

Abstract
Morphinone reductase (MR) is an important model system for studying the contribution of protein motions to H-transfer reactions. In this research, we used quantum mechanical/molecular mechanics (QM/MM) simulation together with transition path sampling (TPS) simulation to study two important topics of current research on MR: the existence of multiple catalytic reaction pathways and the involvement of fast protein motions in the catalytic process. We have discovered two reaction pathways for the wild type and three reaction pathways for the N189A mutant. With the committor distribution analysis method, we found reaction coordinates for all five reaction pathways. Only one wild-type reaction pathway has a rate-promoting vibration from His186, while all of the other four pathways do not involve any protein motions in their catalytic process through the transition state. The rate-promoting vibration in the wild-type MR, which comes from a direction perpendicular to the donor–acceptor axis, functions to decrease the donor–acceptor distance by causing a subtle “out-of-plane” motion of a donor atom. By comparing reaction pathways between the two enzymes, we concluded that the major effect of the N189A point mutation is to increase the active site volume by altering the active site backbone and eliminating the Asn189 side chain. This effect causes a different NADH geometry at the reactant state, which very well explains the different reaction mechanisms between the two enzymes, as well as the disappearance of the His186 rate-promoting vibrations in the N189A mutant. The unfavorable geometry of the NADH pyridine ring induced by the N189A point mutation is the potential cause of multiple reaction pathways in N189A mutants.
Introduction
Understanding the source of the catalytic power of enzymes has been a continuing focus of biochemistry. Several mechanisms have been proposed to explain the catalytic power of enzymes, among which are rate-promoting vibrations (RPVs),1−3 transition-state stabilization,4,5 and electrostatic preorganization.6,7 We stress that these mechanisms are not mutually exclusive and they likely exist in varying proportions in all enzymes. Among these proposals, rate-promoting vibrations (RPVs), which are fast protein motions on the subpicosecond time scale and which contribute to reduce the free-energy barrier of enzymatic reactions, have been proposed to exist in a variety of enzyme systems.8−10 H-transfer reactions have been viewed as good model systems to study RPVs since the contribution of protein dynamics to the catalytic process would be significant in this type of reactions if there is any tunneling contributing to the barrier-crossing process. This study focuses on the morphinone reductase (MR), a member of the old yellow enzyme family. The reductive half-reaction catalyzed by MR includes a hydride transfer reaction from the NADH nicotinamide C4 atom to the flavin mononucleotide (FMN) N5 atom,11 which has been shown to have a heavy involvement of tunneling effect.12,13 The motivation of this study is to shed light on two major aspects of researches on the MR reductive half-reactions that remain uncertain: the existence of multiple reaction pathways and the involvement of protein motions in the catalytic process of MR.
The existence of multiple reaction pathways in the MR N189A mutant is suggested by several experimental researches. Pudney et al. first proposed in an experimental study that the reductive half-reaction in the MR N189A mutant possesses multiple “parallel” reaction pathways through the reaction barrier, while the wild-type MR seems to have only one reaction pathway.14 The origin of multiple reaction pathways was proposed to be the multiple reaction configurations induced by the N189A point mutation.14,15 However, there has not been a rigorous observation of the multiple reaction pathways from a computational perspective, nor has any work managed to directly link the multiple reaction configurations with the experimentally observed reaction pathways.
Whether protein motions are involved in the catalytic process of the MR has been under debate for the past decade. Existing studies seem to give contrast evidence on both sides. Several experimental studies measuring the temperature and pressure dependences of the kinetic isotope effect (KIE) of MR, as well as a molecular dynamic simulation (MD) research under different pressure, seem to support the idea that protein motion is contributing to the catalytic progress of the MR.15−19 Potential effects of the protein motion have been proposed to be compressing the NADH nicotinamide plane closer to the FMN isoalloxazine plane15,17 or as described by Johannissen et al.20 to be an out-of-plane bending motion of the NADH pyridine ring, which sends the donor C4 atom closer to the acceptor N5 atom. The source of neither of these effects, the protein motion itself, is left undetermined since there does not exist any protein residues “on top of” the NADH nicotinamide plane. However, a recent QM/MM simulation study by Delgado et al. with high-order quantum mechanical method did observe a minor conformational change of the NADH and the FMN molecules during the reaction progress, but called into question the existence of any significant protein conformation change between the reactant state and the transition state, indicating that the active site of this enzyme has been well prepared for the hydride transfer reaction.21 Two important caveats to these studies mentioned above are that they either are not capable of describing the chemical step of the reaction with the classical MD simulation or lack a rigorous analysis of causality between the protein motions and their effects. Specifically, the potential of mean force treatment used in the QM/MM simulation by Delgado et al.21 requires a predefined reaction coordinate. The reaction coordinate being utilized, the bond-breaking and bond-forming distances,21 could inherently result in the inability to capture all contributions to the reaction coordinate in the catalytic process and bias the result.
The simulation method we used in this study, the transition path sampling (TPS) method,22,23 can overcome caveats of the current simulation methods. TPS requires no prior knowledge of or choice of the reaction coordinate and is therefore capable of generating unbiased reactive trajectories of the chemical event in this enzyme. Further analysis using the committor distribution analysis method22 allows us to rigorously identify the reaction coordinate of the MR-catalyzed hydride transfer reaction and analyze the contribution of potential protein motions to the reaction coordinate, avoiding the lack of causality. With these methods, we showed in this paper with rigor the answer to the two unresolved questions mentioned above. We sampled and analyzed multiple reaction pathways in the MR N189A mutant. Also, we found a protein motion coupled to the reaction progress in one of the two wild-type MR reaction pathways and identified its contribution to the reaction progress. Further, the influence of the N189A point mutation on the reaction mechanism has also been clearly illustrated.
Methods
System Setup and Simulation Detail
We used the crystal structure of the wild-type MR cocrystallized with the cofactor 1,4,5,6-tetrahydro NADH (NADH4) and the substrate FMN from Pudney et al.14 as the starting structure of the simulation. All MD and QM/MM simulations are performed with the CHARMM molecular dynamic software package,24,25 with the CHARMM36 forcefield.25 CHARMM parameters of the FMN molecule were taken from a previous work by Freddolino et al.,26 while parameters of the NADH molecule were taken from the CHARMM CGenFF.27 The integration step length was set to 1 fs for all simulations. Nonbonding interactions were cut off at 12 Å. Extra hydrogen atoms on the cocrystallized NADH4 molecule were deleted to form the NADH molecule. To simulate the chemical step of the hydride transfer reaction, we treated part of the NADH and the FMN molecules quantum-mechanically, as shown in Figure 1a,b, using the PM3 semiempirical method.28 This selection of the quantum region is similar to the two QM/MM simulations mentioned above.12,21 Specifically, it has been shown in previous studies, where NADH was the hydride donor,29,30 that this atom selection on the NADH molecule is sufficient. The system is solvated in a spherical water droplet with TIP3P water molecules. The size of the droplet is set to be 15 Å from the surface of the protein. The system is then neutralized with potassium ions. To avoid unfavorable geometries in the crystal structure, we performed a two-step minimization on the system. In the first step, the system was minimized for 100 steps using the steepest descent method to avoid clashes between atoms, with harmonic constraint forces (100 kcal mol–1 Å–2) applied to the QM region. Then, a 1000-step adopted-basis Newton–Raphson (ABNR) method minimization was performed, with the constraint force gradually reduced. The minimized system was heated to 300 K and equilibrated for 1500 ps. The N189A mutant structure was created by modifying the wild-type crystal structure in CHARMM and then prepared following the same scheme as the wild-type MR.
Figure 1.

QM region. Panel (a) shows the atoms selected to be treated quantum-mechanically in this study. Panel (b) shows the relative structure of the QM region (blue) and the classical regions of NADH and FMN (red).
Transition Path Sampling
The transition path sampling (TPS) method22,23 was used to generate reactive trajectories capturing the MR-catalyzed hydride transfer reaction. TPS is a Markov chain Monte Carlo method that samples the phase space of reactive pathways between two metastable states—in this case, the reactant and the product. TPS requires several order parameters to rigorously define the two metastable states but does not require prior knowledge of the reaction coordinate.22 In this study, magnitudes of the bond cleavage (donor–hydride) distance and the bond-forming (acceptor–hydride) distance were used to define the reactant state and the product state. The system is defined to be in the reactant state if the donor–hydride distance is shorter than 1.40 Å and the acceptor–hydride distance is longer than 1.40 Å, and in the product state if the donor–hydride distance is longer than 1.40 Å and the acceptor–hydride distance is shorter than 1.40 Å. A previous study by Hay et al. stated that the hydride transfer reaction catalyzed by MR is not pH-dependent,31 indicating that the proton transfer from Arg238 to the reduced FMN molecule is not concerted with the hydride transfer reaction.31 Therefore, the two order parameters mentioned above are sufficient to describe the reactant and the product states of the reductive half-reaction catalyzed by MR. The initial reactive trajectory of the TPS ensemble was generated by applying a harmonic restraint force on the hydride to bias it being transferred to the acceptor atom. The TPS ensemble was generated by performing the following scheme iteratively. We selected randomly a structure and its momentum from the biased trajectory. The momentum is perturbed by adding a random perturbation chosen from the Boltzmann distribution. The total momentum is then rescaled back to the same magnitude as the initial momentum to ensure the conservation of energy. Two shooting trajectories were initiated from this configuration, propagating forwards and backwards in time, respectively, with the perturbed momentum. The new trajectory would be accepted if it connects the reactant state and the product state and rejected if not. The accepted new trajectory was then used as the seed for the next round of TPS. The time length of both shooting moves was set to be 250 fs, making the total length of reactive trajectories in the TPS ensemble 500 fs. It is worth pointing out that, though the MR-catalyzed hydride transfer reaction has been postulated to involve tunneling, a previous study using the quantum TPS methodology on the yeast alcohol dehydrogenase and the human heart lactate dehydrogenase has shown that involving the quantum component does not change the mechanistic detail of these two hydride transfer reactions.32 Therefore, the classical TPS method used in this research is sufficient in sampling the chemical step of the MR catalytic process.
Committor Analysis
In this study, the committor analysis method3,22 was used to determine the transition state of a reactive trajectory in the TPS ensemble. Several slices were selected from a reactive trajectory. From each slice, we initiated 50 shooting trajectories with randomly assigned momentum. The committor value of each slice is defined as the percentage of these shooting trajectories that end in the product state. The slice with a committor value of 0.5, indicating that shooting trajectories from this slice have equal probability to end in either the reactant or the product state, is the transition-state configuration of this trajectory.
Transition states of a TPS ensemble form the stochastic separatrix, from which the reaction coordinate (orthogonal to the separatrix) was determined using the committor distribution analysis method.3 This method has been used in several previous studies33,34 and was able to successfully identify the reaction coordinate of all systems studied. From transition-state structures on the separatrix, we initiated 250 fs trajectories with some degrees of freedom under constraint. The distribution of committor values of slices on these constraint trajectories was used as the metric to determine the reaction coordinate. If all degrees of freedom in the reaction coordinate have been restrained, then these constraint trajectories would stay on the separatrix, resulting in a committor value distribution centered at 0.5. If these trajectories failed to stay on the separatrix, new trajectories were initiated with further degrees of freedom constrained.
Clustering Method
One caveat of the committor distribution analysis method is that it cannot detect the existence of multiple reaction pathways in a TPS ensemble. Therefore, it is essential to separate potential clusters of reaction pathways prior to the committor distribution calculation, with the understanding that different atoms and residues may be part of the reaction coordinate in different reaction clusters. As will be discussed in further detail in the Results and Discussion section, we were able to identify and separate three reaction pathways of the N189A mutant by just measuring the donor–acceptor distance at the transition state. The wild type, however, requires a more advanced method to clearly separate its two reaction pathways. This need motivated us to use the Gaussian mixture model (GMM)35 to analyze the reactive trajectories in TPS ensembles of the wild type. GMM assumes that the observed T independent random variables were generated from a total of M independent K dimensional Gaussian distributions. The probability of any K dimensional vector x in the feature space to be sampled from the M Gaussian distributions, given the current ωi, μi, and ∑i, can be described by the equation below
where ωi is the weight of each normal distribution in the total and g(x|μi,∑i) is the ith K dimensional normal distribution parameterized by the mean vector μi and covariance matrix ∑i. The likelihood of all T independent observations under the current ωi, μi, and ∑i can then be calculated as
The likelihood is then maximized by the expectation maximization (EM) algorithm,36 which is implemented through the Python Scikit-learn package37 in this study. When the EM algorithm reaches convergence, the probability that an observation xi is generated from mi can be computed by
which gives the converged cluster assignment of all observations.
Results and Discussion
Transition Path Ensembles
We have generated 140 decorrelated reactive trajectories for the wild-type MR using the TPS method. In awareness of the fact that there might be multiple reaction pathways for the N189A mutant, we gathered more trajectories for the N189A mutant. A total of 250 decorrelated reactive trajectories for the N189 mutant were generated. Transition states for these trajectories were determined using the committor analysis method. We measured the donor–acceptor distance for all transition-state structures for both the wild type (Figure 2a) and the N189A mutant (Figure 2b). Amazingly, simply calculating this feature for all TPS trajectories reveals the existence of multiple reaction pathways for both enzymes. As shown in Figure 2, there exist two reaction pathways for the wild type (Figure 2a) and three reaction pathways for the N189A mutant (Figure 2b) with different donor–acceptor distance at the transition state. We did not observe frequent transfer between pathways in both enzymes. Actually, in both TPS ensembles, different pathways were naturally separated along the progress of TPS. In other words, there were relatively large free-energy barriers between the different reactive regions of phase space. According to previous experience,34 a committor distribution analysis calculation requires at least 50 decorrelated reactive trajectories. All three N189A mutant reaction pathways have enough population that, even if we discarded the “intermediate” trajectories for which it is hard to assign cluster membership, there are still enough trajectories left in each pathway for later analysis. The first reaction pathway of the wild-type MR, however, is sampled less frequently. A large population of the wild-type trajectories are intermediate trajectories if determined just by looking at the donor–acceptor distance. To more rigorously gather enough trajectory population for further analysis, we used the GMM method to help assign the intermediate trajectories to the two existing pathways. We found that other than the donor–acceptor distance, two other interatomic distances at the active site show different behavior. The NADH ribose ring–NADH nicotinamide distance (dashed line in Figure 2c) reduces significantly directly prior to the transition state of the first reaction pathway (solid black line in Figure 2e). In the second wild-type reaction pathway, however, this distance is reduced but later rises (solid red line in Figure 2e). Similarly, the His186–NADH amide group distance (dashed line in Figure 2d) reduces prior to the transition state in the first reaction pathway but remains largely unchanged during the reaction process of the second pathway (solid blue and green lines in Figure 2e). The different behavior of these two distances made them good descriptors to separate the two wild-type reaction pathways. To quantify this behavior, for each distance, we defined the “magnitude of reduction” of a distance as
where distanceprior is the collection of all values of interatomic distances in the 200 fs time window prior to the transition state. This metric essentially describes the largest magnitude of reduction in distance within the 200 fs time window (see Supporting Information (SI) Figure S1 for a detailed example). We used this treatment for both distances, together with the donor–acceptor distance, as input to a three-dimensional GMM model. This model assigned the first 56 wild-type reactive trajectories to belong to the first reaction pathway (the decision boundary is marked as the red solid vertical line in Figure 2a), while the remaining 84 reactive trajectories to be belonging to the second reaction pathway. Population of both wild-type pathways are abundant for the committor distribution analysis. By examining the two magnitude of reduction variables for both pathways (Figure 2f), we re-emphasize the major difference between the two pathways: the first pathway has larger magnitude of reduction for both distances (black diamonds in Figure 2f), while the second pathway has smaller magnitude of reduction for both distances (red squares in Figure 2f). As will be further justified by later calculation on the reaction coordinate, the two reaction pathways in the wild type behave differently in terms of the rate-promoting vibration from His186.
Figure 2.

Multiple reaction pathways in two TPS ensembles. Donor–acceptor distances at the transition state were calculated for the wild-type MR (a) and the N189A mutant (b) reactive trajectories. Two other important interatomic distances that behave differently between the two pathways were labeled as the black dashed line in (c) (NADH ribose ring O3′–NADH nicotinamide ring C5) and (d) (His186 NE2–NADH amide group O7). Time evolution of these two distances in representative trajectories from both pathways is shown in (e). The black and the red solid lines represent the NADH ribose ring O3′–NADH nicotinamide C5 distance in the first pathway and the second pathway, respectively. The blue and the green solid lines represent the His186 NE2–NADH amide group O7 distance in the first pathway and the second pathway, respectively. Transition-state slice is labeled by the dashed purple line. Panel (f) shows the clustering result of the three-dimensional GMM model, projected onto the two “magnitude of reduce” axes. The black diamonds represent trajectories assigned to the first pathway, while the red triangles represent trajectories assigned to the second pathway. Decision boundary acquired from this GMM model is labeled as the vertical solid red line in (a).
We calculated the average donor–acceptor, donor–hydride, and the acceptor–hydride distances for all five pathways at the transition state, as shown in Table 1. Reaction pathways are numbered in the order of their appearance during the TPS process. Specifically, for the two pathways in the wild type, the three distances we calculated are larger than the result computed by Delgado et al. in previous work.21 We believe this difference is caused by the difference in quantum mechanical methods as well as the method to identify transition states. In this study, it is the relative difference in transition-state structures between two enzymes and between different pathways that conveys more messages.
Table 1.
| wild-type pathway 1 | wild-type pathway 2 | N189A pathway 1 | N189A pathway 2 | N189A pathway 3 | |
|---|---|---|---|---|---|
| donor–acceptor distance (Å) | 2.81 ± 0.02 | 3.00 ± 0.03 | 3.13 ± 0.05 | 2.96 ± 0.03 | 2.79 ± 0.03 |
| donor–hydride distance (Å) | 1.49 ± 0.02 | 1.60 ± 0.02 | 1.70 ± 0.07 | 1.64 ± 0.03 | 1.48 ± 0.03 |
| acceptor–hydride distance (Å) | 1.34 ± 0.01 | 1.40 ± 0.01 | 1.47 ± 0.04 | 1.36 ± 0.03 | 1.33 ± 0.02 |
Though the chemical reaction we studied is a simple hydride transfer reaction, the QM region has a very complex geometry. To simplify the further discussion on relative positions and directions of conformational changes, we define a set of absolute “directions” using Figure 3a, an overlay of transition-state structures of the wild-type MR reaction pathways. The “left”, “right”, “up”, and “down” directions correspond to the left, right, up, and down directions when facing Figure 3a, respectively, while the “front” direction refers to the “out-of-the-screen” direction of Figure 3a and the “back” direction refers to “inside the screen”. The “horizontal axis (left–right)” would be the direction along the nicotinamide plane, while the “perpendicular axis” would be the direction orthogonal to the nicotinamide plane and the FMN isoalloxazine plane.
Figure 3.

Transition-state structures. Transition-state structures of wild-type MR pathway 1 (blue) and pathway 2 (red) are shown in (a). Transition-state structures of N189A mutant pathway 1 (blue), pathway 2 (red), and pathway 3 (yellow) are shown in (b). Panel (c) shows an overlay of the transition state between the wild-type pathway 1 (blue) and the N189A pathway 3 (red).
Since we were unable to calculate the reaction free-energy barrier of each pathway, it is hard to conclude directly from this perspective that any pathway is more favorable than the other. The following discusses this question through a comparison of transition-state structures of different reaction pathways. At transition states of both reaction pathways of the wild-type MR (Figure 3a), the NADH nicotinamide plane and the FMN isoalloxazine plane are parallel to each other, which is an ideal charge-transfer conformation of the MR proposed in earlier research.21,38 Difference in the transition-state donor–acceptor distance is caused by a subtle difference of the donor atom, which locates better “on top” of the acceptor atom and bends slightly out of the nicotinamide plane in the wild-type pathway 1 (colored blue in Figure 3a), compared to that in the wild-type pathway 2 (colored red in Figure 3a). Since the two planes are parallel to each other, we can deduce that wild-type pathway 1 is a more favorable reaction pathway due to its smaller transition-state donor–acceptor distance, which could lead to only one pathway being detectable by experiments. At transition states of N189A mutant reaction pathways (Figure 3b), the NADH nicotinamide plane and the FMN isoalloxazine plane are not parallel to each other. In all three N189A mutant reaction pathways, while the nicotinamide plane remains at a similar position (Figure 3b), the FMN isoalloxazine plane is distorted significantly to the “up-right” direction, sending the acceptor atom to the donor atom. By comparing transition states of N189A pathway 1 (colored blue in Figure 3b), pathway 2 (colored red in Figure 3b), and pathway 3 (colored yellow in Figure 3b), we found that the transition-state donor–acceptor distance is essentially modulated by the magnitude of the isoalloxazine distortion: the larger the isoalloxazine is distorted, the shorter the transition-state donor–acceptor distance. Therefore, we cannot attribute any of the three N189A reaction pathways to be more favorable than the others since a shorter transition-state donor–acceptor distance is gained at the price of further breaking the favorable parallel relationship between the two planes. This might indicate that all of these three pathways are experimentally detectable. The fact that we have discovered two reaction pathways for the wild-type MR (one of which is possibly more favorable) and three reaction pathways for the N189A mutant is in general agreement with the result from Pudney et al. mentioned earlier, in which a single reaction pathway for the wild-type MR and four reaction pathways for the N189A mutant were discovered.14 Pudney et al. were able to resolve hydride transfer rates via kinetic fitting for the wild-type MR and three of the four reaction pathways of the N189A mutant. One of the N189A pathways has a rate faster than that of the wild-type MR, while the other two are significantly slower than the wild type. By comparing the transition-state structures of the wild-type pathway 1 (colored blue in Figure 3c) and the N189A mutant pathway 3 (colored red in Figure 3c), we re-emphasize that the NADH nicotinamide plane and the FMN isoalloxazine plane are not parallel in N189A reaction pathways. Therefore, we cannot make a direct comparison of the hydride transfer rate between the wild-type MR and the N189A mutant since the N189A pathway 3, which has the shortest transition-state donor–acceptor distance, is in an unfavorable charge-transfer geometry.
Reaction Coordinates
Reaction coordinates of the five reaction pathways were determined separately using the committor distribution analysis method. As shown in Figure 4a,e,g,i, besides the wild-type pathway 2, all other four reaction pathways fail to achieve a committor distribution centered at 0.5 with only the QM region under constraint. The wild-type pathway 2, however, achieved such a committor distribution with only the QM region under constraint (Figure 4c). We were able to find a further set of constraints, which are essentially the reaction coordinate of these pathways, that centered the committor distribution of all other four pathways at or about 0.5. Committor distributions of these reaction pathways with the reaction coordinate under constraint are shown in Figure 4b,f,h,j. The reaction coordinate of the wild-type MR pathway 1 includes the QM region and the NADH and the FMN classical regions, Thr32, Gln104, Trp106, Leu147, His186, Asn189, Arg238, and Tyr356. Positions of these residues relative to the QM region are shown in Figure S2. The reaction coordinate of the N189A mutant pathways 1 and 3 includes the QM region and the NADH and the FMN classical regions. The reaction coordinate of the N189A mutant pathway 2 includes the QM region and the NADH and the FMN classical regions, His186. It becomes immediately obvious that, besides that of the wild-type pathway 2, reaction coordinates of all other pathways share a set of atoms: the QM region and the NADH and the FMN classical regions. Applying this set of constraints to the wild-type pathway 2, however, does not seem to improve its committor distribution (Figure 4d).
Figure 4.


Committor distribution of five reaction pathways. Panels (a), (c), (e), (g), and (i) show the committor distribution of wild-type pathway 1, wild-type pathway 2, N189A pathway 1, N189A pathway 2, and N189A pathway 3, respectively, with only the QM region under constraint. Panel (b) shows the committor distribution of the wild-type pathway 1 with the QM region, NADH and FMN classical regions, and eight amino acids under constraint. Panels (d), (f), and (j) show the committor distribution of the wild-type pathway 2, N189A pathway 1, and N189A pathway 3, respectively, with the QM region and NADH and FMN classical regions under constraint. Panel (h) shows the committor distribution of N189A pathway 2 with the QM region, NADH and FMN classical regions, and His186 under constraint.
Conformational Changes and the Rate-Promoting Vibration
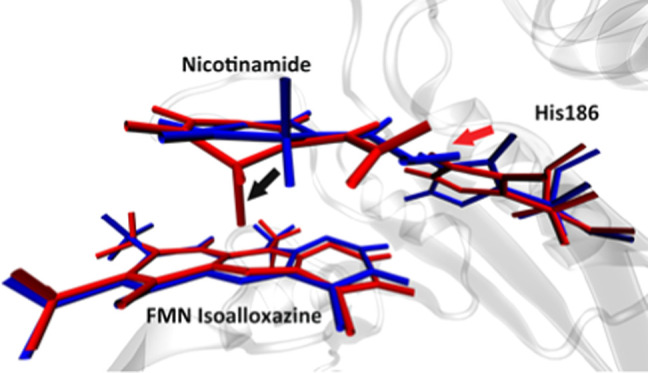
We found one protein motion coupled to the catalytic process in only one pathway, the wild-type MR pathway 1, while all other reaction pathways do not have any protein motion involved in the reaction progress. Due to the complexity of the morphinone reductase active site, the interatomic distance is not always a good descriptor of the conformational change process. Therefore, active site conformational changes in this section will be presented by overlaying multiple structures along the time evolution of reactive trajectories. The rate-promoting vibration in the wild-type pathway 1 comes from His186. As described in Figure 5a, looking from the top of the NADH nicotinamide plane, the side chain of His186 starts to swing toward the NADH nicotinamide, at 300 fs prior to the transition state (colored blue in Figure 5a), and continues to push on the nicotinamide until the transition state (colored red in Figure 5a). The direction of the His186 side-chain motion is labeled by the red arrow in Figure 5a. An apparent effect of this motion is to push the nicotinamide molecule to move along the horizontal axis toward its left side (labeled by the black arrow in Figure 5a). Another important effect of the His186 pushing motion in the wild-type pathway 1 is to induce a conformational change of the NADH nicotinamide ring, which eventually result in the “out-of-plane” bending motion of the donor atom. The NADH classical ribose ring and Tyr356 (both colored gray in Figure 5b), both of which barely move along the progress of the reaction (SI, Figure S3a), together form a solid “wall” to the left (Tyr356) and top-left (NADH ribose ring) sides of the NADH pyridine ring. Recall that the NADH nicotinamide is being pushed to its left side by the His186 side chain from 300 fs prior to the transition state all the way till the transition state. When the pyridine ring moves to impact the wall formed by Tyr356 and the NADH ribose ring (100 fs prior to the transition state, colored red in Figure 5b), its momentum toward the left side functions to bend the middle part of the pyridine ring (specifically the donor atom) outside of its original plane, as shown by the transition-state structure (colored yellow in Figure 5b). An analogue of this effect is to push a piece of paper perpendicular to a wall. If one keeps pushing after the paper has impacted the wall, the paper will bend in its middle inevitably. The fact that both Tyr356 and the NADH ribose ring are in the reaction coordinate of the wild-type MR pathway 1 means that maintaining their geometry is crucial to the progress of the reaction, which side justified the mechanism we proposed. Since the pushing motion from His186 functions to reduce the donor–acceptor distance, though in a complicated way, we can determine it to be a rate-promoting vibration of the wild-type morphinone reductase.
Figure 5.

Conformation change process of the two enzymes. Panel (a) shows the motion from His186 and its effect on the nicotinamide in the wild-type pathway 1, on the start (blue, 300 fs prior to the transition state) and the end (red, the transition state) of the motion, viewing from the top of the nicotinamide ring. The direction of the His186 motion is labeled by the red arrow, and the direction of the effect of the motion is shown by the black arrow. The His186 imidazole ring is labeled by spheres. The direction of the His186 imidazole ring center-of-mass movement is shown as the black dashed line. Panel (b) shows the detailed conformation change of the nicotinamide ring, viewing from the front direction, at 300 fs prior to the transition state (blue), 100 fs prior to the transition state (red), and the transition state (yellow). The NADH ribose ring and Tyr356 are colored gray. The direction of the movement of the acceptor atom is labeled by the black arrow. Panel (c) shows the His186 side-chain center-of-mass root-mean-square deviation (RMSD) using the transition state as a reference, in a 600 fs window centered at each pathway’s transition state. The black, red, blue, green, and orange solid lines represent the wild-type pathway 1, the wild-type pathway 2, the N189A mutant pathway 1, the N189A mutant pathway 2, and the N189A mutant pathway 3, respectively. Panel (d) shows three structures of N189A pathway 3 taken at 400 fs (colored blue) and 200 fs prior to the transition state (colored red and yellow). The NADH classical ribose ring and the Tyr356 residue are colored in dark gray. The donor atom and the acceptor atom are labeled by spheres. The direction of the acceptor atom movement is labeled by the black arrow. The RMSD of the center of mass of the FMN isoalloxazine middle ring is shown in (e), for the wild-type pathway 1 (black), the wild-type pathway 2 (red), the N189A mutant pathway 1 (blue), the N189A mutant pathway 2 (green), and the N189A mutant pathway 3 (yellow), in a 600 fs time window centered at the transition state. Panel (f) shows the overlay of two structures: 100 fs prior to transition state of the wild-type MR pathway 1 (blue) and 200 fs prior to the transition state of the N189A mutant pathway 3 (red). The black and red arrows represent the relative direction between the wall formed by Tyr356 and the NADH ribose ring, and the nicotinamide pyridine, of the wild type and the N189A mutant, respectively.
To quantify the magnitude of the His186 motion, we calculated the center of mass of the His186 imidazole ring (labeled by spheres in Figure 5a). The time evolution of the His186 root-mean-square deviation (RMSD) with respect to the transition state of each reaction pathway essentially shows how much the His186 side chain has moved during the reaction progress. To guarantee that we have covered the whole process of this motion, we acquired 5000 fs long reactive trajectories for each pathway and performed the His186 RMSD calculation on these trajectories. The result of the RMSD calculation centered at a time window 300 fs prior to and after the transition state of each pathway is shown in Figure 5c. The His186 center of mass in the wild-type pathway 1 (solid black line in Figure 5c) moved approximately 1 Å during the 300 fs time window. In all other four pathways, the His186 side chain moves to a far less extent prior to the transition state, meaning that the motion from His186 has disappeared. A very strong support to the rate-promoting vibration from His186 that we proposed earlier is that, with the disappearance of the His186 pushing motion, both effects mentioned above caused by the His186 motion have disappeared in other reaction pathways. First, NADH nicotinamide no longer moves along the horizontal axis in other four reaction pathways, but instead fluctuates along the perpendicular axis, either as a rotation of the pyridine ring (N189A pathway 2 and pathway 3, Figure 5d) or as a translational move along the perpendicular axis (wild-type pathway 2, Figure S3b). Second, no out-of-plane bending motion of the donor atom could be found for these reaction pathways. Simultaneously, His186 has been eliminated from the reaction coordinate, meaning that without the pushing motion, His186 is no longer contributing to the progress of the hydride transfer. Interestingly, Tyr356, which forms the solid wall to the left side of the nicotinamide pyridine ring, has also been eliminated from the reaction coordinate of the other four reaction pathways. This observation further supports the importance of Tyr356 in the conformational change process induced by the rate-promoting vibration in the wild-type pathway 1: without the His186 pushing motion, the wall lost its purpose. There exist two minor exceptions in the other four reaction pathways, but these do not change the general picture of the elimination of His186 pushing motion mentioned above. The first exception is the N189A pathway 1, in which the nicotinamide molecule moves along the horizontal axis (SI, Figure S3c) without the His186 pushing motion. However, since the nicotinamide ring remains at the same location for all N189A reaction pathways, this move of the nicotinamide molecule in the N189A pathway 1 is a fluctuation that corrects an unfavorable geometry. The second exception is the N189A pathway 2. Though His186 is in the reaction coordinate of N189A pathway 2, neither the pushing motion nor its effect (SI, Figure S3d) existed, meaning that His186 is not contributing to the reaction progress through the protein motion.
The existence of two reaction pathways in the wild-type MR, one with a rate-promoting vibration and one without, gives a perfect example to clearly isolate the contribution of a rate-promoting vibration, which is to reduce the transition-state donor–acceptor distance by 0.19 Å, as listed in Table 1. We have also managed to answer an unresolved issue in previous studies: the cause of the out-of-plane bending motion of the donor atom, which has remained a mystery, since there does not exist any amino acids on top of the donor atom. The morphinone reductase is indeed an amazing enzyme that can transfer a rate-promoting vibration along a direction perpendicular to the donor–acceptor axis to eventually compress the active site along the donor–acceptor axis.
When looking at the FMN isoalloxazine plane, we found a distortion of the FMN isoalloxazine molecule, specifically its middle ring, along the reaction progress, which sends the acceptor atom to the up-right direction toward the donor atom. While in wild-type reaction pathways, this distortion is a minor movement of the acceptor N5 atom (Figure 5a), in N189A mutant reaction pathways, the distortion movement of the FMN isoalloxazine is much more significant, as shown by the conformational change process in N189A pathway 3 (Figure 5d). Similar to the idea used previously when discussing the His186 pushing motion, we made use of the center-of-mass RMSD, this time of the FMN isoalloxazine middle ring (labeled by spheres in Figure 5d) to quantify the magnitude of how much the FMN isoalloxazine has moved during the reaction progress. Figure 5e shows the center-of-mass RMSD of the isoalloxazine middle ring, using the transition state of each pathway as reference, in a 600 fs time window centered at the transition state. The magnitude of the distortion ranks from the largest to the smallest in the order of N189A pathway 3 (orange), N189A pathway 2 (green), N189A pathway 1 (blue), wild-type pathway 1 (red), and wild-type pathway 2 (black), which is exactly the same rank as how much the FMN isoalloxazine has been distorted at reaction transition states (Figure 3). The FMN isoalloxazine holds an almost identical planar geometry in all five reaction pathways prior to this distortion (SI, Figure S4), meaning that the FMN isoalloxazine in all five reaction pathways comes from a similar geometry but is distorted with a different magnitude during the reaction progress, which results in its different position at reaction transition states. However, in all five reaction pathways, we did not find any protein residues located “underneath” the isoalloxazine plane to be involved in the reaction coordinate, nor did we found any protein motions that would push the isoalloxazine plane to distort it. The different distortion movements we observed are essentially different regimes of the phase space of the FMN isoalloxazine sampled by TPS, not because of any difference in protein motions.
Further Discussion
Having fully discussed the rate-promoting vibration and its effect, it is crucial to learn how the N189A point mutation influences the reaction progress and eliminates the rate-promoting vibration, which would be a valuable lesson for further tasks in protein design.39,40 A direct intuition of the potential effect of the N189A point mutation would be that it eliminates the side chain of the Asn189, which locates at the “back-right” direction of the NADH pyridine ring, giving the pyridine ring access to more possible conformations. As shown in Figure 5f, an overlay of the wild-type pathway 2 (colored blue in Figure 5f) and the N189A mutant pathway 3 (colored red in Figure 5f) at the maximum of each nicotinamide pyridine’s fluctuation along the perpendicular axis, looking from the top of the nicotinamide plane, indeed there exists more allowable space behind the nicotinamide pyridine ring. The nicotinamide pyridine ring is located more to the back-right direction compared to the wild type and is also further away from the FMN isoalloxazine plane. Aside from this difference, we notice that both Tyr356 and the NADH ribose ring in N189A reaction pathways locate more “forward” compared to those in the wild-type pathways. Together, the relative position of the wall formed by Tyr356 and the NADH ribose ring with respect to the nicotinamide pyridine ring has changed to be more forward in N189A reaction pathways (relative direction labeled by the red arrow in Figure 5f), not directly to the left and top-left sides of the NADH pyridine ring (relative direction labeled by the black arrow in Figure 5f). This change of relative position has two effects. First, it breaks the parallel relationship between the NADH nicotinamide plane and the FMN isoalloxazine plane. In wild-type reaction pathways, the NADH pyridine ring is confined by the NADH ribose ring to remain being parallel to the FMN isoalloxazine. However, in N189A reaction pathways, the NADH ribose ring can only confine the forward corner of the NADH pyridine ring. This change of the confinement results in the fluctuation of the nicotinamide pyridine in N189A reaction pathways to be a rotation that could lean the pyridine plane forward toward the isoalloxazine plane (Figure 5d), instead of a minor translational fluctuation along the perpendicular axis in the wild-type MR (SI, Figure S3b). Second, the change of relative positions of the wall and the nicotinamide pyridine eliminates the His186 rate-promoting vibration. Recalling that in the wild-type pathway 1, the His186 pushing motion causes a latter conformational change of the pyridine ring by pushing it toward a static wall formed by Tyr356 and the NADH ribose ring to the left side of the pyridine ring. Should there exist a His186 pushing motion in N189A pathways that pushes the nicotinamide ring to its left, the wall would no longer be in the opposite direction of the nicotinamide plane movement. Since the pyridine ring in N189A mutant reaction pathways is located further away from the FMN isoalloxazine plane compared to that in the wild type, even at reaction transition states (recall Figure 3c), the FMN isoalloxazine would have to compensate for this gain in donor–acceptor distance by sending the acceptor atom more “upwards” to the donor. This helps to explain the larger magnitude of distortion of the FMN isoalloxazine in N189A reaction pathways.
One immediate follow-up question is, is this difference in the active site geometry caused by the N189A point mutation a regime of the rare event phase space sampled by TPS, which would only occur during the reaction process, or is it a stable change of the active site geometry induced by the N189A mutation? To answer this question, we acquired 50 ps long QM/MM trajectories for each enzyme by shooting backwards in time from reaction transition states and use them as reactant states. We defined three important structure features to characterize the reactant state of both enzymes. The distance between the center of mass of the NADH ribose ring (colored yellow in Figure 6a) and the center of mass of the NADH pyridine ring (colored blue in Figure 6a) describes the relative geometry of the wall and the pyridine. The distance between the center of mass of the pyridine ring (colored blue in Figure 6a) and the center of mass of the isoalloxazine middle ring (colored red in Figure 6a) describes how far away the two planes are from each other. The difference between the NADH C4–FMN N5 distance and the NADH N1–FMN N10 distance (referred to as ddiff in later discussion) is a good measurement of how parallel the two planes are, proposed in a previous study.21 As shown in Figure 6b, when the two planes are parallel to each other, ddiff is small, at 0.90 Å; when the two planes are unparallel to each other, as in Figure 6c, the N1–N10 distance would be significantly larger than the C4–N5 distance, making ddiff significantly larger, in this case at 2.05 Å.
Figure 6.

Important structure features describing the active site geometry. Panel (a) shows the two important interplane distances: the distance between the center of mass of the NADH ribose ring (yellow spheres) and the nicotinamide pyridine ring (blue spheres), and between the nicotinamide pyridine ring and the middle ring of the FMN isoalloxazine. Distances between the center of mass are labeled by the dashed black lines. Panels (b) and (c) show the two bond distances used to describe the parallel relationship between the NADH nicotinamide and the FMN isoalloxazine: the nicotinamide N1–isoalloxazine N10 distance and the nicotinamide C4–isoalloxazine N5 distance. In both figures, these atoms are labeled by spheres, C4–N5 and N1–N10, and the interplane distances are labeled by the black dashed lines. Panels (d) and (e) show the probability density distribution of the NAHD ribose ring–pyridine ring distance and the pyridine ring–isoalloxazine distance of the reactant states, respectively. Panel (f) shows the probability density distribution of ddiff of reactant states. In panels (d)–(f), the probability density distribution of the wild-type MR is labeled in blue, and the probability density distribution of the N189A mutant is labeled in orange.
Pudney et al. suggested in an earlier study from the result of classical MD simulations on reactant states that multiple reaction pathways in the N189A mutant are originated from multiple reactive configurations. The N189A point mutation gave access to these configurations that were not visited by the wild-type MR by eliminating the Asn189–NADH nicotinamide hydrogen bond, not by freeing up more space in the active site.14 We compared the probability density distributions of the three important structure features defined above between reactant states of all five reaction pathways (SI, Figure S5) and found that they are generally identical among reaction pathways belonging to the same enzyme but is significantly different between the two enzymes. Therefore, we cannot draw from our simulations the conclusion that different sampled reaction pathways of either enzyme originate from different reactant states. The origin of multiple reaction pathways induced by the N189A point mutation can be explained by inducing an unfavorable active site geometry at reaction transition states. Since transition-state structures of the N189A mutant are less favorable compared to those of the wild type due to the breaking of the parallel relationship between the two planes, the free-energy landscape crossing the reaction barrier might be more complicated, making it possible for multiple reaction pathways to exist. We did gain an understanding of the difference between the two enzymes from these structure features. The probability density distributions of the three structure features of reactant states from the two enzymes are shown in Figure 6d–f, respectively, in which blue is the wild-type reaction pathways and orange is the N189A mutant. From these probability density distributions, we can conclude that an average reactant state of N189A, compared to that of the wild type, would have a larger NADH ribose ring–pyridine ring distance and a larger interplane distance, with the nicotinamide ring and the FMN isoalloxazine ring being more unparallel to each other. An overlay of “average” reactant structures (structures possessing the average values of the three features mentioned above) of the wild-type pathway (colored blue in Figure 7a) and the N189A mutant pathway (colored red in Figure 7a) is shown in Figure 7a. Clearly, the difference in the active site geometry between the reactant states of the two enzymes is not dissimilar from that during the reaction progress of the two enzymes (Figure 5f). Therefore, the change of relative positions between the wall and the pyridine ring, as well as the breaking of the parallel relationship between the two planes, is indeed a stable change of the active site induced by the N189A point mutation, not rare events sampled by TPS.
Figure 7.

Comparison between the active site of the two enzymes. Panel (a) shows the comparison between reactant state structures with average features, with the wild-type MR structure labeled in blue and the N189A mutant structure labeled in red. Panel (b) shows the comparison of protein residues in the MR active site, with the wild-type MR structure labeled in blue and the N189A mutant structure labeled in red. The distance between the Asn189/Ala189 carbon α and the Gly357 carbon α is marked as the dashed black line. Panel (c) shows the probability density distribution of the Asn189/Ala189 carbon α–Gly357 carbon α distance of the reactant states. The wild type is labeled in blue, and the N189A mutant is labeled in orange.
Asn189 could be hydrogen-bonded to the nicotinamide O7 atom (the black dashed line, Figure 7a); however, this hydrogen bond or its removal does not seem to be the major cause of the change in active site structures, as Figure S6 shows an example structure of the wild-type reactant state, in which this hydrogen bond is entirely broken, but the pyridine ring is still confined. We hereby restate that one effect of the N189A point mutation is to create more free space behind the nicotinamide pyridine, not by eliminating the hydrogen bond. Interestingly, beyond this effect, the protein backbone of the active site is also altered by the N189A point mutation. As shown in Figure 7b, the overlay of the same structures in Figure 7a but viewing from the top of the NADH molecule, the protein backbone of both Gly357 and Ala189, which can be viewed as two barriers that confine the whole NADH molecule, retracts slightly in the N189A mutant compared to that in the wild type. We measured the distance between the carbon α of these two residues in reactant states as a measurement of the size of allowable space between the two barriers that confine the NADH. The probability density distribution of this distance is shown in Figure 7c. Clearly, this distance is significantly larger for the N189A mutant. Essentially, in both enzymes, the NADH ribose ring is folded back to the nicotinamide. The difference in the active site geometry induced by the N189A point mutation, both at reactant states and during the reaction progress, is essentially to “open up” the folded NADH molecule. It is unclear, however, how a point mutation, which only eliminates a potential hydrogen bond to the NADH molecule, could also cause the protein backbone surrounding the active site to be altered so significantly. However, this result does teach us to be careful when designing active site mutations to make better-performing enzymes. A motivation of making a point mutation may result in an unexpected change of the active site protein backbones.
Conclusions
In this study, we successfully answered the two major unresolved questions in the MR catalytic mechanism. We have discovered two reaction pathways (one being possibly dominant) for the wild type and three reaction pathways for the N189A mutant. The N189A point mutation modulated the conformational landscape of the MR active site to induce multiple reaction pathways. In prior works using a similar simulation approach on other enzyme systems, we have not detected the existence of completely distinct reaction pathways. However, a couple of recent studies have found this behavior in many other enzyme systems, including paraoxonase 1,41,42 lactate dehydrogenase,43 and even nanoparticle-catalyzed reactions.44 Therefore, multiple reaction pathways may exist broadly in nature and should be very carefully investigated when studying an enzyme catalysis process.
A rate-promoting vibration from His186 that compresses the donor–acceptor distance was found in the wild-type pathway 1, while all other pathways do not have any protein motions coupled to its catalytic process. Besides, we have learned a couple of valuable lessons from this research. First, a rate-promoting vibration could be perpendicular to the donor–acceptor axis, as in the case of MR, but still function to compress the donor–acceptor axis. Second, an RPV could be eliminated not by changing residues directly involved in the RPV but by altering geometries of the other degrees of freedom that is not directly involved in the RPV itself, but is essential to the conformational change induced by the RPV. Third, unsurprisingly, the influence of even a single point mutation to the active site could be subtle but still important.
Acknowledgments
All computer simulations were performed at the University of Arizona High Performance Computing Center on a Lenovo NeXtScale nx360 M5 supercomputer.
Glossary
Abbreviations
- MR
morphinone reductase
- QM/MM
quantum mechanics/molecular mechanics
- TPS
transition path sampling
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03472.
Illustration of the magnitude of reduction used as GMM inputs; reaction coordinate of the wild-type MR reaction pathway 1; important conformation change process along the reaction progress; overlay of the FMN isoalloxazine plane geometry prior to its distortion of all five reaction pathways; probability density distribution of the three structure features in all five reaction pathways; active site geometry with a broken Asn189–NADH nicotinamide hydrogen bond (PDF)
We acknowledge the support of the National Institute of Health Grant R01GM127594.
The authors declare no competing financial interest.
Supplementary Material
References
- Hay S.; Scrutton N. S. Good vibrations in enzyme-catalysed reactions. Nat. Chem. 2012, 4, 161–168. 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- Schramm V. L.; Schwartz S. D. Promoting vibrations and the function of enzymes. Emerging theoretical and experimental convergence. Biochemistry 2018, 57, 3299–3308. 10.1021/acs.biochem.8b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D.; Schwartz S. D. Protein Dynamics and Enzymatic Chemical Barrier Passage. J. Phys. Chem. B 2011, 115, 15147–15158. 10.1021/jp207876k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Ma S.; Major D. T.; Nam K.; Pu J.; Truhlar D. G. Mechanisms and free energies of enzymatic reactions. Chem. Rev. 2006, 106, 3188–3209. 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. Chemical achievement and hope for the future. Am. Sci. 1948, 36, 51–58. [PubMed] [Google Scholar]
- Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Fried S. D.; Boxer S. G. Electric fields and enzyme catalysis. Annu. Rev. Biochem. 2017, 86, 387–415. 10.1146/annurev-biochem-061516-044432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D.; Ge X. X.; Schramm V. L.; Schwartz S. D. Mass Modulation of Protein Dynamics Associated with Barrier Crossing in Purine Nucleoside Phosphorylase. J. Phys. Chem. Lett. 2012, 3, 3538–3544. 10.1021/jz301670s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davarifar A.; Antoniou D.; Schwartz S. D. The Promoting Vibration in Human Heart Lactate Dehydrogenase Is a Preferred Vibrational Channel. J. Phys. Chem. B 2011, 115, 15439–15444. 10.1021/jp210347h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez S.; Antoniou D.; Schramm V. L.; Schwartz S. D. Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J. Am. Chem. Soc. 2004, 126, 15720–15729. 10.1021/ja0457563. [DOI] [PubMed] [Google Scholar]
- Bruce N. C.; Wilmot C. J.; Jordan K. N.; Trebilcock A. E.; Stephens L. D. G.; Lowe C. R. Microbial degradation of the morphine alkaloids: identification of morphinone as an intermediate in the metabolism of morphine by Pseudomonas putida M10. Arch. Microbiol. 1990, 154, 465–470. 10.1007/BF00245229. [DOI] [PubMed] [Google Scholar]
- Pang J.; Hay S.; Scrutton N. S.; Sutcliffe M. J. Deep tunneling dominates the biologically important hydride transfer reaction from NADH to FMN in morphinone reductase. J. Am. Chem. Soc. 2008, 130, 7092–7097. 10.1021/ja800471f. [DOI] [PubMed] [Google Scholar]
- Pudney C. R.; Johannissen L. O.; Sutcliffe M. J.; Hay S.; Scrutton N. S. Direct Analysis of Donor–Acceptor Distance and Relationship to Isotope Effects and the Force Constant for Barrier Compression in Enzymatic H-Tunneling Reactions. J. Am. Chem. Soc. 2010, 132, 11329–11335. 10.1021/ja1048048. [DOI] [PubMed] [Google Scholar]
- Pudney C. R.; Hay S.; Pang J.; Costello C.; Leys D.; Sutcliffe M. J.; Scrutton N. S. Mutagenesis of morphinone reductase induces multiple reactive configurations and identifies potential ambiguity in kinetic analysis of enzyme tunneling mechanisms. J. Am. Chem. Soc. 2007, 129, 13949–13956. 10.1021/ja074463h. [DOI] [PubMed] [Google Scholar]
- Pudney C. R.; McGrory T.; Lafite P.; Pang J.; Hay S.; Leys D.; Sutcliffe M. J.; Scrutton N. S. Parallel pathways and free-energy landscapes for enzymatic hydride transfer probed by hydrostatic pressure. ChemBioChem 2009, 10, 1379–1384. 10.1002/cbic.200900071. [DOI] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. Incorporation of hydrostatic pressure into models of hydrogen tunneling highlights a role for pressure-modulated promoting vibrations. Biochemistry 2008, 47, 9880–9887. 10.1021/bi8005972. [DOI] [PubMed] [Google Scholar]
- Hay S.; Sutcliffe M. J.; Scrutton N. S. Promoting motions in enzyme catalysis probed by pressure studies of kinetic isotope effects. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 507–512. 10.1073/pnas.0608408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudney C. R.; Hay S.; Levy C.; Pang J.; Sutcliffe M. J.; Leys D.; Scrutton N. S. Evidence to support the hypothesis that promoting vibrations enhance the rate of an enzyme catalyzed H-tunneling reaction. J. Am. Chem. Soc. 2009, 131, 17072–17073. 10.1021/ja908469m. [DOI] [PubMed] [Google Scholar]
- Pudney C. R.; Hay S.; Sutcliffe M. J.; Scrutton N. S. α-Secondary isotope effects as probes of “tunneling-ready” configurations in enzymatic H-tunneling: Insight from environmentally coupled tunneling models. J. Am. Chem. Soc. 2006, 128, 14053–14058. 10.1021/ja0614619. [DOI] [PubMed] [Google Scholar]
- Johannissen L. O.; Scrutton N. S.; Sutcliffe M. J. How does pressure affect barrier compression and isotope effects in an enzymatic hydrogen tunneling reaction?. Angew. Chem., Int. Ed. 2011, 50, 2129–2132. 10.1002/anie.201006668. [DOI] [PubMed] [Google Scholar]
- Delgado M.; Görlich S.; Longbotham J. E.; Scrutton N. S.; Hay S.; Moliner V.; Tuñón I. Convergence of theory and experiment on the role of preorganization, quantum tunneling, and enzyme motions into flavoenzyme-catalyzed hydride transfer. ACS Catal. 2017, 7, 3190–3198. 10.1021/acscatal.7b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis P. G.; Chandler D.; Dellago C.; Geissler P. L. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- Dellago C.; Bolhuis P. G.; Csajka F. S.; Chandler D. Transition path sampling and the calculation of rate constants. J. Chem. Phys. 1998, 108, 1964 10.1063/1.475562. [DOI] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM - A Program For Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Freddolino P. L.; Dittrich M.; Schulten K. Dynamic switching mechanisms in LOV1 and LOV2 domains of plant phototropins. Biophys. J. 2006, 91, 3630–3639. 10.1529/biophysj.106.088609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A. P.; Radom L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Moller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. A 1996, 100, 16502–16513. 10.1021/jp960976r. [DOI] [Google Scholar]
- Dametto M.; Antoniou D.; Schwartz S. D. Barrier Crossing in Dihydrofolate Reductasedoes not involve a rate-promoting vibration. Mol. Phys. 2012, 110, 531–536. 10.1080/00268976.2012.655337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson J. E.; Schwartz S. D. Evolution Alters the Enzymatic Reaction Coordinate of Dihydrofolate Reductase. J. Phys. Chem. B 2015, 119, 989–996. 10.1021/jp506373q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S.; Pudney C. R.; Sutcliffe M. J.; Scrutton N. S. Solvent as a probe of active site motion and chemistry during the hydrogen tunnelling reaction in morphinone reductase. ChemPhysChem 2008, 9, 1875–1881. 10.1002/cphc.200800303. [DOI] [PubMed] [Google Scholar]
- Dzierlenga M. W.; Antoniou D.; Schwartz S. D. Another look at the mechanisms of hydride transfer enzymes with quantum and classical transition path sampling. J. Phys. Chem. Lett. 2015, 6, 1177–1181. 10.1021/acs.jpclett.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga M. J.; Dzierlenga M. W.; Schwartz S. D. Structurally Linked Dynamics in Lactate Dehydrogenases of Evolutionarily Distinct Species. Biochemistry 2017, 56, 2488–2496. 10.1021/acs.biochem.7b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoi I.; Motley M. W.; Antoniou D.; Schramm V. L.; Schwartz S. D. Enzyme homologues have distinct reaction paths through their transition states. J. Phys. Chem. B 2015, 119, 3662–3668. 10.1021/jp511983h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds D. A. Gaussian Mixture Models. Encycl. Biom. 2009, 741 10.1007/978-0-387-73003-5_196. [DOI] [Google Scholar]
- Dempster A. P.; Laird N. M.; Rubin D. B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 1977, 39, 1–22. [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Fox K. M.; Karplus P. A. Old yellow enzyme at 2 Å resolution: overall structure, ligand binding, and comparison with related flavoproteins. Structure 1994, 2, 1089–1105. 10.1016/S0969-2126(94)00111-1. [DOI] [PubMed] [Google Scholar]
- Zoi I.; Antoniou D.; Schwartz S. D. Incorporating Fast Protein Dynamics into Enzyme Design: A Proposed Mutant Aromatic Amine Dehydrogenase. J. Phys. Chem. B 2017, 121, 7290–7298. 10.1021/acs.jpcb.7b05319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoi I.; Suarez J.; Antoniou D.; Cameron S. A.; Schramm V. L.; Schwartz S. D. Modulating Enzyme Catalysis through Mutations Designed to Alter Rapid Protein Dynamics. J. Am. Chem. Soc. 2016, 138, 3403–3409. 10.1021/jacs.5b12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David M.; Elias M.; Filippi J.-J.; Duñach E.; Silman I.; Sussman J. L.; Tawfik D. S. Catalytic versatility and backups in enzyme active sites: the case of serum paraoxonase 1. J. Mol. Biol. 2012, 418, 181–196. 10.1016/j.jmb.2012.02.042. [DOI] [PubMed] [Google Scholar]
- Ben-David M.; Soskine M.; Dubovetskyi A.; Cherukuri K.-P.; Dym O.; Sussman J. L.; Liao Q.; Szeler K.; Kamerlin S. C. L.; Tawfik D. S. Enzyme Evolution: An Epistatic Ratchet versus a Smooth Reversible Transition. Mol. Biol. Evol. 2020, 37, 1133–1147. 10.1093/molbev/msz298. [DOI] [PubMed] [Google Scholar]
- Reddish M. J.; Callender R.; Dyer R. B. Resolution of submillisecond kinetics of multiple reaction pathways for lactate dehydrogenase. Biophys. J. 2017, 112, 1852–1862. 10.1016/j.bpj.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R.; Mao X.; Sun X.; Chen P. Analogy between enzyme and nanoparticle catalysis: a single-molecule perspective. ACS Catal. 2019, 9, 1985–1992. 10.1021/acscatal.8b04926. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.