CreERT2 mouse models, the combination of Apc loss, Pten loss and Kras G12D activation was the most effective inducer of exclusively pyloric (antral) IGC, though lesser combinations also produced tumours, although sole activation of Kras G12D was not tumorigenic. A population of Lgr5+ chief cells has also been unearthed at the base of pyloric glands, though lineage tracing from these cells did not generate fully traced glands. 93 However, fully traced glands were evident after damage caused by a high tamoxifen dose, indicating they are ‘reserve’ stem cells, and after Kras G12D activation, these cells produced SPEM, progressing to IGC within 4 months. It is interesting to note that H pylori‐infected patients with IGC have expanded numbers of Lgr5+ cells in the antral mucosa displaying DNA damage, a probable prelude to mutation. 94
In summary, the two main locations of functional gastric stem cells are the isthmus in the corpus (oxyntic stomach) and towards the base of the antral glands. Numerous markers have been proposed to identify them 95 , 96 and undoubtedly many cells co‐express many of them (Figure 7). Using cell‐specific drivers, it is clear that conditional activation of the likes of Kras G12D and/or inactivation of Apc are potent inducers of IGC and/or the preceding metaplasia.
3.3. Intestines
Small intestinal and colonic crypts are shaped like test tubes, closed at one end. Lined by a simple epithelium, there is a strict hierarchical organization, with ISCs at the base giving rise to transit amplifying cells that move up the crypt, with cells eventually leaving the cell cycle in the top‐third as they undergo terminal differentiation. Like all somatic stem cells, ISC behaviour is thought to be strictly regulated by a stem cell niche, in particular by unique stromal cells beneath the crypt base. 97 In the small intestine, Pdgfra+ telocytes at the crypt:villus junction produce abundant bone morphogenetic proteins (BMPs), while at the crypt base, specialized cells with expansive cytoplasm expressing low levels of Pdgfra, but positive for CD81 (a cell surface protein belonging to the tetraspanin family), express the BMP inhibitor (BMPi) Gremlin 1 (Grem1). These cells are able to expand ISCs to organoids in the absence of exogenous factors such as Wnts and BMPi factors. Subversion of BMP signalling is widely implicated in intestinal tumorigenesis with Grem1 often highly expressed in the stromal cells of many tumours, thus contributing to tumorigenesis by promoting ISC expansion. 98
In murine intestinal inflammation, abnormal T‐cell activation and IFNγ production are potent mediators of ISC injury 99 ; hence, the gut requires a robust stem cell network and, similar to the stomach, appears to have both homeostatic and reserve stem cells. In 2007, genetic lineage labelling indicated that in both mouse small and large intestines, rapidly cycling multipotential homeostatic ISCs were located at the crypt base (crypt base columnar cells, CBCs), expressing Lgr5. 100 Lineage tracing from the Prom1/CD133 locus produced the same result. 101 In the small intestine, the CBCs are intermingled with ‘nurturing’ Wnt‐secreting Paneth cells that constitute part of the stem cell niche. 10 It had been formerly believed that the intestine's homeostatic stem cells were located about 4‐5 cell positions up from the crypt base (so‐called + 4 cells). 102 This was because these cells retained an administered DNA label (label‐retaining cells, LRCs) long after one might have expected the label to have been diluted by repeated divisions, thus rendering it undetectable, the prevailing belief at the time being that all stem cells were slowly cycling, hence LRCs. Subsequent studies now indicate that the + 4 cells, expressing the likes of Bmi1 but not Lgr5, may represent a reserve stem cell compartment in the small intestine that is activated when Lgr5+ CBCs are selectively ablated. 103 Further investigation has revealed that LRCs are progenitors for secretory intestinal lineages but they can be recalled to the stem cell pool after injury. 104 Somewhat counterintuitively, +4 cells are highly expressive of Wip1 (wild‐type p53‐induced phosphatase), a negative regulator of p53. 105 Loss of Wip1 in Apc Min/+ mice severely reduces polyposis in these mice, so while expression of Wip1 in + 4 ISCs might be a survivability factor in the face of genetic damage, it might also make them uniquely sensitive to neoplastic transformation. ISCs may also be regenerated from cells much higher up the crypt. If Lgr5‐GFP‐CreERT2;R26 tdTom mice are injected with tamoxifen to label the progeny of Lgr5+ ISCs, and 4 days later are exposed to an ISC‐ablative dose of irradiation, then 1 day later all crypt cells lack GFP expression, confirming inactivation of the Lgr5GFP‐Cre allele at this time. 106 Subsequently, it was observed that all new ISCs and their resulting crypts emitted red fluorescence indicative of an origin from Lgr5+ cell progeny. The bHLH transcription factor Ascl2 (Achaete scute‐like 2) is an important developmental factor and is normally expressed by the Lgr5+ CBCs. Ascl2 was found to be indispensable for the restoration of ISCs, with expression being initiated by cells 8‐9 tiers above the crypt base after the irradiation as both enterocytes and secretory cells dedifferentiated on their downward trajectory. Thus, the ISC pool can be maintained and regenerated from a number of sources and evidence from humans and GEMMs points to cells located at the bottom of crypts being the predominant cells of origin of intestinal cancer.
Studies of adenomas from FAP patients had suggested that dysplastic cells originated from the surface colonic epithelium and grew down into normal crypt territories, in effect a ‘top‐down’ histogenesis 107 but subsequent investigation of very small monocryptal adenomas clearly favoured a ‘bottom‐up’ mode of spread, with the dysplastic cells, possessing nuclear β‐catenin, sharply demarcated from the overlying normal epithelium (Figure 8). 108 However, when larger adenomas were studied, dysplastic cells from neighbouring crypts could indeed grow down into normal crypts.
Figure 8.

‘Top‐down’ vs ‘Bottom‐up’. (A‐C) In the bottom‐up model, initiation occurs in a basally situated stem cell (red cell), and through neutral competition, the crypt can become monoclonally converted. (D‐F) In the top‐down model, initiation occurs at the top of the crypt and dysplastic cells migrate downwards into normal crypt territories. Photomicrograph of a monocryptal adenoma immunostained for β‐catenin expression. Note the sharp demarcation between the dysplastic crypt epithelial cells expressing nuclear β‐catenin (Wnt signalling on) and the overlying normal epithelia showing membranous β‐catenin immunostaining—a pattern of expression consistent with an upward migration of initiated cells. Data from Refs.107, 108
An elegant study from the Clevers group established, at least in the mouse, that stem cells were the most likely cell of origin of intestinal adenomas. 64 Using a very low dose of β‐naphthoflavone (an agonist of the aryl‐hydrocarbon receptor) to treat Ah‐Cre/Apc flox/flox /Rosa26R mice that almost totally restricted lineage tracing to the more differentiated cells above the stem cell zone just resulted in few persistent microadenomas and very rare macroscopic adenomas over a 284‐day period. On the other hand, aggressively expanding adenomas arose within a month of tamoxifen administration to Lgr5‐EGFP‐IRES‐CreERT2/Apc flox/flox mice. Confirming these findings, activation of endogenous Wnt signalling in Prom1 (CD133)‐positive ISCs by the Cre‐dependent introduction of a mutant allele of Ctnnb1 (Ctnnb1+/lox[ex3]) also leads to neoplastic development. 101 Secretory precursors expressing Mist1 (Bhlha15), located above the ISC zone, are another possible founder cell for neoplastic development, particularly upon sustained Notch signalling in the context of Apc disruption. 109 Clearly, numerous markers (Lgr5, Bmi1, Hopx, Lrig1, OFLM4) are common to many of the cells within the bottom one‐third of the crypt, another one being CK15, and unsurprisingly cells expressing CK15 give rise to adenomas and adenocarcinomas in the small intestine and colon in the setting of Apc loss. 110 Since the phenotype of the tumours was more severe than commonly seen in other models, in particular the adenocarcinoma development, then maybe CK15 expression encompasses a cell type hitherto largely ignored as a cell of origin. One such cell is the tuft cell, a very rare, long‐lived and quiescent secretory cell in the intestine, derived from a Lgr5+ cell, identified by the expression of doublecortin‐like kinase 1 (Dclk1) and located in the putative stem cell niches of both stomach and intestine. 111 Tamoxifen administration to Dclk1‐CreERT‐Apcflox/flox mice had seemingly no long‐term effect, but subsequently inducing dextran sodium sulphate colitis in these mice generated rapidly fatal poorly differentiated colorectal adenocarcinomas. Moving completely outside the classical ISC niche, epithelial Grem1 overexpression in a mouse model of hereditary mixed polyposis syndrome can lead to the formation of intravillus ectopic crypt‐like foci from which dysplasia appears to have its origin. 98 These cells were not Lgr5+ but did have the persistence/reacquisition of progenitor features such as Sox9 expression; thus, we cannot be too dismissive of a top‐down model of tumour histogenesis for some types of bowel cancer!
Studying normal intestinal crypt cell dynamics in Confetti mice, it was concluded that homeostasis was maintained by neutral competition between symmetrically dividing stem cells as crypts drift towards monoclonality (neutral drift dynamics). 25 Further discussion beyond the initiated ISC is not the remit of this review, but accelerated division of mutated ISCs can create a biased drift towards crypt clonality and further spread of mutant crypts can occur by enhanced crypt fission (Figure 9). 112 Neutral competition between ISCs probably protects against the acquisition of deleterious mutations, so when the pool of ISCs is reduced by inhibiting Wnt secretion, there are fewer competing stem cells, speeding up the fixation of mutations and leading to enhanced tumorigenesis. 113 Of course, one consequence of this accelerated crypt fission is the replacement of a field of normal crypts by a field of cancer‐primed crypts that may not yet show any demonstrable phenotype—a process known as ‘field cancerization’. 114 This process can be quite extensive with large fields of even non‐dysplastic cells having the same KRAS and TP53 mutations in inflammatory bowel disease 115 , 116 ; in one case of Crohn's colitis, an identical TP53 mutation had, in 3 years, spread along almost the entire length of the colon. 115
Figure 9.
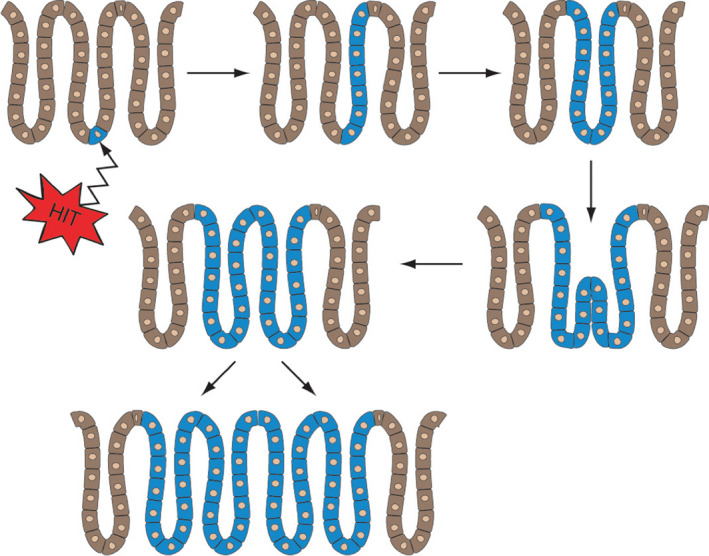
A model for the spread of premalignant cells in the colonic mucosa. Initiation occurs in a basally located stem cell that undergoes biased genetic drift to rapidly occupy the whole crypt. 112 The wholly mutated crypt then undergoes an accelerated rate of crypt fission to occupy a large area of the colon (field cancerization)
In summary, experimental evidence certainly points to the bottom of the crypt for the founder cells of many intestinal cancers, though the existence of ‘reserve’ stem cells and disruption of the morphogen gradients that normally act to maintain the hierarchical organization along the vertical crypt axis may provide alternative cells of origin.
3.4. Liver
Unlike the digestive tract, the liver is not a rapidly renewing cell system and, on average, hepatocytes have a turnover time of about 1 year. 46 The relationship between cell proliferation and the fixation of mutations is clearly apparent in organs such as liver; more than 40 years ago, it was shown that carcinogens such as dimethylnitrosamine were only effective in inducing preneoplastic hepatocytic foci if administered during a time of active organ regeneration. 117 Since most liver cancers arise within a setting of chronic liver damage, regeneration is a fertile ground for the fixation of mutations. While all hepatocytes in young adult healthy livers can re‐enter the cell cycle and thus potentially serve as a cell of origin, there are also sub‐populations of so‐called homeostatic hepatocyte stem cells that, at least in mouse models, become highly clonogenic after inflicted damage. 44 These would include pericentrally located Axin2+ hepatocytes, randomly distributed Tert+ hepatocytes and periportally located hepatocytes expressing high levels of HNF4α, but also low levels of Opn and Sox9; indeed, it is tempting to speculate that TERThigh hepatocytes are founders of many HCCs given that TERT promoter mutations are commonly identified in the early stages of HCC development both in the low‐grade and high‐grade dysplastic lesions, and represent the most frequent mutation in some series of HCC, with an overall frequency of 60%. 118
Primary liver cancers have a spectrum of histological patterns ranging from classic HCC (solid growth pattern), through mixed HCC‐iCCA to iCCA (glandular pattern often accompanied by a marked stroma). The major genetic changes underlying PLCs have been reviewed elsewhere, 54 , 119 but here we should note that tumour phenotype is not necessarily a reliable indicator of histogenesis. Hepatocytes and BECs derive from a common hepatoblast precursor and can readily transdifferentiate into each other, 58 thus belying attempts to ‘guess’ a cell of origin. Moreover, the existence of bipotential HPCs that are activated under the very conditions that most PLCs originate, that is inflammation, hepatocyte loss and regenerative senescence, simply makes for a very complex scenario. 44 , 45 HCC makes up 90% of PLCs, and numerous mouse models can demonstrate an origin from hepatocytes. 119 For example, using a model of liver injury encompassing a single administration of the mutagen diethylnitrosamine (DEN) followed by repeated injections of carbon tetrachloride (CCl4) to AAV‐TBG‐Cre; Rosa26 LSL‐YFP mice results in tumour nodules that were exclusively YFP‐positive, despite the presence of an unlabelled ductular reaction (DR) considered to be derived from HPCs. 120 Noteworthy was the expression of HPC markers such as Sox9 and Epcam in many of the tumour cells despite their hepatocyte origin! Using the same injury model, but this time with Foxl1‐Cre;RosaYFP mice to follow the fate of HPCs, resulted in the same tumour burden, but none were YFP‐positive. As cautioned, 121 though the above studies indicated that hepatocytes can be a cell of origin for HCC, DRs derived from HPCs are more likely to be carcinogen targets when hepatocyte senescence shifts the burden of regeneration onto them, and we might further add that DEN requires metabolic activation, not a function attributed to primitive BECs! Using either Opn (osteopontin) or CK19 Cre drivers for lineage tracking in the same mouse DEN/CCl4 injury model, perhaps not surprisingly, also failed to find HCC derived from HPCs. 122 Likewise, DEN fails to induce HCC from ductular cells lineage traced from the Hnf1β locus in a variety of models that induce extensive DRs, but fail to demonstrate hepatocytic differentiation from them because of the lack of the requisite non‐competitive environment. 123
On the other hand, since hepatitis B virus infection is a major cause of liver cancer and the hepatitis B virus X (Hbx) protein is implicated in the carcinogenic process, it is notable that Epcam+ ductular cells isolated from Hbx transgenic mice with liver injury formed mixed‐lineage tumours when transplanted into immunodeficient NOD/SCID mice. 124 In another model of liver injury, DRs lineage traced from the Epcam locus were subjected to AID (activation‐induced cytidine deaminase)‐mediated genetic alterations and developed HCCs that were mainly HNF4α+ with a minor cholangiocellular component. 55 As the injury model was the feeding of a 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC)‐containing diet that does not provide the requisite hepatocyte growth inhibition for the hepatocytic differentiation of HPCs, the authors were confident that the tumours arose directly from the ductular cells. In many other cases, we cannot be sure where the malignant tumour arose along the HPC/ductular cell/premalignant stage pathway. For example, liver cirrhosis is a premalignant condition with already a substantial mutational burden, 125 and mtDNA analysis shows that many nodules are monoclonal and derived from ductular cells. 126 If HCC arises within a cirrhotic nodule, what is its cell of origin, an HPC or a nodular hepatocyte?
The observation that patients with viral hepatitis often develop iCCA has prompted a more detailed exploration of the origins of this tumour. Though it is likely that most are derived from BECs, 57 experimentally iCCA can be derived from hepatocytes, particularly in the setting of Notch overexpression (Figure 10). 59 , 60 Targeted loss of Tp53 in hepatocytes can lead to HCC, but in the context of DDC‐induced injury, the oncogenically primed hepatocytes are reprogrammed to a cholangiocyte phenotype. 127
The local microenvironment can also determine the tumour phenotype, when hepatocytes with the same oncogenic drivers are exposed to different models of chronic liver disease, then the mode of cell death in the adjacent hepatocytes appears to determine tumour phenotype. 62 , 63 A necroptosis signature selects for CK19+ iCCA, probably under the direction of cytokines from immune cells, activated by damage‐associated molecular patterns (DAMPS) released from necroptotically dying hepatocytes. On the other hand, HNF4α‐positive HCCs were produced when these hepatocytes were surrounded by apoptotic hepatocytes. These observations provide some explanation for why the same liver injuries can produce quite different PLCs.
In summary, it appears that hepatocytes, or possibly a sub‐population of them, are the major founders of HCC. HCC with stem cell features could signify an origin from progenitor cells, but equally well could be simply a measure of dedifferentiation. Most iCCA likely derive from cholangiocytes, but origins from differentiated hepatocytes are most certainly possible.
3.5. Pancreas
Pancreatic ductal adenocarcinomas (PDAC) make up 95% of pancreatic tumours and most arise from one of several precursor lesions, the most common being pancreatic intraepithelial neoplasia (PanIN), followed by intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs). All these preneoplastic have a variable risk of progression to malignancy, thus representing progressive stages of PDAC evolution; hence, their cell of origin is the founder cell of most PDACs.
In mouse models, activating mutations of Kras seem essential for initiating the development of PanINs, and indeed, 90% of human PDACs have activating KRAS mutations, as do most of even the lowest grade of PanIN. 128 , 129 As noted, 128 targeting oncogenic Kras to various pancreatic cell types in GEMMs has shown the malignant potential of duct, endocrine and acinar cells; however, is this a reflection of the human situation? In humans, KRAS mutations are typically followed by loss of CDKN2/p16, while loss of TP53 and SMAD4 is usually only found in high‐grade PanIN. On the other hand, the preneoplastic cystic lesions, IPMN and MCN, appear to have unique GNAS and RNF43 mutations suggestive of a quite different histogenesis.
As PanINs display a duct‐like morphology, it was widely thought that they arise from pancreatic ducts. However, ductal cells are very resistant to mutant Kras‐induced neoplastic transformation, so the consensus is that PanINs evolve from the transdifferentiation of acinar cells, so‐called acinar‐ductal metaplasia (ADM) and then acquire further mutations in a linear fashion, see Figure 1A. 130 , 131 There are many observations to support the existence of ADM. For example, lineage‐traced human pancreatic acinar cells can transdifferentiate to ductal cells in vitro, 132 while the introduction of oncogenic Kras to mouse acinar cells can induce a significant ADM, progressing to PanIN and finally PDAC. All grades of PanIN lesions can be induced through expression of the Kras G12D allele in either the Elastase‐ or Mist1‐expressing mature acinar cell compartments 133 ; however, further progression to PDAC is not seen. However, the introduction of oncogenic Kras G12V into elastase‐expressing cells can produce the complete spectrum of PanINs and PDAC, but only in the context of cerulein‐induced mild chronic pancreatitis. 134 On the other hand, in the absence of an activated oncogene in the cerulein‐induced pancreatitis model using the Elastase‐Cre mouse to heritably label acinar cells, only a minority of metaplastic mucinous ductal lineages are derived from acinar cells, and centroacinar cells or ductal cells are the major cells of origin. 135 PDAC can originate from both acinar cells (via. PanIN) or ductal cells (but without preceding PanIN) when identical driver mutations, for example activation of mutant Kras G12D combined with loss of either Fbw7 or Tp53, are targeted to duct cells using Elastase1‐CreERT2 mice or acinar cells using CK19‐CreER mice. 136 Mice with duct‐derived PDAC had a lower survival rate than those with acinar‐derived tumours. Interestingly, organoids created from duct‐derived PDAC could be distinguished from acinar‐derived lesions by an 800‐fold greater expression of Agr2 mRNA, AGR2 (anterior grade protein 2) being a pro‐oncogenic protein whose high expression is associated with cell migration, cell proliferation and a poorer prognosis in many patients with solid tumours. 137 A similar GEMM targeting Sox9+ duct cells and Ptf1a+ acinar cells with combined mutant Kras G12D and Tp53 loss also observed that duct‐derived PDACs developed more rapidly than acinar‐derived tumours; moreover, the former were associated with high‐grade PanIN, while the latter had low‐grade PanIN lesions. 138
IPMNs and MCNs are preneoplastic lesions distinguished by commonly having activating mutations in GNAS and inactivating mutations in RNF43. 139 GNAS codon 21 mutations are present in two‐thirds of IPMNs and lead to increased adenylyl cyclase activity and cAMP production, while RNF43 encodes a ubiquitin E3 ligase implicated in the inhibition of Wnt signalling. IPMNs are large mucin‐producing lesions located within ducts, from which they are thought to be derived. However, progression towards PDAC does not conform to a selective sweep model, but rather originates as a polyclonal lesion. 140 , 141 , 142 By adopting a single‐cell sequencing approach, it was found that early‐stage IPMNs with low‐grade dysplasia had multiple independent clones, each with distinct mutations in early drivers such as KRAS and GNAS, indicative of a polyclonal origin. 141 With progression to high‐grade dysplasia, there was a reduction in the heterogeneity of the early drivers along with acquisition of additional driver mutations in genes such as RNF43 and TP53; this reduction presumably reflects selection of the ‘fittest’ clones, in effect a polyclonal tumour reverting to a more monoclonal expansion.
We know little about stem cell/progenitor cells within the ductal system, or whether there is a cell especially susceptible to neoplastic transformation? The system can certainly be flexible, for example following large‐scale pancreatic cell loss in the rat, ductal cells in the main pancreatic duct can extensively proliferate concomitant with the loss of expression of the transcription factor Hnf6. 143 Regarding cell of origin, single‐cell RNA sequencing has found that pancreatic MCNs and mucinous ovarian tumours are closely related to primordial germ cells (PGCs), leading to speculation that MCNs arise from PGCs that settled in the pancreas during embryonic development. 144 In a mouse model of pancreatitis, proliferating cells with embryonic/stem cell features migrate from the pancreatic duct glands (PDGs), partly under the influence of trefoil factor family (TFF‐1 and −2) motogenic peptides, to repopulate the ducts; thus, the PDGs can function as a progenitor niche for regeneration. 145 The finding of specific mtDNA mutations common to both particular PDGs and their overlying IPMN in humans strongly supports the suggestion that IPMNs can be the direct descendants of PDGs 146 ; moreover, studies with TFF2 knockout mice observed that TFF2 has a profound suppressive effect on the generation of preneoplastic mucinous lesions and even adenocarcinoma.
Possibly related to the cell of origin of PDAC are some tantalizing observations made in GEMMs. In a mouse model targeting the CK19 ductal population to induce PDAC, a population of cells highly expressive of the tetraspanin CD9 were found in the early stages of tumorigenesis. 147 The level of CD9 expression affected the ability of these cells to form organoids and initiate new tumours, and functionally CD9 interacted with the glutamine transporter ASCT2 to enhance glutamine uptake. Tuft cells, identified by the expression of Dclk1, are another cell type to emerge early in the tumorigenic process in GEMMs, and, like gastrointestinal tuft cells, possessed numerous bundles of microtubules and abundant apical cilia. 148 , 149 , 150 Although these clonogenic and tumorigenic cells were found in abundance in early PanINs, their seeming absence in the normal pancreas would indicate they are not the founder cell of metaplastic lesions. On the other hand, CD9+ cells or tuft cells could be the immediate descendants of the cells of origin of PDAC.
In summary, the specific cell of origin of PDAC is unknown, but acinar cells can give rise to PanINs that progress linearly through increasing grades of dysplasia to culminate in PDAC. PDAC can also arise from pancreatic ducts, and the precursor lesions such as IPMNs can have a polyclonal origin.
4. CONCLUSIONS
This review has summarized information we have on the identity of the founder cells for neoplasms of the digestive tract and its associated organs, the liver and pancreas. Much of this information has come from GEMMs where activating oncogenes or tumour suppressor gene losses can be introduced in a cell‐specific manner. In the continually renewing tissues, it is clear that tumorigenesis can be readily induced in their normal ‘homeostatic’ stem cells, and as cells differentiate, they become increasingly refractory to neoplastic induction. However, as chronic inflammation is a common accompaniment to tumour formation, alternative ‘reserve’ stem cells may be recruited for regeneration and become susceptible to tumour initiation. In the normal liver and pancreas, there is little cell turnover, but hepatitis and pancreatitis are well‐recognized risk factors for cancer development. There is no firm evidence that these two organs harbour specific stem cells, but disparate cell types can be the founders of HCC and PDAC. In the liver, hepatocytes, BECs and HPCs can all give rise to HCC, while in the pancreas, both ductal cells and acinar cells can serve as founders of PDAC.
CONFLICT OF INTEREST
None.
ETHICAL APPROVAL
None required.
Alison MR. The cellular origins of cancer with particular reference to the gastrointestinal tract. Int. J. Exp. Path.. 2020;101:132–151. 10.1111/iep.12364
Funding information
None.
REFERENCES
- 1. Berenblum I, Shubik P. The persistence of latent tumour cells induced in the mouse's skin by a single application of 9:10‐dimethyl‐1:2‐benzanthracene. Br J Cancer. 1949;3:384‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berenblum I. Mechanisms of carcinogenesis: the role of carcinogenic metabolites. Acta Unio Int Contra Cancrum. 1959;15:22‐30. [PubMed] [Google Scholar]
- 3. Potten CS. Epidermal cell production rates. J Invest Dermatol. 1975;65:488‐500. [DOI] [PubMed] [Google Scholar]
- 4. Potten CS. Epidermal transit times. Br J Dermatol. 1975;93:649‐658. [DOI] [PubMed] [Google Scholar]
- 5. Warwick GP. Effect of the cell cycle on carcinogenesis. Fed Proc. 1971;30:1760‐1765. [PubMed] [Google Scholar]
- 6. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 7. Vessoni AT, Filippi‐Chiela EC, Lenz G, Batista LFZ. Tumor propagating cells: drivers of tumor plasticity, heterogeneity, and recurrence. Oncogene. 2020;39:2055‐2068. [DOI] [PubMed] [Google Scholar]
- 8. Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2011;223:147‐161. [DOI] [PubMed] [Google Scholar]
- 9. Schepers AG, Snippert HJ, Stange DE, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730‐735. [DOI] [PubMed] [Google Scholar]
- 10. Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamashita T, Forgues M, Wang W, et al. EpCAM and alpha‐fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451‐1461. [DOI] [PubMed] [Google Scholar]
- 12. Ziskin JL, Dunlap D, Yaylaoglu M, et al. In situ validation of an intestinal stem cell signature in colorectal cancer. Gut. 2013;62:1012‐1023. [DOI] [PubMed] [Google Scholar]
- 13. Theise ND. Hepatic stem cells and cancers: a pathologist's view. Hepat Oncol. 2015;2:329‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia SB, Novelli M, Wright NA. The clonal origin and clonal evolution of epithelial tumours. Int J Exp Pathol. 2000;81:89‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Novelli M, Cossu A, Oukrif D, et al. X‐inactivation patch size in human female tissue confounds the assessment of tumor clonality. Proc Natl Acad Sci USA. 2003;100:3311‐3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Novelli MR, Williamson JA, Tomlinson IP, et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science. 1996;272:1187‐1190. [DOI] [PubMed] [Google Scholar]
- 17. Thirlwell C, Will OC, Domingo E, et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology. 2010;138:1441‐1454. [DOI] [PubMed] [Google Scholar]
- 18. Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. 2013;15:126‐134. [DOI] [PubMed] [Google Scholar]
- 19. Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. BioEssays. 2013;35:253‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das RN, Yaniv K. Discovering new progenitor cell populations through lineage tracing and in vivo imaging. Cold Spring Harbor Perspect Biol. 2020. pii: a035618. 10.1101/cshperspect.a035618. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fellous TG, McDonald SA, Burkert J, et al. A methodological approach to tracing cell lineage in human epithelial tissues. Stem Cells. 2009;27:1410‐1420. [DOI] [PubMed] [Google Scholar]
- 22. Walther V, Alison MR. Cell lineage tracing in human epithelial tissues using mitochondrial DNA mutations as clonal markers. Wiley Interdiscip Rev Dev Biol. 2016;5:103‐117. [DOI] [PubMed] [Google Scholar]
- 23. Gutierrez‐Gonzalez L, Deheragoda M, Elia G, et al. Analysis of the clonal architecture of the human small intestinal epithelium establishes a common stem cell for all lineages and reveals a mechanism for the fixation and spread of mutations. J Pathol. 2009;217:489‐496. [DOI] [PubMed] [Google Scholar]
- 24. Taylor RW, Barron MJ, Borthwick GM, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112:1351‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Snippert HJ, van der Flier LG, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134‐144. [DOI] [PubMed] [Google Scholar]
- 26. Lopez‐Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822‐825. [DOI] [PubMed] [Google Scholar]
- 27. Greaves LC, Preston SL, Tadrous PJ, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci USA. 2006;103:714‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McDonald SA, Greaves LC, Gutierrez‐Gonzalez L, et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134:500‐510. [DOI] [PubMed] [Google Scholar]
- 29. Gutierrez‐Gonzalez L, Graham TA, Rodriguez‐Justo M, et al. The clonal origins of dysplasia from intestinal metaplasia in the human stomach. Gastroenterology. 2011;140:1251‐1260. [DOI] [PubMed] [Google Scholar]
- 30. Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967;58:1468‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J, Kao YR, Sun D. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. 2019;25:103‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xie W, Chen ZN, Wang SA, et al. Lymphoblastic leukemia following myelodysplastic syndromes or myelodysplastic/myeloproliferative neoplasms. Leuk Lymphoma. 2019;60:2993‐3001. [DOI] [PubMed] [Google Scholar]
- 34. Gurbuxani S. Lymphoblastic leukemia following MDS or MDS/MPN: clinical evidence for stem cell origins of these disorders. Leuk Lymphoma. 2019;60:2847‐2848. [DOI] [PubMed] [Google Scholar]
- 35. Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ‐TIF2, but not BCR‐ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587‐596. [DOI] [PubMed] [Google Scholar]
- 36. Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL‐AF9. Nature. 2006;442:818‐822. [DOI] [PubMed] [Google Scholar]
- 37. Ye M, Zhang H, Yang H, et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell. 2015;17:611‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. George J, Uyar A, Young K, et al. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat Commun. 2016;7:12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chopra M, Bohlander SK. The cell of origin and the leukemia stem cell in acute myeloid leukemia. Genes Chromosomes Cancer. 2019;58:850‐858. [DOI] [PubMed] [Google Scholar]
- 40. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. 2014;6(238):238ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dick JE. Tumor archaeology: tracking leukemic evolution to its origins. Sci Transl Med. 2014;6(238):238fs23. [DOI] [PubMed] [Google Scholar]
- 42. Alpar D, Wren D, Ermini L, et al. Clonal origins of ETV6‐RUNX1⁺ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015;29:839‐846. [DOI] [PubMed] [Google Scholar]
- 43. Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6‐22. [PubMed] [Google Scholar]
- 44. Alison MR. The many ways to mend your liver: a critical appraisal. Int J Exp Pathol. 2018;99:106‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alison MR, Lin WR. Regenerating the liver: not so simple after all? F1000Res. 2016;5 pii: F1000 Faculty Rev‐1818 10.12688/f1000research.8827.1. eCollection 2016. [DOI] [Google Scholar]
- 46. Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol. 2009;217:282‐298. [DOI] [PubMed] [Google Scholar]
- 47. Alison MR. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev. 2005;1:253‐260. [DOI] [PubMed] [Google Scholar]
- 48. Lu W, Bird TG, Boulter L, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Bio. 2015;17:971‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raven A, Lu WY, Man TY, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547:350‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Komuta M, Spee B, Vander Borght S, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47:1544‐1556. [DOI] [PubMed] [Google Scholar]
- 51. Brunt E, Aishima S, Clavien PA, et al. cHCC‐CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentiation. Hepatology. 2018;68:113‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Durnez A, Verslype C, Nevens F, et al. . The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology. 2006;49:138‐151. [DOI] [PubMed] [Google Scholar]
- 53. Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410‐416. [DOI] [PubMed] [Google Scholar]
- 54. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer. 2015;15:653‐667. [DOI] [PubMed] [Google Scholar]
- 55. Matsumoto T, Takai A, Eso Y, et al. Proliferating EpCAM‐positive ductal cells in the inflamed liver give rise to hepatocellular carcinoma. Cancer Res. 2017;77:6131‐6143. [DOI] [PubMed] [Google Scholar]
- 56. Li XF, Chen C, Xiang DM, et al. Chronic inflammation‐elicited liver progenitor cell conversion to liver cancer stem cell with clinical significance. Hepatology. 2017;66:1934‐1951. [DOI] [PubMed] [Google Scholar]
- 57. Guest RV, Boulter L, Kendall TJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 2014;74:1005‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Michalopoulos GK. The regenerative altruism of hepatocytes and cholangiocytes. Cell Stem Cell. 2018;23:11‐12. [DOI] [PubMed] [Google Scholar]
- 59. Fan B, Malato Y, Calvisi DF, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911‐2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch‐mediated conversion of hepatocytes. J Clin Invest. 2012;122:3914‐3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alison MR, Lin WR. Liver cancer: intrahepatic cholangiocarcinoma–appearances can be deceiving. Nat Rev Gastroenterol Hepatol. 2013;10:131‐133. [DOI] [PubMed] [Google Scholar]
- 62. Seehawer M, Heinzmann F, D'Artista L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562:69‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pikarsky E. Neighbourhood deaths cause a switch in cancer subtype. Nature. 2018;562:45‐46. [DOI] [PubMed] [Google Scholar]
- 64. Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature. 2009;457:608‐611. [DOI] [PubMed] [Google Scholar]
- 65. Sánchez‐Danés A, Hannezo E, Larsimont JC, et al. Defining the clonal dynamics leading to mouse skin tumour initiation. Nature. 2016;536:298‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barbera M, di Pietro M, Walker E, et al. The human squamous oesophagus has widespread capacity for clonal expansion from cells at diverse stages of differentiation. Gut. 2015;64:11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pan Q, Nicholson AM, Barr H, et al. Identification of lineage‐uncommitted, long‐lived, label‐retaining cells in healthy human esophagus and stomach, and in metaplastic esophagus. Gastroenterology. 2013;144:761‐770. [DOI] [PubMed] [Google Scholar]
- 68. Liu K, Jiang M, Lu Y, et al. Sox2 cooperates with inflammation mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12:304‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Agrawal N, Jiao Y, Bettegowda C, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sánchez‐Danés A, Blanpain C. Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer. 2018;18:549‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Alcolea MP, Greulich P, Wabik A, Frede J, Simons BD, Jones PH. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat Cell Biol. 2014;16:615‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Frede J, Greulich P, Nagy T, Simons BD, Jones PH. A single dividing cell population with imbalanced fate drives oesophageal tumour growth. Nat Cell Biol. 2016;18:967‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Giroux V, Rustgi AK. Metaplasia: tissue injury adaptation and a precursor to the dysplasia‐cancer sequence. Nat Rev Cancer. 2017;17:594‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jiang M, Li H, Zhang Y, et al. Transitional basal cells at the squamous‐columnar junction generate Barrett's oesophagus. Nature. 2017;550:529‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhuang L, Fitzgerald RC. Cancer development: origins in the oesophagus. Nature. 2017;550:463‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shields HM, Zwas F, Antonioli DA, Doos WG, Kim S, Spechler SJ. Detection by scanning electron microscopy of a distinctive esophageal surface cell at the junction of squamous and Barrett's epithelium. Dig Dis Sci. 1993;38:97‐108. [DOI] [PubMed] [Google Scholar]
- 77. Leedham SJ, Preston SL, McDonald SA, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett's oesophagus. Gut. 2008;57:1041‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nicholson AM, Graham TA, Simpson A, et al. Barrett's metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut. 2012;61:1380‐1389. [DOI] [PubMed] [Google Scholar]
- 79. Lavery DL, Nicholson AM, Poulsom R, et al. The stem cell organisation, and the proliferative and gene expression profile of Barrett's epithelium, replicates pyloric‐type gastric glands. Gut. 2014;63:1854‐1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McDonald SA, Lavery D, Wright NA, Jansen M. Barrett oesophagus: lessons on its origins from the lesion itself. Nat Rev Gastroenterol Hepatol. 2015;12:50‐60. [DOI] [PubMed] [Google Scholar]
- 81. Quante M, Bhagat G, Abrams J, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett’s‐like metaplasia. Cancer Cell. 2012;21:36‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Spechler SJ, Merchant JL, Wang TC, et al. A summary of the 2016 James W. Freston Conference of the American Gastroenterological Association: intestinal metaplasia in the esophagus and stomach: origins, differences, similarities and significance. Gastroenterology. 2017;153:e6‐e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Que J, Garman KS, Souza RF, Spechler SJ. Pathogenesis and cells of origin of Barrett's esophagus. Gastroenterology. 2019;157:349‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hayakawa Y, Sethi N, Sepulveda AR, Bass AJ, Wang TC. Oesophageal adenocarcinoma and gastric cancer: should we mind the gap? Nat Rev Cancer. 2016;16:305‐318. [DOI] [PubMed] [Google Scholar]
- 85. Quante M, Abrams JA, Wang TC. The rapid rise in gastroesophageal junction tumors: is inflammation of the gastric cardia the underwater iceberg? Gastroenterology. 2013;145:708‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hata M, Kinoshita H, Hayakawa Y, et al. GPR30‐expressing gastric chief cells do not dedifferentiate but are eliminated via PDK‐dependent cell competition during development of metaplasia. Gastroenterology. 2020;158(6):1650‐1666.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Goldenring JR, Nam KT, Wang TC, Mills JC, Wright NA. SPEM and IM: Time for re‐evaluation of metaplasias and the origins of gastric cancer. Gastroenterology. 2010;138:2207‐2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Choi E, Hendley AM, Bailey JM, Leach SD, Goldenring JR. Expression of activated Ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology. 2016;150:918‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hayakawa Y, Ariyama H, Stancikova J, et al. Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell. 2015;28:800‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sakitani K, Hayakawa Y, Deng H, et al. CXCR4‐expressing Mist1 + progenitors in the gastric antrum contribute to gastric cancer development. Oncotarget. 2017;8:111012‐111025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Barker N, Huch M, Kujala P, et al. Lgr5(+ve) stem cells drive self‐renewal in the stomach and build long‐lived gastric units in vitro. Cell Stem Cell. 2010;6:25‐36. [DOI] [PubMed] [Google Scholar]
- 92. Tan SH, Swathi Y, Tan S, et al. AQP5 enriches for stem cells and cancer origins in the distal stomach. Nature. 2020;578:437‐443. [DOI] [PubMed] [Google Scholar]
- 93. Leushacke M, Tan SH, Wong A, et al. Lgr5‐expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol. 2017;19:774‐786. [DOI] [PubMed] [Google Scholar]
- 94. Uehara T, Ma D, Yao Y, et al. pylori infection is associated with DNA damage of Lgr5‐positive epithelial stem cells in the stomach of patients with gastric cancer. Dig Dis Sci. 2013;58:140‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Han S, Fink J, Jörg DJ, et al. Defining the identity and dynamics of adult gastric isthmus stem cells. Cell Stem Cell. 2019;25:342‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Phesse TJ, Sansom OJ. Lgr5 joins the club of gastric stem cell markers in the corpus. Nat Cell Biol. 2017;19:752‐754. [DOI] [PubMed] [Google Scholar]
- 97. McCarthy N, Manieri E, Storm EE, et al. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell. 2020;26:391‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Davis H, Irshad S, Bansal M, et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med. 2015;21:62‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Takashima S, Martin ML, Jansen SA, et al. T cell‐derived interferon‐γ programs stem cell death in immune‐mediated intestinal damage. Sci Immunol. 2019;4(42):eaay8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003‐1007. [DOI] [PubMed] [Google Scholar]
- 101. Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 2002;115:2381‐2388. [DOI] [PubMed] [Google Scholar]
- 103. Tian H, Biehs B, Warming S, et al. A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature. 2011;478:255‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Buczacki SJ, Zecchini HI, Nicholson AM, et al. Intestinal label‐retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65‐69. [DOI] [PubMed] [Google Scholar]
- 105. Demidov ON, Timofeev O, Lwin HN, Kek C, Appella E, Bulavin DV. Wip1 phosphatase regulates p53‐dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell Stem Cell. 2007;1:180‐190. [DOI] [PubMed] [Google Scholar]
- 106. Murata K, Jadhav U, Madha S, et al. Ascl2‐dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell. 2020;26:377‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shih IM, Wang TL, Traverso G, et al. Top‐down morphogenesis of colorectal tumors. Proc Natl Acad Sci USA. 2001;98:2640‐2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Preston SL, Wong WM, Chan AO, et al. Bottom‐up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003;63:3819‐3825. [PubMed] [Google Scholar]
- 109. Hayakawa Y, Tsuboi M, Asfaha S, et al. BHLHA15‐positive secretory precursor cells can give rise to tumors in intestine and colon in mice. Gastroenterology. 2019;156:1066‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Giroux V, Stephan J, Chatterji P, et al. Mouse intestinal Krt15+ crypt cells are radio‐resistant and tumor initiating. Stem Cell Reports. 2018;10:1947‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Westphalen CB, Asfaha S, Hayakawa Y, et al. Long‐lived intestinal tuft cells serve as colon cancer‐initiating cells. J Clin Invest. 2014;124:1283‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15:62‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Huels DJ, Bruens L, Hodder MC, et al. Wnt ligands influence tumour initiation by controlling the number of intestinal stem cells. Nat Commun. 2018;9:1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. 2018;18:19‐32. [DOI] [PubMed] [Google Scholar]
- 115. Galandiuk S, Rodriguez‐Justo M, Jeffery R, et al. Field cancerization in the intestinal epithelium of patients with Crohn's ileocolitis. Gastroenterology. 2012;142:855‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Leedham SJ, Graham TA, Oukrif D, et al. Clonality, founder mutations, and field cancerization in human ulcerative colitis‐associated neoplasia. Gastroenterology. 2009;136:542‐550. [DOI] [PubMed] [Google Scholar]
- 117. Craddock VM, Frei JV. Induction of liver cell adenomata in the rat by a single treatment with N‐methyl‐N‐nitrosourea given at various times after partial hepatectomy. Br J Cancer. 1974;30:503‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nault JC, Zucman‐Rossi J. TERT promoter mutations in primary liver tumors. Clin Res Hepatol Gastroenterol. 2016;40:9‐14. [DOI] [PubMed] [Google Scholar]
- 119. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152:745‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Shin S, Wangensteen KJ, Teta‐Bissett M, et al. Genetic lineage tracing analysis of the cell of origin of hepatotoxin‐induced liver tumors in mice. Hepatology. 2016;64:1163‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Marquardt JU. Deconvolution of the cellular origin in hepatocellular carcinoma: Hepatocytes take the center stage. Hepatology. 2016;64:1020‐1023. [DOI] [PubMed] [Google Scholar]
- 122. Mu X, Español‐Suñer R, Mederacke I, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125:3891‐3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Jörs S, Jeliazkova P, Ringelhan M, et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J Clin Invest. 2015;125:2445‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang C, Yang W, Yan HX, et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine‐treated HBx transgenic mice. Hepatology. 2012;55:108‐120. [DOI] [PubMed] [Google Scholar]
- 125. Müller M, Bird TG, Nault JC. The landscape of gene mutations in cirrhosis and hepatocellular carcinoma. J Hepatol. 2020;72:990‐1002. [DOI] [PubMed] [Google Scholar]
- 126. Lin W, Lim S, McDonald SAC, et al. The histogenesis of regenerative nodules in human liver cirrhosis. Hepatology. 2010;51:1017‐1026. [DOI] [PubMed] [Google Scholar]
- 127. Hill MA, Alexander WB, Guo B, et al. Kras and Tp53 mutations cause cholangiocyte‐ and hepatocyte‐derived cholangiocarcinoma. Cancer Res. 2018;78:4445‐4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ying H, Dey P, Yao W, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016;30:355‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Matthaei H, Semaan A, Hruban RH. The genetic classification of pancreatic neoplasia. J Gastroenterol. 2015;50:520‐532. [DOI] [PubMed] [Google Scholar]
- 130. Yamaguchi J, Yokoyama Y, Kokuryo T, Ebata T, Nagino M. Cells of origin of pancreatic neoplasms. Surg Today. 2018;48:9‐17. [DOI] [PubMed] [Google Scholar]
- 131. Makohon‐Moore A, Iacobuzio‐Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Houbracken I, de Waele E, Lardon J, et al. Lineage tracing evidence for transdifferentiation of acinar to duct cells and plasticity of human pancreas. Gastroenterology. 2011;141:731‐741. [DOI] [PubMed] [Google Scholar]
- 133. Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci USA. 2008;105:18913‐18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K‐Ras oncogenes in adult mice. Cancer Cell. 2007;11:291‐302. [DOI] [PubMed] [Google Scholar]
- 135. Strobel O, Dor Y, Stirman A, et al. Beta cell transdifferentiation does not contribute to preneoplastic/metaplastic ductal lesions of the pancreas by genetic lineage tracing in vivo. Proc Natl Acad Sci USA. 2007;104:4419‐4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ferreira RMM, Sancho R, Messal HA, et al. Duct‐ and acinar‐derived pancreatic ductal adenocarcinomas show distinct tumor progression and marker expression. Cell Rep. 2017;21:966‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tian SB, Tao KX, Hu J, et al. The prognostic value of AGR2 expression in solid tumours: a systematic review and meta‐analysis. Sci Rep. 2017;7:15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lee AYL, Dubois CL, Sarai K, et al. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut. 2018;68:487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fischer CG, Wood LD. From somatic mutation to early detection: insights from molecular characterization of pancreatic cancer precursor lesions. J Pathol. 2018;246:395‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kuboki Y, Fischer CG, Beleva Guthrie V, et al. Single‐cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J Pathol. 2019;247:347‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fischer CG, Beleva Guthrie V, Braxton AM, et al. Intraductal papillary mucinous neoplasms arise from multiple independent clones, each with distinct mutations. Gastroenterology. 2019;157:1123‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jacob HKC, Banerjee S. Intraductal papillary mucinous neoplasms: attack of the clones. Gastroenterology. 2019;157:929‐932. [DOI] [PubMed] [Google Scholar]
- 143. Li WC, Rukstalis JM, Nishimura W, et al. Activation of pancreatic‐duct‐derived progenitor cells during pancreas regeneration in adult rats. J Cell Sci. 2010;123:2792‐2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Elias KM, Tsantoulis P, Tille JC, et al. Primordial germ cells as a potential shared cell of origin for mucinous cystic neoplasms of the pancreas and mucinous ovarian tumors. J Pathol. 2018;246:459‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yamaguchi J, Liss AS, Sontheimer A, et al. Pancreatic duct glands (PDGs) are a progenitor compartment responsible for pancreatic ductal epithelial repair. Stem Cell Res. 2015;15:190‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yamaguchi J, Mino‐Kenudson M, Liss AS, et al. Loss of trefoil factor 2 from pancreatic duct glands promotes formation of intraductal papillary mucinous neoplasms in mice. Gastroenterology. 2016;151(6):1232‐1244.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang VM, Ferreira RMM, Almagro J, et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat Cell Biol. 2019;21:1425‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014;146:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Delgiorno KE, Hall JC, Takeuchi KK, et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology. 2014;146(1):233‐244.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kopp JL, Sander M. New insights into the cell lineage of pancreatic ductal adenocarcinoma: evidence for tumor stem cells in premalignant lesions? Gastroenterology. 2014;146:24‐26. [DOI] [PubMed] [Google Scholar]
- 151. Sottoriva A, Kang H, Ma Z, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47:209‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Venkatesan S, Swanton C. Tumor evolutionary principles: How intratumor heterogeneity influences cancer treatment and outcome. Am Soc Clin Oncol Educ Book. 2016;35:e141‐149. [DOI] [PubMed] [Google Scholar]
- 153. Cross WCH, Graham TA, Wright NA. New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol. 2016;240:126‐136. [DOI] [PubMed] [Google Scholar]
- 154. McGranahan N, Swanton C. clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. [DOI] [PubMed] [Google Scholar]
- 155. Cross W, Mossner M, Nowinski S, et al. Stabilising selection causes grossly altered but stable karyotypes in metastatic colorectal cancer. bioRxiv preprint. 2020; 10.1101/2020.03.26.007138 [DOI] [Google Scholar]