

Abstract
The subgranular zone of the dentate gyrus is a subregion of the hippocampus that has two uniquely defining features; it is one of the most active sites of adult neurogenesis as well as the location where the highest concentrations of synaptic zinc are found, the mossy fiber terminals. Therefore, we sought to investigate the idea that vesicular zinc plays a role as a modulator of hippocampal adult neurogenesis. Here, we used ZnT3 −/− mice, which are depleted of synaptic‐vesicle zinc, to test the effect of targeted deletion of this transporter on adult neurogenesis. We found that this manipulation reduced progenitor cell turnover as well as led to a marked defect in the maturation of newborn cells that survive in the DG toward a neuronal phenotype. We also investigated the effects of zinc (ZnCl2), n‐acetyl cysteine (NAC), and ZnCl2 plus 2NAC (ZN) supplement on adult hippocampal neurogenesis. Compared with ZnCl2 or NAC, administration of ZN resulted in an increase in proliferation of progenitor cells and neuroblast. ZN also rescued the ZnT3 loss‐associated reduction of neurogenesis via elevation of insulin‐like growth factor‐1 and ERK/CREB activation. Together, these findings reveal that ZnT3 plays a highly important role in maintaining adult hippocampal neurogenesis and supplementation by ZN has a beneficial effect on hippocampal neurogenesis, as well as providing a therapeutic target for enhanced neuroprotection and repair after injury as demonstrated by its ability to prevent aging‐dependent cognitive decline in ZnT3 −/− mice. Therefore, the present study suggests that ZnT3 and vesicular zinc are essential for adult hippocampal neurogenesis.
Keywords: adult neurogenesis, hippocampus, n‐acetyl cysteine, zinc, zinc transporter 3
This study utilized ZnT3 −/− mice with a genetic loss‐of‐function approach to investigate the consequences of vesicular zinc depletion in adult hippocampal neurogenesis. We found that ZnT3 −/− mice displayed reduced hippocampal neurogenesis under physiological conditions even at 3 months of age. We also found that administration of ZnCl2 plus 2NAC (ZN) not only reversed the ZnT3 loss‐associated reduction of neurogenesis, but also improved aging‐dependent cognitive decline in ZnT3 −/− mice.
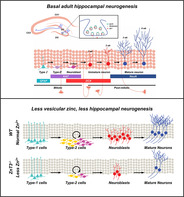
Significance statement.
This study tested the hypothesis that ZnT3 and vesicular zinc serve a critical role in the regulation of adult hippocampal neurogenesis. It was demonstrated that ZnT3 −/− mice display reduced hippocampal neurogenesis under physiological conditions, even at 3 months of age. The findings reveal that ZnT3 has a central function in maintaining adult hippocampal neurogenesis and zinc supplementation by ZN has a beneficial effect on hippocampal neurogenesis and may offer a means to enhance neuroprotection and repair after injury.
1. INTRODUCTION
The divalent cationic zinc (Zn2+) is the second most abundant transition metal in the brain and spinal cord following iron and is a major player in numerous cellular and physiological processes. 1 Most ionic zinc in the brain is tightly bound with proteins such as metallothionein or metalloproteins. 2 Loosely bound or free‐zinc is often found at increased concentrations in synaptic vesicles in brain regions where neurogenesis and neural migration actively occur in the adult brain, such as the hippocampal dentate gyrus (DG) and olfactory bulb. 3 , 4 Zinc is a component of over 300 enzymes and highly involved in the regulation of cell division and DNA synthesis. 5 Zinc deficiency leads to impaired DNA synthesis and growth retardation in animals. 6 Zinc also influences hormonal regulation of cell division, such as that mediated by insulin‐like growth factor‐1 (IGF‐1) or nerve growth factor. 7 Several studies have reported the adverse effects of an insufficient supply of zinc on normal physiological function. Dietary zinc deprivation impairs performance in visual‐attention and short‐term‐memory tasks. 8 Taken together, the evidence suggests that zinc is an essential element for maintaining normal neuronal cell proliferation, development, and cognitive function.
The role of zinc transporter 3 (ZnT3) and vesicular zinc on cognitive function have been investigated by several labs. Adlard et al reported that ZnT3 −/− mice exhibit age‐dependent deficits in learning and memory. 9 Sindreu et al demonstrated that ZnT3 −/− mice have impaired contextual discrimination and spatial working memory. 10 Martel et al showed that ZnT3 −/− mice performed normally at initial learning but showed difficulty finding a second platform location. These ZnT3 −/− mice display enhanced social interaction but were likewise strikingly poor at forming social and object recognition memory. Therefore, it appears that ZnT3 is involved in spatial memory and behavior that relies on the hippocampus and perirhinal cortex. 11
Although the above studies reported that ZnT3 −/− mice showed learning and memory deficits during the aging process the precise mechanism is not clear. Thus, we hypothesized that deletion of ZnT3 gene negatively affects adult hippocampal neurogenesis. To determine whether synaptic zinc is an important factor for modulating hippocampal neurogenesis, the present study also used ZnT3 −/− mice. We demonstrate a severe reduction of progenitor cell proliferation and neuronal differentiation in the DG of ZnT3 −/− mice. We propose a cell‐extrinsic mechanism of ZnT3 deficiency mediated by decreasing levels of IGF‐1 and phosphorylation of both extracellular signal‐regulated kinase (ERK) and cyclic AMP response element binding protein (CREB) in the hippocampus of ZnT3 −/− mice. The present study suggests that ZnT3 −/− mice display reduced hippocampal neurogenesis under physiological conditions even at 3 months of age. Our findings indicate that vesicular zinc plays an important role in adult hippocampal neurogenesis and have implications for cognitive health in aging and disease.
2. MATERIALS AND METHODS
2.1. Animals and ethics statement
ZnT3 +/+ and ZnT3 −/− male mice (a kind gift from Dr Jae‐Young Koh, Department of Neurology, University of Ulsan College of Medicine, Korea; background strains, C57BL/6 and Sv129 hybrid), aged 3 months (25‐30 g) or 15‐18 months (30‐45 g), were bred and maintained in the facility of University of Hallym College of Medicine. The animals were housed in a temperature‐ and humidity‐controlled environment (22°C ± 2°C, 55% ± 5% and a 12 hours light:12 hours dark cycle), supplied with Purina diet (Purina, Gyeonggi, Korea) and water ad libitum. Animal use and relevant experimental procedures were approved by the Institutional Animal Care and Use Committee, Hallym University (Protocol # Hallym 2017‐16). This manuscript was written up in accordance with the ARRIVE (Animal Research: Reporting in vivo Experiments) guidelines. 12 Before all experiments, genotyping for ZnT3 was performed using PCR with a primer set to amplify WT (5′‐GGT ATC CAT GCC CTT CCT CTA GAG‐3′), or common (5′‐ATA GTC ACT GGC ATC CTC CTG TAC C‐3′), or the KO allele (5′‐CCT GTG CTC TAG TAG CTT TAC GG‐3′) as described previously. 13
2.2. Experimental design and 5‐bromo‐2‐deoxyuridine labeling
To test the role of vesicular zinc at different stages of adult neurogenesis, we conducted 4‐phase studies using ZnT3 −/− mice and zinc supplementation. In phase 1, to assess the effects of eliminating vesicular zinc at the early or late phase of adult hippocampal neurogenesis, mice were divided into two groups in each phase: ZnT3 +/+ (n = 7) and ZnT3 −/− (n = 9). The thymidine analog BrdU (50 mg/kg; Sigma Cat# B5002, St. Louis, Missouri) was intraperitoneally injected twice daily for four consecutive days and the brains were harvested at 5 days (early phase) or 6 weeks (late phase) after the initial BrdU injection. In phase 2, to evaluate the effects of zinc supplementation at the early or late phase of adult hippocampal neurogenesis, mice were divided into four groups in each phase: vehicle (Veh; early phase, n = 10; late phase, n = 6), zinc chloride (ZnCl2; early phase, n = 10; late phase, n = 8), n‐acetyl cysteine (NAC; early phase, n = 11; late phase, n = 9), and ZnCl2 plus 2NAC (ZN; early phase, n = 10; late phase, n = 9). Mice were given ZnCl2 (4 mg/kg), NAC (20 mg/kg), or ZN intraperitoneally available once per day for 2 (early phase) or 8 weeks (late phase). BrdU was injected two times per day for four consecutive days starting 10 days after the initial ZN treatment, and then mice were sacrificed after the last dose of ZN. Control mice were injected with equal volumes of saline only (vehicle). In phase 3, zinc supplementation by ZN was initiated at the early or late phase of adult hippocampal neurogenesis and mice were divided into four groups in each phase: vehicle‐treated ZnT3 +/+ mice (early phase, n = 6; late phase, n = 6), ZN‐treated ZnT3 +/+ mice (early phase, n = 9; late phase, n = 8), vehicle‐treated ZnT3 −/− mice (early phase, n = 6; late phase, n = 7), ZN‐treated ZnT3 −/− mice (early phase, n = 7; late phase, n = 6). Mice were given ZN intraperitoneally once per day for 2 or 8 weeks. BrdU was injected two times per day for four consecutive days starting 10 days after initial ZN treatment, then mice were sacrificed at 2 (early phase) or 8 weeks (late phase) after initial ZN treatment. In phase 4, to analysis the effects of zinc supplementation by ZN on cognitive abilities, mice were divided into four groups: (a) vehicle‐treated aged ZnT3 +/+ mice (n = 11), (b) vehicle‐treated aged ZnT3 −/− mice (n = 13), (c) ZN‐treated aged ZnT3 +/+ mice (n = 12), and (d) ZN‐treated aged ZnT3 −/− mice (n = 14). ZN supplementation was performed once per day for 8 weeks. Morris water maze (MWM) test were conducted for six consecutive days starting 50 days after the initial ZN supplementation, then mice were sacrificed after the last dose of ZN.
2.3. Quantitative analysis
To quantify BrdU‐, Ki67‐, DCX‐, and pCREB‐positive cells, every sixth coronal section spanning the septal hippocampus was collected. The number of BrdU‐, Ki67‐, DCX‐, and pCREB‐positive cells was determined in the subgranular zone (SGZ) and granular cell layer (GCL) from both hemispheres using optical fractionators probe unbiased stereology investigation (Stereo Investigator, MicroBrightField, Williston, Vermont). 14 , 15 Counting frames (15 × 15 × 20 μm) were placed at the intersection of a matrix (40 × 40 μm) randomly superimposed onto the region of interest by the program. Cells were counted using a ×63 oil objective. To analyze the phenotype of the BrdU‐positive cells, as mentioned previously, fluorescence signals were detected using a Zeiss LSM 710 confocal microscopy (Carl Zeiss, Oberkochen, Germany) with a sequential scanning mode for Alexa 488 and 594. Stacks of images (1024 × 1024 pixels) from consecutive slices of 0.9 to 1.2 μm in thickness were obtained by averaging eight scans per slice and were processed using ZEN 2 (Carl Zeiss). The number of BrdU+NeuN+ cells was estimated by multiplying the percentages of colocalization (determined by confocal microscopy) to the total number of BrdU‐labeled cells (determined by stereology).
To measure IGF‐1‐immunofluorescence intensity, CA1, CA3, or DG from the brain section images were selected as regions of interest (ROIs) and measured using ImageJ. Briefly, the image was loaded into ImageJ, and changed into 8‐bit via the menu option (Image/Color/Split Channels). The image was binarized, and the menu option (Analyze/Measure) was selected, and then immunofluorescence signal was expressed as the mean gray value.
To analyze the levels of pERK and TSQ intensity, DG and mossy fibers (MF) from the brain section images were selected as ROIs and measured using ImageJ. The image was converted to 8‐bit through the menu options (Image/Type/8‐bit). Next, an image threshold was set as follows (Image/Adjust/Threshold): the type was set to black and white and the bottom slider moved to a value sufficient to show only the pERK immunoreactive area or TSQ signal. The thresholded image was binary and only represented pERK immunoreactivity or TSQ signal. The selected part in the whole image was sorted, and then, the intensity of pERK or TSQ was represented as the mean gray value.
2.4. Statistical analysis
For statistical comparisons between ZnT3 +/+ and ZnT3 −/− mice from phase 1, significance was evaluated using an unpaired Student's t test. The intensity data of immunostaining, Western blot, and TSQ staining from phase 2 to 3 were analyzed by Kruskal‐Wallis test with post hoc analysis using Bonferroni correction to compare the values among four groups since the values did not meet normal distribution. The remaining immunohistochemical data, Western blot and probe trial data from phases 2 to 4 were analyzed by a one‐way analysis of variance (ANOVA; ZnT3 −/− or ZnT3 +/+ mice) × (ZN or vehicle) with a post hoc Bonferroni multiple comparison test using statistical package for the social sciences (SPSS, Chicago, Illinois). Repeated measure ANOVA was used to examine the contribution of treatment and day on the escape latency. All data were expressed as the mean ± SE and P value <.05 were considered significant. We then performed a post hoc power analysis with means, standard deviation, and number of subjects for four groups using G*Power 3.1.9.7. 16 The result revealed that we obtained a power of 0.99. Therefore, our study design had sufficient power to reveal statistical significance in our study.
3. RESULTS
3.1. Vesicular zinc is eliminated in the hippocampus of ZnT3 −/− mice
We determined mouse genotype by PCR. PCR analysis of genomic DNA shows loss of the ZnT3 band (650 bp) and presence of the NEO cassette (400 bp) in the ZnT3 −/− mice (Figure 1A). We also stained brain sections of ZnT3 +/+ and ZnT3 −/− mice with the TSQ, zinc‐specific fluorescent dye, to test whether vesicular zinc regulation is altered in ZnT3 −/− mice. ZnT3 +/+ mice showed high intensity TSQ fluorescence signals in the hippocampal MF area. However, ZnT3 −/− mice showed almost no fluorescence signals in the hippocampus (Figure 1B,B′). After examining TSQ fluorescence, we evaluated ZnT3 immunoreactivity (IR) in both ZnT3 +/+ and ZnT3 −/− mice. ZnT3 localization on vesicle membranes was also lower in the hippocampus of ZnT3 −/− mice. ZnT3‐IR from ZnT3 −/− mice was undetectable in the MFs of hippocampus (Figure 1C,C′).
FIGURE 1.
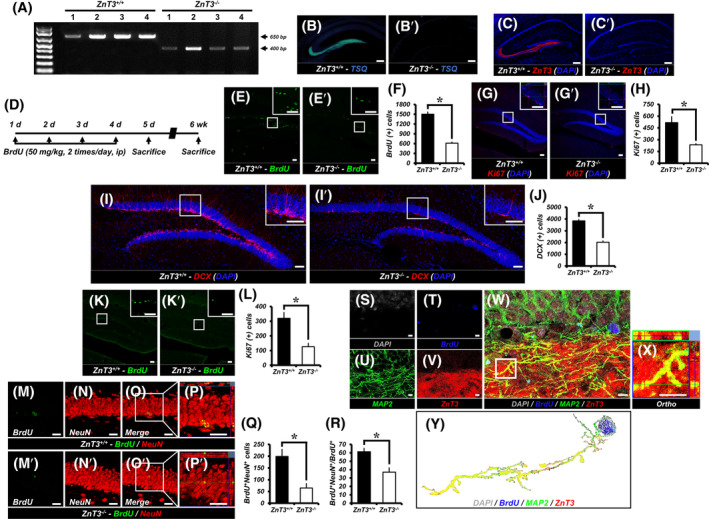
ZnT3 −/− mice reduces adult hippocampal neurogenesis. A, Genotyping with PCR. PCR analysis of genomic DNA demonstrates the absence of the ZnT3 band (650 bp) and presence of the NEO cassette (400 bp) in ZnT3 −/− mice. B‐C′, The distributions of TSQ‐histofluorescence (B,B′) and ZnT3‐immunoreactive fluorescence (C,C′) were detected with TSQ and an anti‐ZnT3 antibody. ZnT3 +/+ mice revealed high intensity of TSQ fluorescence and ZnT3‐immunoreactivity (IR) in the hippocampal mossy fiber area. However, ZnT3 −/− mice did not show TSQ and ZnT3‐IR in the hippocampus. Scale bar = 200 μm. D, Timeline showing the experimental design. BrdU was intraperitoneally administered twice per day for four consecutive days. Mice were then killed at 5 days or 6 weeks after initial injection of BrdU. E,E′, Representative images of the dentate gyrus (DG) showing BrdU (green) staining to ZnT3 +/+ (E) and ZnT3 −/− (E′) mice at 5 days after first injection of BrdU. Scale bar = 50 μm. F, Quantification of BrdU+ cells in the subgranular zone/granule cell layer (SGZ/GCL) (mean ± SEM; ZnT3 +/+: n = 7; ZnT3 −/−: n = 9). *P < .05 (unpaired Student's t test). G,G′, Representative images of Ki67‐ (red) and DAPI‐ (blue) immunopositive cells as merged images to ZnT3 +/+ (G) and ZnT3 −/− (G′) mice. Scale bar = 50 μm. H, Number of progenitor cells (Ki67+DAPI+) in the SGZ/GCL (mean ± SEM; ZnT3 +/+: n = 7; ZnT3 −/−: n = 9). *P < .05 (unpaired Student's t test). I,I′, Representative images of DCX‐ (red) and DAPI‐ (blue) immunopositive cells as merged images to ZnT3 +/+ (I) and ZnT3 −/− (I′) mice. Scale bar = 50 μm. J, Number of immature neurons (DCX+DAPI+) in the SGZ/GCL (mean ± SEM; ZnT3 +/+: n = 7; ZnT3 −/−: n = 9). *P < .05 (unpaired Student's t test). K,K′, Representative images of the DG showing BrdU (green) staining to ZnT3 +/+ (K) and ZnT3 −/− (K′) mice at 6 weeks after first injection of BrdU. Scale bar = 50 μm. L, Quantification of BrdU+ cells in the SGZ/GCL (mean ± SEM; ZnT3 +/+: n = 7; ZnT3 −/−: n = 9). *P < .05 (unpaired Student's t test). M‐P′, Representative images of BrdU‐ (green) (M,M′) and NeuN‐ (red) (N,N′) immunopositive cells as merged images to ZnT3 +/+ (O,P) and ZnT3 −/− (O′,P′) mice. Scale bar = 50 μm. Q,R, Bar graphs showing the number (Q) and percentage (R) of cells that express the markers BrdU and NeuN (newborn neurons) in the SGZ/GCL (mean ± SEM; ZnT3 +/+: n = 7; ZnT3 −/−: n = 9). *P < .05 (unpaired Student's t test). S‐Y, ZnT3 is expressed in newborn neurons. Representative immunofluorescence images showing DAPI‐ (gray) (S), BrdU‐ (blue) (T), MAP2‐ (green) (U), and ZnT3‐ (red) (V) immunoreactive cells in the DG of ZnT3 +/+ mice at 6 weeks after initial BrdU injection as merged image (W). High‐power view (X) showing colocalized ZnT3 immunoreactivity in axon of DAPI+BrdU+MAP2+ cells. Scale bar = 5 μm. Y, Representative image of the combined expression of ZnT3 and individual newborn neuron (DAPI+BrdU+MAP2+) using ImageJ
3.2. Genetic deletion of ZnT3 reduces hippocampal progenitor cell proliferation
To assess whether endogenous ZnT3 affects DG progenitor cell proliferation, mice were injected with BrdU (50 mg/kg) twice per day for four consecutive days in both ZnT3 +/+ and ZnT3 −/− mice (Figure 1D). We found that the BrdU‐ and Ki67‐immunopositive cells were distributed mainly in the SGZ/GCL. A significant decrease in the number of BrdU (Figure 1E,F) and Ki67 (Figure 1G,H) labeled cells was seen in the SGZ/GCL of ZnT3 −/− mice as compared to age‐matched ZnT3 +/+ mice at 5 days after first injection of BrdU. The ZnT3 −/− mice had a 59.2% (BrdU) or 54.8% (Ki67) reduction of the hippocampal progenitor cell proliferation in comparison with controls.
3.3. ZnT3 −/− mice show reduced production of neuroblasts
To test whether ZnT3 influences the presence of immature neurons we performed immunostaining for DCX. Genetic deletion of ZnT3 caused a 47.3% reduction in the number of DCX‐immunopositive cells, the specific marker of immature neurons (neuroblast) (Figure 1I,J).
3.4. Compromised adult neurogenesis in the hippocampus of ZnT3−/− mice
To investigate whether endogenous ZnT3 influences survival of DG newborn cells, we birthdated newly generated cells in ZnT3 +/+ and ZnT3 −/− mice by injecting BrdU twice daily for four consecutive days and investigated their fate at 6 weeks after initial BrdU injection. A significant decrease in the number of BrdU‐labeled cells was seen in the SGZ/GCL of ZnT3 −/− mice (Figure 1K,L). The ZnT3 −/− mice had a 60.6% decrease in survival of newly generated cells in comparison with controls. We also calculated the ratio of the number of BrdU+ cells in the SGZ/GCL at 6 weeks to the number of BrdU+ cells in the DG at 5 days after initial BrdU injection and found that the survival ratio of BrdU‐labeled cells in ZnT3 −/− mice (20.5%) was similar to that of controls (21.2%; data not shown).
Next, we determined the role of ZnT3 on neuronal maturation of cells that survived in the SGZ/GCL at 6 weeks after initial BrdU injection. When calculating the number of adult newborn neurons, genetic deletion of ZnT3 caused a significant decrease in the total number of BrdU+ cells double labeled for the specific marker of mature neurons (NeuN) in the SGZ/GCL (Figure 1M‐Q). In addition, ZnT3 −/− mice showed a 39.52% decrease in the percentage of BrdU+NeuN+ cells in BrdU+ cells (Figure 1R). These results show that deletion of ZnT3 decreases newborn mature neurons in the adult hippocampal DG.
3.5. ZnT3 is expressed in newly generated neurons
Next, we investigated whether ZnT3 is expressed in newborn neurons in addition to mature neurons. To confirm the presence of ZnT3 in newborn neurons, the DG of ZnT3 +/+ mice at 6 weeks after initial BrdU injection were assayed via immunofluorescence staining for BrdU, ZnT3, and MAP2. We detected much stronger expression of ZnT3 and MAP2 in the MF of DG. We also found that ZnT3 protein colocalized with axons of BrdU+MAP2+ cells in the DG (Figure 1S‐Y). This indicates that ZnT3 is expressed in the axonal terminal of newborn neurons.
3.6. ZN increases adult hippocampal neurogenesis
To determine whether ZN supplementation enhances adult hippocampal neurogenesis, we analyzed the effects of ZN supplementation on the rate of cell proliferation and neuronal differentiation within the DG (Figure 2A). Compared with vehicle‐treated mice, the BrdU‐ and Ki67‐labeled cells were not significantly changed in ZnCl2‐treated mice, whereas NAC‐ or ZN‐treated mice exhibited a significant increase in the number of BrdU‐ (Figure 2B‐F) and Ki67‐labeled cells (Figure 2G‐K) in the SGZ. In addition, we also observed a greatly increased number of DCX‐immunopositive cells in ZN‐treated mice compared with vehicle, ZnCl2 or NAC‐treated mice (Figure 2L‐P).
FIGURE 2.
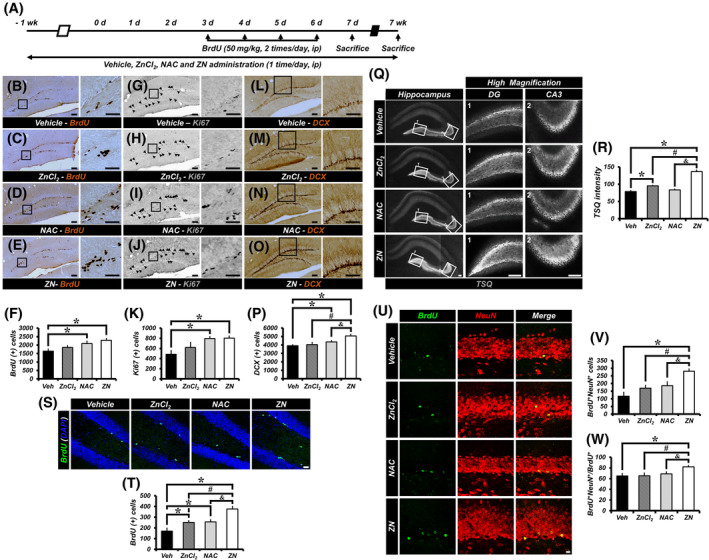
ZN increases adult hippocampal neurogenesis. A, Schematic of the experimental time course. B‐E, Representative images of the DG showing BrdU staining to vehicle‐ (B), zinc chloride‐ (ZnCl2) (C), n‐acetyl cysteine‐ (NAC) (D) or ZnCl2 + 2NAC‐ (ZN) (E) treated groups at 2 weeks after initial ZN supplementation. Scale bar = 50 μm. G‐J, Representative images of Ki67‐immunopositive cells to vehicle‐ (G), ZnCl2‐ (H), NAC‐ (I), or ZN‐ (J) treated groups. Scale bar = 50 μm. L‐O, Representative images of DCX‐immunopositive cells to vehicle‐ (L), ZnCl2‐ (M), NAC‐ (N), or ZN‐ (O) treated groups. Scale bar = 50 μm. F,K,P, Number of progenitor cells (BrdU+ [F] or Ki67+ [K]) and neuroblasts (DCX+ [P]) in the SGZ/GCL (mean ± SEM; Veh, n = 10; ZnCl2, n = 10; NAC, n = 11; ZN, n = 10). *P < .05 vs vehicle‐treated group; #P < .05 vs ZnCl2‐treated group; &P < .05 vs NAC‐treated group (one‐way analysis of variance [ANOVA] followed by Bonferroni post hoc test; F: F = 4.332, P = .01; K: F = 5.022, P = .005; P: F = 6.591, P = .001). Q, Vesicular zinc expression in the mossy fibers (MFs) of hippocampal DG from mice treated with vehicle, ZnCl2, NAC, or ZN as shown by TSQ fluorescence. Scale bar = 100 μm. R, Quantification of TSQ fluorescent intensity from the MFs of DG. Data are mean ± SEM; n = 3 from each group, *P < .05 vs vehicle‐treated group; #P < .05 vs ZnCl2‐treated group; &P < .05 vs NAC‐treated group (Kruskal‐Wallis test followed by Bonferroni post hoc test: Chi square = 9.842, df = 3, P = .02). S, Representative images of the DG showing BrdU‐ (green) and DAPI‐ (blue) immunopositive cells as merged images to vehicle‐, ZnCl2‐, NAC‐, or ZN‐treated mice at 8 weeks after initial ZN supplementation. Scale bar = 20 μm. T, Quantification of BrdU+ cells in the SGZ/GCL (mean ± SEM; vehicle, n = 6; ZnCl2, n = 8; NAC, n = 9; ZN, n = 9). *P < .05 vs vehicle‐treated group; #P < .05 vs ZnCl2‐treated group; &P < .05 vs NAC‐treated group (one‐way ANOVA followed by Bonferroni post hoc test: F = 12.651, P < .001). U, Representative images of BrdU‐ (green) and NeuN‐ (red) immunopositive cells as merged images to vehicle‐, ZnCl2‐, NAC‐, or ZN‐treated mice. Scale bar = 10 μm. V,W, Bar graphs showing the number (V) and percentage (W) of cells that express the markers BrdU and NeuN (newborn neurons) in the SGZ/GCL (mean ± SEM; vehicle, n = 6; ZnCl2, n = 8; NAC, n = 9; ZN, n = 9). *P < .05 vs vehicle‐treated group; #P < .05 vs ZnCl2‐treated group; &P < .05 vs NAC‐treated group (one‐way ANOVA followed by Bonferroni post hoc test; V: F = 9.802, P < .001; W: F = 3.803, P = .021)
Furthermore, we also determined whether ZN supplementation increases levels of vesicular zinc in the hippocampal mossy fibers (MFs). We found that there were no differences in vesicular TSQ intensity between vehicle‐treated and NAC‐treated mice. However, vesicular TSQ intensity was increased in ZnCl2‐treated mice compared with vehicle‐treated mice and ZN‐treated mice showed significantly increased vesicular TSQ intensity in the MFs compared with vehicle‐, ZnCl2‐, or NAC‐treated mice (Figure 2Q,R).
We then explored whether ZN impacts the newly generated cell survival and neuronal maturation of newborn cells in the SGZ/GCL. ZN supplementation was performed once a day for 8 weeks. We injected BrdU twice per day for four consecutive days starting at 10 days after the initial ZN supplementation. The numbers of BrdU+ and BrdU+NeuN+ cells from mice treated with ZN were significantly higher than in sections from vehicle‐, ZnCl2‐, or NAC‐treated mice (Figure 2S‐V). In addition, ZN‐treated mice showed a significant increase in the percentage of BrdU+NeuN+ cells among BrdU+ cells (Figure 2W).
3.7. ZN treatment reversed adult hippocampal neurogenesis in ZnT3−/− mice
Here we sought to ascertain if the decline in progenitor cell proliferation and neuronal differentiation could be rescued by ZN. ZnT3 +/+ and ZnT3 −/− mice were treated with ZN or vehicle (Figure 3A). We found that ZN treatment revealed an increase in the number of BrdU+, Ki67+ or DCX+ cells in the SGZ/GCL of both ZnT3 +/+ and ZnT3 −/− mice compared with vehicle‐treated mice, although ZnT3 −/− mice treated with ZN showed a lower number of BrdU+, Ki67+ or DCX+ cells than ZnT3 +/+ mice treated with ZN (Figure 3B‐G). These results show that ZN reverses the decrease in progenitor cell proliferation and neuroblast production, caused by genetic deletion of ZnT3.
FIGURE 3.
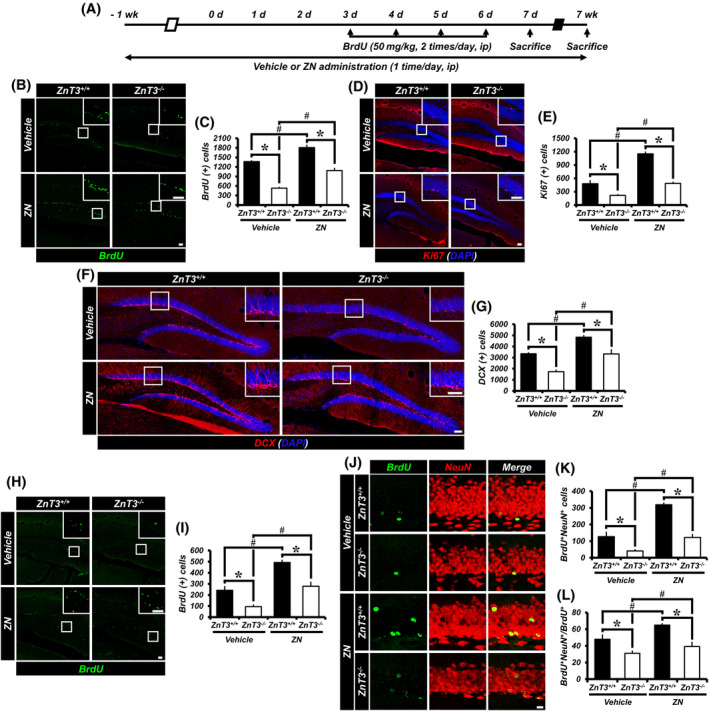
Administration of ZN in ZnT3 −/− mice increases adult hippocampal neurogenesis. A, Timeline showing the experimental design. B, Representative images of the DG showing BrdU (green) staining from vehicle‐ or ZN‐treated mice (either ZnT3 +/+ or ZnT3 −/−) at 2 weeks after initial ZN supplementation. Scale bar = 50 μm. C, Quantification of BrdU+ cells in the SGZ/GCL (mean ± SEM; n = 6‐9 from each group). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test: F = 76.753, P < .001). D, Representative images of Ki67‐ (red) and DAPI‐ (blue) immunopositive cells as merged images from vehicle‐ or ZN‐treated mice (either ZnT3 +/+ or ZnT3 −/−). Scale bar = 50 μm. E, Number of progenitor cells (Ki67+DAPI+) in the SGZ/GCL (mean ± SEM; n = 6‐9 from each group). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test: F = 92.234, P < .001). F, Representative images of DCX‐ (red) and DAPI‐ (blue) immunopositive cells as merged images from vehicle‐ or ZN‐treated mice (either ZnT3 +/+ or ZnT3 −/−). Scale bar = 50 μm. G, Number of immature neurons (DCX+DAPI+) in the SGZ/GCL (mean ± SEM; n = 6‐9 from each group). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test: F = 34.601, P < .001). H, Representative images of the DG showing BrdU (green) staining to vehicle‐ or ZN‐treated mice (either ZnT3 +/+ or ZnT3 −/−) at 8 weeks after initial ZN supplementation. Scale bar = 50 μm. I, Quantification of BrdU+ cells in the SGZ/GCL (mean ± SEM; n = 6‐8). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test: F = 44.071, P < .001). J, Representative images of BrdU‐ (green) and NeuN‐ (red) immunopositive cells as merged images to vehicle‐ or ZN‐treated mice (either ZnT3 +/+ or ZnT3 −/−). Scale bar = 50 μm. K,L, Bar graphs showing the number (K) and percentage (L) of cells that express the markers BrdU and NeuN (newborn neurons) in the SGZ/GCL (mean ± SEM; n = 6‐8). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test; K: F = 60.390, P < .001; L: F = 24.074, P < .001)
We then explored whether ZN also impacts the survival of newly generated cells and the neuronal maturation of newborn cells that survive in the SGZ/GCL. ZN supplementation was performed once a day for 8 weeks. We injected BrdU twice per day for four consecutive days starting at 10 days after the initial ZN supplementation. The numbers of BrdU+ and BrdU+NeuN+ cells in ZnT3 −/− mice were significantly decreased in both vehicle‐ and ZN‐treated groups, as compared to ZnT3 +/+ mice. However, ZN treatment remarkedly increased the numbers of BrdU+ and BrdU+NeuN+ cells in both ZnT3 +/+ and ZnT3 −/− mice, as compared to vehicle‐treated mice (Figure 3H‐K). In addition, ZN treatment revealed an increase in the percentage of BrdU+NeuN+ cells among BrdU+ cells in both ZnT3 +/+ and ZnT3 −/− mice compared with vehicle‐treated mice, although ZnT3 −/− mice treated with ZN or vehicle showed a lower percentage of BrdU+NeuN+ cells among BrdU+ cells than ZnT3 +/+ mice treated with ZN or vehicle, respectively (Figure 3L). In contrast with results obtained after administration of ZN, no significant difference in the ratio of the number of surviving BrdU+ cells in the SGZ/GCL at 47 days to the number of BrdU+ cells in the DG at 5 days after initial BrdU injection in either ZnT3 +/+ and ZnT3 −/− mice treated with ZN vs vehicle‐treated mice, indicating that ZN does not regulate the survival ratio of newly generated cells. However, we noted that ZN significantly increased the number of BrdU+ and BrdU+NeuN+ cells in the SGZ/GCL of both ZnT3 +/+ and ZnT3 −/− mice compared with vehicle‐treated mice. Thus, these results demonstrate that ZN reverses the reduction in adult hippocampal neurogenesis, caused by deletion of the ZnT3 gene.
3.8. ZN Treatment increases IGF‐1 expression and ERK/CREB activation in the hippocampus
The effects of ZnT3 gene deletion and ZN supplementation on the expression of IGF‐1 and the activity of ERK/CREB pathways were investigated to elucidate the mechanisms underlying the effect of vesicular zinc on adult hippocampal neurogenesis. It has is known that IGF‐1 acts to promote adult neurogenesis and that the ERK/CREB signaling pathway also plays an important role in maintaining normal levels neurogenesis. We therefore examined IGF‐1 expression in addition to measuring ERK and CREB phosphorylation (pERK and pCREB), which are the activated forms of these molecules. Western blot revealed a significant decrease in the level of IGF‐1, pERK, and pCREB proteins in the hippocampus of ZnT3 −/− mice as compared with the ZnT3 +/+ mice. However, administration of ZN remarkably increased the protein level of IGF‐1, pERK and pCREB in both ZnT3 +/+ and ZnT3 −/− mice as compared with the vehicle‐treated mice, although ZnT3 −/− mice treated with ZN showed lower IGF‐1, pERK and pCREB protein levels than ZnT3 +/+ mice (Figure 4A‐D). We also observed a significant reduction of IGF‐1‐ and pERK‐IR and the number of pCREB‐positive cells in ZnT3 −/− mice compared with ZnT3 +/+ mice. However, administration of ZN remarkably increased IGF‐1‐ and pERK‐IR and pCREB+ cells in both ZnT3 +/+ and ZnT3 −/− mice, although ZnT3 −/− mice treated with ZN showed a lower IGF‐1‐ and pERK‐IR or pCREB+ cells than ZnT3 +/+ mice treated with ZN (Figure 4E‐P).
FIGURE 4.
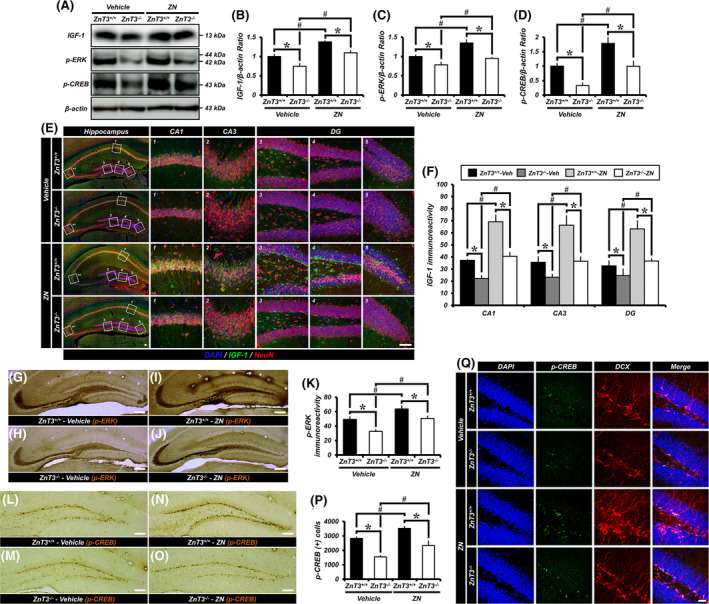
ZN increases IGF‐1 expression and leads to ERK/CREB activation in the hippocampus. A, Western blotting analysis of IGF‐1, pERK or pCREB in the hippocampus from either ZnT3 +/+ or ZnT3 −/− mice treated with vehicle or ZN. B‐D, Quantification of IGF‐1 (B), pERK (C), or pCREB (D) expression from the hippocampus (mean ± SEM; n = 5 from each group). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (Kruskal‐Wallis test followed by Bonferroni post hoc test; B: Chi square = 9.134, df = 3, P = .028; C: Chi square = 14.634, df = 3, P = .002; D: Chi square = 14.429, df = 3, P = .002). E, DAPI‐ (blue), IGF‐1‐ (green), and NeuN‐ (red) immunofluorescence in vehicle‐ and ZN‐treated mice of either ZnT3 +/+ or ZnT3 −/− genotype. Scale bar = 50 μm. F, Quantification of IGF‐1 immunoreactivity in the hippocampal CA1, CA3 and DG (mean ± SEM; n = 5 from each group). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (Kruskal‐Wallis test followed by Bonferroni post hoc test; CA1: Chi square = 12.728, df = 3, P = .005; CA3: Chi square = 11.184, df = 3, P = .011; DG: Chi square = 12.482, df = 3, P = .006). G‐J, Representative images showing pERK‐immunopositive mossy fiber (MF) pathway to vehicle‐treated ZnT3 +/+ (G) and ZnT3 −/− (H) mice or ZN‐treated ZnT3 +/+ (I) and ZnT3 −/− (J) mice daily for 2 weeks. Scale bar = 50 μm. K, Quantification of pERK immunoreactivity in the hippocampal MF (mean ± SEM; vehicle‐treated ZnT3 +/+ mice, n = 7; ZN‐treated ZnT3 +/+ mice, n = 8; vehicle‐treated ZnT3 −/− mice, n = 9; ZN‐treated ZnT3 −/− mice, n = 7). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice. L‐O, Representative images of the DG showing pCREB‐immunopositive cells to vehicle‐treated ZnT3 +/+ (L) and ZnT3 −/− (M) mice or ZN‐treated ZnT3 +/+ (N) and ZnT3 −/− (O) mice daily for 2 weeks. Scale bar = 50 μm. P, Bar graph showing the number of pCREB immunopositive cells in the SGZ/GCL (mean ± SEM; vehicle‐treated ZnT3 +/+ mice, n = 7; ZN‐treated ZnT3 +/+ mice, n = 8; vehicle‐treated ZnT3 −/− mice, n = 9; ZN‐treated ZnT3 −/− mice, n = 7). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test: F = 22.439, P < .001). Q, DAPI‐ (blue), pCREB‐ (green), and DCX‐ (red) immunofluorescence in vehicle‐ and ZN‐treated mice of either the ZnT3 +/+ or ZnT3 −/− genotype. Scale bar = 50 μm. P, phosphorylated
Several studies have demonstrated that the pCREB has been found to be present in the majority of newborn immature neurons in the adult DG and thus CREB signaling is known to play an important role in neuronal activation and in the survival stages of neurogenesis. 17 To confirm the relationship between immature neurons and pCREB‐expressing cells in the SGZ, we performed immunofluorescence staining against pCREB and DCX. Most of the pCREB+ cells were colocalized with DCX+ neuroblasts in their nuclei. We observed a significant decrease of pCREB+DCX+ cells in ZnT3 −/− mice compared with ZnT3 +/+ mice. However, administration of ZN remarkably increased the number of pCREB+DCX+ cells in both ZnT3 +/+ and ZnT3 −/− mice (Figure 4Q). Together, these findings provide further evidence that ERK and CREB activation were both necessary for the effect of vesicular zinc on adult hippocampal neurogenesis.
3.9. ZN treatment improves cognitive function in aged ZnT3+/+ and ZnT3 −/− mice
To investigate whether ZN supplementation improves cognitive abilities in both aged ZnT3 +/+ and ZnT3 −/− mice, mice were subjected to the MWM test for six consecutive days starting 50 days after the initial ZN supplementation. At 24 hours after the last training session, their behavior was recorded for 60 seconds after removing the platform as a probe trial (Figure 5A). The escape latency decreased progressively during 5 training days. ZnT3 −/− mice spent longer periods of time finding the platform, had a decreased number of platform crossings, and had reduced time spent in the target quadrant compared to ZnT3 +/+ mice. However, ZN supplementation resulted in an improved cognitive ability in both ZnT3 +/+ and ZnT3 −/− mice, as proven by a significant reduction in escape latency (Figure 5B) and an increase in the number of platform crossings (Figure 5C) and time spent in the target quadrant (Figure 5D,E) compared to the vehicle‐treated mice. These results demonstrate that ZN reverses aging‐dependent cognitive decline in ZnT3 −/− mice.
FIGURE 5.
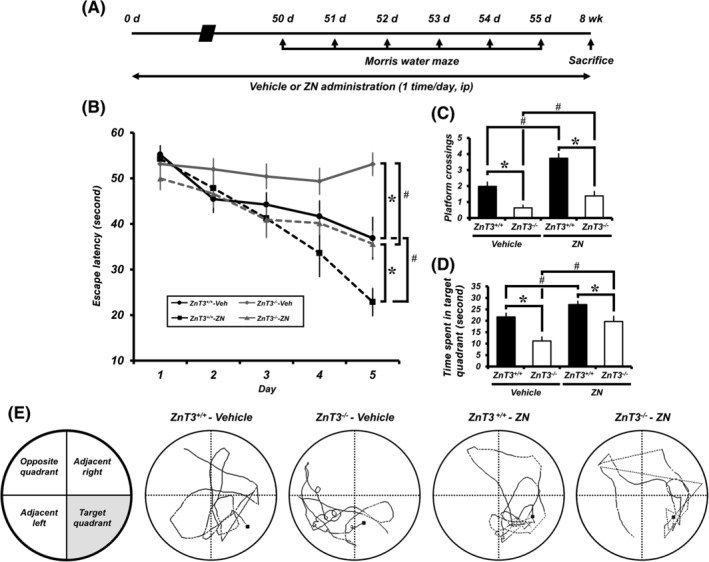
ZN improves cognitive function in aged ZnT3 +/+ and ZnT3 −/− mice. A, Timeline showing the experimental design. B, Morris water maze (MWM) performance. Escape latency of acquisition trial for five consecutive days starting 50 days after the initial ZN supplementation (mean ± SEM; vehicle‐treated ZnT3 +/+ mice, n = 11; ZN‐treated ZnT3 +/+ mice, n = 12; vehicle‐treated ZnT3 −/− mice, n = 13; ZN‐treated ZnT3 −/− mice, n = 14). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (repeated measure ANOVA; Day: F = 74.695, P < .001; Group: F = 7.543, P < .001; Day × Group interaction: F = 11.120, P < .001). C, MWM platform crossing. D, Time spent in target quadrant. E, Schematic diagram of tank and probe trial testing. Representative searching/swimming tracks by mice with each group in the probe trial (mean ± SEM; vehicle‐treated ZnT3 +/+ mice, n = 11; ZN‐treated ZnT3 +/+ mice, n = 12; vehicle‐treated ZnT3 −/− mice, n = 13; ZN‐treated ZnT3 −/− mice, n = 14). *P < .05 vs ZnT3 +/+ mice; #P < .05 vs vehicle‐treated mice (one‐way ANOVA followed by Bonferroni post hoc test; C: F = 26.412, P < .001; D: F = 13.204, P < .001)
4. DISCUSSION
The present study tested our hypothesis that vesicular zinc plays a central role in regulating adult neurogenesis occurring in the hippocampus. To test our hypothesis, we used mice in which vesicular zinc had been depleted by ZnT3 gene deletion. Here we found that ZnT3 gene deletion completely depleted vesicular zinc in the hippocampal MF tract and the number of progenitor cells, neuroblasts, and newborn neurons were remarkably reduced in the ZnT3 −/− mice, even under physiological conditions. In addition, we also found that zinc supplementation with ZN reversed the reduction of adult neurogenesis in ZnT3 −/− mice. This defective adult neurogenesis is due to a cell‐extrinsic mechanism of ZnT3 deficiency and is accompanied by reduced levels of pERK and pCREB in the hippocampus of ZnT3 −/− mice. The present study also demonstrated that IGF‐1 expression is lower in the hippocampus of ZnT3 −/− mice than wild type mice. Reduction of the level of IGF‐1 is reversed by ZN supplementation in both ZnT3 +/+ and ZnT3 −/− mice, which suggests that ZnT3 is involved in IGF‐dependent neurogenesis in the brain. Thus, these findings describe a central function for ZnT3 in maintaining adult hippocampal neurogenesis.
A possible relationship between zinc and hippocampal neurogenesis after brain injury has been reported by our previous studies. Our previous study suggests that decreased vesicular zinc content in the MF terminals of dentate granule neurons may cause impaired progenitor cell proliferation. 15 We also reported that zinc chelation by clioquinol (5‐chloro‐7‐iodo‐8‐hydroxyquinoline; CQ) reduced hippocampal progenitor cell proliferation and neurogenesis after hypoglycemia, traumatic brain injury, and seizure. 15 , 18 , 19 Supporting these findings, we recently demonstrated that enhancing hippocampal vesicular zinc via zinc supplementation with ZC increased hippocampal vesicular zinc concentration and thereby promotes adult neurogenesis under physiological 20 or pathological conditions such as diabetes. 21 These results support the notion that zinc is a critical regulator of adult neurogenesis.
Our previous studies demonstrated that zinc‐deficient diet reduced zinc concentration in the presynaptic hippocampal vesicles in rats fed a zinc‐deficient diet and that these mice showed reduced hippocampal neurogenesis. 15 A separate group also reported that dietary zinc deprivation caused impaired cognitive function. 8 , 22 However, the precise mechanism that leads to cognitive impairment in zinc‐deficient animals is still unknown. Several laboratories have shown that ZnT3 gene deletion mice exhibit age‐dependent deficits in learning and memory, 9 impaired contextual discrimination, and spatial working memory 10 and are deficient in social and object recognition memory. 11 McAllister et al. demonstrated that that ZnT3 −/− mice did not appear to readily develop the usual depression‐like effects that occur after stress but however, were not completely free of stress‐induced dysfunction, 23 as fear memory generation was enhanced by stress in ZnT3 −/− mice. Although the stress response is affected by ZnT3 gene deletion, hippocampal cellular proliferation was not significantly disrupted by stress in CD1 background mice. This group recently demonstrated that ZnT3 −/− mice showed increased basal dendritic length in the cerebral cortex, which is not affected by housing condition. 24 Thus, these studies suggest that ZnT3 and vesicular zinc are involved in certain types of spatial memory and behavior that are dependent on the background strain or on different stress conditions during development. We believe that further study will be needed to evaluate this difference as suggested. 25 , 26
From the above studies, including our works, we hypothesized that a zinc deficient‐induced cognitive impairment may be associated with a decline in hippocampal neurogenesis. Zinc‐deficient diet not only affects hippocampal vesicular zinc concentration but also affects systemic zinc levels, including blood and cerebrospinal fluid (CSF) in the brain. For this reason, we cannot differentiate whether the dietary zinc‐deficient‐induced cognitive impairment is solely affected by decreased hippocampal neurogenesis. Thus, to test our hypothesis we used ZnT3 −/− mice, which contain no vesicular zinc in the presynaptic terminal of hippocampus and cerebral cortex due to an inability to package zinc into synaptic vesicles. 27 , 28 The basal level of BrdU‐, Ki67‐, and DCX‐immunopositive cells was significantly diminished in ZnT3 −/− mice compared with ZnT3 +/+ mice. These results suggest that vesicular zinc originating from synaptic vesicles is necessary for progenitor cell proliferation and neuronal differentiation.
Why ZnT3 −/− mice develop cognitive impairment during aging is not clear. Sindreu et al showed that ZnT3 −/− mice have reduced activation of the ERK1/2 mitogen‐activated protein kinase (MAPK) in hippocampal MF terminals and reduced inhibition of the zinc‐sensitive tyrosine phosphatase MAPK in the setting of hippocampus‐dependent learning. 10 They suggested that a deficit of ERK signaling in ZnT3 −/− mice caused impaired contextual discrimination, which is a possible mechanism of memory impairment of these mice. ERK signaling is also important for hippocampal neurogenesis and presynaptic pERK also modulates MF plasticity. 29 Zinc can promote transcription of the BDNF gene and zinc supplementation increases the expression of BDNF mRNA in the adult rat brain. 30 The zinc finger transcription factor CREB can stimulate transcription of BDNF. 31 Currently, it is unknown how zinc induced BDNF expression occurs. One line of potential evidence to explain this phenomenon is the observation that zinc deficiency leads to a decreased phosphorylation of CREB in mice, 32 which indicates that zinc could modulate CREB activity. ERK1/2 phosphorylates the kinases RSK and MSK, which can directly phosphorylate CREB. 33 Zinc might stimulate CREB to increase the expression of BDNF via activation of ERK1/2, potentially forming a positive feedback loop to further activate local ERK1/2 signaling as an autocrine or paracrine factor. These results may explain why ZnT3 −/− mice showed depressed progenitor cell proliferation and neurogenesis.
Studies using ZnT3 −/− mice have produced mixed results with respect to how BDNF levels in the brain are affected by this manipulation. Several labs have reported that no difference exists between ZnT3 +/+ and ZnT3 −/− mice. 9 , 24 On the other hand, other researchers have demonstrated that ZnT3 −/− mice showed increased levels of hippocampal BDNF. 34 , 35 Therefore, as an alternative line of investigation into whether neurotrophic factors are impacted by manipulation of zinc transporter function, the present study sought to explore IGF‐1 levels in both ZnT3 +/+ and ZnT3 −/− mice treated with or without ZN supplementation. We found that IGF‐1 levels in the hippocampus were reduced in ZnT3 −/− mice, compared to ZnT3 +/+ mice, which was reversed by ZN supplementation. Increased expression of IGF‐1 and ERK phosphorylation during hippocampal neurogenesis has been suggested as potential mechanisms for these effects. 36 , 37 , 38 , 39 , 40 However, subsequent studies are needed to elucidate how the observed decrease in ERK and CREB phosphorylation via IGF‐1 arises from ZnT3 gene deletion.
One factor that can influence cellular differentiation is the redox potential. It is known to dynamically change as cells differentiate; for instance, it has been shown that the level of superoxide radicals increases as cells move from G1 to M phase. 41 Additionally, concentrations of hydrogen peroxide and superoxide play a role in the choice to either enter or exit the cell cycle. 42 In support of these observations, we previously demonstrated that newly generated neurons are exposed to oxidative stress at specific developmental timepoints, potentially explaining effects on hippocampal neurogenesis 43 observed in these animals. To test this, we used ZN which is zinc chloride mixed with NAC in 1:2 M ratio to increase brain zinc and cysteine levels. NAC, a glutathione precursor, is known to be a membrane‐permeant cysteine prodrug with potent antioxidant, anti‐inflammatory properties, and proneurogenesis. Strikingly, we observed increased pERK and pCREB expression in the hippocampus of both ZnT3 +/+ and ZnT3 −/− mice treated with ZN, thereby increasing adult hippocampal neurogenesis. This indicates that ZN supplementation has uniquely beneficial effects in that it can enhance de novo synthesis and also protect against the loss of newborn neurons.
The present study has potential limitations that need to be addressed in future studies. Here we found that zinc supplementation by ZN significantly increased not only hippocampal vesicular zinc levels but also neurogenesis. According to our results, the present study suggests that vesicular zinc is critical for adult hippocampal neurogenesis. However, ZN treatment did not affect vesicular zinc levels in the ZnT3 −/− mice, which completely depleted vesicular zinc in the hippocampal MFs, suggesting the possibility that not only vesicular zinc but also extracellular zinc can promote neurogenesis. So, we cannot exclude the possibility that brain can use extracellular zinc arising from blood and CSF to maintain neurogenesis. Thus, exploring the effect of zinc supplementation by ZN on extracellular zinc concentrations could be a future direction for research that not only vesicular zinc but also extracellular zinc can promote adult hippocampal neurogenesis. The results presented here of defective adult neurogenesis in the DG of ZnT3 −/− mice and decreased pERK and pCREB expression thus raise the intriguing possibility that this adult hippocampal ZnT3‐related phenotype reflects aging‐dependent cognitive decline in ZnT3 −/− mice. However, future investigation will be surely be required to dissect the molecular and cellular events that underlie decreased ERK and CREB phosphorylation seen after genetic deletion of ZnT3.
5. CONCLUSION
Together, the results from this study indicate that ZnT3 has a central function in maintaining adult hippocampal neurogenesis and zinc supplementation by ZN has a beneficial effect on hippocampal neurogenesis and may serve as a therapeutic target for enhanced neuroprotection and repair after injury. Therefore, the present study suggests that vesicular zinc is a critical regulator of adult hippocampal neurogenesis, under physiologic conditions.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
B.Y.C.: conception and design, financial support, collection, and assembly of data, data analysis and interpretation, manuscript writing; D.K.H., J.H.J., B.E.L.: collection and assembly of data; J.Y.K.: data analysis and interpretation, provision of study material or patients; S.W.S.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
This study was supported by funding from the National Research Foundation of Korea (NRF) (NRF‐2019R1A2C4004912) to B.Y.C. Additionally, this work was supported by the Brain Research Program through the NRF, funded by the Ministry of Science, Information and Communication Technology and Future Planning (NRF‐2017M3C7A1028937 and 2020R1A2C2008480) to S.W.S.
Choi BY, Hong DK, Jeong JH, Lee BE, Koh J‐Y, Suh SW. Zinc transporter 3 modulates cell proliferation and neuronal differentiation in the adult hippocampus. Stem Cells. 2020;38:994–1006. 10.1002/stem.3194
Funding information National Research Foundation of Korea, Grant/Award Numbers: NRF‐2017M3C7A1028937, NRF‐2019R1A2C4004912, NRF‐2020R1A2C2008480
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB. Importance of zinc in the central nervous system: the zinc‐containing neuron. J Nutr. 2000;130:1471S‐1483S. [DOI] [PubMed] [Google Scholar]
- 2. Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics. 2010;2:306‐317. [DOI] [PubMed] [Google Scholar]
- 3. Jo SM, Danscher G, Daa Schroder H, et al. Zinc‐enriched (ZEN) terminals in mouse spinal cord: immunohistochemistry and autometallography. Brain Res. 2000;870:163‐169. [DOI] [PubMed] [Google Scholar]
- 4. Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223‐250. [DOI] [PubMed] [Google Scholar]
- 5. MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr. 2000;130:1500S‐1508S. [DOI] [PubMed] [Google Scholar]
- 6. Swenerton H, Shrader R, Hurley LS. Zinc‐deficient embryos: reduced thymidine incorporation. Science. 1969;166:1014‐1015. [DOI] [PubMed] [Google Scholar]
- 7. Stewart GR, Frederickson CJ, Howell GA, Gage FH. Cholinergic denervation‐induced increase of chelatable zinc in mossy‐fiber region of the hippocampal formation. Brain Res. 1984;290:43‐51. [DOI] [PubMed] [Google Scholar]
- 8. Golub MS, Takeuchi PT, Keen CL, Gershwin ME, Hendrickx AG, Lonnerdal B. Modulation of behavioral performance of prepubertal monkeys by moderate dietary zinc deprivation. Am J Clin Nutr. 1994;60:238‐243. [DOI] [PubMed] [Google Scholar]
- 9. Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter‐3 knock‐out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease? J Neurosci. 2010;30:1631‐1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sindreu C, Palmiter RD, Storm DR. Zinc transporter ZnT‐3 regulates presynaptic Erk1/2 signaling and hippocampus‐dependent memory. Proc Natl Acad Sci USA. 2011;108:3366‐3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martel G, Hevi C, Kane‐Goldsmith N, Shumyatsky GP. Zinc transporter ZnT3 is involved in memory dependent on the hippocampus and perirhinal cortex. Behav Brain Res. 2011;223:233‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2012;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi BY, Hong DK, Suh SW. ZnT3 gene deletion reduces colchicine‐induced dentate granule cell degeneration. Int J Mol Sci. 2017;18:2189‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gundersen HJ, Bendtsen TF, Korbo L, et al. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379‐394. [DOI] [PubMed] [Google Scholar]
- 15. Suh SW, Won SJ, Hamby AM, et al. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J Cereb Blood Flow Metab. 2009;29:1579‐1588. [DOI] [PubMed] [Google Scholar]
- 16. Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149‐1160. [DOI] [PubMed] [Google Scholar]
- 17. Nakagawa S, Kim JE, Lee R, et al. Localization of phosphorylated cAMP response element‐binding protein in immature neurons of adult hippocampus. J Neurosci. 2002;22:9868‐9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim JH, Jang BG, Choi BY, et al. Zinc chelation reduces hippocampal neurogenesis after pilocarpine‐induced seizure. PLoS One. 2012;7:e48543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choi BY, Kim JH, Kim HJ, et al. Zinc chelation reduces traumatic brain injury‐induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J Trace Elem Med Biol. 2014;28:474‐481. [DOI] [PubMed] [Google Scholar]
- 20. Choi BY, Kim IY, Kim JH, et al. Zinc plus cyclo‐(his‐pro) promotes hippocampal neurogenesis in rats. Neuroscience. 2016;339:634‐643. [DOI] [PubMed] [Google Scholar]
- 21. Choi BY, Kim IY, Kim JH, et al. Administration of zinc plus cyclo‐(his‐pro) increases hippocampal neurogenesis in rats during the early phase of streptozotocin‐induced diabetes. Int J Mol Sci. 2017;18:73‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda A, Takefuta S, Okada S, Oku N. Relationship between brain zinc and transient learning impairment of adult rats fed zinc‐deficient diet. Brain Res. 2000;859:352‐357. [DOI] [PubMed] [Google Scholar]
- 23. McAllister BB, Wright DK, Wortman RC, et al. Elimination of vesicular zinc alters the behavioural and neuroanatomical effects of social defeat stress in mice. Neurobiol Stress. 2018;9:199‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McAllister BB, Thackray SE, de la Orta BKG, et al. Effects of enriched housing on the neuronal morphology of mice that lack zinc transporter 3 (ZnT3) and vesicular zinc. Behav Brain Res. 2020;379:112336. [DOI] [PubMed] [Google Scholar]
- 25. McAllister BB, Dyck RH. A new role for zinc in the brain. Elife. 2017;6:e31816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McAllister BB, Pochakom A, Fu S, et al. Effects of social defeat stress and fluoxetine treatment on neurogenesis and behavior in mice that lack zinc transporter 3 (ZnT3) and vesicular zinc. Hippocampus. 2019. [DOI] [PubMed] [Google Scholar]
- 27. Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci USA. 1999;96:1716‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT‐3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci USA. 1996;93:14934‐14939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vara H, Onofri F, Benfenati F, Sassoe‐Pognetto M, Giustetto M. ERK activation in axonal varicosities modulates presynaptic plasticity in the CA3 region of the hippocampus through synapsin I. Proc Natl Acad Sci USA. 2009;106:9872‐9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cieslik K, Sowa‐Kucma M, Ossowska G, et al. Chronic unpredictable stress‐induced reduction in the hippocampal brain‐derived neurotrophic factor (BDNF) gene expression is antagonized by zinc treatment. Pharmacol Rep. 2011;63:537‐543. [DOI] [PubMed] [Google Scholar]
- 31. Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850‐860. [DOI] [PubMed] [Google Scholar]
- 32. Gao HL, Xu H, Xin N, Zheng W, Chi ZH, Wang ZY. Disruption of the CaMKII/CREB signaling is associated with zinc deficiency‐induced learning and memory impairments. Neurotox Res. 2011;19:584‐591. [DOI] [PubMed] [Google Scholar]
- 33. Impey S, Obrietan K, Wong ST, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB‐dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869‐883. [DOI] [PubMed] [Google Scholar]
- 34. Helgager J, Huang YZ, McNamara JO. Brain‐derived neurotrophic factor but not vesicular zinc promotes TrkB activation within mossy fibers of mouse hippocampus in vivo. J Comp Neurol. 2014;522:3885‐3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoo MH, Kim TY, Yoon YH, Koh JY. Autism phenotypes in ZnT3 null mice: involvement of zinc dyshomeostasis, MMP‐9 activation and BDNF upregulation. Sci Rep. 2016;6:28548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Kusky JR, Ye P, D'Ercole AJ. Insulin‐like growth factor‐I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci. 2000;20:8435‐8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson MF, Aberg MA, Nilsson M, et al. Insulin‐like growth factor‐I and neurogenesis in the adult mammalian brain. Brain Res Dev Brain Res. 2002;134:115‐122. [DOI] [PubMed] [Google Scholar]
- 38. Lichtenwalner RJ, Forbes ME, Sonntag WE, Riddle DR. Adult‐onset deficiency in growth hormone and insulin‐like growth factor‐I decreases survival of dentate granule neurons: insights into the regulation of adult hippocampal neurogenesis. J Neurosci Res. 2006;83:199‐210. [DOI] [PubMed] [Google Scholar]
- 39. Nieto‐Estevez V, Oueslati‐Morales CO, Li L, et al. Brain insulin‐like growth factor‐I directs the transition from stem cells to mature neurons during postnatal/adult hippocampal neurogenesis. Stem Cells. 2016;34:2194‐2209. [DOI] [PubMed] [Google Scholar]
- 40. Morel GR, Leon ML, Uriarte M, et al. Therapeutic potential of IGF‐I on hippocampal neurogenesis and function during aging. Neurogenesis (Austin). 2017;4:e1259709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Menon SG, Goswami PC. A redox cycle within the cell cycle: ring in the old with the new. Oncogene. 2007;26:1101‐1109. [DOI] [PubMed] [Google Scholar]
- 42. Menon SG, Sarsour EH, Spitz DR, et al. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003;63:2109‐2117. [PubMed] [Google Scholar]
- 43. Choi BY, Won SJ, Kim JH, et al. EAAC1 gene deletion reduces adult hippocampal neurogenesis after transient cerebral ischemia. Sci Rep. 2018;8:6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.