

Abstract
Introduction
Our aim in this study was to identify the prevalence and clinical characteristics of LRP4/agrin‐antibody–positive double‐seronegative myasthenia gravis (DNMG).
Methods
DNMG patients at 16 sites in the United States were tested for LRP4 and agrin antibodies, and the clinical data were collected.
Results
Of 181 DNMG patients, 27 (14.9%) were positive for either low‐density lipoprotein receptor–related protein 4 (LRP4) or agrin antibodies. Twenty‐three DNMG patients (12.7%) were positive for both antibodies. More antibody‐positive patients presented with generalized symptoms (69%) compared with antibody‐negative patients (43%) (P ≤ .02). Antibody‐positive patients’ maximum classification on the Myasthenia Gravis Foundation of America (MGFA) scale was significantly higher than that for antibody‐negative patients (P ≤ .005). Seventy percent of antibody‐positive patients were classified as MGFA class III, IV, or V compared with 39% of antibody‐negative patients. Most LRP4‐ and agrin‐antibody–positive patients (24 of 27, 89%) developed generalized myathenia gravis (MG), but with standard MG treatment 81.5% (22 of 27) improved to MGFA class I or II during a mean follow‐up of 11 years.
Discussion
Antibody‐positive patients had more severe clinical disease than antibody‐negative patients. Most DNMG patients responded to standard therapy regardless of antibody status.
Keywords: agrin, clinical features, LRP4, myasthenia gravis, neuromuscular transmission disorders, seronegative myasthenia gravis
Abbreviations
- AChR
acetylcholine receptor
- CMS
congenital myasthenic syndrome
- DNMG
double‐seronegative myasthenia gravis (negative for AChR and MuSK)
- ELISA
enzyme‐linked immunosorbent assay
- IVIg
intravenous immunoglobulin
- LAPMG
DNMG with positivity for LRP4 and/or agrin antibodies
- LE
lower extremity
- LRP4
low‐density lipoprotein receptor–related protein 4
- MG
myasthenia gravis
- MGFA
Myasthenia Gravis Foundation of America
- MuSK
muscle‐specific kinase
- NS
not statistically significant
- QMG
quantitative myasthenia gravis score
- QNMG
quadruple seronegative myasthenia gravis (negative for AChR, MuSK, LRP4, and agrin)
- TBST
Tris‐buffered saline plus Tween
- UE
upper extremity
1. INTRODUCTION
Myasthenia gravis (MG) is an autoimmune disease in which antibodies directed against the neuromuscular junction cause fatigable weakness. 1 In approximately 80% of patients with generalized MG, antibodies to acetylcholine receptors (AChRs) have been identified. 2 In another 10% of generalized MG patients, antibodies to muscle‐specific kinase (MuSK) have been reported. 3 Although a small percentage of generalized MG patients may have genetic defects (congenital myasthenic syndromes [CMSs]), rather than an immune pathogenesis, most of the remaining 10% of double‐seronegative MG patients (DNMG) are believed to have an autoimmune etiology, as they respond to immunosuppressant and immunomodulatory therapies. 4
Agrin is a proteoglycan utilized by motor neurons to instruct neuromuscular junction formation and maintenance. 5 Agrin binds to low‐density lipoprotein receptor–related protein 4 (LRP4) and activates MuSK. The ensuing intracellular pathways lead to the aggregation of AChR at the neuromuscular junction. 6 , 7 , 8 , 9 Antibodies to LRP4 and agrin have been found in MG patients, particularly DNMG, 10 , 11 , 12 , 13 , 14 and have been shown to produce MG in experimental animals. 15 , 16 , 17 This makes it likely that anti‐agrin and LRP4 antibodies may be pathogenic factors for DNMG. Also, mutations in these proteins have been reported in CMSs. 18 , 19 , 20 , 21 , 22 , 23 To date, there have been 225 patients described in the literature who have MG associated with LRP4 antibodies, 10 , 11 , 12 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 but clinical data were described for only 133 of these patients. Thirty‐six agrin‐antibody–associated MG cases have been described, 13 , 14 only 5 of which have clinical data. 13 The data on DNMG patients with LRP4 and/or agrin antibodies are even more limited.
The prevalence rates vary dramatically in the literature. For example, the prevalence of LRP4 antibodies in MG patients was found to vary from 0.45% to 40%, and in DNMG patients from 0.14% to 50%. 10 , 11 , 12 , 25 , 26 , 27 , 28 , 29 , 30 , 32 The prevalence of anti‐agrin antibodies in MG patients varied from 7.5% to 14.9%, and in DNMG patients fluctuated between 0% and 50%. 13 , 14 , 26 Little is known regarding the clinical value of these antibodies. In this study, we assessed prevalence, clinical features, and prognosis in carefully screened DNMG patients in the United States who have antibodies to LRP4 and/or agrin.
2. METHODS
Patients over the age of 21 seen between May 2016 and June 2018 were enrolled at 16 clinical sites (Figure 1). Eligible patients had clinical signs of MG, either an abnormal repetitive stimulation study and/or single‐fiber electromyogram, and tested negative for both AChR and MuSK antibodies. All patients provided informed consent for participation in the study. Patients were excluded if they received either plasma exchange or intravenous immunoglobulin (IVIg) within 6 weeks of their antibody test or rituximab within 24 weeks of their test. Pregnant patients were also excluded from study participation. Normal control samples were obtained from 106 random blood donors. The study was approved by the institutional review board at each participating clinical site.
FIGURE 1.
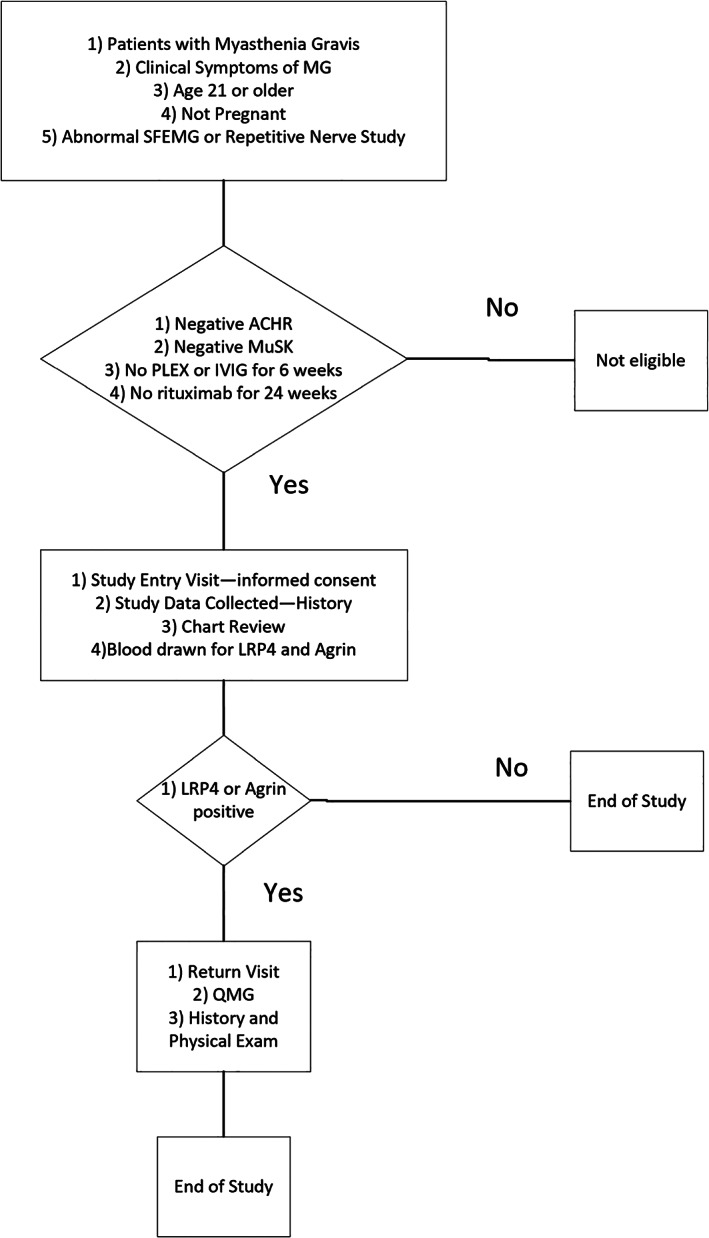
Sequence of study visits and details on which procedures are performed at each visit. Abbreviations: ACHR, acetylcholine receptor; IVIG, intravenous immunoglobulin; LRP4, low‐density lipoprotein receptor–related protein 4; PLEX, plasma exchange; MG, myathenia gravis; MuSK, muscle‐specific kinase; QMG, quantitative myasthenia gravis score
Chart review was performed on patients who met study entry criteria. They were graded according to the Myasthenia Gravis Foundation of America (MGFA) classification during two time periods 34 : (1) on enrollment in the study (study entry); and (2) when their MGFA classification was at its highest (maximum severity). LRP4 and/or agrin‐antibody–positive DNMG patients (LAPMG) were also graded at disease onset. Blood samples were tested for LRP4 and agrin by enzyme‐linked immunoassay (ELISA), as described in what follows. Investigators performing the ELISA were blinded to whether samples were from MG patients or controls and the origin of the samples. LAPMG patients underwent an additional detailed history, physical and neurological examinations, and quantitative myasthenia gravis (QMG) testing 35 during a second return examination (return exam). DNMG patients negative for LRP4 and/or agrin antibodies, quadruple‐negative MG (QNMG), underwent chart review only.
ELISA assays for agrin and LPR4 antibodies were performed as described elsewhere. 12 , 14 Briefly, 96‐well Nunc MaxiSorp plates (Thermo Fisher Scientific, Waltham, Massachusetts) were incubated with recombinant agrin or LRP4 in carbonate buffer (500 mmol/L, pH 9.6) overnight at 4°C. After rinsing with 0.3% Tween in Tris‐buffered saline (TBST) for agrin antibody and 0.1% TBST for LRP4 antibody, wells were incubated in blocking buffer (5% milk in 0.3% TBST) for 2 hours at room temperature. Sera were diluted in 1:100 with the blocking buffer and added to the wells for incubation overnight at 4°C. The wells were washed six times with respective TBST buffers on a shaking platform and then incubated with anti‐human immunoglobulin antibodies (1:2000 dilution in 2.5% milk/0.3% TBST) for 1 or 2 hours at room temperature. After washing, the wells were incubated with substrate buffer containing 10 mol/L diethanolamine, 0.5 mmol/L MgCl2, and p‐nitrophenylphosphate for 5 to 15 minutes at room temperature. Samples were subjected to optical density readings at 405 nm.
Antibody testing was considered abnormal if the value was 2.5 standard deviations above values for normal control samples. Data were entered by each site on the ONCORE database system at Augusta University. Data were reviewed for completeness and analyzed using Excel (Microsoft Corp, Redmond, Washington). Statistical testing was done in Excel with QI Macro (KnowWare International, Denver, Colorado) and SPSS version 25 (IBM, Armonk, New York). Qualitative statistics were evaluated using a two‐tailed Fisher exact test. A two‐tailed t test was also used for quantitative data.
3. RESULTS
Of 181 DNMG patients who were tested for LRP4 and agrin antibodies, 154 (85.1%) were negative for autoantibodies to LRP4 and agrin (QNMG) and 27 (14.9%) were positive for at least one of these two autoantibodies (LAPMG). Twenty‐three (12.7%) patients were positive for both LRP4 and agrin antibodies. Three patients were positive for only agrin antibodies, whereas one patient was positive for only LRP4 antibodies. Because only a small number of patients were positive for only a single antibody, we focused on the 27 patients who were positive for either LRP4 or agrin antibodies (LAPMG) and compared their results with those negative for these antibodies (QNMG).
Table 1 summarizes the basic data of our DNMG population. The majority of our patients were female. There was no significant difference in gender between the LAPMG and QNMG groups, even though there was a slightly higher percentage of females in the QNMG group. Caucasian/white patients made up the bulk of our population, but other ethnic groups were also represented. There was no significant difference in ethnicity between the two groups. The mean age at disease onset was 46.3 years. This did not differ significantly between the LAPMG and QNMG groups. The mean time between disease onset and entry into the study was 11.4 years. This latency did not differ significantly between the LAPMG and QNMG groups. The first, second, and third quartiles for disease duration in the LAPMG group were 3.3, 8.5, and 15.7 years, respectively, and for the QNMG group 3.9, 7.8, and 14.4 years. Most patients had an ocular‐only onset. Significantly fewer LAPMG patients had an ocular onset compared with the QNMG group. Therefore, LAPMG patients are more likely to have generalized MG at disease onset. By the time patients entered the study (study entry), there was no significant difference in the percentage of patients with ocular‐only symptoms between the two groups (Table 1).
TABLE 1.
Features of MG
| QNMG | LAPMG | |||||
|---|---|---|---|---|---|---|
| Number | Percent | Number | Percent | Significance | Percent of group | |
| Clinical features | ||||||
| Age of onset | 46.75 years | 44.15 years | P = .47 (NS) | |||
| Disease duration | 11.4 years | 11.3 years | P = .94 (NS) | |||
| Female | 104 | 67.53% | 16 | 59.26% | P = .40 (NS) | |
| Ocular onset | 88 | 57.14% | 8 | 30.77% | P ≤ .02 * | |
| Ocular study entry | 55 | 35.71% | 6 | 22.22% | P = .19 (NS) | |
| Caucasian | 111 | 72.08% | 21 | 77.78% | 18.92% | |
| Black | 28 | 18.18% | 4 | 14.81% | P = .87 (NS) | 14.29% |
| Asian | 6 | 3.90% | 2 | 7.41% | 33.33% | |
| Hispanic | 4 | 2.60% | 0 | 0.00% | 0.00% | |
| Drug treatments | ||||||
| PLEx | 37 | 24.03% | 8 | 29.63% | P = .63 (NS) | |
| IVIg | 36 | 23.38% | 4 | 14.81% | P = .45 (NS) | |
| Rituximab | 18 | 11.69% | 2 | 7.41% | P = .74 (NS) | |
| Pyridostigmine | 139 | 90.26% | 24 | 88.89% | P = .74 (NS) | |
| Prednisone | 105 | 68.18% | 19 | 70.37% | P = 1.00 (NS) | |
| Azathioprine | 33 | 21.43% | 5 | 18.52% | P = 1.00 (NS) | |
| Mycophenolate mofetil | 49 | 31.82% | 12 | 44.44% | P = .27 (NS) | |
| Methotrexate | 20 | 12.99% | 3 | 11.11% | P = 1.00 (NS) | |
Abbreviations: IVIg, intravenous immunoglobulin; LAPMG, double‐seronegative myasthenia gravis with positivity for low‐density lipoprotein receptor–related protein 4 and/or agrin antibodies; MG, myathenia gravis; NS, not statistically significant; PLEx, plasma exchange; QNMG, quadruple seronegative myasthenia gravis.
Statistically significant (P < .05).
The symptoms at disease onset and during study entry are detailed on Figure 2. Symptoms did not differ significantly between the two groups. LAPMG patients tend to have more bulbar symptoms, but this difference was neither significant at disease onset nor at the time of study entry. At the study entry visit, LAPMG patients tended to have more upper extremity symptoms but this difference was not significant (P = .18, not statistically significant [NS]).
FIGURE 2.
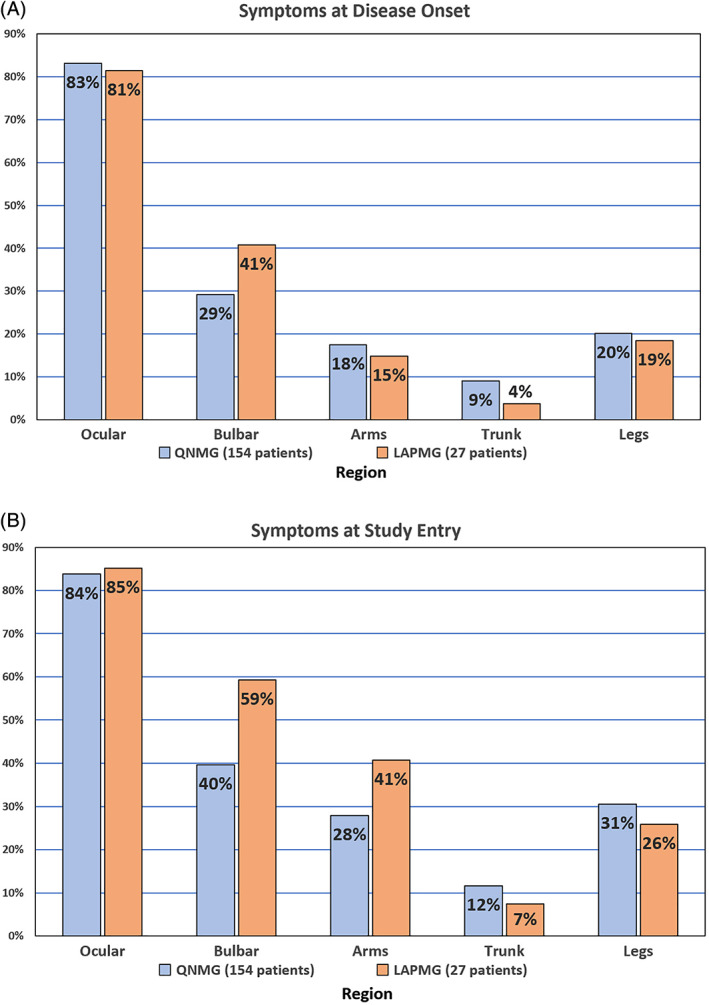
Symptoms of both antibody‐positive and ‐negative patients at two time periods. A, Disease onset. B, Time of study visit (study entry exam). Abbreviations: DNMG, double‐seronegative myasthenia gravis (negative for AChR and MuSK); LAPMG, double‐seronegative myasthenia gravis with positivity for LRP4 and/or agrin antibodies [Color figure can be viewed at wileyonlinelibrary.com]
The MGFA classification of our patients at study entry did not differ between the two groups (Figure 3A,B). Most patients (85.6%) in our total MG population had either ocular or mild, generalized symptoms. Most (81.5%) in the LAPMG group had either ocular or mild disease at the time of study entry. MGFA classification at disease onset was studied only in the LAPMG group (Figure 3E). The majority (69.2%) of LAPMG patients were MGFA class I or II, whereas the others (30.8%) were MGFA class III, IV, or V. One patient presented as MGFA class V.
FIGURE 3.

Percentage of patients in each MGFA class for three time periods. A, Study entry MGFA class for antibody‐negative patients. B, Study entry MGFA class for antibody‐positive patients. C, Maximal MGFA class for antibody‐negative patients. D, Maximal MGFA class for antibody‐positive patients. E, MGFA class at disease onset for antibody‐positive patients. Abbreviations: LAPMG, double‐seronegative myasthenia gravis with positivity for LRP4 and/or agrin antibodies; MGFA, Myasthenia Gravis Foundation of America; QNMG, quadruple seronegative myasthenia gravis (negative for acetylcholine receptor, muscle‐specific kinase, low‐density lipoprotein receptor–related protein 4, and agrin) [Color figure can be viewed at wileyonlinelibrary.com]
At the time of maximum disease severity (Figure 3C,D), 56.4% of all DNMG patients had ocular or mild disease severity (MGFA class I or II). In the LAPMG group, 70.4% of subjects were MGFA class III or higher as compared with 39% in the QNMG group (Figure 4). This difference was highly significant (P = .003) during this time period, but not at the time of study entry (Figure 4).
FIGURE 4.
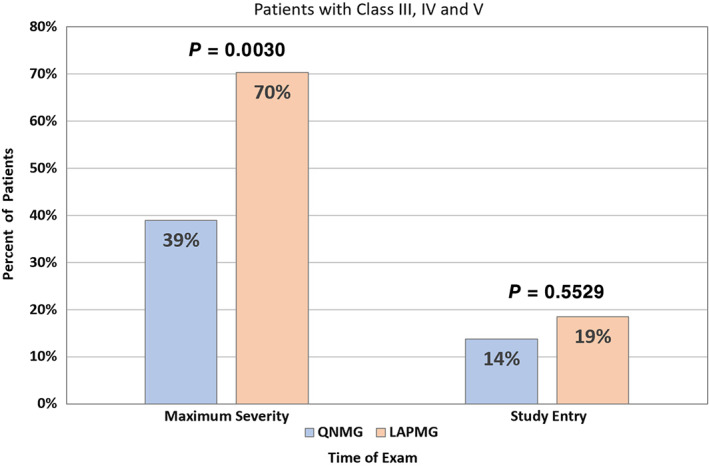
Percentage of patients with MGFA class III or higher at time of maximum disease severity and during the study entry exam. Abbreviations: MGFA, Myasthenia Gravis Foundation of America; QNMG, quadruple seronegative myasthenia gravis (negative for acetylcholine receptor, muscle‐specific kinase, low‐density lipoprotein receptor–related protein 4, and agrin) [Color figure can be viewed at wileyonlinelibrary.com]
MGFA classification at the time of study entry was similar for the LAPMG and QNMG groups (Figure 3A,B) (P = .26, NS). Even when combining a (axial/limb muscle predominance) and b (bulbar muscle predominance) at each classification level, the results were not significant (P = .45, NS). At the time of maximal disease severity (Figure 3C,D), LAPMG patients had more severe disease than QNMG patients. This difference was not significant (P = .0608) when each classification was considered by itself, but if the patients are grouped by severity (combining the a and b groups at each severity level), then the LAPMG group would be significantly worse than the QNMG group (P = .0253). Three patients (11.1%) in the LAPMG group needed mechanical ventilation, which was double the percentage (5.2%) requiring ventilation in the QNMG group, but this difference was not significant (P = .21). There was no significant difference in the number of patients classified as a (axial/limb muscle predominance) vs b (bulbar muscle predominance) between the two groups (P = .22).
The patients were treated with standard therapies for MG, regardless of antibody status (Table 1). There was no significant difference in the medications used by the two groups. Most patients (90.1%) were treated with pyridostigmine. Plasma exchange was used in 24.9%, IVIg in 22.1%, and rituximab in 11.0%. Prednisone was used in 68.5%, azathioprine in 21.0%, mycophenolate mofetil in 33.7%, and methotrexate in 12.7% of patients. The majority (65.4%) of LAPMG patients responded to pyridostigmine, 23.1% worsened, and the remainder (11.5%) had unclear responses. Based on improvement in the MGFA grade between the time of maximum disease severity and study entry (Figure 3), it appears that most LAPMG patients (P < .001) as well as most QNMG patients (P < .001) responded to standard treatments for MG. Although disease fluctuation may account for some of this difference in disease severity between these two time periods, it cannot account for most of this improvement. Furthermore, our subjects’ improvement was better than that described in historical studies before the introduction of immunosuppressant therapies. 36
Figure 5 illustrates the symptoms of LAPMG patients during three time periods: return exam symptoms, initial symptoms, and maximal symptoms. The data obtained for the initial and maximal symptoms are historical, but the return exam symptoms are based on an evaluation after patients were found positive for the antibodies. Ptosis and diplopia are common symptoms and occur in the majority of LAPMG patients during the maximum symptoms period. They are present to a lesser extent during other time periods. Other prominent symptoms in LAPMG patients at maximum disease severity are proximal upper extremity (UE) and lower extremity (LE) weakness, speech, swallowing, chewing, and respiratory and neck weakness. More than half of the LAPMG patients (54.5%) had respiratory symptoms. In addition to the usual bulbar findings seen in MG, LAPMG patients had proximal LE and UE symptoms during other stages of the disease.
FIGURE 5.

Clinical symptoms of LRP4/agrin‐positive patients at three discrete time‐points. Rear bars: maximal symptoms; middle bars: initial symptoms; front bars: return exam symptoms. Abbreviations: Dist, distal; LAPMG, double‐seronegative myasthenia gravis with positivity for LRP4 and/or agrin antibodies; LRP4, low‐density lipoprotein receptor–related protein 4; LE, lower extremity; Prox, proximal; Resp, respiratory; UE, upper extremity [Color figure can be viewed at wileyonlinelibrary.com]
Physical exam findings showed ptosis, diplopia, and mild weakness of the orbicularis oculi in more than 45% of LAPMG patients. More than 20% of LAPMG patients had orbicularis oris, neck flexion, shoulder, and hip weakness. The remainder of the exams, including reflex and sensory exams, were unremarkable. The average QMG score of LAPMG patients was 9.36 (SD, 5.54). Fifteen LAPMG patients (62%) had chest imaging. None had a thymoma. Five patients underwent a thymectomy before study entry. One patient had thymic hyperplasia; the remainder were normal.
4. DISCUSSION
In this study we found that about 15% of DNMG patients had autoantibodies to either LRP4 or agrin. LRP4 autoantibodies were found in 13% of our double‐negative patients, which coincides with the range of 2% to 50% reported in the literature. 10 , 11 , 12 , 25 , 26 , 27 , 28 , 29 , 30 , 32 and is in agreement with a previous report. 12 Agrin autoantibodies were found in 14% of our double‐negative patients compared with the 0% to 50% reported in the literature. 13 , 14 , 26 Most (85%) of our LAPMG patients had autoantibodies to both agrin and LRP4. Because few patients in previous studies were tested for agrin, we do not know whether most of the other patients described in the literature were positive for both antibodies. Although the number of MG patients previously reported with agrin antibodies is too few to make any meaningful comparisons with our data (36 total and 5 with clinical data), 13 , 14 , 26 there are more data on LRP4 patients with MG (225 total and 133 with clinical data). 10 , 11 , 12 , 25 , 26 , 27 , 28 , 29 , 30 , 32 Some of the variability in incidence of LRP4 antibodies may be accounted for by the assay used. Several studies used cell‐based assays, 11 , 25 , 26 , 27 , 28 , 29 , 32 and others, including ours, used ELISA 12 , 13 , 14 , 25 or alternative methods. 10 This cannot explain the varying incidences reported because different rates were reported using the same methods.
Ethnic or racial differences may account for some differing incidences. Lower incidences were reported in Asian populations 10 , 27 , 28 ; however, in our population we found no difference among races, even though our study included relatively few minority patients. In addition, widely varying incidences were reported in two German studies. 11 , 25 Several studies found incidences similar to ours. 29 , 32 The small numbers of patients studied may account for these disparities. We believe an incidence of around 15% reflects the likely proportion of positive patients in a group of DNMG patients, as this is similar to the rates seen in our work and the two largest studies. 29 , 33 It is of interest that most patients in our cohort who were positive for LRP4 were also positive for agrin. Although patients positive for both have been reported previously, 13 this is not a commonly observed finding among the few patients who have been tested for both antibodies.
Average age of onset (44 years) is consistent with that found in other studies. 11 , 29 , 32 Most LAPMG patients were female, also consistent with other studies. 11 , 27 , 29 , 32
Our data on MGFA classification at symptom onset show that 31% of our LAPMG patients were MGFA class III or or higher. LAPMG patients were somewhat more symptomatic than those described by other studies at symptom onset, as most had fewer subjects with MGFA class III or higher and none had MGFA class V status. 25 , 32 A significantly greater percentage of our LAPMG patients had generalized MG at symptom onset compared with the QNMG group. At time of maximal severity, a significantly greater proportion of our LAPMG patients were MGFA class III or higher compared with our QNMG patients. Although some studies have shown only mild symptomatology with positive antibodies, 25 , 27 , 28 , 29 , 31 , 32 others have shown a more severe symptomatology, in agreement with our data. 10 , 11 , 24 , 33 Only the study of Zisimopoulou et al 32 included a sizable population of LRP4‐positive patients. In their study, clinical data were collected on 67 DNMG patients who were LRP4‐positive. Although both of our studies had a similar percentage of MGFA class I patients at disease onset, 31% of our LAPMG patients presented with MGFA class III or higher compared with 15% of their patients. One of our patients presented with MGFA class V, but none did so in the earlier study. The differences in the assay used may account for the differences between studies. The earlier study used a cell‐based assay compared with the ELISA used in the present study. Also, the authors did not truly address maximum severity, whereas we had data on patients who had an average disease duration of 11.3 years. Furthermore, the combination of antibodies to agrin and LRP4 may produce more severe disease than having antibodies only to LRP4. It is unknown whether the LRP4‐positive patients in the Zisimopoulou et al 32 study were also positive for agrin antibodies because testing was not done. Only a single patient in our study was positive for LRP4 alone, and this patient had only mild symptoms. All three patients who were agrin‐positive but LRP4 negative had maximum MGFA scores of III or higher, suggesting that presence of agrin antibodies is associated with more severe disease. At the time of maximum disease severity, a significantly greater number of LAPMG patients were MGFA class III or higher compared with QNMG patients. In fact, 11% of our LAPMG patients were ventilator‐dependent as compared with 5% of our QNMG patients. Thus, we have shown that LAPMG patients have a more severe disease onset as well as a more severe course than QNMG patients.
On their return exam, LAPMG patients had mild ocular, oral, neck, proximal upper extremity, and proximal lower extremity symptoms and signs. Although the timing of the study entry exam was somewhat arbitrary, the percentage of LAPMG patients who were MGFA class III or higher was less at study entry exam (19%) compared with disease onset (31%) and point of maximal symptomatology (70%), which suggests that most LAPMG patients are optimally treated at the time of their study entry exam. The timing of the study entry exam in our comparison QNMG group was similarly arbitrary, as there was no significant difference in disease duration at the time of the study entry exam between the two groups. More than 40% of LAPMG patients had chewing, swallowing, speech, respiratory, neck, and proximal upper and lower extremity weakness at maximum severity. Although we did not obtain comparable data in our QNMG group, based on the difference in MGFA classification, we believe bulbar, respiratory, and limb symptoms are more common in LAPMG patients.
Despite having more severe symptoms initially and during the course of their disease, most LAPMG patients improved with standard immunosuppressive or immunomodulatory therapy. We do not have enough data to determine which therapies are most effective, and this is not a prospective, randomized therapeutic head‐to‐head comparative study, but the presence of these antibodies might guide future therapeutic choices. With the development of complement‐modifying therapies such as eculizumab, identifying what antibodies are present in patients may allow us to tailor therapy and thus improve treatment. 37 Because presence of agrin and LRP4 antibodies is associated with a more severe disease course compared with having with no detectable autoantibodies, finding these antibodies in DNMG patients will allow us to better predict which patients will develop severe disease. According to the data we currently have, we could find no distinguishing clinical characteristics that would allow us to differentiate LAPMG from QNMG. More information will be needed to compare severity and clinical characteristics with those seen in AChR‐ and MuSK‐positive MG. The current data show that many LAPMG patients have severe clinical courses, similar to those seen in many AChR‐ and MuSK‐positive patients. As with all MG patients, current management can lead to clinical improvement in DNMG patients. 38 We suggest ordering LRP4 and agrin antibodies in MG patients who are negative for AChR and MuSK antibodies (DNMG).
The final issue concerns whether these antibodies (LRP4/agrin) produce disease pathology or are just an epiphenomenon. Antibodies to LRP4 and agrin led to pathology in mice that mimics MG in humans. 15 , 16 Furthermore, inherited defects of LRP4 and agrin produce congenital MG. 18 , 19 , 20 , 21 , 22 , 23 Although presence of agrin and LRP4 antibodies has also been associated with amyotrophic lateral sclerosis, 39 , 40 LRP4 with neuromyelitis optica 12 and multiple sclerosis, 32 other disease control patients tested in the literature were antibody‐negative. Based on the location of LRP4 at the neuromuscular junction, its binding to agrin, and its relationship to MuSK activation, it is highly likely that the presence of these antibodies in DNMG is related to their disease pathology. 6 , 7
In conclusion, in this study we found that 15% of DNMG patients had antibodies to either LRP4 or agrin and 13% of our patients had autoantibodies to both. The average age of onset was 44 years and 59% were female. LAPMG patients have a more severe presentation compared with QNMG patients. The presence of these autoantibodies was associated with a more severe disease course, but most patients responded well to standard MG therapy.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
CONFLICT OF INTEREST
M.H.R.: consultant and research support from Alexion and Allergan; research support from UCB, Mallinckrodt, Cytokinetics, Momenta, Shire Takeda, Orion, Biohaven, Catalyst, Grifols, Seikagaku, and Ra Pharmaceuticals. B.M.Q.: research support from Alexion, UCB, Mallinckrodt, Cytokinetics, Momenta, Shire Takeda, Orion, Biohaven, Catalyst, Grifols, Seikagaku, and Ra Pharmaceuticals. J.F.H.: research support from Alexion, Argenx BVBA, Centers for Disease Control and Prevention, Muscular Dystrophy Association (MDA), National Institutes of Health (NIH, including National Institute of Neurological Disorders and Stroke and National Institute of Arthritis and Musculoskeletal and Skin Diseases), and Ra Pharmaceuticals; honoraria from Alexion; and nonfinancial support from Alexion, Argenx BVBA, Ra Pharmaceuticals, and Toleranzia. M.M.D.: consultant for ArgenX, CSL‐Behring, Kezar, Momenta, NuFactor, RMS Medical, Sanofi Genzyme, Shire Takeda, and Spark; grants from Alexion, Alnylam, Amicus, Biomarin, Bristol‐Myers Squibb, Catalyst, CSL‐Behring, US Food and Drug Administration/Office of Orphan Products Development, GlaxoSmithKline, Genentech, Grifols, Mitsubishi Tanabe Pharma, MDA, NIH, Novartis, Sanofi Genzyme, Octapharma, Orphazyme, Sarepta, Shire Takeda, Spark, UCB Biopharma, Viromed, and TMA. C.J.: speaker's bureau for Mitsubishi Tanabe Pharma America, Avanir, CSL Behring, and Strongbridge; research support and consultant fees from Cytokinetics and Mitsubishi Tanabe Pharma America; consultant fees from Argenx, Alexion, and ITF; data safety monitoring board for Mallinckrodt, Brainstorm, and Anelixis. T.V.: speaker and/or consultant for Alexion, ARGX, and UCB; participated in clinical trials sponsored by Alexion, ARGX, Ra Pharmaceuticals, UCB, and Grifols. G.S.: consultant for Alexion. R.P.L.: over past 2 years has been funded for research support from the NIH, National Multiple Sclerosis Society (USA), Genentech, Teva, Novartis, Medimmune, Ra Pharmaceuticals, Argenx, Alexion, Catalyst, and Chugai; served as a consultant to Argenx, Novartis, Mallinckrodt, Catalyst, GLG, Alpha Sites, Insights Consulting, Informa Pharma Consulting, Slingshot Consulting, Schlessinger Group Consulting, Health Choices, Adivo Associates, Haven Consulting, kc2 Medical Communications, and Cello Health Bioconsulting. I.L.: funding support from the Myasthenia Gravis Foundation of America (MGFA) for the MG Registry project. R.J.N.: research support from the NIH, Genentech, Alexion, Ra Pharmaceuticals, MGFA, Momenta, Argenx, Annexon Biosciences, and Grifols; consultant/advisor for Alexion Pharmaceuticals, Argenx, CSL Behring, Grifols, Ra Pharmaceuticals, Roivant, Viela Bio, and Momenta (no conflicts directly related to this study). J.A.F.: funding from Mallinckrodt Pharmaceuticalsand consultant fees from Biogen. Z.S.: research support from Cytokinetics, Mallinckrodt, and Biohaven; consulting fees from Biohaven; payment from Wiley for his position as editor‐in‐chief for Muscle & Nerve. A.S.: research support from Amylyx. R.B.: consultant for NuFactor and Momenta and receives research support from PTC, Ra Pharmaceuticals, Orphazyme, Sanofi Genzyme, FDA/OOPD, NIH, and the Patient‐Centered Outcomes Research Institute. R.B.S.: advisory board for Argenyx. C.G.: editorial board of the journal Neurology. E.U.: research support from NIH/NINDS and Genzyme Sanofi; clinical program support from the MDA; book royalties from Springer Science + Business Media; and has a nonexclusive commercial license (held by Baylor Licensing Group) for immortalized human blood‐nerve barrier endothelial cells. J.C.: research grant funding from Orion and Cytokinetics. M.P.: consultant and on advisory board for TerumoBCT, Alexion, CSL Behring, Momenta, Argenx, and Calalyst. L.M.: recipient of the NIH grant supporting this research project and author of a patent on LRP4 antibodies. The remaining authors declare no potential conflicts of interest.
APPENDIX 1.
The LRP4 Research Group consists of the following participating institutions and individuals: Augusta University (Michael Rivner MD, Brandy Quarles MPH, Kristy Bouchard BS, Hongyan Xu PhD); Case Western Reserve University (Lin Mei MD, PhD, Jin‐Xiu Pan MD, Zheng Yu MD); University of North Carolina at Chapel Hill (James Howard MD, Manisha Chopra MBBS); Johns Hopkins University (Andrea Corse MD, Kristen Riley PhD, Lora Clawson MSN, CRNP, Alpa Uchil MSN, MPH, CRNP); University of Texas Health Science Center at San Antonio (R. Bhavaraju Sanka MD, Carlayne Jackson MD, Pamela Kittrell MSN, Floyd Jones BS); University of Kansas (Mazen M. Dimachkie MD, Richard Barohn MD, Mamatha Pasnoor MD, Constantine Farmakidis MD, Omar Jawdat, MD, Duaa Jabari, MD, Andrew Heim BS CCRP); Allegheny General Hospital (George Small MD, Mary Fetter BS); Wayne State University (Robert Lisak MD, Eva Bernitsas MD, Melody Hackett BS, Kelly Jia MD); University of Alabama at Birmingham (Eroboghene Ubogu MD, Ikjae Lee MD, Sandi Mumfrey‐Thomas BSHA, LaToya Conner LPN, Mia Davis LPN, Sabrina Miller LPN); University of South Florida (Tuan Vu MD, Clifton Gooch MD, Jerrica Farias APRN, Brittany Harvey BA); Yale University (Richard Nowak MD, Joan L. Nye BS); Wake Forest University (Vanessa Baute MD, James Caress MD, Janet Hutchens BS, CCRC); Mount Sinai‐Beth Israel Hospital (Stephen N. Scelsa MD, Emily Ripps RN, BSN); Rutgers Robert Wood Johnson Medical School (Jerry Belsh MD, Patricia Sonsalla PhD); Penn State Health Milton S. Hershey Medical Center (Zachary Simmons MD, Jennifer A. Crossen BS, Heidi Runk CCRC); University of Nebraska Medical Center (J. Americo Fernandes MD, Pariwat Thaisetthawatkul MD, Ezequiel Piccione MD, Nicholas Miller BS); University of Iowa (Andrea Swenson MD, Jeri Sieren RN).
Rivner MH, Quarles BM, Pan J‐X, et al. Clinical features of LRP4/agrin‐antibody–positive myasthenia gravis: A multicenter study. Muscle & Nerve. 2020;62:333–343. 10.1002/mus.26985
The content of this article was presented in part at the scientific session of the Myasthenia Gravis Foundation of America at the 66th annual meeting of the American Association of Neuromuscular and Electrodiagnostic Medicine on October 16, 2019, Austin, Texas.
Funding information National Institutes of Health, Grant/Award Number: 5R01NS090083–06
Contributor Information
Michael H. Rivner, Email: mrivner@augusta.edu.
Lin Mei, Email: lin.mei@case.edu.
REFERENCES
- 1. Conti‐Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843‐2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoch WM, Helms J, Newsom‐Davis S, Melms J, Vincent A. Auto‐antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365‐368. [DOI] [PubMed] [Google Scholar]
- 3. Sanders DB, El‐Salem K, Massey JM, McConville J, Vincent A. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. 2003;60:1978‐1980. [DOI] [PubMed] [Google Scholar]
- 4. Birmanns B, Brenner T, Abramsky O, Steiner I. Seronegative myasthenia gravis: clinical features, response to therapy and synthesis of acetylcholine receptor antibodies in vitro. J Neurol Sci. 1991;102:184‐189. [DOI] [PubMed] [Google Scholar]
- 5. Li L, Xiong WC, Mei L. Neuromuscular junction formation, aging, and disorders. Annu Rev Physiol. 2018;80:159‐188. [DOI] [PubMed] [Google Scholar]
- 6. Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim N, Stiegler AL, Cameron TO , et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magill‐Solc C, McMahan UJ. Motor neurons contain agrin‐like molecules. J Cell Biol. 1988;107:1825‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zong Y, Zhang B, Gu S, et al. Structural basis of agrin‐LRP4‐MuSK signaling. Genes Dev. 2012;26:247‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low‐density lipoprotein receptor‐related protein 4 in myasthenia gravis. Ann Neurol. 2011;69:418‐422. [DOI] [PubMed] [Google Scholar]
- 11. Pevzner A, Schoser B, Peters K, et al. Anti‐LRP4 autoantibodies in AChR‐ and MuSK‐antibody‐negative myasthenia gravis. J Neurol. 2012;259:427‐435. [DOI] [PubMed] [Google Scholar]
- 12. Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein‐related protein 4 in patients with double‐seronegative myasthenia gravis. Arch Neurol. 2012;69:445‐451. [DOI] [PubMed] [Google Scholar]
- 13. Gasperi C, Melms A, Schoser B, et al. Anti‐agrin autoantibodies in myasthenia gravis. Neurology. 2014;82:1976‐1983. [DOI] [PubMed] [Google Scholar]
- 14. Zhang B, Shen C, Bealmear B, et al. Autoantibodies to agrin in myasthenia gravis patients. PLoS One. 2014;9:e91816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan M, Liu Z, Fei E, et al. Induction of anti‐agrin antibodies causes myasthenia gravis in mice. Neuroscience. 2018;373:113‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen C, Lu Y, Zhang B, et al. Antibodies against low‐density lipoprotein receptor‐related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123:5190‐5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mori S, Motohashi N, Takashima R, Kishi M, Nishimune H, Shigemoto K. Immunization of mice with LRP4 induces myasthenia similar to MuSK‐associated myasthenia gravis. Exp Neurol. 2017;297:158‐167. [DOI] [PubMed] [Google Scholar]
- 18. Huze C, Bauche S, Richard P, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karakaya M, Ceyhan‐Birsoy O, Beggs AH, Topaloglu H. A Novel missense variant in the AGRN gene; congenital myasthenic syndrome presenting with head drop. J Clin Neuromuscul Dis. 2017;18:147‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maselli RA, Fernandez JM, Arredondo J, et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non‐neural (z‐) agrin. Hum Genet. 2012;131:1123‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicole S, Chaouch A, Torbergsen T, et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. 2014;137:2429‐2443. [DOI] [PubMed] [Google Scholar]
- 22. Ohkawara B, Cabrera‐Serrano M, Nakata T, et al. LRP4 third beta‐propeller domain mutations cause novel congenital myasthenia by compromising agrin‐mediated MuSK signaling in a position‐specific manner. Hum Mol Genet. 2014;23:1856‐1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Selcen D, Ohkawara B, Shen XM, McEvoy K, Ohno K, Engel AG. Impaired synaptic development, maintenance, and neuromuscular transmission in LRP4‐related myasthenia. JAMA Neurol. 2015;72:889‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beck G, Yabumoto T, Baba K, et al. Double seronegative myasthenia gravis with anti‐LRP4 antibodies presenting with dropped head and acute respiratory insufficiency. Intern Med. 2016;55:3361‐3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cordts I, Bodart N, Hartmann K, et al. Screening for lipoprotein receptor‐related protein 4‐, agrin‐, and titin‐antibodies and exploring the autoimmune spectrum in myasthenia gravis. J Neurol. 2017;264:1193‐1203. [DOI] [PubMed] [Google Scholar]
- 26. Cossins J, Belaya K, Zoltowska K, et al. The search for new antigenic targets in myasthenia gravis. Ann NY Acad Sci. 2012;1275:123‐128. [DOI] [PubMed] [Google Scholar]
- 27. Hong Y, Zisimopoulou P, Trakas N, et al. Multiple antibody detection in 'seronegative’ myasthenia gravis patients. Eur J Neurol. 2017;24:844‐850. [DOI] [PubMed] [Google Scholar]
- 28. Li Y, Zhang Y, Cai G, et al. Anti‐LRP4 autoantibodies in Chinese patients with myasthenia gravis. Muscle Nerve. 2017;56:938‐942. [DOI] [PubMed] [Google Scholar]
- 29. Marino M, Scuderi F, Samengo D, et al. Flow cytofluorimetric analysis of anti‐LRP4 (LDL receptor‐related protein 4) autoantibodies in Italian patients with myasthenia gravis. PLoS One. 2015;10:e0135378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez Cruz PM, Huda S, Lopez‐Ruiz P, Vincent A. Use of cell‐based assays in myasthenia gravis and other antibody‐mediated diseases. Exp Neurol. 2015;270:66‐71. [DOI] [PubMed] [Google Scholar]
- 31. Tsivgoulis G, Dervenoulas G, Kokotis P, et al. Double seronegative myasthenia gravis with low density lipoprotein‐4 (LRP4) antibodies presenting with isolated ocular symptoms. J Neurol Sci. 2014;346:328‐330. [DOI] [PubMed] [Google Scholar]
- 32. Zisimopoulou P, Evangelakou P, Tzartos J, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti‐LRP4 in myasthenia gravis. J Autoimmun. 2014;52:139‐145. [DOI] [PubMed] [Google Scholar]
- 33. Zouvelou V, Zisimopoulou P, Rentzos M, et al. Double seronegative myasthenia gravis with anti‐LRP 4 antibodies. Neuromuscul Disord. 2013;23:568‐570. [DOI] [PubMed] [Google Scholar]
- 34. Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Neurology. 2000;55:16‐23. [DOI] [PubMed] [Google Scholar]
- 35. Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker‐Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology. 2005;64:1968‐1970. [DOI] [PubMed] [Google Scholar]
- 36. Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci. 1981;377:670‐677. [DOI] [PubMed] [Google Scholar]
- 37. Howard JF Jr, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti‐acetylcholine receptor antibody‐positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double‐blind, placebo‐controlled, multicentre study. Lancet Neurol. 2017;16:976‐986. [DOI] [PubMed] [Google Scholar]
- 38. Phillips LH 2nd, Torner JC. Epidemiologic evidence for a changing natural history of myasthenia gravis. Neurology. 1996;47:1233‐1238. [DOI] [PubMed] [Google Scholar]
- 39. Rivner MH, Liu S, Quarles B, et al. Agrin and low‐density lipoprotein‐related receptor protein 4 antibodies in amyotrophic lateral sclerosis patients. Muscle Nerve. 2017;55:430‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tzartos JS, Zisimopoulou P, Rentzos M, et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol. 2014;1:80‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]