

Abstract
A novel approach towards the activation of different arenes and purines including caffeine and theophylline is presented. The simple, safe and scalable electrochemical synthesis of 1,1,1,3,3,3‐hexafluoroisopropanol (HFIP) aryl ethers was conducted using an easy electrolysis setup with boron‐doped diamond (BDD) electrodes. Good yields up to 59 % were achieved. Triethylamine was used as a base as it forms a highly conductive media with HFIP, making additional supporting electrolytes superfluous. The synthesis was optimized using Design of Experiment (DoE) techniques giving a detailed insight to the significance of the reaction parameters. The mechanism was investigated by cyclic voltammetry (CV). Subsequent transition metal‐catalyzed as well as metal‐free functionalization led to interesting motifs in excellent yields up to 94 %.
Keywords: anode, boron-doped diamond, caffeine, C−H functionalization, electrolysis
An alternative route for the activation of different arenes including caffeine and theophylline, optimized by design of experiment techniques is presented. We provide a simple electroorganic protocol using BDD electrodes in undivided cells for the synthesis of aryl 1,1,1,3,3,3‐hexafluoroisopropyl ethers. The value of these fluorinated ethers for subsequent functionalization with a variety of nucleophiles has been proved (see scheme).
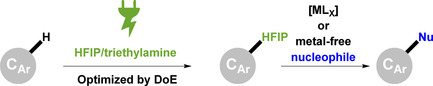
Cross‐coupling reactions represent a very important synthetic tool used for the formation of aryl–carbon or aryl–heteroatom bonds. Substantial efforts have been taken to develop simple and sustainable reactions of this kind, using methods like electrochemistry[1]‐[8] or photoredox catalysis.9 However, transition metal catalysis remains dominant in the field of cross‐coupling reactions,10 despite that often it requires synthesis of precursors to introduce for example, halides or pseudohalides. Furthermore, the costs rise for Rh, Pd or Pt constantly and strongly, which further increases the desire to avoid transition metals in organic synthesis.11 The high selectivity and efficiency of the cross‐coupling reaction itself might be diminished by the lack of selectivity and the use of partly hazardous reagents such as bromine, chlorinating agents, trifluoromethanesulfonic anhydride or tosyl chloride during the pre‐functionalization.12 Besides the risks associated with handling such compounds, they generate stoichiometric amounts of reagent waste. In the case of direct oxidative cross‐coupling reactions, pre‐functionalization is not necessary but stoichiometric amounts of an oxidizer must be used, again resulting in stoichiometric amounts of reagent waste.13 Electro‐organic synthesis, on the other hand, fulfils many of the green chemistry postulates and uses only electrons as an inherently clean reactant, hence minimizing reagent waste to a certain degree.1‐8, 14 Furthermore, it offers safe‐to‐conduct protocols and simple cell setups. Combining the benefits of both worlds we designed an electrochemical protocol for the pre‐functionalization of different aromatic compounds for a subsequent metal‐free or Ni‐ or Pd‐catalyzed cross‐coupling reaction. The electroorganic reactions conducted in simple beaker type cells left many parameters to optimize. Using a simple but not very efficient one‐variable‐at‐a‐time approach (OVAT) does not always lead to satisfying results. Design of Experiment techniques provide high quality information from a comparably low number of experiments.15, 16, 17 In order to make this efficient, an appropriate screening tool is required, providing good quality results with sufficient accuracy.18 In our previous work, benzylic C−H functionalization using HFIP as both solvent and reagent was reported.19‐22 In addition, our group has a long‐standing interest in using HFIP based electrolytes in electro‐organic synthesis, since unique reactivity can be attributed to solvent effects and stabilization of intermediates.5, 23 In the work described here, the scope of the reaction has been successfully expanded to further aromatic compounds using a DoE approach, demonstrating the broad applicability of this method.
The functionalization of xanthine derivatives like caffeine or theophylline is of great interest for the development of pharmaceuticals.24 The examples shown in Scheme 1 are approved drugs used for the treatment of type II diabetes (Linagliptin)25 and Parkinson's disease (Istradefylline)26, 27 or to prevent postoperative vomiting and symptoms of motion sickness (Dimenhydrinate).28, 29 Lei et al. recently demonstrated the electrochemical oxidative functionalization of caffeine.30
Scheme 1.

Xanthine derived pharmaceuticals functionalized in position 8 of the purine scaffold.[25–29]
We present the activation of position 8 of the purine scaffold in caffeine and theophylline, as well as derivatization of naphthalene and aromatic acetamides by installation of the 1,1,1,3,3,3‐hexafluoroisopropoxide moiety (HFIP). Furthermore, the resulting HFIP ethers were amenable to subsequent derivatization by metal‐catalyzed as well as metal‐free nucleophilic substitution reactions. The first electrochemical step is easy to conduct, free from metals, does not require inert conditions and the substrates used are readily available, making this method cost‐efficient, simple and quick (Scheme 2). The screening was conducted in undivided cells made of PTFE equipped with two BDD electrodes. This allows for the parallel operation of 8 independent electrolysis cells. The limited number of electrolysis cells is rewarded by highly accurate electrosynthetic data.18
Scheme 2.
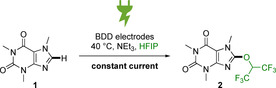
Constant current electrolysis of caffeine. The oxidative peak potentials are 1.80 V for 1 and 1.60 V for 2 vs. Ag/AgCl, respectively (see Supporting Information).
The electrochemical installation of alcohols to arenes involves a major challenge, due to the electron‐releasing properties of the ether moiety. Cyclic voltammetry studies have revealed the mechanism to be of the ECEC type (see Supporting Information) and the products were found to have a lower redox potential than the starting materials. Therefore, over‐oxidation is a significant problem, hence careful optimization of the reaction conditions is needed. The caffeyl HFIP ether synthesis was first optimized in initial screening reactions using an OVAT approach. The isolated yield of 2 was 33 % by these conditions. With the aim of increasing the yield and to obtain detailed information about the importance of the parameters investigated, we turned to a DoE approach and started with a 25–1‐plan with a center point added.15, 17 The yields during the optimization were determined by qNMR using 1,3,5‐trimethoxybenzene as an internal standard. The factors examined and their settings are shown in Table 1.
Table 1.
Factors used in the initial 25–1‐plan.[a]
|
Factor |
− (lower level) |
0 (center point) |
+ (upper level) |
|
|---|---|---|---|---|
|
|
200 |
300 |
400 |
|
|
|
0.15 |
0.20 |
0.25 |
|
|
|
0.10 |
0.15 |
0.20 |
|
|
|
2.00 |
2.25 |
2.50 |
|
|
|
30 |
45 |
60 |
is the stirring rate, and are the concentrations of caffeine and NEt3, is the amount of applied charge and j is the current density.
It was observed that the current density, the stirring rate and the concentration of caffeine were significant for the yield in this area of the experimental space. With the best settings being the low current density, high stirring rate and high concentration. The yield at the center point did not indicate any curvature, so we did not expect to be close to the maximum yet. With the obtained data a second plan was designed with these three significant factors and, considering that the reaction is electrochemically driven, the amount of charge was taken into consideration. A 24–1‐plan was conducted and analyzed. This time the center point did not match the linear model and hence indicated curvature in the yield in this area of the experimental space. Star points were added to convert this plan into a central composite design (CCD).15, 17 From the results it could be seen that a maximum was reached regarding the amount of charge and the current density . The optimal conditions in this area were found using the Response Optimizer in Minitab.
The result shown in Figure 1 indicates that an increase in stirring rate and a decrease in concentration would improve the yield even further. Due to the high stirring rates we experienced a lot of failures, so we used the conditions from this step (conditions b) for all further reactions. This is discussed in more detail in the supporting information. To verify the model, we isolated 2 using these conditions and obtained exactly 42 % yield.
Figure 1.
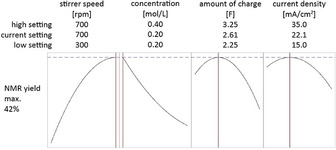
Minitabs Response Optimizer was used to maximize the yield from the model obtained through a CCD plan. The predicted yield was 42 %. The labelling was rearranged for better readability.
Comparing conditions a) and b), significant improvements introduced by the optimization via DoE are apparent. The time needed for the electrolysis dropped to about one third and at the same time, the isolated yield increased by 9 %. The significant influence of the stirring rate on the reaction suggests that convection was crucial. Therefore, the setup was changed to investigate temperature, electrode distance and stirring rate more effectively. With these parameters, a 23‐plan and a subsequent 22‐plan excluding electrode distance (see Supporting Information) was explored. This way we were able to isolate 2 in 45 % yield in a 10 mmol scale. The larger cell setup for these plans demonstrated the scalability of the electrolysis and considering a few parameters during the scale‐up, the yield could even be improved further. Besides using a different batch setup, we tried to bypass the problem of over‐oxidation using a flow setup but the yields obtained could not meet those of the batch electrolyses.31
The scope was extended conducting reactions on a 1.00 to 1.25 mmol scale and both conditions a) and b) (see Table 2) were investigated. Improved results with yields up to 59 % could be achieved (Scheme 3).
Table 2.
Comparison between the results of the optimization processes.
|
|
Conditions a) OVAT optimized |
Conditions b) DoE optimized |
|
|---|---|---|---|
|
|
7.2 |
22.1 |
|
|
|
2 |
2.61 |
|
|
|
300 |
700 |
|
|
|
0.25 |
0.2 |
|
|
|
0.1 |
0.2 |
|
|
electrolysis time |
5 h 10 min |
1 h 45 min |
|
|
product |
0.41 mmol |
0.42 mmol |
|
|
isolated yield |
33 % |
42 % |
Scheme 3.

Scope of the reaction and yields of the isolated products. The conditions working better are displayed.
As shown in previous work, the HFIP moiety can be used as a leaving group.20, 21 We wanted to show that this strategy can also be applied to arenes and therefore various functionalization reactions were conducted (Scheme 4). Cyanides could be installed by transition metal‐catalysis using nickel or palladium in 38 and 60 % yield, respectively. Metal‐free cyanation was not possible in this case. Also, higher yields were achieved in amination reactions with morpholine (11), when Pd was used (94 vs. 75 %). Allylic amine (13) and benzylic amine (12) provided yields up to 76 %. The direct metal‐free reaction with thiophenol (14) and propane‐1‐thiol (16) with 2 gave high yields up to 81 %. Application of the respective oxygen derivatives such as phenol and propan‐1‐ol yielded the desired ethers in up to 15 % yield. When submitting 8 to Kumada‐type couplings only small amounts of desired product could be detected. Other transition‐metal‐catalyzed did not deliver the desired product.
Scheme 4.
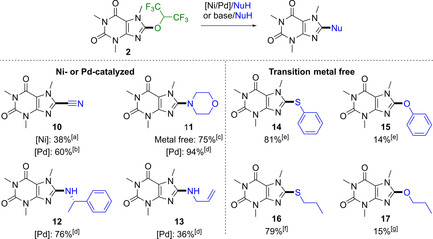
Scope of the reaction of the caffeyl HFIP ether and yields of the isolated products. [a] NiCl2(PPh3)2 (10 mol %), PPh3 (20 mol %), KCN (4 equiv.), Zn (1 equiv.) in DMF 115 °C, 4 h; [b] Pd(OAc)2 (5 mol %), XantPhos (10 mol %), KCN (1.5 equiv), DMF, 85 °C, 14 h; [c] Pd(OAc)2 (5 mol %), XantPhos (10 mol %), amine (2.0–3.0 equiv), DMA, 100 °C, 3–14 h; [d] amine (3.0 equiv), DMA, 100 °C, 14 h; [e] Cs2CO3 (3.0 equiv), phenol/thiophenol (2.0 equiv.), DMF, RT [f] NaOH (15 equiv.) in propan‐1‐ol/water 1/3, 60 °C, 2 h; [g] K2CO3 (3.0 equiv.), propan‐1‐thiol (2.0 equiv.), in DMF, 65 °C, 2 h;.
In conclusion, we expanded the scope of the electroorganic synthesis of aryl HFIP ethers from our previous work to heterocycles. Key for these conversions is the amine‐HFIP electrolyte.19, 20, 21 In addition, the value of these intermediates was demonstrated in the activation within subsequent reactions. A sustainable alternative to common pre‐functionalization using hazardous compounds was presented. A DoE approach led to efficient optimization with mild reaction conditions, and shorter electrolysis times across a range of substrates. The subsequent reactions of the caffeyl HFIP ether gave access to various functionalized caffeine derivatives.
Experimental Section
Detailed information on general procedures, electrolytic conversions and product characterization can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
J. L. Röckl is a recipient of a DFG fellowship through the Excellence Initiative by the Graduate School Materials Science in Mainz (GSC 266). J. Rein is a recipient of an undergraduate Scholarship by the Heinrich Böll Foundation. Support of the Advanced Lab of Electrochemistry and Electrosynthesis—ELYSION (Carl‐Zeiss‐Stiftung) is gratefully acknowledged.
M. Dörr, J. L. Röckl, J. Rein, D. Schollmeyer, S. R. Waldvogel, Chem. Eur. J. 2020, 26, 10195.
References
- 1. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kärkäs M. D., Chem. Soc. Rev. 2018, 47, 5786–5865. [DOI] [PubMed] [Google Scholar]
- 3. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149. [Google Scholar]
- 4. Röckl J. L., Pollok D., Franke R., Waldvogel S. R., Acc. Chem. Res. 2020, 53, 45–61. [DOI] [PubMed] [Google Scholar]
- 5. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706–6765. [DOI] [PubMed] [Google Scholar]
- 6. Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721. [Google Scholar]
- 7. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Y., Lan J., You J., Chem. Rev. 2017, 117, 8787–8863. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 9b. Fukuzumi S., Ohkubo K., Org. Biomol. Chem. 2014, 12, 6059–6071; [DOI] [PubMed] [Google Scholar]
- 9c. Xi Y., Yi H., Lei A., Org. Biomol. Chem. 2013, 11, 2387–2403; [DOI] [PubMed] [Google Scholar]
- 9d. Nicewicz D. A., Nguyen T. M., ACS Catal. 2014, 4, 355–360; [Google Scholar]
- 9e. Hockin B. M., Li C., Robertson N., Zysman-Colman E., Catal. Sci. Technol. 2019, 9, 889–915. [Google Scholar]
- 10.
- 10a. Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062–5085; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150–5174; [Google Scholar]
- 10b. Zeng Q., Zhang L., Zhou Y., Chem. Rec. 2018, 18, 1278–1291. [DOI] [PubMed] [Google Scholar]
- 11.“Precious Metal Trading”, zu finden unter https:/ pm−1-prices.heraeus.com/Heraeus CurrentPrices.aspx, 2020. Zuletzt geprüft am 03.03.2020.
- 12.
- 12a. Wolfe J. P., Wagaw S., Marcoux J.-F., Buchwald S. L., Acc. Chem. Res. 1998, 31, 805–818; [Google Scholar]
- 12b. Hartwig J. F., Acc. Chem. Res. 1998, 31, 852–860; [Google Scholar]
- 12c. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; [Google Scholar]
- 12d. Tamao K., Sumitani K., Kumada M., J. Am. Chem. Soc. 1972, 94, 4374–4376; [Google Scholar]
- 12e. King A. O., Okukado N., Negishi E.-i., J. Chem. Soc. Chem. Commun. 1977, 683; [Google Scholar]
- 12f. Miyaura N., Suzuki A., J. Chem. Soc. Chem. Commun. 1979, 866; [Google Scholar]
- 12g. Hartwig J. F., Pure Appl. Chem. 1999, 71, 1417–1423; [Google Scholar]
- 12h. Miyaura N., Yamada K., Suzuki A., Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar]
- 13.
- 13a. Cheng Y., Wu Y., Tan G., You J., Angew. Chem. Int. Ed. 2016, 55, 12275–12279; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12463–12467; [Google Scholar]
- 13b. Dong J., Long Z., Song F., Wu N., Guo Q., Lan J., You J., Angew. Chem. Int. Ed. 2013, 52, 580–584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 608–612; [Google Scholar]
- 13c. Xi P., Yang F., Qin S., Zhao D., Lan J., Gao G., Hu C., You J., J. Am. Chem. Soc. 2010, 132, 1822–1824. [DOI] [PubMed] [Google Scholar]
- 14. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099. [Google Scholar]
- 15. Lee R., Chem. Ing. Tech. 2019, 91, 191–200. [Google Scholar]
- 16.
- 16a. Möckel R., Hille J., Winterling E., Weidemüller S., Faber T. M., Hilt G., Angew. Chem. Int. Ed. 2018, 57, 442–445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 450–454; [Google Scholar]
- 16b. Murray P. M., Bellany F., Benhamou L., Bučar D.-K., Tabor A. B., Sheppard T. D., Org. Biomol. Chem. 2016, 14, 2373–2384; [DOI] [PubMed] [Google Scholar]
- 16c. Zhang X., Wang R., Yang X., Yu J., Chemom. Intell. Lab. Syst. 2007, 89, 45–50; [Google Scholar]
- 16d. Weissman S. A., Anderson N. G., Org. Process Res. Dev. 2015, 19, 1605–1633; [Google Scholar]
- 16e. Renzi P., Bella M., Synlett 2016, 28, 306–315. [Google Scholar]
- 17. Leardi R., Anal. Chim. Acta 2009, 652, 161–172. [DOI] [PubMed] [Google Scholar]
- 18. Gütz C., Klöckner B., Waldvogel S. R., Org. Process Res. Dev. 2016, 20, 26–32. [Google Scholar]
- 19. Röckl J. L., Hauck A. V., Schollmeyer D., Waldvogel S. R., ChemistryOpen 2019, 8, 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Röckl J. L., Imada Y., Chiba K., Franke R., Waldvogel S. R., ChemElectroChem 2019, 6, 4184–4187. [Google Scholar]
- 21. Imada Y., Röckl J. L., Wiebe A., Gieshoff T., Schollmeyer D., Chiba K., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 12136–12140; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12312–12317. [Google Scholar]
- 22. Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., J. Am. Chem. Soc. 2017, 139, 12317–12324. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Wiebe A., Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 14727–14731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14920–14925; [Google Scholar]
- 23b. Gieshoff T., Schollmeyer D., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 9437–9440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9587–9590; [Google Scholar]
- 23c. Gieshoff T., Kehl A., Schollmeyer D., Moeller K. D., Waldvogel S. R., Chem. Commun. 2017, 53, 2974–2977; [DOI] [PubMed] [Google Scholar]
- 23d. Elsler B., Wiebe A., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Chem. Eur. J. 2015, 21, 12321–12325; [DOI] [PubMed] [Google Scholar]
- 23e. Kehl A., Gieshoff T., Schollmeyer D., Waldvogel S. R., Chem. Eur. J. 2018, 24, 590–593; [DOI] [PubMed] [Google Scholar]
- 23f. Franzmann P., Beil S. B., Schollmeyer D., Waldvogel S. R., Chem. Eur. J. 2019, 25, 1936–1940; [DOI] [PubMed] [Google Scholar]
- 23g. Hollóczki O., Macchieraldo R., Gleede B., Waldvogel S. R., Kirchner B., J. Phys. Chem. Lett. 2019, 10, 1192–1197; [DOI] [PubMed] [Google Scholar]
- 23h. Breising V. M., Gieshoff T., Kehl A., Kilian V., Schollmeyer D., Waldvogel S. R., Org. Lett. 2018, 20, 6785–6788; [DOI] [PubMed] [Google Scholar]
- 23i. Hollóczki O., Berkessel A., Mars J., Mezger M., Wiebe A., Waldvogel S. R., Kirchner B., ACS Catal. 2017, 7, 1846–1852; [Google Scholar]
- 23j. Schulz L., Waldvogel S., Synlett 2019, 30, 275–286; [Google Scholar]
- 23k. Selt M., Mentizi S., Schollmeyer D., Franke R., Waldvogel S. R., Synlett 2019, 30, 2062–2067. [Google Scholar]
- 24. Singh N., Shreshtha A. K., Thakur M. S., Patra S., Heliyon 2018, 4, e00829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forst T., Pfützner A., Expert Opin. Pharmacother. 2012, 13, 101–110. [DOI] [PubMed] [Google Scholar]
- 26. Dungo R., Deeks E. D., Drugs 2013, 73, 875–882. [DOI] [PubMed] [Google Scholar]
- 27. Takahashi M., Fujita M., Asai N., Saki M., Mori A., Expert Opin. Pharmacother. 2018, 19, 1635–1642. [DOI] [PubMed] [Google Scholar]
- 28. Özkan C. K., Taşdemir U., Taş Ç., Savaşer A., Erol H., Özkan Y., Chromatographia 2013, 76, 1521–1525. [Google Scholar]
- 29. Halpert A., Neurosci. Biobehav. Rev. 2002, 26, 61–67. [DOI] [PubMed] [Google Scholar]
- 30. Liang X.-A., Liu D., Sun H., Jiang C., Chen H., Niu L., Mahato K., Abdelilah T., Zhang H., Lei A., Adv. Synth. Catal. 2020, 362, 1138–1143. [Google Scholar]
- 31. Xu F., Qian X.-Y., Li Y.-J., Xu H.-C., Org. Lett. 2017, 19, 6332–6335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary