

Abstract
Calcium oxalate (CaOx) crystal deposition within the tubules is often a perplexing finding on renal biopsy of both native and transplanted kidneys. Understanding the underlying causes may help diagnosis and future management. The most frequent cause of CaOx crystal deposition within the kidney is hyperoxaluria. When this is seen in native kidney biopsy, primary hyperoxaluria must be considered and investigated further with biochemical and genetic tests. Secondary hyperoxaluria, for example due to enteric hyperoxaluria following bariatric surgery, ingested ethylene glycol or vitamin C overdose may also cause CaOx deposition in native kidneys. CaOx deposition is a frequent finding in renal transplant biopsy, often as a consequence of acute tubular necrosis and is associated with poorer long-term graft outcomes. CaOx crystal deposition in the renal transplant may also be secondary to any of the causes associated with this phenotype in the native kidney. The pathophysiology underlying CaOx deposition is complex but this histological phenotype may indicate serious underlying pathology and should always warrant further investigation.
Keywords: Calcium oxalate, Oxalosis, Primary hyperoxaluria, Enteric hyperoxaluria
Introduction
Calcium oxalate (CaOx) crystal deposition within the nephron [1–3], tubular cells [4] or interstitium [5] are sometimes found by the histopathologist examining a renal biopsy. CaOx, along with calcium phosphate (CaP) deposition may lead to nephrocalcinosis [6, 7], although in practice CaOx crystal deposition is often referred to as renal oxalosis or oxalate nephropathy. Bagnasco et al. examined biopsies of both native and transplanted kidneys over the course of 6 years [6]. Overall, 1% of native kidney biopsies and 4% of transplanted kidney biopsies demonstrated CaOx crystal deposition.
The presence of CaOx crystal deposition within a renal biopsy may indicate serious underlying pathology and indicate an underlying diagnosis that may not have previously been considered [7, 8]. Of particular relevance are the primary hyperoxalurias (PH), which may cause end stage kidney disease and may recur following kidney transplantation. The diagnosis of PH has potentially life-changing effects with a broad range of treatment options, up to and including dual kidney and liver transplant [9, 10].
Crystalluria, although associated with hyperoxaluria [11], is an uncommon finding [12–14]. There are no descriptions of the association between CaOx crystalluria and renal oxalosis. Here we aim to explore the causes of CaOx crystal deposition within a renal biopsy and therefore the implications and future management for the patient. We will review the histological appearances, the substrates that are most likely to cause CaOx crystal deposition and the pathophysiology associated with CaOx crystal deposition.
Histology of calcium oxalate deposition
Oxalate crystals precipitate in renal tubules causing tubular injury and in the longer term, interstitial fibrosis and tubular atrophy. They have a clear appearance on light microscopy [15] (Fig. 1a) but are much more easily seen when viewed under polarised light where they show bright birefringence (Fig. 1b). Particularly abundant crystals are typically associated with PH or ethylene glycol ingestion. Lesser degrees of deposition can be seen in a wide variety of conditions, which are discussed below. The main pathological differential diagnosis is 2,8 dihydroxyadenine crystalline nephropathy other cause of polarisable crystals seen in the kidney by the histopathologist. These patients, with biallelic mutations in APRT, have adenine phosphoribosyltransferase deficiency and often develop recurrent nephrolithiasis. Diagnosis can be challenging but the crystals can be distinguished from calcium oxalate crystals by their brown colour on haematoxylin and eosin staining [16].
Fig. 1.
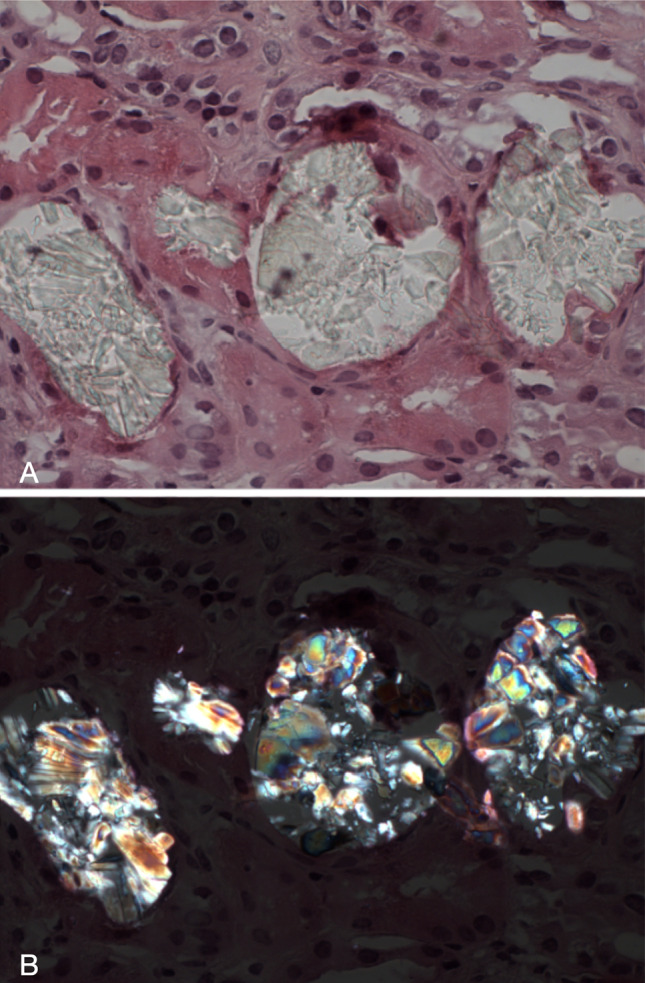
a = Oxalate nephropathy. A transplant kidney biopsy showing calcium oxalate crystals in dilated tubules. The crystals are clear with a refractile quality on routine microscopy (haematoxylin and eosin × 400). b = Oxalate nephropathy. The same calcium oxalate crystals exhibit bright birefringence when viewed under polarised light (polarised haematoxylin and eosin × 400)
Calcium and oxalate: a tale of two substrates
Hypercalciuria and hyperoxaluria are both known to cause crystal deposition within the kidney [17]. In patients with hypercalciuria, the primary crystal deposited is CaP [2], this nidus may form the focus of aggregation for either CaP or CaOx [18] This variable aggregation has been demonstrated in vitro [19], in rat models [17, 20], and observed in humans [2]. However, in patients with hyperoxaluria the predominant crystal type is CaOx [21]; this has again been demonstrated in a rat model [17], in vitro [4, 5, 22] and in humans [2].
Crystal type and the components of subsequent aggregation are dependent upon specific locations along the nephron and degrees of supersaturation. In the urinary space, it seems that a CaP nidus initiates subsequent CaOx aggregation in the in vitro model [19], as in nephrolithiasis.
In the kidney, the type of crystal deposition appears to be different dependent on the location along the nephron. CaP crystals have been observed in the interstitium surrounding the ascending thin limb of the loop of Henle [2], in stone-forming patients with hypercalciuria. CaOx crystal deposition is typically seen more distally, having been observed within the collecting duct and the interstitium surrounding it [1, 14].
The situation therefore appears that in hypercalciuria, CaP crystals are deposited within and around the nephron, especially near the loop of Henle. By contrast, in hyperoxaluria, CaOx crystal deposition is found within collecting duct nephron segments. To test this hypothesis, Khan and Glenton examined hypercalciuric mice with increasing levels of oxaluria [20]. They demonstrated that in the genetic hypercalciuric stone-forming (GHS) rat model before dietary manipulation, only CaP crystals were formed. However, as the oxalate precursor hydroxyproline was added to their diet, CaOx crystals were observed. As hydroxyproline concentrations increased, inducing a hyperoxaluria, the crystal type switched to become entirely CaOx. This suggests that intrarenal CaOx crystal formation is dependent upon hyperoxaluria rather than hypercalciuria.
The mechanism of CaOx deposition within the kidney is subject to several factors. These include supersaturation and precipitation, crystal aggregation and deposition within the tubule/epithelium/interstitium. Several studies have demonstrated that hyperoxaluria induces intratubular precipitation of CaOx crystals located in the collecting duct [1, 23]. There are two potential mechanisms by which crystal passage through the tubule is inhibited (crystal retention). They have either aggregated and become too large [24, 25], or they have adhered to the epithelium [26]. Following either of these mechanisms, CaOx crystals then migrate into the epithelium [27] and interstitium [5]. The process behind this migration is unclear. However, crystal containing macrophages have been observed in both animal [28, 29] and human [30] epithelium/interstitium. Therefore active removal by macrophages is a possible mechanism for this observation, although this has yet to be demonstrated.
Pathologies associated with calcium oxalate crystal deposition
CaOx crystal deposition may be noted in both native and transplanted kidneys, as a consequence of hyperoxaluria. Oxalate has both endogenous and exogenous sources [31, 32] and both are equally able to induce hyperoxaluria (defined as > 40–45 mg per 24 h or > 0.45–0.5 mmol per 24 h). Tubular CaOx deposition leading to acute or chronic tubular injury, interstitial fibrosis and progressive renal insufficiency is termed oxalate nephropathy or renal oxalosis.
Both native and transplanted kidneys are susceptible to hyperoxaluria and subsequent oxalate nephropathy and the causes for hyperoxaluria and crystal deposition differ (Table 1).
Table 1.
Causes of Calcium Oxalate crystal deposition within the native and transplanted kidney
| Calcium oxalate crystal deposition | |
|---|---|
| Native kidney | Transplanted kidney |
| Primary hyperoxaluria – types 1–3 | Causes as per native kidney |
| Secondary hyperoxaluria: | Transient hyperoxaluria due to sudden increase in GFR and previous systemic oxalosis secondary to end stage kidney disease |
| Enteric hyperoxaluria (fat malabsorption) | Acute tubular necrosis |
| High oxalate diet | Chronic allograft nephropathy |
| Ethylene glycol intoxication | |
| Thiamine/Pyridoxine deficiency | |
| Vitamin C overdose (precursor of oxalic acid) | |
| Orlistat use | |
| Alterations in intestinal flora | |
| Genetic variations of oxalate transporters | |
| Acute tubular necrosis | |
On light microscopy 2,8-hydroxyadenine crystals may mimic CaOx crystals under polarized light, because of their high birefringence [15]. However, the finding of 2,8-hydroxyadenine crystals mimicking CaOx crystals can lead to a rare, often missed and important genetic diagnosis being made. Likewise, genuine CaOx deposition can lead to other important diagnoses being made and should never be ignored.
Diabetes mellitus is a common cause of nephropathy and it is unclear whether it is associated with renal oxalosis. Diabetics have demonstrably higher urinary oxalate concentrations than healthy patients [33]. They have also been observed to develop oxalate nephropathy in several case reports [34, 35]. However, in these case reports, the patients had independent risk factors for renal oxalosis including Roux-en-Y bypass and increased dietary oxalate. Moreover, CaOx crystals are not among the number of histological features of diabetic nephropathy [36, 37]. A large study of cadaveric renal biopsies examined risk factors associated with renal oxalosis [38] Diabetes mellitus was shown not to be associated with renal oxalosis. Therefore, if CaOx crystals are seen on renal biopsy of a patient with diabetes, the likely driving factor is hyperoxaluria. The type and cause of hyperoxaluria should therefore be investigated as this may lead to important changes in patient management.
Primary hyperoxaluria
Primary hyperoxaluria is a rare autosomal recessive disorder associated with renal CaOx crystal deposition. Oxalate is an end metabolite for glyoxylate and the three types of primary hyperoxalurias (PH1-3) affect enzymes of glyoxylate metabolism. The enzymes implicated are: alanine glyoxylate aminotransferase (PH1) [39], glycolate reductase/hydroxypyruvate reductase (PH2) [40] and 4-hydroxy-2-ketoglutarate aldolase (PH3) [41, 42]. These disorders tend to present in childhood to early adolescence with severe recurrent nephrolithiasis, although given some may be asymptomatic (especially PH3), they may not present until the development of advanced renal failure. PH may also present in late adult life with calcium oxalate stone formation or insidious chronic kidney disease.
The majority of cases are PH1, which have the most severe disease phenotypes. PH1 and PH2 both cause progressive nephrocalcinosis, nephrolithiasis and renal damage resulting in early end stage renal failure [13, 26–28]. With the progressive decline in renal function comes rising plasma oxalate levels. At a glomerular filtration rate < 45 ml/min/1.73 m2 plasma oxalate concentrations exceed the supersaturation threshold leading to systemic deposition of CaOx (systemic oxalosis) [43]. This leads to early death if left untreated [44].
It is unclear if patients with PH3 have the same natural history as PH1/2 given its rarity and recent description. Recent data has shown children with PH3 show a decline in renal function [45]. However, there remains a lack of long-term follow-up data to allow for an accurate description of its clinical course. It is possible that all types of PH may present with unexplained chronic kidney disease and CaOx crystal deposition on renal biopsy.
Secondary hyperoxalurias
Secondary hyperoxaluria may be due to a number of different causes. The passage of oxalate through the body helps illustrate why differing mechanisms cause hyperoxaluria. There is a large oxalate content in certain foods [46], which is both metabolized by gut commensals (Oxalobacter formigenes) [47] and absorbed into the enterohepatic circulation [48, 49]. Absorbed oxalate is then filtered and excreted in the kidney [48, 49] along with oxalate produced as an end-point of glyoxylate metabolism.
At each of these points, excess oxalate may occur. Case reports describing high intakes of oxalate containing foods [46] or vitamin C [50] (which is catabolized into oxalate) are associated with hyperoxaluria. Deficiencies, dietary or otherwise, in thiamine or pyridoxine [51–54], deliberate ingestion of orlistat [55] or ethylene glycol [56, 57] may also lead to hyperoxaluria. High doses of vitamin C [50], some foods [58–61], excessive dieting [62] and ethylene glycol [56] have been demonstrated to induce acute oxalate nephropathy.
The gut commensal Oxalobacter formigenes, catabolizes oxalate thus diminishing gut absorption [63, 64]. There has been an attempt to exploit this phenomenon for PH, which showed initial promise, but unfortunately failed in phase II/III trials [65]. Although touted as a treatment, there have not been further studies of its effectiveness to treat secondary hyperoxaluria.
Several case reports have associated hyperoxaluria with bariatric surgery [66, 67] as well as chronic pancreatitis [68, 69], with both conditions associated with acute oxalate nephropathy [66, 68]. Increased oxalate absorption is a function of fat malabsorption (enteric hyperoxaluria). In the normal state, oxalate is bound to calcium within the gut. Fat malabsorption leads to free fatty acids binding to calcium, leaving the oxalate in its absorbable, ionised state [49].
Mice and humans with genetic variations of gut oxalate transporters have also been demonstrated to have increased urinary oxalate [70] Deletion of Slc26a6 in mice [71, 72] along with variants V185M in the SLC26A6 transporter in humans [73] have both been associated with hyperoxaluria. None of these studies performed renal biopsies and therefore further study is required to see if these are risk factors for oxalate nephropathy and CaOx deposition.
Transplanted kidneys
Around 4% of transplanted kidneys will display CaOx crystals on biopsy [6]. Crystals can be found early or late, distributed throughout the kidney or only in focal segments.
In the initial post-operative period it is thought that, due to the poor renal function indicating the need for transplant, there is systemic oxalosis. With the improvement in renal function attained by transplantation there is rapid excretion of the excess oxalate. This leads to a transient hyperoxaluria with a small proportion developing subsequent renal precipitation of CaOx [74]. There is debate as to whether or not this initial transient hyperoxaluria is pathological, and long-term outcomes of this have not been proven.
There is more evidence for the implications of CaOx crystals on renal biopsy, albeit conflicting. In the short term, the presence of CaOx crystals on graft biopsy up to 3 months after transplantation seems to be associated with poorer longer term graft survival [75]. Although a later study demonstrated that, although graft function at 1 year was significantly poorer in those with CaOx deposition, there was no statistically significant difference in renal function at 2 years [6]. In this second study however, there was an overall drop in both control and crystal graft function in the second year compared to the first. It is likely that CaOx crystals are a negative prognostic indicator for long-term graft survival in the initial period following transplantation. These patients should be followed-up closely.
Delayed graft function and acute tubular necrosis (ATN) or acute cell‐mediated rejection is associated with focal CaOx deposition [76, 77]. The long-term impact of these acute events is unclear. The majority of transplanted kidneys demonstrated normal function at follow-up [76]. However, these observations were underpowered, lacked follow-up biopsies, and biochemical data for clinical correlation. The authors postulated this observation was due to high oxalate excretion using the mechanism previously described. However, inferring this mechanism from the data is difficult due to the lack of clinical context and small numbers of patients.
In the longer term, CaOx crystals are seen on biopsy of those with chronic allograft nephropathy [76]. In the two patients studied, CaOx crystal deposition was widespread in keeping with chronic renal failure (mechanism discussed below). An earlier study by Memeo et al. of forty allograft nephrectomies showed 87% had widespread CaOx crystals [78]. Again, given the low numbers it is difficult to draw conclusions from these case reports, but they suggest CaOx crystals, identified at any point in time from biopsy, are associated with long-term graft failure.
Transplanted kidneys can also be affected by any of the primary or secondary hyperoxalurias. Failure to diagnose PH prior to transplantation may result in early graft failure [79, 80]. Likewise for secondary hyperoxalurias, failure to recognise may lead to acute kidney injury [81] or even graft failure. There have been graft failure case reports for enteric hyperoxaluria [82, 83] and excessive vitamin C intake [84].
Pathophysiology of renal damage associated with crystal deposition
Severe hyperoxaluria has been demonstrated to be clinically associated with acute or chronic renal failure, although it is unclear which is causative of the other. It is also unclear whether mild to moderate hyperoxaluria, such as that seen in PH3, is associated with renal damage, despite evidence of CaOx crystal deposition in both conditions.
There is a large body of evidence from rat and in vitro models, and human observation that CaOx crystal deposition is associated with renal epithelial damage [4, 5, 85–89]. Differing structures of CaOx crystals can damage renal epithelial cells inducing apoptosis [22]. This body of evidence suggests that epithelial injury and progressive inflammation is caused by CaOx crystals, rather than CaOx crystals forming secondary to renal damage. This explains the findings in PH and severe secondary hyperoxaluria.
The observation that CaOx crystals are only found in focal segments of acute tubular necrosis in transplanted kidneys [76, 77] however, does not fit with the widespread renal damage and CaOx crystals of hyperoxaluria. It implies that CaOx crystal deposition seen in this situation is secondary to focal epithelial damage [4], rather than crystal precipitation and subsequent epithelial damage.
The pathophysiology of renal oxalosis secondary to severe hyperoxaluria has been described. However, the mechanism of focal CaOx crystal deposition in acute tubular necrosis remains unclear. CaOx crystals on renal biopsy should always prompt investigation for serious underlying conditions in both the native and transplanted kidney (Table 1), that could lead to progressive renal failure.
Conclusion
CaOx crystals identified histologically on renal biopsy are indicative of a potential underlying pathology. This finding warrants further investigation to determine the cause, the most serious of which is PH. Much of the clinical literature describing conditions associated with CaOx crystal deposition are case reports. In the long-term there appears to be a potential association between CaOx deposition and increased risk of chronic kidney disease. Larger studies are needed to examine this association in more depth.
Acknowledgements
RG is supported by the National Institute for Health Research. JAS is supported by the Northern Counties Kidney Research Fund and Kidney Research UK.
Compliance with Ethical Standards
Conflicts of interest
The authors have no conflicts of interest to declare.
Statement of human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed written consent was obtained for use of patient biopsy images in this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kok DJ. Crystallization and stone formation inside the nephron. Scan Microsc. 1996;10:471–484. [PubMed] [Google Scholar]
- 2.Evan AP, Lingeman JE, Coe FL, et al. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Invest. 2003;111:607–616. doi: 10.1172/JCI17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evan AP, Coe FL, Lingeman J, et al. Randall’s plaque in stone formers originates in ascending thin limbs. Am J Physiol Renal Physiol. 2018;315:F1236–F1242. doi: 10.1152/ajprenal.00035.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan SR. Calcium oxalate crystal interaction with renal tubular epithelium, mechanism of crystal adhesion and its impact on stone development. Urol Res. 1995;23:71–79. doi: 10.1007/bf00307936. [DOI] [PubMed] [Google Scholar]
- 5.Water R, Noordermeer C, Kwast TH, et al. Calcium oxalate nephrolithiasis effect of renal crystal deposition on the cellular composition of the renal interstitium. Am J Kidney Dis. 1999;33:761–771. doi: 10.1016/s0272-6386(99)70231-3. [DOI] [PubMed] [Google Scholar]
- 6.Bagnasco SM, Mohammed BS, Mani H, et al. Oxalate deposits in biopsies from native and transplanted kidneys, and impact on graft function. Nephrol Dial Transplant. 2009;24:1319–1325. doi: 10.1093/ndt/gfn697. [DOI] [PubMed] [Google Scholar]
- 7.Sayer JA, Carr G, Simmons NL. Nephrocalcinosis: molecular insights into calcium precipitation within the kidney. Clin Sci. 2004;106:549–561. doi: 10.1042/CS20040048. [DOI] [PubMed] [Google Scholar]
- 8.Dickson FJ, Sayer JA. Nephrocalcinosis: a review of monogenic causes and insights they provide into this heterogeneous condition. Int J Mol Sci. 2020;21:369. doi: 10.3390/ijms21010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watts RW, Calne RY, Rolles K, et al. Successful treatment of primary hyperoxaluria type I by combined hepatic and renal transplantation. Lancet. 1987;2:474–475. doi: 10.1016/s0140-6736(87)91791-0. [DOI] [PubMed] [Google Scholar]
- 10.Kotb MA, Hamza AF, Abd El Kader H, et al. Combined liver-kidney transplantation for primary hyperoxaluria type I in children: single center experience. Pediatr Transplant. 2019;23:e13313. doi: 10.1111/petr.13313. [DOI] [PubMed] [Google Scholar]
- 11.Daudon M, Jungers P, Lacour B. Intérêt clinique de l’étude de la cristallurie. Ann Biol Clin. 2004;62:379–393. [PubMed] [Google Scholar]
- 12.Winkens RA, Wielders JP, Degenaar CP, van Hoof JP. Calcium oxalate crystalluria, a curiosity or a diagnostical aid? J Clin Chem Clin Biochem. 1988;26:653–654. [PubMed] [Google Scholar]
- 13.Robert M, Boularan AM, Delbos O, et al. Study of calcium oxalate crystalluria on renal and vesical urines in stone formers and normal subjects. Urol Int. 1998;60:41–46. doi: 10.1159/000030201. [DOI] [PubMed] [Google Scholar]
- 14.Elliot JS, Rabinowitz IN. Calcium oxalate crystalluria: crystal size in urine. JURO. 1980;123:324–327. doi: 10.1016/s0022-5347(17)55918-2. [DOI] [PubMed] [Google Scholar]
- 15.Herlitz LC, D'Agati VD, Markowitz GS. Crystalline nephropathies. Arch Pathol Lab Med. 2012;136:713–720. doi: 10.5858/arpa.2011-0565-RA. [DOI] [PubMed] [Google Scholar]
- 16.Bagai S, Khullar D, Bansal B. Rare crystalline nephropathy leading to acute graft dysfunction: a case report. BMC Nephrol. 2019;20:428–433. doi: 10.1186/s12882-019-1616-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evan AP, Bledsoe SB, Smith SB, Bushinsky DA. Calcium oxalate crystal localization and osteopontin immunostaining in genetic hypercalciuric stone-forming rats. Kidney Int. 2004;65:154–161. doi: 10.1111/j.1523-1755.2004.00396.x. [DOI] [PubMed] [Google Scholar]
- 18.Sayer JA, Simmons NL. Urinary stone formation: Dent’s disease moves understanding forward. Exp Nephrol. 2002;10:176–181. doi: 10.1159/000058344. [DOI] [PubMed] [Google Scholar]
- 19.Xie B, Halter TJ, Borah BM, Nancollas GH. Aggregation of calcium phosphate and oxalate phases in the formation of renal stones. Cryst Growth Des. 2015;15:204–211. doi: 10.1021/cg501209h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan SR, Glenton PA. Calcium oxalate crystal deposition in kidneys of hypercalciuric mice with disrupted type IIa sodium-phosphate cotransporter. Am J Physiol Renal Physiol. 2008;294:F1109–F1115. doi: 10.1152/ajprenal.00620.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467–475. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 22.Sun X-Y, Ouyang J-M, Yu K. Shape-dependent cellular toxicity on renal epithelial cells and stone risk of calcium oxalate dihydrate crystals. Sci Rep. 2017;7:7250–7313. doi: 10.1038/s41598-017-07598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kok DJ. Intratubular crystallization events. World J Urol. 1997;15:219–228. doi: 10.1007/bf01367659. [DOI] [PubMed] [Google Scholar]
- 24.Kok DJ, Khan SR. Calcium oxalate nephrolithiasis, a free or fixed particle disease. Kidney Int. 1994;46:847–854. doi: 10.1038/ki.1994.341. [DOI] [PubMed] [Google Scholar]
- 25.Robertson WG. Potential role of fluctuations in the composition of renal tubular fluid through the nephron in the initiation of Randall's plugs and calcium oxalate crystalluria in a computer model of renal function. Urolithiasis. 2015;43(1):93–107. doi: 10.1007/s00240-014-0737-1. [DOI] [PubMed] [Google Scholar]
- 26.Verkoelen CF, Van Der Boom BG, Kok DJ, et al. Attachment sites for particles in the urinary tract. J Am Soc Nephrol. 1999;10(14):S430–S435. [PubMed] [Google Scholar]
- 27.Ebisuno S, Kohjimoto Y, Tamura M, et al. Histological observations of the adhesion and endocytosis of calcium oxalate crystals in MDCK cells and in rat and human kidney. Urol Int. 1997;58:227–231. doi: 10.1159/000282989. [DOI] [PubMed] [Google Scholar]
- 28.Water R, Noordermeer C, Houtsmuller AB, et al. Role of macrophages in nephrolithiasis in rats an analysis of the renal interstitium. Am J Kidney Dis. 2000;36:615–625. doi: 10.1053/ajkd.2000.16203. [DOI] [PubMed] [Google Scholar]
- 29.Thurgood LA, Sørensen ES, Ryall RL. The effect of intracrystalline and surface-bound osteopontin on the degradation and dissolution of calcium oxalate dihydrate crystals in MDCKII cells. Urol Res. 2012;40:1–15. doi: 10.1007/s00240-011-0423-5. [DOI] [PubMed] [Google Scholar]
- 30.Kusmartsev S, Dominguez-Gutierrez PR, Canales BK, et al. Calcium oxalate stone fragment and crystal phagocytosis by human macrophages. J Urol. 2016;195:1143–1151. doi: 10.1016/j.juro.2015.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. 2001;59:270–276. doi: 10.1046/j.1523-1755.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- 32.Holmes RP, Ambrosius WT, Assimos DG. Dietary oxalate loads and renal oxalate handling. JURO. 2005;174:943–947. doi: 10.1097/01.ju.0000169476.85935.e2. [DOI] [PubMed] [Google Scholar]
- 33.Eisner BH, Porten SP, Bechis SK, Stoller ML. Urolithiasis/endourology diabetic kidney stone formers excrete more oxalate and have lower urine ph than nondiabetic stone formers. JURO. 2010;183:2244–2248. doi: 10.1016/j.juro.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Moutzouris D-A, Skaneli G, Margellos V, et al. Oxalate nephropathy in a diabetic patient after gastric by-pass. Clin Nephrol. 2011;75(1):16–19. [PubMed] [Google Scholar]
- 35.Muji A, Moll S, Saudan P. Oxalate nephropathy: a new entity of acute kidney injury in diabetic patients? Rev Med Suisse. 2015;11:493–496. [PubMed] [Google Scholar]
- 36.Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27:195–207. doi: 10.1016/j.semnephrol.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fioretto P, Barzon I, Mauer M. Is diabetic nephropathy reversible? Diabetes Res Clin Pract. 2014;104:323–328. doi: 10.1016/j.diabres.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Mori S, Beppu T. Secondary renal oxalosis. A statistical analysis of its possible causes. Acta Pathol Jpn. 1983;33:661–669. [PubMed] [Google Scholar]
- 39.Danpure CJ, Jennings PR. Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–34. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 40.Cramer SD, Ferree PM, Lin K, et al. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999;8:2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 41.Williams EL, Bockenhauer D, et al. The enzyme 4-hydroxy-2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol Dial Transplant. 2012;27:3191–3195. doi: 10.1093/ndt/gfs039. [DOI] [PubMed] [Google Scholar]
- 42.Huang A, Burke J, Bunker RD, et al. Regulation of human 4-hydroxy-2-oxoglutarate aldolase by pyruvate and α-ketoglutarate: implications for primary hyperoxaluria type-3. Biochem J. 2019;476:3369–3383. doi: 10.1042/BCJ20190548. [DOI] [PubMed] [Google Scholar]
- 43.Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12:1986–1993. doi: 10.1681/ASN.V1291986. [DOI] [PubMed] [Google Scholar]
- 44.Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int. 2001;59:31–36. doi: 10.1046/j.1523-1755.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 45.Allard L, Cochat P, Leclerc A-L, et al. Renal function can be impaired in children with primary hyperoxaluria type 3. Pediatr Nephrol. 2015;30:1807–1813. doi: 10.1007/s00467-015-3090-x. [DOI] [PubMed] [Google Scholar]
- 46.Noonan SC, Savage GP. Oxalate content of foods and its effect on humans. Asia Pac J Clin Nutr. 1999;8:64–74. doi: 10.1046/j.1440-6047.1999.00038.x. [DOI] [PubMed] [Google Scholar]
- 47.Stewart CS, Duncan SH, Cave DR. Oxalobacter formigenes and its role in oxalate metabolism in the human gut. FEMS Microbiol Lett. 2004;230:1–7. doi: 10.1016/S0378-1097(03)00864-4. [DOI] [PubMed] [Google Scholar]
- 48.Hatch M, Freel RW. Renal and intestinal handling of oxalate following oxalate loading in rats. Am J Nephrol. 2003;23:18–26. doi: 10.1159/000066300. [DOI] [PubMed] [Google Scholar]
- 49.Whittamore JM, Hatch M. The role of intestinal oxalate transport in hyperoxaluria and the formation of kidney stones in animals and man. Urolithiasis. 2017;45:89–108. doi: 10.1007/s00240-016-0952-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong K, Thomson C, Bailey RR, et al. Acute oxalate nephropathy after a massive intravenous dose of vitamin C. Aust N Z J Med. 1994;24:410–411. doi: 10.1111/j.1445-5994.1994.tb01477.x. [DOI] [PubMed] [Google Scholar]
- 51.Tiselius HG, Almgård LE. The diurnal urinary excretion of oxalate and the effect of pyridoxine and ascorbate on oxalate excretion. Eur Urol. 1977;3:41–46. doi: 10.1159/000472053. [DOI] [PubMed] [Google Scholar]
- 52.Hannett B, Thomas DW, Chalmers AH, et al. Formation of oxalate in pyridoxine or thiamin deficient rats during intravenous xylitol infusions. J Nutr. 1977;107:458–465. doi: 10.1093/jn/107.3.458. [DOI] [PubMed] [Google Scholar]
- 53.Gill HS, Rose GA. Mild metabolic hyperoxaluria and its response to pyridoxine. Urol Int. 1986;41:393–396. doi: 10.1159/000281241. [DOI] [PubMed] [Google Scholar]
- 54.Svedruzić D, Jónsson S, Toyota CG, et al. The enzymes of oxalate metabolism: unexpected structures and mechanisms. Arch Biochem Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 55.Solomon LR, Nixon AC, Ogden L. Nair B (2017) Orlistat-induced oxalate nephropathy: an under-recognised cause of chronic kidney disease. BMJ Case Rep. 2017;21:6–23. doi: 10.1136/bcr-2016-218623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ting SMS, Ching I, Nair H, et al. Early and late presentations of ethylene glycol poisoning. Am J Kidney Dis. 2009;53:1091–1097. doi: 10.1053/j.ajkd.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 57.Hanouneh M, Chen TK. Calcium oxalate crystals in ethylene glycol toxicity. N Engl J Med. 2017;377:1467–1467. doi: 10.1056/NEJMicm1704369. [DOI] [PubMed] [Google Scholar]
- 58.Syed F, Mena-Gutierrez A, Ghaffar U. A case of iced-tea nephropathy. N Engl J Med. 2015;372:1377–1378. doi: 10.1056/NEJMc1414481. [DOI] [PubMed] [Google Scholar]
- 59.Getting JE, Gregoire JR, Phul A, Kasten MJ. Oxalate nephropathy Due to “Juicing”: case report and review. Am J Med. 2013;126:768–772. doi: 10.1016/j.amjmed.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 60.Lien Y-HH. Juicing is not all juicy. Am J Med. 2013;126:755–756. doi: 10.1016/j.amjmed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 61.Makkapati S, D'Agati VD, Balsam L. “Green smoothie cleanse” causing acute oxalate nephropathy. Am J Kidney Dis. 2018;71:281–286. doi: 10.1053/j.ajkd.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Khneizer G, Al-Taee A, Mallick MS, Bastani B. Chronic dietary oxalate nephropathy after intensive dietary weight loss regimen. J Nephropathol. 2017;6:126–129. doi: 10.15171/jnp.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mittal RD, Kumar R. Gut-inhabiting bacterium Oxalobacterformigenes: role in calcium oxalate urolithiasis. J Endourol. 2004;18:418–424. doi: 10.1089/0892779041271706. [DOI] [PubMed] [Google Scholar]
- 64.Hatch M, Cornelius J, Allison M, et al. Oxalobacter sp. reduces urinary oxalate excretion by promoting enteric oxalate secretion. Kidney Int. 2006;69:691–698. doi: 10.1038/sj.ki.5000162. [DOI] [PubMed] [Google Scholar]
- 65.Milliner D, Hoppe B, Groothoff J. A randomised Phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis. 2018;46:313–323. doi: 10.1007/s00240-017-0998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nasr SH, D'Agati VD, Said SM, et al. Oxalate nephropathy complicating Roux-en-Y Gastric Bypass: an underrecognized cause of irreversible renal failure. Clin J Am Soc Nephrol. 2008;3:1676–1683. doi: 10.2215/CJN.02940608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sinha MK, Collazo-Clavell ML, Rule A, et al. Hyperoxaluric nephrolithiasis is a complication of Roux-en-Y gastric bypass surgery. Kidney Int. 2007;72:100–107. doi: 10.1038/sj.ki.5002194. [DOI] [PubMed] [Google Scholar]
- 68.Cartery C, Faguer S, Karras A, et al. Oxalate nephropathy associated with chronic pancreatitis. Clin J Am Soc Nephrol. 2011;6:1895–1902. doi: 10.2215/CJN.00010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Demoulin N, Issa Z, Crott R, et al. Enteric hyperoxaluria in chronic pancreatitis. Medicine (Baltimore) 2017;96:e6758. doi: 10.1097/MD.0000000000006758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sayer JA. Progress in understanding the genetics of calcium-containing nephrolithiasis. J Am Soc Nephrol. 2017;28:748–759. doi: 10.1681/ASN.2016050576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freel RW, Hatch M, Green M, Soleimani M. Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G719–G728. doi: 10.1152/ajpgi.00481.2005. [DOI] [PubMed] [Google Scholar]
- 72.Jiang Z, Asplin JR, Evan AP, et al. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet. 2006;38:474–478. doi: 10.1038/ng1762. [DOI] [PubMed] [Google Scholar]
- 73.Clark JS, Vandorpe DH, Chernova MN, et al. Species differences in Cl− affinity and in electrogenicity of SLC26A6-mediated oxalate/Cl− exchange correlate with the distinct human and mouse susceptibilities to nephrolithiasis. J Physiol. 2008;586:1291–1306. doi: 10.1113/jphysiol.2007.143222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Worcester EM, Fellner SK, Nakagawa Y, Coe FL. Effect of renal transplantation on serum oxalate and urinary oxalate excretion. Nephron. 1994;67:414–418. doi: 10.1159/000188014. [DOI] [PubMed] [Google Scholar]
- 75.Pinheiro HS, Câmara NOS, Osaki KS, et al. Early presence of calcium oxalate deposition in kidney graft biopsies is associated with poor long-term graft survival. Am J Transplant. 2005;5:323–329. doi: 10.1111/j.1600-6143.2004.00684.x. [DOI] [PubMed] [Google Scholar]
- 76.Truong LD, Yakupoglu U, Feig D, et al. Calcium oxalate deposition in renal allografts: morphologic spectrum and clinical implications. Am J Transplant. 2004;4:1338–1344. doi: 10.1111/j.1600-6143.2004.00511.x. [DOI] [PubMed] [Google Scholar]
- 77.Olsen S, Burdick JF, Keown PA, et al. Primary acute renal failure (“acute tubular necrosis”) in the transplanted kidney: morphology and pathogenesis. Medicine (Baltimore) 1989;68:173–187. doi: 10.1097/00005792-198905000-00005. [DOI] [PubMed] [Google Scholar]
- 78.Memeo L, Pecorella I, Ciardi A, et al. Calcium oxalate microdeposition in failing kidney grafts. Transplant Proc. 2001;33:1262–1265. doi: 10.1016/s0041-1345(00)02470-2. [DOI] [PubMed] [Google Scholar]
- 79.Bilgin N, Tirnaksiz MB, Moray G, et al. Early recurrence of oxalate deposition after renal transplantation in a patient with primary hyperoxaluria type I. Transplant Proc. 1999;31:3219–3220. doi: 10.1016/s0041-1345(99)00699-5. [DOI] [PubMed] [Google Scholar]
- 80.Riksen NP, Timmers HJLM, Assmann KJM, Huysmans FTM. Renal graft failure due to type 1 primary hyperoxaluria. Neth J Med. 2002;60:407–410. [PubMed] [Google Scholar]
- 81.Suneja M, Kumar AB. Secondary oxalosis induced acute kidney injury in allograft kidneys. Clin Kidney J. 2013;6:84–86. doi: 10.1093/ckj/sfs167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuvelier C, Goffin E, Cosyns J-P, et al. Enteric hyperoxaluria: a hidden cause of early renal graft failure in two successive transplants: spontaneous late graft recovery. Am J Kidney Dis. 2002;40:6. doi: 10.1053/ajkd.2002.33934. [DOI] [PubMed] [Google Scholar]
- 83.Rankin AC, Walsh SB, Summers SA, et al. Acute oxalate nephropathy causing late renal transplant dysfunction due to enteric hyperoxaluria. Am J Transplant. 2008;8:1755–1758. doi: 10.1111/j.1600-6143.2008.02288.x. [DOI] [PubMed] [Google Scholar]
- 84.Parasuraman R, Zhang L, et al. Primary nonfunction of renal allograft secondary to acute oxalate nephropathy. Case Rep Transplant. 2011;2011:876906–876914. doi: 10.1155/2011/876906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lieske JC, Spargo BH, Toback FG. Endocytosis of calcium oxalate crystals and proliferation of renal tubular epithelial cells in a patient with type 1 primary hyperoxaluria. JURO. 1992;148:1517–1519. doi: 10.1016/s0022-5347(17)36954-9. [DOI] [PubMed] [Google Scholar]
- 86.Khan SR, Shevock PN, Hackett RL. Acute hyperoxaluria, renal injury and calcium oxalate urolithiasis. JURO. 1992;147:226–230. doi: 10.1016/s0022-5347(17)37202-6. [DOI] [PubMed] [Google Scholar]
- 87. de Water R, Boevé ER, van Miert PP, et al. Pathological and immunocytochemical changes in chronic calcium oxalate nephrolithiasis in the rat. Scanning Microsc. 1996;10:577–587. [PubMed] [Google Scholar]
- 88.Umekawa T, Chegini N, Khan SR. Oxalate ions and calcium oxalate crystals stimulate MCP-1 expression by renal epithelial cells. Kidney Int. 2002;61:105–112. doi: 10.1046/j.1523-1755.2002.00106.x. [DOI] [PubMed] [Google Scholar]
- 89.Schepers MSJ, van Ballegooijen ES, Bangma CH, Verkoelen CF. Crystals cause acute necrotic cell death in renal proximal tubule cells, but not in collecting tubule cells. Kidney Int. 2005;68:1543–1553. doi: 10.1111/j.1523-1755.2005.00566.x. [DOI] [PubMed] [Google Scholar]