

Abstract
Natural selection on gene expression was originally predicted to result primarily in cis- rather than trans-regulatory evolution, due to the expectation of reduced pleiotropy. Despite this, numerous studies have ascribed recent evolutionary divergence in gene expression predominantly to trans-regulation. Performing RNA-seq on single isofemale lines from genetically distinct populations of the cactophilic fly Drosophila mojavensis and their F1 hybrids, we recapitulated this pattern in both larval brains and whole bodies. However, we demonstrate that improving the measurement of brain expression divergence between populations by using seven additional genotypes considerably reduces the estimate of trans-regulatory contributions to expression evolution. We argue that the finding of trans-regulatory predominance can result from biases due to environmental variation in expression or other sources of noise, and that cis-regulation is likely a greater contributor to transcriptional evolution across D. mojavensis populations. Lastly, we merge these lines of data to identify several previously hypothesized and intriguing novel candidate genes, and suggest that the integration of regulatory and population-level transcriptomic data can provide useful filters for the identification of potentially adaptive genes.
Keywords: cactophilic, local adaptation, pleiotropy, RNA-seq, transcriptional regulation
Significance
Gene expression evolution can be driven by changes in the focal gene itself (cis-regulation) or by changes in the genes that regulate it (trans-regulation). The importance of these two processes, and their contribution to adaptation specifically, remains under debate. Through a novel integration of data on genetic variation in gene expression with data on gene regulatory evolution, we found increased evidence for a primary role of cis-regulation in both total expression evolution as well as adaptive expression evolution. These results inject nuance into the discussion of how regulatory processes influence evolution within species and outline an approach for using expression data to address adaptive hypotheses.
Introduction
Statistical correlations between phenotypes impose fundamental constraints on phenotypic evolution (Lande 1979). As such, selection may disfavor the propagation of especially pleiotropic mutations whose causal effects alter many traits (Otto 2004). This idea has led to considerable speculation on the precise molecular effects of successful mutations. Vigorous debate regarding the relative importance of coding sequence and gene regulatory evolution hinged on claims regarding the respective pleiotropic consequences of these types (Hoekstra and Coyne 2007; Carroll 2008). Within the category of regulatory mutations, however, further distinctions are likely to be relevant in this context. Specifically, trans-regulatory changes, which are primarily a consequence of changes in expression and/or structure of transcription factors, are expected to affect large networks of target genes and therefore be highly pleiotropic (Gibson 1996; Wittkopp 2007; but see Lynch and Wagner [2008]). In contrast, cis-regulatory mutations, occurring in promoters or enhancers of the target genes themselves, might affect only single genes in specific contexts (Stern 2000; Prud’homme et al. 2007). In an early and thorough theoretical treatment of the subject, Wray et al. (2003) did not equivocate in hypothesizing that cis-regulatory evolution should primarily be responsible for the evolution of gene expression phenotypes.
In the years since that prediction, the accumulation of evidence regarding the prevalence of cis- and trans-regulatory effects in evolution has led to a far murkier picture. This may in part reflect the methodological diversity of studies approaching the question (reviewed in Signor and Nuzhdin [2018]). Some experiments, such as chromosomal substitutions (Hughes et al. 2006; Osada et al. 2006), crosses utilizing the diversity of a reference panel (Genissel et al. 2007; Fear et al. 2016; Osada et al. 2017), and eQTL mapping studies (Massouras et al. 2012; King et al. 2014) have generally, but not always (Lemos et al. 2008; Wang et al. 2008) corroborated the hypothesis, finding greater contributions of cis-effects to intrapopulation variation. On the other hand, results from another frequently used experimental design, which we will henceforth call the F1 hybrid design, have consistently led to the opposite conclusion. The F1 hybrid design requires expression data from two parental lines and their F1 hybrids. Cis-regulatory effects are measured using the differential expression of allele-specific reads within the hybrid samples, whereas trans-regulatory effects are calculated by subtracting the cis-regulatory effect from the overall differential expression between the parental lines (Wittkopp et al. 2004). Usage of the F1 hybrid design has repeatedly found that trans-regulation dominates expression variability within species, whereas cis-regulation plays a greater role in interspecific differences (Graze et al. 2009; Wittkopp et al. 2004, 2008; McManus et al. 2010; Suvorov et al. 2013; Coolon et al. 2014; Metzger et al. 2017; Glaser-Schmitt et al. 2018).
Given the power of the F1 hybrid design and its applicability to a wide range of study systems and biological contexts, closer attention to the interpretations stemming from this approach is merited. As such, recent work has begun to approach the F1 hybrid paradigm with increased nuance. Glaser-Schmitt et al. (2018) perform a tissue-specific study, filling an important gap given the focus of previous work on whole-body samples. Taking this one step further, Combs and Fraser (2018) estimate fine-scale spatial variation in allele-specific expression within embryos. From a different angle, two recent commentaries (Fraser 2019; Zhang and Emerson 2019) make salient points regarding potential biases in the estimation of trans-regulatory divergence given that it cannot be estimated independently of cis-regulatory and parental divergence using this approach, and stress the need for replication to mitigate this. Here, we build from these efforts and probe the initial findings from an across-population F1 hybrid study using two simple experiments. First, we conduct a tissue-specific study in parallel with a whole-body study, to directly estimate the effects of sample heterogeneity on the estimation of regulatory type. Second, we supplement our measures of parental divergence with further sampling of genotypes from each parental population, to gain more confidence in patterns of within and between-population variation in transcription.
We apply these experiments to an investigation of gene expression evolution in larval brains across two populations of the cactophilic fly Drosophila mojavensis. This combination of organism and tissue lends itself to a strong hypothesis of predominant cis-regulatory evolution, for two reasons. First, D. mojavensis is predicted to have experienced strong differential selection pressures across populations due to variable ecological conditions. The two populations studied here, from Santa Catalina Island, CA, and the Sonoran Desert (Guaymas, Sonora, Mexico and Organ Pipe National Monument, Arizona), are genetically distinct (Reed et al. 2006) primarily utilize highly divergent cactus species, the prickly pear Opuntia littoralis and the columnar Stenocereus thurberi, respectively (Heed 1978; Ruiz et al. 1990). These host cacti form unique chemical and nutritional environments (Kircher 1982; Starmer and Phaff 1983), and detoxification genes in particular have seen substantial expression and coding sequence evolution across these populations (Matzkin et al. 2006; Allan and Matzkin 2019). In addition to selection from the host, these populations experience vastly different temperature and humidity regimes, which is expected to generate selection broadly on phenology and organismal physiology (Matzkin 2014). We choose to focus on brains here in part because we previously identified larval behavioral differences related to locomotion and pupation (Coleman et al. 2018), indicating the potential for the evolution of expression changes in the brain, as well as muscle and fat body. Second, despite this potential for selection, there are also a priori expectations that transcriptome-wide evolution should actually be reduced. Brain gene expression is highly conserved in many animals, including Drosophila (Brawand et al. 2011; Catalán et al. 2012; Uebbing et al. 2016). Additionally, gene expression in larvae is more conserved than in later developmental stages in Drosophila (Artieri and Singh 2010). The pairing of strong directional and strong stabilizing selection across genes is precisely the scenario that should result in transcriptional fine-tuning due to cis-regulatory evolution. Thus, our expectation was to uncover a greater role for cis-regulatory changes than observed in other intraspecific studies using similar experimental designs.
Materials and Methods
Sample Collection and Sequencing
For initial analysis of population divergence and analyses of allele-specific expression, we used single genome-sequenced isofemale lines of D. mojavensis from Santa Catalina Island, CA (Drosophila 12 Genomes Consortium 2007) and Guaymas, Sonora, Mexico (Allan and Matzkin 2019). These lines have been maintained as isofemale lines without direct inbreeding in the laboratory on banana-molasses media (Coleman et al. 2018) since 2002 and 1999, respectively. We generated F1 hybrids between these two lines by placing 20 virgin genome-line Catalina Island males and 20 virgin genome-line Sonora females in vials containing banana-molasses media, and performed the reciprocal cross in an identical manner. For analyses of genotypic variation in expression, we selected seven additional isofemale lines from Santa Catalina Island and seven isofemale lines from the Sonoran Desert population from Organ Pipe National Monument, AZ, which were collected between 2007 and 2009 (Coleman et al. 2018) and maintained as described earlier.
We collected all samples during the third-instar wandering stage. For whole-body samples, we collected five larvae per replicate, washing each larva in deionized water before storing them on ice in tris–EDTA buffer. For brain samples, we dissected ten brains per replicate in tris–EDTA before storing them on ice. We then froze samples at −80 °C for storage. We collected three biological replicates for each genome line and hybrid (brain and body) and single replicates of each additional isofemale line (brain only). We ground samples in TRIzol (Thermo Fisher, Waltham, MA) and used Qiagen RNEasy columns (Qiagen, Hilden, Germany) to extract RNA, prepared libraries using Illumina TruSeq kits (Illumina, San Diego, CA), and sequenced samples as 150-bp paired-end reads on an Illumina HiSeq. Information on sample identity and sequencing can be found in supplementary table S1, Supplementary Material online.
Bioinformatic Analysis
We removed Illumina adapters and low-quality sequence using Trimmomatic (Bolger et al. 2014) and used NextGenMap (Sedlazeck et al. 2013) with default parameters to separately map all reads to both the original Catalina Island genome (Drosophila 12 Genomes Consortium 2007; FlyBase version r1.04_FB2018_06) and the same genome templated with Sonora genomic reads (Allan and Matzkin 2019). We calculated total read counts at the gene level for each sample using HTSeq-count (Anders et al. 2015), using the reads mapped to the Catalina Island genome for analysis. We then downsampled reads to 11,908,854 reads over 13,410 genes in brain samples and 14,001,634 reads over 13,628 genes in whole-body samples, to match the lowest coverage sample in each tissue. For the additional brain isofemale lines, which were more highly covered, we downsampled to 18,665,415 reads over 13,565 genes. From these gene sets, we analyzed only genes with at least ten total reads in each sample. After comparing the consistency between biological replicates within each group using Spearman’s correlation coefficients, we discarded three samples from the genome lines as outliers: one Sonora brain sample, one Sonora (f)×Catalina Island (m) hybrid body sample, and one Catalina Island (f)×Sonora (m) body sample. In the analysis of genotypic variation in the brain, we discarded an additional Sonora sample as an outlier based on the same criteria. We included only a single randomly chosen replicate from each genome-sequenced line in the analysis of genotypic variation to avoid pseudoreplication.
For allele-specific counts, we used SAMtools mpileup (Li et al. 2009) and VarScan2 (Koboldt et al. 2013) to identify informative variants for allele-specific expression analysis. We first removed all SNPs where we found, in any of the parental genotype samples the allele from the other parental genome at >5% frequency (Glaser-Schmitt et al. 2018). This step helps to avoid analyzing heterozygous sites, which will lead to inaccuracy in the assignment of reads to parental genomes. We then compared the remaining SNPs from the mapping results to both reference genomes and removed sites with substantially differing allele frequencies in the two resulting data sets, following previous work (Benowitz et al. 2019). In this way, we removed sites potentially affected by mapping bias, which, although not a major problem here (supplementary table S1, Supplementary Material online), would result in overestimation of allele-specific expression of genes containing those sites. We then filtered all bam files (mapped to the Catalina Island reference) for informative reads using VariantBam (Wala et al. 2016) and output these reads as text files using sam2tsv (https://lindenb.github.io/jvarkit/Sam2Tsv.html; last accessed July 2020). We then counted allele-specific reads overlapping each informative SNP, generating gene-level counts after accounting for reads overlapping multiple variants in R 3.4 (R Core Team 2018). We ran this pipeline independently for brain and body samples. We randomly downsampled allele-specific reads in each brain sample to 4,541,638 reads, matching the reads in the lowest coverage sample. These reads covered SNPs in 7,933 genes. For the whole-body samples, which covered 8,584 genes, we downsampled all read counts to 4,939,804 total reads, to preserve the ratio of reads per gene between brains and whole bodies. In both data sets, we analyzed only genes containing at least ten total reads in each sample. The same three samples identified as outliers above were also outliers in this data set and we accordingly discarded them for allele-specific expression analysis as well.
Statistical Analysis
We calculated per-gene parental divergence from the total (not allele-specific) expression counts as log2(PCI/PSON), where PCI and PSON are either parental genome-sequenced line means (when comparing single genotypes; in brains and whole bodies) or parental population means (when comparing all genotypes; brains only). We calculated transcriptome-wide differentiation across populations as 1−ρ, where ρ is the Spearman’s correlation of expression divergences between populations. We estimated 95% confidence intervals of 1−ρ from 10,000 bootstrapped replicates.
We calculated cis-regulatory divergence, following previous studies, as log2(HCI/HSON), where HCI and HSON are averages of allele-specific counts across all F1 hybrid replicates. We then calculated trans-regulatory divergence as the difference between parental divergence (between the genome-sequenced lines) and cis-regulatory divergence, log2(PCI/PSON)−log2(HCI/HSON). We independently assessed the contributions of cis- and trans-regulatory divergence to both metrics of parental divergence using Spearman’s correlation coefficient ρ. As above, we assessed 95% confidence intervals from 10,000 bootstrap replicates. We visualized correlations using least-squares regression lines and 95% confidence regions around those regressions using the R package ggplot2.
We estimated differential expression between parental populations using FDR-corrected negative binomial tests using the R package NBPseq (Di et al. 2011). We also used negative binomial tests comparing allele-specific counts to assess the significance of cis-regulatory effects. To estimate the significance of trans-regulatory effects at the gene level, we used Fisher’s exact tests comparing the ratio of allele-specific expression differences to total gene expression differences in the parental samples.
For evolutionary analysis of regulatory evolution in the brain, we used previously published dN/dS values across the four D. mojavensis populations (Allan and Matzkin 2019). To estimate network connectivity, we identified the closest Drosophila melanogaster ortholog for each gene and used the in-degree metric calculated in Marbach et al. (2012) and used previously in a similar analysis in Yang and Wittkopp (2017). Briefly, in-degree quantifies the number of transcription factors found to have significant regulatory interactions with each gene. We analyzed dN/dS and in-degree between genes in different regulatory categories using Mann–Whitney U test with the R function pairwise.wilcox.test, using Holm’s method to correct for multiple comparisons. For these analyses, we defined trans-regulated genes via the brain analysis using multiple parental genotypes. We performed all statistical analyses in R 3.4 (R Core Team 2018).
Results
Analysis of Single Parental Genotypes in Brains and Whole Bodies
Examining a single genotype per population, transcriptome-wide expression differentiation across populations was lower in brains (1−ρ = 0.017; 95% CI = [0.016, 0.018]) than in bodies (1−ρ = 0.036; 95% CI = [0.034, 0.038]), as expected. In contrast, we found evidence for considerably more significantly differentially expressed (DE) genes in brains than in bodies (table 1 and supplementary table S2, Supplementary Material online). The lack of statistical support for many DE genes in bodies despite increased overall expression differences reflects substantially greater intragenotypic variation in whole-body data (supplementary fig. S1, Supplementary Material online).
Table 1.
Numbers of Differentially Expressed, Cis-Regulated, and Trans-Regulated Genes Using Both the Single-Genotype Parental Data Set and the Multigenotype Parental Data Set (brains only)
| Single Parental Genotype |
Multiple Parental Genotypes |
||
|---|---|---|---|
| Whole Bodies | Brains | Brains | |
| DE genes | 231 | 530 | 308 |
| Cis-regulated genes | 20 | 143 | – |
| Trans-regulated genes | 1072 | 467 | 265 |
Note.—The calculation of cis-regulated genes relies only on F1 hybrids, thus there is no recalculation of the number of cis-regulated genes in the multiparent genotype data set.
We then correlated measures of divergence across all genes with measures of cis- and trans-regulatory divergence as estimated from F1 hybrids between these two lines (Coolon et al. 2014; Metzger et al. 2017). This correlation broadly estimates the contributions of each regulatory type to total expression divergence without relying on thresholds of statistical significance. We found that trans-effects were more closely associated with parental divergence than cis-effects to parental divergence in both brains (trans: ρ = 0.577; 95% CI = [0.561, 0.591], cis: ρ = 0.455; 95% CI = [0.438, 0.472], fig. 1A) and whole bodies (trans: ρ = 0.643; 95% CI = [0.629, 0.657], cis: ρ = 0.338; 95% CI = [0.319, 0.356], fig. 1B). Lastly, we found more individual genes displaying evidence of regulation in trans than in cis in both brains and bodies, although this trend was much more dramatic in whole bodies (table 1 and supplementary tables S3 and S4, Supplementary Material online).
Fig. 1.

The relationship between cis- and trans-regulatory divergence and divergence between parental genotypes in (A) brains and (B) whole bodies. Bold points indicate significantly differentially expressed genes in each data set. Trend lines represent least-squares regressions surrounded by 95% confidence intervals.
Analysis of Multiple Parental Genotypes in Brains
The above comparison, using only single parental genotypes, provides a limited estimate of expression evolution across populations. To more confidently assess expression evolution across populations, we analyzed brain RNA-seq data from seven additional Catalina Island genotypes and six additional Sonoran genotypes. Specifically, we expected the inclusion of multiple genotypes to reduce sampling error and result in lower expression differentiation between populations. Indeed, parental divergence across populations was lower in this data set (1−ρ = 0.005; 95% CI = [0.004, 0.005]) and the number of significantly DE genes was reduced (table 1 and supplementary table S2, Supplementary Material online). We then examined correlations between parental divergence using multiple genotypes with the identical cis- and trans-regulatory divergence values calculated above. We now found the opposite result: cis-effects were more related to population divergence as measured by multiple genotypes than trans-effects (cis: ρ = 0.362; 95% CI = [0.343, 0.380], trans: ρ = 0.287; 95% CI = [0.268, 0.306], fig. 2). We also used the multigenotype data set to recalculate the number of trans-regulated genes, and found far fewer than in the single genotype analysis, although still considerably more than the number of cis-regulated genes (table 1 and supplementary tables S3 and S4, Supplementary Material online).
Fig. 2.
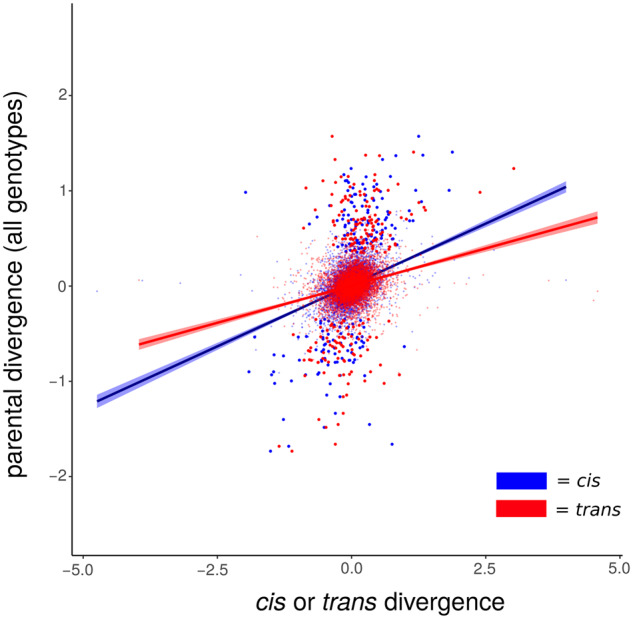
The relationship between cis- and trans-regulatory divergence and divergence between parental genotypes in brains, where parental divergence is measured using all genotypes. Cis- and trans-regulatory divergence data are the same as in figure 1. Bold points indicate significantly differentially expressed genes. Trend lines display least-squares regressions surrounded by 95% confidence intervals.
Evolutionary and Candidate Gene Analysis
We found that cis-regulated genes had higher evolutionary rates within D. mojavensis than did trans-regulated genes (P = 0.0010) or genes that were either conserved or lacking a clear regulatory pattern (P = 0.0018). We also found that the in-degree (number of transcriptional regulators), as inferred from D. melanogaster orthologs, of cis-regulated genes in our data set was lower than that of either trans-regulated genes (P = 0.0034) or those with no identified regulatory type (P = 1.1e−5). The distributions of dN/dS and in-degree values for genes in each regulatory classification are shown in figure 3. To examine potential evolutionary hypotheses on a more granular level, we also compiled a list of candidate genes displaying two criteria: differential expression between the two populations in the multiple genotype brain data set, and a statistically significant pattern of cis- and/or trans-regulatory evolution. About 68 genes met these criteria, of which 27 where cis-regulated, 35 were trans-regulated, and six had significant cis- and trans effects (table 2). Of these six, five showed evidence for compensatory (cis×trans) evolution, whereas only one showed evidence for combined (cis+trans) evolution.
Fig. 3.

Relationships between regulatory classification and gene-level metrics. (A) Comparison of evolutionary rate (dN/dS) among Drosophila mojavensis populations (from Allan and Matzkin [2019]). (B) Comparison of the number of transcriptional regulators (in degree) as inferred from D. melanogaster orthologs (Marbach et al. 2012). P values for significant pairwise comparisons are indicated, all other comparisons are nonsignificant.
Table 2.
The Genes Displaying Both Differential Expression across Populations in the Brain Multigenotype Data Set and Statistical Evidence for Cis- and/or Trans-Regulatory Evolution in the Brain
| Drosophila mojavensis FlyBase ID | Drosophila melanogaster Gene Name | P Value (Parental DE) | P Value (Regulatory Type) | Population with Higher Expression | Regulatory Classification |
|---|---|---|---|---|---|
| FBgn0140302 | Cyp28a5 | 6.75E-30 | 4.3E-10 | SON | cis |
| FBgn0147102 | GstD1 | 1.67E-28 | 8.25E-09 | CI | cis |
| FBgn0139804 | Ugt36Bc | 2.81E-22 | 5.14E-08 | SON | cis |
| FBgn0280601 | NA | 2.62E-19 | 0.00000471 | CI | cis |
| FBgn0143628 | NA | 1.22E-16 | 0.00159 | SON | cis |
| FBgn0147447 | Cyp9f2 | 7.89E-13 | 0.000000667 | CI | cis |
| FBgn0136497 | CG14567 | 2.41E-11 | 5.24E-10 | CI | cis |
| FBgn0133237 | RdhB | 1.82E-08 | 2.58E-08 | CI | cis |
| FBgn0133140 | CG5379 | 6.73E-08 | 0.000332 | SON | cis |
| FBgn0136870 | CG33521 | 0.000000535 | 0.00000103 | SON | cis |
| FBgn0136131 | CG2211 | 0.0000014 | 0.000000357 | CI | cis |
| FBgn0146788 | CG18547 | 0.0000025 | 0.0195 | SON | cis |
| FBgn0140494 | CG31777 | 0.0000444 | 0.0000392 | SON | cis |
| FBgn0139666 | CG5316 | 0.0000474 | 1.67E-09 | SON | cis |
| FBgn0136235 | CG33969 | 0.0000835 | 0.000765 | CI | cis |
| FBgn0142519 | Phr | 0.000162 | 0.00133 | SON | cis |
| FBgn0145297 | CG10550 | 0.000917 | 0.00377 | SON | cis |
| FBgn0140061 | NA | 0.00149 | 2.15E-11 | SON | cis |
| FBgn0147596 | CG34409 | 0.00373 | 0.0169 | CI | cis |
| FBgn0146770 | Cyp12e1 | 0.00447 | 0.000129 | CI | cis |
| FBgn0085888 | ST6Gal | 0.00738 | 0.009 | SON | cis |
| FBgn0142451 | CG9344 | 0.00885 | 0.0111 | CI | cis |
| FBgn0280848 | NA | 0.0199 | 0.0486 | SON | cis |
| FBgn0136999 | CG8086 | 0.0208 | 0.00551 | SON | cis |
| FBgn0132957 | CG10165 | 0.021 | 0.00602 | CI | cis |
| FBgn0141924 | CG3511 | 0.0244 | 0.0322 | CI | cis |
| FBgn0132961 | Acyp2 | 0.0434 | 0.0195 | CI | cis |
| FBgn0134299 | Fbp1 | 3.37E-26 | 1.72E-46 | CI | trans |
| FBgn0145801 | Lsp1beta | 4.18E-24 | 6.82E-35 | CI | trans |
| FBgn0143382 | Lsp2 | 7.92E-18 | 2.74E-22 | CI | trans |
| FBgn0142713 | Sans | 1.53E-16 | 0.00527 | SON | trans |
| FBgn0140799 | Lsp1gamma | 1.71E-16 | 2.23E-08 | CI | trans |
| FBgn0140406 | Fbp2 | 1.35E-12 | 2.53E-15 | CI | trans |
| FBgn0135991 | NA | 2.29E-12 | 0.0000427 | CI | trans |
| FBgn0135183 | CAH2 | 4.69E-12 | 0.000012 | CI | trans |
| FBgn0134843 | CG32037 | 2.93E-10 | 0.0223 | CI | trans |
| FBgn0139371 | GstE2 | 1.4E-09 | 0.00239 | CI | trans |
| FBgn0146682 | TyrRII | 2.96E-09 | 0.00385 | SON | trans |
| FBgn0139800 | Mhc | 3.12E-09 | 0.0346 | SON | trans |
| FBgn0139723 | Cg25C | 8.58E-09 | 0.00847 | CI | trans |
| FBgn0138516 | Sirup | 2.74E-08 | 0.0179 | SON | trans |
| FBgn0147600 | MtnA | 0.000000896 | 4.35E-39 | CI | trans |
| FBgn0142365 | CG7997 | 0.00000124 | 0.000746 | CI | trans |
| FBgn0141762 | CG3520 | 0.0000019 | 0.000176 | CI | trans |
| FBgn0142390 | Bru | 0.0000187 | 0.000000015 | SON | trans |
| FBgn0147345 | CG14291 | 0.0000735 | 0.0226 | SON | trans |
| FBgn0141634 | CG30460 | 0.000217 | 0.00153 | SON | trans |
| FBgn0146346 | NA | 0.000343 | 0.00903 | CI | trans |
| FBgn0142161 | CG13742 | 0.000549 | 0.000399 | CI | trans |
| FBgn0146625 | CG31278 | 0.00111 | 0.0197 | CI | trans |
| FBgn0138139 | Nocte | 0.00196 | 2.85E-33 | CI | trans |
| FBgn0132831 | CG6364 | 0.00233 | 0.00353 | CI | trans |
| FBgn0134066 | CG18081 | 0.00795 | 0.000801 | CI | trans |
| FBgn0146517 | Snap25 | 0.0111 | 0.0000444 | CI | trans |
| FBgn0140557 | CG17124 | 0.0118 | 0.00327 | CI | trans |
| FBgn0138450 | CG14785 | 0.0178 | 0.00243 | SON | trans |
| FBgn0132826 | CG6723 | 0.0179 | 0.0000984 | SON | trans |
| FBgn0143281 | RpL23 | 0.0267 | 0.00682 | CI | trans |
| Fbgn0145013 | CG34377 | 0.027 | 0.000183 | CI | trans |
| FBgn0144093 | NA | 0.0313 | 0.00176 | SON | trans |
| FBgn0145765 | Npc2b | 0.0363 | 0.000000643 | SON | trans |
| FBgn0145063 | Obp99b | 0.0397 | 0.0289 | CI | trans |
| FBgn0146132 | Obp99a | 1.67E-28 | cis: 4.74E-04 | CI | cis + trans |
| trans: 4.30E-06 | |||||
| FBgn0141410 | CG18067 | 2.95E-11 | cis: 7.01E-13 | SON | cis x trans |
| trans: 1.12E-04 | |||||
| FBgn0146495 | Spartin | 0.0000296 | cis: 7.89E-03 | CI | cis x trans |
| trans: 6.74E-11 | |||||
| FBgn0084818 | NA | 0.00145 | cis: 1.02E-05 | CI | cis x trans |
| trans: 7.85E-04 | |||||
| FBgn0147549 | NANS | 0.0039 | cis: 2.13E-03 | SON | cis x trans |
| trans: 1.03E-05 | |||||
| FBgn0141293 | CG15651 | 0.0235 | cis: 4.28E-16 | SON | cis x trans |
| trans: 1.11E-06 |
Discussion
The measurement of allele-specific expression in F1 hybrid offspring has been one of the primary approaches for understanding genome-wide patterns of cis- and trans-regulatory evolution both within and between species. Although other methodologies for quantifying these effects have been used effectively, the advantage of the F1 hybrid approach, in our opinion, is its simplicity and potential applicability to a wide range of study systems and evolutionary contexts. However, as with any other genome-scale approach to evolution, the conclusions stemming from F1 hybrid studies come with biases and limitations. Here, our goal was to investigate how two straightforward modifications to this common experimental design affect the evolutionary interpretations regarding the prevalence of cis- and trans-regulation in natural populations. Furthermore, we aimed to leverage the ecological and evolutionary information from our model system, D. mojavensis, to examine how successfully the integration of complex regulatory data can uncover adaptive gene expression changes across populations.
The Effects of Tissue Specificity on Estimation of Regulatory Type
Most of the original genome-wide studies of cis- and trans-regulatory evolution used gene expression measurements taken from whole organisms. This experimental design may blunt the ability to detect cis-regulatory changes, if those changes are only realized in a subset of tissues. Here, we performed allele-specific expression experiments in both whole bodies and brains of larval D. mojavensis in parallel, to determine if and how much the use of heterogeneous tissue samples affects quantification of regulatory type. We found clear evidence that our analysis of whole-body samples both overestimated trans effects and underestimated cis effects. This is reflected both in correlations between regulatory and parental divergences (fig. 1) as well as in the numbers of genes statistically categorized as cis- or trans-regulated (table 1). We cannot say precisely how much of this difference is due to the problems of using heterogeneous tissue samples and how much is due to regulatory properties specific to the larval brain. A systematic data set of allele-specific expression in multiple tissues and life stages collected would be needed to robustly address this question.
The Effects of Using Multiple Parental Genotypes on Estimation of Regulatory Type
It is well known that the F1 hybrid approach can bias estimates of trans-regulatory evolution, because they cannot be estimated independently of the measurement of parental expression divergence. Fraser (2019) pointed out how this issue, when combined with error in the estimation of allele-specific expression, can lead to overestimation of cis–trans compensatory evolution. By the same logic, we hypothesized that simple errors in the measurement of parental expression evolution might lead to inflation of the degree of trans-regulatory effects.
To address this issue, we simply compared the quantification of cis- and trans-regulatory effects in brains using two measures of parental expression divergence across populations: one measured from only the single genotype utilized in the allele-specific expression experiment, and one using seven additional genotypes from each population. Using population expression values has two obvious potential consequences for each gene. First, it should reduce noise in the estimation of population expression means coming from the small sample size of using only a few samples of a single genotype. This should lead to reduced estimates of trans-regulatory evolution. However, there will also be a subset of genes whose expression in the focal genotype substantially differs from the mean of its population. Thus, for some number of genes, our method should result in the artificial detection of trans-regulatory effects because the parental divergence will be mismatched with the allele-specific expression data.
Despite this, we find that using estimates of parental population divergence using multiple genotypes considerably reduces the genome-wide estimate of trans-regulatory evolution. Our data present mixed results, however, on the question of whether cis- or trans-regulation is primarily responsible for expression evolution between our populations. Although the correlation analysis suggests that cis-regulatory divergence is more closely related to population divergence (fig. 2), our per-gene hypothesis tests maintain nearly twice as many genes with evidence of trans-regulatory divergence (table 1). However, we argue that the numbers of genes displaying evidence for trans-regulation here is an overestimate for three reasons. First, as mentioned above, the inclusion of multiple parental genotypes will induce false positives in cases where intrapopulation variation in gene expression is substantial. Second, it is likely that the difference in power between the methods to detect cis- and trans effects contributes to the number of genes detected in each category (Graze et al. 2009; Coolon et al. 2014; but see Glaser-Schmitt et al. [2018]).
Third, the estimation of parental expression differences, and therefore trans-regulatory effects, are more error-prone due simply to the biology of our samples. We compared larval samples across two populations that develop at different rates (egg-pupation time [h]: CI = 275.20 ± 5.48; SON = 315.21 ± 5.96; Benowitz KM, Unpublished data), making it impossible to guarantee that sampling occurred at precisely the same exact developmental stage. Thus, some ontogenetic or environmental variation in gene expression is inevitable here. The usage of multiple genotypes should mitigate this problem but is unlikely to resolve it completely, and thus we may be generating false positives for this reason. For example, we find significant and consistent population differences in expression of two fat body proteins (Fbp1; Fbp2) and three larval serum proteins (Lsp1beta; Lsp1gamma; Lsp2) that are all clearly attributed to trans-regulatory evolution (table 2). Fat body proteins and larval serum proteins interact in a key pathway for nutritional storage prior to pupation (Burmester et al. 1999), and therefore could lead us to a hypothesis of adaptation via trans-regulation to variable nutritive environments. However, it is also well established that all four of these genes undergo rapid increases in expression during the third-instar wandering stage (Burmester et al. 1999). Our results are therefore equally consistent with the possibility that the expression differences were due to slight variations in the developmental stages sampled, and that expression patterns of these genes have not meaningfully evolved at all. In contrast, the estimation of cis-regulatory effects is completely controlled for any such environmental variability because it is measured within individuals (Pastinen 2010), and is therefore inherently less prone to similar errors.
Integrating Regulatory Data to Address Evolutionary Hypotheses
Recent work has sought to identify the evolutionary and structural properties associated with genes evolving via cis- and trans-regulation. Here, we demonstrate that in D. mojavensis larval brain cis-regulated genes tend to display faster rates of coding evolution. Furthermore, we show that cis-regulated genes also tend to occupy less central positions within transcriptional networks, confirming the results of Yang and Wittkopp (2017) and supporting their generality. Notably, we reached this conclusion using D. melanogaster network data, given that similar data are unavailable for D. mojavensis. Thus, our results suggest that gross network properties may be conserved across significant lengths of evolutionary time. Considered together, our findings linking regulatory type to evolutionary rate and network connectivity indicate that the genes experiencing cis-regulatory evolution are relatively unconstrained compared with trans-regulated genes. This perhaps contrasts with our predictions, which were that cis-regulation should be predominant due precisely to the presence of such constraints. However, it is not clear whether errors regarding the determination of trans-regulation discussed above may be obscuring any potential statistical relationships.
Ideally, the determination of regulatory evolution will also help identify adaptively regulated genes (Fraser 2011; Delbare and Clark 2018). RNA-seq experiments in ecology and evolution nearly always result in hundreds if not thousands of DE genes, many of which are likely false positives (Todd et al. 2016; Bengston et al. 2018). Truly DE genes must have experienced cis- or trans-regulatory evolution; therefore, the corroboration provided by a statistically significant regulatory effect estimated from an independent sample may help weed out noisy or environmentally variable genes. Thus, allele-specific expression data have been used to supplement studies of gene expression adaptation at the genome-wide (Juneja et al. 2016; Verta and Jones 2019) and candidate gene (Bendesky et al. 2017) levels. We thus turned our attention to the identities of the genes displaying clear patterns of both divergence and regulation, and compared between those displaying cis- and trans-regulation.
Previous transcriptomic (Matzkin et al. 2006; Matzkin 2012; Matzkin and Markow 2013; Smith et al. 2013) and genomic (Allan and Matzkin 2019) investigations of D. mojavensis have identified detoxification and chemosensory genes as important classes of genes likely related to adaptation to the alternative chemical environments provided by their hosts. Taking this as an a priori hypothesis, we examined the identities of genes identified here to search for candidates fitting these categories. We are most interested in the cis-regulated genes, which have the cleanest interpretations in this data set. Among the 33 cis-regulated candidate genes are 28 with D. melanogaster orthologs, of which 12 have described functions. Noteworthy among these is GstD1, a detoxification gene with considerable evidence for a functional role in adaptation across D. mojavensis populations (Matzkin et al. 2006; Matzkin 2008). Here, we find that expression differences in GstD1 are clearly attributable to cis-regulatory evolution between these populations, leading to increased expression in the Catalina Island population. Four other genes, including three cytochrome p450s and one UDP-glycosyltransferase, have well-characterized roles in detoxification of plant chemicals as well (Heckel 2014). We also find a single chemosensory gene, Obp99a. Among the 19 characterized genes displaying trans-regulation, we find the detoxification gene GstE1 as well as the chemosensory genes Obp99a (regulated in cis and trans) and Obp99b. Thus, although we are less confident in the trans-regulated gene set as a whole, this confirmation suggests that the regulatory evolution of at least a subset of these genes is accurately represented in this data set.
Although it is unsurprising that chemosensory genes are expressed and have evolved specifically in the brain, it is not as immediately clear why the expression of detoxification genes should be important in brain tissue. Detoxification is usually associated with tissues such as the midgut, Malpighian tubule, and fat body (Chung et al. 2009) and the blood–brain barrier tends to shield the brain from harmful chemicals (Stork et al. 2008; Hindle and Bainton 2014). However, the Drosophila blood–brain barrier is not completely impermeable to xenobiotics (Zhang et al. 2018), and important detoxification processes have been demonstrated in the brain in other insects (Zhu et al. 2010). Thus, it is at least plausible, given the chemical cocktail that D. mojavensis is exposed to within organ pipe and prickly pear necroses (Kircher 1982; Starmer and Phaff 1983), that some compounds may enter the brain. Alternatively, it is possible that we are witnessing the consequences of indirect selection. For example, strong selection on GstD1 expression in the midgut might have resulted in a cis-regulatory change to a binding site for a transcription factor that is also highly expressed in brains. If the resulting change in brain expression is neutral or nearly neutral it may then persist without having any adaptive function.
Given the ability of our approach to recapitulate a priori hypotheses about expression evolution, we also asked whether this approach might lead to novel predictions about phenotypic and genetic adaptation. Among the remaining cis-regulated genes are photorepair (phr) and CG5316 (ortholog of human aprataxin), which function to repair UV-damaged DNA (Boyd and Harris 1987; Hirano et al. 2007). In D. melanogaster, selection has generated adaptive differences in DNA repair mechanisms between tropical and temperate populations, and has resulted in both coding and noncoding genetic changes (Svetec et al. 2016). Here, both phr and CG5316 are upregulated in the Sonoran Desert population, where presumably intense UV exposure is a more pressing environmental challenge than in the cooler and wetter clime of Santa Catalina Island. Interestingly, phr was not among the D. melanogaster candidate genes differentiated in sequence or expression among populations, whereas CG5316 showed evidence of protein-coding but not expression evolution (Svetec et al. 2016). Thus, even when the predictability of DNA repair evolution pathways to similar environmental variables may extend to the gene, the type of genetic change itself may still be unpredictable.
Broadly, our usage of a replicated, tissue-specific data set and requirement of gene to display a clear regulatory pattern, especially one of cis-regulation, has led us to a manageable set of a few dozen highly intuitive genes that may be adaptively regulated across D. mojavensis populations associated with local ecological conditions. We propose that deeper understanding of patterns of regulatory evolution in ecological model systems, where there are strong predictions regarding selection, will be essential for a robust understanding of the differing roles of cis- and trans-regulation in local adaptation and evolution.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
C. Jaworski, F. Diaz, J. Hurtado, T. Shaible, N. Talamantes, and two anonymous reviewers provided helpful comments on the analyses and article. We would like to thank the Catalina Island Conservancy and the United States National Park Service at Organ Pipe National Monument for allowing the original collection of Drosophila utilized to establish the stocks used in this study. This work was supported by the National Science Foundation (IOS-1557697 to L.M.M.).
Author Contributions
K.M.B. and L.M.M. conceived and designed the study. K.M.B. and J.M.C collected samples and performed molecular work. K.M.B and C.W.A performed bioinformatic analysis. K.M.B performed statistical analysis and wrote the article with input from all authors.
Data deposition: This project has been deposited at the NCBI Sequence Read Archive under the Bioproject accession PRJNA574480.
Literature Cited
- Allan CW, Matzkin LM. 2019. Genomic analysis of the four ecologically distinct cactus host populations of Drosophila mojavensis. BMC Genomics 20:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq – a Python framework to work with high throughput sequencing data. Bioinformatics 31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Singh RS. 2010. Molecular evidence for increased regulatory conservation during metamorphosis, and against deleterious cascading effects of hybrid breakdown in Drosophila. BMC Biol. 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendesky A, et al. 2017. The genetic basis of parental care evolution in monogamous mice. Nature 544(7651):434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengston SE, et al. 2018. Genomic tools for behavioural ecologists to understand repeatable individual differences in behaviour. Nat Ecol Evol. 2(6):944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz KM, Coleman JM, Matzkin LM. 2019. Assessing the architecture of Drosophila mojavensis locomotor evolution using bulk segregant analysis. G3 (Bethesda) 9:1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JB, Harris PV. 1987. Isolation and characterization of a photorepair-deficient mutant in Drosophila melanogaster. Genetics 84:527–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478(7369):343–348. [DOI] [PubMed] [Google Scholar]
- Burmester T, Antoniewski C, Lepesant J-A. 1999. Ecdysone-regulation of synthesis and processing of fat body protein 1, the larval serum protein receptor of Drosophila melanogaster. Eur J Biochem. 262(1):49–55. [DOI] [PubMed] [Google Scholar]
- Carroll SB. 2008. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134(1):25–36. [DOI] [PubMed] [Google Scholar]
- Catalán A, Hutter S, Parsch J. 2012. Population and sex differences in Drosophila melanogaster brain gene expression. BMC Genomics. 13(1):654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, et al. 2009. Characterization of Drosophila melanogaster P450 genes. Proc Natl Acad Sci U S A. 106(14):5731–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JM, Benowitz KM, Jost AG, Matzkin LM. 2018. Behavioral evolution associated with host shifts in cactophilic Drosophila larvae. Ecol Evol. 8(14):6921–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs PA, Fraser HB. 2018. Spatially varying cis-regulatory divergence in Drosophila embryos elucidates cis-regulatory logic. PLoS Genet. 14(11):e1007631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ. 2014. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 24(5):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbare SYN, Clark AG. 2018. Allele-specific expression elucidates cis-regulatory logic. PLoS Genet. 14(11):e1007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y, Schafer DW, Cumbie JS, Chang JH. 2011. The NBP negative binomial model for assessing differential gene expression from RNA-seq. Stat Appl Genet Mol Biol. 10(1):24. [Google Scholar]
- Drosophila 12 Genomes Consortium. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed] [Google Scholar]
- Fear JM, et al. 2016. Buffering of genetic regulatory networks in Drosophila melanogaster. Genetics 203(3):1177–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB. 2011. Genome-wide approaches to the study of adaptive gene expression evolution. BioEssays 33(6):469–477. [DOI] [PubMed] [Google Scholar]
- Fraser HB. 2019. Improving estimates of compensatory cis-trans regulatory divergence. Trends Genet. 35(1):3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genissel A, McIntyre LM, Wayne ML, Nuzhdin SV. 2007. Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster. Mol Biol Evol. 25(1):101–110. [DOI] [PubMed] [Google Scholar]
- Gibson G. 1996. Epistasis and pleiotropy as natural properties of transcriptional regulation. Theor Popul Biol. 49(1):58–89. [DOI] [PubMed] [Google Scholar]
- Glaser-Schmitt A, Zeĉić A, Parsch J. 2018. Gene regulatory variation in Drosophila melanogaster renal tissue. Genetics 210(1):287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, McIntyre LM, Main BJ, Wayne ML, Nuzhdin SV. 2009. Regulatory divergence in Drosophila melanogaster and D. simulans, a genomewide analysis of allele-specific expression. Genetics 183(2):547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckel DG. 2014. Insect detoxification and sequestration strategies In: Voelckel C, Jander G, editors. Annual plant reviews. Vol. 47 Chichester (United Kingdom: ): Wiley; p. 77–114. [Google Scholar]
- Heed WB. 1978. Ecology and genetics of Sonoran desert Drosophila In: Brussard PF, editor. Ecological genetics: the interface. New York (NY: ): Springer; p. 109–126. [Google Scholar]
- Hindle SJ, Bainton RJ. 2014. Barrier mechanisms in the Drosophila blood-brain barrier. Front Neurosci. 8:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, et al. 2007. DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann Neurol. 61(2):162–174. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE, Coyne JA. 2007. The locus of evolution: evo devo and the genetics of adaptation. Evolution 61(5):995–1016. [DOI] [PubMed] [Google Scholar]
- Hughes KA, et al. 2006. Segregating variation in the transcriptome: cis regulation and additivity of effects. Genetics 173(3):1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja P, Quinn A, Jiggins FM. 2016. Latitudinal clines in gene expression and cis-regulatory element variation in Drosophila melanogaster. BMC Genomics 17(1):981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EG, Sanderson BJ, McNeil CL, Long AD, Macdonald SJ. 2014. Genetic dissection of the Drosophila melanogaster female head transcriptome reveals widespread allelic heterogeneity. PLoS Genet. 10(5):e1004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher HW. 1982. Chemical composition of cacti and its relationship to Sonoran desert Drosophila In: Barker JSF, Starmer WT, editors. Ecological genetics and evolution: the cactus-yeast-Drosophila model system. New York: Academic: p. 143–158. [Google Scholar]
- Koboldt DC, Larson DE, Wilson RK. 2013. Using VarScan2 for germline variant calling and somatic mutation detection. Curr Protocol Bioinformatics. 44:15.4.1–15.4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R. 1979. Quantitative genetic analysis of multivariate evolution, applied to brain-body size allometry. Evolution 33(1Part2):402–416. [DOI] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Fontanillas P, Hartl DL. 2008. Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci U S A. 105(38):14471–14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Wagner GP. 2008. Resurrecting the role of transcription factor change in developmental evolution. Evolution 62(9):2131–2154. [DOI] [PubMed] [Google Scholar]
- Marbach D, et al. 2012. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 22(7):1334–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massouras A, et al. 2012. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 8(11):e1003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzkin LM. 2008. The molecular basis of host adaptation in cactophilic Drosophila: molecular evolution of a Glutathione-S-transferase gene (GstD1) in Drosophila mojavensis. Genetics 178(2):1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzkin LM. 2012. Population transcriptomics of cactus host shifts in Drosophila mojavensis. Mol Ecol. 21(10):2428–2439. [DOI] [PubMed] [Google Scholar]
- Matzkin LM. 2014. Ecological genomics of host shifts in Drosophila mojavensis. Adv Exp Med Biol. 781:233–247. [DOI] [PubMed] [Google Scholar]
- Matzkin LM., Markow TA. 2013. Transcriptional differentiation across the four cactus host races of Drosophila mojavensis In: Michalak P, editor. Speciation: natural processes, genetics, and biodiversity. Hauppauge (NY: ): Nova Science Publishers; p. 119–136. [Google Scholar]
- Matzkin LM, Watts TD, Bitler BG, Machado CA, Markow TA. 2006. Functional genomics of cactus host shifts in Drosophila mojavensis. Mol Ecol. 15(14):4635–4643. [DOI] [PubMed] [Google Scholar]
- McManus CJ, et al. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res. 20(6):816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger BPH, Wittkopp PJ, Coolon JD. 2017. Evolutionary dynamics underlying gene expression divergence among Saccharomyces species. Genome Biol Evol. 9(4):843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Kohn MH, Wu C-I. 2006. Genomic inferences of the cis-regulatory nucleotide polymorphisms underlying gene expression differences between Drosophila melanogaster mating races. Mol Biol Evol. 23(8):1585–1591. [DOI] [PubMed] [Google Scholar]
- Osada N, Miyagi R, Takahashi A. 2017. Cis- and trans-regulatory effects on gene expression in a natural population of Drosophila melanogaster. Genetics 206(4):2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP. 2004. Two steps forward, one step back: the pleiotropic effects of favoured alleles. Proc R Soc Lond B. 271(1540):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T. 2010. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 11(8):533–538. [DOI] [PubMed] [Google Scholar]
- Prud’homme B, Gompel N, Carroll SB. 2007. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 104(Suppl 1):8605–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing; Available from: https://www.r-project.org/. Accessed April 2020. [Google Scholar]
- Reed LK, Nyboer M, Markow TA. 2006. Evolutionary relationships of Drosophila mojavensis geographic host races and their sister species Drosophila arizonae. Mol Ecol. 16(5):1007–1022. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Heed WB, Wasserman M. 1990. Evolution of the mojavensis cluster of cactophilic Drosophila with descriptions of two new species. J Hered. 81(1):30–42. [DOI] [PubMed] [Google Scholar]
- Sedlazeck FJ, Reschender P, von Haeseler A. 2013. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 29(21):2790–2791. [DOI] [PubMed] [Google Scholar]
- Signor SA, Nuzhdin SV. 2018. The evolution of gene expression in cis and trans. Trends Genet. 34(7):532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, et al. 2013. Transcriptome-wide expression variation associated with environmental plasticity and mating success in cactophilic Drosophila mojavensis. Evolution 67(7):1950–1963. [DOI] [PubMed] [Google Scholar]
- Starmer WT, Phaff HJ. 1983. Analysis of the community structure of yeasts associated with the decaying stems of cactus. Microb Ecol. 9(3):247–259. [DOI] [PubMed] [Google Scholar]
- Stern DL. 2000. Evolutionary developmental biology and the problem of variation. Evolution 54(4):1079–1091. [DOI] [PubMed] [Google Scholar]
- Stork T, et al. 2008. Organization and function of the blood-brain barrier in Drosophila. J Neurosci. 28(3):587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorov A, et al. 2013. Intra-specific regulatory variation in Drosophila pseudoobscura. PLoS One. 8(12):e83547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetec N, Cridland JM, Zhao L, Begun DJ. 2016. The adaptive significance of natural genetic variation in the DNA damage response of Drosophila melanogaster. PLoS Genet. 12(3):e1005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EV, Black MA, Gemmell NJ. 2016. The power and promise of RNA-seq in ecology and evolution. Mol Ecol. 25(6):1224–1241. [DOI] [PubMed] [Google Scholar]
- Uebbing S, et al. 2016. Divergence in gene expression within and between two closely related flycatcher species. Mol Ecol. 25(9):2015–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verta J-P, Jones FC. 2019. Predominance of cis-regulatory changes in parallel expression divergence of sticklebacks. ELife 8:43785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala J, Zhang C, Meyerson M, Beroukhim R. 2016. VariantBam: filtering and profiling of nextgenerational sequencing data using region-specific rules. Bioinformatics 32(13):2029–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-Y, et al. 2008. Complex genetic interactions underlying expression differences between Drosophila races: analysis of chromosome substitutions. Proc Natl Acad Sci U S A. 105(17):6362–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ. 2007. Variable gene expression in eukaryotes: a network perspective. J Exp Biol. 210(9):1567–1575. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430(6995):85–88. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet. 40(3):346–350. [DOI] [PubMed] [Google Scholar]
- Wray GA, et al. 2003. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 20(9):1377–1419. [DOI] [PubMed] [Google Scholar]
- Yang B, Wittkopp PJ. 2017. Structure of the transcriptional regulatory network correlates with regulatory divergence in Drosophila. Mol Biol Evol. 34(6):1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yue Z, Arnold DM, Artiushin G, Sehgal A. 2018. A circadian clock in the blood-brain barrier regulates xenobiotic efflux. Cell 173(1):130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Emerson JJ. 2019. Inferring compensatory evolution of cis- and trans-regulatory variation. Trends Genet. 35(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, et al. 2010. A brain-specific cytochrome P450 responsible for the majority of deltamethrin resistance in the QTC279 strain of Tribolium castaneum. Proc Natl Acad Sci U S A. 107L:557–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.