

Abstract
Background & Aims
Ferroportin disease (FD) and hemochromatosis type 4 (HH4) are associated with variants in the ferroportin‐encoding gene SLC40A1. Both phenotypes are characterized by iron overload despite being caused by distinct variants that either mediate reduced cellular iron export in FD or resistance against hepcidin‐induced inactivation of ferroportin in HH4. The aim of this study was to assess if reduced iron export also confers hepcidin resistance and causes iron overload in FD associated with the R178Q variant.
Methods
The ferroportin disease variants R178Q andA77D and the HH4‐variant C326Y were overexpressed in HEK‐293T cells and subcellular localization was characterized by confocal microscopy and flow cytometry. Iron export and cytosolic ferritin were measured as markers of iron transport and radioligand binding studies were performed. The hepcidin‐ferroportin axis was assessed by ferritin/hepcidin correlation in patients with different iron storage diseases.
Results
In the absence of hepcidin, the R178Q and A77D variants exported less iron when compared to normal and C326Y ferroportin. In the presence of hepcidin, the R178Q and C326Y, but not the A77D‐variant, exported more iron than cells expressing normal ferroportin. Regression analysis of serum hepcidin and ferritin in patients with iron overload are compatible with hepcidin deficiency in HFE hemochromatosis and hepcidin resistance in R178Q FD.
Conclusions
These results support a novel concept that in certain FD variants reduced iron export and hepcidin resistance could be interlinked. Evasion of mutant ferroportin from hepcidin‐mediated regulation could result in uncontrolled iron absorption and iron overload despite reduced transport function.
Keywords: ferroportin disease, hemochromatosis, hepcidin resistance, SLC40A1
Abbreviations
- cpm
counts per minute
- FCS
fetal calf serum
- FD
ferroportin disease
- GFP
green fluorescent protein
- HA
human influenza hemagglutinin
- HCV
hepatitis C virus
- HEK
human embryonic kidney
- HH4
hemochromatosis type 4
- MRI
magnetic resonance imaging
Key Points.
Ferroportin disease is an iron storage disorder caused by inactivating mutations in the iron exporter ferroportin. As iron absorption form the diet, is dependent on normal ferroportin function, it is unclear why such mutations cause iron overload. This study shows that normal ferroportin is more responsive to the natural ferroportin inhibitor hepcidin than mutant ferroportin, which can result in paradoxically increased iron absorption in ferroportin disease.
1. INTRODUCTION
Mutations in SLC40A1 have been identified in patients with hepatic iron overload and are considered the second most common genetic cause of hemochromatosis after the disease‐associated polymorphism C282Y in HFE. 1 , 2 , 3 SLC40A1 mutations can cause two different clinical phenotypes differentiated by transferrin saturation and patterns of iron storage. 4 Mutations leading to reduced iron export function cause ferroportin disease (FD), which presents with low to normal transferrin saturation and splenic and Kupffer cell iron overload, 5 whereas mutations conferring resistance of ferroportin to hepcidin cause haemochromatosis type 4 (HH4). 6 , 7 In the latter, patients present with high transferrin saturation and predominant iron accumulation in the hepatocytes. 8 Although iron overload is a hallmark of both entities it is yet unclear how reduced iron export function in FD leads to systemic iron overload.
Ferroportin is expressed at the basolateral membrane of enterocytes, in macrophages and hepatocytes and its function is crucial for dietary iron uptake and recycling of iron from aged red blood cells. 9 , 10 , 11 , 12 Iron recovered from hemoglobin is taken back to the bone marrow via the blood stream after ferroportin‐mediated export of the metal from macrophages. 12 The concentration of potentially toxic iron in plasma is under homeostatic control by the iron‐hormone hepcidin. 13 , 14 This peptide is produced and secreted from hepatocytes when plasma‐ or hepatic‐iron concentrations increase. 15 Hepcidin then reduces plasma iron and intestinal iron uptake from enterocytes by binding to ferroportin. 16 , 17 Thereby hepcidin can inhibit iron export by direct occlusion of the transporter, 18 ferroportin internalization from the plasma membrane, ubiquitination and degradation. 16
Hepcidin resistance and hepcidin deficiency have similar functional consequences on iron homeostasis, which are: (a increased transferrin saturation, (b) low spleen iron and (c) hepatic iron overload. 19 These are the hallmarks of all hemochromatosis types, including HH4, which can be caused by the C326Y, A69T and R507H variants in SLC40A1. 2
In contrast, a reduced iron export function as previously reported for A77D and R178Q could explain increased iron storage in macrophages and low transferrin saturation, but does not explain total body iron overload. 20 The R178Q mutation has been first identified in in France and Greece where affected patients had FD 21 , 22 , 23 and further characterized in a cohort of 22 patients from six unrelated pedigrees in France, Belgium and Iraq. 24 In vitro experiments also showed a reduced iron export capacity and high serum hepcidin concentration. 24 The clinical presentation of patients with iron overload despite reduced cellular iron export capacity of the mutant transporter and high hepcidin concentrations represents a gap in the understanding of disease biology, because total iron overload is incompatible with reduced iron transport activity, which is essential for intestinal iron absorption. This notion is highlighted by the fact that transgenic animals expressing mutant ferroportin with reduced iron transport activity are iron deficient. 10 , 11
The present study addresses this translational gap in a clinical and molecular study on pathogenic SLC40A1 variants.
2. METHODS
2.1. Expression vector
The full‐length ORF clone pENTR221 (Invitrogen, Lofer, Austria) containing normal human ferroportin (IOH26826) was recombined with the expression vector pcDNA6.2/N‐EmGFP‐DEST (Invitrogen) using the Gateway LR Clonase Kit II (Invitrogen) as indicated by the manufacturer. Before recombining the IOH26826 vector with the expression vector pcDNA6.2/N‐EmGFP‐DEST, the following ferroportin mutations were introduced through site‐directed mutagenesis (SDM) into the ferroportin sequence: R178Q and A77D were used as ‘loss of function’ variants, whereas C326Y as a hepcidin‐resistant control. 6 , 20 , 25 To create a human influenza hemagglutinin (HA) tagged ferroportin, the respective sequence was again inserted through SDM into the expression vector pcDNA6.2/N‐EmGFP‐DEST with a stop codon before the green fluorescent protein (GFP) sequence. SDM was exerted using the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA). The sequence of normal and mutant ferroportin constructs were confirmed by Sanger sequencing.
2.2. Cell culture
HEK (human embryonic kidney) 293T cells (American Type Culture Collection no. 11268; LGC Standards GmbH, Wesel, Germany) were cultivated in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal calf serum (FCS, Invitrogen), penicillin (100 U/mL) and streptomycin (100 μg/mL) and incubated at 37°C in 5% CO2. Ferroportin fused to GFP and/or HA‐tagged ferroportin were transiently overexpressed by transfection of HEK‐293T cells using Effectene (Qiagen, Hilden, Germany), following the manufacturer's instructions. For mock‐treated control, cells were transfected with the expression vector pcDNA6.2/N‐EmGFP‐DEST without ferroportin.
2.3. Confocal microscopy
For confocal microscopy cells were seeded in 0.1% coated 8‐well chambered coverglasses (Nalge Nunc International, Rochester, NY) and transfected as described above. Sixteen hours after transfection, images were acquired on a microlens‐enhanced Nipkow disk‐based confocal system UltraView RS (Perkin Elmer, Wellesley, MA) mounted on an Olympus IX‐70 inverse microscope (Olympus, Nagano, Japan). For image acquisition and processing, the UltraView RS software (Perkin Elmer, Vienna, Austria) was used in combination with a 60 × oil immersion objective.
2.4. 59Fe export assay
For these experiments, 1.6 × 105 HEK‐293T cells were seeded in 12‐well plates and transiently transfected with normal or mutant ferroportin expression vectors. Sixteen hours after transfection, Effectene‐DNA‐complexes were removed and 1 mL fresh medium supplemented with 2.5 × 104 counts per minute (cpm) 59Fe‐holotransferrin (Perkin Elmer, Vienna, Austria). After a 16h‐incubation, cells were washed twice with PBS to remove unbound transferrin and 1 mL fresh medium was added. Six hours later, culture medium was removed and the pellet was collected after two wash cycles with 1mM NaOH to determine iron export and iron internalization by gamma counting (Wizard 2 gamma counter, Perkin Elmer, Vienna, Austria) respectively. Iron export is expressed as a ratio of counts of the supernatant divided by the sum of the counts in the cell pellet and in the supernatant. The results of independent experiments are shown where iron export was calibrated against wild‐type‐transfected HEK‐293T cells and the maximum iron export in cells overexpressing normal ferroportin was set as 100%.
2.5. 125I‐Hepcidin‐25 binding assay
Hepcidin, with an M21Y substitution (Bachem, Bubendorf, Switzerland), was iodinated using the IODOGEN method. Approximately 10 MBq of 125I‐Na (10 µL of a 10 µM NaOH solution pH 10; Perkin Elmer, Vienna, Austria) were added to 50 µL hepcidin (2 µg/µL in H2O with 0.1% TFA) and 100 µL 0.25 M phosphate buffered saline pH 7.4 in a low protein binding Eppendorf cap coated with 150 µg IODOGEN (Sigma‐Aldrich, Vienna, Austria). The solution was allowed to react for 25 minutes at room temperature, and the reaction was terminated by transfer of the solution to another Eppendorf cap. Labelling success was analysed via HPLC (Dionex Ultimate 3000 UHPLC system with manual injection port, Thermo Fischer Scientific, Vienna, Austria) using an acetonitrile (ACN)/water/0.1% TFA gradient (20% to 40% ACN in 20 minutes) and a Jupiter C‐18 polymeric reversed‐phase column (5 µm, 300 Å, 150 x 4,6 mm; Phenomenex, Aschaffenburg, Germany) with a flow rate of 1,5 mL/min. Radiodetection was carried out with a Gabi Star radiometric detector (Raytest, Straubenhardt, Germany). Purification of the peptide was performed with the same HPLC system. Therefore, 120 µL of the reaction mixture was passed through the HPLC and peaks were manually collected. The peak with the highest activity (1.35 MBq) correlates with the radiolabelled hepcidin. Radiochemical purity was >95%.
HEK‐293T cells were seeded and transfected as described above. Sixteen hours after transfection, Effectene‐DNA‐complexes were removed and cells were incubated with 1 mL fresh antibiotic‐free medium with approximately 2.0 × 104 cpm 125I‐hepcidin and different concentrations of non‐labelled hepcidin (1, 10, 100 or 1000 nM). Two hours later, the medium was removed and the wells were washed twice with antibiotic‐free medium. The collected media were gamma counted for the non‐bound radioligand. Next, cells were incubated twice for 5 minutes with an ice‐cold glycine buffer (pH 2.8) in order to remove any non‐specifically membrane‐bound ligand. Finally, cells were lysed and collected after two wash cycles with 1N NaOH to determine internalized radiolabelled hepcidin. Measurements were performed on a Wizard 2 gamma counter (Perkin Elmer, Vienna, Austria). The results of independent experiments are shown, where maximal 125I uptake was observed in cells overexpressing normal ferroportin and set at 100%.
2.6. Flow cytometry and intracellular ferritin quantification
The cells were seeded and transfected as described above. Untransfected cells were included as a control. After 16 hours, the Effectene‐DNA‐complexes were removed and 1 mL fresh serum‐ and antibiotic‐free medium supplemented with 2 mg/mL holotransferrin (BBI Solutions, Crumlin, UK) to prevent cell death due to iron depletion. At this time point, cells were also incubated either with or without hepcidin, in different concentrations (10, 100 and 1000 nM), for 4 or 6h depending on the experiments and then harvested by incubation with Cell Dissociation Solution (Sigma, Vienna, Austria) at room temperature for 10 minutes. For direct cell sorting, the cells were washed three times with a flow cytometry solution (PBS + 0,2M EDTA + 0.5% FCS). For immunostaining, cells were additionally incubated with the PE/Dazzle 594 anti‐HA 11 epitope tag antibody (BioLegend, San Diego, CA, USA) in a concentration of 5µL/106 cells for 30 minutes at 4°C in dark conditions, as indicated by the manufacturer. In this case, cells were again washed three times with the previously described solution.
Cell sorting was then carried out on a CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA, USA). GFP fluorescence was measured using the 488 nm laser and the 525/40 filter, whereas PE/Dazzle fluorescence was measured with the 561 nm laser and the 585/42 filter. Raw data were analysed using the CytExpert 2.2 analysis software (Beckman Coulter, Brea, CA, USA). The number of fluorescent cells was counted and hepcidin effect was expressed as a relative reduction upon incubation with the hormone. The means and standard deviations from independent experiments (100.000 counts each) are shown.
Intracellular ferritin quantification was carried out using the Human Ferritin ELISA Kit (Abnova, Taipei, Taiwan).
2.7. Structural modelling
To study the potential location of the R178Q within the proposed structure of human ferroportin, the primary sequence of the human ferroportin protein (Q9NP59.1) was aligned with the sequence of the homologue iron transporter of the bacterium Bdellovibrio bacteriovorus (BbFPN, PDB: 5AYO) using the multiple sequence alignment tool MUSCLE (Figure S3). 26 This alignment showed that R178 in the human sequence corresponds to R159 in BbFPN. The published BbFPN structure was visualized in CCP4 Molecular Graphics. 27
2.8. Patient cohort for hepcidin/ferritin correlation data
A cohort of unselected patients referred to the outpatient liver clinic at the University Hospital of Innsbruck from 1 September 2007 to 30 November 2017 for the evaluation of hyperferritinemia was investigated by MRI (magnetic resonance imaging) susceptometry using iron‐sensitive sequences (R2*). High serum ferritin was defined as ≥200 μg/l for women and ≥300 μg/l for men. Of the 410 patients who met these inclusion criteria, 32 patients were homozygous for p.Cys282Tyr in HFE and had sera available for hepcidin determination using the DRG Hepcidin 25 ELISA kit according to the manufacturer's instructions (DRG Instruments GmbH, Marburg, Germany). Of all patients with a normal HFE genotype (p.Cys282 and p.His63) an increased hepatic iron concentration on R2* MRI (defined as R2* ≥ 70 sec−1 28 , 29 ) and serum for hepcidin determination was available in 40 patients. Hepcidin was also determined in archival serum specimens of the index patient heterozygous for R178Q (ENST00000261024: c.533G>A/ ENSP00000261024.2 p.Arg178Gln) in SLC40A1. Correlation analysis between serum ferritin and serum hepcidin was independently carried out in both patient cohorts and the serum samples from the index case after log10 transformation of serum ferritin concentration. Regression lines and 95%‐confidence intervals were calculated independently for all three groups using the software GraphPad Prism (GraphPad Software, Inc Version 8.0.1, La Jolla, CA, USA).
The study was approved by the local ethics research committee of the Medical University of Innsbruck (internal registration number UN5093).
3. RESULTS
To investigate the mechanism how recessive ‘loss of function’ mutations cause the phenotypic spectrum of hepatic iron overload associated with SLC40A1 variants, functional studies in cells overexpressing different ferroportin variants were conducted. As shown in Figure 1A, cells expressing normal ferroportin export 8‐10‐fold more 59Fe than untransfected or mock‐transfected cells. In contrast, in cells expressing A77D or R178Q mutant ferroportin iron export is reduced when compared with normal ferroportin. By comparison, the iron transport capacity of the C326Y mutant ferroportin is similar to normal ferroportin. These results confirm that the A77D and R178Q but not the C326Y variant show reduced iron export capacity to a different extent.
Figure 1.
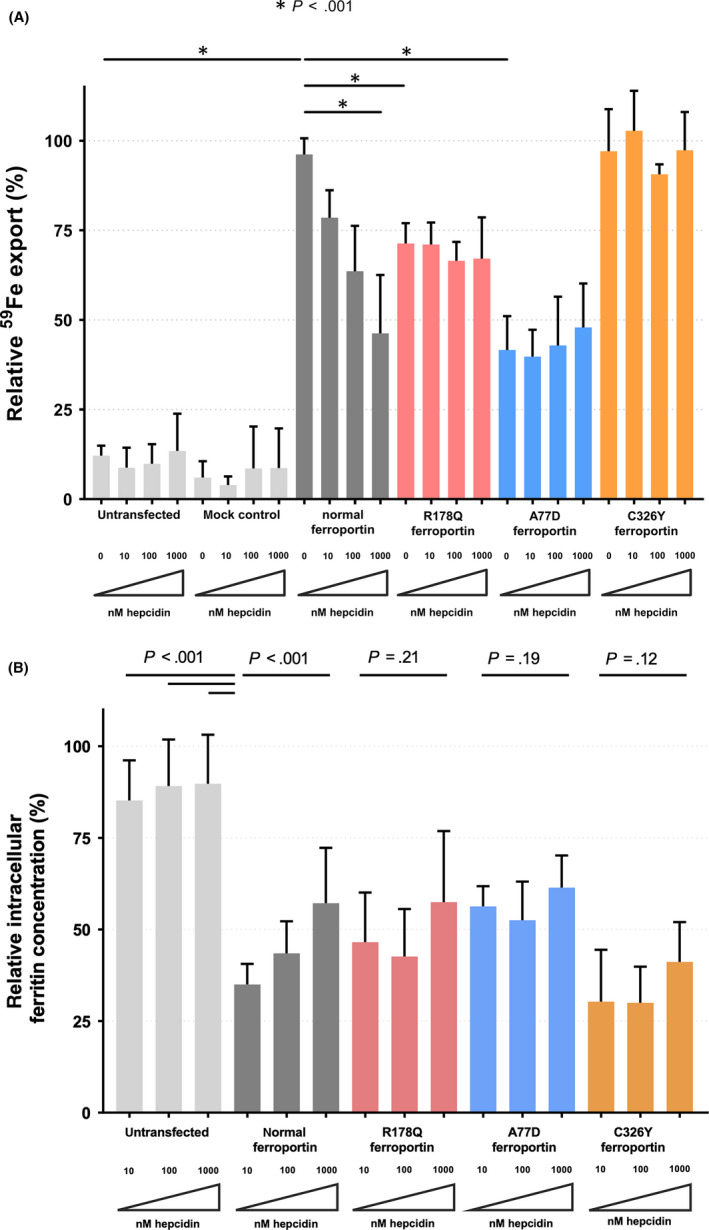
Hepcidin‐dose dependent 59Fe export. (A) 59Fe export within 6 hours is expressed as a fractional of total 59Fe uptake and relative to the maximal iron exported from cells overexpressing normal ferroportin. Mean and standard deviation are represented. The mock‐treated control cells were transfected with a backbone plasmid with GFP but without ferroportin as described in the methods. The overexpression of normal ferroportin caused a 8‐10 fold increase 59Fe export compared to untransfected or mock‐transfected cells. Without incubation with hepcidin, cells overexpressing R178Q and A77D ferroportin exported significantly less iron than normal and C326Y ferroportin expressing cells. Upon incubation with hepcidin, a dose‐dependent decrease on iron export could be observed only in cells expressing normal ferroportin. However, iron export remained similar to baseline in cells expressing R178Q, A77D and C326Y ferroportin, showing therefore resistance to hepcidin (n = 6). (B) Hepcidin‐dose‐dependent intracellular ferritin concentration after 6h incubation. Intracelullar ferritin concentration (ng/mL) is expressed in relation to total protein concentration (mg/mL). Mean and standard deviation are represented. All cells were incubated with 2 mg/mL holotransferrin as described previously. As expected, untransfected cells with holotransferrin showed the highest values and no notable difference under increasing hepcidin concentrations. Cells overexpressing normal ferroportin showed a dose‐dependent increase of intracellular ferritin as an indirect sign of reduced iron export upon hepcidin‐dependent ferroportin deactivation. No such increase could be observed in cells overexpressing the R178Q, A77D and C326Y ferroportin variants (n = 9). Raw p‐values from group comparisons by Student's t‐test are displayed
Next, the effect of hepcidin on different ferroportin variants was tested. A dose‐dependent decrease of iron export was found only in cells overexpressing normal ferroportin. In contrast, iron export was less inhibited by hepcidin in cells overexpressing either of the ferroportin variants tested. Only at high hepcidin concentrations the ferroportin variant R178Q exports more iron than cells expressing normal ferroportin (Figure 1A).
The effect of ferroportin overexpression on cellular iron status was further investigated by measuring intracellular ferritin. As shown in Figure 1B and Figure S1, overexpression of ferroportin was associated with a decrease in intracellular ferritin. Again, intracellular ferritin concentration increased in response to hepcidin in a dose‐dependent manner only in cells overexpressing normal ferroportin. Intracellular ferritin as surrogate of iron retention remained unchanged in response to hepcidin when cells overexpressed either of the ferroportin variants tested (Figure 1B). Although intracellular ferritin measurements showed an overall high variation and so differences did not reach statistical significance, comparing these results with iron release studies (Figure 1A) a reciprocal relationship was found, which confirms a dose‐dependent hepcidin response of normal ferroportin, which is absent in mutant ferroportin.
To analyse if hepcidin resistance was a result of reduced binding of the peptide hormone to ferroportin, competitive radioligand‐binding studies were employed. As shown in Figure 2, hepcidin bound to normal ferroportin with an apparent affinity of 88.4 nM. The apparent binding affinity was reduced to 444.2 nM when cells expressed R178Q mutant ferroportin. In contrast hepcidin‐binding to cells expressing ferroportin harbouring the A77D or the C326Y mutations could not be specifically blocked with increasing doses of unlabelled hepcidin (Figure 2).
Figure 2.
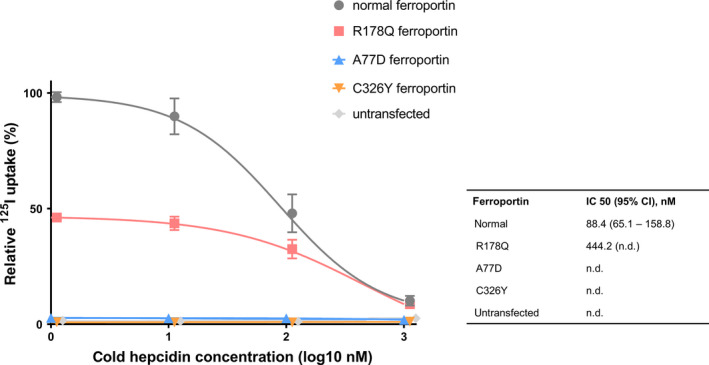
Relative 125I uptake. 125I‐Hepcidin uptake within 2 hours of incubation with 1, 10, 100 or 1000 nM non‐labelled hepcidin is expressed in relation to the maximal 125I uptake observed in cells overexpressing normal ferroportin. Mean and standard deviation are represented and non‐labelled hepcidin concentrations are shown in a logarithmized axis. Untransfected cells and cells overexpressing the A77D and C326Y ferroportin mutants showed only minimal background (non‐specific) uptake of radioactivity, whereas cells transfected with normal and R178Q ferroportin fitted to a competitor‐response curve. The apparent affinity (IC 50) of hepcidin to normal ferroportin was 88.4 nM and much reduced (444.2 nM) in cells expressing the R178Q variant (n = 6)
To test if this difference in hepcidin‐binding was due to altered subcellular localization of ferroportin, cells were studied through confocal microscopy. As shown in Figure S2, the ferroportin‐GFP fusion protein localized to the plasma membrane in cells overexpressing either the normal ferroportin or any of the three variants.
As confocal microscopy showed that only a fraction of overexpressed ferroportin localized to the plasma membrane, surface ferroportin and total ferroportin were independently quantified by flow cytometry of cells co‐transfected with vectors encoding two differently tagged ferroportins. To quantify total ferroportin, a GFP‐fusion protein was used, whereas for cell surface quantification a HA‐tag was used for immunostaining. As shown in Figures 3A,1, 2, 3% of transfected cells expressed ferroportin at the cell surface, although 23%‐26% of transfected cells were GFP‐positive. This model allows differential analysis of ferroportin internalization and degradation. As shown in Figure 3B, internalization and degradation are regulated in lock step in response to hepcidin when cells overexpress normal ferroportin. In contrast, R178Q and A77D overexpressing cells exhibited differential hepcidin response with respect to internalization and degradation of ferroportin. In both variants, degradation of ferroportin‐GFP is apparently higher than internalization of HA‐tagged ferroportin. These findings are in line with the results shown in Figure 1, where all mutant ferroportins tested show some degree of hepcidin resistance.
Figure 3.
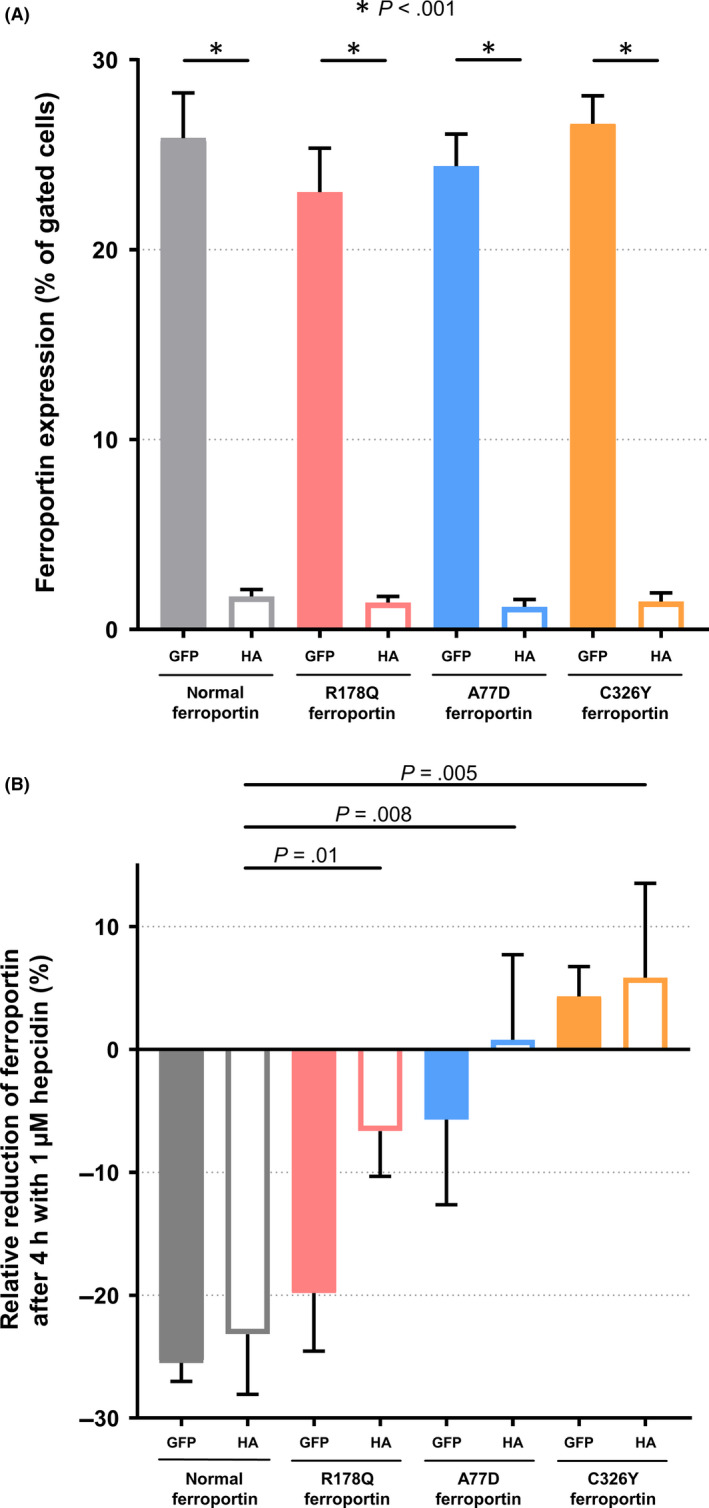
Flow cytometry studies. Cells were co‐transfected with a plasmid encoding for a ferroportin‐GFP fusion protein and simultaneously with another vector encoding a HA‐tagged ferroportin, which upon immunostaining allowed cell surface quantification. (A) Ferroportin expression levels as a percentage of gated cells expressing normal and the different ferroportin mutants. Although 23%‐26% of transfected cells expressed the ferroportin‐GFP fusion protein, only 1%‐3% did actually express ferroportin at the cell surface (n = 9). (B) Time‐dependent downregulation of ferroportin by hepcidin. The relative reduction was calculated after a 4h incubation with 1µM hepcidin. Mean and standard deviation are represented. In cells overexpressing normal ferroportin, incubation with hepcidin lead to a 26% reduction of cells expressing ferroportin‐GFP and 23% of cells expressing HA‐tagged ferroportin. In contrast, in cells overexpressing the C326Y ferroportin variant, an increase of 4 and 6% could be measured in cells expressing the GFP fusion and the HA‐tagged ferroportins respectively. R178Q and A77D overexpressing cells showed a differential hepcidin response to internalization (HA‐tagged ferroportin) and degradation (ferroportin‐GFP fusion protein) with higher degradation than internalization (n = 9)
Taken together, in vitro studies show that all pathogenic ferroportin variants tested in this study cause hepcidin resistance, as potential cause of iron overload. The impairment of iron export function in the absence of hepcidin is variable, where the R178Q mutation causes a milder impairment of iron export than A77D. In accord with the molecular phenotype of R178Q causing reduced iron export function, patients with this mutation were invariably reported with normal transferrin saturation, but marked hyperferritinemia. 24
To further refine description of the FD phenotype caused by the R178Q mutation, clinical, biochemical and radiological findings from a female patient heterozygous for R178Q in SLC40A1 were assessed. At the age of 63 years, the patient presented with hyperferritinemia (1149 µg/L, reference range 30‐200 µg/L) and normal transferrin saturation. Sixteen units of blood (450 mL each) were removed over a period of 12 months (Figure 4A), corresponding to ~ 4 g of mobilizable iron. Five years later, ferritin serum levels had returned to 647 µg/L and, at this point in time, non‐invasive iron quantification by MRI showed a normal hepatic concentration of 20 µmol/g (reference range <36 µmol/g 30 ) but an increased spleen iron concentration according to a T2* value of 8 ms (reference range 20‐80 ms, 29 Figure 4B). Although the patient was positive for hepatitis C virus (HCV) RNA, transaminases were normal and non‐invasive fibrosis markers, Fibroscan and MR‐elastography did not show evidence of hepatic fibrosis. During follow‐up, ferritin concentrations remained significantly elevated (range 540 ‐ 786 µg/L) but transferrin saturation was normal (14%‐45%). Hepcidin was first determined at the age of 72 years and found to be elevated (86 µg/L, reference range 1.5‐41.5 µg/L). At that age, the patient was cured from chronic HCV infection with a 3 month‐course of ritonavir‐boostered paritaprevir, ombitasvir and dasabuvir. This was associated with a rapid decline in serum ferritin and serum hepcidin. (Figure 4A). Accordingly, liver iron decreased at the end of treatment on MRI, which also showed virtually no change in spleen iron concentration despite successful HCV clearance (Figure 4B).
Figure 4.
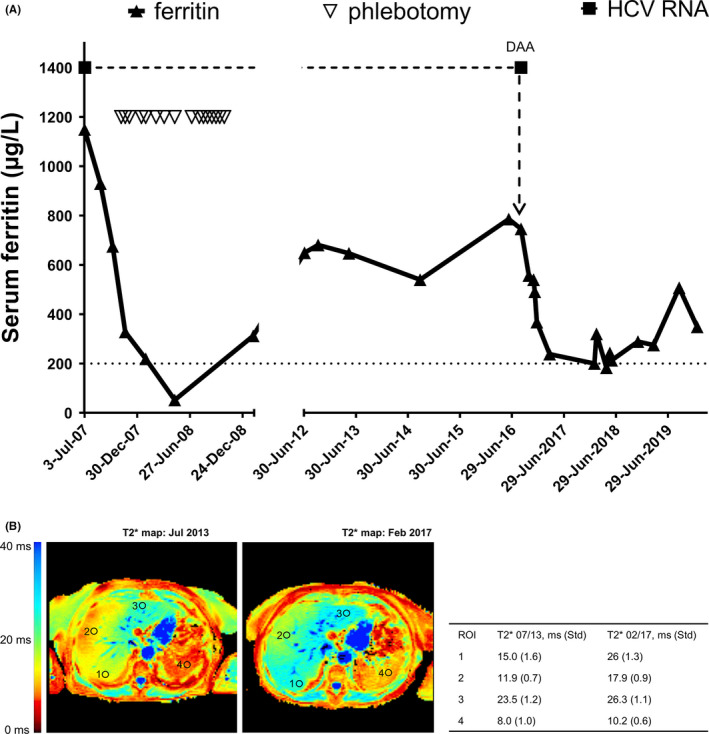
(A) Serum ferritin concentration over 10 years since first observation of hyperferritinemia (July 2007) until last contact on January 2020 in a patient with R178Q variant FD. The time axis is broken, as between October 2008 and June 2012 there is no clinical record. The black upward pointing triangles represent the serum ferritin values expressed in µg/L; the upper cut‐off (200 µg/L) of the normal range is signalized with a dotted line. The white downward pointing triangles represent the 16 phlebotomies (450 mL each) which the patient underwent between November 2007 and October 2008. As shown, ferritin values descent from an initial value of 1149 µg/L, until normal values (52 µg/L) after five venesections. In June 2012, the patient presented again with a high serum ferritin concentration (649 µg/L). After a new increase, the patient underwent a 12‐week therapy with Ombitasvir/Paritaprevir/Ritonavir and Dasabuvir (start with direct‐acting antivirals – DAA – is also marked in the graphic), and had a rapid virological response. Sustained virological response at week 12 after therapy was also achieved. As shown, ferritin levels lowered under and after therapy for chronic HCV infection. (B) Left panel – MRI T2* scan before DAA treatment showing severe splenic iron accumulation (T2* = 8.0 ms, reference range 20 ‐ 80 ms) with normal liver iron concentration (20 µmol/g, reference range 1.5 ‐ 41.5 µmol/g). Right panel – MRI T2* scan after DAA treatment showing less marked iron accumulation in the spleen (T2* = 10.2 ms) and still normal hepatic iron concentration (14 µmol/g)
When regression analysis between log(ferritin) and hepcidin was used as a surrogate of hepcidin regulation in response to iron stores, the R178Q heterozygous patient had an exaggerated response in comparison to a cohort of HFE‐hemochromatosis patients and a cohort of patients with proven high liver iron concentration but normal HFE genotype (Figure 5). This response pattern could per se be interpreted as an indicator of hepcidin resistance in FD.
Figure 5.
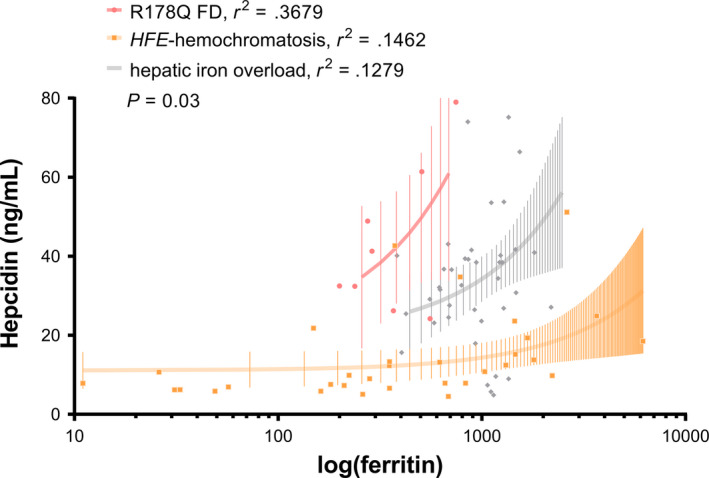
Regression analysis of ferritin and hepcidin. When compared to a control cohort of 32 patients with HFE‐hemochromatosis (homozygosity for the C282Y polymorphism and wild‐type for the H63D polymorphism) and a cohort of 40 patients with normal HFE genotype (wild‐type for both HFE polymorphisms C282Y and H63D) and proven high iron concentration through MR T2*, our patient with R178Q‐associated FD showed a much steeper correlation between serum ferritin and hepcidin concentrations
4. DISCUSSION
In search for the cause of iron overload in patients with FD associated with ‘loss of function’ variants in SLC40A1, A77D and R178Q ferroportin variants were overexpressed and functionally characterized. Cellular studies confirmed the reduced iron export function for both variants. As ferroportin is essential for the transfer of iron across the basolateral membrane of absorptive duodenal enterocytes, its impaired function would not be predicted to result in systemic iron overload. In contrast, variants in ferroportin that cause hepcidin resistance are known to cause iron overload similar to hemochromatosis by constitutively active ferroportin despite high hepcidin. 6 , 7 As shown in detailed hepcidin dose‐response studies reported in this manuscript, the ‘loss of function’ ferroportin variants A77D and R178Q are, however, resistant to inactivation by hepcidin. A previous study has also shown that D181V mutant ferroportin poorly responds to hepcidin, 31 but the present report also shows that, in the presence of high hepcidin concentrations, the ‘loss of function’ variant R178Q exports not less iron than normal ferroportin. This finding provides a potential additional explanation why patients with FD develop a spectrum of iron overload phenotype. A major limitation of this study certainly is that only A77D and R178Q were studied, where R178Q but not A77D exhibited a mixed phenotype of hepcidin resistance and loss of function. Functional studies of other pathogenic SLC40A1 variants will be needed to test if this mixed functional phenotype is a more general molecular pathway in the spectrum of FD.
An important alternative hypothesis to explain macrophage iron overload in FD was recently proposed in a study showing that high iron ‘turnover’ in macrophages leads to failure of ferroportin trafficking to the cell membrane and could account for iron accumulation in these cells.. 32 Findings presented herein are of particular interest in the context of enterocyte function, because inappropriate dietary iron absorption is necessary to develop systemic iron overload. 33 Impaired iron export function by ferroportin can cause iron retention in recycling macrophages, but without compensatory increase in iron absorption, reduced iron recycling alone would result in low transferrin saturation, iron restricted erythropoiesis and anaemia. Although low transferrin saturation is present in some patients with FD, previous studies have shown that the concentration of circulating hepcidin is increased. 1 , 19 , 34 Considering that the rate of intestinal iron absorption is controlled by hepcidin, these findings would be expected to aggravate low plasma iron, reduced transferrin saturation and impaired erythropoiesis. However, most patients with FD present with normal haemoglobin, normal mean corpuscular volume and total body iron overload. 2 Recent data from iron supplementation studies in humans would be in a line with different effects of hepcidin on macrophage iron retention versus enterocyte iron absorption, as those studies demonstrate that inflammation induced hepcidin expression resulted in macrophage iron retention but had only little effects on duodenal iron absorption. 35
The common cause of primary iron overload is uncontrolled intestinal iron absorption, where iron repletion fails to inhibit further duodenal iron uptake. In hemochromatosis this is thought to result from impaired iron sensing and hepcidin production. In HH4, hepcidin resistance has the same net effect as hepcidin deficiency. The physiological relation between iron status and hepcidin can be illustrated with a linear regression between log(ferritin) and hepcidin concentrations. The shallow slope of the regression line in a cohort of HFE‐hemochromatosis patients indicates relative hepcidin deficiency, whereas the steeper than normal slope in our patient with R178‐associated FD suggests an exaggerated hepcidin response compatible with hepcidin resistance. This cross‐sectional approach is only a proxy for investigating hepcidin response in individual patients, which would be best assessed by oral iron tolerance testing and has been previously reported. 36 Limited insight into the dynamics of iron‐dependent hepcidin response in R178Q FD can be gained from the longitudinal study reported here, which suggests that R178Q FD could reflect the changes in iron metabolism seen in dysmetabolic iron overload syndrome. 37
One question would be why apparent hepcidin resistance has not been identified in previous studies as a potential cause of iron overload FD. A potential explanation for this is that most studies have used overexpression of ferroportin fused to fluorescent tags. With this method, total cellular ferroportin rather than cell surface ferroportin is measured. The results presented in this study could be interpreted as an indicator of only a small proportion of ferroportin reaching the plasma membrane. As binding of hepcidin to ferroportin induces its internalization and degradation, these processes were considered to be regulated in lock step. However, our results further indicate that this notion may not apply to the ‘loss of function’ variants tested in this study, R178Q and A77D. Hepcidin paradoxically induces more degradation of total ferroportin than internalization from the plasma membrane for both of these variants. A potential explanation for this finding would be that R178Q, which was previously reported to affect a gating residue, 24 results in a failure to bind iron in the inward open conformation. This notion is supported by the structural model presented in Figure 4. Iron binding induces the conformational change of ferroportin to the outward open structure, which opens the ferroportin binding site. 38 , 39 Homology modelling indeed showed that the Arginine residue corresponding to human R178 is highly conserved among species (Figure S3) and faces the cavity in the ‘inward‐open’ confirmation of ferroportin (Figure S4).
The R178Q variant is therefore expected to have a lower apparent hepcidin affinity, as shown in this manuscript. Reduced affinity and consequent partial hepcidin resistance can be overcome by high hepcidin concentrations frequently used in vitro. This provides a potential explanation why partial hepcidin resistance could have been missed in previous studies. This could also account for supra‐physiological activation of downstream‐signalling and consecutive degradation of intracellular ferroportin independent of its internalization from the plasma membrane. Consulting patients with iron storage disease includes assessment of liver fibrosis risk. Patients with HFE‐hemochromatosis are known to be at higher risk for fibrosis and development of cirrhosis, which is the basis for the recommendation to biopsy patients with ferritin >1000 µg/L, elevated transaminases and hepatomegaly. In contrast, little is known about the risk of fibrosis progression in FD and HH4. The finding that a patient with a pathogenic SLC40A1 variant and chronic HCV infection presented without any evidence of fibrosis at the age of 63 years, supports the notion that FD patients may have a low risk of progressive fibrosis.
In conclusion, comprehensive analysis of ferroportin function and its response to hepcidin at different concentrations suggest that in patients heterozygous for the R178Q variant subtle effects on hepcidin‐ferroportin interaction could contribute to the spectrum of iron overload phenotypes in FD as a consequence of partial hepcidin resistance. Concurrent impairment in the iron export function of the R178Q variant ferroportin results in mild iron overload of questionable clinical relevance and low risk of progressive fibrosis.
CONFLICT OF INTEREST
The authors disclose no conflicts of interest in relation to this work.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Acknowlegements
The authors thank Elizabeta Nemeth and Tomas Ganz and for providing a detailed protocol and support for radiolabeling of hepcidin.
Viveiros A, Panzer M, Baumgartner N, et al. Reduced iron export associated with hepcidin resistance can explain the iron overload spectrum in ferroportin disease. Liver Int. 2020;40:1941–1951. 10.1111/liv.14539
Funding information
André Viveiros and Benedikt Schaefer received a grant from the “Verein zur Foerderung der Wissenschaft in Gastroenterologie & Hepatologie.”
Handling Editor: Luca Valent
REFERENCES
- 1. Mayr R, Griffiths WJH, Hermann M, et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology. 2011;140(7):2056‐2063.e1. [DOI] [PubMed] [Google Scholar]
- 2. Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: a systematic meta‐analysis of clinical and molecular findings. J Hepatol. 2010;53:941‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology. 2007;46:1291‐1301. [DOI] [PubMed] [Google Scholar]
- 4. Viveiros A, Schaefer B, Tilg H, et al. Iron Matryoshka‐Haemochromatosis nested in Ferroportin Disease? Liver Int. 2019;39:1014‐1015. [DOI] [PubMed] [Google Scholar]
- 5. Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131‐138. [DOI] [PubMed] [Google Scholar]
- 6. Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosis‐associated mutations of ferroportin. Blood. 2005;106:1092‐1097. [DOI] [PubMed] [Google Scholar]
- 7. Fernandes A, Preza GC, Phung Y, et al. The molecular basis of hepcidin‐resistant hereditary hemochromatosis. Blood. 2009;114:437‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brissot P, Pietrangelo A, Adams PC, et al. Haemochromatosis. Nat Rev Dis Primers. 2018;4:18016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abboud S, Haile DJ. A novel mammalian iron‐regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906‐19912. [DOI] [PubMed] [Google Scholar]
- 10. Donovan A, Brownlie A, Zhou YI, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776‐781. [DOI] [PubMed] [Google Scholar]
- 11. Fraenkel PG, Traver D, Donovan A, et al. Ferroportin1 is required for normal iron cycling in zebrafish. J Clin Invest. 2005;115:1532‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron‐regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299‐309. [DOI] [PubMed] [Google Scholar]
- 13. Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806‐7810. [DOI] [PubMed] [Google Scholar]
- 14. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783‐788. [DOI] [PubMed] [Google Scholar]
- 15. Lou D‐Q, Lesbordes J‐C, Nicolas Gal, et al. Iron‐ and inflammation‐induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo. Hepatology. 2005;41:1056‐1064. [DOI] [PubMed] [Google Scholar]
- 16. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090‐2093. [DOI] [PubMed] [Google Scholar]
- 17. Theurl I, Aigner E, Theurl M, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood. 2009;113:5277‐5286. [DOI] [PubMed] [Google Scholar]
- 18. Aschemeyer S, Qiao BO, Stefanova D, et al. Structure‐function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 2018;131:899‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105:4103‐4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schimanski LM, Drakesmith H, Merryweather‐Clarke AT, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis‐associated FPN mutations. Blood. 2005;105:4096‐4102. [DOI] [PubMed] [Google Scholar]
- 21. Cunat S, Giansily‐Blaizot M, Bismuth M, et al. Global sequencing approach for characterizing the molecular background of hereditary iron disorders. Clin Chem. 2007;53:2060‐2069. [DOI] [PubMed] [Google Scholar]
- 22. Speletas M, Kioumi A, Loules G, et al. Analysis of SLC40A1 gene at the mRNA level reveals rapidly the causative mutations in patients with hereditary hemochromatosis type IV. Blood Cells Mol Dis. 2008;40:353‐359. [DOI] [PubMed] [Google Scholar]
- 23. Speletas M, Onoufriadis E, Kioumi A, et al. SLC40A1‐R178G mutation and ferroportin disease. J Hepatol. 2011;55(3):730‐731. author reply 731–2. [DOI] [PubMed] [Google Scholar]
- 24. Ka C, Guellec J, Pepermans X, et al. The SLC40A1 R178Q mutation is a recurrent cause of hemochromatosis and is associated with a novel pathogenic mechanism. Haematologica. 2018;103:1796‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Domenico I, Nemeth E, Nelson JM, et al. The hepcidin‐binding site on ferroportin is evolutionarily conserved. Cell Metab. 2008;8:146‐156. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McNicholas S, Potterton E, Wilson KS, et al. Presenting your structures: the CCP4mg molecular‐graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pepe A, Lombardi M, Positano V, et al. Evaluation of the efficacy of oral deferiprone in beta‐thalassemia major by multislice multiecho T2*. Eur J Haematol. 2006;76:183‐192. [DOI] [PubMed] [Google Scholar]
- 29. Schwenzer NF, Machann J, Haap MM, et al. T2* relaxometry in liver, pancreas, and spleen in a healthy cohort of one hundred twenty‐nine subjects‐correlation with age, gender, and serum ferritin. Invest Radiol. 2008;43:854‐860. [DOI] [PubMed] [Google Scholar]
- 30. Henninger B, Alustiza J, Garbowski M, et al. Practical guide to quantification of hepatic iron with MRI. Eur Radiol. 2020;30:383‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Praschberger R, Schranz M, Griffiths WJH, et al. Impact of D181V and A69T on the function of ferroportin as an iron export pump and hepcidin receptor. Biochim Biophys Acta. 2014;1842:1406‐1412. [DOI] [PubMed] [Google Scholar]
- 32. Sabelli M, Montosi G, Garuti C, et al. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology. 2017;65:1512‐1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zoller H, Theurl I, Koch RO, et al. Duodenal cytochrome b and hephaestin expression in patients with iron deficiency and hemochromatosis. Gastroenterology. 2003;125:746‐754. [DOI] [PubMed] [Google Scholar]
- 34. Zoller H, McFarlane I, Theurl I, et al. Primary iron overload with inappropriate hepcidin expression in V162del ferroportin disease. Hepatology. 2005;42:466‐472. [DOI] [PubMed] [Google Scholar]
- 35. Stoffel NU, Lazrak M, Bellitir S, et al. The opposing effects of acute inflammation and iron deficiency anemia on serum hepcidin and iron absorption in young women. Haematologica. 2019;104:1143‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Girelli D, Trombini P, Busti F, et al. A time course of hepcidin response to iron challenge in patients with HFE and TFR2 hemochromatosis. Haematologica. 2011;96:500‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rametta R, Dongiovanni P, Pelusi S, et al. Hepcidin resistance in dysmetabolic iron overload. Liver Int. 2016;36:1540‐1548. [DOI] [PubMed] [Google Scholar]
- 38. Deshpande CN, Ruwe TA, Shawki A, et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat Commun. 2018;9:3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taniguchi R, Kato HE, Font J, et al. Outward‐ and inward‐facing structures of a putative bacterial transition‐metal transporter with homology to ferroportin. Nat Commun. 2015;6:8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material