

Abstract
We herein disclose the first report of a first- row transition metal-catalyzed α,β-dehydrogenation of carbonyl compounds using allyl-nickel catalysis. This development overcomes several limitations of previously reported allyl-palladium-catalyzed oxidation, and is further leveraged for the development of an oxidative cycloalkenylation reaction that provides access to bicycloalke-nones with fused, bridged, and spirocyclic ring systems using unactivated ketone and alkene precursors.
Methodological advancements in the area of transition metal catalysis have enabled challenging C—C bond formations using unstabilized enolates. Among these advancements are α-alkenylation reactions1 that employ a cross coupling strategy (Figure 1A) to unite a vinyl halide and a metal enolate to provide synthetically versatile β,γ-enone products. As an alternative to cross coupling, seminal work by Saegusa2 and Kende3 demonstrated that unactivated alkenes can be employed for the cycloalkenylation4 of ketones using palladium, but required the preactivation of the carbonyls as enoxysilanes (Figure 1B). Additionally, stabilized enolates5 such as 1,3-diketones can also be employed in cycloalkenylation reactions, including using palladium catalysis as reported by Widenhoefer.6
Figure 1.

Strategies for α-alkenylation of ketones.
As existing strategies for α-alkenylation require either activated ketone or alkene partners,5 a direct alkenylation between unactivated ketones and alkenes by an oxidative approach would advantageously allow for the employment of more abundantly available precursors, and contribute to increased step-efficiency for cycloalkenylation reactions, which are broadly used in natural product synthesis for the construction of carbocycles.4b
Given our previous success using allyl oxidants to promote β-hydride elimination in the context of palladium-catalyzed carbonyl dehydrogenation (10),7‘8 we hypothesized that allyltransition metal catalysis could also facilitate our desired dehydrogenative cycloalkenylation. In particular, we were interested in probing the possibility of using allyl-nickel catalysis to facilitate the desired cycloalkenylation reaction due to nickel’s increased propensity to undergo migratory insertion.9
The envisioned oxidative cycloalkenylation reaction involves generation of allyl-nickel enolate 4 by deprotonation of 1 followed by transmetalation with allyl-nickel species 3. A subsequent migratory insertion event with a pendant alkene would generate alkyl-nickel species 5 that then undergoes β-hydride elimination to give the desired cycloalkenylation product (2) and allyl-nickel-hydride (6). Catalyst turnover by reductive elimination of 6 gives propene as the stoichiometric byproduct and Ni(0), which completes the catalytic cycle via oxidative addition to allyl electrophile 9. Additional mechanistic support comes from Tsuji’s observation that allyl-palladium enolates generated by the decarboxylation of allyl β-ketoesters could undergo migratory insertion and β-hydride elimination with electron-deficient alkenes to give cycloalkenylation products.10
Despite the often more challenging β-hydride elimination of first-row transition metals,9 successful development of this methodology would provide a more sustainable and cost- effective alternative for allyl-metal-catalyzed β-hydride elimination in both cycloalkenylation and carbonyl α,β-dehydrogenation contexts. We also aimed to leverage the inherent differences of first- and second-row transition metals to address some of the current limitations of allyl-palladium- catalyzed dehydrogenation chemistry. In particular, for reluctant substrates that give predominantly Tsuji-Trost allylation byproducts, we expected the use of a less electrophilic allyl-nickel species to be more successful.
Unactivated carbonyl compounds can undergo α,β-dehydrogenation by several methods including using metal catalysis.11 Stahl has reported an exciting process for ketone and aldehyde dehydrogenation using palladium catalysis that is beginning to see widespread adoption.12 Dong has also reported a palladium-catalyzed approach for lactams,13 and more recently a method employing platinum to dehydrogenate several substrate classes.14 Methods utilizing first-row metals for dehydrogenation have either focused on activated systems that lead to products with extended conjugation, or these intermediates were not isolated but rather reacted further to form more stable products.15 This report describes the first practical first-row transition metal-catalyzed α,β-dehydrogen- ation of carbonyl compounds using nickel-catalysis. The discovery of nickel-catalyzed dehydrogenation was a starting point for the development of an oxidative cycloalkenylation of ketone enolates for the construction of diverse bicyclic scaffolds.
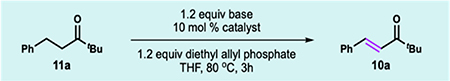
Ketone 11a was used as our model substrate (Table 1) as we previously identified that acyclic ketones were poor substrates for our reported allyl-palladium-catalyzed ketone dehydrogenation protocol (entry 1).8 Several first-row metal catalysts were investigated (entries 2–6), and encouragingly we found that a similar, albeit low yield of enone 10a could be detected using NiBr2(dme) (entry 6), demonstrating the feasibility of a nickel-mediated reaction. Investigation of the base (entries 7–9) revealed that our previously disclosed hindered lithium anilide base, LiCyan, proved to be most effective.7b
Table 1.
Optimization of α,β-Dehydrogenation
 | |||
|---|---|---|---|
| Entry | Base | Catalyst | Yield (%)a |
| 1 | Zn(TMP)2 | [Pd(allyl)Cl]2 | 8 |
| 2 | Zn(TMP)2 | MnBr2 | 0 |
| 3 | Zn(TMP)2 | FeBr2 | 0 |
| 4 | Zn(TMP)2 | CoBr2 | 0 |
| 5 | Zn(TMP)2 | CuBr2 | 0 |
| 6 | Zn(TMP)2 | NiBr2(dme) | 7 |
| 7 | Zn(TMP)2, ZnBr2 | NiBr2(dme) | 12 |
| 8 | LiTMP, ZnBr2 | NiBr2(dme) | 61 |
| 9 | LiCyan, ZnBr2 | NiBr2(dme) | 69 |
1H-NMR yield was determined using 1,3,5-trimethoxybenzene as an internal standard.
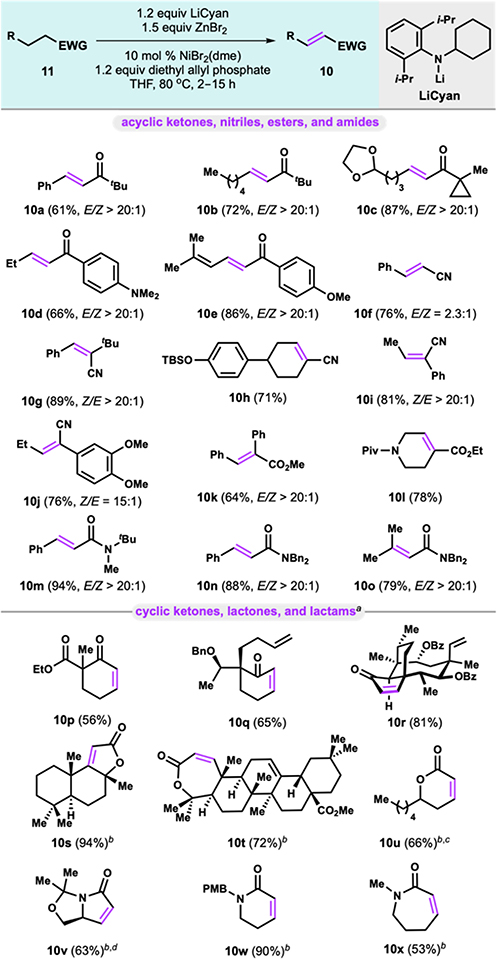
We investigated the scope of the nickel-catalyzed dehydrogenation with particular interest in other substrates that were previously unsuccessful with the palladium-catalyzed system (Table 2). A variety of acyclic ketones, including aliphatic (10a-c) and electron-rich aryl ketones (10d-e) were efficiently dehydrogenated under the optimized conditions, and tolerated acid sensitive acetal and cyclopropane functionality. Primary nitriles (10f) and more hindered secondary nitriles ( 10g—j) were also excellent substrates for this nickel- catalyzed dehydrogenation. Notably, α-aryl nitriles ( 10i—j ), which predominantly gave allylation byproducts when palladium was used, were efficiently dehydrogenated using the less electrophilic allyl-nickel system. Similarly, α-aryl ester 10k was also accessed in good yield. Unsaturated ester 10l containing N-Piv functionality was also obtained in excellent yield. Amides (10m-o) were a viable substrate class for this nickel-catalyzed dehydrogenation, including sterically hindered β,β-disubstituted amide 10o.
Table 2.
Scope of Carbonyl and Nitrile α,β-Dehydrogenation
 |
1.2 equiv Zn(TMP)2 and no ZnBr2 was used.
1,4-Dioxane was used as solvent.
Reaction conducted at 23 °C.
1.0 equiv diethyl allyl phosphate was used.
When we tried our optimized conditions on cyclic ketones, we saw significantly depreciated yields. Instead we found that commercially available Zn(TMP)2 was most effective for cyclic ketones, as previously demonstrated.8 β-ketoester 10p could be dehydrogenated in good yield, despite the possibility of metal chelation by the 1,3-dicarbonyl suppressing reactivity. Ketone 10q containing a pendant alkene and mutilin derivative 10r could also be accessed. Interestingly, we found that Zn(TMP)2 could also be used to efficiently dehydrogenate 5-, 6-, and 7- membered lactones (10s-u) and lactams (10v-x) despite their decreased acidity. Notably, unsaturated lactam 10v is a key intermediate needed to access an important class of IRAK4 inhibitors.16 While LiCyan is still an effective base for lactones and lactams, it was generally observed that Zn(TMP)2 outperformed LiCyan for cyclic carbonyl compounds (see Supporting Information). The use of Zn(TMP)2 also provides the advantage of a more operationally simple protocol. Under our reaction conditions, structural features of the molecule (acyclic or cyclic) seem to dictate the optimal base necessary for efficient dehydrogenation.
Furthermore, the development of a nickel-catalyzed system for carbonyl dehydrogenation overcomes previous limitations in scope, such as acyclic ketones (10a-e) and allylation-prone a-aryl substrates (10i—k) and provides an efficient and cost- effective alternative for carbonyl dehydrogenation, which has only been demonstrated previously with precious metals.
Using our newly established allyl-nickel-based system, we then explored the possibility of an oxidative cycloalkenylation of ketones. It was found that with only a minor modification to the dehydrogenation conditions using Zn(TMP)2 as base, the desired cycloalkenylation could be achieved. The addition of ZnBr2 as an additive was crucial for attaining an efficient reaction and a broad substrate scope (see Supporting Information for a broad investigation of additives), and highlights the remarkable role of salt additives in transformations involving organozinc species.17
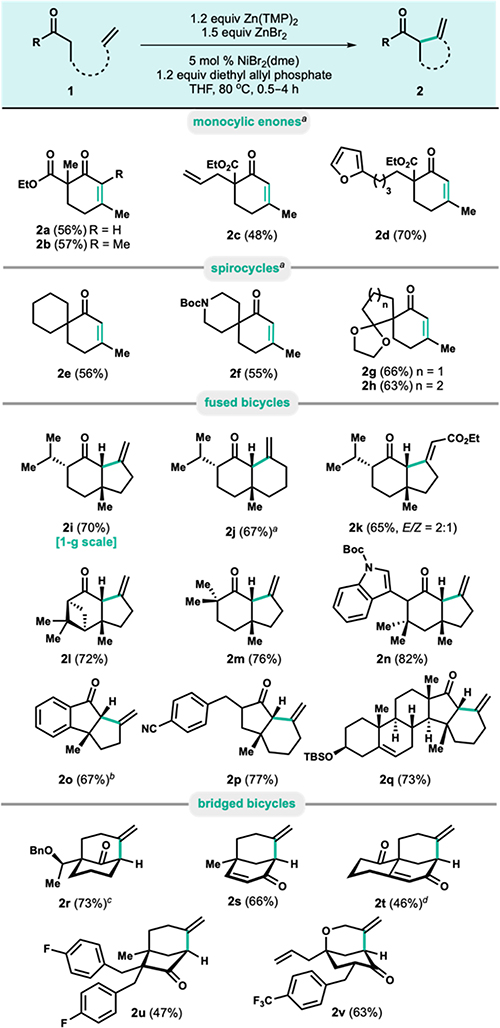
The substrate scope was explored with particular interest in the various carbocyclic architectures that could be accessed (Table 3). Acyclic ketones could efficiently cyclize with a pendant homoallyl group, providing β-methyl cyclohexenone products after alkene isomerization to the more thermodynamically stable α,β-enone products (2a—d). Importantly, ethyl ketones could also be used despite the possibility of dehydrogenation, providing access to tetrasubsituted enone 2b. Enone product 2c highlights that this protocol is selective for cyclization of 6-membered rings rather than 5-membered rings, which is in contrast to previous reports of palladium-mediated cycloalkenylations.4 Gratifyingly, sensitive furan functionality was also well tolerated (2d). Geminally substituted cyclic structures could be used to access spirocyclic motifs in good yields (2e—h) and tolerated the acid sensitive N-Boc (2f) and acetal functionality (2g—h).
Table 3.
Scope of Oxidative Cycloalkenylation
 |
2.0 equiv Zn(TMP)2 was used.
20 mol % NiBr2(dme) and no ZnBr2 was used.
40 °C.
1.5 equiv Zn(TMP)2 was used.
Bicyclo[4.3.0]nonanes and bicyclo[4.4.0]decanes could also be accessed using this methodology as demonstrated by products 2i—q. Access to piperitone derivative 2i highlights that 5-membered ring formation is also possible, and 2i was prepared on 1 g scale without any depreciation in yield. β,γ-enone 2j can be used to access a variety of eudesmane-type sesquiterpene natural products, but was previously reported to be a challenging scaffold to access by existing cycloalkenylation approaches.18 Gratifyingly, pendant alkenes bearing electron withdrawing groups were also tolerated in the substrate scope and could provide α,β-unsaturated ester product 2k as a 2:1 mixture of separable E- and Z-isomers. Cycloalkenylation with olefins bearing electron-donating substituents such as alkyl or ether groups were not tolerated. Other bicyclo[4.3.0]nonanes (2l —n), such as verbenone derivative 2l bearing an acid- sensitive cyclobutane and Boc-protected indole 2n were also excellent substrates under the reaction conditions. Cyclo- alkenylation initiated by cyclopentanones could also be achieved (2o—q) and demonstrated the tolerance of benzonitrile (2p) and TBS-protected alcohol functionality (2q). Indanone 2o was accessed using modified reaction conditions that employed 20 mol % catalyst loading and no ZnBr2 additive, which was found to be an excellent alternative for reluctant substrates. Notably, while 6-membered ring formation under previously reported cycloalkenylation methods with palladium is challenging,2‘4 this allyl-nickel system provides an efficient disconnection for forming cyclohexanes with unactivated alkenes.
Although cycloalkenylation to form fused ring systems was not possible when β-hydride elimination was a competing pathway, bridged bicyclic ring systems such as bicyclo[3.3.l]- nonanes (2r-1) and bicyclo[3.2.l]octanes (2u), which are important core structures in a variety of natural products,19 could be accessed despite the possibility of β-hydride elimination. Access to bicyclo[3.3.l]nonane 2r demonstrates the possibility of bifurcating reactivity using reagent control (Table 2, 10q), although this was not found to be a general phenomenon. Wieland-Miescher ketone derivative 2t containing two ketone functional groups selectively underwent cycloalkenylation via the thermodynamic enolate, and 2v demonstrates selective cyclization with the axially disposed O- tethered alkene to give a bicyclo[3.3.l]nonane rather than with the all-carbon tethered alkene to give a bicyclo[3.2.l]octane.
Under the reported reaction conditions, all substrates were completely selective for the exo-mode of cyclization and gave cis-fused products. No alkene isomerization was observed for substrates that gave fused or bridged bicyclic products, which is an advantage over previously reported palladium-mediated cycloalkenylations of enoxysilanes, which gave isomeric mixtures of alkenes.4
We wanted to probe the identity of the metal responsible for C—C bond formation between the ketone enolate and the pendant olefin (Figure 2). Two mechanistic possibilities could be envisioned: one involves the carbometalation of the pendant alkene20 by a zinc enolate and the other involves the proposed migratory insertion of an alkene to an allyl-nickel enolate. Under the standard conditions, 1i underwent efficient alkenylation (Figure 2a). When substrate 1i was heated with Zn(TMP)2 and ZnBr2, we found that the zinc-mediated cyclization was indeed possible, albeit in low conversion. Subsequent quenching with D2O afforded the fully saturated, deuterated species 12 in 32% yield with 68% deuterium incorporation (Figure 2b) and suggests the intermediacy of an alkylzinc intermediate. Related cyclizations of activated zinc enolates with unactivated olefins have been reported.21
Figure 2.

Mechanistic investigations.
Despite the possibility of a zinc-mediated cyclization, the subsequent addition of nickel and allyl oxidant to effect transmetalation and β-hydride elimination of the alkylzinc species did not provide a significant amount of alkenylation product 2i (Figure 2c). Similarly, treatment of an alkylzinc intermediate prepared from the corresponding alkyl bromide to identical allyl-nickel conditions also provided low yields of alkenylation product (see Supporting Information). These results suggest that while a zinc-mediated anionic cyclization followed by an allyl-nickel-mediated β-hydride elimination is one possible mechanism for cycloalkenylation, the migratory insertion of a nickel enolate cannot be ruled out given additional experiments conducted in the absence of zinc (Figure 2d).
The nickel-mediated cyclization was demonstrated to be plausible by treatment of α-bromo ketone 13 with a stoichiometric quantity of Ni(cod)2. The low yield for this transformation may reflect the importance of the specific ligand sphere accessed through our catalytic manifold and emphasizes the synthetic utility of our methodology. Taken together, these mechanistic investigations highlight the possibility of both zinc and nickel to mediate C—C bond formation between unstabilized enolates and unactivated alkenes. Additional mechanistic investigations of the roles of zinc and nickel are ongoing and will be reported in due course.
We have effectively expanded allyl-metal catalysis for carbonyl α,β-dehydrogenation from palladium to nickel, thereby allowing for the use of a more sustainable and inexpensive first-row transition metal catalyst. Furthermore, key limitations to the substrate specificity and scope of the allyl-palladium-catalyzed α,β-dehydrogenation conditions were overcome using the more general nickel-catalyzed reaction conditions for the dehydrogenation of ketones, nitriles, esters, and amides. The use of allyl-nickel catalysis also enabled an oxidative cycloalkenylation reaction between unstabilized ketone enolates and unactivated alkenes, providing access to diverse bicyclic architectures and a new retrosynthetic C—C bond disconnection for the synthesis of complex polycyclic molecules.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support from Yale University, the Sloan Foundation, Nalas Engineering, Amgen, the NSF (CAREER, 1653793), and the NIH-funded Chemistry/ Biology Interface Training Program (D.H., T32GM067543). Dr. Brandon Mercado is gratefully acknowledged for X-ray crystallography of compound SI-13.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b02552.
Experimental procedures and spectroscopic data for all new compounds including 1H- and 13C NMR spectra (PDF)
X-ray crystallography of compound SI-13 (CIF)
REFERENCES
- (1).(a) For a seminal report of nickel-catalyzed alkenylation of enolates, see: Millard AA; Rathke MW A Nickel Catalyst for the Arylation and Vinylation of Lithium Ester Enolates. J. Am. Chem. Soc. 1977, 99, 4833–4835. [Google Scholar]; (b) For a review on palladium- and nickel- catalyzed α-alkenylation via cross coupling, see: Ankner T; Cosner CC; Helquist P Palladium- and Nickel-Catalyzed Alkenylation of Enolates. Chem. - Eur. J. 2013, 19, 1858–1871. [DOI] [PubMed] [Google Scholar]; (c) For a recent report of copper-catalyzed alkenylation of zinc enolates employing iodonium salts, see: Liu C; Wang Q Alkenylation of C(sp3)-H Bonds by Zincation/Copper-Catalyzed Cross Coupling with Iodonium Salts. Angew. Chem. Int. Ed. 2018, 57, 4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ito Y; Aoyama H; Hirao T; Mochizuki A; Saegusa T Cyclization Reactions via Oxo-n-allylpalladium(II) Intermediates. J. Am. Chem. Soc. 1979, 101, 494–496. [Google Scholar]
- (3).(a) Kende AS; Roth B; Sanfilippo SJ Facile, Palladium(II)- Mediated Synthesis of Bridged and Spirocyclic Bicycloalkenones. J. Am. Chem. Soc. 1982, 104, 1784–1785. [Google Scholar]; (b) Kende AS; Roth B; Sanfilippo PJ; Blacklock TJ Mechanism and Regioisomeric Control in Palladium(II)-Mediated Cycloalkenylations. A Novel Total Synthesis of (±)-Quadrone. J.Am. Chem. Soc. 1982, 104, 5808–5810. [Google Scholar]
- (4).(a) For a review on palladium-mediated cycloalkenylations, see: LeBras J; Muzart J Base-free palladium-mediated cycloalkenylations of olefinic enolic systems. Tetrahedron 2015, 71, 9035–9059. [Google Scholar]; (b) Toyota M; Ihara M Development of Palladium-Catalyzed Cycloalkenylation and its Application to Natural Product Synthesis. Synlett 2002, 1211–1222. [PubMed] [Google Scholar]
- (5).Denes F; Perez-Luna A; Chemla F Addition of Metal Enolate Derivatives to Unactivated Carbon-Carbon Multiple Bonds. Chem. Rev. 2010, 110, 2366–2447. [DOI] [PubMed] [Google Scholar]
- (6).Pei T; Wang X; Widenhoefer RA Palladium-Catalyzed Intramolecular Oxidative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2003, 125, 648–649. [DOI] [PubMed] [Google Scholar]
- (7).(a) Chen Y; Romaire JP; Newhouse TR Palladium- Catalyzed α,β-Dehydrogenation of Esters and Nitriles. J. Am. Chem. Soc. 2015, 137, 5875–5878. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Turlik A; Newhouse TR Amide α,β-Dehydrogenation Using Allyl-Palladium Catalysis and a Hindered Monodentate Anilide. J. Am. Chem. Soc. 2016, 138, 1166— 1169. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y; Chen Y; Newhouse TR Allyl-Palladium Catalyzed α,β-Dehydrogenation of Carboxylic Acids via Enediolates. Angew. Chem., Int. Ed. 2017, 56, 13122–13125. [DOI] [PubMed] [Google Scholar]; (d) Schuppe AW; Huang D; Chen Y; Newhouse TR Total Synthesis of (−)-Xylogranatopyridine B via a Palladium-Catalyzed Oxidative Stannylation of Enones. J. Am. Chem. Soc. 2018, 140, 2062–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Szewczyk SM; Zhao Y; Sakai HA; Dube P; Newhouse TR α,β-Dehydrogenation of esters with free O-H and N-H functionalities with allyl-palladium catalysis. Tetrahedron 2018, 74, 3293–3300. [Google Scholar]
- (8).For allyl-palladium-catalyzed dehydrogenation of ketones, see:; (a) Chen Y; Huang D; Zhao Y; Newhouse TR Allyl-Palladium-Catalyzed Ketone Dehydrogenation Enables Telescoping with Enone α,β-Vicinal Difunctionalization. Angew. Chem., Int. Ed. 2017, 56, 8258–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang D; Zhao Y; Newhouse TR Synthesis of Cyclic Enones by Allyl-Palladium-Catalyzed α,β-Dehydrogenation. Org. Lett. 2018, 20, 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Tasker SZ; Standley EA; Jamison TF Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zweig JE; Kim DE; Newhouse TR Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. [DOI] [PubMed] [Google Scholar]
- (10).Nokami J; Watanabe H; Mandai T; Kawada M; Tsuji J The palladium-catalyzed Michael addition reaction of the ketone enolates generated by the decarboxylation of allyl-β-keto carboxylates under neutral conditions. Tetrahedron Lett. 1989, 30, 4829–4832. [Google Scholar]
- (11).For reviews on carbonyl dehydrogenation, see:; (a) Muzart J One-Pot Syntheses of α,β-Unsaturated Carbonyl Compounds through Palladium-Mediated Dehydrogenation of Ketones, Aldehydes, Esters, Lactones, and Amides. Eur. J. Org. Chem. 2010, 2010, 3779–3790. [Google Scholar]; (b) Stahl SS; Diao T Oxidation Adjacent to C = X bonds by Dehydrogenation. Comp. Org. Synth 2014, 7, 178–212. [Google Scholar]; (c) Turlik A; Chen Y; Newhouse TR Dehydrogenation Adjacent to Carbonyls Using Palladium-Allyl Intermediates. Synlett 2016, 27, 331–336. [Google Scholar]; (d) Iosub AV.; Stahl SS Palladium-Catalyzed Aerobic Dehydrogenation of Cyclic Hydrocarbons for the Synthesis of Substituted Aromatics and Other Unsaturated Products. ACS Catal. 2016, 6, 8201–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hirao T Synthetic Strategy: Palladium- Catalyzed Dehydrogenation of Carbonyl Compounds. J. Org. Chem. 2019, 84, 1687–1692. [DOI] [PubMed] [Google Scholar]
- (12).(a) Diao T; Stahl SS Synthesis of Cyclic Enones via Direct Palladium-Catalyzed Aerobic Dehydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 14566–14569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Diao T; Wadzinski TJ; Stahl SS Direct aerobic α,β-dehydrogenation of aldehydes and ketones with a Pd(TFA)2/4,5-diazafluorenone catalyst. Chem. Sci. 2012, 3, 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Diao T; Pun D; Stahl SS Aerobic Dehydrogenation of Cyclohexanone to Cyclohexenone Catalyzed by Pd(DMSO)2(TFA)2: Evidence for Ligand-Controlled Chemoselec- tivity. J. Am. Chem. Soc. 2013, 135, 8205–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen M; Dong G Direct Catalytic Desaturation of Lactams Enabled by Soft Enolization. J. Am. Chem. Soc. 2017, 139, 7757–7760. [DOI] [PubMed] [Google Scholar]
- (14).Chen M; Rago A; Dong G Platinum-catalyzed Desaturation of Lactams, Ketones and Lactones. Angew. Chem. Int. Ed. 2018, 57, 16205–16209. [DOI] [PubMed] [Google Scholar]
- (15).(a) Ueno S; Shimizu R; Kuwano R Nickel-Catalyzed Formation of a Carbon-Nitrogen Bond at the β-Position of Saturated Ketones. Angew. Chem., Int. Ed. 2009, 48, 4543–4545. [DOI] [PubMed] [Google Scholar]; (b) Tutkowski BM; Grigalunas M; Wiest O; Helquist P A nickel-catalyzed α,β-coupling of saturated ketones. Tetrahedron Lett. 2015, 56, 3468–3472. [Google Scholar]; (c) Jie X; Shang Y; Zhang X; Su W Cu-Catalyzed Sequential Dehydrogenation-Conjugate Addition for β-Functionali- zation of Saturated Ketones: Scope and Mechanism. J. Am. Chem. Soc. 2016, 138, 5623–5633. [DOI] [PubMed] [Google Scholar]; (d) Shang Y; Jie X; Jonnada K; Zafar SN; Su W Dehydrogenative desaturation-relay via formation of multicenter-stabilized radical intermediates. Nat. Commun. 2017, 8, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li H; Jiang Q; Jie X; Shang Y; Zhang Y; Goossen LJ; Su W Rh/Cu-Catalyzed Ketone ^-Functionalization by Merging Ketone Dehydrogenation and Carboxyl-Directed C-H Alkylation. ACS Catal. 2018, 8, 4777–4782. [Google Scholar]
- (16).Lee KL; Ambler CM; Anderson DR; Boscoe BP; Bree AG; Brodfuehrer JI; Chang JS; Choi C; Chung S; Curran KJ; Day JE; Dehnhardt CM; Dower K; Drozda SE; Frisbie RK; Gavrin LK; Goldberg JA; Han S; Hegen M; Hepworth D; Hope HR; Kamtekar S; Kilty IC; Lee A; Lin L-L; Lovering FE; Lowe MD; Mathias JP; Morgan HM; Murphy EA; Papaioannou N; Patny JP; Pierce BS; Rao VR; Saiah E; Samardjiev IJ; Samas BM; Shen MWH; Shin JH; Soutter HH; Strohbach JW; Symanowicz PT; Thomason JR; Trzupek JD; Vargas R; Vincent F; Yan J; Zapf CW; Wright SW Discovery of Clinical Candidate 1-{[2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}−7-methoxyisoquinoline-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J. Med. Chem. 2017, 60, 5521–5542. [DOI] [PubMed] [Google Scholar]
- (17).(a) Achonduh GT; Hadei N; Valente C; Avola S; O’Brien CJ; Organ MG On the role of additives in alkyl-alkyl Negishi cross-couplings. Chem. Commun. 2010, 46, 4109–4111. [DOI] [PubMed] [Google Scholar]; (b) McCann LM; Organ MC On the Remarkably Different Role of Salt in the Cross-Coupling of Arylzincs From That Seen with Alkylzincs. Angew. Chem. Int. Ed. 2014, 53, 4386–4389. [DOI] [PubMed] [Google Scholar]
- (18).(a) Chen K; Ishihara Y; Galan MM; Baran PS Total synthesis of eudesmane terpenes: cyclase phase. Tetrahedron 2010, 66, 4738–4744. [Google Scholar]; (b) Thomas AF; Ozainne M; Decorzant R; Naf F; Lukacs G 10-Epijuneol, a new cis-eudesmane sesquiterpenoid. Tetrahedron 1976, 32, 2261–2264. [Google Scholar]; (c) Toyota M; Asakawa Y An eudesmane-type sesquiterpene alcohol from the liverwort FrulUania tamarisci. Phytochemistry 1990, 29, 3664–3665. [Google Scholar]
- (19).(a) Presset M; Coquerel Y; Rodriguez J Syntheses and Applications of Functionalized Bicyclo[3.2.1]octanes: Thirteen Years of Progress. Chem. Rev. 2013, 113, 525–595. [DOI] [PubMed] [Google Scholar]; (b) Ruiz M; Lopez- Alvarado P; Giorgi G; Menendez JC Domino reactions for the synthesis of bridged bicyclic frameworks: fast access to bicyclo- [n.3.1]alkanes. Chem. Soc. Rev. 2011, 40, 3445–3454. [DOI] [PubMed] [Google Scholar]
- (20).(a) Stadtmuller H; Lentz R; Tucker CE; Studemann T; Dorner W; Knochel P Palladium-Catalyzed Iodine-Zinc Exchange Reactions. A New Palladium-Mediated Intramolecular Carbozincation of Alkenes. J. Am. Chem. Soc. 1993, 115, 7027–7028. [Google Scholar]; (b) Meyer C; Marek I; Courtemanche G; Normant J-F Carbocyclization of Functionalized Zinc Organometallics. Synlett 1993, 1993, 266–268. [Google Scholar]
- (21).Perez-Luna A; Botuha C; Ferreira F; Chemla F Carbometalation of unactivated alkenes by zinc enolate derivatives. New J. Chem. 2008, 32, 594–606. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.