

Abstract
Aim
The aim of the present study was to characterize the role of Smad3 signalling on high glucose‐induced podocyte injury.
Methods
Synchronized conditionally immortalized mouse podocyte cell line (MPC5) cells were treated with either D‐glucose alone or D‐glucose plus the Smad3 inhibitor SIS3. The distribution of F‐actin and transgelin in a high glucose‐induced model of podocyte injury were examined by immunofluorescence. Levels of transgelin and Smad3 signalling proteins in MPC5 cells were determined by Western blot.
Results
A disordered distribution of F‐actin, as well as co‐localization of F‐actin and transgelin, was observed in podocytes exposed to high glucose. Increased levels of transgelin were first observed 10 minutes after treatment with glucose, suggesting that this protein is sensitive to hyperglycaemic injury. Levels of phosphorylated Smad3 and cleaved caspase 3 increased significantly with glucose stimulation. Moreover, expression of the downstream protein c‐Myc, but not JAK1/STAT3, was induced in conditions of high glucose. The Smad3‐specific inhibitor SIS3 prevented the effects of high glucose on Smad3 phosphorylation, expression of transgelin and c‐Myc, caspase 3 cleavage and cytoskeletal organization. Expression of the tumour suppressor protein p15INK4B increased after podocyte injury but was unaffected by Smad3 inhibition, suggesting that Smad3 regulation of high glucose‐induced podocyte injury occurs through a p15INK4B‐independent mechanism.
Conclusion
Smad3 signalling plays a critical role in the modulation of hyperglycaemic injury. Targeted inhibition of the Smad3 pathway may offer a novel route for treatment of podocyte damage, especially in cases of diabetic nephropathy.
Keywords: high glucose, podocyte injury, Smad3, transgelin
SUMMARY AT A GLANCE
Podocyte damage is characteristic of diabetic kidney disease. This study showed that inhibition of Smad‐3 signalling using the specific inhibitor SIS3 in vitro, could alleviate podocyte abnormalities induced by high glucose including actin cytoskeletal rearrangement and actviation of cell death pathways.
Kidney injury is often accompanied by de‐differentiation and apoptosis of podocytes, differentially terminated cells that are involved in kidney diseases such as diabetic nephropathy.1 Podocyte cytoskeletal proteins, actin‐binding proteins and polysaccharide proteins in the apical region of the foot process function together to maintain the integrity of slit diaphragms.2 Podocyte loss, deteriorated microstructure and foot process effacement are frequently observed in diabetic nephropathy.3, 4 Loss of podocyte function is also a key feature of diabetic nephropathy. However, the precise mechanism underlying hyperglycaemic podocyte injury remains unclear. Therefore, understanding the mechanism of high glucose‐induced podocyte injury will contribute to therapeutic strategies for treatment of diabetic nephropathy.
Morphological changes of podocytes are typically accompanied by disruption of the actin cytoskeleton. It has been reported that high glucose induces actin remodelling in a murine podocyte cell line5 as well as in a mouse model.6 In addition, transgelin has also been identified as a marker of cytoskeleton remodelling.7 Transgelin is an actin‐binding protein that is absent in normal rat glomeruli, but highly expressed in podocytes during nephrotoxicity, diabetic nephropathy, IgA nephropathy and membrane kidney diseases. Transgelin is known to mediate morphological changes of podocyte foot processes in a rat model of puromycin aminonucleoside‐induced kidney injury.8 However, the role and location of transgelin during high glucose‐induced podocyte cells injury have yet to be described.
In a model of high fat diet‐induced diabetic nephropathy in rats, the protective effects of Taxus chinensis occurred in part via down‐regulation of TGF‐β1 expression and Smad2/3 phosphorylation.9 In another study, betulinic acid alleviated high glucose‐induced cardiac fibroblasts mainly through inhibition of the TGF‐β1‐Smad signalling pathway.10 The damage to cardiomyocytes via reperfusion injury, aggravated by high glucose, was also reported to be mediated by TGF‐β1‐Smad signalling.11 During high glucose‐induced epithelial‐to‐mesenchymal transition in peritoneal mesothelial cells, Smad3 protein was significantly more phosphorylated than in low‐glucose conditions.12 In addition to its involvement in high glucose stimulation, the Smad signalling pathway is also activated during podocyte injury.13 Podocyte injury was alleviated in obese Smad3‐deficient mice,14 and Smad3 phosphorylation was induced by puromycin aminonucleoside‐stimulated podocyte injury.15 In a mouse podocyte cell line, treatment with gremlin aggravates high glucose‐induced cell injury and also enhances activation of TGF‐β1‐Smad signalling.16 Interestingly, TGF‐β1 was also able to disrupt the cytoskeleton and induce podocyte apoptosis by regulating the interaction of Smad2/3 with other proteins.17 The precise mechanism by which TGF‐β1 signalling regulates high glucose‐induced podocyte injury is still unclear.
In this study, changes in the distribution of F‐actin and transgelin during high glucose‐induced kidney injury were examined, and the expression of transgelin and related Smad3 signalling pathways were analysed. These results provide an experimental basis for clarifying the pathogenesis of kidney injury under conditions of high glucose.
1. MATERIALS AND METHODS
1.1. Podocyte cultures and treatments
The conditionally immortalized podocyte mouse cell line (MPC5) (gifted by Professor Peter Mundel, Mount Sinai Medical School, New York) was cultured at 33°C with recombinant murine γ‐interferon (PEPRO Tech), and then was incubated at 37°C for 1 week to promote differentiation.18 Mature podocytes were seeded onto 60‐mm Petri dishes coated with 10 μg/mL type‐I collagen (Sigma), and cultured in the RPMI‐1640 medium (Gibco) in 37°C. When mature podocytes reached 60% confluency they were synchronized first in the RPMI‐1640 media with 0.5% fetal bovine serum (Gibco) for 12 hours, then treated with 35 or 5 mmol/L D‐glucose or 5 mmol/L D‐glucose (Sinopharm) with 30 mmol/L mannitol for 2, 5, 10, 15 and 30 minutes. Total cell lysates were extracted at different time points to be used for immunoblot assay.
To detect the effect of the Smad3 inhibitor SIS3 (Calbiochem) on high glucose‐induced podocyte injury, MPC5 cells were grouped to receive one of the following five treatments: (a) blank control, (b) osmotic pressure control, (c) SIS3 treatment, (d) high glucose treatment and (e) SIS3 with high glucose treatment. This experiment was repeated at least three times.
1.2. Immunofluorescence staining
When mature podocytes reached 80% confluency, they were synchronized first in the RPMI‐1640 media with 0.5% serum for 6 hours, and then treated with 35 mmol/L D‐glucose for 0, 24, 48 and 72 hours. To examine the effect of SIS3 on cytoskeletal morphology after treatment with high glucose, the synchronized MPC5 cells were incubated with or without 5 μmol/L SIS3 for 30 minutes, followed by 35 mmol/L D‐glucose for 12 hours. These cells were subjected to immunofluorescence staining as previously described.19 Briefly, cells were fixed with 4% paraformaldehyde for a quarter of an hour at room temperature, and then washed several times with phosphate buffer (PBS). After permeabilizing with Triton X‐100 for 15 minutes, podocytes were blocked with 5% bovine serum albumin for half an hour at room temperature. Cells were incubated with primary antibodies (mouse anti‐F‐actin, 1:100, Ab205, Abcam; rabbit anti‐transgelin, 1:100, Ab155272, Abcam) overnight at 4°C, and then washed three times with PBS and incubated with secondary antibodies (donkey anti‐mouse IgG (H + L) Alexa Fluor 546‐conjugated, cat. No. A10036, ThermoFisher Scientific; goat anti‐rabbit IgG (H + L) Alexa Fluor 488‐conjugated, cat. No. A11034, ThermoFisher Scientific) for 1 hour at room temperature, then washed three times with PBS. These cells were then mounted with the Prolong Gold antifade mountant with DAPI (cat. No. P36931, ThermoFisher Scientific). Cellular morphology was observed using laser scanning confocal microscopy (TE300, Nikon), and analysed with the ImageQuant software v5.3. The distribution of F‐actin and transgelin were observed in 10 randomly selected high‐power fields of podocytes (original magnification ×600).
1.3. Western blot
MPC5 cells were treated with D‐glucose, mannitol or SIS3 as described above. Cell lysates were extracted with lysis buffer (P001, UKzybiotech, China) and proteinase inhibitor cocktail (04693116001, Roche). Protein levels were quantified using the BCA Protein Assay Kit (cat. No. 23225, ThermoFisher Scientific). Protein extracts were run through 12% of SDS‐polyacrylamide gel electrophoresis gels, then transferred to polyvinylidene fluoride membranes (0.45 μm pore size, IPVH00010, Millipore). Membranes were first washed with PBS and then blocked with 5% skim milk for 1 hour. Primary antibodies used were as follows: rabbit anti‐Smad3 (1:750, 9523T), rabbit anti‐phospho‐Smad3 (1:1000, 9520T), rabbit anti‐phospho‐JAK (1:1000, 3331S), rabbit anti‐phospho‐STAT3 (1:1000, 9145T), rabbit anti‐caspase 3 (1:1000, 9661T) and rabbit anti‐c‐myc (1:1000, 9402S) antibodies were purchased from the cell signalling technology; rabbit anti‐transgelin (1:1000, Ab155272) and rabbit anti‐p15INK4B (1:1000, Ab53034) antibodies from Abcam; and mouse anti‐beta‐Actin (1:1000, TA‐09) from Zsbio. Peroxidase‐AffiniPure goat anti‐mouse (1:4000, 115‐035‐003) and anti‐rabbit (1:4000, 111‐035‐003) secondary antibodies were purchased from Jackson Laboratory. Immunoblot signals were detected with the Super Signal West Pico‐enhanced chemiluminescent substrate (WBKLS0500, Millipore) and then analysed with the AlphaImager mini gel imaging system.
1.4. Statistical analysis
The grey value of the immunoblotting bands of at least three independent experiments was quantified with the ImageJ software. The data were then compared by one‐way and two‐way analysis of variance (ANOVA) with the least significant difference test (equal variance) or Dunnett's T3 test (unequal variance) where appropriate. P value less than .05 was considered significant.
2. RESULTS
2.1. Transgelin is induced and co‐localized with F‐actin in a model of high glucose‐induced podocyte injury
Control cells displayed intact cytoskeleton, and evenly distributed F‐actin in cytoplasm as well as in podocyte foot processes (Figure 1A). Cells that were treated with high glucose displayed cytoskeletal disruption that increased with the duration of glucose treatment; the most profound foot process retraction and cytoskeletal damage were observed at the 72‐hour time point.
Figure 1.
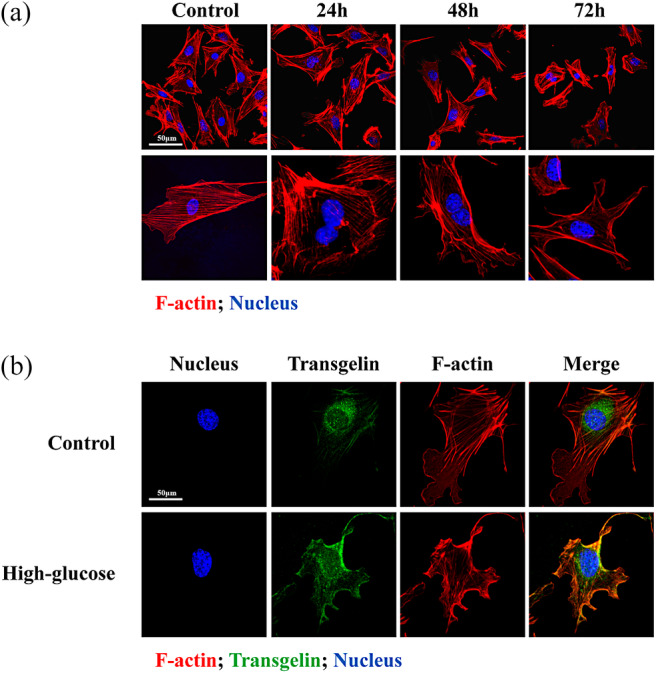
Distribution of cytoskeletal proteins upon treatment of podocytes with high glucose. A, Time‐dependent disruption of F‐actin in MPC5 cells treated with high glucose. B, High glucose induces co‐localization of F‐actin (red) and transgelin (green) in MPC5 cells. Scale bar = 50 μm
Immunofluorescence localization of transgelin showed low levels of the cytoskeletal marker protein in control cells, but high levels after treatment with high glucose for 12 hours (Figure 1B). Levels of F‐actin were high in both groups, although mild disarrangement of cytoskeleton was observed after glucose treatment as described above. Finally, it was observed that F‐actin and transgelin were mostly co‐localized in cells exposed to high glucose.
2.2. The time course of transgelin expression upon high glucose stimulation
As assayed by Western blot, levels of transgelin and the cleaved (activated) form of the apoptosis marker caspase 3 varied significantly after exposure to high glucose. Specifically, transgelin levels increased as early as 10 minutes after exposure to high glucose, and remained at a high level 30 minutes after exposure (Figure 2). In contrast, cells treated with mannitol or low glucose displayed low levels of transgelin at all time points. Levels of the cleaved (activated) apoptosis marker caspase 3 were also notably higher 10 minutes after treatment with high glucose, implying that apoptosis of podocytes had been induced.
Figure 2.
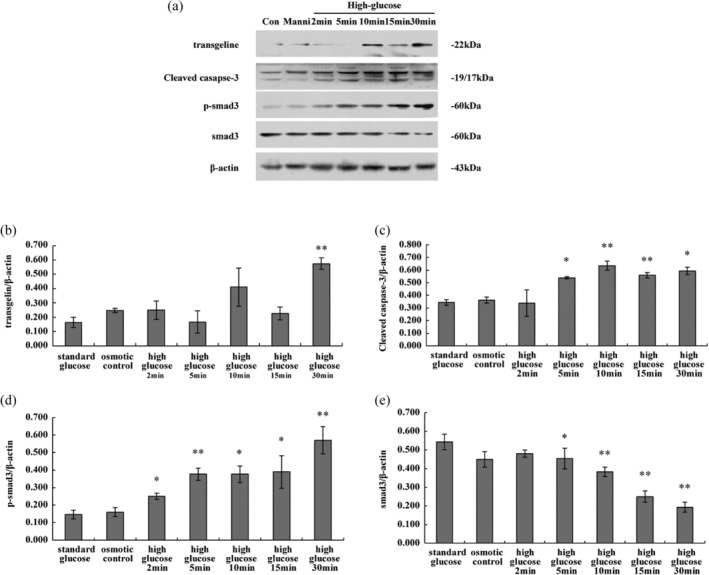
Expression of transgelin and signalling proteins upon treatment of podocytes with high glucose. Protein levels of transgelin, cleaved caspase 3, Smad3 and phospho‐Smad3 protein were assessed by Western blot. β‐Actin was used as an internal control. A, Representative blot, B, Average levels of transgelin, C, cleaved caspase 3, D, phospho‐smad3 and E, total smad3 were normalized with respect to β‐Actin. Data are representative of three independent experiments, and were analysed using one‐way analysis of variance. *P < .05, **P < .01 compared with control or mannitol group
It is well known that TGF‐β signalling is important for the apoptosis of podocyte cells,20, 21 so we next examined whether and when Smad3 was phosphorylated (activated) during high glucose treatment. Smad3 phosphorylation was first observed 10 minutes after treatment with high glucose, and levels continued to be high throughout the observation period. This suggests that activation of TGF‐β signalling may be important for high glucose‐induced apoptosis.
2.3. Inhibition of Smad3 decreases high glucose‐induced podocyte injury and transgelin expression
To further elucidate the relationship between Smad3 signalling and high glucose‐induced apoptosis, the SIS3 was applied to MPC5 cells. As described above, levels of transgelin, phospho‐Smad3 and cleaved caspase 3 increased upon treatment with high glucose; this effect was significantly inhibited by SIS3 based on the treatment with high glucose, suggesting that SIS3 may be able to block apoptosis in podocytes (Figure 3). Proteins downstream of Smad3, c‐myc and p15INK4B, followed a similar trend, but SIS3 had no effect on the glucose‐induced expression of p15INK4B. Levels of phosphorylated JAK and STAT3 in cells exposed to high glucose did not differ from those of the low glucose or mannitol groups, indicating that the JAK/STAT signalling pathway was not affected.
Figure 3.
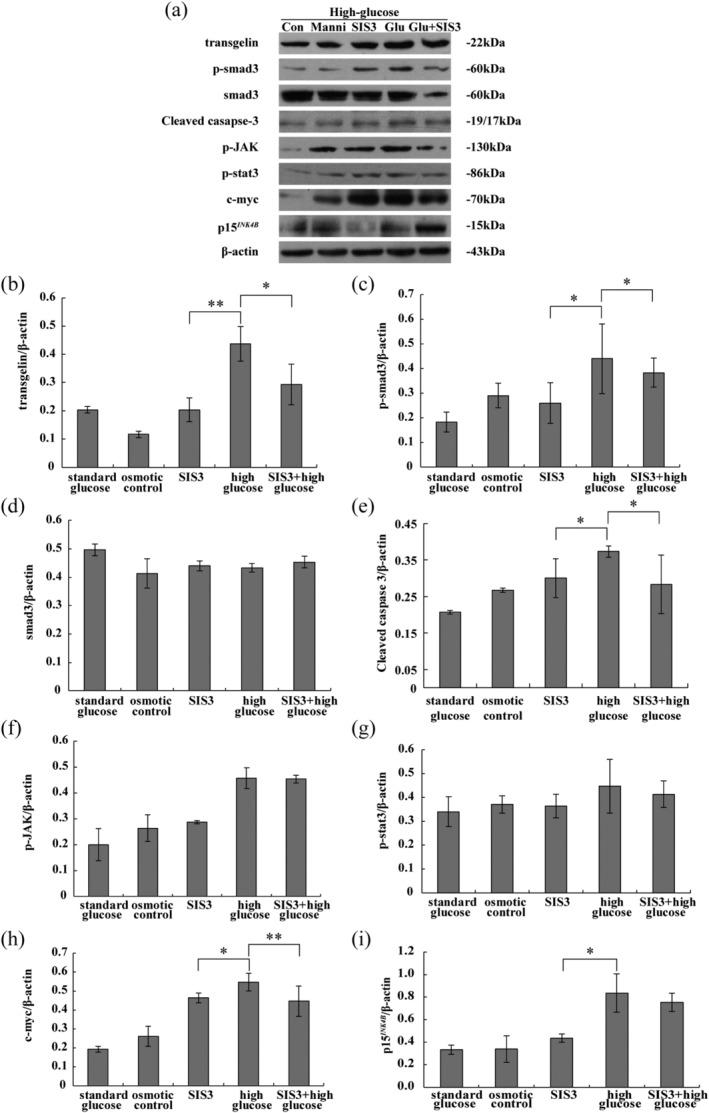
Expression of transgelin and signalling proteins upon treatment of podocytes with high glucose and SIS3. A, Representative Western blot used to quantify protein levels of B, transgelin, C, phospho‐smad3, D, total smad3, E, cleaved caspase 3, F, phospho‐JAK, G, phospho‐STAT3, H, c‐Myc and I, p15INK4B. Protein levels were normalized with respect to β‐Actin. Data are representative of three independent experiments, and were analysed using two‐way analysis of variance. *P < .05, **P < .01
2.4. Smad3 inhibition rescues the morphological changes induced by high glucose stimulation
Complete cytoskeletal filaments were readily observed in the control and SIS3 groups. In the high glucose group, however, the cytoskeleton appeared disordered and there were decreased numbers of podocyte microfilaments (Figure 4A). Cells treated with both high glucose and SIS3 resembled controls, with organized F‐actin and increased numbers of podocyte filaments (Figure 4B).
Figure 4.
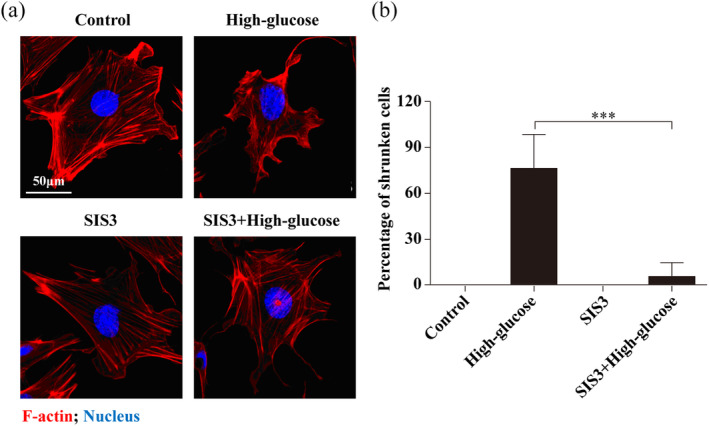
Distribution of F‐actin upon inhibition of Smad3. A, control or SIS3 group (top left): untreated control podocytes display well‐organized F‐actin; high glucose group (top right): disordered cytoskeleton was observed in podocytes treated with high glucose; SIS3 with high glucose group (bottom left): the Smad3‐specific inhibitor had no effect on F‐actin organization, and (bottom right) appeared to block the disruptive effects of high glucose. Scale bar = 50 μm. B, Percentage ratios of shrunken cells to total cells for each group
3. DISCUSSION
In the present study, we investigated changes to the location and expression of newly identified cytoskeleton protein transgelin during high glucose‐induced podocyte injury. High glucose induced a striking disarrangement of cytoskeletal F‐actin and enhanced the expression of transgelin; moreover, F‐actin and transgelin were observed to mostly co‐localize. Concomitant with these changes to F‐actin and transgelin was a significant up‐regulation of Smad3 phosphorylation and its downstream proteins c‐Myc and p15INK4B. Inhibition of Smad3 via SIS3 was protective against cytoskeletal disarrangement and transgelin up‐regulation. These results begin to define the mechanism by which Smad3 signalling affects high glucose‐induced podocyte injury, and provides an experimental basis for novel treatments for diabetic nephrology.
The cytoskeletal protein transgelin was identified in our previous study using a rat model of kidney injury induced by puromycin aminonucleoside.7 Others have reported that transgelin expression can be induced in proteinuric human diseases, including diabetic nephropathy.8 In human skeletal cells, transgelin was reported to be important for differentiation and cytoskeletal integrity.22 In mice with crescentic glomerulonephritis, more podocytes were observed in transgelin knockout mice than in wild‐type controls, suggesting that transgelin may aggravate glomerular cell injury. In the present study, transgelin expression was also increased upon high glucose treatment. This induction occurred as early as 10 minutes after treatment, suggesting that transgelin can be induced quickly during high glucose‐induced podocyte injury.
Localization of transgelin was reported to be different from that of the myofibroblast marker protein αSMA in nephritic rat model.23 The induced transgelin was preferentially located in podocytes, whereas αSMA was mainly expressed in mesangial cells, indicating that transgelin specifically functions in podocytes in this kidney injury model.
In the present study, transgelin protein levels increased after treatment with high glucose, an effect that was blocked by Smad3 inhibition, implying that Smad3 acts as a regulator of transgelin under high glucose conditions. Smad3 may directly regulate expression of transgelin by binding to the transgelin promoter. It has been reported that the promoter region of transgelin is activated by the phosphorylated Smad.24 There is also a TGF‐β control element within the transgelin promoter,25 suggesting that TGF‐β/Smad signalling has a direct role in the regulation and control of transgelin expression. Smad1/3/4 proteins can be activated by TGF‐β to form heterotrimers, which then translocate to cell nuclei to induce transcription of downstream genes. During this process, Smad3 plays a critical role in inducing transgelin transcription, due to the Smad binding site in its promoter region.26
In addition to TGF‐β1/Smad3 signalling, expression of transgelin is also influenced by pro‐inflammatory signals such as interleukin‐1beta (IL‐1β) and TGFβ224 and JNK and p38 MAPK signalling27 in smooth muscle cells. Smad3 can be regulated by TGFβ2 at the mRNA level in chondrocytes28 and in primary human trabecular meshwork cells.29 In addition, reduced levels of Smad3 expression were reported during IL‐1β‐induced chondrocyte injury.30 Together, these data suggest that TGF‐β1/Smad3 signalling may also indirectly regulate the expression of transgelin via cross talk with other signalling pathways such as IL‐1β or TGFβ2.
In the present study, addition of a Smad3 inhibitor attenuated podocyte injury upon treatment with high glucose. However, in a mouse model of diabetic glomerulosclerosis disease, proteinuria and foot process fusion still occur even after Smad3 knockout,31 indicating that there may be other signalling pathways involved.
Smad3 and c‐Myc signalling are known to interact. During liver fibrosis, activated Smad3 interacts with BRD4 to promote its direct binding to the promoter region of c‐Myc.32 Smad3/c‐Myc signalling was also reported to be important for oncogenesis of hepatocellular carcinoma.33 It is also known that TGF‐β can inhibit cell proliferation by down‐regulating expression of c‐Myc and up‐regulating expression of the cyclin‐dependent kinases inhibitor p15INK4B.34 In the present study, c‐Myc was identified to be a downstream target of Smad3 during high glucose‐induced podocyte injury. However, p15INK4B expression was induced by podocyte injury, but unaffected by Smad3 inhibition, suggesting that the involvement of Smad3 in regulating high glucose‐induced podocyte injury was not through p15INK4B. Although JAK/STAT3 signalling has been shown to be activated in other cell types exposed to high glucose conditions,35 we did not observe any such involvement here. Therefore, JAK/STAT3 activation by high glucose may be dependent on cell type. There are likely additional, as‐yet‐unidentified, signalling pathways involved as well. Finally, all cited experiments were conducted at the molecular or cellular level. Further in vivo experiments should be done to confirm these results at a whole‐organism level.
In conclusion, disordered cytoskeleton, foot process retraction and Smad3 activation were observed during high glucose‐induced podocyte injury. In addition, transgelin protein was observed to increase and to co‐localize with the cytoskeletal protein F‐actin. The expression of downstream protein c‐Myc, but not JAK1/STAT3 signalling proteins, was increased by high glucose condition. Addition of SIS3 blocked the effects of high glucose on Smad3 phosphorylation, expression of transgelin and c‐Myc, cleavage of caspase 3 and cytoskeletal structure, implying a critical role for Smad3 signalling in modulating hyperglycaemic injury. Targeted inhibition of the Smad3 pathway may be a novel treatment route for alleviating podocyte damage, especially in cases of diabetic nephropathy.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Data S1: Supporting information
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation of China (Grant No. 81400718) and Research Foundation of Beijing Friendship Hospital, Capital Medical University (Grant No. yyqdkt2018‐21).
Jiang L, Cui H, Ding J. Smad3 signalling affects high glucose‐induced podocyte injury via regulation of the cytoskeletal protein transgelin. Nephrology. 2020;25:659–666. 10.1111/nep.13701
Funding information National Natural Science Foundation of China, Grant/Award Number: 81400718; Research Foundation of Beijing Friendship Hospital, Capital Medical University, Grant/Award Number: yyqdkt2018‐21
Contributor Information
Hong Cui, Email: skybamboo_wsj@163.com.
Jie Ding, Email: djnc_5855@126.com.
REFERENCES
- 1. Mora‐Fernandez C, Dominguez‐Pimentel V, de Fuentes MM, Gorriz JL, Martinez‐Castelao A, Navarro‐Gonzalez JF. Diabetic kidney disease: from physiology to therapeutics. J Physiol. 2014;592:3997‐4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253‐307. [DOI] [PubMed] [Google Scholar]
- 3. Tung CW, Hsu YC, Shih YH, Chang PJ, Lin CL. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology. 2018;23(4):32‐37. [DOI] [PubMed] [Google Scholar]
- 4. Reddy GR, Kotlyarevska K, Ransom RF, Menon RK. The podocyte and diabetes mellitus: is the podocyte the key to the origins of diabetic nephropathy? Curr Opin Nephrol Hypertens. 2008;17:32‐36. [DOI] [PubMed] [Google Scholar]
- 5. Ohigashi M, Kobara M, Takahashi T, Toba H, Wada T, Nakata T. Pitavastatin suppresses hyperglycaemia‐induced podocyte injury via bone morphogenetic protein‐7 preservation. Clin Exp Pharmacol Physiol. 2017;44:378‐385. [DOI] [PubMed] [Google Scholar]
- 6. Zhang H, Luo W, Sun Y, et al. Wnt/beta‐catenin signaling mediated‐UCH‐L1 expression in podocytes of diabetic nephropathy. Int J Mol Sci. 2016;17(9):1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miao J, Fan Q, Cui Q, et al. Newly identified cytoskeletal components are associated with dynamic changes of podocyte foot processes. Nephrol Dial Transplant. 2009;24:3297‐3305. [DOI] [PubMed] [Google Scholar]
- 8. Marshall CB, Krofft RD, Blonski MJ, et al. Role of smooth muscle protein SM22alpha in glomerular epithelial cell injury. Am J Physiol Renal Physiol. 2011;300:F1026‐F1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weng HB, Han WK, Xiong YW, et al. Taxus chinensis ameliorates diabetic nephropathy through down‐regulating TGF‐beta1/Smad pathway. Chin J Nat Med. 2018;16:90‐96. [DOI] [PubMed] [Google Scholar]
- 10. Jiang L, Chen FX, Zang ST, Yang QF. Betulinic acid prevents high glucoseinduced expression of extracellular matrix protein in cardiac fibroblasts by inhibiting the TGFbeta1/Smad signaling pathway. Mol Med Rep. 2017;16:6320‐6325. [DOI] [PubMed] [Google Scholar]
- 11. Zhang H, Cui YC, Li K, et al. Glutamine protects cardiomyocytes from hypoxia/reoxygenation injury under high glucose conditions through inhibition of the transforming growth factor‐beta1‐Smad3 pathway. Arch Biochem Biophys. 2016;596:43‐50. [DOI] [PubMed] [Google Scholar]
- 12. Liu J, Bao J, Hao J, Peng Y, Hong F. HSP70 inhibits high glucose‐induced Smad3 activation and attenuates epithelial‐to‐mesenchymal transition of peritoneal mesothelial cells. Mol Med Rep. 2014;10:1089‐1095. [DOI] [PubMed] [Google Scholar]
- 13. Kim JH, Kim BK, Moon KC, Hong HK, Lee HS. Activation of the TGF‐beta/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 2003;64:1715‐1721. [DOI] [PubMed] [Google Scholar]
- 14. Sun YB, Qu X, Howard V, et al. Smad3 deficiency protects mice from obesity‐induced podocyte injury that precedes insulin resistance. Kidney Int. 2015;88:286‐298. [DOI] [PubMed] [Google Scholar]
- 15. Yu L, Liu Y, Wu Y, et al. Smad3/Nox4‐mediated mitochondrial dysfunction plays a crucial role in puromycin aminonucleoside‐induced podocyte damage. Cell Signal. 2014;26:2979‐2991. [DOI] [PubMed] [Google Scholar]
- 16. Li G, Li Y, Liu S, et al. Gremlin aggravates hyperglycemia‐induced podocyte injury by a TGFbeta/smad dependent signaling pathway. J Cell Biochem. 2013;114:2101‐2113. [DOI] [PubMed] [Google Scholar]
- 17. Liu L, Lin W, Zhang Q, Cao W, Liu Z. TGF‐beta induces miR‐30d down‐regulation and podocyte injury through Smad2/3 and HDAC3‐associated transcriptional repression. J Mol Med. 2016;94:291‐300. [DOI] [PubMed] [Google Scholar]
- 18. Hino M, Nagase M, Kaname S, et al. Expression and regulation of adrenomedullin in renal glomerular podocytes. Biochem Biophys Res Commun. 2005;330:178‐185. [DOI] [PubMed] [Google Scholar]
- 19. Su K, Zeng P, Liang W, et al. FTY720 attenuates angiotensin II‐induced podocyte damage via inhibiting inflammatory cytokines. Mediators Inflamm. 2017;2017:3701385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schiffer M, Bitzer M, Roberts IS, et al. Apoptosis in podocytes induced by TGF‐beta and Smad7. J Clin Invest. 2001;108:807‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Das R, Xu S, Nguyen TT, et al. Transforming growth factor beta1‐induced apoptosis in podocytes via the extracellular signal‐regulated kinase‐mammalian target of Rapamycin complex 1‐NADPH oxidase 4 axis. J Biol Chem. 2015;290:30830‐30842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elsafadi M, Manikandan M, Dawud RA, et al. Transgelin is a TGFbeta‐inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization. Cell Death Dis. 2016;7:e2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakamaki Y, Sakatsume M, Wang X, et al. Injured kidney cells express SM22alpha (transgelin): unique features distinct from alpha‐smooth muscle actin (alphaSMA). Nephrology. 2011;16:211‐218. [DOI] [PubMed] [Google Scholar]
- 24. Maleszewska M, Gjaltema RA, Krenning G, Harmsen MC. Enhancer of zeste homolog‐2 (EZH2) methyltransferase regulates transgelin/smooth muscle‐22alpha expression in endothelial cells in response to interleukin‐1beta and transforming growth factor‐beta2. Cell Signal. 2015;27:1589‐1596. [DOI] [PubMed] [Google Scholar]
- 25. Frick CL, Yarka C, Nunns H, Goentoro L. Sensing relative signal in the Tgf‐beta/Smad pathway. Proc Natl Acad Sci USA. 2017;114:E2975‐E2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Budi EH, Duan D, Derynck R. Transforming growth factor‐beta receptors and Smads: regulatory complexity and functional versatility. Trends Cell Biol. 2017;27:658‐672. [DOI] [PubMed] [Google Scholar]
- 27. Kaplan‐Albuquerque N, Garat C, Van Putten V, Nemenoff RA. Regulation of SM22 alpha expression by arginine vasopressin and PDGF‐BB in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H1444‐H1452. [DOI] [PubMed] [Google Scholar]
- 28. Khaghani SAB, Akbarova G, Soon CF, Dilbazi G. Effect of transforming growth factor‐beta2 on biological regulation of multilayer primary chondrocyte culture. Cell Tissue Bank. 2018;19:763‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kasetti RB, Maddineni P, Patel PD, Searby C, Sheffield VC, Zode GS. Transforming growth factor beta2 (TGFbeta2) signaling plays a key role in glucocorticoid‐induced ocular hypertension. J Biol Chem. 2018;293:9854‐9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He J, Zhang J, Wang D. Down‐regulation of microRNA‐216b inhibits IL‐1beta‐induced chondrocyte injury by up‐regulation of Smad3. Biosci Rep. 2017;37(2):BSR20160588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang A, Ziyadeh FN, Lee EY, et al. Interference with TGF‐beta signaling by Smad3‐knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol. 2007;293:F1657‐F1665. [DOI] [PubMed] [Google Scholar]
- 32. Cai X, Li Z, Zhang Q, et al. CXCL6‐EGFR‐induced Kupffer cells secrete TGF‐beta1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C‐MYC/EZH2 pathway in liver fibrosis. J Cell Mol Med. 2018;22:5050‐5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu R, Tang C, Shen A, et al. IL‐37 suppresses hepatocellular carcinoma growth by converting pSmad3 signaling from JNK/pSmad3L/c‐Myc oncogenic signaling to pSmad3C/P21 tumor‐suppressive signaling. Oncotarget. 2016;7:85079‐85096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz‐1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400‐408. [DOI] [PubMed] [Google Scholar]
- 35. Duan WN, Xia ZY, Liu M, et al. Protective effects of SOCS3 overexpression in high glucoseinduced lung epithelial cell injury through the JAK2/STAT3 pathway. Mol Med Rep. 2017;16:2668‐2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting information