

Abstract
Monocyte‐derived macrophages are the major innate immune cells that provide the first line of cellular defense against infections or injuries. These recruited macrophages at the site of inflammation are exposed to a broad range of cytokines that categorically incite a robust pro‐inflammatory response. However, macrophage pro‐inflammatory activation must be under exquisite control to avert unbridled inflammation. Thus, endogenous mechanisms must exist that rigorously preserve macrophage quiescence and yet, allow nimble pro‐inflammatory macrophage response with precise spatiotemporal control. Herein, we identify the CBP/p300‐interacting transactivator with glutamic acid/aspartic acid‐rich carboxyl‐terminal domain 2 (CITED2) as a critical intrinsic negative regulator of inflammation, which broadly attenuates pro‐inflammatory gene programs in macrophages. Our in vivo studies revealed that myeloid‐CITED2 deficiency significantly heightened macrophages and neutrophils recruitment to the site of inflammation. Our integrated transcriptomics and gene set enrichment analysis (GSEA) studies uncovered that CITED2 deficiency broadly enhances NFκB targets, IFNγ/IFNα responses, and inflammatory response gene expression in macrophages. Using complementary gain‐ and loss‐of‐function studies, we observed that CITED2 overexpression attenuate and CITED2 deficiency elevate LPS‐induced NFκB transcriptional activity and NFκB‐p65 recruitment to target gene promoter in macrophages. More importantly, blockade of NFκB signaling completely reversed elevated pro‐inflammatory gene expression in macrophages. Collectively, our findings show that CITED2 restrains NFκB activation and curtails broad pro‐inflammatory gene programs in myeloid cells.
Keywords: CITED2, inflammation, innate immunity, macrophage
Abbreviations
- BMDMs
bone marrow‐derived macrophages
- ChIP
chromatin immunoprecipitation
- CITED2
Cbp/p300 interacting transactivator with Glu/Asp rich carboxy‐terminal domain 2
- DMEM
Dulbecco's modified Eagle medium
- FBS
fetal bovine serum
- IFNα
interferon‐alpha
- IFNγ
interferon‐gamma
- IL1β
interleukin 1 beta
- LPS
lipopolysaccharides
- NFκB
nuclear factor kappa B
- TNF
tumor necrosis factor
1. INTRODUCTION
Monocyte‐derived macrophages are strategically located throughout the body, where they play an essential role in maintaining tissue homeostasis. 1 Under physiological conditions, macrophages govern tissue repair and regeneration by clearing dead cells, cellular debris, and preventing excessive inflammatory response to subtle environmental changes. 2 However, inflammation cascade initiated by infection or injury predominantly recruits innate immune cells consisting of a large number of activated macrophages. These macrophages mainly utilize pattern recognition receptors (PRRs) to distinguish host‐derived signals from foreign pathogens and orchestrate an appropriate inflammatory response. 3 An important subclass of pattern recognition receptors includes Toll‐like receptors (TLRs) that are highly conserved across species, and play a critical role in the detection of microbial‐associated molecular patterns derived from invading pathogens. 4 The TLRs activation by microbial agents elicits intracellular signaling pathways that relay extracellular signals to the nucleus to facilitate the expression of genes to orchestrate appropriate host protection response. These effects are predominantly facilitated through the activation of a specific set of transcription factors and cofactors. 5 These transcriptional modules promote macrophage pro‐inflammatory activation by elevating genes that are associated with the production of cytokines, chemokines, antimicrobial peptides, extracellular matrix modulating enzymes, and reactive nitrogen and oxygen species. Indeed, TLRs‐mediated macrophage activation is an explicitly robust biological process that is associated with broad alteration in gene expression profiles affecting a substantial part of the cellular genome. 6 Therefore, TLRs‐mediated pro‐inflammatory response must be constantly constrained to prevent molecular, cellular, tissue and organ damage. Moreover, unbridled macrophage‐mediated inflammation is associated with many human ailments, including atherosclerosis, arthritis, inflammatory bowel diseases, oncogenesis, metabolic syndrome, sepsis, and autoimmune diseases. 7 Despite the acknowledged importance of unbridled macrophage activation in the pathogenesis of human disorders, the molecular components that restrain broad inflammatory gene expression and tissue inflammation remain incompletely understood. In this study, we identify myeloid Cbp/p300 interacting transactivator with Glu/Asp rich carboxy‐terminal domain 2 (CITED2) as such a factor.
CITED2 is exclusively a nuclear protein. Previous studies have revealed that mutation in the human CITED2 gene is strongly associated with congenital heart defects. 8 , 9 Concordant with these observations, genetically engineered mice models imitating human CITED2 mutations also replicated these phenotypes. 10 , 11 Deletion of Cited2 gene in mice results in embryonic lethality in mid to late gestation with embryos displaying cardiac malformation, neural tube anomalies, adrenal agenesis, left‐right patterning deformities, and placental defects. 10 , 11 , 12 , 13 , 14 Our previous studies have demonstrated that CITED2 is predominantly expressed in human and murine macrophages. 15 Interestingly, CITED2 deficiency substantially elevated pro‐inflammatory gene expression while diminishing the expression of anti‐inflammatory genes in macrophages. 15 At the molecular level, our previous studies demonstrated that CITED2 cooperates with PPARγ to elevate anti‐inflammatory genes while repressing HIF1α functions to diminish pro‐inflammatory gene expression in macrophages. 15 However, whether myeloid‐CITED2 deficiency alters systemic inflammatory gene programs in macrophages has not been investigated. In this study, we provide evidence that CITED2 represses innate immune cell pathogenic response by modulating broad inflammatory gene programming in macrophages.
2. MATERIALS AND METHODS
2.1. Generation of myeloid‐CITED2 deficient mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and conformed to guidelines established by the American Association for Accreditation of Laboratory Animal Care. All mice were bred and maintained under pathogen‐free conditions, fed standard laboratory chow (Harlan Teklad, Indianapolis, IN), and kept on a 12‐h light/dark cycle. The mouse line expressing lysozyme‐M promoter‐driven Cre recombinase (Lyz2cre) was obtained from The Jackson Laboratory (Bar Harbor, ME). Cited2 flox (Cited2fl/fl) mice were generated as described earlier. 16 Myeloid CITED2‐specific null mice were generated in Dr Mahabeleshwar's laboratory as described in the previous report. 15 Briefly, Cited2 flox mice were crossed with Lyz2cre mice to generate a mouse line harboring the Cited2‐flox and Lyz2cre alleles. These mice were further cross‐bred to generate male and female offspring expressing two Lyz2cre and Cited2‐flox alleles. The mice with two Cited2‐flox and Lyz2cre alleles were used as the CITED2 myeloid‐specific null group (on C57BL/6 background). Mice with only two Lyz2cre alleles were used as the control group.
2.2. Lung inflammation studies
The lung inflammation model was established by intratracheal delivery of 10 mg/kg body weight zymosan or Lipopolysaccharide (LPS) solution to the 8‐10 weeks old Lyz2cre and Cited2fl/fl:Lyz2cre mice. Control groups received saline were served as vehicle controls. These mice were euthanized on day‐3 and intubated with 1 mL of saline to collect bronchoalveolar lavage. The lavages were centrifuged to separate the cellular and liquid components for further analyses. In parallel studies, lung tissues were obtained from Lyz2cre and Cited2fl/fl:Lyz2cre mice on day‐3 after zymosan challenge. These lung tissues were fixed in 10% of buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin. To determining the macrophage and neutrophil infiltration in lung tissue, paraffin‐sections of lung tissues were deparaffinized in xylene and rehydrated in graded ethanol series. Samples were subjected to antigen retrieval steps with antigen unmasking solution. Samples were treated with 0.3% of H2O2 for 30 minutes at room temperature and the nonspecific binding was blocked with blocking buffer. Samples were incubated with rabbit anti‐F4/80 or anti‐Ly6G antibody overnight at 4°C and were subsequently incubated with biotin‐conjugated goat anti‐rabbit IgG or goat anti‐rat IgG, respectively, for 30 minutes at room temperature. Samples were incubated in ABC reagent and the immunostaining was visualized using a DAB reagent. Images were acquired utilizing a microscope and macrophage or neutrophil area were quantified by Image J software.
2.3. Cell culture
RAW264.7 cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% of fetal bovine serum (FBS), 100 U/mL of penicillin, 10 µg/mL of streptomycin, and 2 mM of glutamine in a humidified incubator (5% of CO2 and 37°C). Mice bone marrow‐derived macrophages (BMDMs) were generated by ex vivo differentiation of bone marrow cells. Briefly, bone marrow cells from 8‐week‐old Lyz2cre and Cited2fl/fl:Lyz2cre mice were harvested from the femur and tibia. These bone marrow cells were cultured in DMEM supplemented with recombinant mouse M‐CSF for 7 days. Mice bone marrow‐derived neutrophils (BMDNs) were purified by Percoll‐gradient centrifugation according to the methods described previously. 17
2.4. RNAseq analysis
Total RNA from primary macrophages were obtained by using the High Pure RNA Isolation Kit. Quality control of total RNA samples were assessed using Qubit (Invitrogen) for quantification and Agilent 2100 BioAnalyzer analysis to assess quality using a cut‐off of RIN > 7.0 to select specimens for further analysis. cDNA library for RNAseq was generated from 150 ng of total RNA using the Illumina TruSeq Stranded Total RNA kit with Ribo Zero Gold for rRNA removal according to the manufacturer's protocol. The resulting purified mRNA was used as input for the Illumina TruSeq kit, in which libraries are tagged with unique adapter‐indexes. Final libraries were validated on the Agilent 2100 BioAnalyzer, quantified via qPCR, and pooled at equimolar ratios. Pooled libraries were diluted, denatured, and loaded onto the Illumina NextSeq 550 System using a high output flowcell. STAR Aligner was used for mapping the sequencing reads to the mm10 mouse reference genome. The aligned reads were then analyzed with Cuffdiff to obtain gene‐level expression data using the GENCODE gene annotation for mm10 and reported as fragments per kilobase per million reads mapped (FPKM). Differential expression analysis was also performed using the Cuffdiff package and significantly differentially expressed genes were defined using an adjusted P‐value < .05 (FDR corrected). Gene expression tables for relevant pairwise comparisons were analyzed for gene set enrichment (GSEA) 18 using GenePattern (Broad Institute). We specifically utilized Hallmark pathways data sets for current studies. A gene set was considered to be significantly enriched using an FWER cutoff <0.05. Heatmaps were generated using ClustVis. 19 The accession number for the sequencing data reported in this paper is GEO: GSE147943.
2.5. RNA extraction, Real‐time quantitative PCR, and western blotting
Total RNA was isolated from indicated samples using the High Pure RNA Isolation Kit. One microgram of total RNA was reverse‐transcribed using M‐MuLV reverse transcriptase in the presence of random hexamers and oligo (dT) primers. Real‐time quantitative PCR was performed using Universal SYBR Green PCR Master Mix or TaqMan Universal Master Mix on Applied Biosystems Step One Plus real‐time PCR system in presence of gene‐specific primers.
The cellular protein extracts were subjected to western blot analysis as described below. The indicated cell types were lysed using radioimmunoprecipitation lysis buffer (Sigma‐Aldrich) supplemented with a protease and phosphatase inhibitor mixture tablets (Roche Applied Science) following the specified treatments. Equal quantities of total protein were separated by SDS‐PAGE and transferred to nitrocellulose membrane. These nitrocellulose membranes were incubated with primary antibody against β‐Actin, p‐IκBα, total IκBα, p‐NFκB‐p65 (1:1000 dilution), and HRP‐conjugated secondary antibody (1:5000 dilution).
2.6. Transient transfection, Luciferase reporter, and Chromatin immunoprecipitation (ChIP)
Transfection of RAW264.7 cells and mice BMDMs were performed using Lipofectamine transfection reagents (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Transfected cells were stimulated in vitro with either LPS (100 ng/mL), or PBS (control), and used for experiments. RAW264.7 cells were transfected with luciferase reporter plasmids driven by the NFκB concatemer or were cotransfected with pCMV‐Cited2 plasmid or Cited2‐specific siRNA using Lipofectamine transfection reagent (Invitrogen). These cells were exposed to 100 ng/mL of LPS, 50 ng/mL of zymosan or 20 ng/mL of IFNγ for 18 hours. Luciferase reporter activity was measured and normalized according to the manufacturer's instructions. Results are presented as relative luciferase activity over the control group. Chromatin immunoprecipitation (ChIP) analyses were performed using the EZ‐Magna ChIP G Kit (17‐409; MilliporeSigma) according to the manufacturer's instruction. Briefly, CITED2 overexpressing RAW264.7 cells, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with 100 ng/mL of LPS. Chromatin immunoprecipitations were performed using anti‐NFκB‐p65 or anti‐CITED2 antibody. Chromatin samples from these experiments were evaluated by real‐time quantitative RT‐PCR. DNA levels were first normalized to the internal control region in the first intron of the mouse Actb gene (forward: 5′‐CGTATTAGGTCCATCTTGAGAGTAC‐3′, reverse: 5′‐GCCATTGAGGCGTGATCGTAGC‐3′). Primer pairs flanking the NFκB‐binding site were targeted to amplify the mouse Ccl2 (FW, 5′‐TGGGAATTTCCACGCTCTTA‐3′; REV, 5′‐CTCTGTCAACAAAGGATGTTCTTC‐3′) promoter region. ChIP performed using isotype IgG was used as a negative control. Relative enrichment was calculated by dividing the normalized levels of ChIP DNA to that of input DNA at the corresponding locus.
2.7. Quantification and statistical analysis
All data, unless indicated are presented as the mean ± standard deviation (SD). The statistical significance of differences between the two groups were analyzed by Student's t test or two‐way ANOVA with Bonferroni multiple comparison tests. P < .05 was considered statistically significant.
3. RESULTS
3.1. CITED2 curbs broad pro‐inflammatory gene programs in macrophages
Our previous studies have shown that CITED2 deficient macrophages are highly responsive to pro‐inflammatory agents and express elevated levels of pro‐inflammatory cytokines, and chemokines. 15 However, whether CITED2 deficiency affects the systemic pro‐inflammatory gene programming in macrophages following exposure to an inflammatory agent has not been examined. Previous studies have demonstrated that toll‐like receptors activation by lipopolysaccharides (LPS) elicit broad and potent pro‐inflammatory responses by macrophages. 6 Therefore, we utilized LPS to elicit systemic pro‐inflammatory signaling pathways and gene expression in macrophages. To identify the broadly deregulated pathogenic inflammatory signaling pathways and gene targets associated with CITED2 deficiency, we performed unbiased RNAseq analyses. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with LPS, and total RNA samples were obtained to perform gene expression profiling analyses. The data derived from gene expression profiling studies were subjected to Gene Set Enrichment Analysis (GSEA) to identify CITED2 regulated inflammatory signaling pathways and gene targets. Surprisingly, our analyses show that LPS‐induced gene expression profile from Cited2fl/fl:Lyz2cre mice BMDMs were broadly enriched for inflammatory gene programs such as TNF signaling via NFκB, IFNγ response, IFNα response, IL2‐STAT5 signaling, IL6‐STAT3 signaling, allograft rejection, apoptosis, inflammatory response, and activation of the complement system (Figure 1A‐D). As anticipated, LPS exposure significantly elevated a large number of NFκB‐regulated pro‐inflammatory gene targets in Lyz2cre mice BMDMs (Figure 1E). Interestingly, these pro‐inflammatory gene targets (Serpinb2, Rela, Il1β, Cxcl2, Il12β, Tnf, Plau, Klf6, F3, Il1α, etc) were substantially elevated in Cited2fl/fl:Lyz2cre mice BMDMs following LPS exposure (Figure 1E). Concordantly, LPS exposure significantly elevated a substantial number of IFNγ response gene targets in Lyz2cre mice BMDMs (Figure 1F). Remarkably, CITED2 deficiency significantly heightened the expression of these critical IFNγ response genes (Cxcl9, Il6, Cd86, Cd274, Ccl2, Icam1, etc) following LPS stimulation (Figure 1F). Similarly, LPS stimulation markedly elevated a significant number of IFNα response gene targets in Lyz2cre mice BMDMs (Figure 1G). Surprisingly, these pro‐inflammatory IFNα response gene targets (Irf9, Irf2, Nub1, Cxcl10, Irf1, etc) were substantially elevated in Cited2fl/fl:Lyz2cre mice BMDMs following LPS exposure (Figure 1G). Collectively, our analyses show that CITED2 deficiency broadly elevates pro‐inflammatory signaling pathways and gene targets expression in macrophages.
Figure 1.
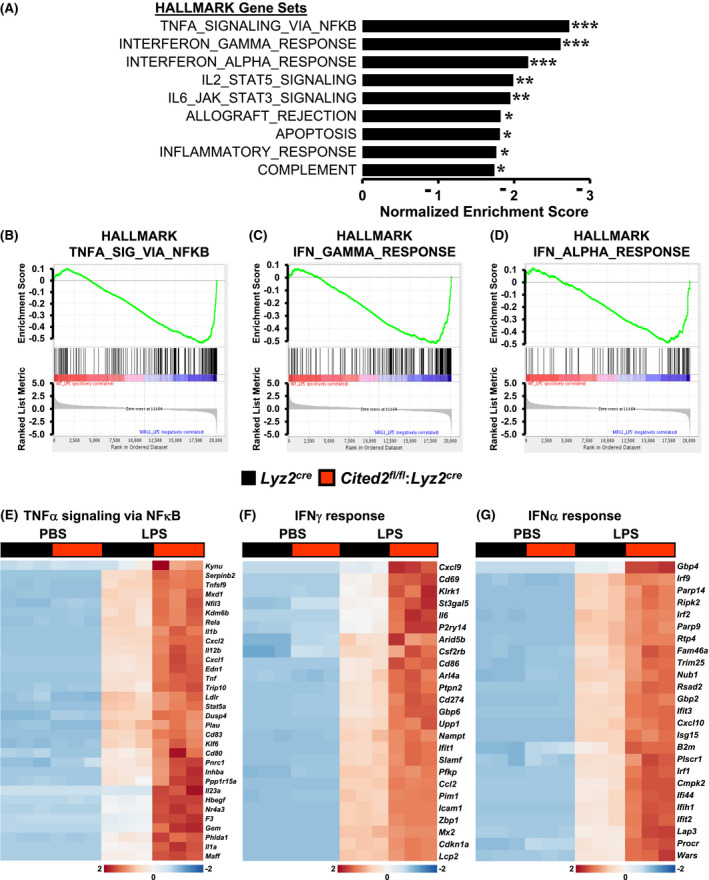
CITED2 curbs broad pro‐inflammatory gene programs in macrophages. A, GSEA of RNAseq data that are altered in Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs following 4 hours of 100 ng/mL of LPS treatment. FWER P‐value less than .05 was considered significant (n = 3). B‐D, Enrichment plots of indicated gene set obtained by GSEA comparing Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs RNAseq data following LPS treatment (n = 3). E‐G, Heatmap of genes involved in TNFα signaling via NFκB (E), IFNγ response (F), and IFNα response (G) that are altered in Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs following LPS stimulation (n = 3). FWER P‐value *P < .05; **P < .01; ***P < .001
3.2. CITED2 restrains myeloid cell response to inflammation in vivo
Our unbiased RNAseq studies have demonstrated that CITED2 curbs unbridled and systemic inflammatory gene response in macrophages against pro‐inflammatory incitements (Figure 1).
Therefore, we hypothesized that CITED2 may calibrate myeloid cell response against nebulous inflammatory stimuli and protects the host from deleterious effects associated with uncontrolled myeloid cell activation. To test this hypothesis, we utilized the zymosan‐induced acute lung inflammation and injury model. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice were challenged by delivering zymosan through the intratracheal route. As anticipated, zymosan exposure markedly elevated immune cell recruitment to Lyz2cre mice lungs (Figure 2A,D). Surprisingly, myeloid‐CITED2 deficient mice exhibited significantly heightened immune cell recruitments to lungs compared to Lyz2cre mice group following the zymosan challenge (Figure 2A,D). Previous studies have demonstrated that macrophages and neutrophils are prominent innate immune cells recruited to lungs in the face of zymosan exposure. 20 , 21 Therefore, we examined whether myeloid‐CITED2 deficiency specifically altered macrophage or neutrophil recruitment to lungs following zymosan challenge. As shown in Figure 2B,E, zymosan challenge significantly elevated macrophage recruitment to lungs in Lyz2cre mice. Remarkably, myeloid‐CITED2 deficient mice challenged with zymosan exhibited significantly heightened macrophage recruitment to lungs compared to Lyz2cre mice group (Figure 2B,E). Next, we assessed whether myeloid‐CITED2 deficiency altered neutrophil recruitment to lungs following zymosan exposure. Interestingly, zymosan‐induced neutrophil accumulation were significantly elevated in myeloid‐CITED2 deficient mice compared to Lyz2cre mice group (Figure 2C,F). Taken together, our studies demonstrate that CITED2 restrains macrophage and neutrophil recruitments to the site of inflammation to protect the host against nebulous inflammatory insults.
Figure 2.
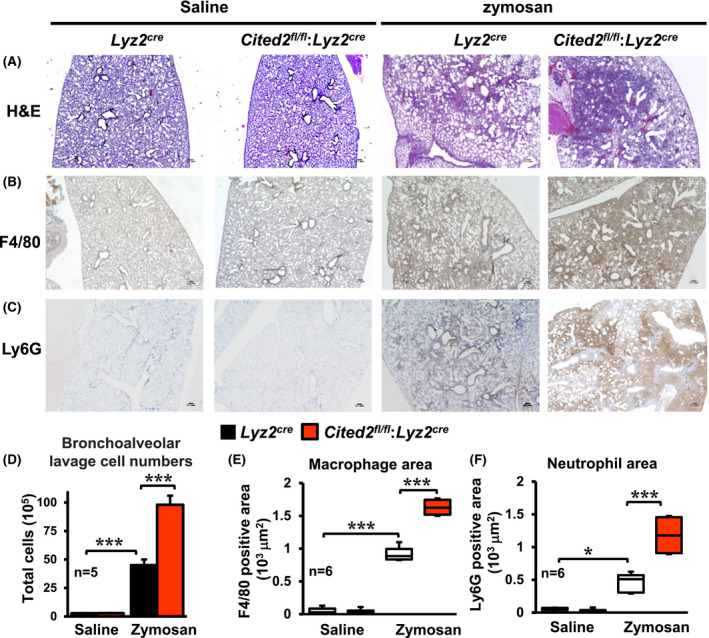
CITED2 restrains myeloid cell response to the site of inflammation in vivo. A‐C, Lung sections of Lyz2cre and Cited2fl/fl:Lyz2cre mice 3‐days after intratracheal delivery of saline or zymosan. These lung sections were stained for hematoxylin‐eosin (A). The macrophages and neutrophils localization within the lung tissue were determined by anti‐F4/80 (B) and anti‐Ly6G (C) antibodies staining, respectively (n = 6 per group). D, Total cell numbers in bronchoalveolar lavage of Lyz2cre and Cited2fl/fl:Lyz2cre mice 3‐days after intratracheal delivery of saline or zymosan (n = 5 per group). E and F, Area of F4/80 (E) and Ly6G (F) positive cells were quantified using ImageJ software (n = 6 per group). The box plot represents the median with first and third quartiles, and whiskers represent minimum/maximum. All other values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni posttesting. *P < .05; **P < .01; ***P < .001
3.3. CITED2 deficiency augments pro‐inflammatory gene expression in vivo
Our studies thus far demonstrated that myeloid‐CITED2 deficiency broadly exaggerates the pro‐inflammatory gene program ex vivo (Figure 1) and pathological inflammatory response in vivo (Figure 2). Next, we intend to examine whether CITED2 deficiency altered pro‐inflammatory gene expression in responding myeloid cells in vivo. To test this notion, we specifically utilized myeloid cells derived from bronchoalveolar lavages of Lyz2cre and Cited2fl/fl:Lyz2cre mice following the zymosan challenge. As shown in Figure 3A‐F, expression of Cxcl1, Cxcl2, Cxcl10, Ccl2, Ccl3, and Ccl4 were significantly elevated in Lyz2cre mice bronchoalveolar myeloid cells following zymosan challenge. Remarkably, expression of these pro‐inflammatory cytokines and chemokines were significantly heightened in myeloid‐CITED2 deficient mice compared to the respective control group (Figure 3A‐F). To this end, our analyses demonstrate that CITED2 restrain myeloid cell inflammatory gene response program ex vivo and in vivo.
Figure 3.
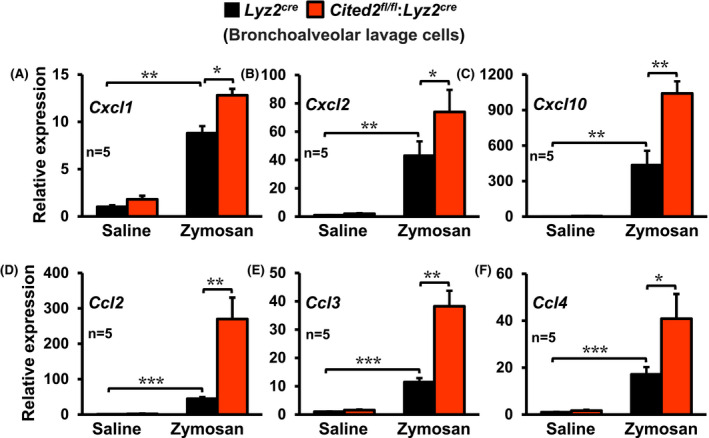
CITED2 deficiency augments pro‐inflammatory gene expression in vivo. A‐F, The Lyz2cre and Cited2fl/fl:Lyz2cre mice were subjected to acute lung inflammation by delivering zymosan or saline through the intratracheal route. The bronchoalveolar lavage cells were collected 3‐days after intratracheal delivery of saline or zymosan. The total RNA samples from these cells were evaluated for expression of Cxcl1, Cxcl2, Cxcl10, Ccl2, Ccl3, and Ccl4 by RT‐qPCR (n = 5 per group). The 36B4 gene was used as a housekeeping gene for RT‐qPCR analyses. These experiments were performed three independent times. All values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni posttesting. *P < .05; **P < .01; ***P < .001
3.4. CITED2 deficiency elevates NFκB gene targets in myeloid cells
Our GSEA of RNAseq studies has demonstrated that CITED2 constrain classical NFκB activation and pro‐inflammatory gene program in macrophages (Figure 1). Therefore, we examined whether CITED2 deficiency altered NFκB regulated pro‐inflammatory gene expression in neutrophils and macrophages following pro‐inflammatory challenge. We have utilized pro‐inflammatory gene targets identified by our GSEA studies (Figure 1) to evaluate this notion. Accordingly, bone marrow‐derived neutrophils (BMDNs) from Lyz2cre and Cited2fl/fl:Lyz2cre mice were stimulated with LPS or IFNγ, and expression of NFκB regulated pro‐inflammatory gene targets were evaluated by RT‐qPCR (Figure 4A,B). As shown in Figure 4A, LPS exposure significantly elevated classical NFκB targets such as Tnf, Il1β, Ccl3, and Ccl4 expression in Lyz2cre mouse BMDNs. Compellingly, LPS‐induced expression of these pro‐inflammatory gene targets were robustly heightened in CITED2 deficient mice BMDMs (Figure 4A). Concordant with these observations, IFNγ stimulation substantially enhanced Tnf, Il1β, F3, and Ccl4 expression in Lyz2cre mouse BMDNs (Figure 4B). Intriguingly, IFNγ‐induced expression of these pro‐inflammatory gene targets were markedly elevated in Cited2fl/fl:Lyz2cre mice BMNMs (Figure 4B). Next, we evaluated whether CITED2 deficiency altered NFκB, IFNγ, or IFNα regulated inflammatory gene expression in macrophages. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with LPS and expression of NFκB, IFNγ, or IFNα response gene targets were evaluated by RT‐qPCR. As shown in Figure 5A‐C, LPS exposure significantly elevated expression of Cxcl2, Tnf, F3, Cxcl1, Ccl2, Zbp1, Cd274, Icam1, Ifi44, Gbp4, Ifit2, and Cxcl10 expression in Lyz2cre mouse BMDMs. Interestingly, the expression of these classical pro‐inflammatory genes were markedly heightened in CITED2 deficient mice BMDMs following the LPS challenge (Figure 5A‐C). Next, we examined whether these observations are recapitulated under in vivo conditions. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice were challenged by delivering LPS or saline through the intratracheal route. The purified bronchoalveolar lavage macrophages were obtained three days after LPS challenge. Total RNA derived from these macrophages were evaluated for the expression of classical pro‐inflammatory genes by RT‐qPCR analyses. As shown in Figure 5D, LPS challenge robustly elevated Il1α, Il1β, Icam1, and Il12b expression in Lyz2cre mice lung macrophages. Interestingly, Cited2fl/fl:Lyz2cre mice lung macrophages maintained significantly heightened these pro‐inflammatory target gene expression (Il1α, Il1β, Icam1, and Il12b) even three days after the LPS challenge (Figure 5D). Taken together, our studies demonstrate that CITED2 deficiency decisively elevates NFκB regulated pro‐inflammatory gene targets in myeloid cells.
Figure 4.
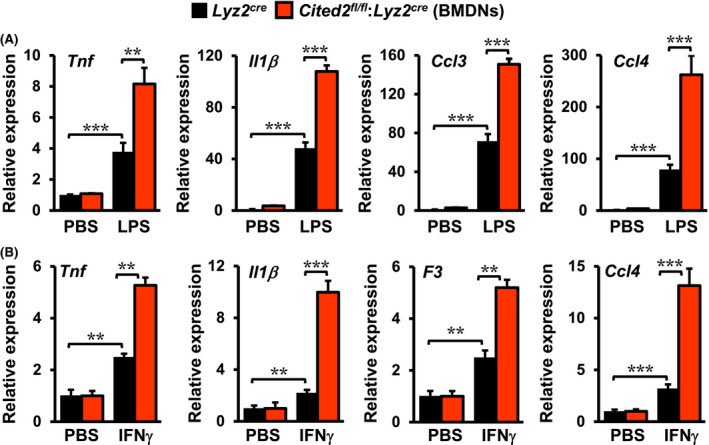
CITED2 deficiency elevates NFκB target gene expression in neutrophils. A, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDNs were stimulated with 100 ng/mL of LPS for 6 hours. Total RNA samples were evaluated for expression of Tnf, Il1β, Ccl3, and Ccl4 by RT‐qPCR (n = 4). B, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDNs were stimulated with 10 ng/mL of IFNγ for 6 hours. Total RNA samples were evaluated for expression of Tnf, Il1β, F3, and Ccl4 by RT‐qPCR (n = 4). The 36B4 gene was used as a housekeeping gene for RT‐qPCR analyses. These experiments were performed three independent times with four replicates. All values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni posttesting. *P < .05; **P < .01; ***P < .001
Figure 5.
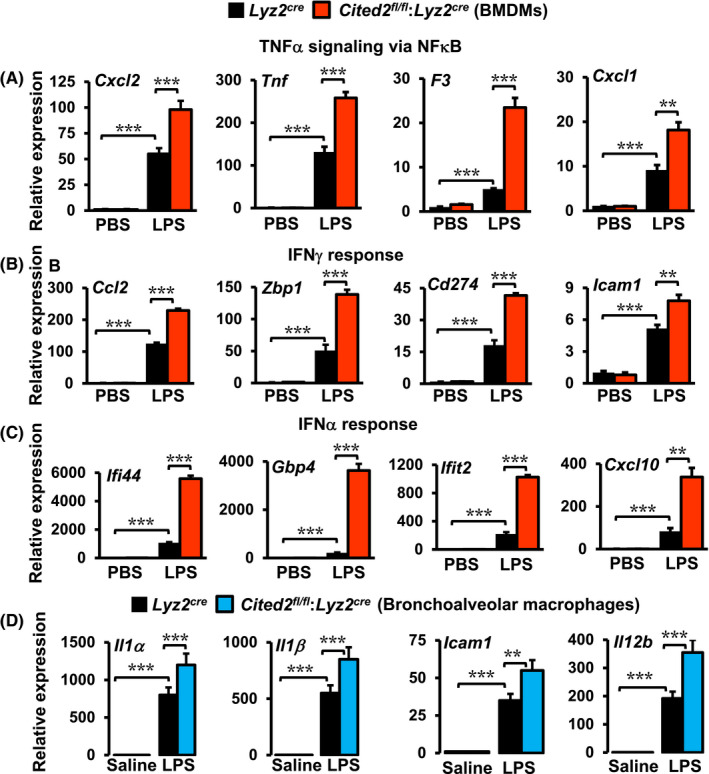
CITED2 deficiency elevates NFκB target gene expression in macrophages. A‐C, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with 100 ng/mL of LPS for 6 hours. Total RNA samples derived from these experiments were evaluated for the expression of indicated inflammatory genes by RT‐qPCR (n = 4). These experiments were performed three independent times. D, The Lyz2cre and Cited2fl/fl:Lyz2cre mice were exposed to acute lung inflammation by delivering LPS or saline through the intratracheal route. The bronchoalveolar lavage macrophages were purified 3‐days after intratracheal delivery of saline or LPS. The total RNA samples from these macrophages were evaluated for expression of Il1α, Il1β, Icam1, and Il12b by RT‐qPCR (n = 5 per group). The 36B4 gene was used as a housekeeping gene for RT‐qPCR analyses. All values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni posttesting. *P < .05; **P < .01; ***P < .001
3.5. CITED2 restrains LPS‐induced NFκB activation in macrophages
Our studies thus far demonstrated that CITED2 curbs NFκB governed inflammatory gene programs and target gene expression in macrophages. Thus, we examined whether altering CITED2 levels modulate LPS‐induced NFκB transcriptional activity in macrophages. Accordingly, RAW264.7 cells were cotransfected with NFκB concatemer‐driven luciferase reporter plasmid in presence of pCMV‐Cited2 or siCited2. These cells were stimulated with inflammatory agents and luciferase activities were recorded. As shown in Figure 6A, over‐expression of Cited2 significantly diminished LPS, zymosan, or IFNγ‐induced NFκB luciferase reporter activity in macrophages. Conversely, CITED2 deficiency significantly elevated LPS‐induced NFκB luciferase reporter activity in macrophages (Figure 6B). Next, we examined whether CITED2 regulates upstream signaling that modulates NκB activation in macrophages. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with LPS, and total protein extracts were examined for IκBα phosphorylation, IκBα degradation, and NFκB‐p65 activation in these macrophages (Figure 6C). Our analyses revealed that CITED2 deficiency did not alter LPS‐induced IκBα phosphorylation (Serine 32/36) or IκBα degradation in macrophages (Figure 6C). Similarly, CITED2 deficiency did not significantly impact activating phosphorylation (Serine‐536) of NFκB‐p65 in these macrophages (Figure 6C). The transcriptionally active NFκB heterodimeric complex consists of p50 and p65 subunits. The transcriptional activation domain (TAD) of the p65 subunit directly interacts with the transcriptional adaptor zinc‐binding 1 (TAZ1) domain of CBP/p300 to initiate the process of effective transcription. 22 Previous studies have demonstrated that the CITED2 transactivation domain binds to the TAZ1 domain of CBP/p300 with very high affinity. 23 , 24 This high‐affinity CITED2 binding could potentially displace NFκB‐p65 interaction with coactivator CBP/p300 and subsequently oust NFκB from the transcriptional complex. Therefore, we examined whether overexpression or deficiency of CITED2 altered NFκB‐p65 enrichment on target gene promoter following LPS stimulation. Accordingly, CITED2 overexpressing RAW264.7 cells, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with LPS, and chromatin immunoprecipitation (ChIP) was performed using anti‐p65 or anti‐CITED2 antibodies (Figure 6D‐F). The isotype IgG antibodies were served as a negative control for ChIP studies. These ChIP samples were analyzed for p65 enrichment on the Ccl2 promoter (−2344 to −2354) regions by utilizing site‐specific primers. Our analysis revealed that CITED2 overexpression significantly attenuated LPS‐induced p65 enrichment on Ccl2 promoter in macrophages (Figure 6D). Concordantly, CITED2 deficiency significantly elevated LPS‐induced p65 recruitment to Ccl2 promoter in primary macrophages (Figure 6E). Interestingly, LPS exposure significantly elevated CITED2 enrichment on Ccl2 promoter (−2344 to −2354) in Lyz2cre mice BMDMs (Figure 6F). However, these effects were not observed in Cited2fl/fl:Lyz2cre mice BMDMs (Figure 6F). Taken together, our analyses demonstrate that CITED2 restrain LPS‐induced NFκB activation and functions in macrophages.
Figure 6.
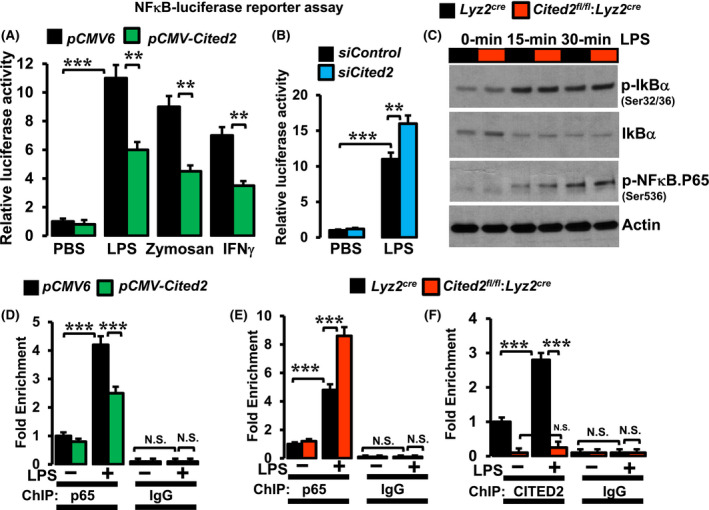
CITED2 restrains LPS‐induced NFκB activation in macrophages. A and B, RAW264.7 cells were transfected with a NFκB concatemer‐driven luciferase reporter construct in the presence of pCMV‐Cited2 plasmid (A) or Cited2‐specific siRNA (B). These cells were stimulated with 100 ng/mL of LPS, 50 ng/mL of zymosan, or 20 ng/mL of IFNγ for 18 hours, and cell lysates were analyzed for luciferase activity (n = 3). C, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with 100 ng/mL of LPS for 0‐30 minutes. Total protein extracts from these studies are evaluated for p‐IκBα, total IκBα, p‐NFκB‐p65 levels by western blot. Actin was used as a loading control (n = 3). D, RAW264.7 cells were transfected with pCMV‐Cited2 plasmid stimulated with 100 ng/mL of LPS for 1 hour and ChIP analysis was performed on Ccl2 promoter (−2344 to −2354) utilizing anti‐NFκB‐p65 antibody (n = 3). E and F, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with 100 ng/mL of LPS for 1 hour, and ChIP analysis was performed on Ccl2 promoter (−2344 to −2354) utilizing anti‐NFκB‐p65 (E) or anti‐CITED2 (F) antibody (n = 3). ChIP analyses performed using isotype IgG were used as a negative control. These experiments were performed three independent times. All values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni posttesting. *P < .05; **P < .01; ***P < .001
3.6. NFκB blockade revert heightened inflammatory response in CITED2‐deficient macrophages
Our studies thus far demonstrated that CITED2 deficiency elevates LPS‐induced NFκB signaling and promotes pro‐inflammatory gene programs in macrophages. Therefore, we evaluated whether heightened pro‐inflammatory gene response in CITED2‐deficient macrophages are NFκB dependent. We utilized pharmacological and genetic tools to block NFκB activation in primary macrophages. Specifically, we have utilized pro‐inflammatory gene targets identified by our RNAseq analyses to evaluate this notion. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were pretreated with a cell‐permeable peptide inhibitor of NFκB (SN50) or control peptide (SN50M). 25 These cells were stimulated with LPS and expression of pro‐inflammatory gene targets were evaluated by RT‐qPCR analyses. As anticipated, CITED2 deficiency significantly elevated LPS‐induced pro‐inflammatory gene targets such as Ccl2, Icam1, Tnf, and Cxcl1 expression in macrophages (Figure 7A‐D). Surprisingly, blockade of NFκB signaling was sufficient to completely reverse elevated levels these pro‐inflammatory gene targets in CITED2 deficient macrophages (Figure 7A‐D). Next, to further validate these observations, we utilized a genetic approach to silence NFκB signaling in primary macrophages. Accordingly, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were transfected with NFκB‐p65‐specific siRNA. These cells were stimulated with LPS, and expression of pro‐inflammatory gene targets were evaluated by RT‐qPCR. As expected, CITED2 deficiency markedly and significantly elevated Ccl2, Icam1, Tnf, and Cxcl1 expression following LPS stimulation (Figure 7E‐H). Intriguingly, genetic inhibition of NFκB signaling was sufficient to completely reverse the heightened expression of these pro‐inflammatory gene targets in CITED2 deficient macrophages (Figure 7E‐H). Collectively, these results demonstrate that CITED2 curb NFκB activation to restrain pro‐inflammatory gene expression in macrophages.
Figure 7.
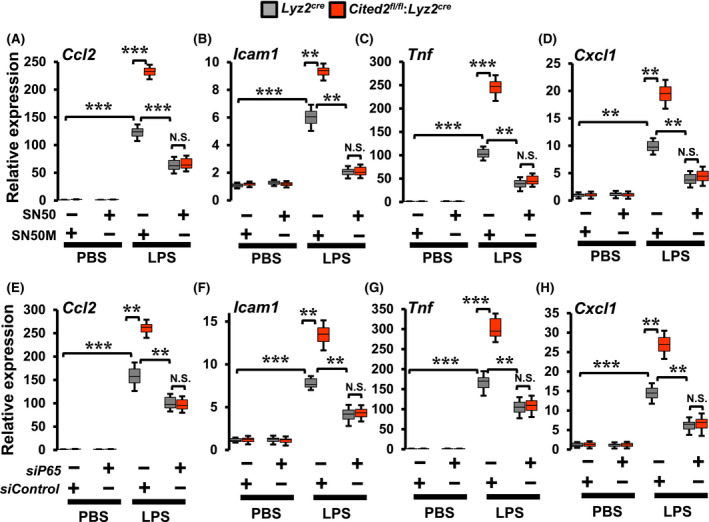
NFκB blockade revert heightened inflammatory gene expression in CITED2‐deficient macrophages. A‐D, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were stimulated with 100 ng/mL of LPS for 6 hours in presence of SN50 or SN50M peptide. Total RNA from these experiments was analyzed for Ccl2, Icam1, Tnf, and Cxcl1 expression by RT‐qPCR (n = 3). E‐H, Lyz2cre and Cited2fl/fl:Lyz2cre mice BMDMs were transfected with p65‐specific siRNA, and these cells were stimulated with 100 ng/mL of LPS for 6 hours. Total RNA from these experiments was analyzed for expression of Ccl2, Icam1, Tnf, and Cxcl1 by RT‐qPCR (n = 3). The 36B4 gene was used as a housekeeping gene for RT‐qPCR analyses. These experiments were performed three independent times with three replicates. The box plot represents the median with first and third quartiles, and whiskers represent minimum/maximum. Data were analyzed by ANOVA followed by Bonferroni posttesting. NS not significant; *P < .05; **P < .01; ***P < .001
4. DISCUSSION
Our studies identify CITED2 as a critical molecular switch that restrains broad pro‐inflammatory gene programs in macrophages and blocks unbridled myeloid cell activation at the site of inflammation. The central findings of our study are as follows: (i) CITED2 curbs broad pro‐inflammatory gene programs in macrophages, (ii) CITED2 restrains myeloid cell response to the site of inflammation in vivo, (iii) CITED2 deficiency augments myeloid cell pro‐inflammatory gene expression in vivo, (iv) CITED2 deficiency elevates NFκB, IFNγ response, and IFNα response target gene expression in myeloid cells, (v) CITED2 restrains LPS‐induced NFκB activation in macrophages, and (vi) NFκB blockade revert heightened inflammatory gene expression in CITED2‐deficient macrophages. Collectively, our findings show that CITED2 restrain inflammatory cytokine‐induced NFκB activation and curtail broad pro‐inflammatory gene expression, and functions in myeloid cells (Figure 8).
Figure 8.
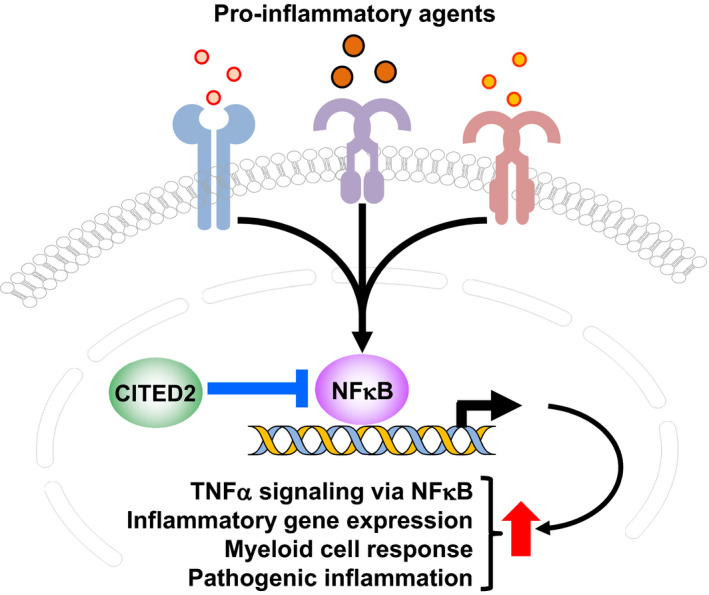
CITED2 bridle pathological inflammation. CITED2 curtails inflammatory agents‐induced NFκB activation and blocks broad pro‐inflammatory gene expression and myeloid cell accumulation at the site of inflammation in vivo
Previous studies have demonstrated that CITED2 plays a very critical role in embryonic and adult hematopoiesis, in mice. 26 Our previous studies have demonstrated that myeloid deficiency of CITED2 did not markedly alter circulating levels of lymphocytes, neutrophils, monocytes, eosinophils, and basophils in adult animals. 15 Recent genetic studies have revealed that single nucleotide polymorphisms in and around the human CITED2 gene are associated with dysregulated erythrocyte phenotypes. 27 Interestingly, myeloid‐CITED2 deficient mice also exhibited significantly altered erythrocyte phenotypes. 15 Our earlier reports have shown that CITED2 interacts with PPARγ to promote IL4‐induced anti‐inflammatory gene expression in macrophages. 15 Further, pro‐inflammatory agent exposure attenuated CITED2 expression macrophages and may provide contexture to elevate subsequent inflammatory gene expressions. It is well established that CITED2 interacts with a wide variety of transcription factors, including hepatocyte nuclear factor 4 alpha (HNF‐4α), 28 peroxisome proliferator‐activated receptor alpha (PPARα), 29 PPAR‐γ, and hypoxia‐inducible factor 1 alpha (HIF1α), 30 and thereby governs gene transcription. In this context, our current transcriptomic studies have shown that CITED2 deficiency significantly elevates NFκB, STAT5, and STAT3 regulated pro‐inflammatory gene expression in myeloid cells. In addition, our studies also have shown that CITED2 deficiency broadly elevated IFNα/IFNγ‐response, allograft rejection, apoptosis, inflammatory response, and complement associated gene expression in myeloid cells. Previous studies have demonstrated that LPS mediated pro‐inflammatory myeloid cell activation is in part mediated through expression and production of interferons. 31 Further, recent studies have demonstrated that cross‐talk between toll‐like receptor (TLR) and IFNγ signaling pathways facilitate the induction of broad pro‐inflammatory gene expression in macrophages. 32 In this context, our GSEA studies have demonstrated that CITED2 abrogates broad pro‐inflammatory gene expression by repressing NFκB activation, IFNγ response, and IFNα response gene expression in macrophages. Interferons mediate its cellular effects through expression and activation of interferon regulatory factors (IRFs). In this context, our GSEA studies have shown that CITED2 deficiency significantly elevates IRF1, IRF2 and IRF9 expression in macrophages (Figure 1G). Therefore, it is possible that CITED2 may exert broad anti‐inflammatory effects through suppressing NFκB as well as IRFs functions in macrophages. The previous reports have revealed that IRFs‐STATs cross‐talk modulate broad inflammatory gene expression in immune cells. 33 In this setting, our studies also show that CITED2 repress IL2‐mediated STAT5 and IL6‐mediated STAT3 signaling and target gene expression in macrophages. Therefore, one could envision that CITED2 broadly abrogates pro‐inflammatory gene programs by repressing NFκB, IRFs, and STATs signaling in myeloid cells.
Our previous studies have shown that myeloid‐CITED2 deficient mice were prone to LPS‐induced sepsis symptomology and mortality. 15 In this context, our current studies show that myeloid‐CITED2 deficient mice are highly susceptible to zymosan‐induced lung inflammation. In addition, myeloid‐CITED2 deficient mice also exhibited heightened levels of macrophage and neutrophil accumulation in lung tissue. Recent studies have demonstrated that resident alveolar macrophages play a critical role in the maintenance of lung tissue homeostasis in health and diseases. 34 In this context, our current studies have shown that CITED2 deficiency significantly elevates recruitment neutrophils and monocyte‐derived macrophages to the sites of inflammation. Therefore, it is possible that CITED2 may contribute to the maintenance of cellular quiescence in alveolar macrophages. Our ex vivo and in vivo gene expression analyses show that CITED2 deficiency elevates pro‐inflammatory gene expression in macrophages and neutrophils. Earlier molecular studies have shown that CITED2 is a competitive inhibitor of HIF1α. Specifically, CITED2 compete for binding to the TAZ1 domain of p300/CBP through their transactivation domains. 35 This will disrupt the HIF1α association with p300/CBP and abolish HIF1α transcriptional activity. It is also well known that the TAD segment of the NFκB‐p65 subunit directly interacts with the TAZ1 domain of CBP/p300 to initiate the process of effective transcription. 22 However, whether CITED2 deficiency altered NFκB‐p65 recruitment to target gene promoters in primary macrophages was not been investigated. In this context, our analyses show that CITED2 deficiency did not alter inflammatory agent‐induced IκBα phosphorylation, IκBα degradation, and NFkB‐p65 activating phosphorylation in macrophages. Interestingly, inflammatory cytokine exposure significantly heightened CITED2 protein recruitment to pro‐inflammatory gene promoters in macrophages. However, CITED2 deficiency elevates NFκB‐p65 recruitment to target gene promoters and heighten NFκB transcriptional activity in macrophages. Therefore, we postulated that CITED2 represses NFκB activation by blocking its recruitment to target gene promoters. It is possible that CITED2 could modulate other signaling mechanisms as well as affect mitochondrial functions to modulate NFκB activation in macrophages. 36 However, blockade of NFκB signaling completely reversed elevated levels of pro‐inflammatory gene expression in CITED2 deficient macrophages. Collectively, our current studies demonstrate that CITED2 bridle broad inflammatory gene programs in myeloid cells and protect the host from pathogenic inflammation.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
G.H. Mahabeleshwar conceived and designed the study. G.H. Mahabeleshwar, H. Pong Ng, and G.D. Kim performed the experiments. G.H. Mahabeleshwar, H. Pong Ng, G.‐D. Kim, E. Ricky Chan, and S.L. Dunwoodie analyzed and interpreted the data. G.H. Mahabeleshwar wrote and edited the manuscript, and it was approved by all authors.
Supporting information
Table S1‐S2
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant HL126626, HL141423, and Crohn's and Colitis Foundation Senior Research Award 421904 (to G. H. M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
Pong Ng H, Kim G‐D, Ricky Chan E, Dunwoodie SL, Mahabeleshwar GH. CITED2 limits pathogenic inflammatory gene programs in myeloid cells. The FASEB Journal. 2020;34:12100–12113. 10.1096/fj.202000864R
REFERENCES
- 1. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392‐404. [DOI] [PubMed] [Google Scholar]
- 2. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805‐820. [DOI] [PubMed] [Google Scholar]
- 4. Fitzgerald KA, Kagan JC. Toll‐like receptors and the control of immunity. Cell. 2020;180:1044‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll‐like receptors. Annu Rev Cell Dev Biol. 2006;22:409‐437. [DOI] [PubMed] [Google Scholar]
- 6. Das A, Yang CS, Arifuzzaman S, et al. High‐resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co‐factor networks in classically and alternatively activated macrophages. Front Immunol. 2018;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Volcik KA, Zhu H, Finnell RH, Shaw GM, Canfield M, Lammer EJ. Evaluation of the Cited2 gene and risk for spina bifida and congenital heart defects. Am J Med Genet A. 2004;126a:324‐325. [DOI] [PubMed] [Google Scholar]
- 9. Sperling S, Grimm CH, Dunkel I, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. 2005;26:575‐582. [DOI] [PubMed] [Google Scholar]
- 10. Weninger WJ, Lopes Floro K, Bennett MB, et al. Cited2 is required both for heart morphogenesis and establishment of the left‐right axis in mouse development. Development (Cambridge, England). 2005;132:1337‐1348. [DOI] [PubMed] [Google Scholar]
- 11. Bamforth SD, Braganca J, Farthing CR, et al. Cited2 controls left‐right patterning and heart development through a Nodal‐Pitx2c pathway. Nat Genet. 2004;36:1189‐1196. [DOI] [PubMed] [Google Scholar]
- 12. Bamforth SD, Braganca J, Eloranta JJ, et al. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co‐activator. Nat Genet. 2001;29:469‐474. [DOI] [PubMed] [Google Scholar]
- 13. Withington SL, Scott AN, Saunders DN, et al. Loss of Cited2 affects trophoblast formation and vascularization of the mouse placenta. Dev Biol. 2006;294:67‐82. [DOI] [PubMed] [Google Scholar]
- 14. Lopes Floro K, Artap ST, Preis JI, et al. Loss of Cited2 causes congenital heart disease by perturbing left‐right patterning of the body axis. Hum Mol Genet. 2011;20:1097‐1110. [DOI] [PubMed] [Google Scholar]
- 15. Kim GD, Das R, Rao X, et al. CITED2 restrains proinflammatory macrophage activation and response. Mol Cell Biol. 2018;38(5):e00452‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Preis JI, Wise N, Solloway MJ, Harvey RP, Sparrow DB, Dunwoodie SL. Generation of conditional Cited2 null alleles. Genesis. 2006;44:579‐583. [DOI] [PubMed] [Google Scholar]
- 17. Swamydas M, Luo Y, Dorf ME, Lionakis MS. Isolation of mouse neutrophils. Curr Protoc Immunol. 2015;110:3.20.1‐3.20.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015;43:W566‐W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Newson J, Stables M, Karra E, et al. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood. 2014;124:1748‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Segal BH, Han W, Bushey JJ, et al. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF‐kappaB‐driven transcription. PLoS Biol. 2013;11:e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu WT, Chu X, Wang J. Investigations of the underlying mechanisms of HIF‐1alpha and CITED2 binding to TAZ1. Proc Natl Acad Sci U S A. 2010;117:5595‐5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Guzman RN, Martinez‐Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB‐binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non‐identical binding sites. J Biol Chem. 2004;279:3042‐3049. [DOI] [PubMed] [Google Scholar]
- 25. Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF‐kappa B by a synthetic peptide containing a cell membrane‐permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255‐14258. [DOI] [PubMed] [Google Scholar]
- 26. Chen Y, Haviernik P, Bunting KD, Yang YC. Cited2 is required for normal hematopoiesis in the murine fetal liver. Blood. 2007;110:2889‐2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet. 2009;41:1191‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qu X, Lam E, Doughman YQ, et al. Cited2, a coactivator of HNF4alpha, is essential for liver development. EMBO J. 2007;26:4445‐4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB‐binding protein/p300‐interacting protein CITED2 as a peroxisome proliferator‐activated receptor alpha coregulator. J Biol Chem. 2004;279:24053‐24063. [DOI] [PubMed] [Google Scholar]
- 30. Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF‐1. Genes Dev. 1999;13:64‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Varma TK, Lin CY, Toliver‐Kinsky TE, Sherwood ER. Endotoxin‐induced gamma interferon production: contributing cell types and key regulatory factors. Clin Diagn Lab Immunol. 2002;9:530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao J, Kong HJ, Li H, et al. IRF‐8/interferon (IFN) consensus sequence‐binding protein is involved in Toll‐like receptor (TLR) signaling and contributes to the cross‐talk between TLR and IFN‐gamma signaling pathways. J Biol Chem. 2006;281:10073‐10080. [DOI] [PubMed] [Google Scholar]
- 33. Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front Immunol. 2018;9:2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kopf M, Schneider C, Nobs SP. The development and function of lung‐resident macrophages and dendritic cells. Nat Immunol. 2015;16:36‐44. [DOI] [PubMed] [Google Scholar]
- 35. Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia‐inducible factor‐1alpha by CITED2. Nat Struct Biol. 2003;10:504‐512. [DOI] [PubMed] [Google Scholar]
- 36. Li YP, Atkins CM, Sweatt JD, Reid MB. Mitochondria mediate tumor necrosis factor‐alpha/NF‐kappaB signaling in skeletal muscle myotubes. Antioxid Redox Signal. 1999;1:97‐104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2