

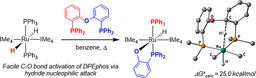
Abstract
C−O bond activation of DPEphos occurs upon mild heating in the presence of [Ru(NHC)2(PPh3)2H2] (NHC=N‐heterocyclic carbene) to form phosphinophenolate products. When NHC=IEt2Me2, C−O activation is accompanied by C−N activation of an NHC ligand to yield a coordinated N‐phosphino‐functionalised carbene. DFT calculations define a nucleophilic mechanism in which a hydride ligand attacks the aryl carbon of the DPEphos C−O bond. This is promoted by the strongly donating NHC ligands which render a trans dihydride intermediate featuring highly nucleophilic hydride ligands accessible. C−O bond activation also occurs upon heating cis‐[Ru(DPEphos)2H2]. DFT calculations suggest this reaction is promoted by the steric encumbrance associated with two bulky DPEphos ligands. Our observations that facile degradation of the DPEphos ligand via C−O bond activation is possible under relatively mild reaction conditions has potential ramifications for the use of this ligand in high‐temperature catalysis.
Keywords: C−O bond activation, density functional calculations, hydride ligands, N-heterocyclic carbenes, phosphines
Hydride ligands: Under mild conditions (25→90 °C), [Ru(N‐heterocyclic carbene)2(PPh3)2H2]/DPEphos or [Ru(DPEphos)2H2] undergo C−O bond activation of this catalytically important diphosphine ligand by attack of the nucleophilic Ru−H ligands.
Since their introduction ca. 20 years ago,1 wide‐angle phosphines such as xantphos and DPEphos (Scheme 1) have become indispensable ligands for a range of catalytic reactions.2 Their usage stems from two advantageous properties; firstly, the availability of highly flexible bite angles that allow cis‐ and trans‐, as well as hemilabile P‐O‐P coordination modes, to be adopted3 and, secondly, resistance to the types of P−C degradation reactions reported in tertiary phosphine metal complexes.4 This latter property has promoted the use of xantphos and DPEphos in reactions that require high temperatures.2c, 2g, 2l, 5
Scheme 1.
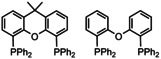
Structures of xantphos and DPEphos.
Any suggestion that such phosphines might be susceptible to degradative reactions, particularly under relatively mild conditions, could therefore have important ramifications for their applications in catalysis. While xantphos has been reported to be susceptible to P−C bond activation at room temperature,6 cleavage of DPEphos appears to be restricted to a single example of high temperature C−O bond activation reported by Weller and Willis.7 In the course of studies on [Rh(η6‐ortho‐xylene)(DPEphos)]+ catalysed carbothiolation of alkynes, they reported that heating the Rh complex together with ortho‐MeSC6H4C(O)Me at 120 °C in the absence of any alkyne led to C−O cleavage of DPEphos to afford a catalytically inactive Rh complex with chelating phosphine aryloxide and bidentate phosphine arylthioether ligands. Herein, we demonstrate that C−O activation of DPEphos can take place even at room temperature in the presence of ruthenium dihydride complexes. DFT calculations reveal that such processes involve attack of highly nucleophilic hydride ligands on the aryl carbon on the C−O bond.
In the course of studies to investigate the substitution chemistry of the all trans‐dihydride complex [Ru(IMe4)2(PPh3)2H2] (1, Scheme 2),8 1 was treated with 1.1–1.5 equiv DPEphos in benzene. No immediate reaction was observed at room temperature, but upon heating to 90 °C for ca. 12 h, a single ruthenium‐containing product 2 (Scheme 2) was formed. An X‐ray crystal structure (Figure 1) revealed the presence of a phosphinophenolate ligand generated upon C−O activation of DPEphos.9 The P,O‐termini of the ligand were trans to PPh3 and Ru−H respectively. The coordination sphere was completed by two mutually trans IMe4 ligands, each of which displayed an N‐Me group with a short C−H⋅⋅⋅O contact to the phosphinophenolate ligand (Supporting Information). The trans H‐Ru‐O arrangement led to both a long Ru−O distance (2.2720(16) Å)10 and a low frequency (δ=−18.40 ppm) hydride resonance.11
Scheme 2.
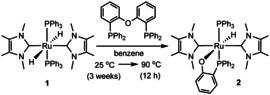
C−O activation of DPEphos by 1 to give 2.
Figure 1.
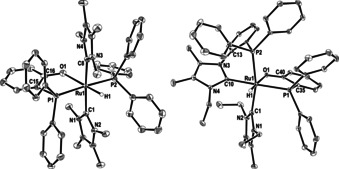
Molecular structures of (left) 2 and (right) 4. Thermal ellipsoids are shown at 30 % level. All hydrogen atoms, except for Ru−H have been omitted for clarity.
The formation of 2 was achieved under even milder conditions, although at the expense of longer reaction times (e.g. 6 days at 70 °C), and a low (5 %) yield could even be formed at room temperature, albeit only over 3 weeks.12 No simple substitution product arising from replacement of the two PPh3 ligands by DPEphos was observed under these conditions (vide infra). Treatment of 1 with the more‐electron rich cyclohexyl diphosphine Cy2P(C6H4)O(C6H4)PCy2 also resulted in C–O activation, although the reaction failed to reach completion, even after heating at 120 °C for 2 days. There was no evidence for C−O activation of xantphos by 1.13
Replacing 1 by the N‐Et substituted carbene derivative cis, cis, trans‐[Ru(IEt2Me2)2(PPh3)2H2] (3, Scheme 3) led to an even more unexpected reaction with DPEphos. Heating in toluene at 90 °C gave the phosphinophenolate complex 4 (Scheme 3), in which the {Ph2P(C6H4)} moiety generated upon C−O cleavage had combined with a C−N activated IEt2Me2 ligand to generate a Ru‐bound N‐phosphino‐functionalised carbene ligand.14 The X‐ray structure of 4 (Figure 1) showed the presence of a distorted octahedral ruthenium centre with a cis‐arrangement of the two carbenes and two phosphines and the same trans H‐Ru‐OAr arrangement as in 1 (Ru−O=2.265(2) Å). The phosphino moiety appended to N3 exhibited a considerable cone‐tilt, with Ru‐P‐Cipso angles ranging from 102° to 132°. In support of the C−N cleavage process, the 1H NMR spectrum showed just three NCH2CH3 methyl and six NCH2CH3 methylene resonances. The Ru−H resonance (δ=−17.7 ppm) was coupled to the two inequivalent phosphorus nuclei (δ=59 and 55 ppm) with cis‐2 J(H,P) coupling constants of 20 and 15 Hz.
Scheme 3.
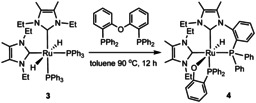
C−O and C−N activation to yield 4.
C−N activation of a metal‐bound NHC ligand has been described previously,15 including in studies on Ru‐NHC complexes related to those employed here.16 However, this process has only rarely been observed alongside the activation of another ligand,17 and, certainly not as a route to the formation of a phosphinocarbene.18
The C−O activation of DPEphos was not restricted to NHC‐containing ruthenium hydride precursors. The reaction of [Ru(PPh3)4H2] with DPEphos gave the isolable cis‐dihydride complex [Ru(DPEphos)2H2] (5; Supporting Information),19 which upon heating to 80 °C overnight underwent C−O activation of one of the DPEphos ligands to afford [Ru(DPEphos)(Ph2PC6H4O)H] (6, Scheme 4).20 This was characterized by the presence of a quartet Ru−H resonance at δ=−14 ppm with a 2 J(H,P) splitting (22 Hz) indicative of hydride cis to all three phosphorus nuclei and a 31P{1H} NMR spectrum which showed a triplet at δ=77 ppm (2 J(P,P)=30 Hz), together with a broad, featureless signal at δ=50 ppm. We attribute the latter to the intact DPEphos ligand switching rapidly between κ 2‐P,P and κ 3‐P,O,P coordination. At −15 °C, this signal resolved into two doublets, the two ends of the DPEphos ligand becoming inequivalent as a result of the oxygen now staying bound to Ru. Although an X‐ray structure of 6 proved elusive, crystals of the chloride derivative 7 were isolated from CH2Cl2/pentane solutions of 6, affording a structure (Figure 2) which confirmed the coordination modes at ruthenium.
Scheme 4.
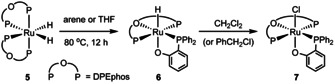
Formation of the C−O activated DPEphos complex 6 and chloride derivative 7.
Figure 2.
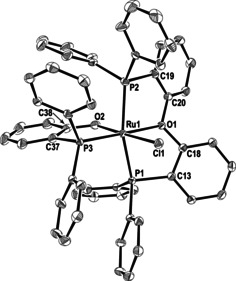
Molecular structure of 7. Thermal ellipsoids are shown at 30 % probability. Cl1 is disordered with a hydride ligand in a 75:25 ratio. Hydrogen atoms and all minor disordered components have been for clarity.
DFT calculations21 have been used to explore the mechanism of the C−O bond cleavage reactions in 1 and 5 and the factors promoting them. For 1, no intermediates are observed experimentally and so all free energies are quoted relative to this species plus free DPEphos. PPh3 substitution in 1 by DPEphos gives [Ru(IMe4)2(DPEphos)H2], 8, for which the all‐cis isomer, 8ccc (+3.6 kcal mol−1), and the cis, cis, trans‐isomer, 8cct (+4.2 kcal mol−1) are most stable.22
The accessibility of the trans dihydride isomer 8cct suggested a hydride nucleophilic attack mechanism may be involved, similar to that characterised for the hydrodefluorination of (hetero)aromatics at trans‐[Ru(NHC)4H2] complexes.23, 24 Figure 3 shows the computed reaction profiles for this process in 8cct and 8ccc. For 8cct the trans hydride arrangement gives a long Ru−H1 bond (1.70 Å) and NBO calculations indicate significant hydridic character (−0.21). Nucleophilic attack proceeds via TS(8‐2)cct at +25.0 kcal mol−1, with a short H1⋅⋅⋅C1 distance of 1.56 Å and Ru⋅⋅⋅H1 stretching to 1.84 Å. The C1−O bond also lengthens to 1.48 Å and elongated C1−C2 and C1−C6 distances in the aryl ring suggest a Meisenheimer‐type structure consistent with nucleophilic aromatic substitution. Hydride attack is also accompanied by a conformational change in the 8‐membered Ru−P−C=C−O−C=C−P ring, from a distorted twist‐boat conformation in 8cct to a boat conformation in the transition state,25 similar to the DPEphos fac‐κ 3‐P,O,P binding mode.26 IRC calculations confirm that TS(8‐2)cct links directly to 2cct in which H2 is trans to the phosphinophenolate oxygen. The lowest energy conformation of 2cct is at −31.5 kcal mol−1.27
Figure 3.
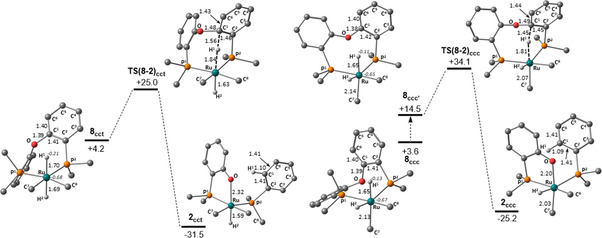
Computed free energy profiles (kcal mol−1, BP86(benzene, D3BJ)) for hydride attack in 8cct and 8ccc, with selected distances in Å. Energies are relative to 1 plus free DPEphos and NBO charges at Ru and H1 are indicated in italics for dihydride precursors. For clarity, IMe4 ligands are truncated at the C2 position (i.e. C7 and C8 in the Figure) and phenyl substituents at the ipso carbons. DPEphos hydrogens are also omitted. 8ccc’ is a conformer of 8ccc that lies directly on the pathway for C−O cleavage (see text for details).
The equivalent reaction of 8ccc involves an initial conformational change of the Ru−P−C=C−O−C=C−P ring to form 8ccc’ at +14.5 kcal mol−1. C−O bond cleavage then proceeds via TS(8‐2)ccc at +34.1 kcal mol−1 with similar geometric changes to those described above for TS(8‐2)cct. The shorter Ru‐H1 distances in 8ccc and 8ccc’ (1.65 Å) and lower NBO charges (ca. −0.12) indicate that H1 is now less nucleophilic than in 8cct, and this reflects the change in the trans ligand, from a hydride in 8cct to IMe4 in 8ccc. This also correlates with C−O bond cleavage being less kinetically accessible in 8ccc. TS(8‐2)ccc leads to 2ccc at −25.2 kcal mol−1, substantially less stable than 2cct as this structure lacks the favourable trans‐H‐Ru‐O arrangement.28
C−O bond cleavage was also modelled for [Ru(DPEphos)2H2] and the most accessible pathway is shown in Figure 4. The all‐cis isomer, 5ccc, reacts via 5ccc’ and TS(5–9)ccc at +29.9 kcal mol−1 to give a phosphinophenolate product, 9ccc, at −23.0 kcal mol−1. The short Ru−H1 distance in 5ccc (1.60 Å) and low NBO charge on H1 (−0.02) indicate reduced hydride nucleophilicity compared to 8ccc, although the barrier in the bis‐DPEphos system is actually lower (see below). In stark contrast to 8cct, the trans dihydride isomer of [Ru(DPEphos)2H2] 5cct, has a large barrier of +48.5 kcal mol−1. This difference is due in part to the higher energy of 5cct (+13.8 kcal mol−1) and the reduced charge on H1 (ca. −0.08 cf. −0.21 in 8cct). The latter result highlights how the NHC ligands also serve to enhance hydride nucleophilicity. Differential steric effects in the transition states may also be a factor, as probed by calculations on 5ccc and 5cct in which the PPh2 groups were replaced by PH2. This model system gave a similar relative energy for 5cct (+12.6 kcal mol−1), but a reduced barrier for the subsequent nucleophilic attack (i.e. from 5cct to TS(5–9)cct: 30.2 kcal mol−1 cf. 34.7 kcal mol−1 in the full system). In contrast, the computed barrier for 5ccc with the small model is 38.6 kcal mol−1, 8.7 kcal mol−1 higher than the full model.
Figure 4.
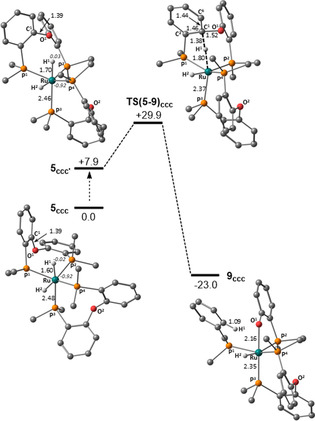
Computed free energy profile (kcal mol−1, BP86(benzene, D3BJ)) for hydride attack in 5ccc, with selected distances in Å and computed NBO charges at Ru and H1 in italics for the dihydride precursors. For clarity, phenyl substituents are truncated at the ipso carbons and DPEphos hydrogens are omitted.
Computed geometries show significant distortions in the full model: in 5ccc the trans‐P‐Ru‐P angle is 142° with the bulky PAr3 moieties tilting over the hydride ligands. As this angle is only 160° in the small model, we speculate that the greater distortion of the full model enables nucleophilic attack.
Comparing [Ru(IMe4)2(DPEphos)H2] and [Ru(DPEphos)2H2] shows C−O bond cleavage via 8cct (ΔG ≠=25.0 kcal mol−1) is more accessible than in 5ccc (ΔG ≠=29.9 kcal mol−1) and this is consistent with the lower reactivity of the bis‐DPEphos system observed experimentally. Lower barriers are computed with higher trans influence ligands (H>IMe4) trans to the hydride nucleophile. The mixed NHC/DPEphos systems appear particularly vulnerable to C−O bond cleavage as the strongly donating NHC ligands both enhance hydride nucleophilicity and render 8cct, the key trans dihydride precursor, accessible. The hydride attack mechanism described here has similarities to the “asynchronous oxidative addition” pathway described by Crimmin and co‐workers where a RuII metal centre acts as a nucleophile prior to C−O bond cleavage.12 A similar asynchronicity is seen here, with C−H bond formation in the transition state being far advanced of either C−O bond cleavage or Ru−O bond formation.
In summary, we have characterised the surprisingly facile C−O bond activation of DPEphos ligands in the presence of nucleophilic hydrides. Ligand exchange of all‐trans‐[Ru(IMe4)2(PPh3)2H2] with DPEphos results in the formation of phosphinophenolate complex, 2, while with cis, cis, trans‐[Ru(IEt2Me2)2(PPh3)2H2], C−O bond cleavage is accompanied by C−N activation of the NHC to form the N‐phosphino‐functionalised carbene complex 4. DFT calculations indicate that C−O activation involves a nucleophilic pathway in which a hydride ligand attacks the aryl carbon of the DPEphos C−O bond. This process is promoted by the accessibility of a trans dihydride intermediate that features highly nucleophilic hydride ligands. C−O bond activation also occurs upon heating cis‐[Ru(DPEphos)2H2], a process that DFT calculations indicate is promoted by the steric encumbrance of the mutually cis DPEphos ligands. This undesirable ligand degradation of DPEphos is of particular note given the wide use of this ligand in high temperature homogeneous catalysis. Indeed, degradation of the Rh‐DPEphos system described by Weller and Willis is also thought to involve nucleophilic attack, in this case by a thiolate ligand.7 On a more constructive note, the hydride nucleophilic attack mechanism proposed here has already been shown to operate in catalytic C−F functionalization,23c, 23d and so may also be an effective means of promoting C−O bond activation of the type required for the valorization of lignin and of its highly oxygenated monomers.29
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the EPSRC for financial support (DTP studentships to M.K.C. and N.A.B.) and Dr. Mark Crimmin (Imperial College London) for valuable discussions.
M. K. Cybulski, N. A. Beattie, S. A. Macgregor, M. F. Mahon, M. K. Whittlesey, Chem. Eur. J. 2020, 26, 11141.
We dedicate this paper to the memory of friend, colleague and collaborator Professor Jonathan M. J. Williams
Contributor Information
Prof. Stuart A. Macgregor, Email: s.a.macgregor@hw.ac.uk.
Prof. Michael K. Whittlesey, Email: m.k.whittlesey@bath.ac.uk.
References
- 1.
- 1a. Kranenburg M., van der Burgt Y. E. M., Kamer P. C. J., van Leeuwen P. W. N. M., Goubitz K., Fraanje J., Organometallics 1995, 14, 3081–3089; [Google Scholar]
- 1b. van Leeuwen P. W. N. M., Kamer P. C. J., Catal. Sci. Technol. 2018, 8, 26–113; [Google Scholar]
- 1c. Adams G. M., Weller A. S., Coord. Chem. Rev. 2018, 355, 150–172. [Google Scholar]
- 2.For representative examples;
- 2a. Wagaw S., Yang B. H., Buchwald S. L., J. Am. Chem. Soc. 1999, 121, 10251–10263; [Google Scholar]
- 2b. Utsunomiya M., Kuwano R., Kawatsura M., Hartwig J. F., J. Am. Chem. Soc. 2003, 125, 5608–5609; [DOI] [PubMed] [Google Scholar]
- 2c. Ahmed M., Seayad A. M., Jackstell R., Beller M., J. Am. Chem. Soc. 2003, 125, 10311–10318; [DOI] [PubMed] [Google Scholar]
- 2d. Moxham G. L., Randell-Sly H. E., Brayshaw S. K., Woodward R. L., Weller A. S., Willis M. C., Angew. Chem. Int. Ed. 2006, 45, 7618–7622; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7780–7784; [Google Scholar]
- 2e. Johns A. M., Utsunomiya M., Incarvito C. D., Hartwig J. F., J. Am. Chem. Soc. 2006, 128, 1828–1839; [DOI] [PubMed] [Google Scholar]
- 2f. Ohshima T., Miyamoto Y., Ipposhi J., Nakahara Y., Utsunomiya M., Mashima K., J. Am. Chem. Soc. 2009, 131, 14317–14328; [DOI] [PubMed] [Google Scholar]
- 2g. Dudle B., Rajesh K., Blacque O., Berke H., J. Am. Chem. Soc. 2011, 133, 8168–8178; [DOI] [PubMed] [Google Scholar]
- 2h. Xu K., Thieme N., Breit B., Angew. Chem. Int. Ed. 2014, 53, 7268–7271; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7396–7399; [Google Scholar]
- 2i. Hanley P. S., Clark T. P., Krasovskiy A. L., Ober M. S., O′Brien J. P., Staton T. S., ACS Catal. 2016, 6, 3515–3519; [Google Scholar]
- 2j. Jang W. J., Lee W. L., Moon J. H., Lee J. Y., Yun J., Org. Lett. 2016, 18, 1390–1393; [DOI] [PubMed] [Google Scholar]
- 2k. Khakyzadeh V., Wang Y.-H., Breit B., Chem. Commun. 2017, 53, 4966–4968; [DOI] [PubMed] [Google Scholar]
- 2l. Hori K., Motohashi H., Saito D., Mikami K., ACS Catal. 2019, 9, 417–421. [Google Scholar]
- 3.
- 3a. van Leeuwen P. W. N. M., Kamer P. C. J., Reek J. N. H., Dierkes P., Chem. Rev. 2000, 100, 2741–2769; [DOI] [PubMed] [Google Scholar]
- 3b. Kamer P. C. J., van Leeuwen P. W. N. M., Reek J. N. H., Acc. Chem. Res. 2001, 34, 895–904; [DOI] [PubMed] [Google Scholar]
- 3c. Freixa Z., van Leeuwen P. W. N. M., Dalton Trans. 2003, 1890–1901. [DOI] [PubMed] [Google Scholar]
- 4. Garrou P. E., Chem. Rev. 1985, 85, 171–185. [Google Scholar]
- 5.
- 5a. Yin J., Zhao M. M., Huffman M. A., McNamara J. M., Org. Lett. 2002, 4, 3481–3484; [DOI] [PubMed] [Google Scholar]
- 5b. Broutin P. E., B̆ern̆a I., Campaniello M., Leroux F., Colobert F., Org. Lett. 2004, 6, 4419–4422; [DOI] [PubMed] [Google Scholar]
- 5c. Jiang D., Peng J., Chen Y., Org. Lett. 2008, 10, 1695–1698; [DOI] [PubMed] [Google Scholar]
- 5d. Ackermann L., Kornhaass C., Zhu Y., Org. Lett. 2012, 14, 1824–1826; [DOI] [PubMed] [Google Scholar]
- 5e. Liu H., Laurenczy G., Yan N., Dyson P. J., Chem. Commun. 2014, 50, 341–343. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Dimmer J.-A., Hornung M., Wuetz T., Wesemann L., Organometallics 2012, 31, 7044–7051; [Google Scholar]
- 6b. Johnson H. C., Weller A. S., Angew. Chem. Int. Ed. 2015, 54, 10173–10177; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10311–10315. [Google Scholar]
- 7. Hooper J. F., Chaplin A. B., González-Rodríguez C., Thompson A. L., Weller A. S., Willis M. C., J. Am. Chem. Soc. 2012, 134, 2906–2909. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Davies C. J. E., Lowe J. P., Mahon M. F., Poulten R. C., Whittlesey M. K., Organometallics 2013, 32, 4927–4937; [Google Scholar]
- 8b. Cybulski M. K., Nicholls J. E., Lowe J. P., Mahon M. F., Whittlesey M. K., Organometallics 2017, 36, 2308–2316. [Google Scholar]
- 9.For examples of phosphinophenolate complexes made by alternative routes;
- 9a. Pietsch J., Braunstein P., Chauvin Y., New J. Chem. 1998, 22, 467–472; [Google Scholar]
- 9b. Dahlenburg L., Herbst K., Kühnlein M., Z. Anorg. Allg. Chem. 1997, 623, 250–258; [Google Scholar]
- 9c. Trzeciak A. M., Ziółowski J. J., Lis T., Choukroun R., J. Organomet. Chem. 1999, 575, 87–97; [Google Scholar]
- 9d. Rogers C. W., Patrick B. O., Rettig S. J., Wolf M. O., J. Chem. Soc. Dalton Trans. 2001, 1278–1283. [Google Scholar]
- 10.
- 10a. Hsu S. C. N., Hu S.-C., Wu Z.-S., Chiang M. Y., Hung M.-Y., J. Organomet. Chem. 2009, 694, 1912–1917; [Google Scholar]
- 10b. Malan F. P., Noh J.-H., Naganagowda G., Singleton E., Meijboom R., J. Organomet. Chem. 2016, 825–826, 139–145. [Google Scholar]
- 11. Lund C. L., Sgro M. J., Cariou R., Stephan D. W., Organometallics 2012, 31, 802–805. [Google Scholar]
- 12.For a recent example of room temperature C−O activation of ketone-functionalised aryl ethers at Ru; Lau S., Ward B., Zhou X., White A. J. P., Casely I. J., Macgregor S. A., Crimmin M. R., Organometallics 2017, 36, 3654–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.NMR analysis of a C6D6 solution of 1 heated with 5 equiv xantphos for 3 days at 70 °C showed formation of a second-order hydride resonance at δ=−8.6 ppm, and a singlet in the 31P{1H} NMR spectrum at δ=27 ppm, suggestive of the formation of trans, cis, cis-[Ru(IMe4)2(xantphos)H2].
- 14.Conducting the reaction in refluxing Et2O gave a better yield of 4 as a result of direct precipitation of crystalline material suitable for X-ray diffraction.
- 15.
- 15a. Caddick S., Cloke F. G. N., Hitchcock P. B., de K. Lewis A. K., Angew. Chem. Int. Ed. 2004, 43, 5824–5827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 5948–5951; [Google Scholar]
- 15b. Wang X., Chen H., Li X., Organometallics 2007, 26, 4684–4687; [Google Scholar]
- 15c. Hu Y.-C., Tsai C.-C., Shih W.-C., Yap G. P. A., Ong T.-G., Organometallics 2010, 29, 516–518; [Google Scholar]
- 15d. Xiang L., Xiao J., Deng L., Organometallics 2011, 30, 2018–2025; [Google Scholar]
- 15e. Day B. M., Pugh T., Hendriks D., Guerra C. F., Evans D. J., Bickelhaupt F. M., Layfield R. A., J. Am. Chem. Soc. 2013, 135, 13338–13341. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Burling S., Mahon M. F., Powell R. E., Whittlesey M. K., Williams J. M. J., J. Am. Chem. Soc. 2006, 128, 13702–13703; [DOI] [PubMed] [Google Scholar]
- 16b. Häller L. J. L., Page M. J., Erhardt S., Macgregor S. A., Mahon M. F., Naser M. A., Vélez A., Whittlesey M. K., J. Am. Chem. Soc. 2010, 132, 18408–18416. [DOI] [PubMed] [Google Scholar]
- 17. Zhong W., Fei Z. F., Scopelliti R., Dyson P. J., Chem. Eur. J. 2016, 22, 12138–1214. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Brown C. C., Plessow P. N., Rominger F., Limbach M., Hofmann P., Organometallics 2014, 33, 6754–6759; [Google Scholar]
- 18b. Brown C. C., Rominger F., Limbach M., Hofmann P., Inorg. Chem. 2015, 54, 10126–10140; [DOI] [PubMed] [Google Scholar]
- 18c. Marchenko A., Koidan G., Hurieva A. N., Vlasenko Y., Kostyuk A., Biffis A., Organometallics 2016, 35, 762–770; [Google Scholar]
- 18d. Mosaferi E., Pan L., Wang T., Sun Y., Pranckevicius C., Stephan D., Dalton Trans. 2016, 45, 1354–1358. [DOI] [PubMed] [Google Scholar]
- 19.A cis-hydride geometry in 5 was apparent from the AA′MM′XX′ pattern of the hydride resonance at δ −9.8 ppm in the 1H NMR spectrum (see, Lenero K. A., Kranenburg M., Guari Y., Kamer P. C. J., van Leeuwen P. W. N. M., Sabo-Etienne S., Chaudret B., Inorg. Chem. 2003, 42, 2859–2866). The X-ray structure of 5 is shown in the Supporting Information. [DOI] [PubMed] [Google Scholar]
- 20.Chaudret, van Leeuwen and co-workers (ref. [19]) prepared a series of cis-[Ru(P,O,P)2H2] complexes (P,O,P=homoxantphos, sixantphos, thixantphos) by treatment of [Ru(cod)(cot)] with the diphosphine and H2 under forcing conditions (150 °C, 16 h). No degradative C−O activation was reported. The inertness of xantphos is in line with our findings (ref. [13]). Similarly, there is no C−O activation of xantphos reported in the catalytic intermolecular C−O activation of aroyl halides with a Pd(xantphos) system. Lee Y. H., Morandi B., Nat. Chem. 2018, 10, 1016–1022.30082881 [Google Scholar]
- 21.Calculations were run with Gaussian 09 and employed the BP86 functional with SDD pseudopotentials and basis sets on Ru and P (with polarisation added to the latter) and 6-31 g** basis sets on other atoms. Free energies are corrected for solvation (benzene, PCM approach) and dispersion (BJD3) effects. All geometries were obtained after extensive conformational searching following our published protocol (ref. [16 b]). See Supporting Information for full details.
- 22.Reaction via alternative isomers proved higher in energy. See the Supporting Information, Figure S16.
- 23.
- 23a. Panetier J. A., Macgregor S. A., Whittlesey M. K., Angew. Chem. Int. Ed. 2011, 50, 2783–2786; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2835–2838; [Google Scholar]
- 23b. Macgregor S. A., McKay D., Panetier J. A., Whittlesey M. K., Dalton Trans. 2013, 42, 7386–7395; [DOI] [PubMed] [Google Scholar]
- 23c. McKay D., Riddlestone I. M., Macgregor S. A., Mahon M. F., Whittlesey M. K., ACS Catal. 2015, 5, 776–787; [Google Scholar]
- 23d. Cybulski M. K., McKay D., Macgregor S. A., Mahon M. F., Whittlesey M. K., Angew. Chem. Int. Ed. 2017, 56, 1515–1519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1537–1541. [Google Scholar]
- 24.Alternative mechanisms based on oxidative addition were also assessed but were ruled out by the high energy of the ligand dissociations required to form the reactive 5-coordinate intermediates (see the Supporting Information, Figures S15 and S16).
- 25.Nomenclature is related to that employed for 1,4-cyclooctadiene, with the aromatic C=C bonds in the 1- and 4-positions (Anet F. A. L., Yavari I., J. Am. Chem. Soc. 1977, 99, 6986–6991). We have not attempted to locate barriers for these conformation changes, but assume such process are more accessible than the C−O bond cleavage transition state. [Google Scholar]
- 26.
- 26a. Venkateswaran R., Mague J. T., Blakrishna M. S., Inorg. Chem. 2007, 46, 809–817; [DOI] [PubMed] [Google Scholar]
- 26b. Dallanegra R., Chaplin A. B., Weller A. S., Organometallics 2012, 31, 2720–2728; [Google Scholar]
- 26c. Pawley R. J., Huertos M. A., Lloyd-Jones G. C., Weller A. S., Willis M. C., Organometallics 2012, 31, 5650–5659. [Google Scholar]
- 27.All structures located via IRC calculations are provided in the ESI. These are discussed in the main text if a significant conformational change of the DPEphos ligand is involved (as is the case for 8ccc/8ccc′).
- 28.The computed energy of 2ttt, the species characterised crystallographically, is −31.3 kcal mol−1.
- 29. Sun Z., Fridrich B., de Santi A., Elangovan S., Barta K., Chem. Rev. 2018, 118, 614–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary