

Summary
Recent advances in sequencing technologies have allowed for the identification of recurrent mutations in acute myeloid leukaemia (AML). The transcription factor CCAAT enhancer binding protein alpha (CEBPA) is frequently mutated in AML, and biallelic CEBPA‐mutant AML was recognised as a separate disease entity in the recent World Health Organization classification. However, CEBPA mutations are co‐occurring with other aberrations in AML, and together these lesions form the clonal hierarchy that comprises the leukaemia in the patient. Here, we aim to review the current understanding of co‐occurring mutations in CEBPA‐mutated AML and their implications for disease biology and clinical outcome. We will put emphasis on patterns of cooperation, how these lesions cooperate with CEBPA mutations and the underlying potential molecular mechanisms. Finally, we will relate this to patient outcome and future options for personalised medicine.
Keywords: molecular haematology, co‐occurring mutations, disease modelling, CEBPA biallelic acute myeloid leukaemia
Acute myeloid leukaemia (AML) is a heterogeneous group of aggressive haematological cancers that displays extensive variation in their clinical courses and in response to therapy. AML is characterised by an expansion of immature myeloid precursors at the expense of normal haematopoiesis, eventually leading to bone marrow failure. It is initiated by the step‐wise accumulation of genetic alterations affecting proliferation and/or differentiation in haematopoietic stem or progenitor cells. Sequencing efforts in AML, e.g. by The Cancer Genome Atlas (TCGA) and BEAT AML studies (Cancer Genome Atlas Research Network et al., 2013; Tyner et al., 2018), have identified an extensive catalogue of somatic mutations where the recurrent mutations show a high degree of overlap, while mutations with a low frequency are highly divergent. This suggests that the recurrent mutations are the ‘true’ oncogenic driver mutations causing the disease, whereas low‐frequency mutations are likely passenger mutations that do not affect AML biology.
Recent work has shown that AML development constitutes a continuous evolutionary process, where genetic changes are acquired at distinct stages during the course of the disease. Clonal haematopoiesis of indeterminate potential (CHIP) and age‐dependent clonal haematopoiesis (ARCH) describe the expansion of haematopoietic clones in healthy individuals and frequently occur in the elderly (Genovese et al., 2014; Jaiswal et al., 2014). Expanded clones harbour lesions in genes that are frequently mutated in haematological malignancies and are particularly enriched for mutations in epigenetic regulators. These lesions have been reported to increase the self‐renewal potential of haematopoietic stem cells (HSCs) and constitute the first step along the disease trajectory (Moran‐Crusio et al., 2011; Jeong et al., 2018). Whereas the vast majority of CHIP/ARCH individuals do not progress to haematological malignancies, clonal haematopoiesis has been reported to be associated with a tenfold increased risk of developing these diseases, and it therefore constitutes a pre‐leukaemic condition (Jaiswal et al., 2014). Additional mutations may drive progression toward cytopaenia, such a clonal cytopaenia of undetermined significance (CCUS), which may eventually develop into myelodysplastic syndrome (MDS) or full‐blown AML. Finally, several clones coexist in the patient, which are under continuous selective pressure, not only from treatment approaches, but potentially also from factors influencing their environment, such as infections.
Co‐occurring mutations in AML
Co‐mutation and mutual exclusivity of recurrent mutations can reveal patterns of mutational co‐segregation, and therefore suggest a potential biological cooperation between certain mutated genes (Tyner et al., 2018). One hypothesis of leukaemogenesis is the so‐called ‘two‐hit’ model where mutations conferring a proliferation/survival benefit cooperate with mutations blocking differentiation (Dash & Gilliland, 2001). However, cooperation may also lie in the selective importance of individual mutations at distinct stages of disease progression, e.g. mutations in epigenetic regulators during clonal haematopoiesis and growth receptors during later disease stages (Shlush et al., 2014; Abelson et al., 2018).
The combination of mutations in AML can be classified as: ‘co‐mutated’, i.e. found together more frequently than would be expected by each gene’s individual frequency; ‘neutral’, i.e. found together in similar frequency as would be expected by each gene’s individual frequency; and ‘mutually exclusive’, i.e. found together less frequently than would be expected by each gene’s individual frequency (Papaemmanuil et al., 2016). Co‐mutated lesions are usually non‐redundant in function and/or cooperate to induce leukaemogenesis, whereas mutually exclusive lesions are either redundant in function or have opposing functions. Mutually exclusive lesions might also arise due to synthetic lethality, if the combination of lesions leads to cell death, whereas the single lesions alone do not.
The fact that two (or more) mutations are found in the same patient does not necessarily mean that they co‐occur in the cells, and mutational co‐occurrence data of leukaemic cohorts need to be interpreted with caution. Due to clonal heterogeneity, several AML clones harbouring different mutations may exist in a patient, and sequencing of bulk AML cells may reveal co‐occurrence of mutations that exist in different clones. That said, approaches using variant allele frequency have been used to establish mutational co‐occurrence in individual cells, although these should be interpreted with some caution for the reasons mentioned above. Future efforts into sequencing AML at the single cell level are needed to fully resolve this issue and to firmly establish clonal hierarchies within individual leukaemias.
Oncogenic collaboration is not restricted to mutated genes, but may also involve genes that are subject to transcriptional deregulation, potentially driven by epigenetic changes. In addition, a genetic lesion may even ‘collaborate’ with a gene that is not subject to transcriptional changes. The latter has been framed non‐oncogenic addiction, where a mutation in gene A induces a cellular state in which the cell is dependent on gene B for its survival (Solimini et al., 2007; Luo et al., 2009). Examples of this class include genes in various stress pathways.
Finally, oncogenic cooperation is frequently discussed in terms of response to therapy. However, mutations are generally not under a selective pressure with regards to response to therapy during their evolution in the primary tumour. One exception is tumours that develop secondary to other cancers. Thus, differences in response to therapy may not necessarily reflect oncogenic cooperation.
CEBPA biallelic AML
The gene encoding the transcription factor, CCAAT enhancer binding protein alpha (CEBPA), is biallelically mutated (CEBPAbi) in 2–15% (average 5%) of de novo AML patients, with a higher incidence in Asian (6–15%; average 12%) compared to Caucasian (2–6%; average 4%) populations (Table 1). CEBPAbi AML has been classified as a novel AML entity in the 2017 revised World Health Organization (WHO) classification of tumours of haematopoietic and lymphoid tissues, and is a favourable prognostic marker in AML, with increased overall survival (OS) and event‐free survival (EFS) of patients with CEBPAbi compared to CEBPA‐wild‐type (CEBPAwt) or monoallelic CEBPA‐mutated patients. When we mention mutations in CEBPA in this review, we will be referring to mutations in the context of CEBPAbi AML.
Table 1.
Data of co‐occurrence of mutations in CEBPA biallelic AML reported in this review come from the listed studies, which were identified during a literature survey for reported CEBPAbi AML cases. Cited references: (Dufour et al., 2010; Chou et al., 2011; Metzeler et al., 2011; Taskesen et al., 2011; Greif et al., 2012; Cancer Genome Atlas Research et al., 2013; Green et al., 2013; Grossmann et al., 2013; Fasan et al., 2014; Kihara et al., 2014; Krauth et al., 2015; Ahn et al., 2016; Lavallee et al., 2016; Metzeler et al., 2016; Papaemmanuil et al., 2016; Theis et al., 2016; Wakita et al., 2016; Wang et al., 2016; Lin et al., 2017; Mannelli et al., 2017; Rose et al., 2017; Konstandin et al., 2018; Su et al., 2018; Tien et al., 2018; Zhang et al., 2019a; Zhang et al., 2019b).
|
Publication Author |
Year | All cases, n | Age, years, median (range) | Proportion de novo AML, % | Proportion CN‐AML, % | CEBPAbi cases, n (%) |
|---|---|---|---|---|---|---|
| Ahn | 2016 | 404 | 52 (15–84) | N.A. | 100 | 51 (13) |
| Chou | 2011 | 486 | 52 (15–90) | 100 | 45 | 45 (9) |
| Cancer Genome Atlas Research | 2013 | 200 | 55 ± 16* | 100 | 47 | 6 (3) |
| Dufour | 2010 | 467 | 61 (17–85) | 71 | 100 | 20 (4) |
| Fasan | 2014 | 2296 | 68 (15–100) | 89 | 76 | 104 (5) |
| Green | 2012 | 1427 | N.A. | N.A. | N.A. | 55 (4) |
| Greif | 2012 | 160 | N.A. | N.A. | 100 | 33 (21) |
| Grossmann | 2013 | 95 | 58 (15–87) | 98 | 95 (–) | |
| Kihara | 2014 | 197 | N.A. (15–64) | 100 | 37 | 19 (10) |
| Konstandin | 2018 | 48 | 57 (20–84) | N.A. | 100 | 48 (–) |
| Krauth | 2015 | 3157 | 67 (17–100) | 85 | 62 | 110 (3) |
| Lavallée | 2016 | 415 | 58 (17–87) | 94 | 32 | 14 (3) |
| Lin | 2017 | 112 | 43 (11–79) | N.A. | N.A. | 7 (6) |
| Mannelli | 2017 | 251 | 57 (16–81) | 91 | 47 | 16 (6) |
| Metzeler | 2011 | 220 | N.A. (60–83) | N.A. | 100 | 11 (5) |
| Metzeler | 2016 | 664 | 57 (18–86) | 86 | N.A. | 27 (4) |
| Papaemmanuil | 2016 | 1540 | † (18–84) | 91 | N.A. | 66 (4) |
| Rose | 2017 | 4373 | 67 (18–100) | 100 | 54 | 136 (3) |
| Su | 2018 | 553 | N.A. | 100 | N.A. | 81 (15) |
| Taskensen | 2011 | 1182 | 48 (16–60) | N.A. | 100 | 91 (8) |
| Theis | 2016 | 113 | N.A. (20–76) | 96 | N.A. | 113 (–) |
| Tien | 2018 | 693 | 55 (15–94) | 100 | N.A. | 65 (9) |
| Wakita | 2016 | 184 | N.A. (17–86) | 100 | N.A. | 16 (9) |
| Wang | 2016 | 95 | 45 (12–88) | N.A. | N.A. | 13 (14) |
| Zhang | 2019a | 259 | 23 (2–68) | N.A. | N.A. | 26 (10) |
| Zhang | 2019b | 609 | 23 (1–75) | N.A. | N.A. | 76 (12) |
De novo AML refers to patients with no prior history of myeloid diseases or exposure to leukaemogenic agents, i.e. excluding secondary and therapy‐related AML.
CN‐AML, cytogenetically normal AML; N.A., data not available.
Mean ± SD.
Three included studies: HD98A (n = 627) median age 47 (18–65) years, HD98B (n = 173) median age 66 (58–84) years, and 07/04 (n = 740) median age 49 (18–61) years.
The functions of CEBPA in normal and malignant haematopoiesis have been extensively reviewed previously (Ohlsson et al., 2016; Avellino & Delwel, 2017; Pulikkan et al., 2017). Briefly, CEBPA is a key myeloid transcription factor that is both important for HSC self‐renewal and for driving transcriptional programmes during myeloid differentiation (Hasemann et al., 2014; Avellino et al., 2016; Pundhir et al., 2018). CEBPA comes in two forms, the full‐length 42 kDa (p42) and the N‐terminally truncated 30 kDa isoform (p30). In normal cells, p42 is dominant, but stop and frameshift mutations within the 5′‐end of the one‐exon CEBPA gene, sustain p30 expression at the expense of p42. These N‐terminal lesions are frequently combined with C‐terminal mutations (although biallelic N‐terminal combinations also occur), which either block CEBPA dimerisation with itself and other CEBP family members or block DNA binding. In either event, biallelic CEBPA mutations converge at the expression of CEBPA‐p30 homodimers as the only form of functional CEBPA in the mutated cells.
Importantly, CEBPAbi AML has successfully been modelled in the mouse with either a mimic of the human N‐terminal CEBPA mutant in both alleles (Lp30 or L/L), C‐terminal CEBPA mutant in both alleles (K/K) or a combination of C‐ and N‐terminal mutations (K/L) (Kirstetter et al., 2008; Bereshchenko et al., 2009). These models have demonstrated that the sole expression of CEBPA‐p30 leads to a block in myeloid differentiation, promotes transcriptional deregulation and is associated with cell cycle defects (Kirstetter et al., 2008). Despite these insights, we still have an incomplete understanding of the mechanistic basis for CEBPA‐p30‐induced AML, specifically in terms of the functional interactions between CEBPA lesions and other genetic and epigenetic aberrations found in human AML.
Co‐occurrence of mutations in CEBPA biallelic AML
In CEBPAbi AML, CEBPA lesions co‐occur with mutations in numerous genes with diverse cellular functions. As for other AML subtypes, the recurrently mutated genes belong to epigenetic regulators, transcription factors, cell‐signalling factors, splicing factors, members of the cohesin complex and tumour suppressors. In contrast, CEBPA lesions do not appear to co‐occur with classical aberrations such as inversions [e.g. inv(16), inv(3)] and translocations [e.g. t(15;17), t(8;21), t(6;9) and mixed‐lineage leukaemia (MLL)‐fusions]. Specifically, in the large study by Papaemmanuil et al. only two cases out of 66 CEBPAbi AML did co‐occur with the 280 reported cases of AML with classical cytogenetic aberrations (Papaemmanuil et al., 2016). Thus, for the purpose of the present review, we therefore focused on mutations in protein‐coding genes. In order to obtain more quantitative data for the co‐occurrence of mutations in CEBPAbi AML, we surveyed the literature and identified 26 studies with reported CEBPAbi AML cases (Table 1), including four large next‐generation sequencing (NGS)‐based studies, with one representing cytogenetically normal (CN‐)AML, and three de novo AML (Papaemmanuil et al., 2016; Konstandin et al., 2018; Su et al., 2018; Zhang et al., 2019b) (Fig 1). From this, it is apparent that mutational burden differs between studies which, in turn, might be due to choice of methodology, i.e. panel vs. exome sequencing, and/or differences in patient cohorts with respect to both the type of AML and population characteristics. In the reviewed literature, data regarding clinical outcomes, i.e. survival and relapse frequency, are sparse; however, when available, the impact of co‐occurring mutations on clinical outcome have been mentioned.
Figure 1.
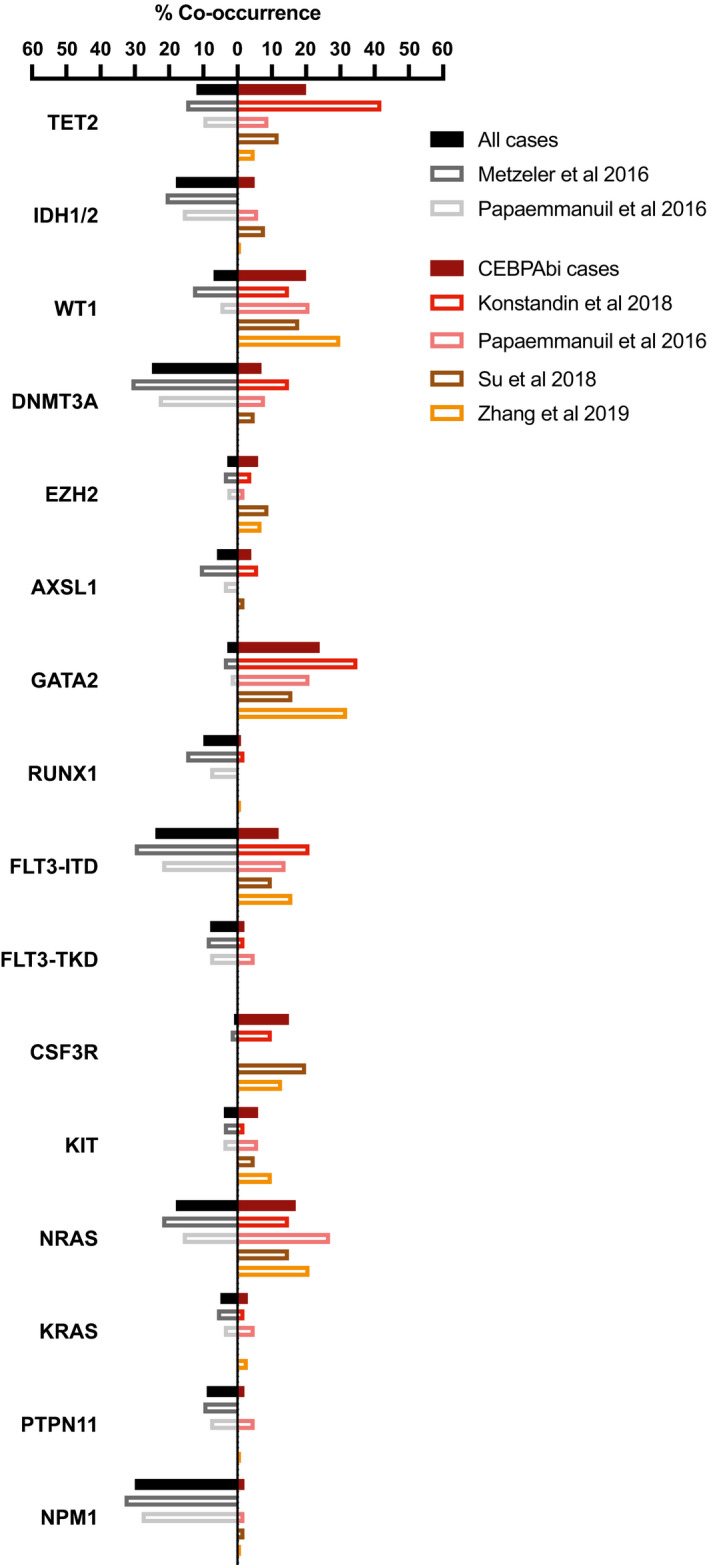
Data on mutational co‐occurrence from four large next‐generation sequencing (NGS) based studies with Konstandin et al. (2018) (n = 48) representing cytogenetically normal (CN‐)AML and Papaemmanuil et al. (2016) (n = 66), Su et al. (2018) (n = 81) and Zhang et al. (2019b) (n = 76) representing de novo AML. For comparison, data from the same cohorts representing all cases have been added when available; Metzeler et al., (2016) (n = 664) CN‐AML cases and Papaemmanuil et al. (2016) (n = 1540) de novo AML cases. Data from the individual studies are represented by non‐filled bars. Solid bars represent a calculated average from the studies included (percent of cases with co‐occurring mutations in all samples (left) or in CEBPAbi cases (right).
Epigenetic regulators
Epigenetic dysregulation is central to AML development and biology, and include aberrant DNA methylation, histone methylation and histone acetylation (Gallipoli et al., 2015). Hence co‐occurrence of mutations in genes coding for epigenetic factors and CEBPAbi mutations is frequent.
DNA methylation
The ten–eleven translocation (TET) family of proteins catalyse the conversion of 5‐methylcytosine (5mC) to 5‐hydroxymethylcytosine (5hmC), thus leading to DNA demethylation. Co‐occurrence of TET2 mutations in CEBPA‐mutated AML is prevalent with up to 40% of the CEBPAbi cases being reported to have mutated TET2 (Table 1). Konstandin et al. (2018) reported TET2 mutations and CEBPA to be co‐mutated, i.e. found together more frequently than would be expected by each gene’s individual frequency. When analysing clinical outcomes in the context of CEBPAbi AML, TET2‐mutated cases had shorter OS and EFS than TET2wt cases (Grossmann et al., 2013; Konstandin et al., 2018).
Moreover, the isocitrate dehydrogenase 1 and 2 (IDH1/2) enzymes, which catalyse the oxidative decarboxylation of isocitrate to α‐ketoglutarate, are both recurrently mutated in CEBPAbi AML. The leukaemogenic effect of mutant IDH1/2 is thought to be the result of inhibition of the TET enzymatic function by aberrant production of the oncometabolite 2‐hydroxyglutarate (2‐HG). These gain‐of‐function mutations are co‐occurring with CEBPAbi in around 10% of the cases (Table 1). IDH1/2 and CEBPA mutations have been reported to be mutually exclusive, i.e. found together less frequently than would be expected by each gene’s individual frequency (Fasan et al., 2014; Konstandin et al., 2018). Additionally, IDH1/2 mutations have been shown not to have an impact on OS or EFS (Grossmann et al., 2013).
Mutations in the WT1 gene encoding the transcription factor and tumour suppressor gene Wilms tumour 1 (WT1; discussed here due to its impact on TET2 function) co‐occur with CEBPA lesions in up to 30% of the CEBPAbi cases (Table 1). WT1 has been shown to recruit TET2 to its target genes and WT1 mutations result in loss of function of TET2, and, in line with this, mutations in TET2, IDH1/2 and WT1 are reported as mutually exclusive in AML (Rampal et al., 2014; Wang et al., 2015). WT1 and CEBPA are significantly co‐mutated in CEBPAbi AML, but effects on EFS are divergent, with Grossmann et al. and Zhang et al. reporting no significant impact, while Su et al. reported shorter EFS (Grossmann et al., 2013; Fasan et al., 2014; Krauth et al., 2015; Lavallee et al., 2016; Papaemmanuil et al., 2016; Su et al., 2018; Zhang et al., 2019b). Nonetheless, WT1 mutations had no impact on OS in any of these studies (Grossmann et al., 2013; Su et al., 2018; Zhang et al., 2019b).
Finally, DNA methyltransferase 3 alpha (DNMT3A), an enzyme responsible for de novo DNA methylation, is also prevalently mutated in CEBPAbi AML cases, where up to 15% carry DNMT3A mutations (Table 1). Further, DNMT3A and CEBPAbi are classified as mutually exclusive (Ahn et al., 2016; Metzeler et al., 2016; Papaemmanuil et al., 2016; Konstandin et al., 2018). DNMT3A mutations have no impact on OS or EFS in the context of CEBPAbi AML (Grossmann et al., 2013).
Collectively, genetic lesions predicted to either reduce (DNMT3A) or enhance (TET2, IDH1/2, WT1) DNA methylation are frequent in CEBPAbi AML. However, co‐mutation seems to be restricted to TET2 and WT1. Concordantly, TET2‐ and WT1‐mutated AML shared nearly the same methylation profile, where the affected sites constitute a subset of those affected in IDH1/2‐mutant AML (Rampal et al., 2014). Whereas the functional consequences of this remain unknown, future experiments in mice promise to uncover the mechanistic basis for how TET2 and WT1 mutations collaborate with CEBPA lesions in the context of CEBPAbi AML.
Histone modification
The lysine methyltransferase 2 family (KMT2) mediates transcriptional activation, and mutations in the genes encoding KMT2A (KMT2A/MLL1), KMT2C (KMT2C/MLL2) and KMT2D (KMT2D/MLL4) have been reported to co‐occur with CEBPA lesions in a few cases (Table 1). KMT2A partial tandem duplication (KMT2A‐PTD) and CEBPA lesions have been reported to be mutually exclusive, and several large studies have found no overlap between the two aberrations (Dufour et al., 2010; Greif et al., 2012; Grossmann et al., 2013; Fasan et al., 2014; Metzeler et al., 2016; Su et al., 2018).
KMT2A is part of a large protein complex involved in transcriptional activation, and although mutations in KMT2 members are infrequent in CEBPAbi AML, the KMT2A complex was recently identified as a potential druggable target in this AML subtype (Liedtke & Cleary, 2009; Schmidt et al., 2019). Specifically, the authors demonstrated that CEBPA‐p30 and KMT2A interacted physically on chromatin. Deletion of KMT2A or mutations within functionally important KMT2A domains, such as the WD repeat domain 5 (WDR5)‐interacting domain or the Menin‐binding‐motif, all showed anti‐proliferative effects, whereas targeting the catalytic SET domain had no impact. Moreover, a small‐molecule inhibitor, which disrupts the Menin‐KMT2A interaction, displayed high activity in CEBPA‐mutated cell lines. This, along with an earlier report demonstrating that a small‐molecule inhibitor targeting the WDR5‐KMT2A interaction also affected proliferation of CEBPA‐mutant cell lines, points to the KMT2A complex being a potential target in CEBPAbi AML (Grebien et al., 2015).
Several genes involved in epigenetic repression via the polycomb repressive complexes (PRC) 1 and 2 have been found to be recurrently mutated in CEBPAbi AML cases. Enhancer of zeste homologue 2 (EZH2) is a histone methyltransferase responsible for depositing the trimethyl mark on lysine 27 of histone 3 (H3K27me3), and, as a part of PRC2, the enzyme mediates transcriptional repression. EZH2 is mutated in 6% of CEBPAbi AML cases (Table 1). Likewise, mutations in additional sex combs like‐1 (ASXL1), encoding a protein which is also part of the PRC2 complex as well as the polycomb repressive deubiquitinase complex, are found to co‐occur with CEBPA lesions in 4% of the cases (Table 1). Further, the genes encoding the BCL6 corepressor (BCOR) and its homologue, BCOR‐like1 (BCORL1), are both recurrently mutated in AML (Tiacci et al., 2012). BCOR is part of the PRC1.1 complex, which also mediates transcriptional repression. BCOR and BCORL1 mutations have been reported to co‐occur with CEBPA lesions in two CEBPAbi cases each (Table 1). Collectively, these finding suggest that lesions in PRC1/2 complexes collaborate with CEBPA mutations in the context of CEBPAbi AML.
Finally, mutations in the genes encoding histone acetyl transferases have also been reported in CEBPAbi AML, including the CREB binding protein (CREBBP or CBP) and E1A binding protein p300 (EP300) (Lavallee et al., 2016; Papaemmanuil et al., 2016). Specifically, EP300 lesions were found to be significantly co‐mutated with CEBPA, as it appeared in four cases (6%) of CEBPAbi AML cases compared to an overall frequency of EP300 mutations of 1·5% in AML (Papaemmanuil et al., 2016).
Collectively, mutations in genes affecting histone modifiers are co‐occurring in CEBPAbi AML, and some of these likely have functional ramifications, although the mechanistic details are lacking. In particular, the KMT2A complex appears to be of functional importance, and it constitutes a potential therapeutic target in this AML subtype.
Transcription factors
Several transcription factors, including important haematopoietic linage‐specific transcription regulators, are recurrently mutated in AML, and therefore also in CEBPAbi AML.
GATA binding protein 2 (GATA2), an important transcription factor for the haematopoietic lineage, is abundantly and heterozygously (when reported) mutated in 15–35% of CEBPAbi AML cases, and indeed GATA2 and CEBPA are significantly co‐mutated (Table 1). As for clinical outcome, GATA2‐mutated CEBPAbi AML cases have been shown to have longer OS and EFS compared to GATA2wt CEBPAbi cases (Greif et al., 2012; Fasan et al., 2013; Grossmann et al., 2013; Hou et al., 2015; Tien et al., 2018). As for a specific functional interaction between GATA2 and CEBPA lesions, mutations within the N‐terminal zinc finger (ZF1) of GATA2 reduced CEBPA‐p30‐dependent transcriptional activation in a reporter assay (Greif et al., 2012). This finding raises the possibility that GATA2 ZF1 mutations may collaborate with CEBPA lesions in order to deregulate the expression of downstream target genes. Interestingly, CRISPR/Cas9‐mediated deletion of GATA2 leads to reduced proliferative capacity and induction of myeloid differentiation in CEBPA‐p30 expressing cells (Schmidt et al., 2019). This finding suggests that, whereas the heterozygous mutations of GATA2 found in patients with CEBPAbi lesions promote leukaemogenesis, the complete loss of GATA2 is incompatible with leukaemic growth. We hypothesise that this is due to the importance of GATA2 in maintaining self‐renewal, a property of GATA2 clearly established in HSCs (Lim et al., 2012).
Runt‐related transcription factor 1 (RUNX1) is important for differentiation of haematopoietic cells. RUNX1 mutations have been reported to co‐occur with CEBPA lesions in AML at low frequencies, and in some studies as not co‐occurring at all, despite the fact that RUNX1 lesions are found in 9% of overall AML cases (Table 1). Thus, RUNX1 and CEBPA lesions are mutually exclusive. However, this does not necessarily apply to translocations involving the RUNX1 locus such as RUNX1‐ETO and RUNX1‐ETV6 (Fasan et al., 2014; Papaemmanuil et al., 2016).
Experimentally, RUNX1 has been reported to control the expression of CEBPA, which could potentially render CEBPA lesions irrelevant in a RUNX1‐mutated context (Guo et al., 2012). Indeed, the expression of CEBPA is reduced in RUNX1‐mutated AML (Grossmann et al., 2012).
Finally, work in mice has shown that the SRY‐box transcription factor 4 (SOX4), which is repressed by wt CEBPA, is increased in the leukaemic stem cell compartment in a mouse model of CEBPA‐mutant AML and that its repression restored myeloid differentiation (Zhang et al., 2013). These findings suggest that mutated CEBPA loses its normal ability to repress SOX4 expression and that targeting SOX4 in the context of CEBPAbi AML may constitute a potential treatment option.
Collectively, CEBPA is co‐mutated with GATA2 and mutually exclusive to mutations in RUNX1, thus highlighting the differential impact that transcription factor mutations has on AML biology.
Cell‐signalling factors
Oncogenic signalling in the receptor tyrosine kinase (RTK)/RAS‐signalling pathway is frequently activated in cancer, and AML is no exception. Thus, mutations in genes encoding proteins involved in this pathway are found in more than half of all AML cases, and these are often gain‐of‐function mutations (Papaemmanuil et al., 2016).
Lesions in the gene encoding the FMS‐like tyrosine kinase 3 (FLT3) occur in approximately 30% of all AML cases, where the internal tandem duplication (ITD) is most frequent, followed by mutations in the tyrosine kinase domain (FLT3‐TKD). FLT3‐ITD and FLT3‐TKD co‐occur with CEBPA aberrations in around 15% and 2% of cases, respectively (Table 1). Moreover, both FLT3‐ITD and FLT3‐TKD are considered mutually exclusive with CEBPA lesions, but for FLT3‐TKD, this negative association was only reported in two studies (Dufour et al., 2010; Taskesen et al., 2011; Greif et al., 2012; Fasan et al., 2014; Kihara et al., 2014; Ahn et al., 2016; Papaemmanuil et al., 2016; Konstandin et al., 2018). FLT3‐ITD has no impact on OS or EFS (Grossmann et al., 2013; Zhang et al., 2019b). However, OS is significantly shorter in FLT3‐ITD than in FLT3‐wt cases if only CN‐CEBPAbi AML cases are considered (Zhang et al., 2019b).
FLT3‐ITD results in the constitutive activation of FLT3, but this is not sufficient to promote AML development in mice. Still, in the context of the mouse model of CEBPAbi AML (K/L), FLT3‐ITD accelerates leukaemic development, and this is associated with an increase in granulocyte‐monocytic progenitors (GMPs) during the pre‐leukaemic phase (Reckzeh et al., 2012). Moreover, gene expression analysis of leukaemic GMPs revealed that FLT3‐ITD was associated with a more immature phenotype and increased self‐renewal. The mutual exclusiveness of FLT3 and CEBPA lesions in human AML, and the observed acceleration of CEBPA‐mutant AML in the context of the murine model, appears conflicting at a first glance. Nonetheless, FLT3 activating mutations have been shown to selectively impede the function of CEBPA‐p42, but not CEBPA‐p30, via phosphorylation of a site (S21) present only in CEBPA‐p42 (Radomska et al., 2006; Radomska et al., 2012). Conversely, CEBPA‐p30 over‐expression has been shown to increase FLT3 expression (Alachkar et al., 2015). These reciprocal interactions between FLT3 and CEBPA are likely to reduce the selective pressure for lesions in both genes during leukaemic initiation, which could explain their mutual exclusivity. Yet, when the two lesions are combined in the context of the mouse model, their combination may accelerate leukaemic growth during the expansion phase.
Mutations in the genes encoding the cytokine receptors colony stimulating factor 3 receptor (CSF3R; Cluster of Differentiation 114 [CD114] or granulocyte colony stimulating factor receptor [G‐CSF‐R]) and KIT (CD117 or stem cell factor receptor [SCF‐R]) are co‐occurring with CEBPA lesions in 10–20% and 2–10% of CEBPAbi AML cases, respectively (Table 1). CSF3R and CEBPA are co‐mutated; however, the clinical significance remains controversial (Lavallee et al., 2016; Konstandin et al., 2018; Zhang et al., 2018). Whereas Su et al. reported both EFS and OS to be shorter in CSF3R‐mutated cases of CEBPAbi AML, similar effects were not identified in another study (Su et al., 2019; Zhang et al., 2019b). Likewise, KIT mutations have been shown to affect EFS adversely when restricting the analysis to CN‐AML, while OS and EFS are not changed when comparing to all CEBPAbi AML cases (Zhang et al., 2019b).
CSF3R predominantly signals through the Janus kinase (JAK)‐signal transducers and activators of transcription (STAT) pathway and, interestingly, CEBPAbi AML has been reported to be more sensitive to JAK inhibitor treatment ex vivo than CEBPAwt AML, with CSF3R‐mutated CEBPAbi AML samples being particularly sensitive to JAK‐inhibition (Lavallee et al., 2016).
Additionally, mutations in JAK2, a non‐receptor tyrosine kinase, have been reported to co‐occur with CEBPA aberrations in six cases, and JAK3 in seven cases of CEBPAbi AML (Kihara et al., 2014; Lin et al., 2017; Konstandin et al., 2018; Su et al., 2018; Zhang et al., 2019b).
Mutations in NRAS (20%) and KRAS (5%) are commonly found in AML and CEBPAbi AML cases mimic the overall mutation frequencies in AML with NRAS and KRAS aberrations co‐occurring with lesions in CEBPA in 10–30% and 2–5% of cases, respectively (Table 1). One study has reported NRAS and CEBPA as significantly co‐mutated, but NRAS mutations have no impact on OS or EFS (Grossmann et al., 2013; Papaemmanuil et al., 2016; Zhang et al., 2019b).
Last among members of the RTK/RAS‐signalling pathway, the gene encoding the enzyme tyrosine‐protein phosphatase non‐receptor type 11 (PTPN11) is mutated in 10% of AML cases. PTPN11 mutations have been reported in CEBPAbi AML at variable frequencies from 0% to 5%, but PTPN11 and CEBPA lesions have not been reported to be mutually exclusive (Table 1).
Finally, Zhang et al. investigated the significance of mutations in RTK/RAS pathway genes (i.e. NRAS, KRAS and PTPN11 mutated) in CEBPAbi AML, and found no significant effects on OS or EFS as compared to non‐mutated CEBPAbi cases, neither when comparing all de novo cases nor the CN‐AML subgroup. Whereas mutations in tyrosine kinase genes (i.e. FLT3‐ITD, CSF3R, KIT and JAK3 mutated) resulted in significantly shorter OS and EFS in CEBPAbi CN‐AML as compared to non‐mutated cases; however, this difference was no longer seen when comparing all CEBPAbi AML (Zhang et al., 2019b).
In summary, whereas mutations in the RTK/RAS pathway are frequent in CEBPAbi AML, they do not seem to cooperate extensively with lesions in CEBPA.
Splicing factors
Splicing factors are recurrently mutated in AML, with lesions found in around 10% of patients with AML, including the genes encoding the serine and arginine rich splicing factor 2 (SRSF2), the splicing factor 3b subunit 1 (SF3B1) and the U2 small nuclear RNA auxiliary factor 1 (U2AF1) (Zhou & Chng, 2017).
In CEBPAbi AML, a few cases of co‐occurring mutations in splicing‐factor genes have been reported; SRSF2 was found to co‐occur with CEBPA in 10 cases of CEBPAbi AML, SF3B1 in one case and U2AF1 in two cases (Table 1). In addition, the zinc finger CCCH‐type, RNA binding motif and serine/arginine rich 2 gene (ZRSR2), which encodes a protein that is associated with the U2 small nuclear ribonucleoprotein (snRNP) complex, was mutated in one case (Lavallee et al., 2016). Hence, splicing‐factor mutations are not common in CEBPAbi, and, moreover, SRSF2 and CEBPA mutations have been shown to be mutually exclusive (Papaemmanuil et al., 2016).
Functional approaches have also been undertaken to study the importance of de‐regulation of splicing in the context of CEBPAbi AML. Using an in vivo short‐hairpin RNA (shRNA) screen, our laboratory recently reported the identification of the non‐mutated splicing regulator, RNA binding motif protein 25 (RBM25), as a novel tumour suppressor in CEBPAbi AML (Ge et al., 2019). Specifically, we found that RBM25 downregulation accelerates leukaemogenesis and shortens survival of mice transplanted with CEBPA‐mutant AML. The effects of RBM25 were not restricted to CEBPAbi AML, and, mechanistically, we demonstrated that RBM25 controls splicing of the genes encoding the apoptotic regulator B‐cell lymphoma‐extra large (BCL‐X) and bridging integrator 1 (BIN1), an inhibitor of the universal oncogene MYC. Thus, decreased RBM25 levels inhibited apoptosis and promoted the expression of MYC target genes, thereby driving oncogenic expansion. In accordance, patients with AML with low RBM25 expression displayed inferior outcomes.
In summary, whereas mutations in splicing factors do not co‐occur frequently with CEBPA lesions, transcriptional de‐regulation of splicing factors may constitute an overlooked driver of AML development.
Cohesins, nucleophosmin 1 (NPM1) and tumour protein p53 (TP53)
The cohesin complex plays multiple roles in the context of chromatin such as promoting chromosome segregation during mitosis, maintaining chromatin boundaries, as well as facilitating the looping between different gene regulatory regions. Members of the cohesin complex are often mutated in AML, and the genes encoding stromal antigen 2 (STAG2) and RAD21 are found to be mutated in 6% and 2–9% of CEBPAbi AML cases, respectively (Table 1). Similarly, the cohesin‐complex components structural maintenance of chromosomes 1A and 3 (SMC1A/SMC3) have been reported to be mutated in CEBPAbi AML, albeit few cases have been identified (Lavallee et al., 2016; Konstandin et al., 2018). Correspondingly, the TCGA research network reported cohesion mutations in one of 16 CEBPAbi cases (Cancer Genome Atlas Research Network et al., 2013). As of yet, no experimental studies have addressed the functional importance of cohesion complex lesions in the context of CEBPAbi AML.
Despite being one of the most frequent molecular abnormalities in AML, nucleophosmin 1 (NPM1) gene mutations are rarely found in CEBPAbi AML cases. Several studies have reported either no or only a modest overlap been NPM1 and CEBPA in the range of 2–3% of CEBPAbi cases (Table 1). Hence, NPM1 and CEBPA mutations are mutually exclusive, with several studies reporting a negative association (Verhaak et al., 2005; Dufour et al., 2010; Taskesen et al., 2011; Fasan et al., 2014; Kihara et al., 2014; Ahn et al., 2016; Metzeler et al., 2016; Papaemmanuil et al., 2016; Konstandin et al., 2018).
In a recent study, Gu et al. (2018) have elucidated the mechanism for the leukaemogenic effects of mutated NPM1, and these results might give clues as to why these mutations are mutually exclusive with CEBPA lesions. Mutant NPM1 aberrantly accumulates in the cytoplasm and also promotes the nuclear export of the myeloid transcription factor PU.1. The loss of nuclear PU.1 leads to a change in the repertoire of interaction partners for CEBPA and RUNX1, from coactivators to corepressors; hence, resulting in the shift towards repression of myeloid differentiation genes. Taken together, this suggests at least partly redundant functions for mutant NPM1 and CEBPA lesions in AML, i.e. a block in myeloid differentiation that may explain their mutual exclusivity in AML.
Finally, tumour protein p53 (TP53) is a tumour suppressor that is mutated in 6% of de novo AML cases, and TP53 mutations co‐occurred with CEBPA lesions in six CEBPAbi cases (Table 1).
The most commonly co‐mutated and mutually exclusive genes and their effect on OS/EFS are summarised in Table 2.
Table 2.
Summary of the recurrently co‐mutated or mutually exclusive co‐occurring mutations in CEBPAbi cases and their effects on clinical outcomes.
| Mutation | Frequency in CEBPAbi, % (min–max) | Classification | Effect on OS | Effect on EFS |
|---|---|---|---|---|
| GATA2 | 24 (14–39) | Co‐mutated | ↑ | ↑ |
| WT1 | 20 (14–30) | Co‐mutated | ↔ | ↓/↔ |
| TET2 | 20 (5–42) | Co‐mutated | ↓ | ↓ |
| CSF3R | 15 (10–20) | Co‐mutated | ↓/↔ | ↓/↔ |
| FLT3 | 14 (3–23) | Mutually exclusive | ↓/↔ | ↔ |
| DNMT3A | 7 (0–15) | Mutually exclusive | ↔ | ↔ |
| IDH1/2 | 5 (0–14) | Mutually exclusive | ↔ | ↔ |
| NPM1 | 2 (0–4) | Mutually exclusive | N.A. | N.A. |
| RUNX1 | 1 (0–6) | Mutually exclusive | N.A. | N.A. |
↑, increased; ↔, unaltered; and ↓, decreased. N.A., data not available; OS, overall survival; and EFS, event free survival.
Additional insights from experimental work
Mechanism‐based studies have been instrumental in providing novel insights into AML biology, and a number of these have been highlighted throughout the present review. It is clear from this work that myeloid progenitors are subject to a strong selective pressure for genetic or transcriptional changes that increase the ratio of CEBPA‐p30 to CEBPA‐p42. Apart from mutations and activation of FLT3, the p30/p42 ratio may also change by the expression of tribbles pseudokinase 2 (TRIB2). TRIB2 is highly expressed in a subgroup of patients with AML who display features of CEBPAbi AML, but without mutations in CEBPA (Keeshan et al., 2006). Recent work has shown that TRIB2 facilitates the specific degradation of the CEBPA‐p42 isoform via ubiquitination of lysine 48, a residue not present in CEBPA‐p30, and consequently that TRIB2‐mediated leukaemogenesis is dependent on the presence of the CEBPA‐p42 isoform (O'Connor et al., 2016). At present, the underlying driver responsible for the transcriptional upregulation of TRIB2 is not known, but inhibition of the TRIB2‐mediated degradation of CEBPA may constitute a novel therapeutic option in CEBPAbi AML.
Mechanism‐based studies have also yielded insights into the downstream targets playing functional roles in CEBPAbi AML. Apart from SOX4 mentioned above, our laboratory recently discovered that the ectoenzyme CD73 (5'‐nucleotidase ecto [NT5E]) is upregulated in human CEBPAbi AML, as well as in the mouse model of CEBPA‐mutant AML. We demonstrated that this up‐regulation was due to the specific binding of the CEBPA‐p30 isoform to a normally silenced enhancer of the NT5E gene (Jakobsen et al., 2019). Knockdown of NT5E delayed AML development in CEBPA‐mutant AML in mice, both by targeting NT5E and its enhancer. CD73 catalyses the conversion of AMP to adenosine, and we find that this sets up a tumour‐promoting adenosinergic autocrine signalling loop, which also appears to be operative in human CEBPAbi AML. Interestingly, the combined inhibition of CD73 and the adenosine receptor, A2AR, resulted in increased survival time of mice transplanted with CEBPA‐mutated leukaemic cells. Hence, blocking the CD73/A2AR axis represents a potential new druggable target in CEBPAbi AML.
Clonal evolution in CEBPA biallelic AML
Clonal evolution is an important aspect of AML biology and we therefore surveyed the limited number of reports describing paired analyses of diagnostic and relapse CEBPAbi samples (Tiesmeier et al., 2003; Shih et al., 2006; Grossmann et al., 2013; Tawana et al., 2015; Li & Su, 2019). Given the different technologies used in these studies, and the limited sample numbers, it was not possible to discern any patterns of changes in the presence of co‐occurring mutations. However, if we focus on the mutations in CEBPA itself an interesting pattern emerges. Specifically, we find that whereas the overwhelming majority of somatic CEBPAbi cases harbour the same CEBPA mutations at both diagnosis and relapse, the pattern is markedly different in familial cases. Here relapse is associated with the acquisition of new C‐terminal lesions, which in combination with the germline N‐terminal mutation, drive the development of a new leukaemic clone. In future studies it will be important to more adequately address the patterns of co‐occurrence mutations during clonal evolution in CEBPAbi AML.
Implications for personalised medicine
The recent progress in NGS has given rise to a new challenge in AML treatment, i.e. how to translate the knowledge of the mutational status of a patient into tailor‐made therapy targeting the specific aberrations of a given leukaemia. The first steps towards personalised medicine have already been taken, where identification of oncogenic driver mutations and the understanding of how they sustain disease development and maintenance are heavily researched.
During the last couple of years, several targeted therapies against AML carrying specific driver mutations have been approved: FLT3‐mutated AML (Midostaurin; Rydapt® & Gilteritinib; Xospata®), and IDH1/2‐mutated AML (IDH1: Ivosidenib; Tibsovo® & IDH2: Enasidenib; Idhifa®) (DiNardo & Perl, 2019). Thus, following the clinical outcomes for patients with CEBPAbi AML carrying FLT3 and/or IDH1/2 mutations treated with these new targeted therapies will be interesting.
Hopefully, this is the start of a new era in AML treatment that will improve survival and quality of life for patients with AML, aided by large studies investigating drug sensitivity in conjunction with mutational status (e.g. the BEAT AML study (Tyner et al., 2018)). Albeit finding a druggable target for specific driver mutations might become more challenging if these mutations are in, for example, transcription factors, commonly viewed as non‐druggable. Basic research might shed light over new druggable targets in cell or animal models for AML with specific mutational patterns. Recent publications have indicated the CD73/A2AR axis, the KMT2A complex and the CSF3R‐JAK/STAT‐signalling pathway as potential druggable targets in CEBPA‐mutated AML. Still, the future therapeutic opportunities for these targets remain to be tested clinically (Grebien et al., 2015; Lavallee et al., 2016; Jakobsen et al., 2019; Schmidt et al., 2019).
The high degree of mutations in druggable tyrosine kinase genes (e.g. CSF3R, KIT and JAK2/3) should suggest that this subgroup of CEBPAbi patients would be responsive to tyrosine kinase inhibitors (such as Tofacitinib; Xeljanz® or Ruxolitinib; Jakavi®), which are already in clinical use for other diagnoses. As mentioned above, Lavallee et al. found CEBPAbi AML to be highly sensitive to JAK inhibitors ex vivo (Lavallee et al., 2016). Moreover, it will be interesting to follow how patients with CEBPAbi AML respond to newly approved treatment regimens such as BCL‐2 inhibitor (Venetoclax; Venclyxto®) in combination with DNA methyltransferase inhibitor (Azacitidine; Vidaza® and Decitabine; Dacogen®). These treatments might be efficient, especially in TET2‐mutated CEBPAbi cases and, by extension, in IDH1/2‐ and WT1‐mutated cases (Duy et al., 2019).
Conclusions
Here we have reviewed the current knowledge of mutational patterns in the context of CEBPAbi AML, excluding classical inversions and translocations that do not co‐occur with CEBPA lesions. We note that relatively few data sets are available and that these deal mainly with point mutations in protein encoding genes. Thus, co‐occurrences involving structural variations affecting gene regulation (e.g. enhancer hijacking etc.) are, to date, essentially unknown and therefore constitute an additional potential layer of biological complexity likely to influence the biology of, and clinical response in, CEBPAbi AML.
While CEBPAbi AML is generally associated with a good prognosis, this may vary widely depending on the co‐occurrence of aberrations in other genes. Indeed, CEBPA is frequently co‐mutated with lesions in GATA2, WT1, TET2 and CSF3R, and this is generally associated with a worsened outcome. In contrast, mutations in factors such as DNMT3A, FLT3, IDH1/2, RUNX1 and, in particular, NPM1 are mutually exclusive with lesions in CEBPA. Mechanistic studies promise to uncover therapeutic vulnerabilities resulting from these functional interactions. In the future, with the introduction of clinical genomics in routine diagnosis, data from large cohorts will allow further stratification based on less frequent mutations, as well as on sex and ethnicity. We believe that this holds great potential for tailoring treatment and thus improving patient outcomes in the near future.
Conflicts of interest
The authors have no conflicting interests to disclose.
Author contributions
Anna S. Wilhelmson and Bo T. Porse wrote the manuscript and critically revised the whole manuscript.
Acknowledgements
This work was supported through a centre grant from the Novo Nordisk Foundation (Novo Nordisk Foundation Center for Stem Cell Biology, DanStem; Grant Number NNF17CC0027852) and is also part of the Danish Research Center for Precision Medicine in Blood Cancers funded by the Danish Cancer Society (Grant number R223‐A13071) and Greater Copenhagen Health Science Partners.
In addition, this work was also supported by the Swedish Research Council's International Postdoc Grant (Anna S. Wilhelmson: 2015–00517).
References
- Abelson, S. , Collord, G. , Ng, S.W.K. , Weissbrod, O ., Mendelson Cohen, N. , Niemeyer, E. , Barda, N. , Zuzarte, P.C. , Heisler, L. , Sundaravadanam, Y. , Luben, R. , Hayat, S. , Wang, T.T. , Zhao, Z. , Cirlan, I. , Pugh, T.J. , Soave, D. , Ng, K. , Latimer, C. , Hardy, C. , Raine, K. , Jones, D. , Hoult, D. , Britten, A. , Mcpherson, J.D. , Johansson, M. , Mbabaali, F. , Eagles, J. , Miller, J.K. , Pasternack, D. , Timms, L. , Krzyzanowski, P. , Awadalla, P. , Costa, R. , Segal, E. , Bratman, S.V. , Beer, P. , Behjati, S. , Martincorena, I. , Wang, J.C.Y. , Bowles, K.M. , Quiros, J.R. , Karakatsani, A. , La Vecchia, C. , Trichopoulou, A. , Salamanca‐Fernandez, E. , Huerta, J.M. , Barricarte, A. , Travis, R.C. , Tumino, R. , Masala, G. , Boeing, H. , Panico, S. , Kaaks, R. , Kramer, A. , Sieri, S. , Riboli, E. , Vineis, P. , Foll, M. , Mckay, J. , Polidoro, S. , Sala, N. , Khaw, K.T. , Vermeulen, R. , Campbell, P.J. , Papaemmanuil, E. , Minden, M.D. , Tanay, A. , Balicer, R.D. , Wareham, N.J. , Gerstung, M. , Dick, J.E. , Brennan, P. , Vassiliou, G.S. & Shlush, L.I. (2018) Prediction of acute myeloid leukaemia risk in healthy individuals. Nature, 559, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, J.S. , Kim, J.Y. , Kim, H.J. , Kim, Y.K. , Lee, S.S. , Jung, S.H. , Yang, D.H. , Lee, J.J. , Kim, N.Y. , Choi, S.H. , Minden, M.D. , Jung, C.W. , Jang, J.H. , Kim, H.J. , Moon, J.H. , Sohn, S.K. , Won, J.H. , Kim, S.H. & Kim, D.D. (2016) Normal karyotype acute myeloid leukemia patients with CEBPA double mutation have a favorable prognosis but no survival benefit from allogeneic stem cell transplant. Annals of Hematology, 95, 301–310. [DOI] [PubMed] [Google Scholar]
- Alachkar, H. , Mutonga, M. , Malnassy, G. , Park, J.H. , Fulton, N. , Woods, A. , Meng, L. , Kline, J. , Raca, G. , Odenike, O. , Takamatsu, N. , Miyamoto, T. , Matsuo, Y. , Stock, W. & Nakamura, Y. (2015) T‐LAK cell‐originated protein kinase presents a novel therapeutic target in FLT3‐ITD mutated acute myeloid leukemia. Oncotarget, 6, 33410–33425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avellino, R. & Delwel, R. (2017) Expression and regulation of C/EBPalpha in normal myelopoiesis and in malignant transformation. Blood, 129, 2083–2091. [DOI] [PubMed] [Google Scholar]
- Avellino, R. , Havermans, M. , Erpelinck, C. , Sanders, M.A. , Hoogenboezem, R. , van de Werken, H.J. , Rombouts, E. , van Lom, K. , van Strien, P.M. , Gebhard, C. , Rehli, M. , Pimanda, J. , Beck, D. , Erkeland, S. , Kuiken, T. , de Looper, H. , Groschel, S. , Touw, I. , Bindels, E. & Delwel, R. (2016) An autonomous CEBPA enhancer specific for myeloid‐lineage priming and neutrophilic differentiation. Blood, 127, 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereshchenko, O. , Mancini, E. , Moore, S. , Bilbao, D. , Mansson, R. , Luc, S. , Grover, A. , Jacobsen, S.E. , Bryder, D. & Nerlov, C. (2009) Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia‐initiating cells in C/EBPalpha mutant AML. Cancer Cell, 16, 390–400. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Ley, T.J. , Miller, C. , Ding, L. , Raphael, B.J. , Mungall, A.J. , Robertson, A. , Hoadley, K. , Triche, T.J. Jr , Laird, P.W. , Baty, J.D. , Fulton, L.L. , Fulton, R. , Heath, S.E. , Kalicki‐Veizer, J. , Kandoth, C. , Klco, J.M. , Koboldt, D.C. , Kanchi, K.L. , Kulkarni, S. , Lamprecht, T.L. , Larson, D.E. , Lin, L. , Lu, C. , McLellan, M.D. , McMichael, J.F. , Payton, J. , Schmidt, H. , Spencer, D.H. , Tomasson, M.H. , Wallis, J.W. , Wartman, L.D. , Watson, M.A. , Welch, J. , Wendl, M.C. , Ally, A. , Balasundaram, M. , Birol, I. , Butterfield, Y. , Chiu, R. , Chu, A. , Chuah, E. , Chun, H.J. , Corbett, R. , Dhalla, N. , Guin, R. , He, A. , Hirst, C. , Hirst, M. , Holt, R.A. , Jones, S. , Karsan, A. , Lee, D. , Li, H.I. , Marra, M.A. , Mayo, M. , Moore, R.A. , Mungall, K. , Parker, J. , Pleasance, E. , Plettner, P. , Schein, J. , Stoll, D. , Swanson, L. , Tam, A. , Thiessen, N. , Varhol, R. , Wye, N. , Zhao, Y. , Gabriel, S. , Getz, G. , Sougnez, C. , Zou, L. , Leiserson, M.D. , Vandin, F. , Wu, H.T. , Applebaum, F. , Baylin, S.B. , Akbani, R. , Broom, B.M. , Chen, K. , Motter, T.C. , Nguyen, K. , Weinstein, J.N. , Zhang, N. , Ferguson, M.L. , Adams, C. , Black, A. , Bowen, J. , Gastier‐Foster, J. , Grossman, T. , Lichtenberg, T. , Wise, L. , Davidsen, T. , Demchok, J.A. , Shaw, K.R. , Sheth, M. , Sofia, H.J. , Yang, L. , Downing, J.R. & Eley, G. (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine, 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, W.C. , Chou, S.C. , Liu, C.Y. , Chen, C.Y. , Hou, H.A. , Kuo, Y.Y. , Lee, M.C. , Ko, B.S. , Tang, J.L. , Yao, M. , Tsay, W. , Wu, S.J. , Huang, S.Y. , Hsu, S.C. , Chen, Y.C. , Chang, Y.C. , Kuo, Y.Y. , Kuo, K.T. , Lee, F.Y. , Liu, M.C. , Liu, C.W. , Tseng, M.H. , Huang, C.F. & Tien, H.F. (2011) TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate‐risk cytogenetics. Blood, 118, 3803–3810. [DOI] [PubMed] [Google Scholar]
- Dash, A. & Gilliland, D.G. (2001) Molecular genetics of acute myeloid leukaemia. Best Pract Res Clin Haematol, 14, 49–64. [DOI] [PubMed] [Google Scholar]
- Dinardo, C.D. & Perl, A.E. (2019) Advances in patient care through increasingly individualized therapy. Nat Rev Clin Oncol, 16, 73–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour, A. , Schneider, F. , Metzeler, K.H. , Hoster, E. , Schneider, S. , Zellmeier, E. , Benthaus, T. , Sauerland, M.C. , Berdel, W.E. , Buchner, T. , Wormann, B. , Braess, J. , Hiddemann, W. , Bohlander, S.K. & Spiekermann, K. (2010) Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. Journal of Clinical Oncology, 28, 570–577. [DOI] [PubMed] [Google Scholar]
- Duy, C. , Teater, M. , Garrett‐Bakelman, F.E. , Lee, T.C. , Meydan, C. , Glass, J.L. , Li, M. , Hellmuth, J.C. , Mohammad, H.P. , Smitheman, K.N. , Shih, A.H. , Abdel‐Wahab, O. , Tallman, M.S. , Guzman, M.L. , Muench, D. , Grimes, H.L. , Roboz, G.J. , Kruger, R.G. , Creasy, C.L. , Paietta, E.M. , Levine, R.L. , Carroll, M. & Melnick, A.M. (2019) Rational targeting of cooperating layers of the epigenome yields enhanced therapeutic efficacy against AML. Cancer Discovery, 9, 872–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasan, A. , Eder, C. , Haferlach, C. , Grossmann, V. , Kohlmann, A. , Dicker, F. , Kern, W. , Haferlach, T. & Schnittger, S. (2013) GATA2 mutations are frequent in intermediate‐risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia, 27, 482–485. [DOI] [PubMed] [Google Scholar]
- Fasan, A. , Haferlach, C. , Alpermann, T. , Jeromin, S. , Grossmann, V. , Eder, C. , Weissmann, S. , Dicker, F. , Kohlmann, A. , Schindela, S. , Kern, W. , Haferlach, T. & Schnittger, S. (2014) The role of different genetic subtypes of CEBPA mutated AML. Leukemia, 28, 794–803. [DOI] [PubMed] [Google Scholar]
- Gallipoli, P. , Giotopoulos, G. & Huntly, B.J. (2015) Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther Adv Hematol, 6, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, Y. , Schuster, M.B. , Pundhir, S. , Rapin, N. , Bagger, F.O. , Sidiropoulos, N. , Hashem, N. & Porse, B.T. (2019) The splicing factor RBM25 controls MYC activity in acute myeloid leukemia. Nature Communications, 10, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, G. , Kahler, A.K. , Handsaker, R.E. , Lindberg, J. , Rose, S.A. , Bakhoum, S.F. , Chambert, K. , Mick, E. , Neale, B.M. , Fromer, M. , Purcell, S.M. , Svantesson, O. , Landen, M. , Hoglund, M. , Lehmann, S. , Gabriel, S.B. , Moran, J.L. , Lander, E.S. , Sullivan, P.F. , Sklar, P. , Gronberg, H. , Hultman, C.M. & McCarroll, S.A. (2014) Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. New England Journal of Medicine, 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien, F. , Vedadi, M. , Getlik, M. , Giambruno, R. , Grover, A. , Avellino, R. , Skucha, A. , Vittori, S. , Kuznetsova, E. , Smil, D. , Barsyte‐Lovejoy, D. , Li, F. , Poda, G. , Schapira, M. , Wu, H. , Dong, A. , Senisterra, G. , Stukalov, A. , Huber, K.V.M. , Schonegger, A. , Marcellus, R. , Bilban, M. , Bock, C. , Brown, P.J. , Zuber, J. , Bennett, K.L. , Al‐Awar, R. , Delwel, R. , Nerlov, C. , Arrowsmith, C.H. & Superti‐Furga, G. (2015) Pharmacological targeting of the Wdr5‐MLL interaction in C/EBPalpha N‐terminal leukemia. Nature Chemical Biology, 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, C.L. , Tawana, K. , Hills, R.K. , Bodor, C. , Fitzgibbon, J. , Inglott, S. , Ancliff, P. , Burnett, A.K. , Linch, D.C. & Gale, R.E. (2013) GATA2 mutations in sporadic and familial acute myeloid leukaemia patients with CEBPA mutations. British Journal of Haematology, 161, 701–705. [DOI] [PubMed] [Google Scholar]
- Greif, P.A. , Dufour, A. , Konstandin, N.P. , Ksienzyk, B. , Zellmeier, E. , Tizazu, B. , Sturm, J. , Benthaus, T. , Herold, T. , Yaghmaie, M. , Dorge, P. , Hopfner, K.P. , Hauser, A. , Graf, A. , Krebs, S. , Blum, H. , Kakadia, P.M. , Schneider, S. , Hoster, E. , Schneider, F. , Stanulla, M. , Braess, J. , Sauerland, M.C. , Berdel, W.E. , Buchner, T. , Woermann, B.J. , Hiddemann, W. , Spiekermann, K. & Bohlander, S.K. (2012) GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood, 120, 395–403. [DOI] [PubMed] [Google Scholar]
- Grossmann, V. , Bacher, U. , Kohlmann, A. , Butschalowski, K. , Roller, A. , Jeromin, S. , Dicker, F. , Kern, W. , Schnittger, S. , Haferlach, T. & Haferlach, C. (2012) Expression of CEBPA is reduced in RUNX1‐mutated acute myeloid leukemia. Blood Cancer J, 2, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann, V. , Haferlach, C. , Nadarajah, N. , Fasan, A. , Weissmann, S. , Roller, A. , Eder, C. , Stopp, E. , Kern, W. , Haferlach, T. , Kohlmann, A. & Schnittger, S. (2013) CEBPA double‐mutated acute myeloid leukaemia harbours concomitant molecular mutations in 76.8% of cases with TET2 and GATA2 alterations impacting prognosis. British Journal of Haematology, 161, 649–658. [DOI] [PubMed] [Google Scholar]
- Gu, X. , Ebrahem, Q. , Mahfouz, R.Z. , Hasipek, M. , Enane, F. , Radivoyevitch, T. , Rapin, N. , Przychodzen, B. , Hu, Z. , Balusu, R. , Cotta, C.V. , Wald, D. , Argueta, C. , Landesman, Y. , Martelli, M.P. , Falini, B. , Carraway, H. , Porse, B.T. , Maciejewski, J. , Jha, B.K. & Saunthararajah, Y. (2018) Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. The Journal of Clinical Investigation, 128, 4260–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H. , Ma, O. , Speck, N.A. & Friedman, A.D. (2012) Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood, 119, 4408–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasemann, M.S. , Lauridsen, F.K. , Waage, J. , Jakobsen, J.S. , Frank, A.K. , Schuster, M.B. , Rapin, N. , Bagger, F.O. , Hoppe, P.S. , Schroeder, T. & Porse, B.T. (2014) C/EBPalpha is required for long‐term self‐renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genetics, 10, e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, H.A. , Lin, Y.C. , Kuo, Y.Y. , Chou, W.C. , Lin, C.C. , Liu, C.Y. , Chen, C.Y. , Lin, L.I. , Tseng, M.H. , Huang, C.F. , Chiang, Y.C. , Liu, M.C. , Liu, C.W. , Tang, J.L. , Yao, M. , Huang, S.Y. , Ko, B.S. , Hsu, S.C. , Wu, S.J. , Tsay, W. , Chen, Y.C. & Tien, H.F. (2015) GATA2 mutations in patients with acute myeloid leukemia‐paired samples analyses show that the mutation is unstable during disease evolution. Annals of Hematology, 94, 211–221. [DOI] [PubMed] [Google Scholar]
- Jaiswal, S. , Fontanillas, P. , Flannick, J. , Manning, A. , Grauman, P.V. , Mar, B.G. , Lindsley, R.C. , Mermel, C.H. , Burtt, N. , Chavez, A. , Higgins, J.M. , Moltchanov, V. , Kuo, F.C. , Kluk, M.J. , Henderson, B. , Kinnunen, L. , Koistinen, H.A. , Ladenvall, C. , Getz, G. , Correa, A. , Banahan, B.F. , Gabriel, S. , Kathiresan, S. , Stringham, H.M. , McCarthy, M.I. , Boehnke, M. , Tuomilehto, J. , Haiman, C. , Groop, L. , Atzmon, G. , Wilson, J.G. , Neuberg, D. , Altshuler, D. & Ebert, B.L. (2014) Age‐related clonal hematopoiesis associated with adverse outcomes. New England Journal of Medicine, 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen, J.S. , Laursen, L.G. , Schuster, M.B. , Pundhir, S. , Schoof, E. , Ge, Y. , D'Altri, T. , Vitting‐Seerup, K. , Rapin, N. , Gentil, C. , Jendholm, J. , Theilgaard‐Monch, K. , Reckzeh, K. , Bullinger, L. , Dohner, K. , Hokland, P. , Fitzgibbon, J. & Porse, B.T. (2019) Mutant CEBPA directly drives the expression of the targetable tumor‐promoting factor CD73 in AML. Science Advances, 5, eaaw4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, M. , Park, H.J. , Celik, H. , Ostrander, E.L. , Reyes, J.M. , Guzman, A. , Rodriguez, B. , Lei, Y. , Lee, Y. , Ding, L. , Guryanova, O.A. , Li, W. , Goodell, M.A. & Challen, G.A. (2018) Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Reports, 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan, K. , He, Y. , Wouters, B.J. , Shestova, O. , Xu, L. , Sai, H. , Rodriguez, C.G. , Maillard, I. , Tobias, J.W. , Valk, P. , Carroll, M. , Aster, J.C. , Delwel, R. & Pear, W.S. (2006) Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell, 10, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara, R. , Nagata, Y. , Kiyoi, H. , Kato, T. , Yamamoto, E. , Suzuki, K. , Chen, F. , Asou, N. , Ohtake, S. , Miyawaki, S. , Miyazaki, Y. , Sakura, T. , Ozawa, Y. , Usui, N. , Kanamori, H. , Kiguchi, T. , Imai, K. , Uike, N. , Kimura, F. , Kitamura, K. , Nakaseko, C. , Onizuka, M. , Takeshita, A. , Ishida, F. , Suzushima, H. , Kato, Y. , Miwa, H. , Shiraishi, Y. , Chiba, K. , Tanaka, H. , Miyano, S. , Ogawa, S. & Naoe, T. (2014) Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia, 28, 1586–1595. [DOI] [PubMed] [Google Scholar]
- Kirstetter, P. , Schuster, M.B. , Bereshchenko, O. , Moore, S. , Dvinge, H. , Kurz, E. , Theilgaard‐Monch, K. , Mansson, R. , Pedersen, T.A. , Pabst, T. , Schrock, E. , Porse, B.T. , Jacobsen, S.E. , Bertone, P. , Tenen, D.G. & Nerlov, C. (2008) Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia‐initiating cells. Cancer Cell, 13, 299–310. [DOI] [PubMed] [Google Scholar]
- Konstandin, N.P. , Pastore, F. , Herold, T. , Dufour, A. , Rothenberg‐Thurley, M. , Hinrichsen, T. , Ksienzyk, B. , Tschuri, S. , Schneider, S. , Hoster, E. , Berdel, W.E. , Woermann, B.J. , Sauerland, M.C. , Braess, J. , Bohlander, S.K. , Klein, H.G. , Hiddemann, W. , Metzeler, K.H. & Spiekermann, K. (2018) Genetic heterogeneity of cytogenetically normal AML with mutations of CEBPA. Blood Advances, 2, 2724–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauth, M.T. , Alpermann, T. , Bacher, U. , Eder, C. , Dicker, F. , Ulke, M. , Kuznia, S. , Nadarajah, N. , Kern, W. , Haferlach, C. , Haferlach, T. & Schnittger, S. (2015) WT1 mutations are secondary events in AML, show varying frequencies and impact on prognosis between genetic subgroups. Leukemia, 29, 660–667. [DOI] [PubMed] [Google Scholar]
- Lavallee, V.P. , Krosl, J. , Lemieux, S. , Boucher, G. , Gendron, P. , Pabst, C. , Boivin, I. , Marinier, A. , Guidos, C.J. , Meloche, S. , Hebert, J. & Sauvageau, G. (2016) Chemo‐genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood, 127, 3054–3061. [DOI] [PubMed] [Google Scholar]
- Li, Y. & Su, L. (2019) Clonal evolution of acute myeloid leukemia with CEBPA double mutations after long‐term remission: case report and a literature review. Turkish Journal of Haematology, 36, 128–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke, M. & Cleary, M.L. (2009) Therapeutic targeting of MLL. Blood, 113, 6061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.C. , Hosoya, T. , Brandt, W. , Ku, C.J. , Hosoya‐Ohmura, S. , Camper, S.A. , Yamamoto, M. & Engel, J.D. (2012) Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. The Journal of Clinical Investigation, 122, 3705–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, P.H. , Li, H.Y. , Fan, S.C. , Yuan, T.H. , Chen, M. , Hsu, Y.H. , Yang, Y.H. , Li, L.Y. , Yeh, S.P. , Bai, L.Y. , Liao, Y.M. , Lin, C.Y. , Hsieh, C.Y. , Lin, C.C. , Lin, C.H. , Lien, M.Y. , Chen, T.T. , Ni, Y.H. & Chiu, C.F. (2017) A targeted next‐generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practice. Cancer Medicine, 6, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, J. , Solimini, N.L. & Elledge, S.J. (2009) Principles of cancer therapy: oncogene and non‐oncogene addiction. Cell, 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannelli, F. , Ponziani, V. , Bencini, S. , Bonetti, M.I. , Benelli, M. , Cutini, I. , Gianfaldoni, G. , Scappini, B. , Pancani, F. , Piccini, M. , Rondelli, T. , Caporale, R. , Gelli, A.M. , Peruzzi, B. , Chiarini, M. , Borlenghi, E. , Spinelli, O. , Giupponi, D. , Zanghi, P. , Bassan, R. , Rambaldi, A. , Rossi, G. & Bosi, A. (2017) CEBPA‐double‐mutated acute myeloid leukemia displays a unique phenotypic profile: a reliable screening method and insight into biological features. Haematologica, 102, 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler, K.H. , Becker, H. , Maharry, K. , Radmacher, M.D. , Kohlschmidt, J. , Mrozek, K. , Nicolet, D. , Whitman, S.P. , Wu, Y.Z. , Schwind, S. , Powell, B.L. , Carter, T.H. , Wetzler, M. , Moore, J.O. , Kolitz, J.E. , Baer, M.R. , Carroll, A.J. , Larson, R.A. , Caligiuri, M.A. , Marcucci, G. & Bloomfield, C.D. (2011) ASXL1 mutations identify a high‐risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood, 118, 6920–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler, K.H. , Herold, T. , Rothenberg‐Thurley, M. , Amler, S. , Sauerland, M.C. , Gorlich, D. , Schneider, S. , Konstandin, N.P. , Dufour, A. , Braundl, K. , Ksienzyk, B. , Zellmeier, E. , Hartmann, L. , Greif, P.A. , Fiegl, M. , Subklewe, M. , Bohlander, S.K. , Krug, U. , Faldum, A. , Berdel, W.E. , Wormann, B. , Buchner, T. , Hiddemann, W. , Braess, J. , Spiekermann, K. & AMLCG Study Group (2016) Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood, 128, 686–698. [DOI] [PubMed] [Google Scholar]
- Moran‐Crusio, K. , Reavie, L. , Shih, A. , Abdel‐Wahab, O. , Ndiaye‐Lobry, D. , Lobry, C. , Figueroa, M.E. , Vasanthakumar, A. , Patel, J. , Zhao, X. , Perna, F. , Pandey, S. , Madzo, J. , Song, C. , Dai, Q. , He, C. , Ibrahim, S. , Beran, M. , Zavadil, J. , Nimer, S.D. , Melnick, A. , Godley, L.A. , Aifantis, I. & Levine, R.L. (2011) Tet2 loss leads to increased hematopoietic stem cell self‐renewal and myeloid transformation. Cancer Cell, 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, C. , Lohan, F. , Campos, J. , Ohlsson, E. , Salome, M. , Forde, C. , Artschwager, R. , Liskamp, R.M. , Cahill, M.R. , Kiely, P.A. , Porse, B. & Keeshan, K. (2016) The presence of C/EBPalpha and its degradation are both required for TRIB2‐mediated leukaemia. Oncogene, 35, 5272–5281. [DOI] [PubMed] [Google Scholar]
- Ohlsson, E. , Schuster, M.B. , Hasemann, M. & Porse, B.T. (2016) The multifaceted functions of C/EBPalpha in normal and malignant haematopoiesis. Leukemia, 30, 767–775. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil, E. , Dohner, H. & Campbell, P.J. (2016) Genomic classification in acute myeloid leukemia. New England Journal of Medicine, 375, 900–901. [DOI] [PubMed] [Google Scholar]
- Pulikkan, J.A. , Tenen, D.G. & Behre, G. (2017) C/EBPalpha deregulation as a paradigm for leukemogenesis. Leukemia, 31, 2279–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pundhir, S. , Bratt Lauridsen, F.K. , Schuster, M.B. , Jakobsen, J.S. , Ge, Y. , Schoof, E.M. , Rapin, N. , Waage, J. , Hasemann, M.S. & Porse, B.T. (2018) Enhancer and transcription factor dynamics during myeloid differentiation reveal an early differentiation block in Cebpa null progenitors. Cell Reports, 23, 2744–2757. [DOI] [PubMed] [Google Scholar]
- Radomska, H.S. , Basseres, D.S. , Zheng, R. , Zhang, P. , Dayaram, T. , Yamamoto, Y. , Sternberg, D.W. , Lokker, N. , Giese, N.A. , Bohlander, S.K. , Schnittger, S. , Delmotte, M.H. , Davis, R.J. , Small, D. , Hiddemann, W. , Gilliland, D.G. & Tenen, D.G. (2006) Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. Journal of Experimental Medicine, 203, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska, H.S. , Alberich‐Jorda, M. , Will, B. , Gonzalez, D. , Delwel, R. & Tenen, D.G. (2012) Targeting CDK1 promotes FLT3‐activated acute myeloid leukemia differentiation through C/EBPalpha. The Journal of Clinical Investigation, 122, 2955–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal, R. , Alkalin, A. , Madzo, J. , Vasanthakumar, A. , Pronier, E. , Patel, J. , Li, Y. , Ahn, J. , Abdel‐Wahab, O. , Shih, A. , Lu, C. , Ward, P.S. , Tsai, J.J. , Hricik, T. , Tosello, V. , Tallman, J.E. , Zhao, X. , Daniels, D. , Dai, Q. , Ciminio, L. , Aifantis, I. , He, C. , Fuks, F. , Tallman, M.S. , Ferrando, A. , Nimer, S. , Paietta, E. , Thompson, C.B. , Licht, J.D. , Mason, C.E. , Godley, L.A. , Melnick, A. , Figueroa, M.E. & Levine, R.L. (2014) DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Reports, 9, 1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckzeh, K. , Bereshchenko, O. , Mead, A. , Rehn, M. , Kharazi, S. , Jacobsen, S.E. , Nerlov, C. & Cammenga, J. (2012) Molecular and cellular effects of oncogene cooperation in a genetically accurate AML mouse model. Leukemia, 26, 1527–1536. [DOI] [PubMed] [Google Scholar]
- Rose, D. , Haferlach, T. , Schnittger, S. , Perglerova, K. , Kern, W. & Haferlach, C. (2017) Subtype‐specific patterns of molecular mutations in acute myeloid leukemia. Leukemia, 31, 11–17. [DOI] [PubMed] [Google Scholar]
- Schmidt, L. , Heyes, E. , Scheiblecker, L. , Eder, T. , Volpe, G. , Frampton, J. , Nerlov, C. , Valent, P. , Grembecka, J. & Grebien, F. (2019) CEBPA‐mutated leukemia is sensitive to genetic and pharmacological targeting of the MLL1 complex. Leukemia, 33, 1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, L.Y. , Liang, D.C. , Huang, C.F. , Wu, J.H. , Lin, T.L. , Wang, P.N. , Dunn, P. , Kuo, M.C. & Tang, T.C. (2006) AML patients with CEBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: a comparative analysis on paired diagnosis and relapse samples. Leukemia, 20, 604–609. [DOI] [PubMed] [Google Scholar]
- Shlush, L.I. , Zandi, S. , Mitchell, A. , Chen, W.C. , Brandwein, J.M. , Gupta, V. , Kennedy, J.A. , Schimmer, A.D. , Schuh, A.C. , Yee, K.W. , McLeod, J.L. , Doedens, M. , Medeiros, J.J. , Marke, R. , Kim, H.J. , Lee, K. , McPherson, J.D. , Hudson, T.J. , HALT Pan‐Leukemia Gene Panel Consortium , Brown, A.M. , Yousif, F. , Trinh, Q.M. , Stein, L.D. , Minden, M.D. , Wang, J.C. & Dick, J.E. (2014) Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature, 506, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini, N.L. , Luo, J. & Elledge, S.J. (2007) Non‐oncogene addiction and the stress phenotype of cancer cells. Cell, 130, 986–988. [DOI] [PubMed] [Google Scholar]
- Su, L. , Tan, Y. , Lin, H. , Liu, X. , Yu, L. , Yang, Y. , Liu, S. , Bai, O. , Yang, Y. , Jin, F. , Sun, J. , Liu, C. , Liu, Q. , Gao, S. & Li, W. (2018) Mutational spectrum of acute myeloid leukemia patients with double CEBPA mutations based on next‐generation sequencing and its prognostic significance. Oncotarget, 9, 24970–24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, L. , Gao, S. , Tan, Y. , Lin, H. , Liu, X. , Liu, S. , Yang, Y. , Sun, J. & Li, W. (2019) CSF3R mutations were associated with an unfavorable prognosis in patients with acute myeloid leukemia with CEBPA double mutations. Annals of Hematology, 98, 1641–1646. [DOI] [PubMed] [Google Scholar]
- Taskesen, E. , Bullinger, L. , Corbacioglu, A. , Sanders, M.A. , Erpelinck, C.A. , Wouters, B.J. , van der Poel‐van de Luytgaarde, S.C. , Damm, F. , Krauter, J. , Ganser, A. , Schlenk, R.F. , Lowenberg, B. , Delwel, R. , Dohner, H. , Valk, P.J. & Dohner, K. (2011) Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood, 117, 2469–2475. [DOI] [PubMed] [Google Scholar]
- Tawana, K. , Wang, J. , Renneville, A. , Bodor, C. , Hills, R. , Loveday, C. , Savic, A. , van Delft, F.W. , Treleaven, J. , Georgiades, P. , Uglow, E. , Asou, N. , Uike, N. , Debeljak, M. , Jazbec, J. , Ancliff, P. , Gale, R. , Thomas, X. , Mialou, V. , Dohner, K. , Bullinger, L. , Mueller, B. , Pabst, T. , Stelljes, M. , Schlegelberger, B. , Wozniak, E. , Iqbal, S. , Okosun, J. , Araf, S. , Frank, A.K. , Lauridsen, F.B. , Porse, B. , Nerlov, C. , Owen, C. , Dokal, I. , Gribben, J. , Smith, M. , Preudhomme, C. , Chelala, C. , Cavenagh, J. & Fitzgibbon, J. (2015) Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood, 126, 1214–1223. [DOI] [PubMed] [Google Scholar]
- Theis, F. , Corbacioglu, A. , Gaidzik, V.I. , Paschka, P. , Weber, D. , Bullinger, L. , Heuser, M. , Ganser, A. , Thol, F. , Schlegelberger, B. , Gohring, G. , Kohne, C.H. , Germing, U. , Brossart, P. , Horst, H.A. , Haase, D. , Gotze, K. , Ringhoffer, M. , Fiedler, W. , Nachbaur, D. , Kindler, T. , Held, G. , Lubbert, M. , Wattad, M. , Salih, H.R. , Krauter, J. , Dohner, H. , Schlenk, R.F. & Dohner, K. (2016) Clinical impact of GATA2 mutations in acute myeloid leukemia patients harboring CEBPA mutations: a study of the AML study group. Leukemia, 30, 2248–2250. [DOI] [PubMed] [Google Scholar]
- Tiacci, E. , Grossmann, V. , Martelli, M.P. , Kohlmann, A. , Haferlach, T. & Falini, B. (2012) The corepressors BCOR and BCORL1: two novel players in acute myeloid leukemia. Haematologica, 97, 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien, F.M. , Hou, H.A. , Tsai, C.H. , Tang, J.L. , Chiu, Y.C. , Chen, C.Y. , Kuo, Y.Y. , Tseng, M.H. , Peng, Y.L. , Liu, M.C. , Liu, C.W. , Liao, X.W. , Lin, L.I. , Lin, C.T. , Wu, S.J. , Ko, B.S. , Hsu, S.C. , Huang, S.Y. , Yao, M. , Chou, W.C. & Tien, H.F. (2018) GATA2 zinc finger 1 mutations are associated with distinct clinico‐biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer Journal, 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiesmeier, J. , Czwalinna, A. , Muller‐Tidow, C. , Krauter, J. , Serve, H. , Heil, G. , Ganser, A. & Verbeek, W. (2003) Evidence for allelic evolution of C/EBPalpha mutations in acute myeloid leukaemia. British Journal of Haematology, 123, 413–419. [DOI] [PubMed] [Google Scholar]
- Tyner, J.W. , Tognon, C.E. , Bottomly, D. , Wilmot, B. , Kurtz, S.E. , Savage, S.L. , Long, N. , Schultz, A.R. , Traer, E. , Abel, M. , Agarwal, A. , Blucher, A. , Borate, U. , Bryant, J. , Burke, R. , Carlos, A. , Carpenter, R. , Carroll, J. , Chang, B.H. , Coblentz, C. , D'Almeida, A. , Cook, R. , Danilov, A. , Dao, K.T. , Degnin, M. , Devine, D. , Dibb, J. , Edwards, D.K.T. , Eide, C.A. , English, I. , Glover, J. , Henson, R. , Ho, H. , Jemal, A. , Johnson, K. , Johnson, R. , Junio, B. , Kaempf, A. , Leonard, J. , Lin, C. , Liu, S.Q. , Lo, P. , Loriaux, M.M. , Luty, S. , Macey, T. , Macmaniman, J. , Martinez, J. , Mori, M. , Nelson, D. , Nichols, C. , Peters, J. , Ramsdill, J. , Rofelty, A. , Schuff, R. , Searles, R. , Segerdell, E. , Smith, R.L. , Spurgeon, S.E. , Sweeney, T. , Thapa, A. , Visser, C. , Wagner, J. , Watanabe‐Smith, K. , Werth, K. , Wolf, J. , White, L. , Yates, A. , Zhang, H. , Cogle, C.R. , Collins, R.H. , Connolly, D.C. , Deininger, M.W. , Drusbosky, L. , Hourigan, C.S. , Jordan, C.T. , Kropf, P. , Lin, T.L. , Martinez, M.E. , Medeiros, B.C. , Pallapati, R.R. , Pollyea, D.A. , Swords, R.T. , Watts, J.M. , Weir, S.J. , Wiest, D.L. , Winters, R.M. , McWeeney, S.K. & Druker, B.J. (2018) Functional genomic landscape of acute myeloid leukaemia. Nature, 562, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak, R.G. , Goudswaard, C.S. , van Putten, W. , Bijl, M.A. , Sanders, M.A. , Hugens, W. , Uitterlinden, A.G. , Erpelinck, C.A. , Delwel, R. , Lowenberg, B. & Valk, P.J. (2005) Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood, 106, 3747–3754. [DOI] [PubMed] [Google Scholar]
- Wakita, S. , Yamaguchi, H. , Ueki, T. , Usuki, K. , Kurosawa, S. , Kobayashi, Y. , Kawata, E. , Tajika, K. , Gomi, S. , Koizumi, M. , Fujiwara, Y. , Yui, S. , Fukunaga, K. , Ryotokuji, T. , Hirakawa, T. , Arai, K. , Kitano, T. , Kosaka, F. , Tamai, H. , Nakayama, K. , Fukuda, T. & Inokuchi, K. (2016) Complex molecular genetic abnormalities involving three or more genetic mutations are important prognostic factors for acute myeloid leukemia. Leukemia, 30, 545–554. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Xiao, M. , Chen, X. , Chen, L. , Xu, Y. , Lv, L. , Wang, P. , Yang, H. , Ma, S. , Lin, H. , Jiao, B. , Ren, R. , Ye, D. , Guan, K.L. & Xiong, Y. (2015) WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Molecular Cell, 57, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Liu, Y. , Hou, G. , Wang, L. , Lv, N. , Xu, Y. , Xu, Y. , Wang, X. , Xuan, Z. , Jing, Y. , Li, H. , Jin, X. , Deng, A. , Wang, L. , Gao, X. , Dou, L. , Liang, J. , Chen, C. , Li, Y. & Yu, L. (2016) Mutational spectrum and risk stratification of intermediate‐risk acute myeloid leukemia patients based on next‐generation sequencing. Oncotarget, 7, 32065–32078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Alberich‐Jorda, M. , Amabile, G. , Yang, H. , Staber, P.B. , di Ruscio, A. , Welner, R.S. , Ebralidze, A. , Zhang, J. , Levantini, E. , Lefebvre, V. , Valk, P.J. , Delwel, R. , Hoogenkamp, M. , Nerlov, C. , Cammenga, J. , Saez, B. , Scadden, D.T. , Bonifer, C. , Ye, M. & Tenen, D.G. (2013) Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell, 24, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, F. , Chen, X. , Zhang, Y. , Wang, M. , Liu, H. , Cao, P. , Ma, X. , Wang, T. , Zhang, J. , Zhang, X. , Lu, P. & Liu, H. (2018) CSF3R Mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer, 124, 3329–3338. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, F. , Chen, X. , Liu, W. , Fang, J. , Wang, M. , Teng, W. , Cao, P. & Liu, H. (2019a) Mutation profiling of 16 candidate genes in de novo acute myeloid leukemia patients. Frontiers in Medicine, 13, 229–237. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, F. , Chen, X. , Zhang, Y. , Wang, M. , Liu, H. , Teng, W. , Cao, P. , Nie, D. , Ma, X. , Wang, T. , Lu, P. & Liu, H. (2019b) Companion gene mutations and their clinical significance in AML with double mutant CEBPA. Cancer Gene Therapy. 10.1038/s41417-019-0133-7 [DOI] [PubMed] [Google Scholar]
- Zhou, J. & Chng, W.J. (2017) Aberrant RNA splicing and mutations in spliceosome complex in acute myeloid leukemia. Stem Cell Investig, 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]